








Three highly fluorescent dilactone bridged terphenyls with crankshaft architectures have been synthesized.This general class of compounds is relatively unexplored. These compounds have been characterizedby fluorescence and UVevis spectroscopy. For all three compounds, a direct correlation between
the rigidity of the terphenyl system and the strength of absorption and emission of light has been observed. Preliminary studies have indicated that compounds with this architecture have the potential to be useful as pH-driven molecular switches and/or sensors with instant fluorescence attenuation at high pH values.
方案详情

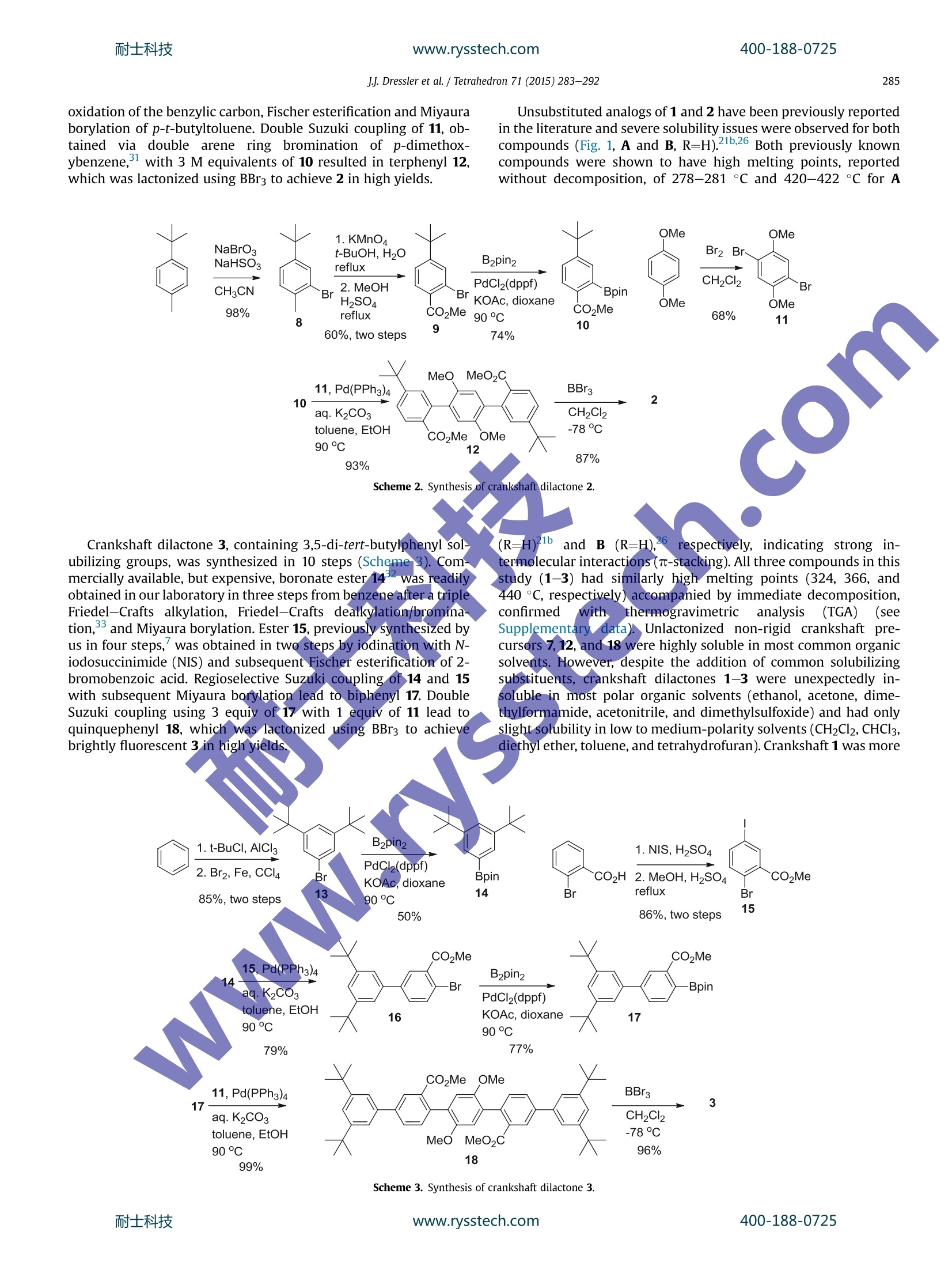
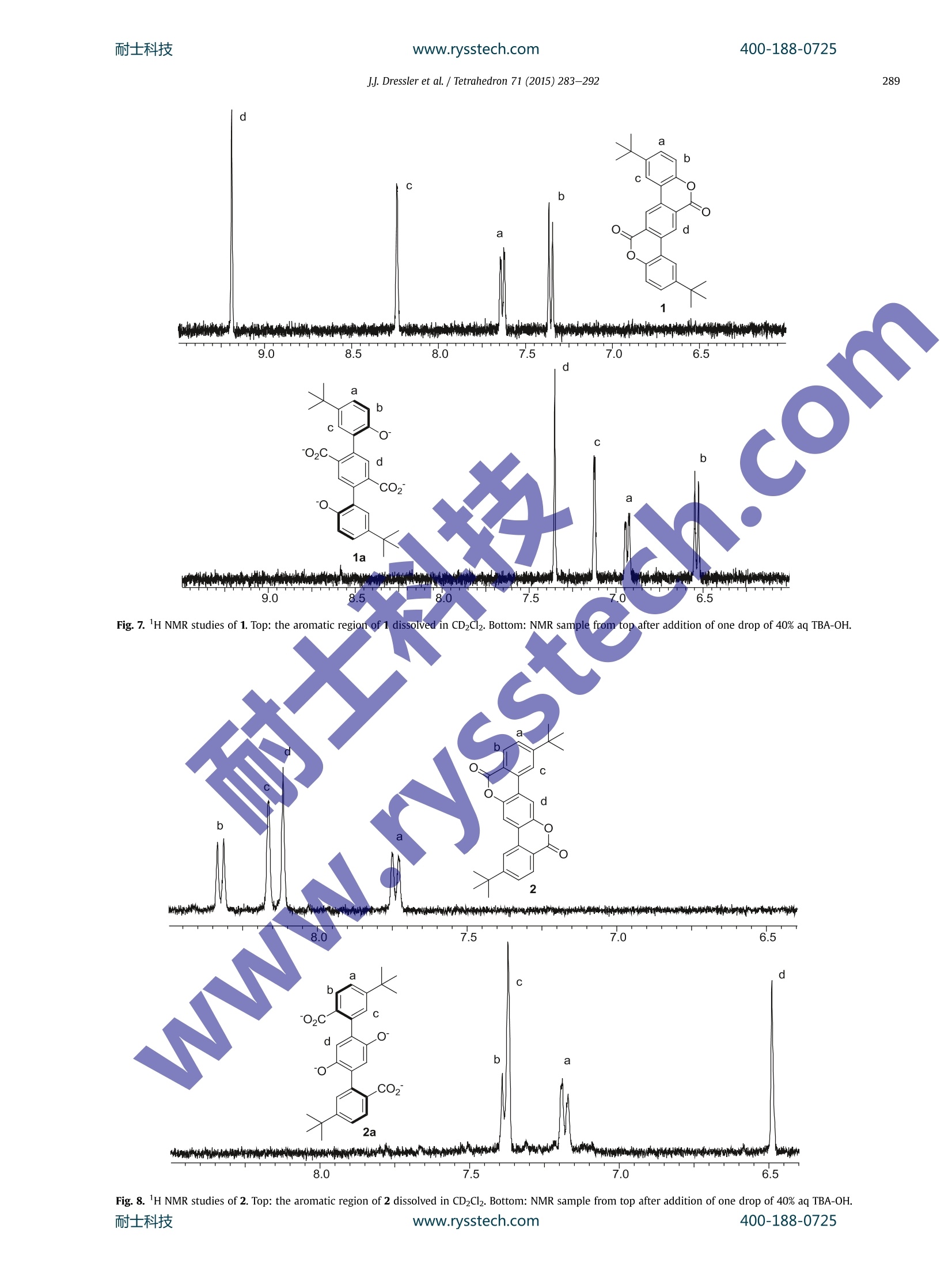
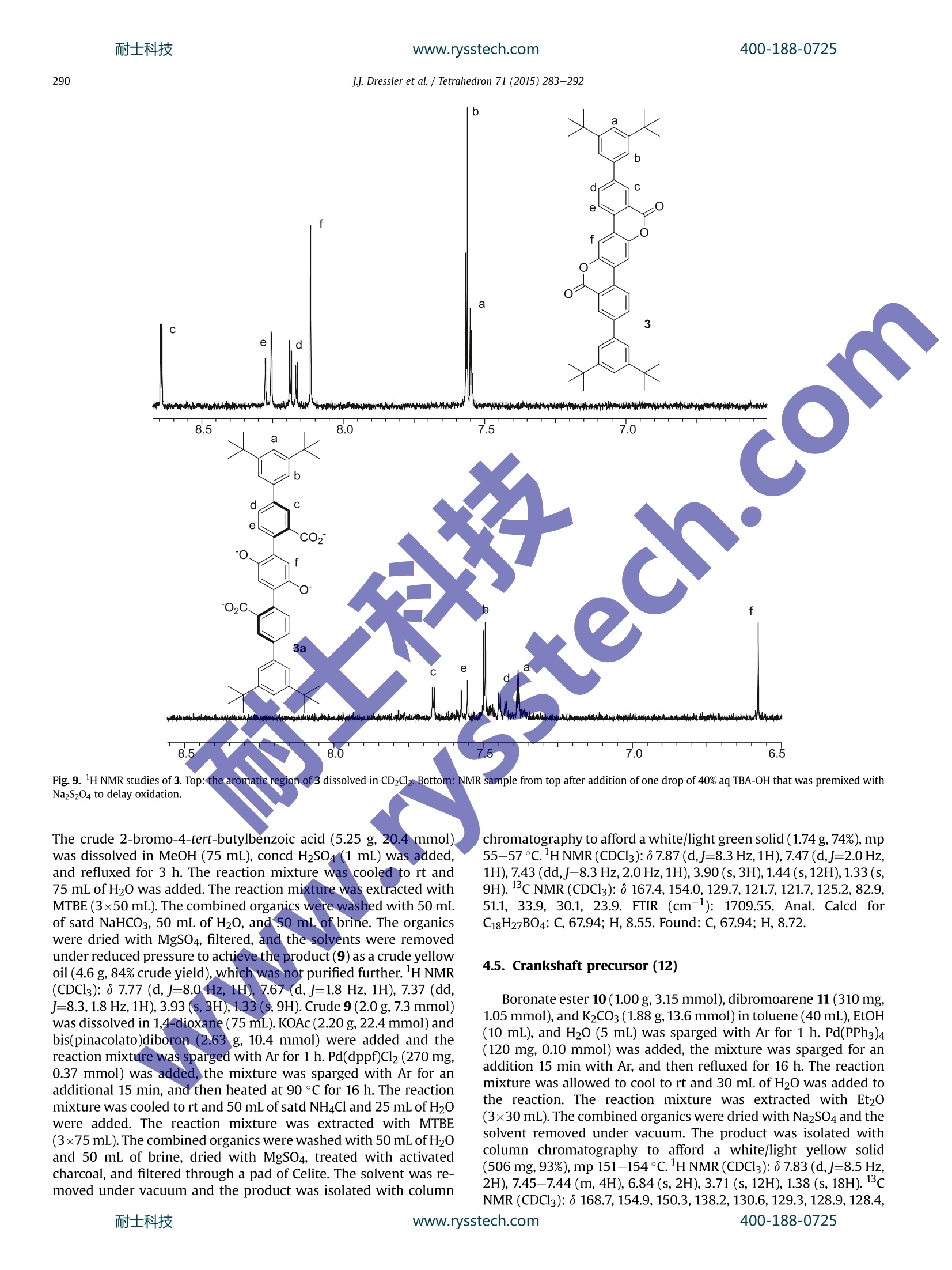
耐士科技400-188-0725www.rysstech.comTetrahedron 71 (2015) 283-292 耐士科技400-188-0725www.rysstech.comJJ. Dressler et al./ Tetrahedron 71 (2015)283-292284 Tetrahedron ELSEVIER journal homepage:www.elsevier.com/locate/tet Synthesis of dilactone bridged terphenyls with crankshaftarchitectures Justin J. Dressler, Sarah A. Miller, Brian T. Meeuwsen, Asia Marie S. Riel, Bart J. Dahl*Department of Chemistry,University of Wisconsin-Eau Claire, 105 Garfield Avenue, Eau Claire, WI 54702, USA ARTICLEINFO ABSTRACT Article history:Received 29 September 2014Received in revised form 19 November 2014Accepted 20 November 2014Available online 25 November 2014 Three highly fluorescent dilactone bridged terphenyls with crankshaft architectures have been synthe-sized. This general class of compounds is relatively unexplored. These compounds have been charac-terized by fluorescence and Uy-vis spectroscopy. For all three compounds, a direct correlation betweenthe rigidity of the terphenyl system and the strength of absorption and emission of light has been ob-served. Preliminary studies have indicated that compounds with this architecture have the potential tobe useful as pH-driven molecular switches and/or sensors with instant fluorescence attenuation at high Keywords:Highly conjugated materialsFluorescent compounds The opticall and electronic?properties of biphenyl and oligo-phenyl containing compounds are highly dependent on the tor-sional angle between the arene subunits. Coplanarity and rigidity ofthe arene rings often lead to significantly higher efficiency in theabsorption and emission of light whereas steric congestion pre-venting planarity or higher aryl-aryl bond rotational freedom leadsto attenuation of these properties. Since simple unsubstituted bi-phenyl and oligophenyl containing compounds are not planar orrigid,rigidity is typically achieved synthetically by building inchemical bridges between the arene subunits to afford, in the caseof oligophenylenes, ladder-type compounds. These highly r-con-jugated compounds are of current interest as they can be attractivecandidates for further development in dye, sensor, organic light-emitting diode (OLED), and photovoltaic technologies.However,the rigidity of these compounds often results in strong in-termolecular r-stacking interactions causing insolubility in mostsolvents, thus making processing and characterization difficult.This can often be addressed by appending solubilizing substituents,such as alkyl groups, to lessen the propensity for T-stacking. We have previously reported a strategy of employing lactone-bridging units to reversibly alter the rigidity of simple biphenyl-containing donor-acceptor (D-A) compounds with pH as thestimulus. The lactone bridge can be cleaved and subsequently re-formed upon addition of base and acid, respectively. When the ( *Corresponding author. Tel.: +1 7 15 8 36 4179; fax: + 1 715 836 4 9 79; e - mail address: d ahl bj @uwec.edu (B . J. Dahl). ) bridge is cleaved these compounds see dramatic attenuation ofoptical absorption and emission due to the increased steric con-gestion at the ortho positions of the biphenyl system and a higherdegree of aryl-aryl rotational freedom. These properties return toprevious high levels upon acid-induced lactonization, re-inducingrigidity in the system. We were interested in extending this basic concept to terphenyldilactone systems (Fig. 1) by first exploring C2h symmetric(crankshaft'-architecture) terphenyl analogs (A and B) of ourpreviously studied D-A biphenyl lactones. It is known that in-creasing the number of conjugated arene units in unsubstituted p-oligophenylenes causes a bathochromic shift in both absorbanceand emission spectra and also causes a dramatic increase in fluo-rescence quantum yield.However, there is a general convergenceobserved for these types of system after which additional benzeneunits (typically around 4-5 total units) have minimal effect onthese properties. In 4,4'-D-A oligophenylenes this convergence isoftenrealizeddmoree lrapidly, sometimeseven resultinggir Fig. 1.Two possible bridge orientations for terphenyl dilactones with crankshaftarchitectures. a hypsochromic shift in absorption spectra, as the cumulativephenyl-phenyl torsions become increasingly dominant, thus dis-rupting electronic communication between the D-A end groups.Due to their expected rigidity, terphenyl dilactones should havegreatly enhanced optical absorption and emission relative to un-bridged analogs. Numerous crankshaft-type C2h symmetric terphenyls withC(R2),"N(R),and c=o1single-atom bridges containing five-membered ring fusions have been reported. Less common crank-shaft terphenyls with single-atom bridging groups including C=C(R)2,13gJj,14 o,4n,15 Si(R2),16 P=O(R),4c.d s,l’ s(=0)2,18 Se,lc (c=N-NH2)14 have also been synthesized. With the exception ofdibenz[a,h]anthracene and other closely related derivatives con-taining alkenyl (C=C) bridges,C2h symmetric terphenyls withtwo-atom bridges containing six-membered ring fusions are quiteuncommon relative to those with one-atom bridges. There are re-ports of crankshaft-type terphenyls with two-atom bridges con- 2. Results and discussion We have synthesized three crankshaft-shaped terphenyl