








Some 5,6-dihydropyrazolo/pyrazolo[1,5-c]quinazoline derivatives were rationally designed, synthesized and evaluated for in vitro xanthine oxidase inhibitory activity for the first time. Some notions about structure activity relationships are presented. The compounds 6g, 6h and 6e were found to be significantly active against XO. The compound 6g emerged as the most potent XO inhibitor as compared to allopurinol and free radical scavenger. The molecular docking of 6g into the XO active site highlighted its mode of binding and important interactions such as hydrogen bonding, p–p stacking with amino acid residues
like Ser876, Thr1010, Phen914, Phe1009 and Phe649 and its close proximity to dioxothiomolybdenum (MOS).
方案详情

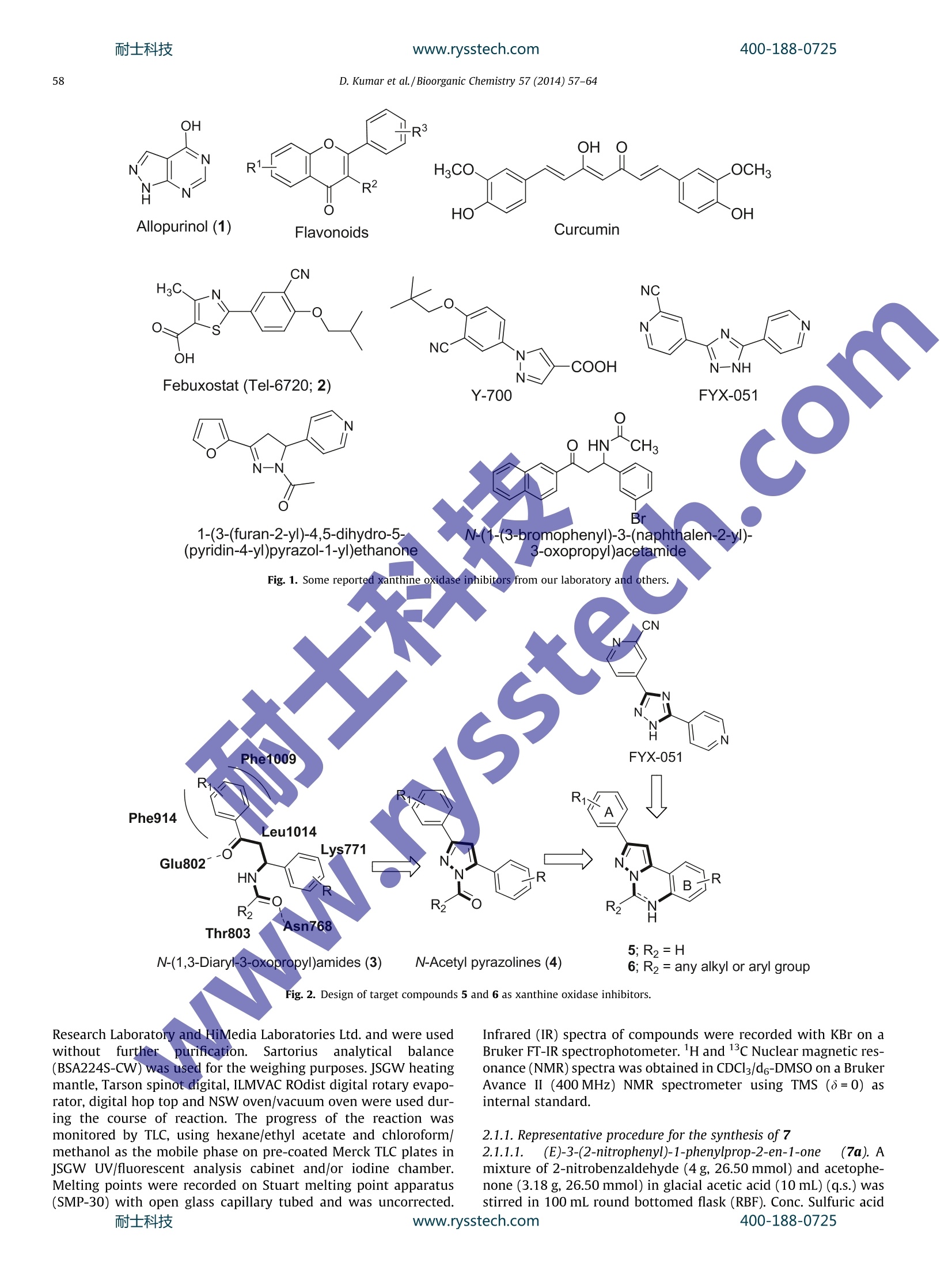
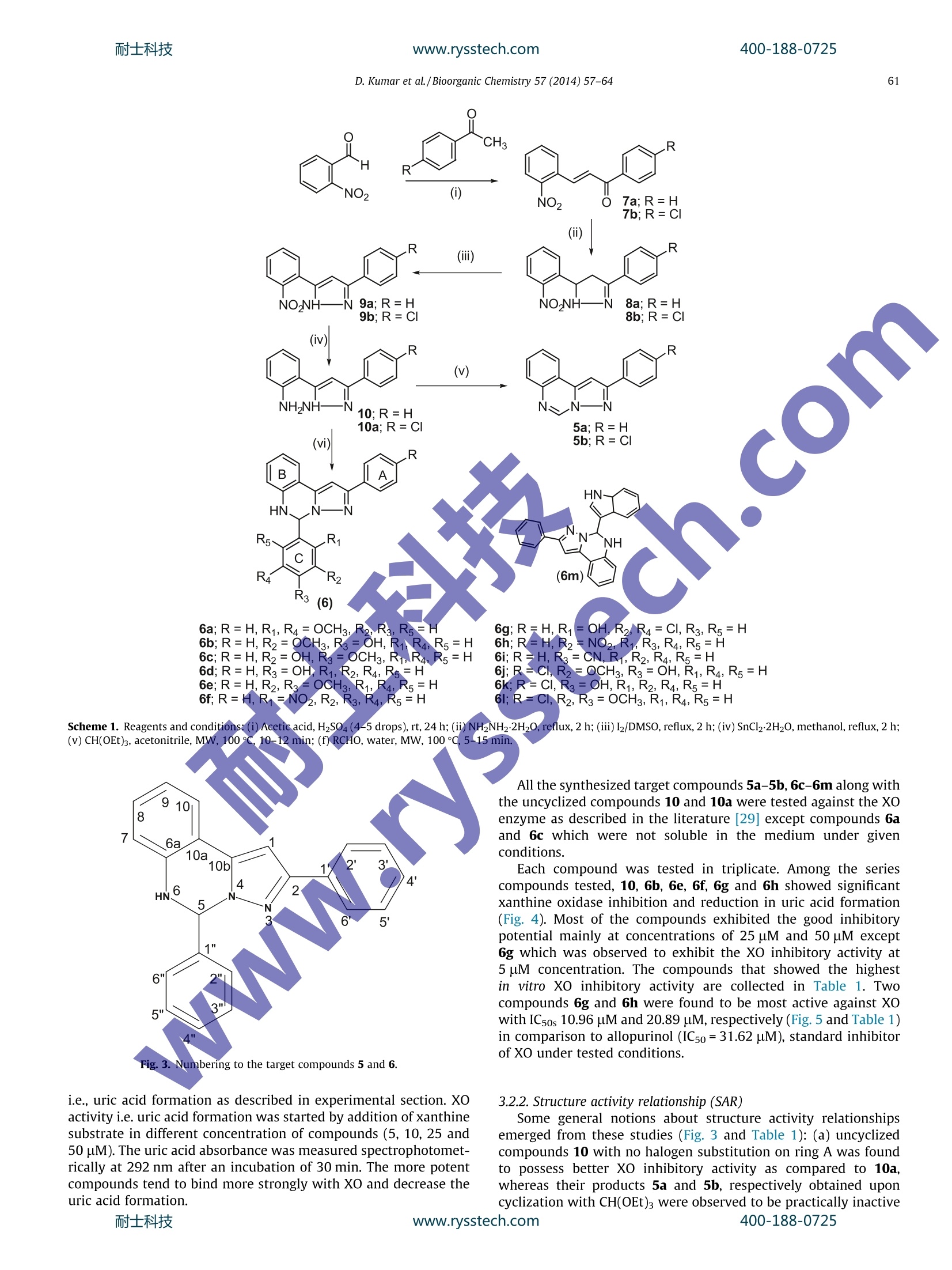
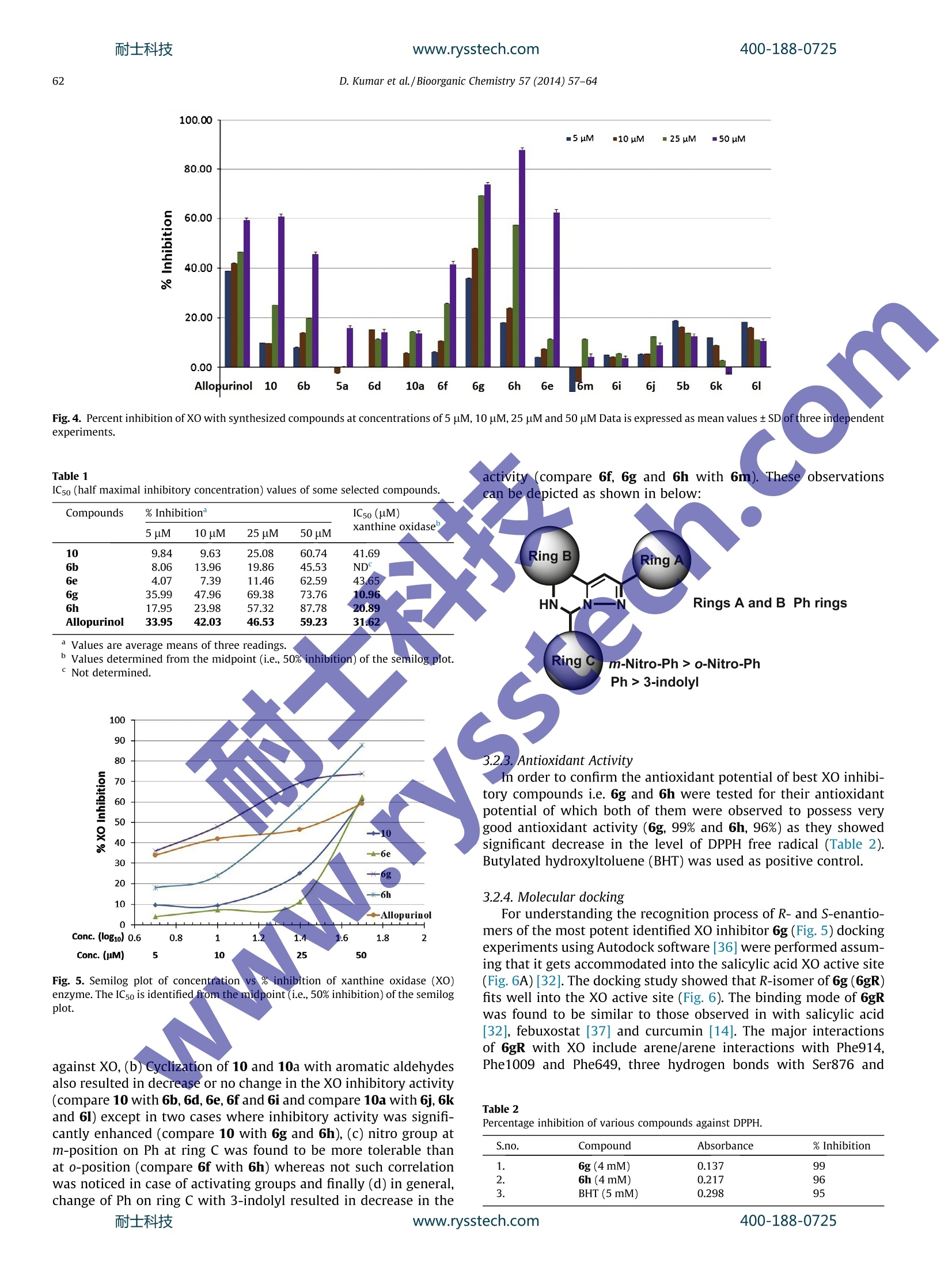
耐士科技www.rysstech.comBioorganic Chemistry 57 (2014)57-64400-188-0725 400-188-0725耐士科技www.rysstech.com58 Contents lists available at ScienceDirect Bioorganic Chemistry E LSEVIER journalhomepage: www.elsevier.com/locate/bioor g Synthesis and xanthine oxidase inhibitory activity CrossMark of 5,6-dihydropyrazolo/pyrazolo[1,5-c]quinazoline derivatives Deependra Kumar , Gagandeep Kaur , Arvind Negi, Sanjeev Kumar", Sandeep Singh, Raj Kumara Laboratory for Drug Design and Synthesis, Centre for Chemical and Pharmaceutical Sciences, School of Basic and Applied Sciences, Central University ofPunjab, Bathinda 151001, India " Centre for Biosciences, School of Basic and Applied Sciences, Central University of Punjab, Bathinda 151 001, India ‘Centre for Genetic Diseases and Molecular Medicine, Central University of Punjab, Bathinda 151 001, India ARTICLE INF O ABSTRACT Article history: Some 5,6-dihydropyrazolo/pyrazolo[1,5-c]quinazoline derivatives were rationally designed, synthesized Received 27 May 2014 and evaluated for in vitro xanthine oxidase inhibitory activity for the first time. Some notions about struc- Available online 30 August 2014 ture activity relationships are presented. The compounds 6g, 6h and 6e were found to be significantly active against XO. The compound 6g emerged as the most potent XO inhibitor as compared to allopurinol Keywords: and free radicall scavenger. The molecular docking of 6g into the XO active site highlighted its mode of Xanthine oxidase binding andimportant interactions such as hydrogen bonding, n-n stacking with amino acid residues 5,6-Dihydropyrazolo/pyrazolo[1,5- like Ser876,Thr1010, Phen914, Phe1009 and Phe649 and its close proximity to dioxothiomolybdenum c]quinazoline(MOS) Synthesis Molecular modeling@ 2014 Elsevier Inc. All rights reserved. Antioxidant Xanthine oxidase (XO; EC 1.17.3.2) is well characterizeddruggable target [1-3] for the treatment and management of dis-ease conditions involving high uric acid level; hyperuricemia andgout [4,5] due its significant role in catalytic oxidative hydroxyl-ation of hypoxanthine and xanthine to produce uric acid. FurtherXO is also associated and unregulated in pathological conditionsinvolving inflammation, metabolic disorders, cellular aging,reper-fusion damage, atherosclerosis,hypertension [6,7] and carcinogen-esis as it generates superoxide anions, H202 and highly reactivespecies (ROS) during the catalytic process2Thus the selectiveinhibition of XO may result in broad spectrum therapeutics forgout, cancer, inflammation and oxidative damage [2-4]. In general XO inhibitors haye been classified into purine andnon-purines based on their scaffolds mimicking to naturally occur-ring purines (Fig. 1). Allopurinol (1) was the first purine based XOinhibitor approved by the US FDA in 1966 for treatment of gout andhyperuricemia [8]. Due to the reported hypersensitivity (Stevens-Johnsons) syndrome induced by allopurinol use [9,10], researchersinitiated their search for new XO inhibitors derived non-purineskeleton [4,11-16] and a great success has been achieved in thisdirection which is:eevident by a recent US FDA approval of ( * C orresponding author.F a x: +91 1 6 4 2240555. ) ( E -mail a