








The 4-exo-dig and 5-exo-dig cyclocarbopalladations have been efficiently used to produce molecular complexity in a straightforward manner. Strained 1,2-cyclobutanediols are rapidly obtained under microwave irradiation in high yields. In many cases, the cyclocarbopalladation cascade reaction is associated with a 6 or 8p electrocyclic reaction. During the process of the 5-exo-dig cyclocarbopalladation on benzosuberone derivatives, an aromatic C–H activation leads to vinylic substituted aromatics. Polycyclic skeletons of natural products of the family of Ophiobolin and Aleurodiscal can be prepared in few steps from simple starting material.
方案详情

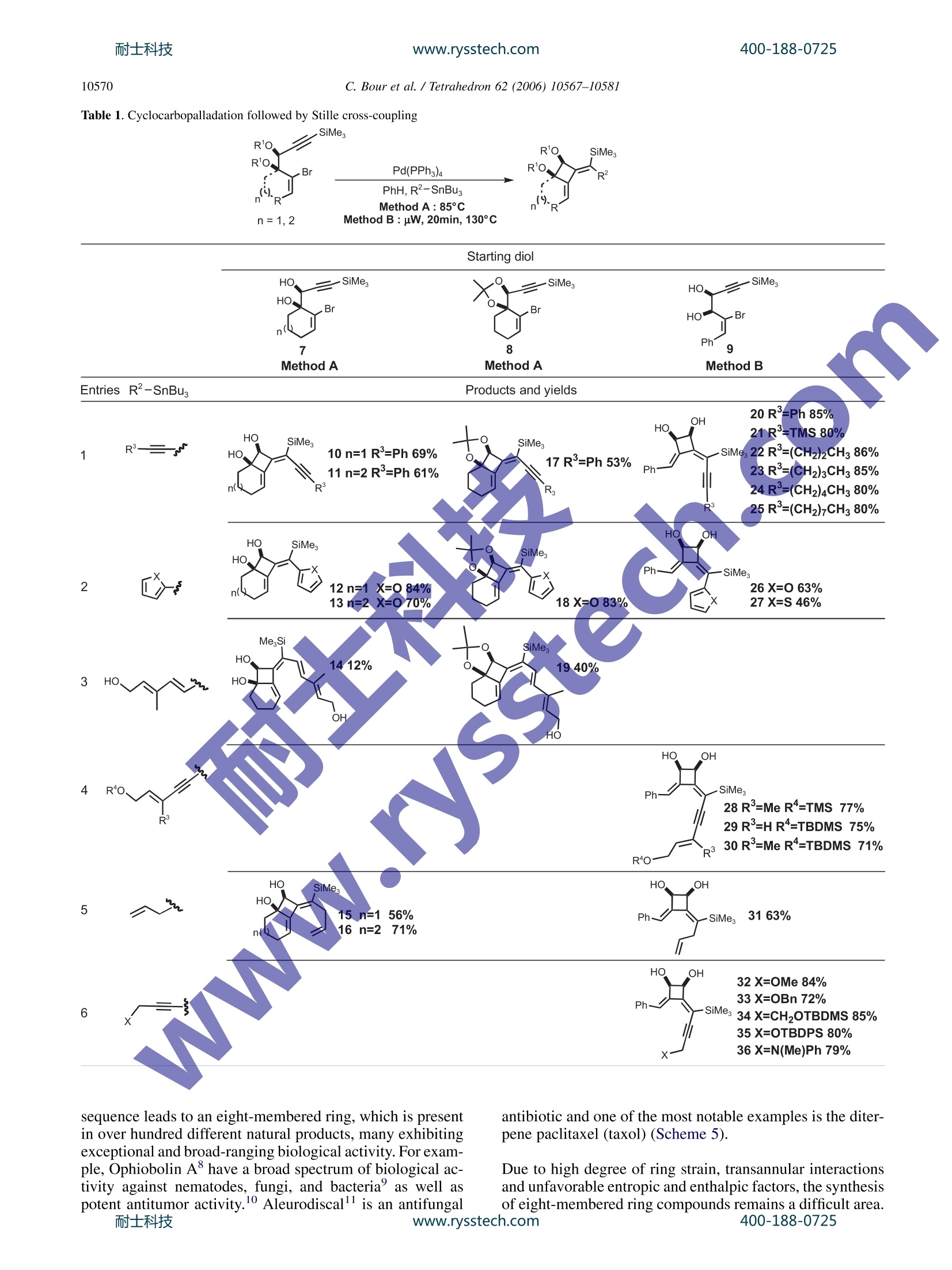
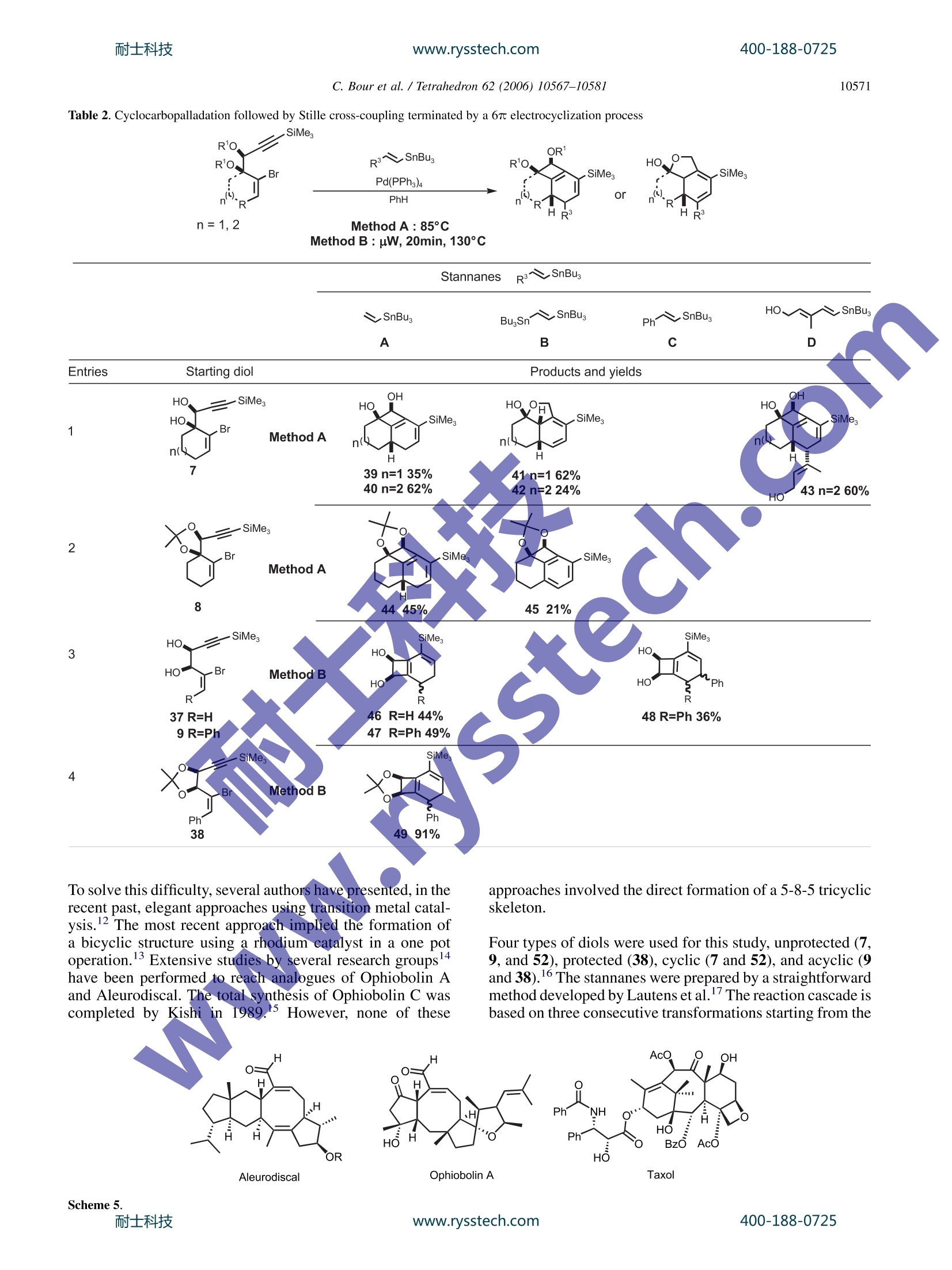
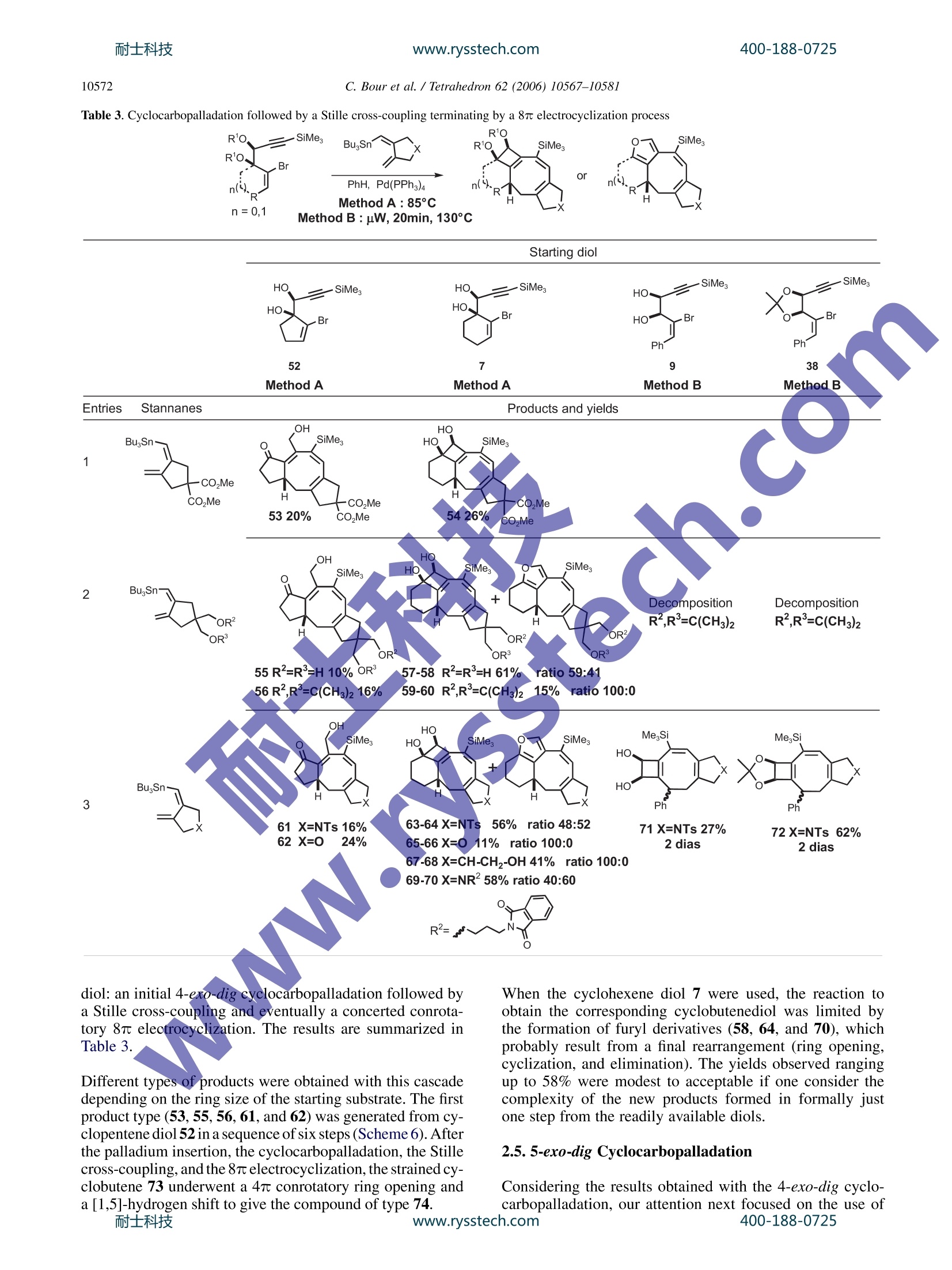
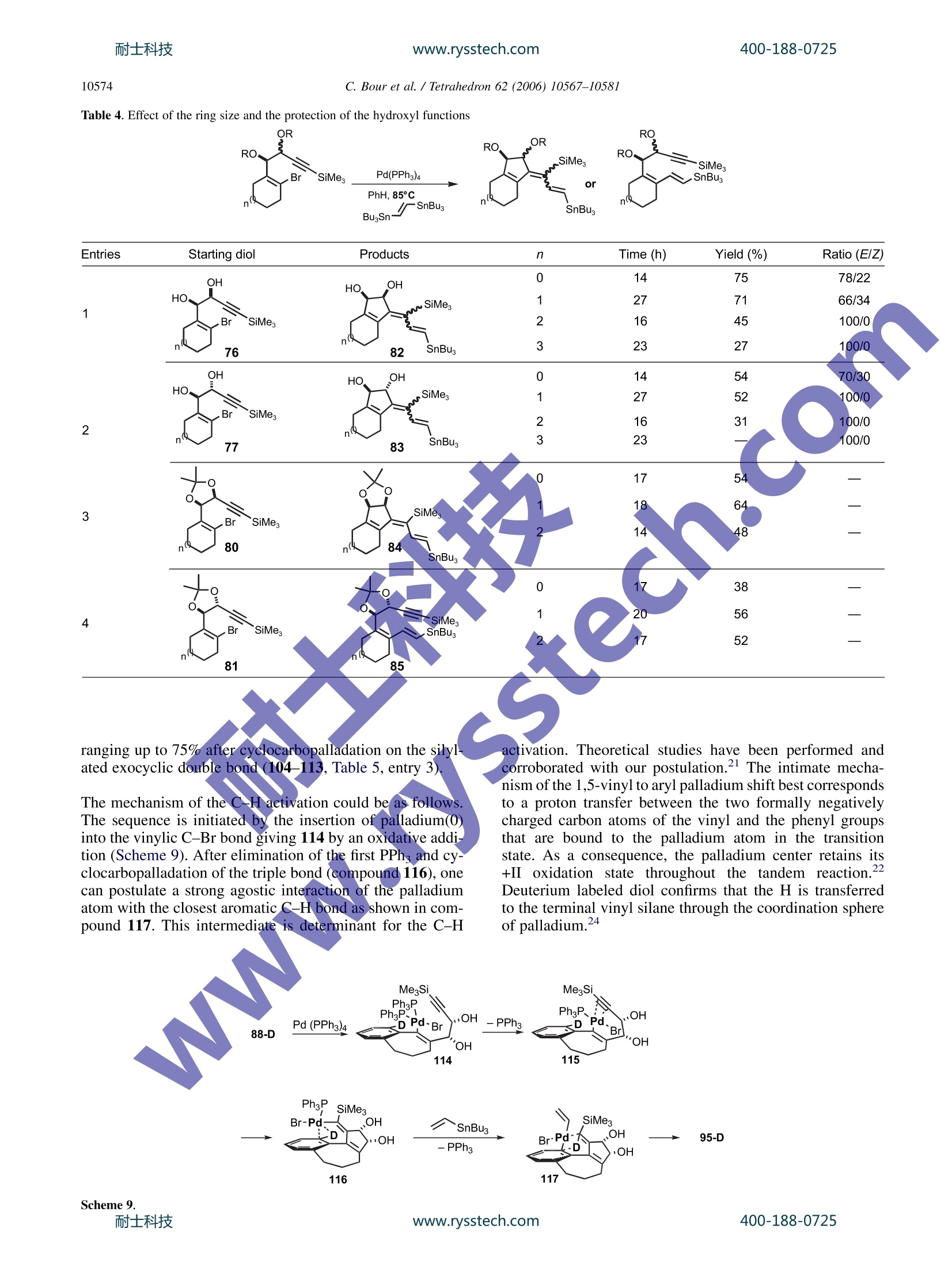
耐士科技400-188-0725www.rysstech.comAvailable online at www.sciencedirect.comTetrahedron 62 (2006) 10567-10581 400-188-0725耐士科技www.rysstech.com10568 Tetrahedron 4-exo-dig and 5-exo-dig Cyclocarbopalladations: an expeditioussolution toward molecular complexity? Christophe Bour, Gaelle Blond, Bahaa Salem and Jean Suffert Universite Louis Pasteur de Strasbourg (LC1-UMR 7175 CNRS/ULP), Faculte de Pharmacie, 74 route du Rhin,67401 Illkirch-Cedex, France Received 27 January 2006; revised 5 May 2006; accepted 27 June 2006Available online 14 August 2006 Abstract-The 4-exo-dig and 5-exo-digcyclocarbopalladations have been efficiently used to produce molecular complexity in a straightfor-ward manner. Strained 1,2-cyclobutanediols are rapidly obtained under microwave irradiation in high yields. In many cases, the cyclocarbo-palladation cascade reaction is associated with a 6 or 8r electrocyclic reaction. During the process of the 5-exo-dig cyclocarbopalladation onbenzosuberone derivatives, an aromatic C-H activation leads to vinylic substituted aromatics. Polycyclic skeletons of natural products of thefamily of Ophiobolin and Aleurodiscal can be prepared in few steps from simple starting material. 1.Introduction The potential of palladium-catalyzed process in organic syn-thesis has not yet been fully explored. The usefulness ofstrategies based on palladium cyclization cascades hasbeen demonstrated in the past leading to polycyclic frame-works in regio- and stereoselective manner. One of themost efficient cyclization process that has been used in theliterature concerns the intramolecular attack of an organo-palladium activated species on a tethered triple bond. Mostof the time, an initial 5-exo-dig,6-exo-dig,or 7-exo-dig cy-clocarbopalladation is involved followed by a terminatingcross-coupling reaction with CO or various organometallicreagents. Significant recent studies in our laboratory are directed atthe development of cyclocarbopalladation to design andelaborate complex molecules from simple starting material.In this context, we report herein our investigations in thestdy of cascade reactions involving 4sexo-dig and 5-exo-dig cyclocarbopalladations 2. Results and discussion 2.1. An unprecedented 4-exo-dig cyclocarbopalladation Our initial studies focused on a rare 4-exo-dig cyclocarbo-palladation through a palladium catalysis using a vinyl or al-kynylstannane as the terminating trapping species. To the ( * Corresponding author. Tel.:+33 3 90 24 42 30; fax: + 3 3 3 9 0 24 43 10 ; e-mail: jean.suffe rt @ p harma.u-strasbg.fr ) best of our knowledge, before our preliminary work in thisfield, there was no report on the preparation of related cyclo-butanes by this process Diols anti-2 and syn-3 were selected as starting materialsand prepared in large scale by addition of a properly pro-tected metalated propargylic alcohol 4 onto bromoalkenones1, followed by deprotection and chromatographic separationof the two diastereomers anti-2 and syn-3 (Scheme 1). Scheme 1. With the diols in hands, we decided to explore the scope andthe limitation of the 4-exo-dig cyclocarbopalladation. Twopossible mechanistic pathways can be envisaged (Scheme 2): -pathway A: favoring a direct Stille cross-coupling givingcompounds of type 5. -pathway B: favoring a 4-exo-dig cyclocarbopalladationfollowed by a Stille cross-coupling giving compoundsof type 6. The 4-exo-dig cyclizations are known to be disfavored pro-cess according to Baldwin’s rules if one considers the orga-nolithium or organomagnesium cyclizations.Of course this C. Bour et al. / Tetrahedron 62 (2006) 10567-1058I type of forbidden reaction can be revisited if in place of a lith-ium or magnesium intermediate species a transition metalactivated route is used. In an effort to favor this process,we examined the effect of several different palladium cata-lysts, various inorganic and organic bases, and the phosphineligands.4 Palladium(II) catalysts such as PdCl2(PhCN)2,PdCl2(CH,CN)2, and PdCl2(AsPh3)2 were used, leadingonly to decomposition of the starting material. The catalyticsystem Pd(OAc)2/PPh, is frequently used in carbopallada-tive processes. Applying these conditions to our substrates,we observed the exclusive formation of the Stille product5. With the same catalyst system, when additives such asEt NCl, silver salts, or EtN were used only the Stille prod-uct 5 was formed and with K2CO3 or Na2CO3 in benzene,black palladium precipitates rapidly and decomposition ofthe substrate occurred. When Pd(PPh3)4 was used, a singleproduct 6 was formed resulting from a 4-exo-dig cyclocarbo-palladation followed by a Stille cross-coupling. It is remark-able to note that, to this date, the only efficient catalyst forthis process is Pd(PPh3)4. Any other Pd(0) sources gavea trace of the cyclobutane ring system. Different solventswere tested: THF,DMF,NMP,and CH,CN cause the decom-position of the starting material. Benzene or toluene seems togive the best results for the 4-exo-dig cyclocarbopalladation. Eventually, this investigation led to the optimized reactionprocedure: 10% mol Pd(PPh3)4 with 1.3 equiv of stannylatedreagent in benzene or toluene. Next, we explored this route to 1,2-cyclobutanediols by ex-amining the influence of the different structural parametersof the substrate in order tofavor the cyclocarbopallada-tion-Stille cross-coupling cascade (Scheme 3). First the influence of the size of the ring bearing the vinylbromide function was studied. We noticed that in the case of the five-membered ring the reaction proceeded only viaan exclusive direct Stille cross-coupling, and decompositionlowered the yield. This result can be explained by the highring strain induced by the presence of the cyclobutanediolconnected to a smaller ring. Decomposition of the startingmaterial occurred with the eight-membered ring, probablycaused by transannular