








方案详情
文
Novel C-aryl glucoside SGLT2 inhibitors containing 1,3,4-thiadiazole moieties were designed and synthesized. Among the compounds tested, biaryl-type compounds containing pyrazine 59, 2-furan 61, and 3-thiophene 71 showed the best in vitro inhibitory activities to date (IC50 = 3.51–7.03 nM) against SGLT2. A
selected compound 61, demonstrated reasonable blood glucose-lowering effects, indicating that the information obtained from the SAR studies in this 1,3,4-thiadiazolylmethylphenyl glucoside series might help to design more active SGLT2 inhibitors that are structurally related.
方案详情

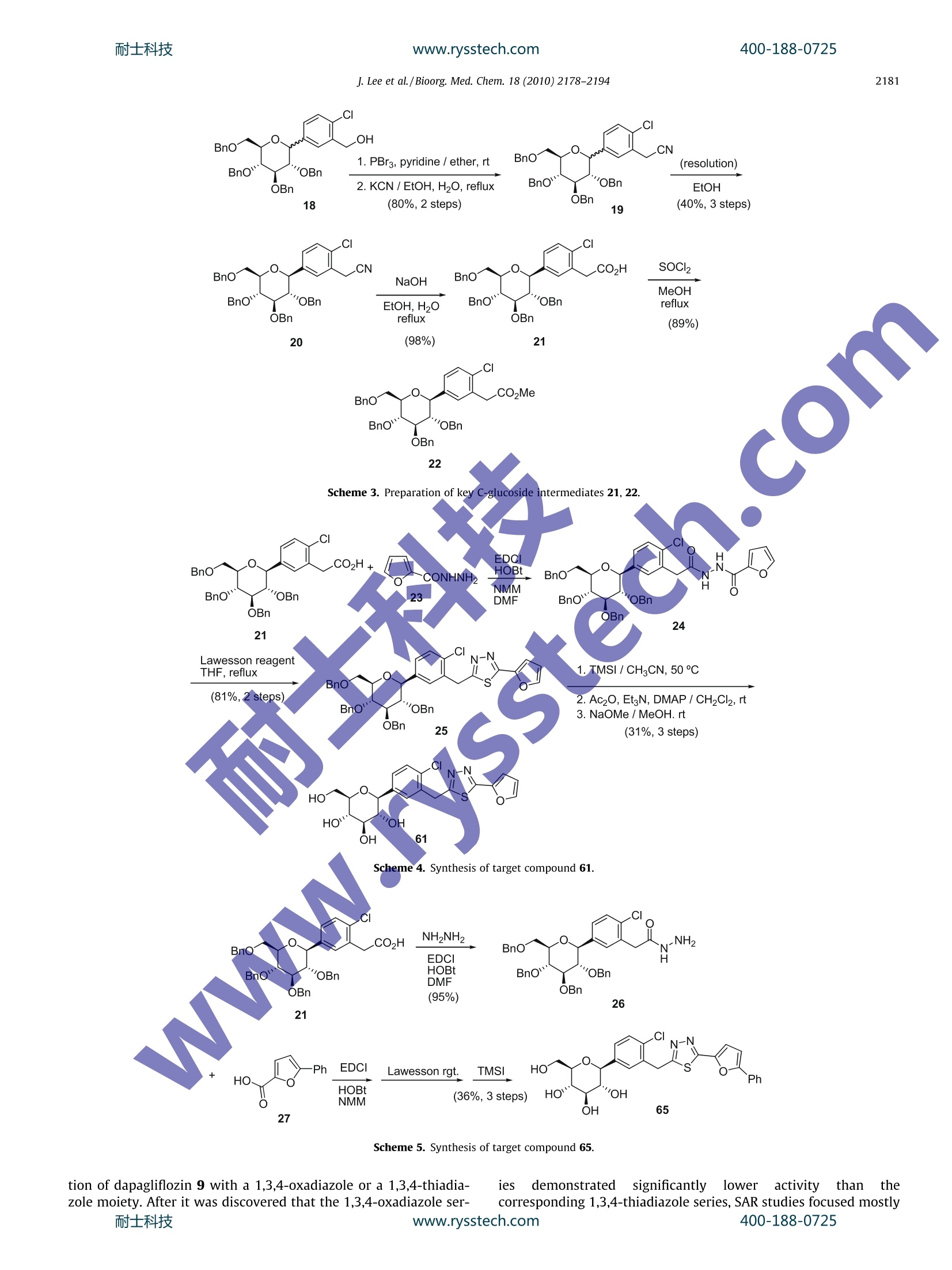
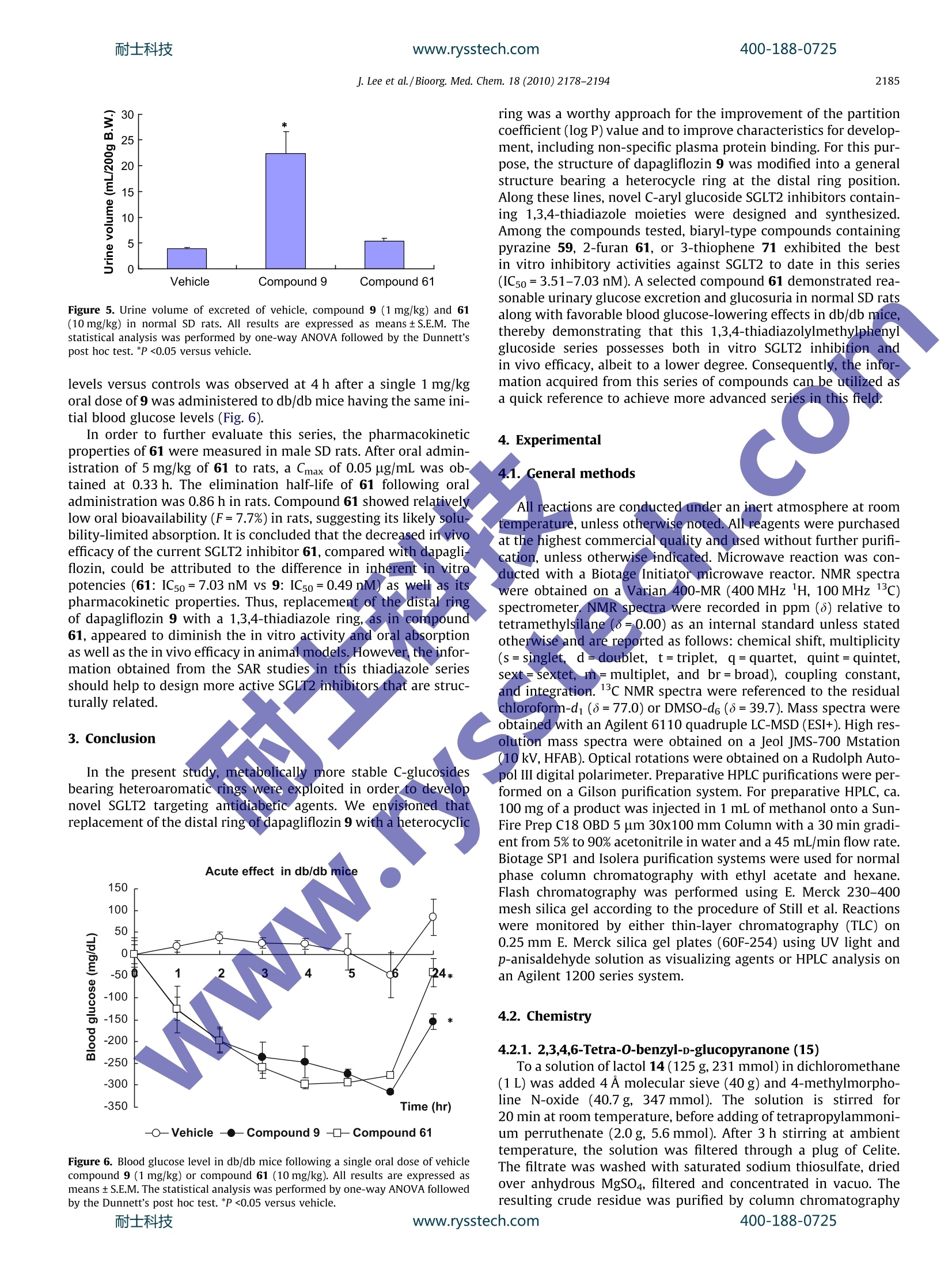
www.rysstech.comBioorganic & Medicinal Chemistry 18 (2010)2178-2194耐士科技400-188-0725 耐士科技400-188-0725www.rysstech.com2179 Contents lists available at ScienceDirect Bioorganic & Medicinal Chemistry ELSEVI ER journal homepage: www.elsevier.com/locate/bmc Novel C-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents:1,3,4-Thiadiazolylmethylphenyl glucoside congeners Junwon Lee, Sung-Han Lee, Hee Jeong Seo, Eun-Jung Son, Suk Ho Lee, Myung Eun Jung, MinWoo Lee, Ho-Kyun Han, Jeongmin Kim,Jahyo Kang tb.*, Jinhwa Lee.* Central Research Laboratories, Green Cross Corporation, 303 Bojeong-Dong, Giheung-Gu, Yongin, Gyeonggi-Do 446-770, Republic of KoreaDepartment of Chemistry, Sogang University, 1 Shinsu-Dong, Mapo-Gu, Seoul 121-742, Republic of Korea ARTICLE IN F O ABSTRAC T Article history:Received 14 December 2009Revised 22 January 2010Accepted 23 January 2010Available online 4 February 2010 > 1. Introduction Diabetes has become an increasing concern to the world’s popp-ulation. In 2007, approximately 246 million people were consid-ered diabetic, with an additional 7 million people diagnosed withthe disease every year. Diabetes is a chronic metabolic disorderthat is defined by the body's inability to generate insulin or to re-spond adequately to circulating insulin. There are two identified,forms of diabetes: type 1 diabetes is distinguished as an autoim-mune disease involving pancreatic β-cells, while type 2e diabetesis defined by B-cell dysfunction and insulin resistance. Type 2 dia-betes is the most common disorder of glucose hhomeostasis.accounting for nearly 90-95% of all cases of diabetes. Sodium-dependent glucose cotransporters (SGLTs) couple thetransport of glucose against a concentration gradient with thesimultaneous transport of Naatdcown a concentration gradient.'Two important SGLT isoforms have been cloned and identified asSGLT1 and SGLT2.4 SGLT1 is located in the gut, kidney, and heartwhere its expression regulates cardiac glucose transport. SGLT1is a high-affinity,low-capacity transporter and therefore accountsfor only a small fraction of renal glucose reabsorption. In contrast,sGLT2 is a low-affinity, l, high-capacity transporter located exclu- ( * C orresponding authors. Tel.: +82 3 1 260 9892; fax: +8 2 31 260 9020 (J.L . ), tel.: +82 2 705 8439; fax:+ 8 2 2 701 0967 (J.K.). ) ( E-mail addresses: k angj@sogang.ac.kr (J. Kang), jinh w alee@greencross.com ( J . Lee). ) sively at the apical domain of the epithelial cells in the early prox-imal convoluted tubule. It is estimated that 90% of renal glucosereabsorption is facilitated by SGLT2; the remaining 10% is likelymediated by SGLT1 in the late proximal straight tubule. SinceSGLT2 appears to be responsible for the majority of renal glucosereabsorption based on human mutation studies, it has become atarget of therapeutic interest. Phlorizin 1, which was isolated formthe root bark of the apple tree, demonstrated antidiabetic activity,lowering plasma glucose and improving insulin resistance byincreasing renal glucose excretion. However, phlorizin was notdeveloped as a drug for the treatment of diabetes because the com-pound was found to be metabolically unstable.This instability isdue to B-glucosidase cleavage in the intestinal tract and preventedoral administration (Fig. 1). In addition, enzymatic release of theaglycone phloretin 2, a micromolar inhibitor of sodium-indepen-dent facilitative glucose transporters (GLUTs), could potentially in-hibit GLUT-mediated cellular uptake of glucose.915 Subsequently, T-1095 3, by Tanabe Seiyaku, was reported as thefirst orally absorbable SGLT2 inhibitor, overcoming the disadvan-tage of phlorizin 1 (Fig. 2).10 3 was absorbed in the intestine andconverted to an active form, T-1095A 4. Following the discoveryof 3, O-glucosides such as sergliflozin 5 and remogliflozin 7 ad-vanced furthest in clinical trials. Compound 5 was disclosed by Kis-sei in 2003 as a potential treatment for diabetes and obesity. In2007, Kissei, in collaboration with GlaxoSmithKline, initiated thedevelopment of 7, a pyrazole-based O-glucoside.2Again, concernregarding gut β-glucosidase-mediated degradation, resulted in J. Lee et al./Bioorg. Med. Chem. 18 (2010)2178-2194 Figure 1. Structure and β-glucosidase-mediated cleavage of phlorizin (1). developing sergliflozin A 6 and remogliflozin A 8 being adminis-tered as the ethyl carbonate prodrugs 5 and 7, respectively. Subsequent endeavors to identify SGLT2 inhibitors suitable fororal administration without the need for a prodrug led to the dis-covery of C-glucoside-derived SGLT2 inhibitors. C-glucosides ap-pear to have drug-like properties withenhanced chemicalstability of the glucosidic bond. Extensive SAR studies by Bristol-Myers Squibb identified dapagliflozin 9, a potent, selective SGLT2inhibitor for the treatment of type 2 diabetes.13-15 At present,dapagliflozin is the most advanced SGLT2 inhibitor in clinical trialsand is believed to be the first SGLT2 inhibitor to go to market. Onthe other hand, Mitsubishi Tanabe Pharma, in collaboration withJohnson & Johnson, is developing canagliflozin 10, another novelC-glucoside-derived SGLT2 inhibitor. In August 2009, a phase 3study was reportedly initiated to evaluate the safety and efficacyof 10 in patients with type 2 diabetes. In the present study, metabolically more stable C-glucosidesbearing a heteroaromatic ring were exploited in order to developnovel SGLT2 targeting antidiabetic agents. We envisioned thatreplacement of the distal ring of dapagliflozin 9 with a heterocyclicring would be a worthy approach for the improvement of the par-tition coefficient (log P) value, to potentially decrease plasma pro-tein binding. For this purpose, the structure of dapagliflozin 9 wasmodified into 11, bearing a heterocyclic ring as shown in Figure 3.Among a variety of heterocycles, we decided to screen thiadiazole Figure 2. Structures of SGLT2 inhibitors. 13 as our initial efforts. Herein, we report the design, synthesis andbiological evaluation of 1,3,4-thiadiazolylmethylphenyl glucosidecongeners. 2. Results and discussion 2.1. Chemistry Synthesis of the target compounds commenced with the prepa-ration of the lactone 15. Commercially available perbenzylatedgluconolactol 1420 was treated with TPAP (tetrapropylammoniumperruthenate) in the presence of NMO (4-methylmorpholine N-oxide) to provide the requisite perbenzylated gluconolactone 15in high yield as shown in Scheme 1. As shown in Scheme 2, the reduction of commercially available5-bromo-2-chlorobenzoic acid (16) with a borane-dimethyl sulfidecomplex, and subsequent silylation of the corresponding alcoholwith TIPSCl(triisopropylsilyl chloride) in the presence of imidazoleand DMAP (4-(dimethylamino)pyridine) generated bromide 17 in95% yield over two steps. Lithium-halogen exchange, followed bythe addition of the nascent lithiated aromatic compound to perb-enzylated gluconolactone 15, produced a mixture of the corre-sponding lactols. The lactols were reduced using triethylsilaneand BF3 etherate, desilylated and afforded alcohol 18 in 98% yieldover the three steps. J. Lee et al./Bioorg. Med. Chem. 18 (2010)2178-2194 Figure 3. Exploration of C-glucoside bearing heteroaryl group at the distal ring. Scheme 1. Oxidation of lactol 14 to lactone 15. The preparation of a key intermediate 21 is described inScheme 3. Thus, alcohol 18 was converted to bromide using PBr3in the presence of pyridine. The bromide was then treated witlKCN in refluxing aqueous EtOH to generate cyanide 19 in 80%yieldover two steps. A mixture of the two isomers 19 was separatedthrough recrystallization from ethanol to produce the requiredbeta-isomer 20 in about 40% yield. Hydrolysis of 20 with sodiumhydroxide in aqueous ethanol generated the carboxylic acid 21 inquantitative yield. Treatment of 21 with thionyl chloride in refluxingmethanol produced the corresponding methylester 22 in 89% yield. A thiazole-containing C-glucoside 61 was synthesized as shownin Scheme 4. Coupling of acid 21 and a hydrazide, such as furan-2-carbohydrazide (23), using EDCI (1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide), HOBt (1-hydroxybenzotriazole), and NMM(4-methylmorpholine) in the presence of DMF (N,N-dimethylfora-mide) yielded the corresponding acylhydrazide 24, which was cy-clized using Lawesson's reagent to generate thiadiazole 25 in 81%over two steps. Deprotection of the four benzyl groups using TMSI(trimethyliodosilane) followed by peracetylation using aceticanhydride and purification by either column chromatography orrecrystallization generated the corresponding tetraacetate. The tet-raacetate was hydrolyzed with NaOMe in methanol to yield thetarget compound 61 in 31% yield over three steps. Alternatively, another target compound was prepared as shownin Scheme 5.Thus, the acid 21 was converted to the hydrazide 26 J. Lee et al./ Bioorg. Med. Chem. 18 (2010)2178-2194 .CL CI mo22 c. 27 Scheme 5. Synthesis of target compound 65. tion of dapagliflozin 9 with a 1,3,4-oxadiazole or a 