ddresses: r a j .khunger@gmail.com, r a jcps@cup.ac.in ( R . Kumar).C U PB Li b rary C o mmunication Number: P128/14. ) febuxostat [17](2) having lesser and non-life threatening sideeffects as compared to allopurinol [18-20]. In continuation to our earlier successful attempts made towardsthe search for new xanthine oxidase inhibitors based on N-(1,3-diaryl-3-oxopropyl)amides [11] (3) and N-acetyl pyrazolines [12](4), we thought of designing and synthesizing 5,6-dihydro pyrazol-o[1,5-c]quinazolines (5 and 6) via structural modification of 3, 4and FYX-051 keeping in mind the following considerations(Fig. 2): (i) two aryl/heteroaryl rings (preferably joined by threeatoms)oriented in such a way so that favorable arene-arene inter-actions with Phe914 and Phe1009 amino acid residues may takeplace; (ii) a site for hydroxylation near molybdenum metal and(iii) bioisosteric replacement of free C=0 with C=N/C-NH fol-lowed by ring cyclization might enhance activity and target speci-ficity through hydrogen bond formation with Ser876 and/orThr1010 amino acid residues of XO active site. Pyrazolo[1,5-c]quinazolines have previously been reported topossess bioactivities such as benzodiazepine binding ligands [21],Gly/NMDA receptor, excitatory amino acid antagonists [22,23],IKK [24], phosphodiesterase 10A [25] inhibitors, AMPA and kainatereceptor antagonists [26] and adenosine receptor antagonists [27]. 2. Materials and methods 2.1. Chemistry All the reagents were of AR/GR quality and were purchasedfrom Sigma-Aldrich,Loba-Chemie Pt. Ltd., SD Fine Chemicals, Sisco D. Kumar et al./Bioorganic Chemistry 57(2014)57-64 Flavonoids Curcumin CN H3C. NC S N NC OH -COOH N-NH N: Febuxostat (Tel-6720; 2) Y-700 FYX-051 N O HN^CH3 moN-N c Br 1-(3-(furan-2-yl)-4,5-dihydro-5- N-(1-(3-bromophenyl)-3-(naphthalen-2-yl)- (pyridin-4-yl)pyrazol-1-yl)ethanone 3-oxopropyl)acetamide Fig. 1. Some reported xanthine oxidase inhibitors from our laboratory and others. Fig. 2. Design of target compounds 5 and 6 as xanthine oxidase inhibitors. Research Laboratory and HiMedia Laboratories Ltd. and were usedwithouttfurther purification. Sartoriusanalyticalbalance(BSA224S-CW) was used for the weighing purposes. JSGW heatingdict ,mantle, Tarson spinot digital, ILMVAC ROdist digital rotary evapo-rator, digital hop top and NSW oven/vacuum oven were used dur-ing the course of reaction. The progress of the reaction wasmonitored by TLC, using hexane/ethyl acetate and chloroform/methanol as the mobile phase on pre-coated Merck TLC plates inJSGW UV/fluorescent analysis cabinet and/or iodine chamber.Melting points were recorded on Stuart melting point apparatus(SMP-30) with open glass capillary tubed and was uncorrected. Infrared (IR) spectra of compounds were recorded with KBr on aBruker FT-IR spectrophotometer. H and 13c Nuclear magnetic res-onance (NMR) spectra was obtained in CDCl3/d6-DMSO on a BrukerAvance ⅡI (400 MHz) NMR spectrometer using TMS (8=0) asinternal standard. 2.1.1. Representative procedure for the synthesis of 72.1.1.1. (E)-3-(2-nitrophenyl)-1-phenylprop-2-en-1-one (7a). Amixture of 2-nitrobenzaldehyde (4g, 26.50 mmol) and acetophe-none (3.18 g, 26.50 mmol) in glacial acetic acid (10 mL) (q.s.) wasstirred in 100 mL round bottomed flask (RBF).Conc. Sulfuric acid400-188-0725 (4-5 drops) was added to the reaction mixture in ice cold conditionand further stirred at rt for 24 h. After the completion of thereaction (TLC), the reaction mixture was poured on ice cold waterand filtered. The solid was washed with water, dried and wasrecrystallized from methanol to afford the pure compound. Yield:85%; Creamy white solid; MP: 85-87C; IR (KBr, cm-): 1666(C=0), 1610 (C=C), 1510 (N=0), 1342 (N=0). 'H NMR (CDCl3, 300 MHz, 8 with TMS=0): 8.12(1H, d,J=15.9 Hz), 8.02 (3H, bs), 7.69-7.74(2H, m),7.50-7.57 (4H, m),7.34 (1H, d, J=15.9Hz); 3C NMR (CDCl3, 75MHz, s withTMS=0): 190.22,148.34,140.03,137.20, 133.56,133.09,131.08,130.31, 129.13,128.62,126.97,124.86. Similar protocol wasutilized for the synthesis of (E)-1-(4-chlorophenyl)-3-(2-nitro-phenyl)prop-2-en-1-one (7b). The above compounds were usedfor next steps. 2.1.1.2. 4,5-Dihydro-5-(2-nitrophenyl)-3-phenyl-1H-pyrazole (8a). Amixture of 7a(3.3g,113.04mmol) andhydrazine hydrate(0.63 mL, 13.04 mmol) was refluxed under methanol for 2 h. Aftercompletion of the reaction (TLC), the reaction mixture was cooledto rt and the precipitates (8a) so obtained were collected, dried andused for the next step for further reaction. Yield:97%; Orange crys-talline solid; MP: 140-142 ℃; IR (KBr, cm-): 3310 (N-H), 1518(N=0), 1333 (N=0), 1299(C=N); 1HNMR(CDCl3, 400 MHz, 8 withTMS=0): 7.96 (2H, t,J=8.4 Hz), 7.65 (3H, m), 7.41 (4H, m), 6.05(D20 exchangeable NH, 1H, s), 5.42 (1H, t,J=13.2Hz), 3.80, (1H,q,J=14.2 Hz), 3.01(1H,q,J=13.2 Hz). 13C NMR(CDCI, 100 MHz8 with TMS=0): 151.12,148.54, 138.04, 133.87,132.43,129.06,128.59,128.53,128.36,126.04,124.65,59.83,41.46. Similar proto-col was utilized for the synthesis of 3-(4-chlorophenyl)-5-(2-nitro-phenyl)-4,5-dihydro-1H-pyrazole (8b). 2.1.1.3. Synthesis of 5-(2-nitrophenyl)-3-phenyl-1H-pyrazole (9a). 8a(3.1 g,11.70 mmol) was refluxed with catalytic amount of molecu-lar iodine in DMSO at 130-140 °C for 2 h. After the completion ofreaction (TLC), the reaction mixture was poured in ice cold waterand was extracted using ethyl acetate (20 mL_×3).Organics werewashed with brine (10 mLx3),, ddried over sodium sulfate andevaporated under reduced pressure using rotary evaporator toobtain the product (9a))wwhich was used for next step. Yield;92%;Brownish color solid:MP:: 194-196℃; IR (KBr, cm ): 3456(N-H), 1664 (C=N), 1524 & 1348 (N=0), 1217 (C-N), H NMR(CDCl3, 400 MHz, 8 with TMS=0): 7.67-7.76 (3H, m), 7.56-7.63(3H. m), 7.33-7.51(5H, m);; 13c NMR (CDCl3, 100 MHz, 8 withTMS=0): 149.02, 146.37,146.16,132.09,130.97,129.04,128.74,128.04,126.47,125.62,124.56,123.82, 102.59. Similar protocolwas utilized for the synthesis of 5-(2-nitrophenyl)-3-phenyl-1H-pyrazole (9b). 2.1.1.4. Representative procedure for the synthesis of 2-(3-Phenyl-1H-pyrazol-5-yl)aniline (10). 9a (2.75 g, 10.38 mmol) was dissolved inmethanol and refluxed with 3 equivalent of stannous chloridedihydrate for 2 h. After the completion of the reaction (TLC), meth-anol was evaporated under reduced pressure using rotary evapora-tor followed by neutralization of reaction mixture using 5% NaOHsolution. Solid product was than extracted with ethyl acetate(10mL×3). Organics were dried over anhydrous sodium sulfateand evaporated under reduced pressure using rotary evaporatorto obtain the product 10a which was used for next step. IR (KBr,cm-): 3360 (N-H), 3285 (N-H), 1613 (C=N), 1579 (C=C), 1248(C-N); H NMR (CDCl3, 400 MHz, 8 with TMS=0): 13.23 (D20exchangeable NH, 1H, bs), 7.81 (1H, s), 7.79 (1H, s), 7.31-7.54(4H, m), 7.00 (2H, s), 6.75 (1H, d, J=7.91Hz), 6.61 (1H, t,J=7.32 Hz), 6.22 (D2O exchangeable NH2,2H, bs); 13CNMR (CDCl3,100 MHz, 8 with TMS=0): 149.11,148.13,145.37,133.05,129.13,129.10, 128.57,127.77, 125.14,119.23,115.63,115.29,99.58. MS (ESI): m|z=236.1[M+1]*. All the remaining reactions were carriedout using this general procedure [28]. 2.1.1.5. Representative procedure for the synthesis of 2-phenylpyraz-olo[1,5-c]quinazoline (5a). To a solution of 10 (1 mmol, 1.0 equiv)in MeCN (2 mL) was added CH(OEt)3(1.1 equiv) and was kept ina vial, sealed and put in Biotage Initiator Microwave synthesizer.The reaction mixture was then heated at 100℃ for 10 min (TLC)under MW. After the completion of the reaction, the reaction mix-ture was left to cool. The precipitates were filtered, dried andwashed with n-hexane (5 mL ×3) to afford the pure product 5a.Yield: 91%, Creamy solid: Mp 135-136℃; IR (KBr, cm-): 3019(CH), 1620 (C=N), 1539 (C=C), 1215 (C-N); 1H NMR (DMSO-do,600 MHz, 8 with TMS=0): 9.42(1H, s), 8.30 (1H, d,J=7.8Hz),8.09 (2H, d,J=8.4 Hz), 7.93 (1H, m), 7.87 (1H, s), 7.75(2H, m)7.6 (2H, m), 7.5(1H, m); 13c NMR (DMSO-d6, 150 MHz, 8 withTMS=0):155.27,140.34, 139.96,139.67,132.35,130.57,129.81,129.54,128.85,128.73,126.81,124.21,119.98, 96.65; MS (ESI):m/z=246 [M+1]t. All the remaining reactions werecarried outusing this general procedure [28]. 