dilac-tones (Fig. 2) with tert-butyl or 3,5-di-tert-butylphenyl arene sub-stituents (1-3). Both tert-butyl and 3,5-di-tert-butylphenyl groupsare common effective solubilizing substituents for otherwise in-soluble highly r-conjugated compounds. Attempts to incorporatemesityl groups as alternative solubilizing substituents onto thecrankshaft terphenyl dilactones resulted in unpractically low yieldsunder multiple conditions and thus were not pursued further.Compounds 1 and 2 are isomeric with the lactone bridges flippedrelative to each other while 3 contains the same bridge orientationas 2, but different arene substituents at the para position of theterphenyl as opposed to the meta position. taining less rigid sphybridized atoms, such as alkyls (CR2-CR2)20and ethers (CR2-O),2 and more rigid two-atom sp²-based systemscontaining lactam ((C=O)NR),” imine (C(R)=N), azo,anddiketone (C=0)251bridges as well. However, unlike lactone-bridging groups, none of the previously reported bridging groupShave the potential to allow for pH-driven rigidity switching.Crankshaft terphenyls containing bridging lactones are very rare.Unsubstituted analogs of the proposed compounds (A and B, R=H)have been reported in 195926 and 2006,21b respectively, and ananalog containing thiophene substituents has also been recentlyreported. The extremely poor solubility of both unsubstitutedanalogs has been documented in those reports,21b.26 with unsub-stituted B being only soluble in concd H2SO4 and alcoholic NaOH,26presumably due to ring-opening of the lactones. This phenomenon Scheme 1. Synthesis of crankshaft dilactone 1. Each compound (1-3) was synthesized efficiently using a simi-lar strategy of a double Suzuki coupling followed by BBr3-promoteddouble lactonization21lb aafter building the individual arene frag-ments from the readily available and very inexpensive startingmaterials:p-tert-butylanisole, p-xylene, p-tert-butyltoluene, p-dimethoxybenzene, benzene, and 2-bromobenzoic acid. Crankshaft dilactone 1 was synthesized in seven steps (Scheme1). Boronate ester 52 was obtained after bromination and sub-sequent Miyaura borylation of p-t-butylanisole. Terephthalate 63was synthesized from p-xylene via double bromination of thearene ring,KMnO4 promoted oxidation of the benzylic carbons, andesterification of the resulting carboxylic acids. Double Suzuki cou-pling with 3 M equivalents of 5 resulted in terphenyl 7, which waslactonized using BBr3 to achieve 1 in high yields. is not unique to dilactones as a dilactam structurally analogous tounsubstituted terphenyl dilactone A was also reported to be diffi-cult to characterize because of its poor solubility..2222Of the Bridge-flipped crankshaft dilactone 2 was synthesized in sevensteps in a similar fashion to isomeric 1 (Scheme 2). Boronate ester10 was obtained after arene ring bromination, KMnO4 promoted oxidation of the benzylic carbon, Fischer esterification and Miyauraborylation of p-t-butyltoluene. Double Suzuki coupling of 11, ob-tained via doublee arene ring bromination of p-dimethox-ybenzene,with 3 M equivalents of 10 resulted in terphenyl 12,which was lactonized using BBrs to achieve 2 in high yields. Unsubstituted analogs of 1 and 2 have been previously reportedin the literature and severe solubility issues were observed for bothcompounds (Fig. 1, A and B, R=H).21b.26 Both previously knowncompounds were shown to have high melting points, reportedwithout decomposition, of 278-281°C and 420-422 °C for A Scheme 2. Synthesis of crankshaft dilactone 2. Crankshaft dilactone 3, containing 3,5-di-tert-butylphenyl sol-ubilizing groups, was synthesized in 10 steps (Scheme 3). Com-mercially available, but expensive, boronate ester 14was readilvobtained in our laboratory in three steps from benzene after a tripleFriedel-Crafts alkylation, Friedel-Crafts dealkylation/bromina-tion, and Miyaura borylation. Ester 15, previously synthesized byus in four steps, was obtained in two steps by iodination with N-iodosuccinimide (NIS) and subsequent Fischer esterification of 2-bromobenzoic acid. Regioselective Suzuki coupling of 14 and 15with subsequent Miyaura borylation lead to biphenyl 17. DoubleSuzuki coupling using 3 equiv of 17 with 1 equiv of 11 lead toquinquephenyl 18, which was lactonized using BBrs to achievebrightly fluorescent 3 in high yields. www.rysstech.com soluble in all solvents compared to lactone bridge-flipped 2 and 3.Fortunately, crankshafts 1-3 were soluble enough to fully charac-terize by NMR spectroscopy. In all cases, H NMR analysis stronglysupports the rigidity of compounds 1-3. The chemical shifts in thearomatic region of 1, 2, and 3 all display a significant downfield shiftdue, in part, to increased steric compression resulting from lacto-nization relative to 7, 12, and 18 (see Supplementary data). Thegeneral downfield chemical shift in the aromatic region of the HNMR spectra due to steric compression has been observed in ourprevious work and for numerous highly conjugated compoundsrelative to their uncyclized precursors.13g.21b,34 Compounds 1-3also exhibit bright fluorescence in the solid state upon irradiationwith a UV lamp while compound 3 displays visible fluorescenceunder direct sunlight in solution. Terphenyl dilactones 1-3 and their unlactonized precursors 7,12, and 18 were analyzed by UV-vis spectroscopy (Fig. 3) andfluorescence spectroscopy (Fig.4). As expected, the rigidity of 1, 2,and 3 results in a dramatic increase in the molar absorptivity (Fig. 3and Table 1) relative to their unlactonized precursors, 7,12, and 18.Quinquephenyl 3 also displays a bathochromic shift and increasedmolar absorptivity relative to terphenyls 1 and 2 resulting from thelonger conjugation pathway. The absorption spectra of 1-3 aretypical of polyaromatic hydrocarbons with low rotational freedomin that multiple sharp peaks are observed due to the distinct 7→transitions, whereas the absorption spectra of 7,12, and 18 displaybroader peaks,typical of a compound with a higher degree of bondrotational freedom. Compounds 1, 2, 3, 7, 12, and 18 are all fluo-rescent (Fig. 4 and Table 1) when excited at 350 nm. However, di-rect comparison of the cyclized 1-3 with their non-lactonizedprecursors (7, 12, and 18) illustrates how the rigidity of thesecompounds affects relative fluorescence quantum yield (TableAll three crankshaft compounds have much higher relative quan-tum yields (1=0.24,2=0.23, and 3=0.92) compared to their muchless rigid precursors (7=0.05,12=0.04,18=0.09). These quantumyield values of 1-3 are similar to other crankshaft bridged ter-phenyls with different bridging groups. 