1,3,4-thiadia- ies demonstrated significantly lower activity than thezole moiety. After it was discovered that the 1,3,4-oxadiazole ser- corresponding 1,3,4-thiadiazole series, SAR studies focused mostly耐士科技 www.rysstech.com 400-188-0725 J. Lee et al./Bioorg. Med. Chem. 18 (2010)2178-2194 Scheme 6. Synthesis of target compound 47. Scheme 7. Synthesis of target compound 59. Table 2 shows the structure-activity relationship upon alter-ation of the substituent at the distal 1,3,4-thiadiazole ring employ-ing only the s-anomer.Compounds bearing aliphatic chains at thedistal 1,3,4-thiadiazole ring were prepared and tested. These com-pounds showed only moderate hSGLT2 inhibitory activities (42-45, IC50=122-379nM). Both trifluoromethyl substituted 48 andtrifluoroethyl substituted 49 did not improve in vitro activityagainst hSGLT2. Substitution with a carbocycle, such as cyclopentyl50, demonstrated suboptimal activity (IC5o=445 nM). However, asdescribed in the initial SAR studies (Table 1), the diaryl-type com-pound containing a phenyl group, 51, showed good in vitro inhib-itory activity against hSGLT2 (IC50=16.6 nM). At this juncture, avariety of different aromatic rings were explored. Five-memberedheterocycles, including 2-furan 61, 3-furan 62, 2-thiophene 70,and 3-thiophene 71, were shown to be highly active, demonstrat-ing low nanomolar inhibitory activity against hSGLT2. Compared tophenyl 51 (IC5o=16.6nM), the incorporation of 3-furan 62 or 2-thiophene 70 resulted in similar levels of inhibitory activity (62,IC5o=17.7 nM; 70, IC5o=14.4 nM). Surprisingly, varying the bond-ing position, for example, 3-furan 62 to 2-furan 61 or 2-thiophene Compound X R IC50(nM) 6.25±0.62 0.49±0.04 31 H 6910 32 H >10,000 33 H 1420 34 H 1230 35 C1 542 36 Cl 103 37 Cl 24.9 38 Cl 45.3 39 Cl 53.0 40 Cl 2390 41 Cl >10,000 CI The ICso values of 6 and 9 were obtained by in-house assay. These data were obtained by single determinations. Table 2In vitro screening data for hSGLT inhibitory activityab CI -N -R HO HO 'OH Compound R IC5o(nM) Compound R ICso (nM) 6 6.25±0.62 60 190 9 0.49±0.04 61 7.03 42 214 62 17.7 43 379 63 106 44 353 64 58.3 45 10.6 46 OMe 249 47 KSMe N-O 257 48 119 49 3280 50 70 `S 14.4 51 16.6 71 S 3.51 52 126 72 762 53 30.3 73 C S 712 54 638 74 -CI 18.2 55 N 97.4 75 S 44.5 (continued on next page) Table 2 (continued) Compound R IC50(nM) Compound R IC50(nM) 56 人 36.4 76 N= S 21.8 N N 57 81.6 77 72.6 S N 58 562 78 >10,000 59 4.06 79 >110,000 These data were obtained by single determinations. 70 to 3-thiophene 71, improved the inhibitory activity by approx-imately 2.5-fold or fourfold, respectively (61, IC50=7.03 nM; 71,IC5o=3.51 nM). Substitution at the C5 position of 2-furan 61 or2-thiophene 70 with methyl 64, phenyl 65, or chloride 74 resultedin a decreased potency. Moreover, addition of substituents at C3 of2-furan 61 or 2-thiophene 70 resulted in significantly lowerhSGLT2 inhibitory activities (63, 72, and 73), likely suggesting thateven slightly increased steric hindrance is not tolerated in the re-gion. Other heteroaryl groups such as pyrrole (68,IC50=119nM)benzofuran (66, IC5o=249 nM), quinoline (58, IC50=562 nM), ancidpyridazine (60, IC50=190 nM) were not tolerated. Meanwhile, pyr-idine 53, isoquinoline 57, benzothiophene 75, and thiazoles 76 and77were found to be somewhat tolerableereplacements(IC50=21.8-81.6nM), but none appeared to be more potent thanthe simple phenyl group. Interestingly, six-membered heteroarylcompounds with two para heteroatoms, such! as pyrazine 59,proved to have highly potent inhibitory activity against hSGLT2(IC50=4.06nM). Thus, the results indicate that the addition ofsmall heteroaryl rings with a favorabIe orientation of the hetero-atom at the distal ring might be able to further increase hSGLT2inhibitory activity. It is noteworthy that any substitution at the arylor heteroaryl ring connected with the distal thiadiazole ring re-sulted in products with significantly lower hSGLT2 inhibitory activ-ities, except furanophenyl 65 (IC50=10.6nM). Di-substitution atthe heteroaryl ring on the distal thiadiazole moiety further deteri-orated hSGLT2 inhibitory activities, as shown for compounds 78and 79, indicating very limited tolerance in thespecific area.Among the tested compounds, as shown in Table 2, biaryl-typecompounds containing 3-thiophene 71, pyrazine 59, or 2-furan61 showed the best in vitro inhibitory activities (IC50=3.51 nM,4.06 nM, 7.03 nM, respectively). This implies that the 1,3,4-thiadi-azole heterocycle at the distal ring could become a reasonable sur-rogate for the phenyl group of dapagliflozin 9, and a properlyoriented small heteroaryl substituent at the distal ring might becapable of augmenting in vitro hSGLT2 inhibitory activity. A selected promising compound 61 was tested on animal modelsfor in vivo efficacy. Urinary glucose excretion was evaluated in nor-mal Sprague-Dawley (SD) rats.24-26 Male SD rats weighing 250-300 g (7-8 weeks old) were purchased from Orient-Bio LaboratoryAnimal Research Center Co. (Gyeonggi-do, Korea). They werehoused in a temperature (25±2℃) and moisture (55±10%) con-trolled room, exposed to a controlled 12 h cycle of light and dark-ness, and allowed free access to food and water. All animals wereacclimated for one week prior to the experiment. For glucosuriaassessment, overnight-fasted SD rats were placed into metabolism cages for baseline urine collection over 24 h. Rats were weighed,randomized into three groups (n=3), dosed orally with single dosesofvehicle or drug (9@1 mg/kg, 61@10 mg/kg), and subsequentlydosed orally with 50% aqueous glucose solution (2 g/kg). Immedi-ately after dosing, rats were returned to the metabolism cages for24 h urine collection and re-fed at 1 h after the glucose challenge.The urine glucose and urine volume data were normalized per200 g of body weight. As shown in Figure 4, a single oral dose ofthiadiazole 61 increased urinary glucose excretion in normal SDfats, resulting in a 100-fold elevation in glucose disposal relativeto vehicle controls. Urinary glucose excretion of compounds 9 and61 were 1648±228 mg/200 g body weight and 195±84 mg/200 gbody weight, respectively. Urine volume excreted in normal SD ratsare also shown in Figure 5. Dapagliflozin 9 caused increased urinevolume over vehicle by 5.7-fold,while thiadiazole 61 increased ur-ine volume merely 1.4-fold (vehicle: 3.9±0.23,9:22.36±4.18,61:5.33±0.67ml/200 mg body weight, respectively). For assessment of acute blood glucose effects in diabetic db/dbmice, the animals were weighed and randomized into two groups(n=4). Mice were dosed with vehicle or drug (9@1 mg/kg,61@10 mg/kg), and blood glucose levels were measured immedi-ately before dosing and at 1, 2, 4, 5, 6, and 24 h post-dose. The ani-mals were allowed to re-feed after the 6-h time point. In thisexperiment, a 72.8% reduction in blood glucose levels versus con-trols were observed at 4 h after a single 10 mg/kg oral dose of 61was administered to db/db mice with an initial blood glucose levelof 430 mg/dL. By comparison, a 61.3% reduction in blood glucose Figure 4. Urinary glucose excretion test of vehicle, compound 9(1 mg/kg) and 61(10 mg/kg) in normal SD rats. All results are expressed as means±S.E.M. Thestatistical analysis was performed by one-way ANOVA followed by the Dunnett’spost hoc test. *P<0.05 versus vehicle. Figure 5. Urine volume of excreted of vehicle, compound 9 (1 mg/kg) and 61(10 mg/kg) in normal SD rats. All results are expressed as means±S.E.M. Thestatistical analysis was performed by one-way ANOVA followed by the Dunnett'spost hoc test. *P <0.05 versus vehicle. ring was a worthy approach for the improvement of the partitioncoefficient (log P) value and to improve characteristics for develop-ment, including non-specific plasma protein binding. For this pur-pose, the structure of dapagliflozin 9 was modified into a generalstructure bearing a heterocycle ring at the distal ring position.Along these lines, novel C-aryl glucoside SGLT2 inhibitors contain-ing 1,3,4-thiadiazole moieties were designed and synthesized.Among the compounds tested, biaryl-type compounds containingpyrazine 59, 2-furan 61, or 3-thiophene 71 exhibited the bestin vitro inhibitory activities against SGLT2 to date in this series(IC50=3.51-7.03nM). A selected compound 61 demonstrated rea-sonable urinary glucose excretion and glucosuria in normal SD ratsalong with favorable blood glucose-lowering effects in db/db mice,thereby demonstrating that this 1,3,4-thiadiazolylmethylphenylglucoside series possesses both in vitro SGLT2 inhibition andin vivo efficacy, albeit to a lower degree. Consequently, the infor-mation acquired from this series of compounds can be utilized asa quick reference to achieve more advanced series in this field. levels versus controls was observed at 4 h after a single 1 mg/kgoral dose of 9 was administered to db/db mice having the same ini-tial blood glucose levels (Fig. 6). 3. Conclusion In the present study, metabolically more stable C-glucosidesbearing heteroaromatic rings were exploited in order to developnovel SGLT2 targeting antidiabetic agents. We envisioned thatreplacement of the distal ring of dapagliflozin 9 with a heterocyclic -o-Vehicle --Compound 9 --Compound 61 Figure 6. Blood glucose level in db/db mice following a single oral dose of vehiclecompound 9 (1 mg/kg) or compound 61 (10 mg/kg). All results are expressed asmeans ±S.E.M. The statistical analysis was performed by one-way ANOVA followedby the Dunnett's post hoc test. *P <0.05 versus vehicle. 