interactions in the cyclooctene. Thesix- and seven-membered rings led to compounds via acyclocarbopalladation process followed by a Stille cross-coupling. The presence of the diol functionality seems to be essentialin the competition between the direct Stille cross-couplingandthe cyclocarbopalladation-Stille cross-coupling cas-cades. We next turned our attention to examine the role ofeach of the hydroxyl groups at positions 1 and 2 (Scheme3). Applying the same catalyst system described previously,t appeared that the propargylic hydroxyl group (position 1)controls the feasibility of the 4-exo-dig cyclocarbopallada-tion. Indeed,without this hydroxyl group in the propargylicposition, the direct Stille cross-coupling was the onlyobserved pathway. A mixture of the Stille product 5 andthe cyclobutanediol 6 was isolated when the hydroxyl in thehomopropargylic position (position 2) was removed. There-fore, to favor the exclusive 4-exo-dig cyclocarbopalladation,the presence of these two oxygen atoms is necessary. Moreover, the stereochemistry of the diol is also importantby directing the triple bond more or less into close proximityof the vinyl bromide. Thus, the experiments on the syn-propargylic diol 3 showed a versatile reactivity: a mixtureof Stille product 5 and cyclobutanediol derivatives 6 wasobtained unlike the reaction with the anti diol 2, whichgave only cyclobutanediol derivatives. As a consequence,the anti diol 2 was selected as a starting material, in all cases,for the rest of the studies. To complete the studies on the different parameters related tothe starting material, the substitution of the triple bond wasvaried. With the unprotected triple bond decomposition oc-curred, and even Sonogashira coupling was not observed.With a terminal methyl group, the substrate underwenta 4-exo-dig cyclocarbopalladation in moderate yields com-pared to the triple bond substituted by the trimethylsilylgroup. In addition changing the terminal silyl group fromtrimethylsilyl to triethylsilyl group did not improve thereaction. The trimethylsilyl group turned out to be the mostappropriate for protection of the triple bond, which may bedue to an electronics effect. In summary, the two oxygens in the y-bromopropargylicdiols in an anti configuration turned out to be essential for thereaction and the trimethylsilyl group is the most appropriatesubstituent on the triple bond for high reaction efficiency. 2.2. 4-exo-dig Cyclocarbopalladation followed by Stillecross-coupling derived from thiophene and furan, it is possible to accessthe new heteroaromatic substrates (12, 13, 18, 26, and 27,entry 2). 2.3. The 6r electrocyclization process In several examples, depending on the stannylated reagents,the original reaction of cyclocarbopalladation followed byStille cross-coupling reaction is terminated by a 67e electro-cyclization process. Thermal conditions and microwave irradiation were alsoused with protected (8-38), unprotected (7, 9, 37), cyclic(7, 8), and acyclic (9, 37,38) diols (Table2). This process gave tricyclic or tetracyclic structures 39-45bearing a strained cyclobutene ring fused to the two otherrings and an anti-Bredt double bond is shared by three cycles(entries 1 and 2). The previous study of different parameters of the reaction(catalyst, solvent, and temperature) and the starting mate-rials allowed optimization of the conditions to favor thecyclocarbopalladation process. Thus, different diols (cyclic 7,8, and acyclic 9) were treatedaccording to the conditions described above in the presenceof alkynes (entries 1, 4, and 6), heteroaromatics (entry 22)vinyl (entry 3), and allyltributylstannanes (Table 1, entry 5).Two methods of activation of the reaction were used: activa-tion by heating the reaction mixture at 85 °C (method A) orby using microwave irradiation as a source of molecularactivation for the catalytic process (method B). In each case, the only product isolated resulted from a 4-exo-dig cyclocarbopalladationr coupling following by a Stilletermination couplingand lOotfrom a direct Stille cross-coupling reaction in keeping with the previous study. Inmajority of the cases, the isolated compounds are stableand were purified by chromatography on silica gel withoutany precaution. The bicyclic and tricyclic cyclobutanediols 10-19 wereobtained in moderate to good yields (12-84%) from cyclicdiols 7 and 8, and highly substituted cyclobutanedienynes20-36 were prepared in high yields (46-86%) with acyclicdiols 9. In the presence of stannylated aromatic heterocycles With acyclic diols, bicyclic compoundss 46-49 were ob-tained, and the double bond in the bicycle system could beselectively oxidized to give functionalized cyclooctenes. To explain the formation of compounds 41 and 42, in thecase of use of bis-stannane B with unprotected diols 7, thenon-isolated strained tricyclic compounds 50 underwent asubsequent elimination of the three alkyl tin group followed,by an opening of the 1,2-cyclobutendiol (Scheme 4, Table 2,entry 1). A final attack of the allylic oxygen on ketone 51afforded the hemiketals 41. It is important to note that the absence of a tributylstannanegroup in molecule of type 50 completely prevents the open-ing of the cyclobutenediol through a 4r electron openingelectrocyclic reaction. In the same manner, this eliminationof the tin is not feasible with the protected diol because ofthe dioxolane protection that also prevents the opening ofthe cyclobutenediol. Thus, after the electrocyclization, adehydrostannation event provides the unusual aromatic tetra-cyclic dioxolane 45 (Table 2, entry 2). 2.4. The 8r electrocyclization process During the studies toward the extension of this new method-ology to the synthesis of other polycyclic structures, wedecided to examine the feasibility of a conrotatory 8 elec-trocyclization in the same reaction sequence.This cascade www.rysstech.com 10570 C. Bour et al./Tetrahedron 62 (2006)10567-1058l Table 1. Cyclocarbopalladation followed by Stille cross-coupling sequence leads to an eight-membered ring, which is presentin over hundred different natural products, many exhibitingexceptional and broad-ranging biological activity. For exam-ple, Ophiobolin A have a broad spectrum of biological ac-tivity against nematodes, fungi, and bacteria’ as well aspotent antitumor activity.10 Aleurodiscal" is an antifungal antibiotic and one of the most notable examples is the diter-pene paclitaxel (taxol) (Scheme5). Due to high degree of ring strain, transannular interactionsand unfavorable entropic and enthalpic factors, the synthesisof eight-membered ring compounds remains a difficult area.400-188-0725 耐士科技 C. Bour et al. / Tetrahedron 62 (2006) 