3f1ofIncreasing the conju-gation length also increases the quantum yield as crankshaft 3 andquinquephenyl 18 have the highest quantum yields relative to 1, 2and 7, 12, respectively. Overall, the two bridging lactone units inbetween the arene rings results in a 3-fold to 5-fold increase inmolar absorptivity and 5-fold to 10-fold increase in fluorescencequantum yield of 1-3 compared to 7,12, and 18. Fig. 3. Absorption spectra ofcompounds 1, 2, 3, 7, 12, and 18 dissolved in CH2Cl2. We were interested in exploring how crankshaft compounds1-3 would behave as pH-driven rigidity switches (Fig. 5). Based onour previous studies, cleavage of the lactone bridges with an or-ganic soluble base, such tetrabutylammonium hydroxide(TBA-OH),should result in significantly less rigid anionic ring-opened analogs Fig. 4. Normalized emission spectra of compounds 1, 2, 3, 7, 12, and 18 dissolved inCH2Cl2. All compounds were excited at 350 nm. Table 1Electronic absorption and emission data for 1, 2, 3, 7, 12, and 18 Compd Lowest E abs Em Amax [nm] Amax [nm](e[M-1cm-1]) 335(37,983) 439 0.24 364 (36,832) 385 0.23 384(65,080) 394 0.92 319(7713) 414 0.05 326 (10,609) 409 0.04 337(22,801) 424 0.09 UV-vis spectral data from compounds dissolved in CH2Cl2. Emission spectral data from compounds dissolved in CH2Cl2 and excited at350 nm. Calculated relative to quinine sulfate dissolved in 0.5 M H2SO4 and excited at350 nm.36 (1a-3a) with attenuated photophysical properties relative to 1-3.Re-acidification with an organic soluble acid, such as trifluoroaceticacid (TFA), should result in a return to 1-3. The interconversion of 1and 1a should be unaffected by the presence of oxygen during theswitching process. However, spontaneous oxidation of aryl lactonessimilar to 2 and 3 has been reported upon base-promoted lactonecleavage resulting in immediate quinone formation.Thereforecompounds 2a and 3a could become readily oxidized to 2b and 3b,halting the reversibility of the switching in 2 and 3. Our previous studies of the pH-driven geometry switching inanalogous D-A biaryl lactones were performed with the biaryllactones dissolved in acetonitrile while employing TBA-OH and TFAas switching stimuli. In these studies, both the rigid lactonizedswitch states and the ring-opened anionic switch states were sol-uble in acetonitrile, thus lending to characterization of theswitching process with UV-vis spectroscopy, 1H NMR spectros-copy, and by visible fluorescence monitoring. In the current study, crankshaft compounds 1-3 are completelyinsoluble in acetonitrile and thus preliminary switching studieswere required to be performed in dichloromethane. The differingsolubility of both 1-3 and 1a-3a in dichloromethane resulted ininconclusive results while monitoring the switching with UV-visspectroscopy, even when attempting additional solvents, includingbiphasic solvent systems. In all cases, addition of TBA-OH to 1-3resulted in partial precipitation and/or decomposition of theproducts making quantitative UV-vis measurements unreliable.However, the samples irradiated with an UV lamp display directlyvisible fluorescence switching upon addition of TBA-OH followedby TFA (Fig.6). All three compounds instantly become non-fluorescent upon addition of TBA-OH. However, only crankshaftcompounds 1 and 2 displayed fluorescence switching after TBA-OHtreatment and a subsequent TFA addition. Unexpectedly, after Fig. 5. Potential pH-driven geometry switching in 1-3. Compounds 2b and 3b are possible oxidative decomposition products of 2a and 2b. addition of TBA-OH to crankshaft compound 3 the subsequent TFAaddition did not result in the return of visible fluorescence. Wereasoned that 3a could be unstable under the switching conditionsand was likely converted to 3b, leading to an irreversibility of theswitching process. This issue was temporarily addressed by dissolving a small amount of sodium dithionite as an oxygen scaven-ger in the TBA-OH prior to addition, resulting in a visible return tofluorescence after subsequent TFA addition. Further analysis ofcompound 2 also showed decomposition after TBA-OH addition butat a much slower rate (several hours) relative to 3. Compound 1showed no signs of decomposition after switching to 1a, even after24 h. All three compounds had a finite ability to reversibly cycle thefluorescence switching process as further subsequent additions ofTBA-OH and TFA caused 1-3 to become insoluble. However, thesepreliminary studies indicate that compounds with this architecturehave the potential to be useful as pH-driven molecular switchesand/or sensors with instant and dramatic fluorescence attenuationat high pH values. 