2.1.1.6. Representative procedure for the synthesis of 5-(2,5-dime-thoxyphenyl)-2-phenyl-5,6-dihydropyrazolo [1,5-c]quinazoline(6a)..AAImixture of 10 (235 mg, 1 mmol, 1.0 equiv) and 2,5-dime-thoxy benzaldehyde (1.0 equiv) in water(2 mL) was taken in a vial,sealed and kept in Biotage Initiator Microwave synthesizer. Thereaction mixture was then heated at 100 ℃ for 15 min (TLC) underMW. After the completion of the reaction, the reaction mixture wasleft to cool. The precipitates were filtered, dried and washed withn-hexane (10mL×3) to afford the pure product 6a. Yield: 80%,Light yellow solid, Mp 230-232℃; IR (KBr, cm-): 1644 (C=N),1587 (C=C), 1220 (C-N), 1123 (C-O); H NMR (CDCl3, 400 MHz, swith TMS=0): 15.60 (D20 exchangeable NH, 1H, bs), 8.76 (1H, d,J=8.0Hz), 8.37 (1H, d, J=8Hz), 8.04 (2H, m), 7.55(2H, d,J=8.0Hz),7.19 (5H, m),7.11 (3H, m), 7.27 (1H, d, J=8.27 Hz),3.70)(3H, s), 3.28 (3H, s); 13C NMR (CDCl3, 100MHz, 8 withTMS=0): 153.51,152.15,149.94,139.68,139.25,133.39,129.38,128.60,127.85,125.83,124.06,123.01,119.52,115.31,113.80,113.16,112.70,111.30, 96.62,67.99,56.05, 55.43; HRMS (TOF-ESI) Calcd for C24H21N302, 383.1634(M)*; observed: 384.1638(M+H). The remaining reactions were carried out using this general procedure [28]. We have recently reported the synthesis and the physical dataof 10-10a, target compounds 5a-5b and 6b-6m [28]. 2.2. Biological evaluation Xanthine oxidase enzyme as lyophilized powder was purchasedfrom SRL Pt. Ltd. and stored at temperature below 4C. Allopurinolwas purchased from SRL Pt. Ltd. Potassium phosphate monobasic(KH2PO4) and dibasic (K2HPO4), DMSO of AR grade were purchasedfrom Loba Chemie Pt. Ltd. Methanol was purchased from SDFCL.Analytical balance (JB1603-c/FACT) and pH meter (Mettler Toledo)were used for the purpose of weighing and measuring the pH,respectively. REMI laboratory refrigerator and ice flaking machine(MSW-136) by Macro scientific works were used for storing solu-tions. Double-beam UV-Visible spectrophotometer (Shimadzu)for measuring absorbance was used. 2.2.1. Evaluation of XO inhibitory activity 2.2.1.1. Principle of the XO assay. Xanthine oxidase assay is an enzy-matic reaction in which xanthine oxidase produces uric acid duringoxidation of xanthine. Uric acid can be easily analyzed with anabsorption wavelength of 292 nm quantifying xanthine oxidaseactivity [29]. 2.2.1.2. Preparation of reagents. ·Phosphate buffer (50mM) For the preparation of 50 mM phosphate buffer at pH 7.5,9.4mL (1M) KH2PO4 (Potassium phosphate monobasic) and40.6 mL (1M) K2HPO4 (Potassium phosphate dibasic) was dilutedup to 1000 mL with deionised water. ·Xanthine solution (0.10 mM) Stock solution of 0.1 (M) xanthine was prepared by dissolving15.2 mg of xanthine in 1 mL of(1M) NaOH solution. Working solu-tion was made in accordance with 1 pL/mL in deionised water andpH was maintained with 1 N HCl. · XO enzyme solutions (0.1-0.2 unit/mL) Stock solution of 16U/mL was prepared by dissolving 1 mg oflyophilised powder of xanthine oxidase enzyme in 1 mL of coldphosphate buffer reagent. For working solution stock solutionwas diluted up to required volume. 2.2.1.3. Procedure. The inhibitory effect on XO was measured spec-trophotometrically at 292 nm under aerobic condition. Allopurinolwas used as a positive control for the inhibition test. The reactionmixture consisted of 1.5 mL of 50 mM potassium phosphate buffer(pH 7.5), 1 mL test sample solution (5, 10,25,50, and 100 uM) wasdissolved in DMSO, 0.5 mL of freshly prepared XO enzyme solution(0.2 units/mL of xanthine oxidase in phosphate buffer). The assaymixture was pre-incubated at room temperature for 15 min. Then,1 mL of substrate solution (0.10 mM of xanthine) was added intothe mixture. The mixture was incubated at room temperature for30 min. Next, the reaction was stopped with the addition of 1 mLof 1 M HCl. The absorbance was measured using UV-VIS spectro-photometer against a blank prepared in the same way but thexanthine was replaced with the phosphate buffer. Readings weretaken in triplicate. 