10567-10581 Table 2. Cyclocarbopalladation followed by Stille cross-coupling terminated by a 6r electrocyclization process SiMe. n= 1,2 Method A: 85°C Method B : uW, 20min, 130°C To solve this difficulty, several authors have presented, in therecent past, elegant approaches using transition metal catal-ysis. The most recent approach implied the formation ofa bicyclic structure using a rhodium catalyst in a one potoperation.13 Extensive studies by several research groups 14have been performed to reach analogues of Ophiobolin Aand Aleurodiscal. The total synthesis of Ophiobolin C wascompleted by Kishi in198915 However, none of these approaches involved the direct formation of a 5-8-5 tricyclicskeleton. Four types of diols were used for this study, unprotected (7,9, and 52), protected (38), cyclic (7 and 52), and acyclic (9and 38).16The stannanes were prepared by a straightforwardmethod developed by Lautens et al.The reaction cascade isbased on three consecutive transformations starting from the Scheme 5.耐士科技 www.rysstech.com C. Bour et al./Tetrahedron 62 (2006)10567-1058I Table 3. Cyclocarbopalladation followed by a Stille cross-coupling terminating by a 8r electrocyclization process R'O, SiMe, Bu,Sn' R'O. .Br diol: an initial 4-exo-dig cyclocarbopalladation followed bya Stille cross-coupling and eventually a concerted conrota-tory 8 electrocyclization. The results are summarized inTable 3. Different types of products were obtained with this cascadedepending on the ring size of the starting substrate. The firstproduct type (53,55, 56, 61, and 62) was generated from cy-clopentene diol 52 in a sequence of six steps (Scheme 6). Afterthe palladium insertion, the cyclocarbopalladation, the Stillecross-coupling, and the 8r electrocyclization, the strained cy-clobutene 73 underwent a 4r conrotatory ring opening anda [1,5]-hydrogen shift to give the compound of type 74. When the cyclohexene diol 7 were used, the reaction toobtain the corresponding cyclobutenediol was limited bythe formation of furyl derivatives (58, 64, and 70), whichprobably result from a final rearrangement (ring opening,cyclization, and elimination). The yields observed rangingup to 58% were modest to acceptable if one consider thecomplexity of the new products formed in formally justone step from the readily available diols. 2.5.5-exo-dig Cyclocarbopalladation Considering the results obtained with the 4-exo-dig cyclo-carbopalladation, our attention next focused on the use of400-188-0725 C. Bour et al./ Tetrahedron 62 (2006)10567-10581 another type of reaction: the 5-exo-dig cyclization, a favoredprocess according to Baldwin’s rules, starting from a sub-strate containing a propargylic diol function and the vinylbromide included in an aliphatic ring. The starting diolsanti-76 and syn-77 were again easily prepared in good yieldsby addition of the protected metalated propargylic alcohol 4on bromo-aldehydes 75 followed by deprotection and chro-matographic separation of the two anti- and syn-diastereo-mers (Scheme 7).l8 The impact of the stereochemistry of the starting diol andthe size of the ring bearing the vinyl bromide function wasexamined with the same conditions and catalytic systemdescribed before for the 4-exo-dig cyclocarbopalladation:at 85C in benzene in the presence of a catalytic amountof Pd(PPh3)4 (10 mol%). The trans-bis(tributylstannyl)-ethylene was used to explore the competition between thetwo reactions: a direct Stille reaction (pathway A, compound78) or cyclocarbopalladation followed by a Stille termina-tion (pathway B, compound 79) (Scheme 8). It turns out that the diols anti-76 and syn-77,irrespective ofcycle size, afforded only the products resulting from a 5-exo-dig cyclocarbopalladation, 82 and 83 (Table 4, entries 1 and2), generally with acceptable to good yields. However, whenthe diols are protected (entries 3 and 4, 80 and 81) as a di-oxolane, the direct Stille cross-coupling competes signifi-cantly. Independent of considerations of the size of the ring, we distinguished the cis-dioxolane 80 that proceededvia a cyclocarbopalladation process to obtain compound84, from the trans dioxolane 81 that led exclusively to the di-rect Stille cross-coupling product 85. In the later case, thecyclocarbopalladation did not proceed at all due to thehighly strained tricyclic derivatives that should be obtainedin theory. 2.6.5-exo-dig Cyclocarbopalladation and CH activation Considering the results obtained from the 5-exo-dig cyclo-carbopalladation, our attention focused on the use of new,original anti-propargylic-1,2-diols 86, 87, 88, 89 derived,re-spectively, from acetophenone, indole,tetralone, or benzo-suberone (Table 5) IIn the presence of vinylic (entry 1),allylic, or heteroaromatic stannane derivatives (entry 2),5-exo-dig cyclocarbopalladation under classical conditions(85°C, PhH, 10% mol Pd(PPh3)4) is efficient and productswere isolated ranging up to 70% yields. The use of diol 88gave the expected products 93 and 94 after the 5-exo-digcy-clocarbopalladation and the Stille cross-coupling. The five-membered ring diol 87, under the same reaction conditions,led to the decomposition of starting materials. However,when acyclic diol 86 and benzosuberone derivative diol 89are employed under these conditions, none of the expectedproducts were obtained but only the styrene derivatives 90,95-103 were observed. The formation of these productsresulted from a C-H palladative activation. Some recent pa-pers describe a new C-H activation process of nonactivatedaromatic derivatives using metal catalysis.2 In the case ofthe diol 87, this C-H activation was not observed probablybecause of the low flexibility of the ring. Thus, the regiose-lectivity of the final vinylic stannane partner is totally depen-dent of the substrate. On the other hand, when alkynylstannanes were used, onlythe Stille cross-coupling product was observed with yields C. Bour et al./ Tetrahedron 62 (2006) 10567-1058l Table 4. Effect of the ring size and the protection of the hydroxyl functions OR RO. OR RO. SiMe, SiMes3 Pd(PPh3)4 or ranging up to 75% after cyclocarbopalladation on the silylated exocyclic double bond (104-113, Table 5, entry 3) The mechanism of the C-H activation could be as follows.The sequence is initiated by the insertion of palladium(0)into the vinylic C-Br bond giving 114 by an oxidative addi.tion (Scheme 9). After elimination of the first PPh and cy-clocarbopalladation of the triple bond (compound 116), onecan postulate a strong agostic interaction of the palladiumatom with the closest aromatic C-H bond as shown in com-pound 117. This intermediate is determinant for the C-H activation. Theoretical studies have been performed andcorroborated with our postulation.21 The intimate mecha-nism of the 1,5-vinyl to aryl palladium shift best correspondsto a proton transfer between the two formally negativelycharged carbon atoms of the vinyl and the phenyl groupsthat are bound to the palladium atom in the transitionstate. As a consequence, the palladium center retains its+II oxidation state throughout the tandem reaction.Deuterium labeled diol confirms that the H is transferredto the terminal vinyl silane through the coordination sphereof palladium.24 Scheme 9.