'H NMR spectroscopy studies were all indicative of 1a-3a beingformed during upon addition of TBA-OH (Figs. 7-9) to 1-3.Crankshafts 1-3 were dissolved in CD2Cl2, a drop of TBA-OH wasadded, and H NMR spectroscopy with analysis in the aromaticregion of the spectra was immediately performed. Addition of TBA-OH to both 1 and 2 clearly resulted in formation of 1a and 2a, re-spectively, in the HNMR spectra.The upfield chemical shifts of thesignals in the aromatic region relative to 1 and 2 resulting fromrelief of steric compression and increase in electron density (due tothe formation of phenoxide anions) is indicative of 1a and 2a. Ad-dition of TFA to the samples resulted in reformation of 1 and 2, bothbeing detected by 1H NMR (see Supplementary data). In the case of3, decomposition was apparent in the spectra after the addition ofTBA-OH with only small amounts of 3a being detected. The aro-matic region in the lH NMR spectrum was largely unreadable (see Supplementary data), likely due to the spontaneous conversion of3a into quinone 3b, followed by rapid decomposition. Simple 1,4-benzoquinone has long been known to be sensitive toward strongbases resulting in spontaneous decomposition.38 Treatment of TBA-OH with sodium dithionite before addition resulted in enoughformation of 3a to clearly detect in the'H NMR spectrum, although3a still decomposed rapidly. Once again, the upfield chemical shiftof the signals in the aromatic region relative to 3 is indicative of 3a.Small amounts of 3 could be detected by lH NMR upon re-p.acidification by the addition of TFA to the NMR sample (seeSupplementary data). Interestingly, 2a appears to be much morestable than 3a as evidenced by both visual inspection of fluores-cence switching and 'H NMR data. However, 2a does degrade over24 h (see Supplementary data) to 2b without significant furtherdecomposition. In all cases, H NMR data supports the initialproducts off base-promoted switching of 1-3 being the expected products,1a-3a. 3. Conclusion Terphenyl dilactones 1, 2, and 3 with crankshaft architectureswere readily synthesized from common and inexpensive startingmaterials. Molecules containing the dilactone terphenyl structureare relatively unexplored. Compounds 1 and 2 have the same sol-ubilizing groups but reversed lactone bridge orientation and com-pound 3 has the same bridge orientation as 2 but differentsolubilizing groups. All three compounds were slightly soluble inorganic solvents allowing for purification and characterization bytypical methods. All three compounds were also highly fluorescentin both the solid state and in solution. The crankshaft dilactoneswere shown to have much stronger absorption and emission rela-tive to the unlactonized precursors 7, 12, and 18 thus indicating theeffect of rigidity on these desirable properties. Preliminary pH- 400-188-0725 Fig. 6. Photographs of visual fluorescence switching of 1, 2, and 3 via irradiation withan UV lamp. Top: solutions of 1, 2, and 3 dissolved in CH2Cl2. Middle: same solutionsfrom top after addition of one drop of 40% aq TBA-OH treated with Na2S204. Bottom:same solutions from middle after re-acidifying with TFA. driven switching studies indicate promise for application as mo-lecular switches and/or sensors but both solubility (1-3) and sta-bility (2 and especially 3) in the ring-opened state are problematic.We are currently exploring different solubilizing groups to helpaddress these issues as well as the synthesis of donor-acceptoranalogs to fine tune the photophysical properties of this un-explored class of molecules. 4. Experimental section 4.1. General comments All starting materials and solvents were purchased commer-lly and uusseedd wvithout further purification. All 1H and 13c NMR mixtuispectra were recorded on a Bruker Avance II 400 (100) MHz in-strument. Chemical shifts are reported in parts per million (ppm)and referenced to the appropriate residual solvent peak. All IRspectra were recorded with a Thermo Scientific Nicolet I5S FT-IRspectrometer with ATR. UV-vis spectroscopy was performedwith a Cary 50 spectrophotometer using 1 cm quartz cells. Fluo-rescence spectroscopy was performed with a Perkin-Elmer LS55spectrofluorometer using 1 cm quartz cells. Purification by column chromatography was performed using a Biotage Isolera One flashchromatography system and Biotage SNAP KP-Sil cartridges. Thinlayer chromatography was performed using silica gel 60 F254 platesand visualized with an UV lamp. Melting points were obtained witha Mel-Temp electrothermal melting point apparatus. TGA wasperformed with a TA Instruments Q50 thermogravimetric analysisinstrument. Mass spectrometry was performed with a MALDI massspectrometer by the University of Minnesota Mass SpectrometryLab in Minneapolis, MN.Elemental analysis was performed byMicro Analysis, Inc. in Wilmington, DE. 4.2. Crankshaft precursor (7) Dibromoarene 6 (370 mg, 1.05 mmol), boronate ester 5 (850 mg,2.93 mmol), K2CO3 (1.76 g, 12.7 mmol) in toluene (40 mL), EtOH(10 mL), and H20 (5 mL) was sparged with Ar for 1 h. Pd(PPh3)4(0.15 g, 0.13 mmol) was added, the mixture was sparged with Ar foran additional 15 min, and then refluxed for 48 h. The reactionmixture was cooled to rt and 50 mL of Et20 and 50 mL of H20 wereadded. The layers were separated and the aqueous layer wasextracted with Et20 (3×50 mL). The combined organics werewashed with 100 mL of brine, dried with MgSO4, and the solventwas removed under vacuum. The product was isolated with columnchromatography to afford a white solid!(0.51 g, 68%), mp223-225 PC.HNMR(CDCl3): 6 7.82 (s,2H), 7.36 (dd,J=8.5,2.3 Hz,/22H), 7.32(d,J=2.3 Hz, 2H), 6.85(d,J=8.5Hz, 2H), 3.74 (s, 6H), 3.67(s,6H), 1.35 (s, 18H). 13c NMR (CDCl3):6 168.2,153.9, 143.4, 137.7,134.2, 132.2,128.6,127.4,125.6,109.7,55.3,51.8,34.2,31.6. FTIR(cm):1729.72. UV-vis (CH2Cl2) lowest E abs Amax [nm] (e[M- cm-1]): 319 (7713). Em.(CH2Cl2) Amax [nm]: 414. Anal. Calcdfor C32H3806: C,74.11; H, 7.39. Found: C, 74.06; H, 7.64. 4.3. Crankshaft (1) Crankshaft precursor 7 (220 mg, 0.420 mmol) was dissolved inCH2Cl2(25mL) and chilled to -78°Cunder an Ar atmosphere. BBr3(1.0M) in CH2Cl2 (1.7 mL,1.7 mmol) was added and the cooling bathwas removed after 5 min. The reaction mixture was stirred at rt for1 h. The reaction was quenched with 25 mL of H20 and the layerswere separated. The aqueous layer was extracted with CH2Cl2(2x50mL). The combined organics were dried with Na2SO4 and thesolvents removed under vacuum. The product was purified overa pad of silica to afford a pale yellow powder (170 mg, 94%), mp324℃ dec. H NMR (CDCl3): 6 9.21 (s, 2H), 8.21 (s, 2H), 7.62 (d,J=9.0Hz, 2H), 7.38 (d,J=8.5 Hz, 2H), 1.46 (s,18H).13CNMR(CDCl3):; 160.6,149.1,148.5,134.4,129.1,126.9,124.7,119.6,117.5,116.3,39.9,31.5. FTIR(cm-1): 1734.34. UV-vis (CH2Cl2) lowest E abs Amax [nm](e[M-1cm-1]): 335 (37,983). Em. (CH2Cl2) Amax [nm]: 439. Ms(MALDI) for C26H2804 [M+] calcd: 426.2, found 426.2. Anal. Calcdfor C28H2604: C, 78.85; H, 6.14. Found: C, 78.53; H, 6.16. 