4.1. General methods All reactions are conducted under an inert atmosphere at roomtemperature, unless otherwise noted. All reagents were purchasedat the highest commercial quality and used without further purifi-cation, unless otherwise indicated. Microwave reaction was con-ducted with a Biotage Initiator microwave reactor. NMR spectrawere obtained on a Varian 400-MR (400 MHz 1H, 100 MHz 13C)spectrometer. NMR spectra were recorded in ppm (8) relative totetramethylsilane (8=0.00) as an internal standard unless statedotherwise and are reported as follows: chemical shift, multiplicity(s= singlet, (d= doublet,t=triplet, q=quartet, quint=quintet,sext= sextet, m=multiplet, and br=broad), coupling constant,and integration.13c NMR spectra were referenced to the residualchloroform-d1(8=77.0) or DMSO-d6(8=39.7). Mass spectra wereobtained with an Agilent 6110 quadruple LC-MSD (ESI+). High res-olution mass spectra were obtained on a Jeol JMS-700 Mstation(10kV, HFAB). Optical rotations were obtained on a Rudolph Auto-pol III digital polarimeter. Preparative HPLC purifications were per-formed on a Gilson purification system. For preparative HPLC, ca.100 mg of a product was injected in 1 mL of methanol onto a Sun-Fire Prep C18 OBD 5 pm 30x100 mm Column with a 30 min gradi-ent from 5% to 90% acetonitrile in water and a 45 mL/min flow rate.Biotage SP1 and Isolera purification systems were used for normalphase column chromatography with ethyl acetate and hexane.Flash chromatography was performed using E. Merck 230-400mesh silica gel according to the procedure of Still et al. Reactionswere monitored by either thin-layer chromatography (TLC) on0.25 mm E. Merck silica gel plates (60F-254) using UV light andp-anisaldehyde solution as visualizing agents or HPLC analysis onan Agilent 1200 series system. 4.2. Chemistry 4.2.1. 2,3,4,6-Tetra-O-benzyl-D-glucopyranone (15) To a solution of lactol 14 (125 g, 231 mmol) in dichloromethane(1L) was added 4 A molecular sieve (40 g) and 4-methylmorpho-line N-oxide (40.7 g, 347 mmol). The solution is stirred for20 min at room temperature, before adding of tetrapropylammoni-um perruthenate (2.0 g, 5.6 mmol). After 3 h stirring at ambienttemperature, the solution was filtered through a plug of Celite.The filtrate was washed with saturated sodium thiosulfate, driedover anhydrous MgSO4, filtered and concentrated in vacuo. Theresulting crude residue was purified by column chromatography (5-40%EtOAc/hexanes) to yield the title compound (106 g, 86%) asa colorless oil. H NMR (400 MHz, CDCl3) 8 7.39-7.16 (m, 20H),4.98 (d,J=11.2 Hz, 1H), 4.74-4.44 (m, 8H), 4.12 (d,J=6.4Hz,1H), 3.97-3.89(m, 2H), 3.72 (dd,J=10.8, 2.4Hz, 1H), 3.67 (dd,J=10.8, 3.2 Hz, 1H). 4.2.2. (5-Bromo-2-chlorobenzyloxy)triisopropylsilane (17) Toaa solutioni5-bromo-2-chlorobenzoic acid 16 (100g,425 mmol) in THF (500 mL) at 0℃ was added borane-dimethylsulfide complex (170 mL, 1,700 mmol). The resuting mixture wasstirred with gradual warming to ambient temperature over 12 h,re-cooled to 0℃, and quenched with MeOH and then water. Themixture was extracted with EtOAc. The organic layer was washedwith brine, dried over anhydrous MgSO4, filtered and evaporatedin vacuo to yield a white solid, which was carried on to the nextstep without further purification. To a solution of the benzyl alcohol in DMF(400 mL) was addedimidazle (57.9g, 850 mmol),4-(dimethylamino)pyridine (2.6g,21 mmol), and triisopropylsilyl chloride (136 mL, 683 mmol). Theresulting solution was stirred at ambient temperature for 12 h, di-luted with a saturated ammonium chloride and extracted withEtOAc. The organic layer was washed with water then brine, driedover anhydrous MgSO4, filtered and concentrated in vacuo. Thecrude residue was purified by column chromatography (3-10%EtOAc/hexanes) to yield the title compound (152 g,95%) as a color-less oil. 1H NMR (400 MHz, CDCl3) 8 7.77 (d,J=2.4 Hz, 1H), 7.31(dd,J=8.4, 2.4 Hz, 1H), 7.16 (d,J=8.4Hz, 1H), 4.83 (s, 2H),1.25-1.17(m, 3H), 1.11 (d,J=6.8 Hz, 18H). To a stirred -50°C solution of the lactols in dichloromethane(500 mL) was added triethylsilane (63 mL, 396mmol) followedby boron trifluoride diethyl etherate (50 mL, 396 mmol) at a ratesuch that the reaction temperature was maintained between -40and -50°C. The solution was allowed to warm to -10C over2 h prior to quenching with saturated potassium carbonate. Afterremoval of organic volatiles under reduced pressure, the residuewas partitioned between EtOAc and water. Following extractionof the aqueous layer with ethyl acetate, the combined organic lay-ers were washed with water prior to drying over anhydrousMgSO4. Filtration and concentration under reduced pressureyielded ayellow oil, which was carried on to the next step withoutfurther purification. To a solution of the silyl ether in THF (500 mL) was added tet-rabutylammonium fluoride (1.0M in THF, 594mL, 594 mmol)and the reaction mixture stirred at ambient temperature for 2 h.After removal of organic volatiles under reduced pressure, the res-idue was partitioned between EtOAc and saturated ammoniumchloride. The organic layer was separated and the aqueous layerwas extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered and con-centrated in vacuo. The crude residue was purified on columnchromatography (10-60% EtOAc/hexanes) to yield the title com-pound (130g, 98%, ca. 2:1 :a) as a white solid. H NMR(400MHz, CDCl) B-anomer: 8 7.50 (s, 1H), 7.38-7.17 (m, 20H),6.94-6.92 (m, 2H), 4.96-4.46 (m, 9H), 4.23 (d, J=9.2 Hz, 1H),3.86(d,J=10.4Hz, 1H), 3.81-3.67(m, 4H), 3.61-3.59 (m, 1H),3.44 (t,J=9.2Hz, 1H); a-anomer: 8 7.78 (s, 1H), 7.63 (dd,J=8.4,1.6Hz, 1H), 7.38-7.17 (m, 19H), 7.13-7.10 (m, 2H), 5.15 (d,J=3.6 Hz, 1H), 4.96-4.46 (m, 8H), 4.01-3.95 (m, 2H), 3.83-3.64(m, 5H), 3.55-3.51 (m, 1H); MS (ESI) m/z 682 (M+NH4), 687(M+Na). 4.2.4. 2-(2-Chloro-5-((3S,4R,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2-yl)phenyl)acetonitrile (19) To a solution of 2-chlorobenzyl alcohol 18 (130 g, 195 mmol) inether (600 mL) at 0 ℃ was added pyridine (0.79 mL, 9.8 mmol) andphosphorus tribromide (6.4 mL, 68 mmol). The reaction was al-lowed to slowly warm to room temperature over 15 h and refluxedfor 1 h. After cooling to room temperature, the reaction mixturewas diluted with EtOAc and washed with water then brine. The or-ganic extract was dried over anhydrous MgSO4, filtered, and evap-orated in vacuo to yield a yellow solid, which was carried on to theext step without further purification. To a solution of the 2-chlorobenzyl bromide in ethanol(260 mL)and water (130mL) was addeddpotassium cyanide (31.8g,488 mmol). The reaction mixture was refluxed overnight. Aftercooling to room temperature, the mixture was diluted with waterand extracted with EtOAc. The organic layer was washed withbrine,dried over anhydrous MgSO4, filtered and evaporated in va-cuo. Purification was accomplished by chromatography to give thetitle compound (105 g, 80%, ca. 2:1 p:o) as a colorless oil. H NMR(400 MHz, CDCl3) B-anomer: 8 7.48 (s, 1H), 7.37-7.17(m, 20H),6.93-6.91 (m, 2H), 4.93 (s, 2H), 4.86 (d,J=10.8 Hz, 1H), 4.63 (d,J=10.8 Hz, 1H), 4.62 (d,J=12.4 Hz, 1H), 4.56 (d,J=12.4Hz, 1H),4.52 (d,J=10.8Hz, 1H), 4.22 (d, J=9.6Hz, 1H), 3.93 (d,J=10.8 Hz, 1H), 3.84-3.68 (m, 6H), 3.62-3.58(m, 1H), 3.43 (t,J=8.8 Hz, 1H); a-anomer: 8 7.83 (d,J=1.6Hz, 1H), 7.64 (dd,J=8.4, 1.6 Hz, 1H), 7.37-7.19 (m, 19H), 7.13-7.10 (m, 2H), 5.11(d, J=2.8 Hz, 1H), 4.93-4.46(m, 8H), 3.96-3.92 (m, 2H), 3.79-3.71(m,3H), 3.70-3.62 (m, 2H), 3.55-3.51(m,1H); MS (ESI) m|z696 (M+Na). 4.2.5. 2-(2-Chloro-5-((2S,3S,4R,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2- yl)phenyl)acetonitrile (20) The mixture of cyanides 19 (105 g, 156 mmol, ca. 2:1 β:a) wasslurried in ethanol (1L) and heated to reflux with stirring. Thereaction mixture was held at reflux for 1 h to insure that all of solu-tion had homogenized. It was then cooled evenly at 15℃/h toambient temperature and stirred overnight at this temperature.The resulting solid was isolated by filtration and dried in vacuoto yield the title compound (53 g, 51%, ca. 75% separation yield)as a white solid. [a21-10.6 (c 1.01, chloroform); 1H NMR(400 MHz, CDCl3) 8 7.48 (s, 1H), 7.37-7.17 (m, 20H), 6.93-6.91(m, 2H), 4.93 (s, 2H), 4.86 (d,J=10.8Hz, 1H), 4.63 (d,J=10.8 Hz,1H), 4.62 (d,J=12.4Hz, 1H), 4.56 (d,J=12.4Hz, 1H), 4.52 (d,J=10.8 Hz, 1H), 4.22 (d,J=9.6 Hz, 1H), 3.93 (d,J=10.8 Hz, 1H),3.84-3.68 (m, 6H), 3.62-3.58 (m, 1H), 3.43 (t, J=8.8Hz, 1H); 13cNMR (100MHz, CDCl3)8 129.49, 128.86,128.45,128.38,128.26,128.08, 128.01,127.87,127.82,127.70,127.68,127.67,127.64,127.61,139.09,138.52,138.23,138.07,137.41,132.97,129.49,128.86,128.45,128.38,128.26, 128.08,128.01,127.87, 127.82,127.70, 127.68,127.67,127.64,127.61,116.57,86.87,83.59,80.47, 79.41,78.28,75.67,75.16,74.96, 73.47,69.07,22.12; MS 4.2.6. 2-(2-Chloro-5-((2S,3S,4R,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2-yl)phenyl)aceticacid (21) To a solution of cyanide 20 (53 g, 79 mmol) in EtOH (300 mL)was added aq NaOH solution (8.0 N, 300 mL, 2.4 mol). The reactionmixture was refluxed overnight. After cooling to room tempera-ture, hydrochloric acid(3.0N) was added to neutralize the reactionmixture. The mixture was diluted with water and extracted withEtOAc. The organic layer was washed with water and brine priorto drying over anhydrous MgSO4. Filtration and concentration un-der reduced pressure yielded the title compound, which was usedin the next step without further purification.[o] -4.7 (c 1.10,chloroform); lH NMR (400 MHz, CDCl;)8 7.37-7.14 (m, 21H),6.93-6.90 (m, 2H), 4.94 (d,J=10.8 Hz, 1H), 4.90 (d,J=10.8 Hz,1H), 4.86 (d,J=11.2Hz, 1H), 4.63 (d,J=10.8 Hz, 1H), 4.62 (d,J=14.0 Hz, 1H), 4.55 (d,J=12.4Hz, 1H), 4.43 (d,J=10.4Hz, 1H),4.20 (d,J=9.6Hz, 1H), 3.88 (d,J=10.8 Hz, 1H). 