2.2.1.4. Calculation of inhibition. The: degree of XO inhibitoryactivity was calculated by following formula %Inhibition=[(Ac - As)/Acx100 where Ac=Absorbance of control (Sample in absence of XO inhibi-tors), As=Absorbance ofsample(Sample withpotentialXOinhibitors). 2.2.2. Evaluation of antioxidant activity Reactive oxygen species can harm various biomolecules includ-ing DNA and consequences are oxidative stress in the biologicalsystems. Antioxidants are known to act via reducing the oxidativestress by scavenging of the free radicals and therefore, preventmany diseases including cancer. Hence, we performed DPPH assayto study the antioxidant activity of our compounds in vitro. DPPHassay was used to determine the total antioxidant activity by scav-enging of the stable DPPH free radical. Antioxidant activity wasobserved to escalate inthe concentration dependent manner[30,31]. 2.2.2.1. Principle of DPPH assay. The principle of the assay is the useof the stable free-radical for estimating antioxidant activity 2,2-diphenyl-1-picrylhydrazyl (DPPH) which gets reduced and thusstabilized in presence of a hydrogen donating substance [30]. 2.2.2.2. Procedure. 2 mL of the different concentrations of com-pounds in methanol were added to 2 mL of 0.135 mM methanolicsolution of DPPH. The mixture was shaken vigorously and was leftto stand for 30 min in the dark. Absorbance was recorded at 517 nm against the blank. Blank was prepared without the addi-tion of DPPH. BHT was used as standard. 2.2.2.3. Calculation of inhibition. The ability to scavenge DPPH rad-ical was calculated by the following equation: DPPH radical scavenging activity(%)=[(Ac-As)/Ac]×100 where Ac=Control (Absorbance of DPPH radical+methanol),As=Sample (Absorbance of DPPH radical + compound). 2.2.3. Molecular docking The coordinates of the bovine milk XO complexed with salicylicacid were obtained from the protein data bank (PDB entry: 1FIQ[32]. The ligands were drawn in ChemDraw and subjected toenergy minimization via MM2 force field, integrated in Chem3DUltra. The ligands were docked into the active sites of xanthineoxidase using the Autodock 4.2.5.1, performs Lamarckian geneticalgorithm based ligand docking to optimize the conformation ofligand at the receptor binding site. We utilized the grid size60×60×60 with a centroid of 26.782×10.121×112.992 andspacing 0.375. It utilizes energy score fitness function to evaluatethe various conformation of ligand at the binding site. This is com-prised of various components such as binding energy, ligand effi-ciency, inhibition constant (uM), inter molecular energy, van derWaals desolvation energy, electrostatic energy, total energy, tor-sional energy, unbound energy. Each docking experiment was setwith 50 different runs with termination after 25,000 energy evalu-ation. Population size was set out 100. The pose was rankedaccording to its total energy Score fitness function. 3. Results and discussion 3.1. Chemistry For the synthesis of target compounds, we followed the route asdepicted in Scheme 1. Briefly, the 2-nitrobenzaldehyde was con-densed with aryl ketones to afford nitro substituted 1,3-diarylpropenones (7) under acid catalyzed conditions. Introduction ofpyrazoline ring system (8) was made feasible by refluxing 7 withhydrazine hydrate in methanol [33]. The structure of 8 was con-firmed by the characteristic ABX pattern with three double dou-blets in H NMR spectrum. Dehydrogenation of 8 to affordpyrazole structure (9) was carried out by refluxing with iodineand DMSO [34]. HNMR showed the disappearance of ABX patterndue to aromatization and appearance of c NMR signal to 100-103 ppm due to C-1 further confirmed the formation of 9. Reduc-tion of 9 using SnCl22H20 in methanol under refluxing afforded10. [35] IR spectra confirmed the formation of 10 as a sharp char-acteristic peak of primary amine (NH2) at 3350-3380 cm- wasobtained. The 10 was eventually cyclocondensed with triethylorthoformate and different aldehydes under microwave irradia-tions to yield target compounds 5 and 6, respectively [28]. A gen-eral observation on appearance