








方案详情
文
2-(3-(Naphthalen-2-yl)propanamido)cyclohex-1-enecarboxylic acid and its 6-hydroxynaphthalen-2-ylanalogue are well-known hydroxyl-carboxylic acid (HCA) receptor HCA2 agonists. A series of novel aryl derivatives of 2-amidocyclohex-1-ene carboxylic acid that contained rigidity elements, such as an E-double bond, triple bond, and trans or cis-substituted cyclopropane rings, instead of the saturated ethane linker
in the amide part of the molecules were designed and synthesized, and the derivatives’ potency for the activation of HCA1, HCA2, and HCA3 receptors by 30–50-cyclic adenosine monophosphate (cAMP) assay were evaluated. The SAR studies revealed that the rigidifying of appropriate molecules enabledmodulation of the potency and selectivity of the HCA2 receptor activation.
方案详情

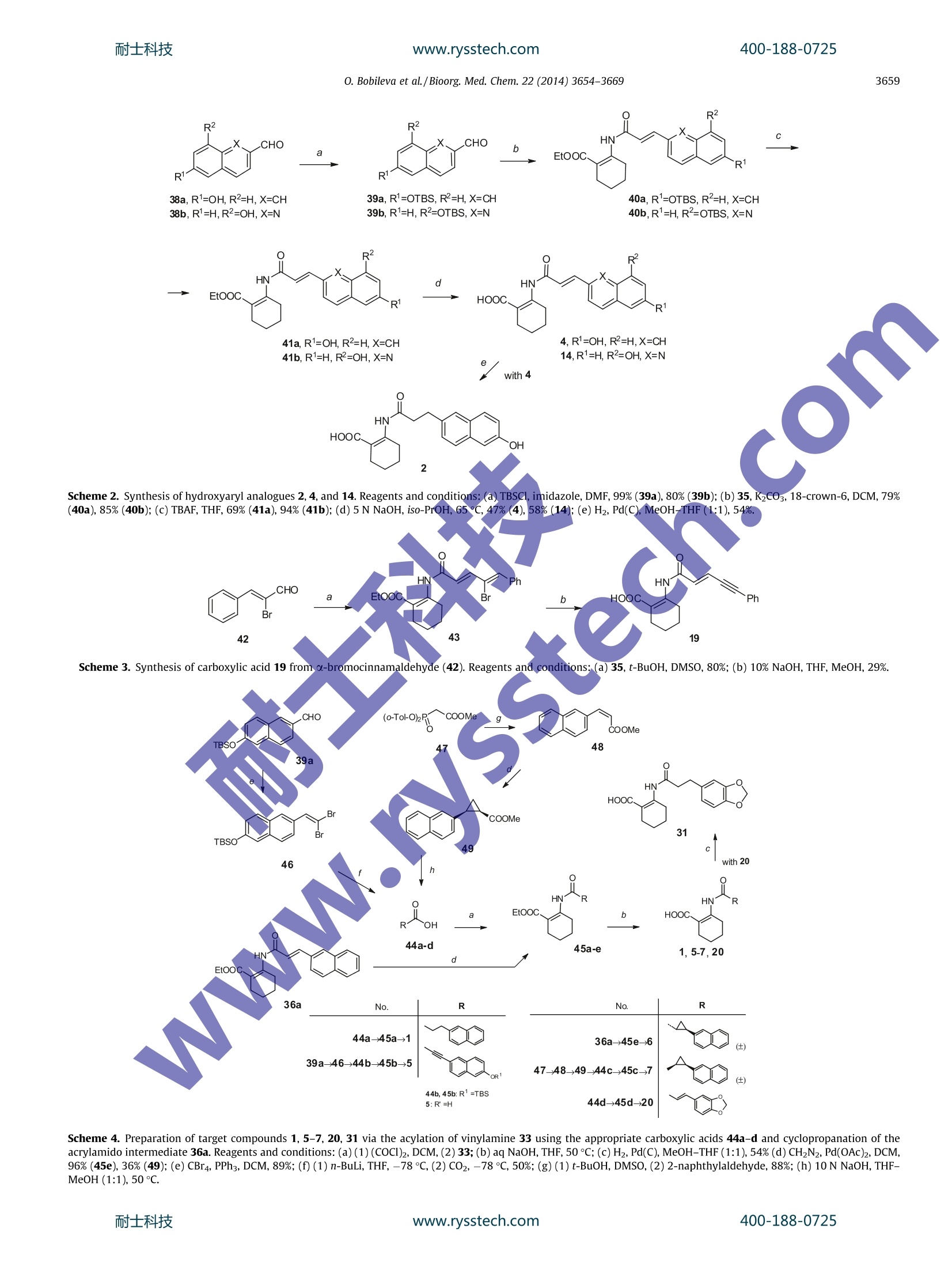
www.rysstech.comBioorganic & Medicinal Chemistry 22 (2014)3654-3669耐士科技400-188-0725 耐士科技400-188-0725www.rysstech.comO. Bobileva et al./Bioorg. Med. Chem. 22 (2014)3654-3669.3655 Bioorganic & Medicinal Chemistryjournal homepage: www.elsevier.com/locate/bmc Synthesis and evaluation of (E)-2-(acrylamido)cyclohex-1-enecarboxylic acid derivatives as HCA1, HCA2, and HCA3 receptoragonists CrossMark Olga Bobileva , Rasma Bokaldere,Vija Gailite, Ilze Kaula , Martins Ikaunieks, Gunars Duburs,Ramona Petrovska, Ilona Mandrika, Janis Klovins, Einars Lozaa* Latvian Institute ofOrganic Synthesis, Aizkraukles 21, Riga LV-1006, Latvia "Biomedical Research and Study Centre, Ratsupites 1, Riga LV-1067, Latvia ABSTRACT 2-(3-(Naphthalen-2-yl)propanamido)cyclohex-1-enecarboxylic acid and its 6-hydroxynaphthalen-2-ylanalogue are well-known hydroxyl-carboxylic acid (HCA) receptor HCA2 agonists. A series of novel arylderivatives of 2-amidocyclohex-1-ene carboxylic acid that contained rigidity elements, such as an E-dou-ble bond, triple bond, and trans or cis-substituted cyclopropane rings, instead of the saturated ethane lin-ker in the amide part of the molecules were designed and synthesized, and the derivatives' potency forthe activation of HCA1, HCA2, and HCA3 receptors by 3'-5'-cyclic adenosine monophosphate (cAMP)assay were evaluated. The SAR studies revealed that the rigidifying of appropriate molecules enabledmodulation of the potency and selectivity of the HCA2 receptor activation. ARTICLEINFO Article history:Received 9 December 2013Revised 15 April 2014Accepted 9 May 2014Available online 17 May 2014 Keywords:GPR109AHCA2NiacinDyslipidemiaAgonistcAMP 2-Acrylamidocyclohex-1-ene 1. Introduction The beneficial role of niacin (nicotinic acid, one of the vitaminBg forms) in the regulation of lipid profiles and its anti-athero-sclerotic properties has been recognized since 1955 due to apublication by Altschul et al. Human clinical trials showed thatniacin treatment decreases the morbidityand mortality ofpatients with coronary diseases and slows the progression ofatherosclerosis.2-4 Niacin reduces free fatty acid (FFA), very low-density lipopro-tein (VLDL) cholesterol, low-density lipoprotein (LDL) cholesterol,triglyceride (TG), and lipoprotein a (Lp-a) levels in plasma, whileeffectively increasing the high-density lipoprotein (HDL) choles-terol in plasma.5-9 ( Abbreviations: FF A, free f atty acid; V LDL, very l o w-density lipoprotein; LDL , low-density l ipoprotein; TG , t riglyceride ; Lp-a, lipoprotei n a; HDL, h igh-density l ipoprotein; cAMP, 3'-5'-cyc l ic adenosine m onophosphate; SAR, structure-activity relationship; GPCR, G-protein c oupled receptor; D IPEA, diisopropylethylamine; P y ,pyridine; TM S , trimethylsilyl; TB S , tert-butyltrimethylsilyl. ) * Correspondingauthor. Tel./fax: +371 67014883. E-mail address: einars@osi.lv (E. Loza). In 2003, several groups identified GPR109A as a high affinityreceptor of the antidyslipidemic drug niacin, which is expressedin adipocytes, keratinocytes and immune cells. The highly relatedGPR109B, which shares 95% identity and is only expressed inhumans and chimpanzees, was also identified as a low affinityreceptor for niacin.10-12 GPR109A and GPR109B, together with GPR81, which shares52% identity with GPR109A, form a G-protein coupled receptor(GPCR) subfamily. Recently, for all these receptors, endogenousligands have been described. Because a common feature of theseligands is that they are hydroxyl-carboxylic acid metabolites,naminggtthe corresponding receptors hydroxyl-carboxylicacid (HCA) Ireceptorswasisproposed. The HCA1 receptor(GPR81) is activated by 2-hydroxy-propionate (lactate), theHCA2 (GPR109A) by 3-hydroxy-butyrate, and the HCA3 receptor(GPR109B) is activated by 3-hydroxy-octanoate. The HCA1,HCA2, and HCA3 receptors are predominantly expressed in adi-pocytes and they mediate antilipolytic effects through couplingto Gi-type G proteins. Moreover, the HCA2 and HCA3 receptorsexpressed in immune cells reduces proinflammatory functionby inhibiting proinflammatory cytokine and chemokineproduction.14 Activation of HCA2 in adipose tissue mediates an antilipolyticresponse by lowering the intracellular 3'-5'-cyclic adenosinemonophosphate (cAMP) levels, leading to reduced protein kinaseA activity, which in turn results in a decrease in lipase activityand a reduction in intracellular TG hydrolysis. Reduction inintracellular lipolysis decreases FFA secretion into plasma and ulti-mately reduces the hepatic FFA available for TG synthesis. It hasbeen postulated that the decreased availability of FFA in the liverdirectly reduces the production of hepatic TG and VLDL.15,16 Themechanism by which niacin increases HDL cholesterol is not fullyunderstood, but it seems that it could affect the apolipoprotein A1levels1’ and/or limit the cholesterol ester transfer proteinmediated exchange of cholesterol from HDL cholesterol to VLDLcholesterol and of TG from VLDL to HDL.18 However, the attractive properties of niacin are spoiled due tothe intense cutaneous flushing that accompanies niacin treatmentat therapeutic concentrations. It is postulated that a possible causeof the flushing could be that HCA2 in epidermal Langerhans cellsmediates the