耐士科技 C. Bour et al. / Tetrahedron 62 (2006)10567-1058l Table 5. Synthesis of Polycycles via palladium CH activation MegSi SiMe, Br R'SnBu3 .OH m0H PhH,Pd(PPh3)4 OH or ( OH Method A:85°C,t n=1,2,3 Method B : uW, t, 130°C Starting diol EntriesR1SnBu. Products R2 SiMes OH R2、 SiMes R R2 SiMes R SiMes OH SnBus moc 'OH OHOHOH90 R²=H, 30 min, 43%, B 91 R²=H, 20 min, 0%,B 9)33 R²=H,20 min,59%, B 95R²=H,8h, 70%,A92 R²=SnBu, 20 min,0%, B 94 R=SnBu3, 20 min, 55%, B96 R=SnBu3, 5h,58%,A97 R=Ph, 10 min, 45%,B98 R²=SiMe3,18 min, 70%, B99 R=(3-hydroxymethyl)phenyl, 18h,61%,ASiMesOH xx. etssiMe.iR2-OH R OH R1一SnBus 100 R1=pyr,20h, 53%, A 101 R'=thio, 54h, 48%, A 102 R'=allyl, 20h, 52%, A 103 R'=fur, 30 min,53%, B SiMe, SiMe OH .OH R4 "OH R-三SnBBu. OH OH 104 R²=SiMe, 38 min, 63%, B 105 R=Ph, 20 min, 0%, B 107 R²=Ph, 20 min, 49%, B 109 R²=Ph, 12h,58%, A 106 R=SiMe3, 20 min, 0%, B 108 R=SiMe3, 20 min,56%, B 110 R=SiMe3, 5h, 45%, A 111 R=Pr, 18h,75%,A 112 R=TBSO(CH2)3, 8h, 54%, A 113 R=TBSO(CH2)2, 8h, 48%,A In conclusion, we have shown that different sequences in-cluding a 4-exo-dig cyclization can be realized from y-bro-mopropargylic diols under palladium catalysis. We firstsummarized the study of different parameters of the reaction(catalyst, solvent, and temperature) and the starting mate-rials giving the optimal conditions to favor the cyclocarbo-palladation process. These conditions were applied torealize different cascades: 4-exo-dig cyclocarbopalladationfollowed by a Stille cross-coupling and a 6r or 8 electro-cyclizations. The same kind of cascades has been realized includinga 5-exo-dig cyclocarbopalladation and Stille cross-coupling.In some cases, with acetophenone and benzosuberone diols,a C-H palladative activation after the Stille cross-couplingwas observed and styrene derivatives were isolated. We have shown a major advancement in the field of cyclo-carbopalladation cascades for the consecutive formation ofseveral new carbon-carbon bonds in a single-step process.These approaches have already allowed impressive and eco-nomical constructions of polycyclic compounds with highmolecular complexity. Some applications to the synthesis400-188-0725 C. Bour et al. /Tetrahedron 62 (2006)10567-10581 4. Experimental 4.1. General Reactions were run under an atmosphere of argon in oven-dried glassware using a standard syringe, cannula, and septaapparatus.Et2O and THF were distilled from sodium benzo-phenone. Benzene and DMF were distilled from CaH2, andCH2Cl2 was distilled from P205.EtNsand i-PrNH were dis-tilled from KOH. Crude products were purified by flash col-umn chromatography on Merck 230-400 mesh silica gel.For some compounds, 5% Et N treated silica gel was usedto avoid decomposition. Analytical TLC was carried outon Merck (Kieselgel 60F254) silica gel plates. HNMR spec-tra were recorded at 200 or 300 MHz using the residualsolvent signal as internal reference (CDCl3, 7.26, C6D6,7.16 ppm). Chemical shifts are quoted in parts per million,coupling constants (J) are given in hertz. The followingabbreviations are used to describe peak patterns when appro-priate: s (singlet), d (doublet), t (triplet), q (quartet), quint(quintet), m (multiplet), ap (apparent), br (broad).NMR spectra were recorded at 50 or 75 MHz at ambienttemperature in CDCl3 at 77.0 as internal reference. Multiplicities were determined in some cases byTJJmodpulsesequence. Melting points were determined withi a glasscapillary apparatus and were uncorrected. IR were deter-mined with a Perkin-Elmer 2000 apparatus. Mass spectrawere obtained from the Service de spectrometrie de Masseof the Chemistry Institute of Strasbourg (France). High-Resolution Mass Spectral Analysis (HRMS) was performedusing a Mariner ESI-Tof instrument from Applied Bio-System Perkin-Elmer. n-BuLi was titrated using pivaloylo-toluidine followingthe Suffertt method.23 Microwaveirradiations have been performed using BIOTAGE SmithCreator. Stannanes derivatives were prepared according tothe literature methodssor commercially available. 4.2. Method A: general procedure: 4-exo-dig or 5-exo-dig cyclocarbopalladation under classical conditions The reaction was carried out in an oven-dried 25 mL two-necked flask, equipped with a reflux condenser, under argonatmosphere. To a solution of the diol (1 equiv) in driedbenzene was added Pd(PPh3)4 (0.1 equiv) followed by thestannylated reagent (1.3 equiv). The reaction mixture wasstirred for 1-17 h in a preheated 85°C oil bath. The reactionis followed by TLC. Then, the reaction mixture was con-centrated in vacuo and immediately purified by flash chro-matography. 4.3. Method B: general procedure: 4-exo-dig or 5-exo-dig cyclocarbopalladation under microwave irradiation(BIOTAGE system initiator) The reaction was carried out in a special BIOTAGE flask,under argon atmosphere. To a solution of the diol (1 equiv)in dried benzene (2mL) was added Pd(PPh3)4 (0.1 equiv)followed by the stannylated reagent (1.3 equiv). The mixturewas purged 20 min with argon and heated at 130 °C under microwave irradiation. The reaction was followed by TLC.After the reaction time, the mixture was cooled, dilutedwith Et2O (10 mL), treated with active charcoal, filtratedover Celite, and concentrated to dryness. The reaction mix-ture was purified by flash chromatography (in the majority ofthe cases two chromatographies are necessary). 4.3.1. Cyclocarbopalladation 4-exo-dig following bya Stille cross-coupling. Compounds 10-19 are already de-scribed in Ref. 2b. 4.3.2. (1S,2R,3Z,4Z)-3-Benzylidene-4-(3-phenyl-1-(tri-methylsilyl)prop-2-ynylidene)cyclobutane-1,2-diol (20)Preparation by method B on 0.295 mmol scale with a reac-tion time of 20 min. Purification by flash chromatographyeluting with Et2O/Hept (20/80) afforded the product (90 mg,85%) as a yellow solid. R: 0.36 (Et2O/Hept:30/70).H NMR (300 MHz, CDCl3) ; 7.81 (d, 1H, J=1.5Hz,ArH), 7.63(d, 2H, J=7.1 Hz, ArH), 7.48(d,1H.J=1.5 Hz, ArH), 7.45 (d, 1H, J=1.5 Hz, ArH), 7.38-7.31(m,6H, ArH, Ph-CH), 4.99 (t, 1H, J=5.6 Hz, HO-CH-CH-OH),4.81 (t, 1H, J=6 Hz, HO-CH-CH-OH), 3.74 (br s,1H,OH),:3.61 (br s, 11H.,QH), 0.32 (s, 9H, SiMe3).13cNMR(75 MHz,,CDCl3)8158.8,142.8,135.8,131.2,129.7,129.5,128.6,128.5,128.3.128.1,124.2,121.0,103.4,91.9,72.4,71.3,-1.0.HRMS (ESI, positive ion180eV) calcd for (C23H24Q SiNa)*: 383.1441; found:383.1502. IR (FTIR, film): v=3400 (br),2118,1646,1485,1249,1047,976,844,756cm. 4.3.3.