of chemical shift values in 13cNMR spectra ranging from 66-72 8 and 140-145 8 was noticedfor C-5 in case of 6 and 5, respectively (Fig. 3). All the final productswere fully characterized by mp, IR, NMR and HRMS spectral data. 3.2. Biological evaluation 3.2.1. XO inhibitory activity In order to evaluate the biological activities of synthesized com-pounds, in vitro biochemical screening of the compounds was car-ried out using xanthine oxidase enzyme withUV-Visiblespectrophotometer at 292 nm. Four different concentrations ofcompounds were used for inhibiting the xanthine oxidase enzyme 6c;R=H,R2=OH,R3=OCH3,R R4. :,R5=H 6d;R=H, R3=OH,R1,R2,R4,R6=H 6e; R=H, R2, R3=OCH3,R1,F 6f;R=H,R1=NO2,R2, R3, R4, R5= 6h;R=H,R2=NO2,R,R3,R4,R5=H6i;R=H,R3=CN,R,R2,R4,R5=H6j;R=CI,R2=0CH3, R3=OH,R,R4,R5=H6k;R=CI, R3=OH,R1, R2, R4,R5=H6I;R=CI, R2,R3=OCH3,R1,R4, R5=H Scheme 1. Reagents and conditions: (i)Acetic acid,H2SO4 (4-5 drops), rt, 24 h;(ii)NH2NH22H20,reflux, 2 h; (iii) I2/DMSO, reflux, 2 h;(iv) SnClz2H20, methanol, reflux, 2 h;(v) CH(OEt)3, acetonitrile, MW, 100 ℃, 10-12 min; (f) RCHO, water, MW, 100℃,5-15 min. Fig. 3. Numbering to the target compounds 5 and 6. i.e., uric acid formation as described in experimental section. XOactivity i.e. uric acid formation was started by addition of xanthinesubstrate in different concentration of compounds (5, 10,25 and50 uM). The uric acid absorbance was measured spectrophotomet-rically at 292 nm after an incubation of 30 min. The more potentcompounds tend to bind more strongly with XO and decrease theuric acid formation. All the synthesized target compounds 5a-5b, 6c-6m along withthe uncyclized compounds 10 and 10a were tested against the XOenzyme as described in the literature [29] except compounds 6aand 6c which were not soluble in the medium under givenconditions. Each compound was tested in triplicate. Among the seriescompounds tested, 10, 6b, 6e, 6f, 6g and 6h showed significantxanthine oxidase inhibition and reduction in uric acid formation(Fig. 4). Most of the compounds exhibited the good inhibitorypotential mainly at concentrations of 25 uM and 50 uM except6g which was observed to exhibit the XO inhibitory activity at5 pM concentration. The compounds that showed the highestin vitro XO inhibitory activity are collected in Table 1. Twocompounds 6g and 6h were found to be most active against XOwith ICsos 10.96 uM and 20.89 uM, respectively (Fig. 5 and Table 1)in comparison to allopurinol (IC50=31.62 uM), standard inhibitorof XO under tested conditions. 3.2.2. Structure activity relationship (SAR) Some general notions about structure activity relationshipsDSemerged from these studies (Fig. 3 and Table 1): (a) uncyclizedcompounds 10 with no halogen substitution on ring A was foundto possess better XO inhibitory activity as compared to 10a,whereas their products 5a and 5b, respectively obtained uponcyclization with CH(OEt)3 were observed to be practically inactive Fig.4. Percent inhibition of XO with synthesized compounds at concentrations of 5 uM, 10 uM, 25 uM and 50 uM Data is expressed as mean values ±SD of three independentexperiments. IC5o (half maximal inhibitory concentration) values of some selected compounds. Compounds % Inhibition IC50 (pM) 5 pM 10 uM 25 uM 50 pM xanthine oxidase 9.84 9.63 25.08 60.74 41.69 8.06 13.96 19.86 45.53 ND 4.07 7.39 11.46 62.59 43.65 35.99 47.96 69.38 73.76 10.96 17.95 23.98 57.32 87.78 20.89 Allopurinol 33.95 42.03 46.53 59.23 31.62 Values are average means of three readings.Values determined from the midpoint (i.e., 50% inhibition) of the semilog plot.Not determined. Fig. 5. Semilog plot of concentrationn vs % inhibition of xanthine oxidase (XO)enzyme. The ICso is identified from the midpoint (i.e., 50% inhibition) of the semilogplot. against XO, (b) Cyclization of 10 and 10a with aromatic aldehydesalso resulted in decrease or no change in the XO inhibitory activity(compare 10 with 6b,6d, 6e,6f and 6i and compare 10a with 6j, 6kand 61) except in two cases where inhibitory activity was signifi-cantly enhanced (compare 10 with 6g and 