3.81-3.70 (m,6H), 3.59-3.56 (m, 1H), 3.43 (t, J=8.8 Hz, 1H); 13C NMR(100 MHz, DMSO-d6) 8 171.84, 139.10, 138.69, 138.66, 138.17,133.72,133.49,131.99,129.18,128.68,128.66,128.45,128.26,128.19, 128.10,128.00,127.92, 127.87,127.84,86.25, 83.5380.14, 78.77,78.55,74.97,74.51,74.30,72.75,69.39,39.19; MS(ESI) m/z 710(M+NH4), 715 (M+Na)*. 4.2.7. 2-(2-Chloro-5-((2S,3S,4R,5R,6R)-3,4,5-tris-(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2-yl)phenyl)-acetate(22) To a solution of acid 21 (10 g, 14 mmol) in MeOH(140 mL) was added thionyl chloride (2.1mL, 29 mmol). The reaction mixturewas refluxed overnight and cooled to room temperature. After re-moval oforganic volatiles under reduced pressure, the residue waspartitioned between EtOAc and water. The organic layer was sep-arated and the aqueous layer was extracted with EtOAc. The com-bined organic: layerswere washedwithsaturated sodiumbicarbonate and brine, dried over anhydrous MgSO4, filtered andconcentrated in vacuo. The crude residue was purified by columnchromatography (5-40%EtOAc/hexanes)) to yield the title com-pound (9.1 g, 89%) as a white solid. H NMR (400 MHz, CDCl) 7.36-7.18(m, 21H), 6.94-6.92 (m, 2H), 4.96 (d,J=11.2 Hz, 1H),4.90 (d, J=11.2Hz, 1H),4.86((d,J=10.8Hz,1H), 4.63(d.J=11.6 Hz, 2H), 4.55 (d,J=12.0 Hz, 1H), 4.45 (d,J=10.8Hz, 1H)4.20 (d,J=9.2Hz, 1H), 3.90 (d,J=10.8 Hz, 1H), 3.82-3.67 (m,6H), 3.67 (s, 3H), 3.60-3.57 (m,1H), 3.47 (t,J=8.8Hz, 1H); MS(ESI) m|z 729 (M+Na). 4.2.8.2-(2-Chloro-5-((2S,3S,4R,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2-yl)benzyl)-5-(furan-2-yl)-1,3,4-thiadiazole (25) To a mixture of acid 21 (500 mg, 0.72 mmol), 2-furoic hydrazide23 (119 mg, 0.94 mmol), EDCI(207 mg, 1.08 mmol), and HOBt hy-drate (224 mg, 1.44 mmol) in DMF (10 mL) was added NMM(0.24 mL, 2.16 mmol). The resulting mixture was stirred at ambienttemperature overnight. After dilution with EtOAc, the organic layerwas subsequently washed with water, hydrochloric acid (1.0 N),saturated sodium bicarbonate, and brine prior to drying over anhy-drous MgSO4. Filtration and removal of the volatiles under reducedpressure yielded a glassy off-white amorphous solid, which wascarried on to the next step without further purification. To a solution of the acylhydrazide 24 in THF (15 mL) was addedLawesson reagent (874 mg,2.16 mmol). The reaction mixture wasrefluxed overnight. After cooling to room temperature, the reactionmixture was diluted with EtOAc and washed with saturated so-dium bicarbonate. The organic layer was dried over anhydrous MgSO4, filtered and concentrated in vacuo. The resulting crude res-idue was purified by column chromatography (10-60%EtOAc/hex-anes) to yield the title compound (468 mg, 81%) as a yellow oil. HNMR (400 MHz, CDCl3) 8 7.50 (d,J=1.6 Hz, 1H), 7.46 (d,J= 1.6 Hz,1H), 7.39 (d,J=8.0 Hz, 1H), 7.35-7.13 (m,19H), 7.08(d,J=3.6 Hz,1H), 6.90-6.88(m, 2H), 6.53-6.52(m, 1H), 4.90 (s, 2H), 4.85(d,J=10.8 Hz, 1H), 4.63-4.51(m, 4H), 4.45 (d,J=10.8Hz, 1H), 4.20(d, J=9.6 Hz, 1H), 3.88 (d,J=10.8Hz, 1H), 3.81-3.72 (m, 5H),3.60-3.57 (m, 1H), 3.43 (t,J=8.8 Hz, 1H); MS (ESI) m/z 799(M+H),821(M+Na)*. 4.2.9. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(furan-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (61) To a solution of perbenzylated 25 (468 mg, 0.58 mmol) in aceto-nitrile (5 mL) was added iodotrimethylsilane (5 mL). The resultingreaction mixture was heated to 50℃ overnight. After cooling to0°C, the reaction was quenched with methanol and concentratedin vacuo. Peracetylation was achieved by addition of acetic anhy-dride (3mL) and DMAP (10 mg, 0.08 mmol) to a solution of thisresidue in dichloromethane (10mL) and triethylamine (3 mL).After 2h stirring at ambient temperature, the reaction wasquenched by addition of water, whereupon the resulting mixturewas’ extracted with dichloromethane. The organic layer waswashed with hydrochloric acid (1.0N) and brine prior to dryingover anhydrous MgSO4. After filtration through a plug of silicagel and concentration under reduced pressure, the residue was car-ried on to the next step without further purification. To a solution of the tetraacetate in MeOH (10 mL) was added so-dium methoxide (25 wt.% in MeOH, 0.5 mL). The reaction mixturewas stirred at ambient temperature for 2 h. Amberlite IR120hydrogen form was then added to neutralize the reaction mixture.The resin was filtered off and the filtrate was concentrated in va-cuo. The resulting crude residue was purified by reverse phase pre-parative HPLC toprovide the title compound (78 mg, 31%) as awhite solid. o+21.4 (c 0.21, methanol); 1H NMR (400 MHz,DMSO-d6)8 7.93 (d,J=1.6Hz, 1H), 7.50 (d,J=2.0 Hz, 1H), 7.42(d,J=8.0 Hz,1H), 7.30 (dd,J=8.0, 2.0Hz, 1H),7.23 (d,J=3.6 Hz,1H), 6.70 (dd,J=3.6, 1.6 Hz, 1H), 5.00 (br, 2H), 4.88(d,J=5.6 Hz,1H), 4.59 (d,J=16.0Hz, 1H), 4.53 (d,J=16.0Hz, 1H), 4.43 (t,J=5.6 Hz, 1H), 4.01 (d,J=9.6 Hz, 1H), 3.68-3.65(m, 1H), 3.46-3.40 (m, 1H), 3.26-3.06(m,4H); 13c NMR (100 MHz, DMSO-d6)8167.62, 158.77,146.61,144.94,140.68,134.60,132.39,131.23,129.35, 129.26, 113.23,112.67,81.67,80.89,78.71,75.19, 70.68,61.75, 33.77, MS (ESI) m|z 439 (M+H); HRMSS ((FAB) m|z439.0730 [calcd for C19H20CIN206S(M+H)* 439.0731] 4.2.10. (2S,3R,4R,5S,6R)-2-(4-Chlor0-3-((5-(furan-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (31) Compound 31 was synthesized from de-chlorinated analogue of21 (ca. 2:1(β:o) mixture of anomers) and butyrohydrazide accord-ing to the procedure used to prepare 61. A white solid. Yield 17%.H NMR (400 MHz, CD30D) 8 7.40 (s, 1H), 7.35-7.28 (m, 2H),7.24-7.22 (m, 1H), 4.39 (s, 2H), 4.11 (d,J=9.2 Hz, 1H), 3.87 (d,J=11.2Hz, 1H), 3.71-3.66 (m, 1H), 3.48-3.30 (m, 4H), 3.00 (t,J=7.6 Hz, 2H), 1.75 (sext, J=7.6 Hz, 2H), 0.97 (t, J=7.6Hz, 3H);MS (ESI) m|z 381 (M+H)*. 4.2.11. (3R,4R,5S,6R)-2-(3-((5-tert-Butyl-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (32) Compound 32 was synthesized from de-chlorinated analogue of21 (ca.2:1(β:a) mixture of anomers) and pivalohydrazide accord-ing to the procedure used to prepare 61. A white solid. Yield 20%.H NMR (400MHz, CD3OD) 8 7.42 (s, 1H), 7.37-7.31(m, 2H), 7.27-7.25 (m, 1H), 4.40 (s, 2H), 4.13 (d,J=9.6 Hz, 1H), 3.88 (d,J=11.6Hz, 1H), 3.72-3.64 (m, 1H), 3.47-3.32(m, 4H), 1.43 (s,9H); MS (ESI) m/z 395 (M+H)*, 417 (M+Na)*. 4.2.12. (2R,3S,4R,5R)-2-(Hydroxymethyl)-6-(3-((5-phenyl-1,3,4-thiadiazol-2-yl)methyl)phenyl)-tetrahydro-2H-pyran-3,4,5-triol(33) Compound 33 was synthesized from de-chlorinated analogue of21 (ca. 2:1 (β:o) mixture of anomers) and benzoic hydrazideaccording to the procedure used to prepare 61. A white solid. Yield30%.1H NMR(400 MHz,CD30D)8 7.88(d,J=7.6Hz,2H), 7.01-7.45(m, 4H), 7.37-7.29 (m, 3H), 4.47 (br, 1H), 4.13 (d,J=9.6 Hz, 1H),3.88-3.85 (m 1H), 3.68 (dd,J=12.0, 5.2Hz, 1H), 3.48-3.38 (m,3H), 3.35-3.30 (m, 2H); MS (ESI) m/z 415(M+H)*, 437 (M+Na)*. 4.2.13. (2R,3S,4R,5R)-2-(Hydroxymethyl)-6-(3-((5-(pyridin-3-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-tetrahydro-2H-pyran-3,4,5-triol (34) Compound 34 was synthesized from de-chlorinated analogue of21 (ca. 2:1(β:a) mixture of anomers) and nicotinohydrazideaccording to the procedure used to prepare 61. A white solid. Yield6.5%. HNMR (400 MHz, CD30D) 89.05 (s, 1H), 8.65 (d,J=4.8 Hz,1H),8.34-8.31 (m, 1H), 7.56 (dd,J=8.0, 5.2 Hz,1H), 7.46 (s, 1H),7.37-7.31(m, 3H), 4.51 (s,2H), 4.13 (d,J=9.6Hz, 1H), 3.86 (dJ=11.6 Hz, 1H), 3.68 (dd,J=11.6, 5.2 Hz, 1H), 3.48-3.32(m,4H);MS (ESI) m/z 416 (M+H),438 (M+Na)*. 4.2.14. (3R,4R,5S,6R)-2-(4-Chloro-3-((5-cyclohexyl-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (35) Compound 35 was synthesized from an anomeric mixture of 21(ca. 2:1 β:a) and cyclohexanecarbohydrazide according to the pro-cedure used to prepare 61. A white solid. Yield 25%.. HNMR(400MHz, DMSO-d6) 8 7.45 (d,J=2.0Hz, 1H), 7.39 (d.J=8.4Hz,1H), 7.28 (dd,J=8.4, 2.0 Hz, 1H), 5.07-4.89)(br, 3H), 4.48 (d,J=15.6Hz, 1H), 4.43 (d, J=15.6 Hz, 1H),4.42 (br, 1H), 3.99 (d,J=9.6 Hz, 1H), 3.69-3.65 (m, 1H), 3.46-3.40(m, 1H), 3.24-3.00(m, 5H), 1.99-1.94 (m, 2H), 1.73-1.67(m,,2H), 1.64-1.59 (m,1H), 1.46-1.28 (m, 3H), 1.24-1.14 (m,1H); MS (ESI) m|z 455(M+H),477 (M+Na)*. 4.2.15.(3R,4R,5S,6R)-2-(4-Chloro-3-((5-p-tolyl-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (36) Compound 36 was synthesized from an anomeric mixture of 21(ca.2:1 p:d) and 4-methylbenzohydrazide according to the proce-dure used to prepare 61. A white solid.LYYield 34%. 1H NMR(400 MHz, CD30D)8 7.76 (d,J=8.0Hz, 2H), 7.56 (d,J=2.0 Hz,1H), 7.43-7.37 (m, 2H), 7.29 (d,J-8.0Hz, 2H), 4.58 (s, 2H), 4.14(d,J=9.6 Hz, 1H), 3.87 (dd,J=12.0, 2.0 Hz, 1H), 3.70 (dd,J=12.0,5.2 Hz, 1H), 3.48-3.38 (m, 3H), 3.26 (d, J=9.2 Hz, 1H), 2.37 (s,3H); M:5 (ESI) m|z 463 (M+H), 485 (M+Na). ( 4.2.16. (3R,4R,5S,6R)-2- ( 4- C hloro-3 - ((5-(4-propylphenyl)-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol ( 3 7 ) ) Compound 37 was synthesized from an anomeric mixture of 21(ca. 2:1 p:a) and 4-propylbenzohydrazide according to the proce-dure used to prepare 61. A white solid. Yield 12%. H NMR(400 MHz, CD3OD) 8 7.79 (d,J=8.0Hz, 2H), 7.57 (d,J=1.6 Hz,1H), 7.43-7.37 (m, 2H), 7.30 (d,J=8.0 Hz, 2H), 4.14 (d,J=9.6 Hz,1H), 3.87 (dd,J=12.0, 2.0 Hz, 1H), 3.70 (dd,J=12.0, 5.2 Hz, 1H),3.48-3.38(m, 3H), 3.33 (s, 1H), 3.26 (d,J=9.2Hz, 1H), 2.63 (t,J=7.6Hz, 2H), 1.65 (sext,J=7.6Hz, 2H), 0.93 (t, J=7.6 Hz, 3H);MS (ESI) m/z 491 (M+H), 513 (M+Na)*. 4.2.17. (3R,4R,5S,6R)-2-(4-Chloro-3-((5-(pyridin-3-yl)-1,3,4- ( thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro- 2H-pyran-3,4,5-triol (38) ) ( Compound 38 was synthesized from an anomeric mixture of 21 (ca. 2:1 β:d) and nicotinohydrazide according to the procedure used to prepare 61. A white solid . Yield 21%. H NMR (400 MHz, DMSO-d6) 8 9 . 07 (d,J=1.6 Hz, 1H), 8.80 (dd,J= 4 .8, 1. 