niacin-induced release of prostaglandins D2 andE2.15.19 The activation of the HCA1 and HCA3 receptors possessesthe antilipolytic activity but avoids the flushing side effect ofniacin, suggesting that these receptors could be a new target fordyslipidemia treatment.20.21 Since the discovery of these niacin pharmacologic targets,extensive efforts have been devoted to the search for HCA2receptor agonists as new drugs with improved pharmacologicalproperties, including reduced flushing.22-25 Merck chemists developed tetrahydroanthranilic acid deriva-tives as potent and selective high affinity HCA2 full iagonists.26They demonstrated that a 2-propanamidocyclohex-1-enecarboxy-lic acid scaffold can be used to develop compounds superior to nia-cin for HCA2 activation with more attractive pharmacologicalprofiles, for example, 2-(3-(naphthalen-2-yl)propanamido)cyclo-hex-1-enecarboxylic acid (I) possesses a good pharmacokineticprofile and in vitro activity, and these parameters can be improvedby placing an extra hydroxyl group at the C-6 position of the naph-thalene ring, as in compound II (Fig. 1).26 Based on the 2-propa-namidocyclohex-1-enecarboxylic acidscaffold,appreclinicalcandidate biaryl cyclohexene carboxylic acid MK-6892 (Fig. asa potent and selective high affinity HCA2 receptor full agonist withreduced flushing profiles in animals was developed.The authorsdemonstrated that the biological activity profile of 3-substitutedpropanamidocyclohex-1-enecarboxylic acids can bemodulatedby adding extra substituents on the cyclohexene ringand the saturated side linker.27 Because the incorporation of rigidity elements in the side linkercould help to pre-organize the molecule in a favorable bioactiveconformation, we were interested to see what type of biologicalresponse could exert a change in the saturated and flexible linkerpart of similar molecules to more rigid and constrained linker con-structs, for example, unsaturated bonds or 1,2-cyclopropyl groupsin compounds II (Fig.1). With the aim of testing the possible biological responses, a seriesof 2-amidocyclohex-1-enecarboxylic acid analogues of structure IIIcontaining E-double bonds, triple bond, or 1,2-disubstituted cis-andtrans-cyclopropane cycle were prepared (Table 1). To evaluate the possible agonist properties of the synthesizedcompounds towards human HCA1, HCA2, and HCA3 receptors, for-skolin-stimulated cAMP accumulation assays were arranged. Inthese assays, Flp-In-293 cells, which express the recombinanthuman HCA1,HCA2, and HCA3, were utilized. 2. Chemistry Most compounds in Table 1 were synthesized according to thegeneral strategy shown in Scheme 1. Commercial ethyl 2-oxocyclo-hexanecarboxylate (32) was treated with 7 N ammonia solution inmethanol and converted into ethyl 2-aminocyclohex-1-enecarb-oxylate (33), which was further acylated using 2-bromoacetylbromide to yield 2-bromoacetamido derivative 34. Compound 34was refluxed with triphenylphosphine in benzene to give stable,crystalline triphenylphosphonium bromide 35. The salt 35 wasused for in situ preparation of the corresponding phosphorane,which was condensed with the appropriate aldehydes to give theexpected E-alkenes 36a-p. The ethyl ester groups of compounds36a-p were hydrolyzed to afford the corresponding target carbox-ylic acids 3,8-13,15-18, and 21-26 (Scheme 1 and Table 1). In general, the applied reaction conditions for transformationsdid not change the structure of the aldehyde part R. However, inthe case of 2-chloroquinoline-3-carbaldehyde upon alkaline hydro-lysis of the intermediate ester 36n and in the presence of metha-nol, an exchange of a 2-Cl atom to a methoxy group in thequinoline part was observed, and the 2-methoxy-3-quinolinyl-acrylamide derivative 16 was isolated from the reaction mixture.Only by avoiding methanol upon hydrolysis were we able to pre-pare target compound 17 with the 2-Cl atom intact (Scheme 1). Although the Wittig olefination reaction of compound 35 withaldehydes in allcases formed E-alkenes 36 in overwhelmingexcess, the detectable amounts of the corresponding Z-isomers ofthe ester intermediates 36n, o (E:Z=87:13, based on analysis ofthe H NMR spectra) were observed. After the hydrolysis stepand purification procedures were performed on compounds 36n,o, the target carboxylic acids 17 and 18 were free of the Z isomer. The substituted ethyl acrylamidocyclohex-1-ene carboxylates36j, k,l, o were selectively hydrogenated in a THF-MeOH(1:1) mix-ture in the presence of 10% Pd (C) catalyst into the correspondingethyl propanamidocyclohex-1-ene carboxylates 37a-d. The subse-quent hydrolysis of compounds 37a-d using 10N NaOH-THF-MeOH solutions yielded the expected propanamidocyclohex-1-enecarboxylic acid derivatives 27-30 (Scheme 1 and Table 1). The synthesis sequence of hydroxyaryl acrylamides 4 and 14, incontrast to the general strategy outlined in Scheme 1, includedextra protection and deprotection steps of the hydroxy groups(Scheme 2). Commercial 6-hydroxy-2-naphthaldehyde (38a) and8-hydroxyquinoline-2-carbaldehyde (38b) using TBS-chloride/imidazole treatment were converted into known TBS ethers39a3and 39b, which were condensed using triphenylphospho-nium salt 35 in the presence of K2CO3 to give the expected amido-cyclohex-1-ene carboxylates 40a, b. After the removal of the TBSprotection with tetrabutylammonium fluoride and alkaline hydro-lysis of the ester function of the hydroxy derivatives 41a, b, thetarget carboxylic acids 4 and 14 were obtained. Figure 1. Structures of known HCA2 receptor agonists I, Il, and MK-6892 and envisaged modifications IlI of the linker part.www.rysstech.com 400-188-0725 O. Bobileva et al./Bioorg. Med. Chem. 22 (2014) 3654-3669 Table 1Potency of the synthesized compounds in the HCA1, HCA2, and HCA3 receptor cAMP assay HN^ R HOOC、 0. Bobileva et al./Bioorg. Med. Chem. 22 (2014) 3654-3669 Table 1 (continued) NA-not active up to the 50 pM concentration tested. Each value represents the mean±SD calculated from at least three experiments, each performed in duplicate.Inhibition (%) of forskolin-stimulated intracellular accumulation of