(1S,2R,3Z,4Z)-3-Benzylidene-4-[1,3-bis(trimethyl-silyl prop-2-ynylidene]cyclobutane-1,2-diol (21). Prepa-ration by method B on 0.295 mmol scale with a reactiontime of 20 min. Purification by flash chromatography elutingwith Et2O/Hept (20/80) afforded the product (108 mg, 80%)as a yellow solid. R: 0.30 (Et2O/Hept: 30/70).H NMR(300 MHz, CDCl3) 6 7.81 (d, 1H, J=1.5 Hz, ArH), 7.57 (d,2H,J=6.8 Hz, ArH), 7.35-7.29 (m,3H,ArH, Ph-CH), 4.89(brt, 1H, HO-CH-CH-OH), 4.68 (t, 1H,J=6.2 Hz, HO-CH-CH-OH), 4.40 (br s,1H, OH), 4.22 (br s, 1H, OH), 0.24 (s,9H, SiMe3), 0.22 (s, 9H, SiMe3). 13C NMR (75 MHz,CDCl3) 6 160.1,142.6,135.8,129.9,129.5,128.7,128.3,121.3,109.35,106.9,72.3,71.3,0.1,-1.0.HRMS (ESI, pos-itive ion 180 eV) calcd for (C20H28O2SizNa)*: 379.1526;found: 379.1519. IR (FTIR, film): v=3410 (br), 2116,1639,1487,1245,1045,975,842,752cm-. 4.3.4.(1S,2R,3Z,4Z)-3-Benzylidene-4-[1-(trimethylsilyl)-hex-2-ynylidene]cyclobutane-1,2-dioll (22). Preparationby method B on 0.295 mmol scale with a reaction time of20 min. Purification by flash chromatography eluting withEt2O/Hept (20/80) afforded the product (83 mg, 86%) asa yellow powder. R:0.30 (Et2O/Hept: 30/70). H NMR(300 MHz, CDCl3) 8 7.70 (s, 1H, ArH), 7.58 (d, 2H,J=7.8 Hz, ArH), 7.35-7.25 (m, 3H, ArH, Ph-CH), 4.90 (t,1H, J=5.9 Hz, HO-CH-CH-OH), 4.72 (t, 1H, J=5.9 Hz,HO-CH-CH-OH), 3.89 (br s, 1H, OH), 3.69 (br s, 1H,OH), 2.50 (t, 2H, J=6.8 Hz, CH2), 1.65 (q, 2H, J=7.2 Hz,CH2), 1.05 (t, 3H, J=7.2 Hz, CH3), 0.23 (s, 9H, SiMe3).IC NMR (75 MHz, CDCl3) 6 157.7,142.8,136.0, 129.3,128.6,128.3,128.0,122.6,105.4,82.7,72.2,71.1, 22.5,13.7, -1.1. HRMS (ESI, positive ion 180 eV) calcd for(C20H26O2SiNa)+: 349.1600; found: 349.1595. 400-188-0725 4.3.5. (1S,2R,3Z,4Z)-3-Benzylidene-4-[1-(trimethylsilyl)-hept-2-ynylidene]cyclobutane-1,2-diol (23). Preparationby method B on 0.295 mmol scale with a reaction time of20 min. Purification by flash chromatography eluting withEt2O/Hept (20/80) afforded the product (85 mg, 85%) asa yellow powder. R: 0.32 (EtzO/Hept: 30/70). H NMR(300 MHz, CDCl3) ; 7.70 (s, 1H, ArH), 7.58 (d, 2H,J=7.8Hz, ArH), 7.36-7.26 (m, 3H, ArH, Ph-CH), 4.92 (t,1H, J=5.3 Hz, HO-CH-CH-OH), 4.74 (t, 1H, J=6.24 Hz,HO-CH-CH-OH), 3.62 (d, 1H, J=6.8 Hz, OH), 3.44 (d,1H,J=7.5 Hz, OH), 2.53 (t, 2H, J=6.8 Hz, CH2), 1.64-1.27 (m, 4H, 2×CH2), 0.94 (t, 3H, J=7.2 Hz, CH3), 0.24(s, 9H, SiMe3). 3c NMR (75MHz, CDCl3) 8 157.6,142.7,136.0,129.3,128.6,128.3,128.0,122.7,105.6,82.6,72.1,71.1,31.1,22.1,20.1,13.6,-1.1.HRMS (ESI,positiveion 180 eV) calcdffor(C21H2802SiNa)+:363.1756: found: 363.1760. 4.3.6. (1S,2R,3Z,4Z)-3-Benzylidene-4-[1-(trimethylsilyl)-oct-2-ynylidene]cyclobutane-1,2-diol (24). Preparation bymethod B on 0.295 mmol scale with a reaction time of20 min. Purification by flash chromatography eluting withEt2O/Hept (20/80) afforded the product (83 mg, 80%)as ayellowVpowder. Rr:0.33 (Et2O/Hept: 30/70)'H NMR (300 MHz, CDCl3) 8 7.71 (s, 1H, ArH), 7.56 (d,2H, J=6.8 Hz, ArH), 7.33-7.24 (m, 3H, ArH, Ph-CH),4.85 (br d, 1H, J=5.2 Hz, HO-CH-CH-OH), 4.68 (br m1H, HO-CH-CH-OH, OH), 2.51 (t, 2H, J=7.2 Hz,CH21.68-1.57 (m, 2H, CH2), 1.46-1.27 (m, 4H,2xCH2), 0.94(t, 3H, J=7.2 Hz, CH3), 0.19 (s, 9H, SiMes), 'CNMR(75MHz, CDCl3) 157.6,142.8,136.0,129.3,128.3,128.0,122.7,105.7,82.6,72.2,71.1,31.2,28.7,22.2, 20.4,13.9,13.6,-1.1. HRMS (ESI, positive ion180 eV) calcd for (C22H30Q SiNa)::3377.1913; found:377.1829. 4.3.7. (1S,2R,3Z,4Z)-3-Benzylidene-4-[1-(trimethylsilyl)-undec-2-ynylidene]cyclobutane-1,2-diol (25). Preparationby method B on 0.295 mmol scale with a reaction time of20 min. Purification by flash chromatography eluting withEtzO/Hept (20/80) afforded the product (84 mg, 80%) asa yellow oil. R: 0.39 (Et)O/Hept: 30/70). H NMR(300 MHz, CDCl3) ; 7.70 (s, 1H, ArH), 7.59 (d,2H.J=7.1 Hz, ArH), 7.35-7.25 (m, 3H, ArH, Ph-CH), 4.92 (t,1H, J=7.2 Hz, HO-CH-CH-OH), 4.73 (t, 1H, J=7.2 Hz,HO-CH-CH-OH), 3.63 (d, 1H, J=6.2 Hz, OH), 3.45 (d,1H, J=7.2 Hz, OH), 2.52 (t, 2H, J=6.9 Hz, CH2), 1.68-1.58 (m, 2H, CH2), 1.48-1.27 (m, 10H, 5xCH2) 0.93 (t,3H, J=7.2 Hz, CH3), 0.24s,9H, SiMe3). 13C NMR(75 MHz, CDCl3) 8 157.6, 143.0,136.0,129.4,128.5,128.2,127.9,122.5,105.6,82.7,72.2,71.2,31.8, 29.1,29.0,22.6,20.4,17.3,14.1,13.6,-1.1.HRMS (ESI, positiveion 180 eV) calcd for (C25H3602SiNa)*: 419.2382; found:419.2377. ( 4.3.8. (1S,2R,3Z, 4 Z) - 3-Benzylidene-4-[2-furyl(trimethyl-silyl)methylene]cyclobutane-1,2-diol (26). Preparation by method B on 0 . 295 mmol scale with a reaction time of 20 min. Purification by flash chromatography e luting with AcOEt/Hept ( 2 0/80) afforded t he product (61 mg, 63%) a sa yellow oil. R: 0.40 (AcOEt/Hept: 30/70). H NMR(300 MHz, CDCl3)8 7.49 (d,2H,J=7.2 Hz,ArH), 7.44 (s,1H, ArH), 7.33-7.24 (m,3H, ArH, Ph-CH),6.48 (br s, 1 H, ) ( furyl-H), 6.46 (t, 1H, J=3.2 H z, furyl-H), 6 .24 (d, 1 H, J=3.2 Hz, furyl-H), 4.93 (d , 1H , J=5.6 Hz, HO-CH-CH-OH), 5 .82 (d, 1H, J=5.6 Hz, HO-CH-CH-OH), 3.2 9 (br s, 2H,OH), 0.24 (s, 9H, SiMe3). 13c NMR (75 MHz, CDCl3) ; 154.3 , 153.9, 1 41.5, 1 40.9,135.9,1 3 0.8,1 2 9.3, 1 29.2, 128.6, 128.1 , 111.3,107.9,71.8, 7 0.5, -0.3. MS (ESI,positive ion 180 eV) calcd for (C19H2203SiNa)*:349.12; found: 349.13. ) 4.3.9. (1S,2R,3Z,4Z)-3-Benzylidene-4-[thien-2-yl(trime-thylsilyl)methylene]cyclobutane-1,2-diol (27). Prepara-tion by method B on 0.295 mmol scale with a reactiontime of 30 min. Purification by flash chromatography elutingwith AcOEt/Hept (20/80) afforded the product (47 mg,46%)as a yellow oil. R: 0.31 (AcOEt/Hept: 30/70). H NMR(300 MHz,CDCl3) 8 7.39-7.21 (m, 6H, ArH, Ph-CH), 7.05(dd, 1H, J=3.1, 1.5 Hz, thionyl-H), 6.66 (d, 1H, J=3.1 Hz,thionyl-H), 5.98 (d, 1H, J=1.5 Hz, thionyl-H), 4.95 ((d,1H, J=5.3 Hz, HO-CH-CH-OH), 4.84 (d,1H, J=6.5 Hz,HO-CH-CH-OH), 2.90 (br s, 2H,OH), 0.21 (s, 9H, SiMe3).I'C NMR (75MHz, CDCl;) 6 155.2, 153.5,141.0,140.9,135.8.130.2, 129.1,129.0,127.9,127.6,110.9,107.5,71.6,70.1, -0.2. MS (ESI, positive ion 180 eV) calcd forC19H22O2SSiNa)+: 365.10; found: 365.12. 4.3.10. (1S,2R,3Z,4Z)-3-Benzylidene-4-{(4E)-4-methyl-1-(trimethylsilyl)-6-[(trimethylsilyl)oxy]hex-4-en-2-ynyl-idene}cyclobutane-1,2-diol (28). Preparation by method Bon 0.295 mmol scale with a reaction time of 20 min. Purifi-cation by flash chromatography eluting with Et2O/Hept(20/80) afforded the product (97 mg, 77%) as a yellowpowderr.. R0.36 (Et2O/Hept: 30/70). H NMR (300 MHz,CDCL)67.67((s, 1H, Ph-CH), 7.59 (d, 2H, J=7.8 Hz,ArH), 7.38-7.19 (m, 3H, ArH), 5.98 (t, 1H, J=6.3 Hz,CH-CH), 4.93 (dd, 1H, J=7.8, 6.8 Hz HO-CH-CH-OH),4.77 (dd,1H, J=7.1, 6.8 Hz, HO-CH-CH-OH), 4.28 (d,J=6.3 Hz, O-CH2), 3.35 (d,1H, J=7.1 Hz, OH), 3.23(d, 1H, J=7.1 Hz, OH), 1.92 (s, 3H, CH3), 0.27 (s, 9H,O-SiMe3), 0.15 (s, 9H, SiMe3). 