6 Hz, 1H),8.30-8.27 ( m, 1 H), 7 .55-7.51 (m, 2H), 7.43 ( d , J=8.4Hz, 1H ) , 7.32 (dd, J=8.4, 2 .0Hz, 1H), 5 .02-4.84 ( (br, 3H), 4.63 (d, J =16.0Hz, 1H), 4.58 (d,J=16.0 Hz, 1H) , 4.41 (br , 1H), 4.02 (d, J =9.6Hz, 1 H ), 3.69-3.66 ( m , 1 H ), 3.46-3.41 (m, 1 H ),3.23-3.06 (m,4H);MS (ESI) m/z 450(M+H), 472(M+Na). ) ( 4.2.18. (3R,4R,5S,6R)-2-(4-Chloro-3-((5-(pyridin-3-yl)-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro O- 2H-pyran-3,4,5-triol (39) ) ( Compound 39 was synthesized from an anomeric mixture of 21 (ca. 2:1 p:d) and isonicotinohydrazide a c cording to t he procedure used to prepare 61. A white solid . Yield 22%. H NMR (400 MHz,CD30D) 8 8 . 76 (d, J=6.4 Hz, 2 H ), 8.12 (d,J=6. 4 H z , 2H) , 7. 5 9 ( d, J =1.2 Hz, 1 H), 7 .44-7.39 (m,2H). 4.68 (s, 2 H), 4.14(d,J = 9.6Hz, 1 H) , 3 .87 (dd,J=1 2 .0, 2 .0 Hz, 1H), 3.70 ( d d, J =1 2 .0, 5 .2 Hz, 1H), 3.48- 3. 38 (m, 3 H), 3 .34 (s, 1H), 3.26 (d , J=8.8 Hz, 1H); MS (ESI) m / z 450 (M+H), 472 (M+Na). ) ( 4.2.19. (3R,4R,5S,6R)-2-(4-Chloro-3-(( 5 -(4- c hlorobenzyl)-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6- ( hydroxymethyl)-tetrahydro- 2H-pyran-3,4,5-triol(40) ) ( C ompound 40 was synthesized from an anomeric mixture of 21 (ca.2:1 p:d) and 2 - (4 -chlorophenyl)acetohydrazide according to t he procedure used to p repare 61. A white s o lid. Yield 14%. HNMR (400MHz, CD O D) 8 87.49 (s,1H), 7.37-7.25 (m, 6 H ), 4.47 (s, 2H), 4. 3 5 (s , 2H) , 4.10 ( d ,J=9.6Hz, 1 H),3 . 85 (dd,J=12.0, 1. 6 Hz, 1H), 3.68 ( dd , J =1 2 .0, 5.2 Hz, 1 H), 3.44-3.29( m , 3H), 3 . 24 ( t , J =9. 2 Hz, 1 H) ; M S ( E SI) m/z 497 ( M+H), 519(M+Na) . ) ( 4 .2.20. (3R,4R,5S,6R)-2-(4-Chloro-3-((5-(2-(4-chlorophenyl)propan-2-y l )-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- ) (hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (41) ( Compound 41 was synthesized from an anomeric mixture of 21(ca. 2:1 β:a) and 2-(4-chlorophenyl)-2-methylpropanehydrazideaccording to the procedure used to prepare 61. A white solid. Yield 58%.H NMR (400 MHz, CD0D) 8 7.49 (s,1H), 7.38-7.34(m, 2H), 7.29 ( s, 4H), 4.49 (s, 2H), 4.1 1 (d,J=9.2 Hz, 1 H), 3.85 (dd,J=12. 4 , 2.0 Hz, 1H), 3.68 (dd,J=12.4, 5.6 H z , 1 H ), 3.47-3.31 (m,3H) , 3.2 4 (t,J=8.8Hz,1H),1.80(s,6H); MS(ESI)m|z525(M+H)*,547(M+Na)*. ) 4.2.21. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-propyl-1,3,4- ( thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (42) ) ( Compound 42 was synthesized from an anomeric mixture of 21 (ca. 2:1 p:a) and butyrohydrazide according to the procedure used to prepare 61. The small amount of the o-anomer was removed by reverse phase preparative HPLC. A white solid. Yield 12%. H NMR(400 MHz, CD3OD)8 7.51(d,J=1.6Hz,1H),7.41-7.36(m,2H), 4.52(s, 2H), 4.12 (d,J=9.2 Hz, 1H), 3.86 (dd,J=12.0, 1.6 Hz, 1H), 3.69 (dd, J= 1 2.0, 5.2 Hz, 1H), 3.47-3.37 (m, 3H), 3.25 (d,J=8.8 Hz, 1H), 3 .01 (t, J=7.6Hz, 2 H ), 1.76 (s e xt, J=7.6 Hz, 2H), 0.9 7 (t, J=7.6 Hz, 3H);MS (ESI) m/z 415 (M+H) . ) ( 4.2.22. (2S,3R,4R,5S,6R)-2-(3-((5-Butyl-1,3,4-thiadiazol-2- yl)methyl)-4-chlorophenyl)-6-(hydroxymethyl)-tetrahydro-2H- pyran-3,4,5-triol(43) ) Compound 43 was synthesized from an anomeric mixture of 21(ca.2:1 p:d) and pentanehydrazide according to the procedureused to prepare 61. The small amount of the o-anomer was re- moved by reverse phase preparative HPLC. A white solid.Yield 19%.'H NMR (400 MHz, DMSO-d6) 8 7.45 (d,J=2.0 Hz, 1H), 7.39 (d,J=8.4 Hz, 1H), 7.28(dd,J=8.4,2.0Hz, 1H), 5.04 (br, 2H), 4.87 (d,J=5.6 Hz, 1H), 4.48 (d,J=16.0 Hz, 1H), 4.43 (d,J=16.0 Hz, 1H),4.43 (br, 1H), 3.99 (d, J=9.2 Hz, 1H), 3.68-3.65 (m, 1H), 3.46-3.40 (m,1H), 3.26-3.04(m,4H), 2.97 (t,J=7.6 Hz, 2H), 1.61 (quint,J=7.6 Hz,2H), 1.29(sext,J=7.6 Hz,2H),0.85(t,J=7.6 Hz, 3H); MS(ESI) m/z 429 (M+H)*, 451(M+Na). 4.2.23. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-pentyl-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (44) Compound 44 was synthesized from an anomeric mixture of 21(ca.2:1 p:a) and hexanehydrazide according to the procedure usedto prepare 61. The small amount of the o-anomer was removed byreverse phase preparative HPLC. A white solid. Yield 20%.H NMR(400 MHz, DMSO-d6) 8 7.45 (d,J=2.0 Hz, 1H), 7.40 (d,J=8.0 Hz,1H), 7.28 (dd,J=8.0, 2.0 Hz, 1H), 4.90 (br, 2H), 4.80 (d, 1H), 4.49(d, J=15.6 Hz, 1H), 4.43 (d,J=15.6 Hz, 1H), 4.39 (t, J=6.0 Hz,1H), 4.00 (d, J=9.2 Hz, 1H), 3.69-3.65 (m, 1H), 3.45-3.40 (m,1H), 3.24-3.04(m,4H), 2.96 (t,J=7.6 Hz, 2H), 1.67-1.60 (m, 2H),1.30-1.22 (m, 4H), 0.82 (t, J=7.6 Hz, 3H); MS (ESI) m/z 443.(M+H),465 (M+Na)*. 4.2.24. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-heptyl-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (45) Compound 45 was synthesized from an anomeric mixture of 21(ca.2:1 p:d) and octanehydrazide according to the procedure usedto prepare 61. The small amount of the o-anomer was removed byreverse phase preparative HPLC. A white solid. Yield 38%.HNMR(400 MHz, CD3OD)8 7.52(d,J=1.6 Hz,1H), 7.40-7.36(m,2H), 4.12(d, J=9.2 Hz, 1H), 3.86 (dd,J=12.0, 2.0 Hz, 1H), 3.69(dd,J=12.0,5.2Hz, 1H), 3.47-3.38 (m, 3H), 3.25 (d,J=8.8 Hz, 1H), 3.03 (t,J=7.6Hz, 2H), 1.72 (quint,J=7.6Hz, 2H), 1.38-1.24 (m, 8H),0.87 (t,J=7.6 Hz, 3H); MS (ESI) m/z 471 (M+H)*,493 (M+Na)*. 4.2.25. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-cyclopentyl-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-trioI (50) Compound 50 was synthesized from 21 and cyclopentanecarbo-hydrazide according to the procedure used to prepare 61. A whitesolid. Yield 46%. H NMR (400MHz, DMSO-d6)87.45(d,J=1.6 Hz,1H), 7.40(d,J=8.4Hz,1H), 7.29(dd,J=8.4,2.0 Hz, 1H), 4.96-4.93(m, 2H), 4.85 (d, J=5.6Hz, 1H), 4.50-4.41 (m, 3H), 4.00 (d,J=9.2 Hz, 1H), 3.69-3.65 (m, 1H), 3.47-3.40(m, 2H), 3.26-3.03(m, 4H), 2.09-2.04(m, 2H), 1.69-1.57(m, 6H); MS (ESI) m/z 441(M+H), 463 (M+Na)*. 4.2.26. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-phenyl-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (51) Compound 51 was synthesized from an anomeric mixture of 21(ca. 2:1 :a) and benzoic hydrazide according to the procedureused to prepare 61. The small amount of the o-anomer was re-moved by reverse phase preparative HPLC. A white solid. Yield27%.'H NMR (400 MHz, DMSO-d6) 8 7.89(dd,J=7.2, 2.0 Hz, 2H),7.53-7.42 (m, 5H), 7.32 (dd,J=8.0, 2.0 Hz, 1H), 5.03 (br, 2H),4.94 (br, 1H), 4.59 (d,J=16.0Hz, 1H), 4.54 (d,J=16.0 Hz, 1H),4.50 (br, 1H), 4.03 (d, J=9.6 Hz, 1H), 3.69 (d,J=11.2Hz, 1H),3.46-3.42 (m, 1H), 3.28-3.13 (m, 3H), 3.09 (t,J=8.8Hz, 1H); 13cNMR (100MHz, DMSO-d6)8168.75,140.69,134.65, 132.43,131.72,131.31,129.94,129.87,129.38,129.26,127.99,81.70,80.89,78.68,75.18,70.62,61.71, 34.08; MS (ESI) m/z 449(M+H), 471(M+Na). 4.2.27. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(4-chlorophenyl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (52) Compound 52 was synthesized from an anomeric mixture of21 (ca. 2:1 p:o) and 4-chlorobenzohydrazide according to theprocedure used to prepare 61.The small amount of the o-anomerwas removed by reverse phase preparative HPLC. A white solid.Yield 50%. 1H NMR (400 MHz, DMSO-d6) 8 7.92 (d,J=8.4Hz,2H), 7.56 (d, J=8.4 Hz, 2H), 7.51 (d, J=2.0Hz, 1H), 7.43 (d,J=8.0 Hz, 1H), 7.31 (dd,J=8.0, 2.0 Hz, 1H), 4.96-4.93 (m, 2H),4.86 (d, J=5.6Hz, 1H),4.60 (d, J=16.0Hz, 1H), 4.55 (d,J=16.0Hz, 1H), 4.43 (t, J=5.6 Hz, 1H), 4.01 (d,J=9.6Hz, 1H),3.68-3.65 (m, 1H), 3.26-3.07 (m, 4H); MS (ESI) m/z 483 (M+H)*505(M+Na). 4.2.28. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(pyridin-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (53) Compound 53 was synthesized from 21 and picolinohydrazideaccording to the procedure used to prepare 61.A white solid. Yield18%.HNMR (400 MHz, DMSO-d6) 8 8.62 (dd,J=3.6,0.8 Hz, 1H),8.19 (d,J=7.6 Hz, 1H), 7.98 (td, J=7.6, 1.6 Hz, 1H), 7.54-7.50(m,2H), 7.43 (d,J=8.4 Hz, 1H), 7.31 (dd,J=8.0, 1.6 Hz, 1H), 4.94-4.91 (m, 2H), 4.84 (d,J=5.6 Hz, 1H), 4.59(d,J=16.0Hz, 1H),4.54(d,J=16.0 Hz, 1H),,4.42(t,J=5.6Hz, 1H), 4.02 (d,J=9.6Hz,1H), 3.69-3.65 (m, 1H), 3.44-3.41(m, 1H), 3.27-3.07 (m, 4H);MS (ESI) m/z 450 (M+H),472 (M+Na). 4.2.29. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(6-methylpyridin-3-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (55) Compound 55Vwassynthesizedfrom21 andd6-meth-ylnicotinohydrazide according to the procedure used to prepare61. A white solid. Yield 17%.H NMR (400 MHz, DMSO-d6)8 8.92(d,J=2.0Hz, 1H), 8.16 (dd, J=8.0 Hz, 2.4 Hz, 1H), 7.52 (d,J=1.6 Hz, 1H), 7.48-7.37(m,2H), 7.32(dd,J=8.4 Hz, 2.0 Hz, 1H),4.99-4.90 (m, 2H), 4.86 (d,J=5.6 Hz, 1H), 4.65-4.53 (m, 2H),4.43 (t,J=6.0Hz, 1H), 4.01(d,J=9.2 Hz, 1H), 3.71-3.63 (m, 1H),3.47-3.05(m, 5H), 2.50(s, 3H); MS (ESI) m/z 464(M+H). 4.2.30. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(isoquinolin-3-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)- tetrahydro-2H-pyran-3,4,5-triol (57) Compound 57 was synthesized from 21 and isoquinoline-3-car-bohydrazide according to the procedure used to prepare 61. Awhite solid. Yield 27%. H NMR (400 MHz, DMSO-d6)8 9.36 (s,1H), 8.72 (s, 1H), 8.18 (d, J=8.8 Hz, 2H), 7.86 (td, J=7.6 Hz,1.2 Hz, 1H), 7.76 (td,J=8.0 Hz, 1.2 H, 1H), 7.55 (d,J=2.0Hz, 1H),7.44 (d,J=8.0Hz, 1H), 7.32 (dd,J=8.0 Hz, 2.0Hz, 1H), 4.66-4.52(m, 3H), 4.03 (d,J=9.2 Hz,1H), 3.68(d,J=10.8Hz, 1H), 3.44 (dd,J=11.6 Hz, 5.6 Hz, 2H), 3.33-3.05(m, 6H); MS (ESI) m/z 500(M+H)*. 4.2.31. ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(quinolin-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (58) Compound 58 was synthesized from 21 and quinoline-2-carbo-hydrazide according to the procedure used to prepare 61. A whitesolid. Yield 25%.1H NMR (400 MHz, DMSO-d6)88.56 (d,J=8.4 Hz,1H),8.31 (d,J=8.8 Hz, 1H), 8.04 (t,J=9.4Hz,2H), 7.81 (td,J=8.0,1.6 Hz, 1H), 7.67 (td,J=7.2, 1.2Hz,1H), 7.56(d,J=2.0Hz,1H), 7.45(d,J=8.4Hz, 1H), 7.33 (dd,J=6.4,2.0 Hz, 1H), 4.99-4.90 (m, 2H),4.86 (d,J=5.6Hz, 1H), 4.67-4.55 (m, 2H), 4.43 (t,J=5.6 Hz, 1H),4.04 (d,J=9.6 Hz, 1H), 3.67 (dd,J=7.6, 3.6 Hz, 1H), 3.49-3.38 (m,1H), 3.29-3.07 (m, 4H); MS (ESI) m/z 500 (M+H). 