4.4. Methyl4-(tert-butyl)-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (10) Arene 8 (6.46 g, 28.4 mmol) was dissolved in t-BuOH (50 mL)and H20(50 mL). KMnO4 (9.2 g, 58 mmol) and Celite (10 g) wereadded and the reaction mixture was refluxed for 16 h. The reactionmixture was cooled to rt and filtered through a pad of Celite. Thefiltrate was acidified with concd HCl and extracted with MTBE(3x75 mL). The combined organics were extracted with 5%aq NaoH (3×50 mL). The combined aqueous layers were acidifiedwith concd HCl and extracted with MTBE (3×75mL). The combinedorganics were dried with MgSO4. The solvents were removed undervacuum to achieve a crude white solid (5.25 g, 72% crude yield),which was not purified further. H NMR(CDCl;): 6 7.98 (d,J=8.3 Hz,1H), 7.72 (d,J=1.5 Hz, 1H), 7.42 (dd,J=8.3, 1.5 Hz, 1H), 1.35 (s,9H). d a b c c b d a o. 1州州圳州州州州哪中帅州9.0 8.5 8.0 7.5 7.0 6.5 d a b ) 02C. d moCO2 c. 1a wwk T 9.0 6.5 Fig. 7. 1H NMR studies of 1. Top: the aromatic region of 1 dissolved in CD2Cl2. Bottom: NMR sample from top after addition of one drop of 40% aq TBA-OH. Fig. 8. lH NMR studies of 2. Top: the aromatic region of 2 dissolved in CD2Cl2. Bottom: NMR sample from top after addition of one drop of 40% aq TBA-OH.耐士科技 www.rysstech.com 400-188-0725 Fig. 9. 1H NMR studies of 3. Top: the aromatic region of 3 dissolved in CD2Clz. Bottom: NMR sample from top after addition of one drop of 40% aq TBA-OH that was premixed withNa2S204 to delay oxidation. The crude 2-bromo-4-tert-butylbenzoic acid (5.25 g, 20.4 mmol).was dissolved in MeOH (75 mL), concd H2SO4 (1 mL) was added,and refluxed for 3 h. The reaction mixture was cooled to rt and75 mL of H20 was added. The reaction mixture was extracted withMTBE (3x50 mL). The combined organics were washed with 50 mLof satd NaHCO3, 50 mL of H20, and 50 mL of brine. The organicswere dried with MgSO4, filtered, and the solvents were removedunder reduced pressure to achieve the product (9) as a crude yellowoil (4.6 g,84% crude yield), which was not purified further. H NMR(CDCl3): ; 7.77 (d,J=8.0 Hz, 1H), 7.67 (d,J=1.8 Hz, 1H), 7.37 (dd,J=8.3, 1.8 Hz,1H),3.93 (s,3H),1.33 (s,9H). Crude 9 (2.0 g, 7.3 mmol)was dissolved in 1,4-dioxane (75 mL).KOAc (2.20 g, 22.4 mmol)andbis(pinacolato)diboron (2.63 g, 10.4 mmol) were added and thereaction mixture was sparged with Ar for 1 h. Pd(dppf)Cl2 (270 mg,0.37 mmol) was added, the mixture was sparged with Ar for anadditional 15 min, and then heated at 90 °C for 16 h. The reactionmixture was cooled to rt and 50 mL of satd NH4Cl and 25 mL of H20were added. The reaction mixture was extracted with MTBE(3×75mL). The combined organics were washed with 50 mL of H20and 50 mL of brine, dried with MgSO4, treated with activatedcharcoal, and filtered through a pad of Celite. The solvent was re-moved under vacuum and the product was isolated with column chromatography to afford a white/light green solid (1.74g,74%), mp55-57°C.HNMR(CDCl3): 6 7.87 (d,J=8.3Hz,1H), 7.47 (d,J=2.0Hz,1H), 7.43 (dd,J=8.3 Hz, 2.0 Hz, 1H), 3.90 (s, 3H), 1.44(s,12H), 1.33 (s,9H).1CNMR(CDCl3): 167.4,154.0,129.7,121.7,121.7,125.2,82.9,51.1, 33.9, 30.1, 23.9. FTIR (cm-): 1709.55. Anal. Calcd forC18H27BO4:C, 67.94; H, 8.55. Found: C, 67.94; H, 8.72. 4.5. Crankshaft precursor (12) Boronate ester 10 (1.00 g, 3.15 mmol), dibromoarene 11 (310 mg,1.05 mmol),and K2CO3(1.88 g,13.6 mmol) in toluene(40 mL),EtOH(10 mL), and H20 (5 mL) was sparged with Ar for 1 h. Pd(PPh3)4(120 mg, 0.10 mmol) was added, the mixture was sparged for anaddition 15 min with Ar, and then refluxed for 16 h. The reactionmixture was allowed to cool to rt and 30 mL of H20 was added tothe reaction. The reaction mixture was extracted with Et20(3x30mL). The combined organics were dried with Na2SO4 and thesolvent removed under vacuum. The product was isolated withcolumn chromatography to afford a white/light yellow solid(506 mg,93%), mp 151-154°C. 1H NMR (CDCl3): 6 7.83 (d,J=8.5 Hz,2H), 7.45-7.44 (m, 4H), 6.84 (s,2H), 3.71 (s, 12H), 1.38 (s,18H). 1cNMR(CDCl3): ; 168.7,154.9,150.3,138.2,130.6,129.3,128.9,128.4, 124.3, 113.0, 56.0,51.6,35.0, 31.18. FTIR (cm-1): 1725.25. UV-vis(CH2Cl2) lowest E abs Amax [nm](e[M-1cm-1]): 326 (10,609). Em.(CH2Cl2)max [nm]: 409. Anal. Calcd for C32H3806: C, 74.11;H, 7.39.Found: C, 73.98; H, 7.38. 4.6. Crankshaft (2) Crankshaft precursor 12 (518 mg, 1.00 mmol) was dissolved inCH2Cl2 (25 mL) and cooled to-78°Cunder an Ar atmosphere. BBr3(1.0 M) in CH2Cl2 (4.0 mL, 4.0 mmol) was added and the coolingbath was removed after 5 min. The reaction mixture was stirred atrt for 1 h. The reaction was quenched with 50 mL of H20 and 75 mLof CH2Cl2 was added. The layers were separated and the aqueouslayer was extracted with CH2Cl2 (2×100 mL). The combined or-ganics were dried with Na2SO4 and the solvents removed undervacuum. The product was purified over a pad of silica to afforda pale yellow powder (370 mg, 87%), mp 366°C dec. 1H NMR(CDCl3):6 8.39(d,J=8.5 Hz, 2H), 8.12 (s, 2H), 8.08 (s, 2H), 7.73 (d,j=9.0 Hz, 2H), 1.48 (s, 18H). 13c NMR (CDCl3): 6 160.6,159.4,133.4,139.7,127.8,120.3,119.0,118.6,111.1,35.8, 31.1. FTIR(cm-1): 1713.05.UV-vis (CH2Cl2) lowest E abs Amax [nm] (e [M-1 cm-1]): 364(36,832). Em. (CH2Cl2) Amax [nm]: 385. MS (MALDI) for C26H2804[M+] calcd: 426.2, found 426.2. Anal. Calcd for C28H2604: C,78.85;H, 6.14. Found: C, 77.04;H, 6.01. K2CO3 (1.13 g, 8.19 mmol), boronate ester 17 (800 mg,1.78 mmol), and dibromoarene 11 (176 mg, 0.593 mmol) in toluene(20mL), EtOH (4 mL) and H20 (2 mL) was sparged with Ar for 1 h.Pd(PPh3)4 (73 mg, 0.063 mmol) was added, the mixture wassparged with Ar for an additional 20 min, and then heated 90 °C for16 h. The reaction mixture was cooled to rt and diluted with 50 mLof H2O. The reaction mixture was extracted with Et20(3×50 mL).The combined organics were washed with 150 mL of brine, driedwith Na2SO4, and the solvents were removed under vacuum. Theproduct was isolated via column chromatography as a white solid(433 mg, 99%), mp248-250°C. HNMR(CDCl3): ò 8.09(d,J=1.8Hz,2H), 7.80 (dd,J=8.0, 1.8 Hz,2H), 7.53(d,J=8.0 Hz, 2H), 7.49 (s,6H),6.91 (s, 2H), 3.76 (s, 12H), 1.41 (S,36H). 