13cNMR (75 MHz, CDCl3); 158.0,142.5,141.5,134.8,129.3,128.6,128.1,121.5,109.8, 102.0,92.1,73.2,72.6,65.5,57.3,17.7, 0.2,-1.3.MS (ESI, positive ion 180 eV) calcd for (C24H34O3SiNa)*:449.19; found: 449.23. 4.3.11.(1S,2R,3Z,4Z)-3-Benzylidene-4-[(4E)-6-{[tert-bu-tyl(dimethyl)silyl]oxy}-1-(trimethylsilyl)hex-4-en-2-ynyl-idene]cyclobutane-1,2-diol (29). Preparation by method Bon 0.295 mmol scale with a reaction time of 20 min. Purifi-cation by flash chromatography eluting with Et2O/Hept (20/80) afforded the product (98 mg, 75%) as a yellow oil.R:0.40 (Et2O/Hept: 30/70). H NMR (200 MHz, CDCl3); 7.69 (s, 1H, Ph-CH), 7.60 (d, 2H, J=6.8Hz, ArH),7.35-7.29 (m, 3H, ArH), 6.21 (dt, 1H, J=15, 4.4 Hz,O-CH-CH=CH), 6.06 (d, 1H, J=15 Hz, O-CH2-CH=CH), 4.91 (t, 1H, J=5.9 Hz,HO-CH-CH-OH), 4.72 (t, 1H,J=5.9 Hz, HO-CH-CH-OH), 4.30 (d, 2H, J=4.4 Hz,O-CH2), 4.05 (br s, 1H,OH), 3.88 (br s, 1H,OH), 0.94 (s,9H, O-Si-tButyl), 0.244(s, 9H, SiMe3), 0.10 (s, 6H,O-SiMe2). I3C NMR (75MHz, CDCl3) 6 158.5,142.7,141.6, 135.9,129.4,128.7,128.2,121.5,109.9,102.0,92.1,72.2,71.2,63.2,25.9,18.4,-1.1, -5.3. MS (ESI,positive ion 180 eV) caled for (C23H3803Si2Na)*: 477.23;found: 477.22. 4.3.12. (1S,2R,3Z,4Z)-3-Benzylidene-4-[(4E)-6-{[tert-bu-tyl(dimethyl)silyl]oxy}-4-methyl-1-(trimethylsilyl)hex-4-en-2-ynylidene]cyclobutane-1,2-diol (30). Preparation bymethod B on 0.295 mmol scale with a reaction time of20 min. Purification by flash chromatography eluting withEt2O/Hept (20/80) afforded the product (98 mg, 71%) asa yellow powder. R: 0.41 (EtzO/Hept: 30/70). 'H NMR(300 MHz, CDCl3) ; 7.62 (s, 1H, Ph-CH), 7.58 (d, 2H,J=7.7 Hz, ArH) 7.39-7.20(m, 3H, ArH), 6.01 (t, 1H,J=6.2 Hz, CH2-CH), 4.98 (dd, 1H, J=7.3, 6.8 Hz, HO-CH-CH-OH), 4.77 (dd, 1H, J=7.3, 6.8 Hz, HO-CH-CH-OH), 4.19 (d, 2H, J=6.2 Hz, O-CH2), 3.60 (d, 1H,J=7.3 Hz, OH), 3.46 (d, 1H, J=7.3 Hz,OH), 1.92 (s, 3H,CH3), 0.96 (s, 9H, O-Si-tButyl), 0.23 (s, 9H, SiMe3), 0.12(s, 6H, O-SiMe2). 13c NMR (75 MHz, CDCl3) 6 158.4,142.4,141.2,137.0,136.0,129.2,128.7,128.3,121.5,119.7,106.3,102.0,89.1,72.0,71.1,60.2,26.8,18.3,-1.0, -5.1. MS (ESI, positive ion 180 eV) calcd for(C27H4003Si2Na)+: 491.24; found: 491.20. 4.3.13.(1S,2R,3Z,4Z)-3-Benzylidene-4-[1-(trimethyl-silyl)but-3-enylidene]cyclobutane-1,2-diol (31). Prepara-tion by method B on 0.295 mmol scale with a reactiontime of 30 min. Purification by flash chromatography elutingwith Et2O/Hept (20/80) afforded the product (60 mg, 63%)as a yellow oil. R: 0.30 (Et2O/Hept: 30/70).H NMR(200 MHz, CDCl3) 8 7.58 (d, 2H, J=7.6 Hz, ArH),),7.38-7.25 (m, 3H, ArH), 6.75 (s, 1H, Ph-CH),le5.97-5.78(m, 1H, CH2=CH), 5.15-5.05 (m, 2H, CH22=CH),4.87(ap t, 1H, J=7.3 Hz, HO-CH-CH-OH), 4.73 (apt,1H,J=6.8 Hz, HO-CH-CH-OH), 3.17 (d, 2H, J=5.6 Hz, CH),2.97 (d, 1H, J=6.6 Hz, OH), 2.72 (d, 1H, J=7.3 Hz,OH),0.22 (s,9H, SiMe3). Ic NMR (50 MHz, CDCl) 151.9,142.3,141.1,136.2,135.3,128.8,128.6,127.9, 115.7,71.5,70.4,36.4,-0.6. HRMS (ESI, positive ion 180 eV)calcd for (C18H24OSiNa)t:323.1438; found: 323.1421. 4.3.14. (1S,2R,3Z,4Z)-3-Benzylidene-4-[4-methoxy-1-(trimethylsilyl)but-2-ynylidene]cyclobutane-1,2-diol(32). Preparation by method B on 0.295 mmol scale witha reaction time of 20 min. Purification by flash chromato-graphy eluting with Et2O/Hept (20/80) afforded the product(86 mg, 84%) as a yellow oil. R: 0.39 (Et2O/Hept: 30/70)H NMR (300 MHz, CDCl3) 8 7.70 (s, 1H, ArH), 7.57 (d,2H,J=7.5 Hz, ArH),7.33-7.266 (m, 3H, ArH, Ph-CH),4.87 (d, 1H, J=6.5 Hz, HO-CH-CH-OH), 4.68 (d, 1H,J=6.5 Hz, HO-CH-CH-OH), 4.40 (s, 2H, CH2-OMe), 3.43(s, 3H, OMe), 0.21 (s, 9H, SiMe)13c NMR (75 MHz,CDCl3) 6 159.5, 142.1,135.8,129.5,129.3,128.7,128.4,120.5, 98.8,88.1,72.0,71.1,61.0,57.6, -1.0. MS (ESI,positive ion 180 eV) calcd for (C19H2403SiNa)*: 351.56;found: 351.56. 4.3.15. (1S,2R,3Z,4Z)-3-Benzylidene-4-[4-benzyloxy-1-(trimethylsilyl)but-2-ynylidene]cyclobutane-1,2-diol(33). Preparation by method B on 0.295 mmol scale witha reaction time of 20 min. Purification by flash chromato-graphy eluting with Et2O/Hept (20/80) afforded the product(90 mg,72%) as a yellow oil. R:0.45 (Et2O/Hept: 30/70).H NMR (200 MHz, CDCl3) 8 7.56-7.34 (m, 11H, ArH,Ph-CH), 5.31 (s, 2H, HO-CH-CH-OH), 4.53 (s, 2H,Ph-CH2-O), 4.49 (s, 2H, Ph-CH2-O-CH2), 0.39 (s, 9H,SiMe3).13c NMR (75 MHz, CDCl3) 158.8,149.0,142.3, 135.7,130.9,129.5,129.2,129.1,128.5,128.1,127.3,120.8,118.1,114.1,99.3,85.7,72.0,71.1,43.7,38.7,-1.2.MS (ESI, positive ion 180 eV) calcd for (C25H28O3SiNa)+:427.17; found: 427.18. 4.3.16. (1S,2R,3Z,4Z)-3-Benzylidene-4-[5-{[tert-butyl(di-methyl)silyl]oxy}-1-(trimethylsilyl)pent-2-ynylidene]-cyclobutane-1,2-diol (34). Preparation by method B on0.295 mmol scale with a reaction time of 20 min. Purificationby flash chromatography eluting with Et2O/Hept (20/80→30/70) afforded the product (111 mg, 85%) as a yellowpowder. R: 0.29 (EtzO/Hept: 30/70). HNMR (300 MHz,CDCl3) 8 7.69 (s, 1H, ArH), 7.57 (d, 2H,J=7.2 Hz,ArH)7.33-7.26 (m, 3H, ArH, Ph-CH), 4.84 (br s, 2H, HO-CH-CH-OH, OH), 4.66 (br s, 2H, HO-CH-CH-OH,OH),3.82(t,2H, J=7.6Hz, O-CHz), 2.74(t, 2H, J=7.6Hz,O-CH2-CH2), 0.90 (s, 9H, Si-tButyl), 0.19 (s, 9H, SiMe33),0.08 (s, 6H, SiMe).13c NMR (75 MHz, CDCl3) 6 158.1,142.8,129.5,128.6,128.0,122.0,101.4,83.7,72.2,71.1,62.2,25.8,24.8,18.2,-1.1, -5.3. MS (ESI, positive ion180 eV) calcd for (C25H38O3SizNa)+:475.22; found: 475.18. 4.3.17.(1S,2R,3Z,4Z)-3-Benzylidene-4-[4-{[tert-butyl(di-phenyl)silyl]oxy}-1-(trimethylsilyl)but-2-ynylidene]-cyclobutane-1,2-diol (35). Preparation by method B on0.295 mmol scale with a reaction time of 20 min. Purificationby flash chromatography eluting with Et2O/Hept(20/80)afforded the product (101 mg,80%) as a yellow powder.R: 0.33 (Et)O/Hept: 30/70). H NMR (300 MHz, CDCl;)87.61-7.30 (m, 16H, ArH, Ph-CH), 4.86-4.81 (m,2H, HO-CH-CH-OH), 4.62 (s,2H, O-CH2), 0.91 (s, 9H, Si-tButyl),0.31 (s, 9H, SiMes). 13C NMR (75 MHz, CDCl3) 8 151.6,144.5,138.2,137.8,136.7,136.5,136.1,135.6,135.5,134.7,134.6,133.5,129.7,129.6,129.4,128.8,128.5,128.4,128.1,127.6,127.4,73.5,72.9,63.8,26.7,19.2, -0.9. MS (ESI,positive ion 180 eV) calcd for (C34H40O3SiNa)+: 575.25;found: 575.32. 4.3.18.(1S,2R,3Z,4Z)-3-Benzylidene-4-[4-[methyl(phenyl)-amino]-1-(trimethylsilyl)but-2-ynylidene]cyclobutane-1,2-diol(36). Preparation by method B on 0.295 mmol scalewith a reaction time of 20 min. Purification by flash chro-matography eluting with Et2O/Hept (20/80) afforded theproduct (94 mg, 79%) as a yellow powder. R:0.18 (Et2O/Hept: 30/70). H NMR (200 MHz, CDCl3) 6 7.56-7.11(m, 11H, ArH, Ph-CH), 5.11 (d, 1H,J=6.8 Hz, HO-CH-CH-OH), 4.86 (d, 1H, J=6.8 Hz, HO-CH-CH-OH), 3.89(s, 2H,N-CH2), 3.05 (s, 3H, N-CH3), 0.22 (s, 9H, SiMe3).C NMR (50 MHz, CDCl3) 151.6,148.9,147.0,139.5,138.9,137.8,128.7,128.4,127.5,127.3,127.0,117.3,95.1, 88.6,72.2,69.8,51.0,38.9,-1.3.MS (ESI, positiveion 180 eV) calcd for (C25H2902SiNa)*: 403.19; found:403.22. 