4.2.32. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(furan-3-yl)-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (62) Compound 62 was synthesized from 21 and 3-furoic hydra-zide according to the procedure used to prepare 61. A whitesolid. Yield 32%. H NMR (400 MHz, DMSO-d6) 8 8.44 (dd,J=1.2, 0.4Hz, 1H), 7.83 (t,J=2.0Hz, 1H), 7.50 (d, J=1.6 Hz,1H), 7.42 (d,J=8.4Hz, 1H), 7.31 (dd,J=8.4, 2.0Hz, 1H), 6.96(dd, J=2.0, 0.8 Hz, 1H), 4.96-4.93 (m, 2H), 4.85 (d, J=6.0Hz,1H), 4.56 (d,J=16.0Hz, 1H), 4.51 (d,J=16.0Hz, 1H), 4.44 (t,J=6.0Hz, 1H), 4.0101(d,J=9.6 Hz, 1H), 33.70-3.655 (m, 1H),3.45-3.39 (m, 1H), 3.27-3.04((m, 4H);MS(ESI))1m|z 439(M+H), 461 (M+Na). 4.2.33. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(3-methylfuran-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (63) Compound 63 was synthesized from 21 and 3-methylfuran-2-carbohydrazide according to the procedure used to prepare 61. Awhite solid. Yield 28%. 'H NMR (400 MHz, DMSO-d6) 8 7.80 (d,J=1.2Hz, 1H), 7.49 (d,J=2.0 Hz, 1H), 7.41 (d,J=8.4 Hz, 1H),7.30 (dd,J=8.0, 2.0 Hz, 1H), 6.61 (d,J=1.2 Hz, 1H), 4.97-4.80(m, 2H), 4.83 (d,J=5.2 Hz, 1H), 4.60-4.50(m, 1H),4.47-4.39(m, 1H), 4.01 (d,J=9.6Hz, 1H), 3.71-3.62 (m, 1H), 3.48-3.37(m, 1H), 3.33-3.03(m, 4H),2.32 (s, 3H); MS (ESI) m/z 453(M+H)*. 4.2.34. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(5-methylfuran-2-yl)-1,3,4-thiadiazo1-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (64) Compound 64 was synthesized from 21 and 5-methylfuran-2-carbohydrazide according to the procedure used to prepare 61. Awhite solid. Yield 30%. H NMR (400MHz, DMSO-d6) 8 7.49 (d,J=2.0 Hz, 1H), 7.42 (d,J=8.4 Hz, 1H), 7.30 (dd,J=8.4, 2.0 Hz,1H), 7.11 (d,J=3.6 Hz, 1H), 6.32 (d,J=3.6Hz, 1H), 4.94-4.91 (m,2H), 4.83 (d, J=5.6Hz, 1H), 4.56 (d, J=16.0Hz, 1H), 4.51 (d,J=16.0 Hz, 1H), 4.42 (t, J=5.6Hz, 1H), 4.01 (d, J=9.6 Hz, 1H),3.70-3.65(m, 1H), 3.45-3.40 (m, 1H), 3.27-3.04 (m, 4H), 2.32 (s,3H); MS (ESI) m|z 453 (M+H) 4.2.35. (2S,3R,4R,5S,6R)-2-(3-((5-(Benzofuran-2-yl)-1,3,4- thiadiazol-2-yl)methyl)-4-chlorophenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol(66) Compound 52 was synthesized from an anomeric mixture of21 (ca. 2:1 p:o) and benzofuran-2-carbohydrazide according tothe procedure used to prepare 61. The small amount of the a-anomer was removed by reverse phase preparative HPLC. A whitesolid. Yield 50%. H NMR (400 MH[zH,z ,C DC3D03DO)D) 8 7.71(d,J=8.0 Z.1H), 7.60-7.52 (m, 3H), 7.44-7.39 (m, 3H),7.31 (t, J=7.6Hz,2H), 4.15 (d, J=9.2 Hz, 1H), 3.87 (dd,J=12.0, 1.6 Hz, 1H), 3.70(dd,J=12.0,5.2 Hz,3.48-3.38(m, 3H), 3.33 (s, 2H); MS (ESI)m/z 489 (M+H)*. 4.2.36. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(5-methylisoxazol-3-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (67) Compound 67 was synthesized from 21 and 5-methylfuran-2-carbohydrazide according to the procedure used to prepare 61.Awhite solid. Yield 16%. H NMR (400 MHz, DMSO-d6) 8 7.52 (d,J=2.0Hz, 1H), 7.43 (d,J=8.0 Hz, 1H), 7.31 (dd,J=8.0, 1.6 Hz,1H), 6.87 (s, 1H), 4.93 (t,J=4.8Hz, 2H), 4.85 (d,J=5.6 Hz, 1H),4.64 (d, J=16.0Hz, 1H), 4.59 (d, J=16.0Hz, 1H), 4.42 (t,J=6.0 Hz, 1H), 4.02 (d,J=9.2 Hz, 1H), 3.69-3.65 (m, 1H), 3.46-3.39 (m, 1H), 3.29-3.05 (m, 4H), 2.48 (s, 3H); MS (ESI) m(z 4541(M+H), 476 (M+Na). 4.2.37. ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(1-methyl-1H-pyrrol-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (68) Compound 68 was synthesized from 21 and 1-methyl-1H-pyr-role-2-carbohydrazide according to the procedure used to prepare61. A white solid. Yield 18%.H NMR (400 MHz, DMSO-d6) 8 7.46(d,J=2.0 Hz, 1H), 7.42 (d,J=8.4Hz, 1H), 7.30 (dd,J=8.0,2.0 Hz,1H), 7.02 (t, J=2.0 Hz, 1H), 6.65-6.60 (m, 1H), 6.13-6.07 (m, 1H),4.57-4.42(m, 4H), 4.01 (d,J=9.6 Hz, 1H), 3.80 (s, 3H), 3.67 (dd,J=11.6, 1.6Hz, 1H), 3.47-3.38 (m, 1H),3.27-3.05 (m, 6H); MS(ESI) m|z 452 (M+H). 4.2.38. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(1-methyl-1H-indazol-3-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- (hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (69) Compound 69 was synthesized from 21 and 1-methyl-1H-inda-zole-3-carbohydrazide according to the procedure used to prepare61. A white solid. Yield 26%. 1H NMR (400 MHz, DMSO-d6)8 8.26(d,J=8.4 Hz, 1H), 7.74 (d,J=8.8Hz, 1H), 7.54-7.49((m,2H),7.43(d,J=8.4Hz, 1H), 7.36-7.30 (m, 2H), 4.94-4.91 (m, 2H), 4.85(d,J=5.6 Hz, 1H), 4.61 (d,J=15.6 Hz, 1H), 4.56 (d,J=15.6Hz, 1H),4.43 (t, J= 5.6 Hz, 1H), 4.10(s, 3H), 4.03 (d,J=9.2Hz, 1H), 3.70-3.66 (m, 1H), 3.43 (quint,J=5.6 Hz, 1H), 3.27-3.06 (m, 4H); MS(ESI) mz 503 (M+H)*. 4.2.39.(2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(thiophen-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (70) Compound 70 was synthesized from 21 and thiophene-2-carbo-hydrazide according to the procedure used to prepare 61. A whitesolid. Yield 47%. H NMR (400 MHz, DMSO-d6) 8 7.78 (dd,J=4.8,1.2 Hz, 1H), 7.69(dd,J--3.6,1.2 Hz, 1H), 7.51 (d,J=2.4Hz, 1H),7.43 (d,J=8.0Hz, 1H),7.31 (dd,J=8.0, 2.0Hz, 1H), 7.16 (dd,J=8.0, 4.0 Hz, 1H), 4.97-4.93 (m, 2H), 4.86 (d, J=6.0Hz, 1H),4.577 (d, J=16.0Hz, 1H), 4.52 (d,J=15.6Hz, 1H), 4.45 (t,J=5.6 Hz, 1H), 4.02 (d,J=9.2 Hz, 1H), 3.70-3.65 (m, 1H), 3.46-3.39 (m, 1H), 3.27-3.05 (m, 4H); MS (ESI) m/z 455 (M+H), 477(M+Na). 4.2.40. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(thiophen-3-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (71) Compound 71 was synthesized from 21 and thiophene-3-carbo-hydrazide according to the procedure used to prepare 61. A whitesolid. Yield 66%. HNMR (400 MHz, DMSO-d6)8 8.22(dd,J=2.8,1.2 Hz, 1H), 7.72-7.69(m,1H), 7.58 (dd,J=4.8, 1.2 Hz, 1H), 7.51(d,J=1.6Hz, 1H), 7.42 (d,J=8.0 Hz, 1H), 7.31 (dd,J=8.0, 1.6 Hz,1H), 4.94-4.91(m,2H), 4.844(d, J=6.0Hz, 1H), 4.57 (d,J=16.0 Hz, 1H), 4.52 (d,J=16.0Hz, 1H), 4.42 (t, J=6.0 Hz, 1H),4.01 (d,J=9.6Hz, 1H), 3.70-3.65 (m, 1H), 3.46-3.40 (m, 1H),3.25-3.05 (m, 4H); 13C NMR (100 MHz, DMSO-d6)8 166.40,162.06,139.16,133.14,130.89,129.85, 129.74,127.85,127.68,127.36, 126.69,125.08,80.14, 79.35,77.19,73.65,69.18,60.24,32.47; MS (ESI) m|z 455 (M+H), 477 (M+Na); HRMS (FAB) m/z455.0501 [calcd for C19H20ClN205S2(M+H)* 455.0502]. 4.2.41. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(3-methylthiophen-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (72) Compound 72 was synthesized from 21 and 3-methylthioph-ene-2-carbohydrazide according to the procedure used to prepare61. A white solid. Yield 45%. 1HNMR (400 MHz, DMSO-d6) 8 7.67(d,J=5.2 Hz, 1H), 7.50 (d,J=1.6 Hz, 1H), 7.42 (d,J=8.4 Hz, 1H),7.30 (dd,J=8.4, 1.6 Hz, 1H), 7.06 (d,J=5.2 Hz, 1H), 4.96(m, 2H),4.85 (d, J=5.6Hz,1H), 4.59 (d,J=16.0Hz, 1H),4.55 (d,J=16.0Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 4.01 (d, J=9.2 Hz, 1H),400-188-0725 3.69-3.65(m, 1H), 3.45-3.39(m, 1H), 3.27-3.04(m, 4H), 2.37 (s,3H); MS (ESI) m/z 489(M+H). 4.2.42. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(3-chlorothiophen-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (73) Compound 73 was synthesized from 21 and 3-chlorothiophene-2-carbohydrazide according to the procedure used to prepare 61. Awhite solid. Yield 49%. 1H NMR (400 MHz, DMSO-d6)8 7.90 (d,J=5.2 Hz, 1H), 7.51 (d,J=2.0Hz, 1H), 7.43 (d, J=8.4 Hz, 1H),7.31 (dd,J=8.0, 2.0 Hz, 1H), 7.27 (d,J=5.6 Hz, 1H), 4.92-4.90 (m,2H), 4.82 (d,J=5.6Hz, 1H), 4.62 (d,J=16.0Hz, 1H), 4.58 (d,J=16.0Hz, 1H), 4.40 (t, J=6.0 Hz, 1H), 4.02 (d, J=9.2Hz, 1H),3.70-3.65 (m, 1H), 3.46-3.40 (m, 1H), 3.25-3.05 (m, 4H); MS(ESI) m|z 489 (M+H). 4.2.43. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(3-chlorothiophen-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (74) Compound 76 was synthesized from 21 and thiazole-4-carbo-hydrazide according to the procedure used to prepare 61. A whitesolid. Yield 27%. 'HNMR(400 MHz,DMSO-d6)5 9.23 (d,J=2.4 Hz,1H), 8.49 (d,J=2.0Hz, 1H), 7.51 (d, J=2.0Hz, 1H), 7.42 (d,J=8.4Hz, 1H), 7.30 (dd,J=8.0, 2.0Hz, 1H), 4.93-4.91 (m, 2H),4.83 (d, J=5.6Hz, 1H),,4.59(d,J=15.6Hz, 1H),4.54 (d,J=15.6Hz, 1H), 4.42 (t, J=5.6Hz, 1H), 4.01 (d, J=9.2 Hz, 1H),3.69-3.64 (m, 1H), 3.45-3.39 (m, 1H), 3.26-3.05(m, 4H); MS(ESI) m|z 456(M+H),478(M+Na). 4.2.46. ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(2-(pyridin-4-yl)thiazol-5-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- (hydroxylmethyl)-tetrahydro-2H-pyran-3,4,5-triol (77) Compound 77 was synthesized from 21 and 2-(pyridin-4-yl)thi-azole-4-carbohydrazide according to the procedure used to pre-pare 61. A white solid. Yield 19%. 1H NMR (400 MHz,DMSO-d6)88.73 (d,J=6.0 Hz, 2H), 8.68 (s, 1H), 7.96 (d,J=4.4Hz, 2H), 7.54(d,J=2.0Hz, 1H), 7.44 (d,J=8.4 Hz, 1H), 7.32 (dd,J=8.0, 1.6Hz,1H), 4.67-4.51(m, 5H), 4.02 (d, J=9.2 Hz, 1H), 3.67 (d,J=10.4 Hz, 1H), 3.49-3.39 (m, 1H), 3.27-3.07 (m, 5H); MS (ESI)m|z 533 (M+H)*. 4.2.47. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(3,5- dimethylisoxazol-4-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (78) Compound 78 was synthesized from 21 and 3,5-dimethylisoxa-zole-4-carbohydrazide according to the procedure used to prepare61. A white solid. Yield 9.5%. 1H NMR (400 MHz, DMSO-d6)8 7.50(d,J=2.0 Hz, 1H), 7.42 (d,J=8.4 Hz, 1H), 7.30 (dd,J=8.4, 2.0 Hz,1H), 4.74 (br, 4H), 4.61 (d,J=16.0Hz, 1H), 4.56 (d,J=16.0 Hz,1H), 4.01 (d,J=9.6 Hz, 1H), 3.67 (dd,J=11.6,1.6 Hz, 1H), 3.44-3.40 (m, 1H), 3.26-3.19 (m, 2H), 3.16-3.11(m, 1H), 3.07 (t,J=9.2 Hz, 1H),2.60 (s, 3H), 2.39 (S, 3H); MS (ESI) m/z 468(M+H). 4.2.48. ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(3-ethyl-5-methylisoxazol-4-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol(79) Compound 79 was synthesized from 21 and 3-ethyl-5-methy-lisoxazole-4-carbohydrazide according to the procedure used toprepare 61. A white solid. Yield 12%.H NMR (400 MHz, DMSO-d6)8 7.50 (d,J=2.0Hz, 1H), 7.42 (d,J=8.4Hz, 1H), 7.30 (dd,J=8.4,2.0Hz, 1H), 4.75 (br, 4H), 4.61(d,]=16.0 Hz, 1H), 4.57 (d,J=16.0Hz, 1H), 4.01 (d, J=9.2 Hz, 1H), 3.67 (d,J=10.0Hz, 1H),3.44-3.40 (m,1H), 3.26-3.19 (m, 2H), 3.16-3.11(m, 1H), 3.07 (t,J=9.2 Hz, 1H), 2.85 (q, J=7.2 Hz, 2H),2.59 (s, 3H), 1.75 (t,J=7.2Hz, 3H); MS (ESI) m|z 468 (M+H)*. 4.2.49. 2-(2-Chloro-5-((2S,3S,4R,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2- yl)phenyl)acetohydrazide (26) To a mixture ofacid 21 (693mg, 1.0mmol), EDCI (249 mg,1.3 mmol), and HOBthydrate (233 mg,1.5mmol) in DMF(10 mL) was added hydrazine monohydrate (2 mL). The resultingmixture was stirred! at ambient temperature for 12 h and thenpoured into water. The resulting solid was isolated by filtrationand dried in vacuo to yield (674 mg, 95%) a white solid. 1H NMR(400 MHz, DMSO-d6) 8 9.17 (s, 1H), 7.39-7.14(m, 21H), 6.88-6.86 (m, 2H), 4.80 (d,J=11.2Hz, 1H). 4.77 (d,J=11.2 Hz, 1H),4.70 (d,J=13.6Hz, 1H), 4.54 (d, J=10.8Hz, 1H), 4.51 (d,J=12.4 Hz, 1H), 4.44 (d,J=12.4Hz, 1H), 4.32 (d,J=10.8 Hz, 1H),4.26(d,J=9.6Hz, 1H), 3.83 (d,J=10.8 Hz, 1H), 3.73 (t,J=8.8 Hz,1H), 3.66-3.45 (m, 7H); MS (ESI) m/z 707 ([M+H]*), 729 (M+Na). 4.2.50. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(5-phenylfuran-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (65) To a mixture of hydrazide 32 (674 mg, 0.95 mmol),5-phenyl-2-furoic acid (233 mg, 1.24 mmol), EDCI (273 mg, 1.43 mmol), andHOBt hydrate (295 mg, 1.90 mmol) in DMF (10 mL) was addedNMM (0.31 mL, 2.85 mmol). The resulting mixture was stirred atambient temperature overnight. After dilution with EtOAc, the or-ganic layer was subsequently washed with water, hydrochloricacid (1.0 N), saturated sodium bicarbonate, and brine prior todrying over anhydrous MgSO4. Filtration and removal of the vola-tiles under reduced pressure yielded a glassy yellow amorphoussolid, which was carried on to the next step without furtherpurification. To a solution of the acylhydrazide in THF (20 mL) was addedLawesson reagent (1.2 g,2.9 mmol). The reaction mixture was re-fluxed overnight. After cooling to room temperature, the reactionmixture was diluted with EtOAc and washed with saturated so-dium bicarbonate. The organic layer was dried over anhydrousMgSO4, filtered through a plug of silica gel and concentrated in va-cuo. The resulting crude residue was carried on to the next stepwithout further purification. To a solution of the perbenzylated thiadiazole in acetonitrile(5mL) was added TMSI (5 mL). The resulting reaction mixturewas heated to 50°C overnight. After cooling to 0℃, the400-188-0725 reaction quenched with methanol and concentrated in vacuo.The resulting crude residue was purified by reverse phase pre-parative HPLC to provide the title compound (176mg, 36%) as awhite solid. H NMR (400 MHz, DMSO-d6) 8 7.79-7.77 (m, 2H),7.52 (d, J=2.0Hz, 1H), 7.46-7.31 (m, 6H), 7.20 (d, J=3.6 Hz,1H), 4.95 (dd, J=6.8, 4.8 Hz, 2H), 4.87 (d,J=5.6Hz, 1H), 4.61(d,J=16.0Hz, 1H), 4.56 (d,J=16.0 Hz, 1H), 4.50 (t, J=6.0 Hz,1H). 4.02 (d,J=9.6Hz, 1H), 3.68-3.62 (m, 1H), 3.43-3.40 (m,1H), 3.27-3.06 (m, 4H); 13c NMR(100MHz, DMSO-d6))0167.63,158.60,155.97,144.51, 143.71,140.72,134.63,132.43,131.30,129.52,129.41,129.29,129.14,124.34, 114.87,109.08,81.68, 80.90, 78.73, 75.19,70.71,61.78,33.88; MS (ESI) mz515 (M+H)*. 4.2.51. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(trifluoromethyl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (48) Compound 48 was synthesized from 26 and trifluoroacetic acidaccording to the procedure used to prepare 65. A white solid. Yield56%.HNMR(400 MHz, DMSO-d6)8 7.53(d,J=2.0Hz, 1H), 7.43(d,J=8.4Hz, 1H), 7.33 (dd,J=8.4, 2.4 Hz, 1H), 4.95-4.91 (m,2H), 4.85(d,J=5.6Hz, 1H), 4.71(d,J=16.4 Hz, 1H), 4.67 (d,J=16.4Hz, 1H),4.41(t,J=6.0Hz, 1H), 4.01 (d,J=9.2 Hz, 1H), 3.69-3.65 (m, 1H),3.46-3.40 (m, 1H), 3.25-3.04(m, 4H); MS (ESI) m|z 441 (M+H)463(M+Na)*. 4.2.52. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(2,2,2- trifluoroethyl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6- (hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (49) To a solution of acid 21 (2.8 g, 4.0 mmol) in 1,4-dioxane (30 mL)was added thiosemicarbazide 28 (0.40 g, 4.4 mmol). The solution isstirred for 1 h at 90C, before adding of phosphorus oxychloride(0.41 mL, 4.4 mmol). After overnight stirring at 110 °C, the reactionwas concentrated in vacuo to yield a glassy off-white amorphoussolid, which was carried on to the next step without furtherpurification. To a mixture of the thiadiazole amine and copper (0.04 g,0.62 mmol) in c-hydrochloric、acid((6mL) and acetic acid(30 mL) at 0°℃ was slowly added sodium nitrite (2.8 g, 40 mmol)while maintaining an internal reaction temperature below 10℃.After 1 h stirring at 0℃, the resulting mixture was stirred withgradual warming to ambient temperature over 2 h. The mixturewas diluted with water and extracted with EtOAc. The organiclayer was dried over anhydrous MgSO4, filtered and evaporatedin vacuo. Purification was accomplished by column chromatogra-phy (10-60% EtOAc/hexanes) to yield the title compound (0.66g,022%) as a yellow oil. 1H NMR (400 MHz, CDCl;) 8 7.48 (s, 1H),7.37-7.17 (m, 20H), 6.93-6.91 (m, 2H), 4.90 (S, 2H), 4.85(d,J=10.8 Hz, 1H), 4.63-4.51(m, 4H), 4.45 (d,J=10.8Hz, 1H), 4.20(d,J=9.6Hz, 1H), 3.88 (d,J=10.8Hz, 1H), 3.81-3.72 (m, 5H),3.60-3.57 (m, 1H), 3.43 (t, J=8.8 Hz, 1H); MS (ESI) m/z 767(M+H), 789 (M+Na)*. 4.2.54. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(methylthio)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (47) To a solution of chlorothiadiazole 29 (419 mg, 0.55 mmol in THF(10 mL) was added sodium methanethiolate (77 mg, 1.1 mmol).The resulting mixture was stirred at ambient temperature over-night. After cooling to 0 °C, the reaction was quenched with hydro-chloric acid (1.0N) and diluted with EtOAc and water. The organiclayer was separated and the aqueous layer was extracted withEtOAc. The combined organic layers were washed with brine, driedover anhydrous MgSO4, filtered and concentrated in vacuo. Theresulting residue was carried on to the next step without furtherpurification. To a solution of the perbenzylated thiadiazole in acetonitrile(5mL) was added TMSI (5 mL). The resulting reaction mixturewas heated to 50C overnight. After cooling to 0 ℃, the reactionquenched with methanol and concentrated in vacuo. The resultingcrude residue was purified by reverse phase preparative HPLC toyield the title compound (58 mg, 25%) as a white solid.IH NMR(400 MHz, DMSO-d6)8 7.45(d,J=2.0Hz, 1H), 7.40 (d,J=8.0 Hz,1H), 7.29 (dd,J=8.0, 2.0 Hz, 1H), 4.49 (d,J=15.6Hz, 1H), 4.44 (d,}=15.6 z, 1H), 3.99 (d,J=9.6Hz, 1H), 3.67 (dd, J=12.0,2.0 Hz,1H), 3.42 (dd,J=11.6, 5.6 Hz, 1H), 3.25-3.11(m, 3H),3.06 (t,J-9.2Hz,1H), 2.67 (s, 3H); MS (ESI) m/z 419(M+H), 441(M+Na)*. 4.2.55.(2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-methoxy-1,3,4- thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (46) Compound 46 was synthesized from 29 and sodium methoxideaccording to the procedure used to prepare 47. A white solid. Yield57%.H NMR (400 MHz, DMSO-d6)8 7.42-7.39 (m, 2H), 7.29 (dd,J=8.4, 2.0 Hz, 1H), 4.97-4.94(m, 2H), 4.85 (d,J=5.6 Hz, 1H),4.444 (t,J=5.6Hz,,11H),4.37 (d, J=15.6Hz, 1H),4.32 (d,J=15.6Hz, 1H), 4.01(d,J=9.2Hz, 1H), 3.69-3.65(m, 1H), 3.45-3.40 (m, 1H), 3.27-3.03 (m, 4H), 3.39(s, 3H); MS (ESI) m/z 403(M+H), 425(M+Na)t 4.2.56. 2-(2-Chlor0-5-((2S,3S,4R,5R,6R)-3,4,5-tris(benzyloxy)-6-(benzyloxymethyl)-tetrahydro-2H-pyran-2-yl)benzyl)-5-(pyrazin-2-yl)-1,3,4-thiadiazole (59) To a mixture of acid 21 (1.00 g, 1.44 mmol), pyrazine-2-carbo-hydrazide 30 (259mg, 1.88 mmol), EDCI (415 mg, 2.16 mmol),and HOBt hydrate (448 mg, 2.89 mmol) in DMF (10 mL) was addedNMM (0.476 mL, 4.33 mmol). The resulting mixture was stirred atambient temperature overnight. After dilution with EtOAc, the or-ganic layer was subsequently washed with water, hydrochloricacid (1.0N), saturated sodium bicarbonate, and brine prior to dry-ing over anhydrous MgSO4. Filtration and removal of the volatilesunder reduced pressure yielded a glassy off-white amorphous so-lid, which was carried on to the next step without furtherpurification. To a solution of the acylhydrazide in THF (20 mL) was addedLawesson reagent (874mg, 2.16 mmol). The reaction mixturewas refluxed overnight. After cooling to room temperature, thereaction mixture was diluted with EtOAc and washed with satu-rated sodium bicarbonate. The organic layer was dried over anhy-drous MgSO4, filtered through a plug of silica gel and concentratedin vacuo. The resulting crude residue was carried on to the nextstep without any purification. To a solution of perbenzylated pyrazine at -50℃ in Ac20(10 mL) was slowly added a solution of trimethylsilyl trifluoro-methanesulfonate (1.5 mL, 8.4 mmol) in Ac20 (5 mL). The resultingmixture was stirred with gradual warming to ambient temperatureover 12 h, cooled to 0C, and quenched with saturated sodiumbicarbonate. After dilution with EtOAc, the organic layer waswashed with sodium bicarbonate and brine prior to drying over anhydrous MgSO4. Filtration and removal of volatiles under re-duced pressure yielded peracetylated compound as a yellow amor-phous solid. Deacetylation was achieved by addition of sodiummethoxide (25 wt.