cAMP using 50 uM solution of the tested compound. The forskolin-stimulated cAMP levels were set to100%. ° Maximal inhibition (%) of forskolin-stimulated cAMP. Br P*Phs Br The carboxylic acid 4 was converted into propanamido ana-logue 2 (Scheme 2 and Table 1) via the selective, Pd-catalyzedhydrogenation of the linker double bond. The target acrylamide with an extra triple bond 19 wasprepared from a-bromocinnamaldehyde(42) using a similar reac-tion sequence to Scheme 1. The resulting ester 43 was convertedinto 19 via the alkaline hydrolysis conditions, which eliminatedHBr (Scheme 3). Another set of compounds in Table 1 was prepared using astrategy similar to the one used by Raghavan et al.26 which wasbased on acylation reactions of vinylamine 33 (Scheme 4). Theappropriate carboxylic acids 44a-d were converted in situ intothe corresponding chlorides using oxalyl chloride and werecondensed with vinylamine 33 to give the substituted ethylamidocyclohex-1-ene carboxylates 45a-d. Saponification of ethylesters 45a-d using a 10 N NaOH-THF-MeOH solution yieldedcompounds 1, 5, 7, and 20. Carboxylic acids 44a and 44d, which are necessary for thepreparation of target compounds 1 and 20, respectively, wereobtained from commercial sources. Carboxylic acids 44b and 44c, precursors for the synthesis of target compounds 5 and 7,respectively,were prepared as follows: The reaction of the aboveprepared TBS ether 39a with CBr4-PPh3 yielded geminal dib-romoalkene 46. Treatment of alkene 46 with n-BuLi was usedfor the in situ generation of lithium acetylenide, which was fur-ther quenched with solid CO2 to provide the necessary carboxylicacid 44b (Scheme 4). The Z-selective Horner-Emmons reaction of methyl 2-(bis(o-tolyloxy)phosphoryl)acetate47 with 2-naphthaldehyde provided(Z)-methyl acrylate 48(Z:E=100:8). The palladium-mediatedcyclopropanation reaction34 of the (Z)-acrylate 48 with diazometh-ane afforded cis-(±) cyclopropane carboxylate 49, which was freeof the minor E-isomer product after chromatographic purification.Compound 49 was hydrolyzed to the desired carboxylic acid 44c(Scheme 4). The: palladium-mediated(cyclopropanationreaction34of3-naphthyl substituted acrylamidocyclohexene carboxylate 36a(synthesis shown in Scheme 1) with diazomethane yieldedtrans-(±) cyclopropane carboxamido derivative 45e,which washydrolyzed as above to the desired product 7 (Scheme 4). 十 EtOOC, OH Scheme 4. Preparation of target compounds 1, 5-7, 20, 31 via the acylation of vinylamine 33 using the appropriate carboxylic acids 44a-d and cyclopropanation of theacrylamido intermediate 36a. Reagents and conditions: (a)(1)(COCl)2, DCM,(2)33; (b) aq NaOH, THF, 50℃; (c) H2, Pd(C), MeOH-THF(1:1), 54%(d) CH2N2, Pd(OAc)2, DCM,96% (45e), 36%(49);(e) CBr4, PPh3, DCM, 89%; (f)(1) n-BuLi, THF, -78℃,(2) CO2, -78 ℃, 50%;(g)(1) t-BuOH, DMSO,(2) 2-naphthylaldehyde, 88%;(h) 10 N NaOH, THF-MeOH (1:1),50C. The carboxylic acid 20 was converted into propanamido ana-logue 31 via the selective Pd-catalyzed hydrogenation of the linkerdouble bond (Scheme 4 and Table 1). 