4.3.19. The 6r electrocyclization process. Compounds 39-45 are already reported in Ref. 2b. 4.3.20. (7R,8S)-2-(Trimethylsilyl)bicyclo[4.2.0]octa-1(6),2-diene-7,8-diol (46). Preparation by method B on0.758 mmol scalewith a reaction time of 30 min. Purificationby flash chromatography eluting with Et2O/Hept (30/70)afforded the product (65 mg, 44%) as a white solid. R:0.28 (Et2O/Hept: 30/70). H NMR (200MHz, CDCl3) 400-188-0725 6.06 (t, 1H, J=4.9 Hz, CH-CH2), 4.69 (d, 1H, J=4.4 Hz,HO-CH-CH-OH), 4.28 (d, 1H, J=4.4 Hz, HO-CH-CH-OH), 2.25-1.98 (m, 6H, 2xCH2, 2×OH), 0.18 (s, 9H,SiMe3). 1cNMR(75 MHz, CDCl;) 6 148.8, 146.2,138.4,132.2,74.4,73.6,24.2,19.4, -1.5. MS (ESI, positive ion180 eV) calcd for (Ci1H1802SiNa)*: 233.09; found: 233.13. 4.3.21.(7R,8S)-5-Phenyl-2-(trimethylsilyl)bicyclo[4.2.0]-octa-1(6),2-diene-7,8-diol (47). Preparation by method Bon 0.295 mmol scale with a reaction time of 30 min. Purifi-cation by flash chromatography eluting with AcOEt/Hept(20/80) afforded the product (65 mg,49%) as a white solid.R: 0.36 (Et2O/Hept: 30/70). 'H NMR (200 MHz, CDCl3)7.34-7.21 (m, 3H, ArH), 7.17-7.12 (m, 2H, ArH,Ph-CH), 6.10 (t, 1H, J=4.9 Hz, ArH), 4.85 (d, 1H,J=3 Hz, HO-CH-CH-OH), 4.73 (d, 1H, J=3 Hz, HO-CH-CH-OH), 3.70 (dd, 1H, J=11,5Hz, CH-CH2), 2.94-2.45(m, 4H, CH2, 2×OH), 0.18 (s, 9H, SiMe3). 13C NMR(75 MHz, CDCl3); 149.2,147.2,137.8,132.1,128.6,127.3,126.6,73.0,73.0,36.2,35.4,-1.5. HRMS (ESI,positive ion 180 eV) calcd for (C17H2202SiNa)+: 309.1287;found: 309.1253. IR (FTIR, film): v=3410 (br), 2924,2845, 1652,1490,1448,1250,1107,1062,893,749 cmn method B on 0.33 mmol scale with a reaction time of38 min. Purification by flash chromatography eluting withAcOEt/Hept (5/95→40/60) afforded two diastereoisomers(47 mg, ratio 1/1, 41%) as yellow oils. R: 0.30,0.35(AcOEt/Hept: 40/60). Diastereoisomer A:H NMR(200 MHz, CDCl3) 6.05 (s, 1H,=CH), 4.52 (s, 1H,CH-OH), 3.56 (d, 1H, J=6.4 Hz, CH2-OH), 2.57-2.44(m, 12H, 4×CH2, CH, 3×OH), 2.31-1.64 (m, 6H), 0.21(SiMe3).C NMR (75 MHz, CDCl3) 8 154.2, 144.3, 140.5,136.7,135.6,134.9,133.1,79.6,75.5,67.10,41.3,40.0,38.6,36.8,34.8,33.4,26.7,22.4,-0.29 (SiMe3). HRMS(ESI, positive ion 180 eV) calcd for (C20H3003SiNa)*:369.1862; found: 369.2182. Diastereoisomer B: H NMR(200 MHz, CDCl3) 6 6.0 (s, 1H,=CH), 4.4 (s, 1H,CH-OH), 3.5 (d, 1H, J=4.9 Hz, CH2-OH), 2.8 (br s, 3H,3×OH), 2.7-1.6 (m, 15H), 0.20 (SiMe3). C NMR(75 MHz, CDCl3) 154.3,144.2,139.6,136.3,135.6,134.8,133.1,80.7,75.9,67.3,41.2,39.8,38.4,36.8,34.9,33.5, 26.7, 22.4, -0.3. HRMS (ESI, positive ion 180 eV)calcd for (C20H3003SiNa)+: 369.1862; found: 369.1934. 4.3.24. (10aS)-4,8,8-Tris(hydroxymethyl)-5-(trimethyl-silyl)-1,7,8,9,10,10a-hexahydrodicyclopenta[a,d][8]annu-len-3(2H)-one (55).Preparationby method A on 0.265 mmolscale with a reaction time of 9 h. Purification by flash chro-matography eluting with Et2O/Hept (20/80) afforded product(19 mg, 10%) as yellow oil. R: 0.13 (AcOEt/Hept: 30/70).H NMR (200 MHz, CDCL) 8 6.00 (s, 1H, =CH), 4.48(d, 1H, J=13.3 Hz, CH2a-OH), 4.07 (d, 1H, J=13.3 Hz,CH2b-OH),3.63((s, 2H,(CH2-OH), 3.58 (s, 2H, CH2-OH),3.40-3.21 (m, 1H, CH), 33.01-2.71 (br s, 2H,2×OH),2.62-1.89 (m, 7H), 0.15 (s,9H, SiMe3). c NMR (75 MHz,CDCl3)8 209.4, 156.3,144.0,138.8,136.7,136.1,131.6,69.8,69.7,62.3,45.5,45.0,44.6,40.0,37.5,37.0,26.7, -0.5. HRMS (ESI, positive ion 175 eV) calcd for(C20H36O4Si)*: 363.1992; found: 363.1999. ( 4.3.25.(7R,12S)-2-(Hydroxymethyl)-5-(trimethylsilyl)-1,2,3,8,9,10,10a,11-octahydro-7H-6,7-methanobenzo[a]-cyclopenta[d][8]annulene-7,12-diol (67). P r eparation b y ) Preparation by method B on1.64 mmol scale with a reactiontime of 32 min.Purification by flash chromatography elutingwith AcOEt/Hept (20/80--80/20) afforded two products:furan derivative 69 (28 mg, 34%) as yellow oil, R: 0.30(AcOEt/Hept: 40/60) and diol derivative 70 (20 mg, 24%)as yellow oil, R: 0.05 (AcOEt/Hept: 40/60);58% as globalyield. 4.3.26.2-{3-[(7S,10aS,12S)-7,12-Dihydroxy-10a-methyl-5-(trimethylsilyl)-1,3,7,8,9,10,10a,11-octahydro-2H-6,7-methanobenzo[4,5]cycloocta[1,2-c]pyrrol-9(1H)-yl]propyl}-1H-isoindole-1,3(2H)-dione (69). H NMR(300 MHz, CDCl3) 8 7.86-7.83 (m, 2H, ArH), 7.73-7.71(m, 2H, ArH), 5.96 (s, 1H,=CH), 4.48 (s, 1H, CH-OH),3.81-3.72 (ap t, 2H, J=7.2 Hz, N-CH2), 3.64-3.43 (m,4H),, 2.74 (m, 3H), 2.53-2.46(m, 1H), 2.04-1.83 (m,10H), 1.63 (m, 1H),0.21 (SiMe3). 13c NMR (75MHz,CDCl3) ; 188.0,143.4,139.1,135.6,134.3,133.9,131.0,123.2,79.5,75.1,65.8,64.5,53.4,35.9,34.7,30.3,29.7,29.4,27.0,22.3,21.5,-0.5. MS (ESI, positive ion180 eV) calcd for (C29H36N2O4SiNa)+: 527.23; found:527.22. ( 4.3.27. 2 -{3-[(11aS)-11a-Methyl-6-(trimethylsilyl)-2,3,8,10,1 1 ,11a-hexahydro[1]benzofuro[3',4':5,6,7]cyclo-octa[1,2-c]pyrrol-9(1H)-yl]propyl}-1H-isoindole-1,3(2H)-dione (70).'HNMR (300 MHz, CDCl3) ; 7.82-7.80 (m, 2H, ArH), 7.72-7.68 ( m , 2H, Ar H ), 7.05 (s, 1H, furyl-H), 6. 0 5 (s, 1H , =CH), 3.81-3.72 (ap t, 4H, J=7.2Hz,2xN-CH2), 3.51-3,43 (m, 2H), 2.83-2.70 (m, 2H), 2.60-2.54(m, 4H),2.48-2.38(m, 1 H ), 2.21-2.17 ( m, 1 H), 2 .04-1.83 (m, 4H), 1.82 (m, 1H), 0.11 (s, 9H , SiMe3) . 13 c NMR ( 75 MHz, CDCl3) 6 1 88.1,149.1,1 3 6.5,1 3 5.6,135.4,13 3 .9,13 2 .1, 125.2, 1 23.2, 1 20.7,65.7,64.5,53.4,36.0,34.8,30.3,2 9 .7, 29. 3 ,27.0,23.0,19.3,- 1 .2. MS (ESI, p o sitive ion 180 eV)calcd for (C29H34N203SiNa)*: 509.22; found: 509.22. ) 4.3.28. (6R,7S)-2[(4-Methylphenyl)sulfonyl]-5-phenyl-8-(trimethylsilyl)-2,3,4,5,6,7-octahexahydro-1H-cyclo-buta[5,6]cycloocta[1,2-c]pyrrole-6,7-diol (71). Prepara-tion by method B on 0.59 mmol scale with a reaction timeof 38 min. Purification by flash chromatography eluting400-188-0725 with AcOEt/Hept (5/95→40/60) afforded two diastereoiso-mers (36 mg, 1/1, 27%) as yellow oil. R: 0.30, 0.35(AcOEt/Hept:40/60). HNMR (200MHz, CDCl3). DiastereoisomerA: H NMR (200 MHz, CDCl3) 6 7.68 (d, 2H, J=8.1Hz,ArH), 7.40-7.17 (m, 7H, ArH), 6.00 (s,1H, =CH), 4.53(d, 1H, J=6.2 Hz, CH-OH), 4.47 (d, 1H, J=5.3 Hz, CH-OH), 4.15 (s, 2H, CH2-N), 4.05-4.00 (br s, 2H, 2×OH),3.63-3.50 (br s, 2H, CH2-N), 2.58 (d, 1H, J=7.2 Hz,CHa), 2.45 (s,3H, Ar-CH3), 2.34 (d, 1H,J=7.2 Hz, CH,),1.66 (s, 1H, CH-Ar), 0.21 (SiMe3). 3C NMR (75MHz,CDCl3) ; 149.0,147.5,143.7,143.4,142.7,136.6,133.1,129.7,128.9,127.8,127.5,127.2,73.0,71.9,58.4, 57.1,44.0, 33.4, 26.8,-0.59. HRMS (ESI, positive ion 180 eV)calcd for (C28H33NO4Si2Na)*: 507.19; found: 507.19. Dia-stereoisomer B: HNMR (200 MHz, CDCl3) 6 7.76-7.71(m, 2H, ArH),7.40-7.23 (m, 7H, ArH), 6.02 (s, 1H,=CH), 4.71 (br s, 1H,OH), 4.30-4.15 (m,5H), 4.05-4.00(d, 2H, J=7.1 Hz), 3.52-3.47 (m, 2H,CH2-N), 2.43 (s, 3H,Ar-CH3), 1.62 (s, 1H, CH-Ar), 0.26 (SiMe3). 