6h), (c) nitro group atm-position on Ph at ring C was found to be more tolerable thanat o-position (compare 6f with 6h) whereas not such correlationwas noticed in case of activating groups and finally (d) in general,change of Ph on ring C with 3-indolyl resulted in decrease in the Ph > 3-indolyl 3.2.3. Antioxidant Activity In order to confirm the antioxidant potential of best XO inhibi-tory compounds i.e. 6g and 6h were tested for their antioxidantpotential of which both of them were observed to possess verygood antioxidant activity (6g, 99% and 6h, 96%) as they showedsignificant decrease in the level of DPPH free radical (Table 2).Butylated hydroxyltoluene (BHT) was used as positive control. 3.2.4. Molecular docking For understanding the recognition process of R- and S-enantio-mers of the most potent identified XO inhibitor 6g (Fig.5) dockingexperiments using Autodock software [36] were performed assum-ing that it gets accommodated into the salicylic acid XO active site(Fig.6A) [32]. The docking study showed that R-isomer of 6g (6gR)fits well into the XO active site (Fig. 6). The binding mode of 6gRwas found to be similar to those observed in with salicylic acid[32], febuxostat [37] and curcumin [14]. The major interactionsof 6gR with XO include arene/arene interactions with Phe914,Phe1009 and Phe649, three hydrogen bonds with Ser876 and Table 2Percentage inhibition of various compounds against DPPH. S.no. Compound Absorbance %Inhibition 6g (4mM) 0.137 6h (4mM) 0.217 BHT (5 mM) 0.298 D. Kumar et al./Bioorganic Chemistry 57 (2014) 57-64 Fig. 6. (A) Docking poses of R-isomer of 6g at the salicylic acid XO binding site (R-isomer: green color; salicylic acid: blue color) and, (B) Binding interactions of R-isomer of 6gwith the amino acid residues. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.) Thr1010 (Fig.6B). The other amino acids such as Glu802, Asn768and Lys771 also lie in close proximity of the compound. The phenylrings of Phe914 and Phe1009 lay parallel and perpendicular,respectively to the plane ofphenyl ring (ring C) of 6gR(d=4.59A and d=3.62A). The phenyl residue of Phe649 lies per-pendicular to phenyl ring (ring B) of 6gR (d=3.67 A). This kind ofarrangement ofarene-arene interactions has been previouslyobserved in the co-crystal structures of XO with salicylic acid[32] and febuxostat [37]. This conservation plays an important rolein stabilizing the binding positions of aromatic substrates andmight well represent one of the key features of the substrate recog-nition. Three hydrogen bonds, one between NH of the inhibitor andfree OH group of Ser876, second between OH of the inhibitor andOH of Ser876 and third between OH of the inhibitor and OH ofThr1010 were observed and provided the rationale for binding ofinhibitor. The ring C of the inhibitor gets positioned towards the dioxoth-iomolybdenum (MOS) moiety at a distance of 4.70 A indicatingthat it might block the activity of the enzyme from binding to sub-strate via hydroxylation. The docked conformation of S-enantiomerwas differing from the R-enantiomer. The favorable bindingconformation and higher interaction energy of R-enantiomersuggests its dominant role in XO inhibitory activity. 4. Conclusions We have rationally designed, synthesized and evaluated the XOinhibitory activity of some dihydropyrazolo/pyrazolo[1,5-c]quina-zolines first time. The SAR study disclosed that: (i) nature of ringsA, B and C, (ii) nature of substituents on the rings in particular onring C, and (iii) unsaturation i.e. 5,6-dihydropyrazolo[1,5-c]quina-zolines were found to affect the XO inhibitory activity. Compound 6g was found to be potent XO inhibitor as compared to allopurinoland capable of scavenge the free radicals in DPPH free radicalassay. In silico studies highlighted the role of amino acid residuesinvolved in interactions with 6g and the results are in compliancewith the previous reported studies. Further lead optimization on6g such as synthesis of compounds with diverse permutation andcombinations on ring A, B and C is under progress and will be pub-lished in due course Acknowledgment RK and AN thank University Grant Commission (UGC), NewDelhi, India for the financial assistance (F.No.42-676/2013(SR)). ( References ) ( D . P ar k s, D. Granger, Acta P hysiol . Scand. 