2.1. Biological activity and structure-activity relationship (SAR) The known structures I (h-H-niacin IC5o=38nM, hGTPySEC5o=690 nM)26 and II(h-H-Niacin IC50=8 nM, hGTPyS EC50=76-nM)29 were used as reference compounds for the comparison in ourstudies. The corresponding EC5o values for (1 in Table 1) and II (2in Table1) in our cAMP assay were 8.3±2.4 and 1.9±0.9 uM, respec-tively. Thus, 6-OH derivative 2 exhibited increased activity in allassays compared to the unsubstituted analogue 1. Merk scientists recognizedthat the activity of anthranilides inboth the binding and functional assays can be greatly reduced inthe presence of 4% human serum. In our work, all of the cAMPassays were performed in the presence of 2% bovine serum albu-min (BSA). We have also performed some additional experimentswhere compounds 3, 5,6, 19, 20, and 30 (Table 1) were preincu-bated with 5% BSA for 30-45 min and then used in the cAMP assayin a buffer with 2%BSA. We did not observe significant differencesin the cAMP level inhibition between the experiments. (E)-2-(3-Substitued acrylamido)cyclohex-1-enecarboxylic acids:Encouraged by the finding that a change of the saturated linkerin reference molecules 1 and 2 to the E-double bond leads toHCA2-active 2-naphthalenyl analogues 3 and 4, brief studies toobtain SARs for diverse (E)-2-(3-substitued acrylamido)cyclohex-1-enecarboxylic acids were undertaken. Limited studies of the impact of the substituents attached to thenaphthalene ring of compound 3 were performed. As it was alreadystated, the presence of a 6-hydroxyl group on the naphthalene ringis beneficial for potency and increases the activation of HCA2 by 4.In contrast,the placement of a methoxy group at the C-6 positionin compound 8 is detrimental to the activation of HCA2 and HCA3,while maintaining weak agonist activity towards HCA1 only (26%cAMP decrease by a 50-uM solution of 8). To increase the diversity of (E)-2-(3-aryl acrylamido)cyclohex-1-enecarboxylic acids, a series with various quinolines at the 3-position of the linker were synthesized and tested for activity(compounds 9-18). Among the prepared 4-quinolinyl (9), 2-quin-olinyl (10), 8-quinolinyl (11), and 6-quinolinyl (12) derivatives,only compound 10 exhibits considerable potency with HCA2(EC50=4.1±1.4 uM), while its activity towards HCA1 and HCA3is negligible. Further attempts to modulate the activity by attach-ing additional functional groups to the quinoline heterocyclerevealed that the prepared molecules are very sensitive to minorchanges in the structure.Thus, 6-quinolinyl compound 12 com-pletely lost its weak activity towards HCA2 (68% cAMP decreaseby a 50-uM solution) due to the addition of an extra methyl groupnext to the quinoline N-atom (compound 13). An extra hydroxylgroup at the C-8 position of the 2-quinolinyl cycle reduced theactivity of compound 10 (EC50=4.1±1.4 uM) to 27% cAMPdecrease by a 50-uM solution of compound 14. 