13C NMR(75 MHz, CDCl3) 6 148.9,147.4,143.4,142.9, 142.7,135.6, 133.1,130.1,128.8,127.6,127.5,127.2,73.1,71.9,58.4,57.1,44.0,33.4,26.8,-0.7.MS (ESI, positive ion180 eV) calcd for (C28H33NO4Si2Na)*: 507.19; found:507.21. 4.3.29. (3aR,10bS)-2,2-Dimethyl-7-[(4-methylphenyl)sul-fonyl]-4-phenyl-10-(trimethylsilyl)-4,5,6,7,10b-hexahy-dro-3aH-[1,3]dioxolo[3',4]cycloocta[1',2:5,6]cycloocta-[1,2c]pyrrole (72). Preparation by method B on 0.264 mmolscale with a reaction time of 30 min. Purification by flashchromatography eluting with AcOEt/Hept (20/80→40/60)afforded two nonseparable diastereoisomers (91 mg,1/1.62%) as yellow solid. R: 0.33, 0.35 (AcOEt/Hept: 40/60).H NMR (300 MHz, CDCl3) 8 7.72 (d, 2H, ArH), 7.67-7.20 (m, 5H, ArH), 7.07(m, 2H, ArH), 6.04 (s, 1H,=CH), 5.18 (d, 1H,J=3.4 Hz.CH-OH), 4.65 (d, 1HJ=3.4 Hz, CH-OH),4.30-4.06,(m,. 4H, 2xCH2-N), 3.47(dd, 1H,J=8.7,3.I Hz,CH-Ar), 2.58-2.49 (m, 2H,CH2),2.43 (s, 3H, Ar-CH3),1.27 (br s, 6H,2×CH3), 0.25 (s, 9H,SiMe3).13c NMR (75MHz, CDCl3) 8 150.8, 147.1,143.4,141.2,140.1,132.9,131.0,129.8,129.7,128.8,128.4128.3,127.4,127.0,114.8,81.5,78.2,58.0,57.2,41.2,32.9, 29.2,28.3,21.4,-0.7. MS ESI, positive ion180 eV) calcd for (C31H37NO4SiNa)t:570.21; found:570.20. 4.3.30.5-exo-dig Cyclocarbopalladation. Compounds 74-84 and 85-112 are already described in Refs. 18 and 19. Acknowledgements The work was financially supported by the French Ministryof Education and the Centre National de la Recherche Scien-tifique. References and notes ( 1. (a) Mizoroki, T.; Mori, K.; Ozaki, A. Bull. Chem. Soc. Jpn.1971,44, 581;(b ) He c k, R. F; Nolley, J. P. J. O r g. Chem1972, 37,2320;(c ) Burns, B. ; Grigg, R.; Sridharan, V . ;Worakun, T. Tetrahedron Lett. 1988,29,4325;(d) Burns, B.; ) ( 1 . B H our, C.; Suffert, J.Eur. J. O rg. Chem., i n press. ) ( 8 . L 1 a ue r , U.; Anke, T.; S heldrick, W. S. J. Antibiot . 1989,42,875. ) ( 9..(( a) Sugawara,F.; Stroble , G . ; S trage, R . N .; Siedow,J . N.; V a nDuyne, G. D.; Clardy,J . Proc.Nat l . A cad. Sci. U.S.A. 1987, 84,3081; (b) Au, T. K. ; Ch i ck, W.S . H .; Leung,P. C. Life Sci.2000,67 , 733;(c) H anson, J . R . N a t. P rod. Rep. 1986, 3, 123 and1992,9,481. ) ( 10. Shen,X.;Krasnoff,S. B. ; L u, S.-W.; Dunbar, C. D.;O’Neal, J.;Turgeon, B. G.; Y od e r, O. C.; G ibson, D. M.; H a mann, M. T. . Nat. Prod . 1999 , 62,895. ) ( 1 1 . ( a ) Nozoe, S . ; M o risaki, M.; T suda, K .; I itaka, Y.; Takahashi, N .; . Tamu r a, S.; Ishibashi, K.; Shirasak a , M. J. Am . Chem. Soc . 1 1 965, 8 7 ,49 6 8; (b ) Erguang, L.; Cl a rk, A. M.;Rot e lla, D . P ;H u fford, C. D. J. Nat. Prod. 1995,58, 74 . ) 12. (a) Wender, P. A.; Ihle, N. C. J. Am. Chem. Soc. 1986, 108, ( 4678; (b) W ender, P . A . ; S n apper, M . L. Tetrahedron Let t .1987, 2 8, 2 451;(c) W e nder, P. A. ; Te b be, M. J. S ynthesis 1991 , 1 089; (d) Jorand-Lebrun, C .; F e nsterbank, L .: Malacria, M . Tetrahedron Lett. 1 9 95, 36, 6 4 47; (e ) Wender, P. A.; Nuss, J . M .; S m ith, D. B. ; Su a rez-Sobrino, A.;Vagberg,J.; Decosta, D.; Bordner, J. J.Org. Chem. 1997, 62, 4908; (f) S napper, M . L. ; Tallarico, J . A . ; R a ndall, M. L. J. Am. C hem. Soc. 1 997,119, 1478; (g) W e nder, P. A . ;Correa, A. G.; Sato, Y.;Sun, R.J. Am. Chem. Soc.2000, 122,7815; ( h) E vans, P. A .; Robinson, J. E.; B aum, E . W . ; Fazal,A.N. J. Am. Chem. Soc. 2002,124,8782. ) ( 1 3. (a) Wender, P . A.; Gamber, G. G.; Hubbard, R. D.; Zhang, L.J. Am. Chem. S o c. 2002, 12 4 ,287 6 ; (b) Gilbertson, S. R .; DeBoef, B. J. Am. Chem. S oc. 2002, 124,8734. ) ( 14. (a) Coates, R. M.; Muskopf, J. W.; Senter, P. A. J. Org. Chem. 1985, 50,3541; ( b ) Rowley, M.; Ki s hi, Y . T e trahedron Lett. 1988,2 9 ,49 0 9; (c ) Pa q uette, L. A. ; Li a ng, S. ; Galatsis, P . Synlett 1990, 663; (d) Snider, B. M.; Yang, K. J. Org. Chem. 1992, 5 7, 3 615; (e) L o, P. C.-K.; Snapper, M . L. Or g . Le t t.2001, 3,2819;(f ) Ruprah, P. K.; Cros, J.-P.; P ease, J . E.;Whittingham, W. G.; Williams, J. M . J . Eur J . O rg. C hem. 2002,3145. ) ( 1 5. Rowley,M.;Tsukamoto, M.;Kishi, Y. J. Am. Chem. So c . 1989, 111,2735. ) ( 16. Salem, B.; Suffert, J. Angew. Chem., Int. Ed. 2004,43,2826. ) ( 17. Lautens, M.; Smith,N. D .; Ostrovsky, D. J. Org. Chem. 1997,62,8970. ) 18. Salem, B.; Delort, E.; Klotz, P.; Suffert, J. Org. Lett. 2003, 5,2307. ( 19.B o ur, C.; Suffert, J. Org. Lett. 2005, 7, 653. ) 20. For a few recent examples of 1,4-Pd shift, see for instance: (a)Wang,L.;Pan, Y.;Jiang, X.; Hu, H. Tetrahedron Lett. 2000,41,725; (b) Karig, G.; Moon, M.-T.; Thasana, N.; Gallagher, T.Org. Lett. 2002,4,3115;(c) Campo, M. A.; Larock, R. C.J. Am. Chem. Soc. 2002,124,14326;(d) Campo, M. A.;Huang, Q.; Yao, T.; Tian, Q.; Larock, R. C. J. Am. Chem.Soc. 2003, 125,11506; (e) Huang, Q.; Fazio, A.; Dai, G.;Campo, M. A.; Larock, R. C. J. Am. Chem. Soc. 2004,126,7460; (f) Zhao,J.; Larock, R. C. Org. Lett. 2005,7, 701. 21. Mota, A. J.;Dedieu, A.; Bour, C.; Suffert, J. J. Am. Chem. Soc.2005,127,7171. 22. For a few recent examples of tandem or cascade reactions thatmay involve Pd(IV) palladacycles, see for instance: (a)Catallani, M.; Frignani, F; Rangoni, A. Angew. Chem., Int.Ed. 1997, 36, 119; (b) Catellani, M. Pure Appl. Chem. 2002,74, 63; (c) Catellani, M. Synlett 2003, 298; (d) Motti, E.; Mignozzi, A.; Catellani, M. J. Am. Chem. Soc. 2004,126,78; (e) Motti, E.; Rossetti, M.; Bocelli, G.; Catellani, M.J. Organomet. Chem.2004,689,3741;(f) Grigg, R.;Loganathan, V.; Stevenson, P.; Sukirthalingam, S.; Worakun,T. Tetrahedron 1996, 52, 11479;(g) Dyker, G. Angew. Chem.,Int. Ed. Engl. 1992, 31, 1023; (h) Dyker, G. Angew. Chem.,Int. Ed. Engl. 1994,33,103;(i) Dyker, G. Angew. Chem., Int.Ed. 1999, 38, 1699; (j) Albrecht, K.; Reiser, O.; Weber, M.;Knieriem, B.;de Meijere, A. Tetrahedron 1994,50, 383; (k)de Meijere, A.; Meyer, F. E. Angew. Chem., Int. Ed. Engl.1994, 33, 2379; (1) Huang, Q.; Campo, M. A.; Yao, T.; Tian,Q.; Larock, R. C. J. Org. Chem. 2004, 69,8251. 23. Suffert, J. J. Org. Chem. 1989, 54, 509. 24. (a) Brakta,M.;Doyle Daves, G., Jr.J. Chem. Soc., Perkin Trans.11992,1883;(b) Betzer, J. F.; Ardisson, J.; Lallemand,J.Y.;Pancrazi, A. Tetrahedron Lett. 1997,38,2279; (c) Betzer,J. F.; Delaloge, F.; Muller, B.; Pancrazi, A.; Prunet, J. J.OrgChem. 1997, 62,7768;(d) Rim, C.; Son, D. Y. Org. Lett.2003,5,3443. ww.rysstech.com耐士科技 Scheme .耐士科技
确定










还剩13页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中特殊物质和基团检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中特殊物质和基团检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多