1C NMR (CDCl3): 6 169.0,151.3,150.3,141.6,136.8,132.2,131.6,130.3,129.9,128.4,121.8,121.7,112.8,56.0,51.8,35.0, 31.5. FTIR (cm-): 1720.79.UV-vis (CH2Cl2)lowest E abs Amax [nm](e[M-1cm-]): 337 (22,801). Em. (CHzCl2)Amax [nm]: 424.Anal. Calcd for C52H6206: C,79.76;H, 7.98. Found: C,77.41; H,8.18. 4.7. Methyl 4-bromo-3',5'-di-tert-butyl-[1,1'-biphenyl]-3-carboxylate (16) 4.8. Methyl 3',5'-di-tert-butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-[1,1'-biphenyl]-3-carboxylate (17) Bis(pinacolato)diboron (820 mg, 3.2 mmol), KOAc (680 mg,6.9 mmol), and aryl halide 16 (930 mg, 2.3 mmol) in dioxane(25 mL) was sparged with Ar for 30 min. Pd(dppf)Cl2 (84 mg,0.12 mmol) was added, the mixture was sparged with Ar for anadditional 10 min, and then heated at 90 °C for 16 h. The reactionmixture was cooled to rt and diluted with 25 mL of satd NH4Cl and25 mL of H2O. The reaction mixture was extracted with MTBE(3×30 mL). The combined organics were washed with 25 mL of H2Oand 25 mL of brine, dried with MgSO4,and treated with activatedcharcoal. The mixture was filtered through a pad of Celite and thesolvent was removed under vacuum. The product was isolated viacolumn chromatography as a white/light green solid (800 mg, 77%),mp 46-48°C. 1H NMR (CDCl3): 6 8.15 (d,J=1.5 Hz, 1H), 7.75 (dd,J=7.7, 1.8 Hz, 1H), 7.59 (d,J=7.5 Hz, 1H), 7.47 (d,J=1.5 Hz, 1H), 7.42(d,J=1.8 Hz, 2H), 3.95 (s, 3H), 1.44 (s, 12H),1.39 (s,18H). 13CNMR(CDCl3): 168.6,151.3,139.5,138.6,134.2,132.8,130.6,127.6,122.0,121.6,84.0, 52.3, 35.0, 31.4, 24.9. FTIR (cm-1): 1718.91. Anal. Calcdfor C28H39BO4: C,74.66; H, 8.73. Found: C, 73.18; H, 8.90. Crankshaft precursor 18 (783 mg, 1.00 mmol) was dissolved inCH2Cl2(25 mL) and cooled to-78°C under Ar. BBr3 (1.0 M) inCH2Cl2 (4.0 mL, 4.0 mmol) was added and the cooling bath wasremoved after 5 min. The reaction mixture was stirred at rt for 1 h.The reaction was quenched with 50 mL of H20 and diluted with75 mL of CH2Cl2. The layers were separated and the aqueous layerwas extracted with CH2Cl2 (2×100 mL). The combined organicswere dried with Na2SO4 and the solvents removed under vacuum.The product was purified over a pad of silica to afford a pale white/yellow powder (663mg, 96%), mp 440°C dec. 'H NMR (CDCl3):6 8.69 (s,2H), 8.21(d,J=8.0 Hz, 2H), 8.17 (d,J=8.0 Hz, 2H), 8.08 (s,2H), 7.55 (s,6H), 1.43 (s,36H).13CNMR(CDCl3): 6 160.8,151.8,147.8,144.3,138.3,134.3,132.0,128.97,122.8,121.7,121.6,120.0,111.1, 35.1,32.5. FTIR (cm-1): 1739.29. UV-vis(CH2Cl2) lowest E abs Amax [nm](s[M-1 cm-1]): 384 (65,080). Em. (CH2Cl2) Amax [nm]: 394.MS(MALDI) for C26H2804 [M+] calcd: 690.4, found 690.4. Anal. Calcdfor C48H5004: C, 83.44;H, 7.29. Found: C, 82.84; H, 7.45. Acknowledgements Acknowledgment is made to the Research Corporation for Sci-ence Advancement Cottrell College Science Award (ID: 22480) andto the donors of the American Chemical Society Petroleum Re-search Fund (ACS PRF 51259-UNI1) for support of this research. Weare also grateful for the Student Blugold Commitment DifferentialTuition funds through the University of Wisconsin-Eau Claire Fac-ulty/Student Research Collaboration Grants program for financialsupport. Supplementary data Detailed synthetic procedures for 4-6, 8, 13-15. Copies of lHand 13c NMR spectra for 1-3, 7, 10, 12, 16-18. H NMR spectra ofacidified products of 1a, 2a,and 3a. 1H NMR spectrum for 2a after24 h. 1H NMR spectrum for 3 after addition of TBA-OH withoutNa2S204. Thermogravimetric analysis (TGA) data for 1, 2, and 3.Supplementary data associated with this article can be found in theonline version, at http://dx.doi.org/10.1016/j.tet.2014.11.055. References and notes ( 1 . For examples s e e: ( a) Fahrn i , C . J. ; Cody, J. Tetrahedron 2004, 60 , 110 9 9 - 11107; ) ( ( b) Sherwood, D. W.; C a lv i n, M. J. Am. C hem. S o c. 1 942, 6 4,1 3 5 0-1353; ( c ) ) McFarland, S. A.; Finney, N. S. J. Am. Chem. Soc. 2001, 123,1260-1261;(d) Ef-fenberger, F; Agster, W.; Fischer, P.; Jogun, K. H.; Stezowski,J.J; Daltrozzo,E.;Kollmanssberger-von Nell, G. J. Org. Chem. 1983, 48,4649-4658;(e) Cheng, L.-T.; Tam, W.; Marder, S. R.; Stiegman, A. E.; Rikken, G.; Spangler, C. W. J. Phys.Chem. 1991, 95, 10643-10652. 2.(a)Venkataraman,L.; Klare,J. E.; Nuckolls, C.; Hybertsen, M. S.; Steigerwald, M.L. Nature 2006,442,904-907; (b) Mishchenko, A.; Vonlanthen, D.; Meded, V.;Burkle, M.; Li, C.; Pobelov, I. V.; Bagrets, A.; Viljas, J. K.; Pauly,F.; Evers,F.; Mayor,M.; Wandlowski, T. Nano Lett.2010,10,156-163;(c) Mischenko, A.; Zotti, L. A.;Vonlanthen, D.; Burkle,M.; Pauly, F.;Cuevas,J. C.; Mayor, M.; Wandlowski, T. J.Am. Chem. Soc. 2011, 133,184-187;(d) Sakano, T.; Higashiguchi, K.; Matsuda,K.Chem. Commun. 2011. 8427-8429. ( 3. ( a ) Eaton, V. J .; Ste e le, D. J . Chem. So c ., Faraday Trans . 2 197 3 , 69,1601 - 1 6 0 8;(b ) Akiyama, M.; Watanabe, T. ; K a kihana, M. J. P hys. C hem. 1986, 9 0, 1 752-1 7 5 5. ) ( 4. F or r ecent examples see: ( a ) Vonl a nthen , D.; R o t zler, J; N euburg e r, M . ; M a yor, M. E u r . J. Org . Che m . 2010, 120-133; ( b) Vonlanth e n, D . ; Rudnev , A . ; Mis- h chenko, A.; K a e slin, A . ; R o tzler, J . ; Neuburger, M . ; W a ndlowski, T.; M a y o r, M. C hem.-Eur. J. 2011, 1 7 , 7 23 6 -7250;(c) Ha n i fi , D .; P u n , A.; Li u , Y. Chem. Asian J. 2012, 7 , 2 615-2 6 20;(d ) Fu r ukawa, S.; H a g a , S. ; K o b ayashi, J ; K a washima, T. Or g . L ett. 2014 , 16 , 3228 -3 231; ( e) C hen, D.-M.; Qin, Q; S u n, Z . - B .; P e n g , Q ; Zhao, C . - H. Chem. Commun. 