4,5,6-Trimeth-oxy-2-quinolinyl analogue 15 possesses weak activation of allthree receptors HCA1, HCA2, and HCA3. We had starting materialsfor the synthesis of 3-quinolinyl derivatives with extra substitu-ents only. Thus, the weak activity of 2-methoxy-3-quinolinyl deriv-ative 16 (59% cAMP decrease by a 50-uM solution) is improved byplacing a chlorine atom instead of the methoxy group at the C-2position(compound 17, EC50=27.4±3.5uM) and even betterpotency is achieved in the case of 2-chloro-6-methoxy-3-quinoli-nyl analogue 18 (EC50=6.3±4.1(uM). Interestingly, (E)-2-acrylamidocyclohex-1-enecarboxylic acidderivative 19 containing a phenylethynyl group attached to the E-double bond exhibitscomparable activation of HCA2 (EC50=3.6±1.6 uM) to bicyclic 2-napthalenyl analogue 3 (EC50=4.5±1.2 uM). Benzo[1,3]dioxole analogue 20 exhibits a strong preference forHCA2 activation (EC50=5.8±4.2 uM). A change in the methylenebridge of the adjoining oxa groups in compound 20 to ethyleneproduces the equally potent compound 21 (EC50=6.8±3.7 uM);however, the activity sharply drops upon addition of extra chlorineor bromine atoms at the 6-position of the benzole ring (compounds24 and 25) or disconnecting the bridge, as in the case of 3,4-dime-thoxyy derivative 22 (51% cAMP decrease by a 50-uM solution).Although there is some increase in the activity upon addition ofan extra chlorine atom at the C-2 position of 3,4-dimethoxyphenol(compound 23, EC50=26.3±4.5uM),a bromine atom substitutedat the C-5 position is not beneficial (compound 26). Because we are interested in the activity of 2-naphthenylsubstituted propanamido-cyclohexene, carboxylic acids 1 and 2can be favorably modified in terms of activation and selectivitytowards HCA2 by replacing the saturated propanamido group ofthe molecules with an (E)-acrylamido linker; in this way, anattempt to evaluate the universality of this transformation wasperformed. Thus, (E)-2-acrylamido cyclohexene carboxylic acids11-13 and 20 were selectively hydrogenated into the correspond-ing saturated propanamido derivatives 27-31, respectively. In thecase of the compound pair 20-31, the ability to activate HCA2 sig-nificantly decreased for the saturated structure 31. The activities ofthe pairs 18→30 and 12-28 do not change, and the initially inac-tive compounds were transformed to weakly or moderately activecompounds by hydrogenation in the case of the pairs 11→27 and13→29. 3. Conclusions We designed and synthesized a series of novel aryl derivativesof 2-amidocyclohex-1-ene carboxylic acids, which contained rigid-ity elements of E-double bonds, triple bonds, and trans- orcis-substituted cyclopropane rings in the amide part of the mole-cules that connects the cyclohexene and aromatic moieties. Also, we evaluated their potency for the activation of HCA1, HCA2 andHCA3 receptors using a cAMP assay. The SAR studies revealed thatin the case of naphthalen-2-yl derivatives, the potency andselectivity towards HCA2 receptors increases in the order: cis-(±)-cyclopropane(7)<
确定















还剩14页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中主要物质含量分析检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多