% in methanol, 0.5 mL) to a solution of this resi-due in methanol (5mL). The reaction mixture was stirred at ambi-ent temperature for 2 h. Amberlite IR120 hydrogen form was thenadded to neutralize the reaction mixture. The resin was filtered offand the filtrate was concentrated in vacuo. The resulting crude res-idue was purified by reverse phase preparative HPLC to yield thetitle compound (47 mg, 10%) as a white solid. H NMR (400 MHz,DMSO-d6)8 9.39 (d, J=1.6 Hz, 1H), 8.78 (d,J=2.4Hz, 1H), 8.73(dd, J=2.4, 1.6 Hz, 1H), 7.53 (d,J=1.6Hz, 1H), 7.43 (d,J=8.4 Hz,1H), 7.31 (dd,J=8.4,1.6 Hz, 1H), 4.50 (br, 2H), 4.87 (d,J=5.6Hz,1H), 4.64 (d,J=16.4 Hz, 1H), 4.59 (d,J=16.4Hz, 1H), 4.43 (t,J=5.6 Hz, 1H), 4.02 (d, J=9.2Hz, 1H), 3.69-3.65 (m, 1H), 3.44-3.41 (m, 1H), 3.26-3.07 (m, 4H); 13c NMR (100 MHz, DMSO-d6)s171.09,168.42,147.00,145.34,144.40,141.88,140.72, 134.59,132.40, 131.24,129.35,129.32, 81.67,80.92,78.71,75.21,70.70,61.76, 34.10, MS (ESI) m/z 451(M+H); HRMS(FAB) m|z451.0842 [calcd for C19H20CIN405S (M+H)*451.0843] 4.2.57. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(6-methylpyridin-2-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (54) Compound 54wassynthesized from 21 and16-meth-ylpicolinohydrazide according to the procedure used to prepare59. A white solid. Yield 31%. 1H NMR (400 MHz, DMSO-d6)87.99(d,J=7.6Hz, 1H), 7.86 (t, J=7.6 Hz, 1H), 7.52 (d,J=1.6Hz, 1H)7.43 (d,J=8.4Hz, 1H), 7.38 (d, J=7.6Hz, 1H), 7.31 (dd,=8.4,1.6Hz, 1H), 4.96-4.93 (m, 2H), 4.86 (d,J=5.6Hz, 1H), 4.583((d,J=15.6Hz, 1H), 4.53 (d,J=15.6Hz, 1H), 4.44 (t,J=5.6Hz, 1H),4.02 (d,J=9.6 Hz,1H), 3.69-3.65 (m,1H),3.45-3.40(m,1H),3.27-3.05 (m,4H), 2.47 (s, 3H); MS (ESI)m/z 464(M+H),486(M+Na)*. 4.2.58. (2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(2-methylpyridin-4-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol (56) Compound 56 wassynthesizedfrom21aand 22--mmeth-ylisonicotinohydrazide according to the procedure used to prepare59. A white solid. Yield 45%6.1HNMR1(400MHz, DMSO-d6)8 8.56(d, J=4.8Hz, 1H), 7.74 ((S,1H), 7.67 (d,J=4.8 Hz, 1H), 7.53 (d,J=1.6Hz, 1H), 7.44 (d,J=8.0Hz, 1H), 7.32 (dd,J-8.0, 1.6 Hz,1H), 4.98-4.94(m,2H),4.87(d, J=6.0 Hz,1H),544.64 (d,J=16.0 Hz, 1H), 4.53 (d,J=16.0Hz, 1H), 4.44 (t, J=5.6 Hz, 1H)4.01 (d,J=9.2Hz, 1H), 3.68-3.66 (m, 1H), 3.45-3.39 (m, 1H),3.27-3.05(m, 4H), 2.56 (s, 3H); MS (ESI) m/z 464 (M+H), 486(M+Na). 4.2.59.(2S,3R,4R,5S,6R)-2-(4-Chloro-3-((5-(pyridazin-4-yl)-1,3,4-thiadiazol-2-yl)methyl)phenyl)-6-(hydroxymethyl)-tetrahydro-2H-pyran-3,4,5-triol(60) Compound 60 was synthesized from 21 and pyridazine-4-carbo-hydrazide according to the procedure used to prepare 59. A whitesolid. Yield 21%. H NMR(400MHz, DMSO-d6) 8 9.71 (d,J=2.4 Hz, 1H), 9.51 (d,J=5.6Hz, 1H),8.15(dd,J=5.6,2.4Hz, 1H),7.54 (d, J=1.6 Hz, 1H), 7.44 (d,J=8.4Hz, 1H), 7.32 (dd,J=8.4,1.6 Hz, 1H), 5.03-4.98 (m, 2H), 4.91 (d,J=5.6Hz, 1H), 4.68 (d,J=16.0 Hz, 1H), 4.63 (d,J=16.0Hz, 1H), 4.45 (t, J=5.6 Hz, 1H),4.02 (d, J=9.6 Hz, 1H), 3.69-3.65 (m, 1H), 3.44-3.41 (m, 1H),3.26-3.06 (m, 4H); MS (ESI) m|z 451(M+H),473(M+Na)*. 4.3. In vitro assav 4.3.1. Cloning and cell line construction for human SGLT2 Human SGLT2 (hSGLT2) gene was amplified by PCR from cDNA-Human Adult Normal Tissue Kidney (Invitrogen, Carlsbad, CA). The hSGLT2 sequence was cloned into pcDNA3.1(+) for mammalianexpression and were stably transfected into chinese hamster ovary(CHO) cells. SGLT2-expressing clones were selected based on resis-tance to G418 antibiotic (Geneticin@, Invitrogen, Carlsbad, CA) andactivity in the 14c-a-methyl-p-glucopyranoside (14C-AMG) uptakeassay. 4.3.2. Inhibitory effects on human SGLT2 activities For sodium-dependent glucose transport assay, cells expressinghSGLT2 were seeded into a 96-well culture plate at a density of5×104 cells/well in RPMI medium 1640 containing 10% fetal bo-vine serum. The cells were used 1 day after plating. They wereincubated in pretreatment buffer (10 mM HEPES, 5 mM Tris,140 mM choline chloride, 2 mM KCl, 1 mM CaCl2, and 1 mM MgCl2,pH 7.4) at 37 ℃ for 10 min. They were then incubated in uptakebuffer (10 mM HEPES, 5 mM Tris, 140 mM NaCl, 2mM KCl,1 mMCaCl2,1 mM MgCl2, and 1 mM 14c-nonlabeled AMG pH 7.4) con-taining 14c-labeled (8uM) and inhibitor or dimethyl sulfoxide(DMSO) vehicle at 37℃ for 2h. Cells were washedd twice withwashing buffer (pretreatment buffer containing 10 mM AMG atroom temperature) and then the radioactivity was measured usinga liquid scintillation counter. ICso was determined by nonlinearregression analysis using GraphPad PRISM.24,25 4.4. Normal and diabetic animal studies 4.4.1. Animals Male SD rats and diabetic db/db mice were purchased byCharles River Laboratory.All animals were housed at 23±2C un-der a 12-h light/dark cycle (light on 7:00) and were fed a standardchow and water ad libitum. 4.4.2. Urinary glucose excretion in normal animal24-26 For glucosuria assessment,overnight-fasted SD rats (5 weeks ofages) were placed into metabolism cages for baseline urine collec-tion over 24 h. Rats were weighted, randomized into experimentalgroups (n=4) and orally administered with 50% aqueous glucosesolution(2g/kg) and drugs. Rats were returned to metabolismcages for 24 h urine collection. After the urine volume had beenmeasured, the glucose concentration in the urine was determinedusing a LabAssayTM (Wako Pure Chemicals). These data were nor-malized per 200 g body weight 4.4.3. Blood glucose effects in diabetic db/db mice24-26 For assessment of acute blood glucose effects in db/db mice, theanimals were weighed and randomized into two groups (n=4).Mice were dosed with vehicle or drug (9@1 mg/kg, 61@10 mg/kg)and blood glucose level were measured immediately before dosingand at 1, 2, 4, 5, 6, and 24 h post-dose using blood glucose meter(Lifescan, a Johnson & Johnson). The animals were allowed to re-. feed after the 6-h time point. Acknowledgments This paper is dedicated to the memory of Mr. Young-Sup Huh,Chairman, CEO, Green Cross Corporation (GCC). We appreciateDr. Chong-Hwan Chang for his leadership and dedication duringhis tenure of office as Head of GCC R&D. Also we are grateful toDr. Eun Chul Huh for his leadership and supports during his tenureas Head of GCC R&D Planning and Coordination. References and notes ( 1. International D iabetes Federation Diabetes Atlas, 3rd ed.; International Diabetes F ederation: Brussels, B elgium, 2006. ) ( 2. Dwarakanathan, A. J. Insur. Med. 2006, 38,20 . ) 3. Mackenzie, B.; Loo, D. D. J. Biol. Chem. 1996, 20,32678. ( 4. Wright, E . M. Am. J. P hysiol. 2001,280, F1 0 . ) 5. Z2hou, L.; Cryan, E.V.; D'Andrea, M. R.; Belkowski,S.; Conway, B. R.; Demarest, K.T.J. Cell. Biochem. 2003, 90, 339. 6.E1vans, S.; Grasset, E.; Heyman,M.; Dumonitier, A. M.; Beau,J. P.; Desjeux,J.-F.J.Pediatr. Gastroenterol. Nutr. 1985,4,878. 7. Moe,O. W.; Berry, C. A.; Rector, F. C. In The Kidney; Brenner, B. M.,Rector, F.C.,Eds., 5th ed.; WB Saunders Co.: Philadelphia, 2000; pp 375-415. 8.v\an den Heuvel,L. P.; Assink, K.; Willesen, M.; Monnens, L. Hum. Genet. 2002,111,544. 9. E1hrenkranz, J. R. L.; Lewis, N. G.; Kahn, C. R.; Roth, J. Diabetes Metab. Res. Rev.2005,21,31. 10.T1sujihara, K.; Hongu, M.; Saito, K.; Kawanishi, H.; Kuriyama, K.; Matsumoto,M.; Oku,A.; Ueta, K.; Tsuda, M.; Saito, A. J. Med. Chem. 1999,42,5311. 11. Fushimi, N.; Ito, F.; Isaji, M. PCT Int. Appl. WO2003011880,2004. 12.FuIjikura, H.; Fushimi, N.; Nishimura, T.; Nakabayashi, T.; Isaji, M. PCTInt. Appl.WO2002053573,2002. 13.E1llsworth, B. A.; Meng, W.;Patel, M.; Girotra, R. N.; Wu, G.; Sher, P.; Hagan, D.;Obermeier, M.; Humphreys, W. G.; Robertson, J. G.; Wang, A.; Han, S.; Waldron,T.; Morgan, N. N.; Whaley,J. M.; Washburn, W.N. Bioorg. Med. Chem. Lett. 2008,18,4770. 14. Meng, W.; Ellsworth, B. A.; Nirschl, A. A.; McCann, P. J.; Patel, M.; Girotra,R. N.;Wu, G.; Sher, P. M.; Morrison, E. P.; Biller, A. A.; Zahler, R.; Deshpande, P. P.;Pullockaran, A.; Hagan, D. L.; Morgan, N.; Taylor, J. R.;Obermeier, M. T.;Humphreys, W. G.; Khanna, A.; Discenza, L.;Robertson, J. G.; Wang, A.; Han,S.;Wetterau, J. R.; Janovitz, E. B.; Flint, O. P.; Whaley,J. M.; Washburn, W. N. J.Med. Chem.2008, 51,1145. 15. Washburn, W. N. J. Med. Chem. 2009, 52,1785. 16.NNomura, S.; Sakamaki, S.; Hongu, M.; Kawanski, E.; Kog1a, Y.; Sakamoto, T.;Yamamoto, Y.; Ueta, K.; Kimata, H.; Nakayama, K.; Tsuda-Tsukimoto, M.Presented at the 238th American Chemical Society National Meeting andExposition, Washington, DC, August 16-20, 2009; Medi-151. 17. Johnson & Johnson Pharmaceutical, www.clinicaltrials.gov, NCT00968812,2009. 18. According to a report from Business Wire on October 02, 2009, dapagliflozinstudy demonstrated significantly improved glycemic control and weightreduction in type 2 diabetes patients inadequately controlled with metformin. 19. EEllsworth, B. A.; Doyle, A. G.; Patel, M.; Caceres-Cortes, J; Meng, W.;Deshpande, P. P.; Pullockaran, A.; Washburn, W. N. Tetrahedron: Asymmetry2003,14,3242. 20.).((a) Kraus, G. A.;Molina, M. T.J.Org. Chem. 1998,53,752; (b) Xie,J.; Durrant, F.;Valery, J.-M. J. Org. Chem. 2003,68,7896. ( 21. J ung, M. E.; L yster, M. A. J . Org. Chem. 1997,42,3761. ) 22.C(hauvire, G.; Bouteille, B.; Enanga, B.; de Albuquerque, C.; Croft, S. L.; Dumas,M.; Pri, J.J. Med. Chem. 2003, 46, 427. ( 23. A ngibeaud, P.; Ut i le, J.-P. Synthesis 1 991, 737. ) ( 24. H F an, S.; Hagan, D. L.; Ta y lor, J. R. ; Xin, L. ; Meng, W.; Biller, S. A.; Wetterau,J. R .; W ashburn, W . N.; Whaley, J. M . Diabetes 2008,57,1723. ) ( 25. K atsuno, K.; Fujimori, Y.; T akemura, Y . ; H i ratochi,M.; It o h, F . : Komatsu, Y . ; ) ( F ujikura, H .; I saji, M. J . Pharmacol. Exp. Ther.2007, 320,323. ) ( 26. A A rakawa, K.; I shihara, T .; Oku, A.; Nawano, M.; U e ta, K . ; K i t amura, K . ; ) ( Matsumoto, M. ; Sa i to, A . Br. J . Pharmacol. 2001,1 3 2,578. ) S- see front matter @ Elsevier Ltd. All rights reserved.doi:j.bmc.ww.rysstech.com耐士科技 士科技www.rysstech.com
确定















还剩15页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中主要物质含量分析检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多