2014, 7 82-7 8 4; ( f) P odu v al, M. K. K.; Hah n , S . J; K im, T. - H . Bull . Korean Chem. 1 . S o S c. 2012, 3 3, 2 040-2042; ( g ) Poriel, C.; R au l t- B erthelot,J ; Thirion, D . J. Org. Chem. 2013, 7 8 ,88 6 -898 ; (h) Z h u , X.; Tsu j i , H.; L o pez N a varrete, J . T . ; C a sado,J ; N a kamura, E. J . Am. Ch e m. Soc . 20 1 2 , 134 , 1 9 254-1 9 259; ( i ) Wu, Y .; Zhang, J; B o , Z . Org. L et t .2007, 9 , 4435 - 4 4 38; ( j ) X u, C.; Wakamiya, A . ; Yamag u chi , S. J. Am . Chem. So c. 2005, 1 27,1 6 38-1 6 3 9; ( k ) B r u ch, A .; Fukazawa,A. ; Y amaguchi, E.; Y am aguchi, s.; Stu d er , A. A ngew. Chem., I nt. Ed . 2011, 50 , 1 2094- 1 2098;(I ) Ka i , H.; O hs h ita,j . ; O h ar a , S . ; Nakayama, N. ; K unai, A. ; L ee, I . -S .; Kwak,Y.- W . j .Organomet . Chem. 2008,693, 3490-3 4 94;(m ) R en, Y.; Baumgar t ner , T. J. Am. Chem. So c . 20 1 1,13 3 ,1328 - 1 340; ( n ) K awaguchi, K .; Nakano, K .; N ozaki, K . J. Org. Chem. 2 007, 7 2, 5119-5128;( o ) Gu , R. ; H a - meurlaine, A.; Dehaen, W. J . Org . Chem. 2007 , 72,7207- 7 2 13; ( p ) Dienes, D urben, S.; K arpati, T.; Neumann , T . ; E nglert, U .; Ny ul aszi, L.; Ba um gartner, Chem. -E ur. J . 2007, 1 3,7487 - 7500. ) ( 5. F or r ecent r eviews s e e: ( a) Gr i msda l e, A . C .; M ul l e n, K. M acromol. R a p id l Co m - 一 As i an mun. 2007, 28,1676- 1 702;( b ) F u kazawa,A.; Yamaguchi, S. Chem. Asin 20 09. 4, 1 386 - 1400; ( c ) Scherf , U . J . Mater. Chem.1999,9, 1853-1 8 64; ( d ) Ab bb e l. S chen n ing, A. P. H. J . ; Mei j er, E . W. J . Polym. Sci., Part A : Po l ym . Chem. 2 0 0 9 , 47. 4215 - 4 2 33; ( e ) Anthony, J. E. Chem. Rev . 2006 , 106 , 50 2 8 -5 048 ) ( 6. N ehe r ,D . M acromol. Rapid Commun. 2001,22,1365-1385. ) ( 7 . C ar l son, E .J.; Riel , A . M . S .; Dahl, B . J. Tetrahed r on L e tt.20 1 2,5 3 ,624 5 6 2 49. 8 Z hou, H .; Wue s t, J. D . La ng muir 2013, 29 , 7229- 7 238. 9 ) ( Y amaguchi, Y.; Matsubara, Y.; Ochi, T.; Wakami ya,T T: Yos h ida, Z . - I. J . Am . C he m . Soc . 2008,13 0 ,13867-13869. ) 10.(a) Kim, S.; Oehlhof, A.; Beile, B.; Meier, H.Helv.Chim. Acta 2009,92,1023-1033; (b) Meier, H. Angew. Chem., Int. Ed. 2005,5,44,2482-2506. 11. For recent examples see: (a) Chen,J: Onogi, S.; Hsieh, Y.-C.:: HHsiao, C.-C.;Higashibayashi, S. i; Sakurai, H.; Nu, Y.-T. Adv. Synthh. Catal. 2012,354,1551-1558; (b) Chaurasia, S.; Che Chou, H.-H; Wen, Y.-S.; Lin, J. T.rahedron 2012, 68,7755-7762 Y;1Lee,J.H.; Jung, D. H.; Liu, S.-H.;Lin, Y.-H.; Chen, L.-Y.; Wu, C.-( . Chem. 2010,20,5930-5936; (dZheng, Q.; Gupta, S. K.; He, G rasad, P.N. Adv. Funct.Mater. 2008.18,2770-2779;(e) Yen, W.-C S.; Hung, Y.-C.; Lin, S.-T.; Chao,,C.Y.;Su, W.-F. J. Polym. Sci., Part 2009,47,5044-5056; (f) Lin, T.C.; Hsu, C.-S.; Hu. C.-L.; Chen, W.J. Tetrahedron Lett. 2009,. 50.182-185;(g)Setayesh, S.; Marsitzky, D; Muellen, K. Macromolecules 2000, 332016-2020. 12. For recent examples see: (a) Irgashev, R. A.; Teslenko, A. Y.; Zhilina, E. F;Schepochkin, A. V.; El’tsov, O. S.;Rusinov, G. L.; Charushin, V. N. Tetrahedron2014, 70,4685-4696; (b) Levick,M. T.; Grace, I.; Dai, S.-Y; Kasch, N. S., Muryn,C.;Lambert, C.; Turner, M. L.; Procter, D. J. Org. Lett 2014,16,2292--22295;(c)Yu,J; Luo,J; Chen, Q; He, K.; Meng, F; Deng, X.; Wang, Y;Tan, H.;Jiang, H.; ZhuW. Tetrahedron 2014, 70,1246-1251; (d) Jia, W. Wang, H.-W.; Yang, L.-M.;Lu, H.-B.; Kong, L.; Tian, Y.-P.; Tao, X.-T.; Yang, J. r. Chem. C 2013,1,7092-7101;(e) Chen, S.; Wei,J.; Wang,g. K., Wan Chen,D.; Liu, Y.; Wang,Y.J.Mater. Chem. C 2013,1, 6594-6602;(f)Rivoal, M.: Bekere, L.; Gachet, D.; Lok-shin, V.; Marine, W.; Khodorkovsky, V. Tetrahedron 2013, 69, 3302-3307;(g)Shi, H.-P.; Shi,L.-W.; Dai,J.-X.; Xu, I;Wang.M.-H.; Wu,X.-H.; Fang, L.; Dong, C.;Choi, M. M. F. Tetrahedron 2012, 6l )788-9794;(h) Curiel, D.; Mas-Montoya,M.; Usea, L.; Espinosa, Orenes, R. A., Molina, P. Org. Lett. 2012, 14,3360-3363; (i) Simokaitiene, tanislovaityte, E.; Grazulevicius, J. V.; Jan-kauskas, V.; Gu, R.;.co Dehaen, W.; Hung, Y.-C.; Hsu, C.-P.J. Org. Chem.2012,77,4924-4931. ( 13. For recent examples see: ( a ) Z h a ng, X . ; Ji , X . ; Su, R . ; W eeks, B . L . ; Z hang , .; D e ng, S. Chem P l usC h em 20 1 3,7 8 , 7 0 3-711; (b ) C h ase, D . T.; Fi x , A. G .; Rose, B. D. ; Weber, C . D . ; N obusu e , S . ; Stockwell, C . E . ; Z akharov, L . N.; L onergan,M. C . ; H aley, M . M . A n gew. Ch e m ., I n t . Ed. 2011, 50, 111 0 3 -11 10 6; ( c) P ark , Y . -I . ; L e e , J. S .; K i m, B . J .; Kim, B .; L ee,J; Kim , D . H .; O h , S .-Y.; Cho, J . H . ; Park, J.-W. Chem. M ater. 2 011, 23, 4 038-4044; ( d ) Tak e d a , T.; I n u kai , K. ; T a hara, K.; T o b e, Y. J. Or g . Chem. 2 011,76,9 1 16-91 2 1 ; (e) Cha s e, D. T .; Rose, B. D . ; McClintock, S. P.; Za- k har o v, L . N .; Haley, M . M.Angew. Chem., Int. Ed . 2 011,50,1 1 27-1130; ( f ) R o se, ) ( B . D .; Chase, D . T ; W ebe r , C. D . ; Z a kharov, L. N . ; L o nergan, M . C.; Ha l ey, M. M. O rg. L ett. 2011, 13,2106- 21 09;(g ) Us t a, H. ; Ri s ko, C. ; W a ng, Z.; Hu a ng, H.; D el i omeroglu, M . K. ; Zhukho v itskiy , A .; F a c chet t i, A. ; Ma r k s, T. J . J . A m . Ch e m.Soc. 2 009, 1 3 1 , 5 5 86-5608; (h) Coch e rel , N . ; P ori e l , C. ; Rau l t-B e r t helo t , J ; B arri e re, F . ; Audeb r and, N . ; Slawin, A . M . A.; V i gnau, L . C h em.-Eur. J.2 0 0 8 , 14, 1 1328-1 1 3 42 ; (i) N ak a gawa, T.; K u m a k i , D.; Nishi d a, J.-I.; Tok i to, S.; Ya m ashita, Y . Chem. M ater. 2 008,20,26 1 5- 2 617;(j)U s ta , H.; Fa c c h e tt i , A.;Ma r ks, T. J . J. Am. Chem. Soc. 2008, 130 , 8580- 8 581 . ) ( 14. . ( a) F r ank, W.; Gomppe r , R . Tetrahedron Lett. 1987, 28, 3083-3086;( b ) B r u n etti , F . G. ; Varot t o, A.; Bat a ra, N. A.; Wudl, F. Chem.- Eu r. J. 2 011 , 1 7,8604-86 0 8;(c) D uerr, H. ; Schommer, C.; Muenzmay, T. Angew. Chem. 1986, 9 8,565-5 6 7. ) ( 15 . . Ponce G onzale z ,J; Edga r , M. ; E l s egood, M . R. J ; We a ver, G . W . O rg . Bio m ol. C hem. 2 011, 9, 2294-2305. ) ( 16. . ( a) Shint a ni, R.; Otomo, H.; O ta, K .; H ayashi , T .J . Am. C hem. S o c. 2 0 12, 1 3 4, 7305-7308; ( b ) F u ruk a wa, S. ; K o b a yashi,J. ; Kawas h ima, T. Dalton Trans. 