5 48 (1986)87. ) ( P. P ac h er, A. Niv o rozhkin,C. Szabo, Pharmacol. R ev.58 (2006)87-11 4 . ) F. Borges, E. Fernandes,F. Roleira, Curr. Med. Chem. 9 (2002) 195-217. ( R . K umar, D a rpan,S. Sharma, R. Singh, Expert Opin . T h er. Pat.21 (2011) 1 071-11 0 8 . ) ( [5] H .-Y. Yuan,X . -H . Zhang , X.-L. Z h ang, J . - F . Wei, L . M eng, E xpert Opin. T h er. Pat. (2014) 1 - 1 8. ) ( [6 ] M . B oban, G. K o cic , S. R a de n kovic, R. P a v lovic, T . Cvetko v ic, M. De l ja n in-I l ic,S . I lic, M.D. Boba na , B. D jindjic , D. Stoj a n o vi c, R en . F a il . (2014)1- 6 . ) ( 1 J . Dawson, M . Walte r s, B r i t. J. Cl i n. Pharmacol. 6 2 ( 2 006) 63 3 -644. o1 1 ) ( j . R . Klinenb e rg, S .E. G oldfinger, J. E . S e e gmiller, Ann . I n tern. M e d. 62 (19 6 5 639-647. ) ( [9] J .Z. Si n ger, S.L . Wallace, Arthri t is Rheum. 29 ( 1 986 ) 82 - 87. ) ( [16] T . Sato, N. As h izawa, K . Matsumoto, T . I w anaga, H. Nakam u ra,T. I n ou e , O. Na g ata, Bio org . Med. Chem. Lett . 1 9 (2009)6225-6229. ) [17] K.-H. Yu, Recent Pat. Inflammation Allergy Drug Discovery 1 (2007) 69-75. ( 181 M .A. Schumacher, H.R. B e cker Jr . , R.L. Wor tm ann, P.A. MacD o nald, D . E ustace, W.A. P alo, J. Stre i t, N. Joseph-Ridge, N . En g l . J . Me d .353(2005)2450-2 4 61 . ) ( [19] H.R. Schumacher, M. A . B ecke r , R.L. Wort m ann, P.A. MacDona l d, B. H u n t, J.Streit , C. L ademacher, N. J oseph- R idge, Arthritis Care Res. 59 (2008) 1 540- 1 548. ) ( [20 ] Y . T a kano,K . Ha s e-Aoki, H . H o r iuchi, L. Zhao, Y. Kasahara, S. Kondo, M. A . B ecker, Life Sci. 7 6 (2005)1835- 1 8 47. ) ( [21] V . Colo t ta , D . C a tarzi, F. V a rano, G. Fil a cchioni, L. C e c c hi, A. Gal l i, C. C os t agli,J. M ed. Ch e m. 39 ( 1 996)291 5 -2921. ) ( [22] F . V ara n o, D. Ca t arzi, V . Co l ott a , G . F ilacc h ioni, A. Gall i , C. Costagli, V. C a rl a , J. M ed. C h e m. 45 (2002)1035-1 0 44. ) [23] F. Varano, D. Catarzi, V. Colotta, F.R. Calabri, O. Lenzi, G. Filacchioni, A. Galli, C. ( C ost a gli, F . Deflo r ian , S . Moro, Bioor g . Med. Chem. 1 3 ( 20 0 5)5536-5 5 49. ) ( [24] F . B eau l ieu,C . Ouellet, E.H. Ruediger, M. Be l em a , Y . Q i u ,X. Yan g , J. Ba n ville, J R. B urke,K . R. Gregor, J . F. MacMaster, Bi o o rg. M e d . Chem. Lett. 1 7 (2007) 1 2 3 3 1237. ) ( [25] B . Asproni, G. Murineddu , A. Pau, G.A. Pinna,M. La n ggard, C.T. Christ o ffe r s en , J. ) ( N iel s en,J. Keh l er, Bioorg. M ed. Ch em . 19 (2011)642-649. ) ( 261 F . V arano, D. Catarzi, V. Co l otta, O. Len z i, G. F il a cch i oni, A . G a ll i , C. C o s t a gli, Bioorg. Med. Chem. 1 6 (2008) 2617-2626. ) ( [27 D ] . Catarzi,V. C olotta, F . Varano,D . Poli, L. Sq u arcialupi, G. F i lac c h io n i, K . Var an i, F . Vin c enzi, P .A. B orea, D . D al B en,C. La m bertucci , G. C ri s t alli , Bioor g. M ed. Chem. 2 1 (201 3 )283-294. ) ( [28] D . Kumar, R . K umar, M i crowave-a s sisted s yn t h esi so f p yra z o l o[1, 5 - ) quinazolines and their derivatives Tet Lett 55 (2014) 2679-2683. ( [ 2 9] : S . D . B eedkar, C.N. Kho b ragade , S.S. Chobe, B . S. D a wane , O. Y e m u l , Int . J. B i ol. M a crom ol. 50(2012)947-956. ) ( [ 3 0 ] K .I . Mu s a, A. Abdu l lah, B. K u sw a ndi, M. Amrun H idaya t, Food C hem . 1 4 1 2 0 1 1 3) 4 102 - 4106. ) ( M . Sz abo,c. . ld i toiu, D. C h ambr e, A . L u p ea, Chem . P a p. 61 (2007) 21 4 -216. ) ( 3 2 C . Enrot h , B.T. E ger, K.Okamoto,T . Nish i no , T . Nis h i no, E.F. Pai, Proc. Natl. Acad. Sc i . . 97 ( 20 0 0) 1 0 723-10728. [ 33 ] A . Le vai, Arkivoc 9 (2005 ) 344 - 352 ) http://dx.doi.org/j.bioorg.士科技www.rysstech.com www.rysstech.com耐士科技
确定





还剩6页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《通过Biotage微波合成仪黄嘌呤氧化酶抑制活性的研究以及喹唑啉衍生物的合成by耐士科技》,该方案主要用于其他中特殊物质和基团检测,参考标准--,《通过Biotage微波合成仪黄嘌呤氧化酶抑制活性的研究以及喹唑啉衍生物的合成by耐士科技》用到的仪器有
相关方案
更多
该厂商其他方案
更多