2010, 3 9,9329-9336;(c ) U r esh i no,T.; Y o shida,T.;K u ninobu, Y.; Ta k ai, K. J. Am. Chem. Soc. 2010, 132 , 14324- 1 4 326; ( d) Furukawa, S .; Kobayashi,J; Kawashim a , T A m. C h em. Soc. 2009, 131, 14 1 92-141 9 3 ; (e) L i , L . ; X u, C.; Li, S. M a cromol. chen P hys. 2 009,2 1 0,10 9 7-110 3; (f ) Li, L. ; X i ang, ; X u , C. O rg . Le t t . 2 0 0 7 , 9 4877 - 4 8 79; ( g ) M a tsuda, T. ; K a dowaki, S.; Goya, T . ; Murakami, M . O r g . L e tt . 2007, 9 , 1 33- 1 36. ) ( 17. ( a) Pan, Z .; L i u, Y.; Fan, F .; Ch e n, Y . ; L i, Y.; Zhan, X . ; Song , Y. Chem.-Eur j. 201 3 , 1 9,9771-9 7 74; ( b ) Wang, Y .; Park i n, S . R.; Gierschner,J; W a tson , M. D . O r g . Lett. 2008,10,3307-3 3 10;(c ) Ebata,H . ;M iy azaki, E.;Y a mamoto,T. ; T a k im iya, K.O r g . L ett . 2007,9,4499-45 0 2;( d) Z h ou, Y.; L i u, W.J . ; M a, Y . ; W an g , H .; Qi, L.; g. ; Cao, Y . : Wang,J; P e i,J.J . Am. Chem. S oc. 2007, 129, 12386-12 3 8 7; ( e) W ang, r C - H .; Hu. R .- R .; Liang, S . ; Che n ,J. - H.; Ya n g, Z.; Pei , J. Tetrahe d ron Lett. 2005,46,81 53- 8 157. ) ( 18. ( a ) ) Nandakumar, M . ; Karun ak aran,J; Mo h ana k rishn a n , A . K . O r g . Le t t. 2 0 14, 1 6 , 306 8-3 0 7 1;(b) S p anopoulos, I.; Xydias, P.; M a lliakas, C. D . , Trik al i tis, P. N . I n-org . C hem . 201 3 ,52,855 - 862. ) ( F or r ecent e xamples see: ( a ) W a kabayashi, R . ; K u rah a s hi , T . ; M a tsubara, S . n le 13 , 2297- 2 301; ( b )S h e n, H.- C . C ; . ; T a Ta n n g g , , J J . . - - M M . . ; ; C Chang,H.-K. ; Yang, C .-W.; Li u , R R .-S. J . Org . Chem . 2005, 7 0, 1 0 1 1 3- -1 0 116;(c ) B onif a cio, M . C .; Robertson,C. ) R.;Jung,J.-Y.; King, B. T.J. Org. Chem2005,70,8522-8526. ( 20. ( a ) t ) Pla t t, K. L.; S eti a budi , F . JCh e m. S o c . , Pe r k i n Trans . 1 1992 , 2005- 2 0 0 9; ( b) M il ler D.W.; Herren o -Sae n z, D.; H uang t . K. H . ; Hei n ze, T . M . ; Fu , P. P. J . O r g. Ch em. 1992, 57 , 3746-374 8. ) ( 2 1 (a ) Kim, I . ; Yo o , M.; Kim , T . - H . T et r a hed r on 2007, 6 3,9476-94 8 1;(b) K im , I .; Kim, T .-H . ; Kang , Y ; Lim , , Y .-B . T e tr a he d ron L ett. 2006, 47,8689- 8 692. ) ( 2 2. L a mba,J. J. S .; T o u r, I T J MM . 小 A m . Che m. Soc.1994,116, 11 723 - 11736 . ) ( 23. (a ) Kl e mm , L. H . : ; We i s e e r t, A. J . Heterocy c l.Chem . 1965 , 2,140-1 4 3; ( b) K lemm, L. H. ; J o hnson, W. 0 Wei ser t , A. J . Heterocycl. Chem. 197 1 ,8,763 - 7 68. ) ( 24. B arto n , J. W. , R o we, I Te t rahedron Lett. 198 3 ,24 , 299 - 3 02. ihedi ) ( 25. (a) La Budde, J . A. ; H eid e lberger, C. J . A m . Chem . So c . 1958,80,1225-12 3 6; ( b ) Ste ph en s on , E. F . M. J . Chem. S oc. 1949, 2620- 26 23. ) ( . B ern at ek, E . Acta C hem. Scand . 1959,1719- 1 720. ) ( . Le e , M . -G .; Hahn, S.J.; K i m, T. - H . Bull. Korean Ch e m. S oc. 2 0 1 3, 3 4,249 5 -2 4 98 . M all o ry, F . B .; M allor y , C. W.;R e gan , C . K . ; A spden, R . J ; Ricks, A . B . ; Ra- ) ( (a cowsk i i . j J . . M . ; Nash, A. I . ; Gi b bons, A. V . ; C a rroll, P. J . ; B ohen, J . M . J. O rg.C h em. 2 01 3, 7 8 , 2 0 40-20 4 5;(b) W u, T .-L.; Chou, H. - H . ; H uang, P.- Y . ; C heng, C.- H . ; L iu, R.-S .J. O r g. Chem. 2014, 7 9,267-2 74 ;(c) L i, H .; Zh o u, F.; Ta m , T . L. D.; L a m,Y. M . ; ) ( Mha i salkar, S . G. ; Su, H . ; Gri m sdale, A. C. J . Mater . Chem. C 2013, 1, 1745 - 1 752; (d ) L i, J . ; ji a o , C. ; H ua ng , K.-W. ; W u, j . Chem. -E ur. J. 20 1 1, 17,14672-1 4 680;(e) ) ( Z h a ng, X .; L i, J. ; Qu, H . ; C h i, C.; Wu, J . Org . Lett . 2010 , 12, 3946- 39 49; ( f ) C o n- s table, E. C. ; Hostettler, N . ; Hou s ecroft, C . E .; Kopecky, P .; Neuburger, M . ;Zamp e se, J . A. Dalton Trans. 2012,41 , 2890- 2 897; ( g)Gri m me, J; S cherf, U. M acromol. Chem. P hys. 1996, 197, 2297-2304 . ) ( 29. T u r lington, M.; DeB e rardi n is, A. M. ; Pu, L. Or g . Lett. 20 0 9,11,244 1 -2444 . ) ( 3 0. V an Humbeck , J. F . ; M cDonal d , T. M. ; J i ng, X . ; Wiers , B . M.; Z h u, G. ; Long, J. R.J . A m. C hem. S oc. 2014 . 136,2432-2440. ) ( 31. . 1 Lopez-Alva r ado , P.; Ave n dano, C.; Menendez, J. C . Synth. Commun. 2002, 32 , 3233-3239 . ) ( 32. F inke , A. D.; Moore,J. S. O rg. Lett. 2008, 1 0, 4851 - 4854. 33. Fr ampton, M. J ; A k das, H.; C o w le y , A. R.; R og e rs, J. E.; S l a g le, J . E.; Fl e itz , P. A. ; ) Drobizhev,M.; Rebane, A.; Anderson, H. L. Org. Lett.2005,7,5365-5368. ( 34. F or recent examples see: (a) Nav a le, T. S.; Thakur,K.; Rat h or e , R. O rg . Lett. 2011, 1 3, 1634- 16 37;(b ) Ki n g , B. T.;Kr o uli k , J; R o bertson, C. R. ; R e mpal a , P.; Hilton, C. L .; Kor i nek, J . D.; Gortari, L. M. J . Org . Chem. 2007, 7 2, 2 279-2288;(c) C hen , T . -A .; L ee, T . J .; L in, M . - Y.; S oh el , S. M . A . ; D i au , E. W . -G.;Lus h , S .-F ; Li u , R.-S . Chem.- E u r .J. 2010 , 1 6 ,1 8 26-1 8 3 3;(d) Maj u mda r ,K. C. ; C h akravorty, S.; D e, N. Tetrahedron Lett . 2008, 49 , 34 1 9- 3 422; ( e) G omez-Lo r , B . ; E chavarren, A . M. O rg . Lett . 2004, 6 , 2993 - 2996. ) ( 35. C alcula t ed u s ing the meth od fro m : Williams , A. T.R.; Winfield,S. A. ; M ill e r, J . N . Analyst 1983, 108,1067 - 1 071. ) ( 36. B rouwer, A. M . Pure Appl. Chem. 2 011, 83, 2213- 2 228. 3 7. ( a) Qabaja, G. ; J o nes,G . B . J. Org. Chem. 2 000, 6 5 , 7187-719 4 ; (b ) De Frutos,O.; ) ( A tien z a, C .; Ec h avar r en, A. M. Eur. J . Org. Chem. 2001 , 1 , 1 63 -17 1 ; (c)Mbala, B. M .; J acobs, J; C l aes , P. ; Mudogo, V.; D e K impe, N . T e trahedron 2011, 67,8747-8756. ) ( 3 8. L e ighto n , P . A.; F orbes, G. S . J . Am. Chem. S oc. 1 929, 51, 3 549-3 5 5 9. ) http://dx.doi.org/j.tet.Olsevier Ltd. All rights reserved.士科技www.rysstech.com www.rysstech.com耐士科技
确定







还剩8页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中主要物质含量分析检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多