








方案详情
文
Clandestine laboratories constantly produce new synthetic cannabinoids to circumvent legislative efforts,complicating toxicological analysis. No extensive synthetic cannabinoid quantitative urinary methodsare reported in the literature. We developed and validated a liquid chromatography–tandem mass spec-trometric (LC–MS/MS) method for simultaneously quantifying JWH-018, JWH-019, JWH-073, JWH-081,JWH-122, JWH 200, JWH-210, JWH-250, JWH-398, RCS-4, AM-2201, MAM-2201, UR-144, CP 47,497-C7,CP 47,497-C8 and their metabolites, and JWH-203, AM-694, RCS-8, XLR-11 and HU-210 parent com-pounds in urine. Non-chromatographically resolved alkyl hydroxy metabolite isomers were consideredsemi-quantitative. Glucuronidase hydrolyzed urine was extracted with 1 ml Biotage SLE+ columns.Specimens were reconstituted in 150 L mobile phase consisting of 50% A (0.01% formic acid in water)and 50% B (0.01% formic acid in 50:50 methanol:acetonitrile). 4 and 25 L injections were performed toacquire data in positive and negative ionization modes, respectively. The LC–MS/MS instrument consistedof a Shimadzu UFLCxr system and an ABSciex 5500 Qtrap massspectrometer with an electrospray source.Gradient chromatographic separation was achieved utilizing a Restek Ultra Biphenyl column with a0.5 ml/min flow rate and an overall run time of 19.5 and 11.4 min for positive and negative mode methods,respectively. Quantification was by multiple reaction monitoring with CP 47,497 compounds and HU-210ionized via negative polarity; all other analytes were acquired in positive mode. Lower and upper limitsof linearity were 0.1–1.0 and 50–100 g/l (r2> 0.994).
方案详情

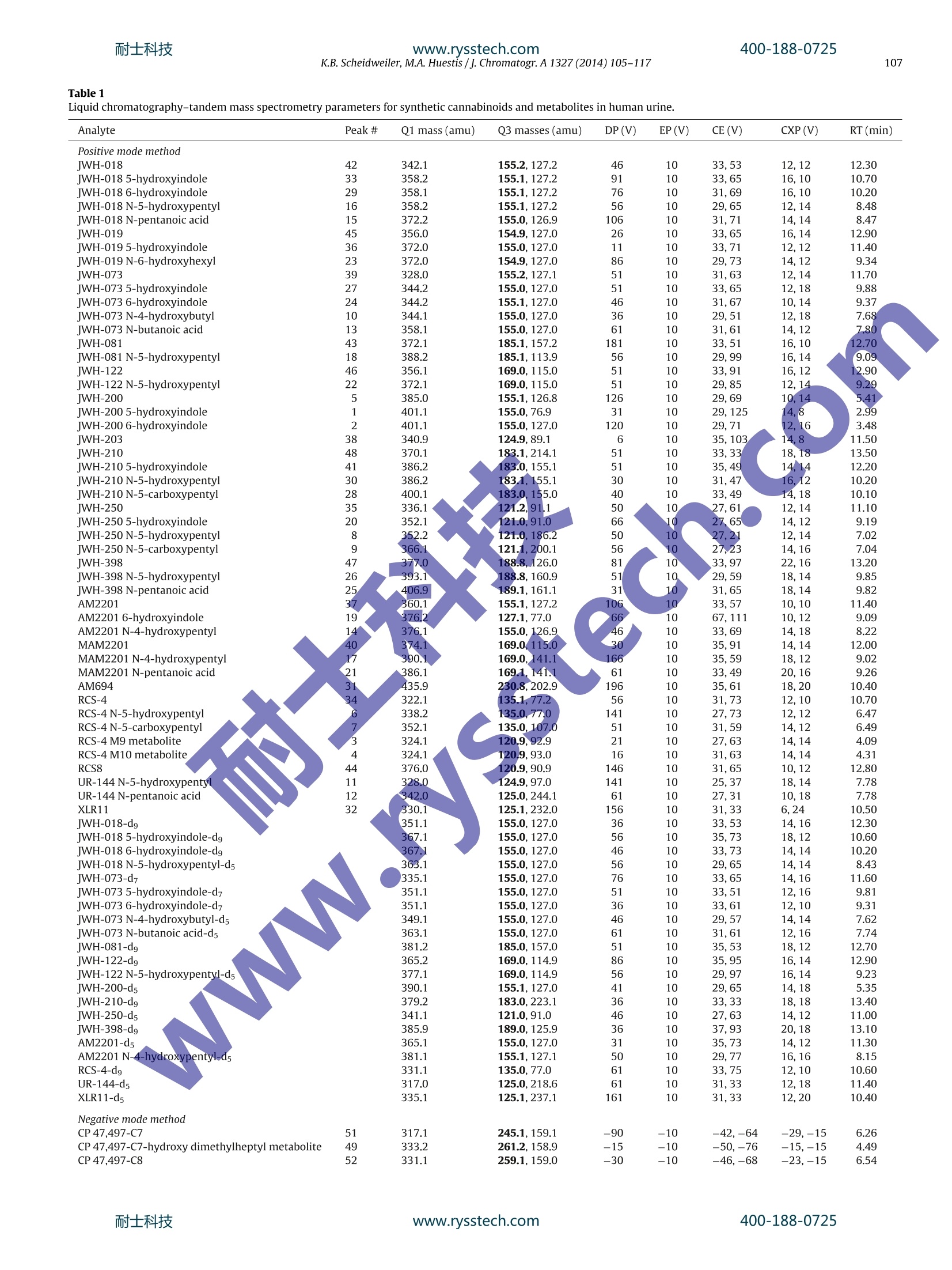
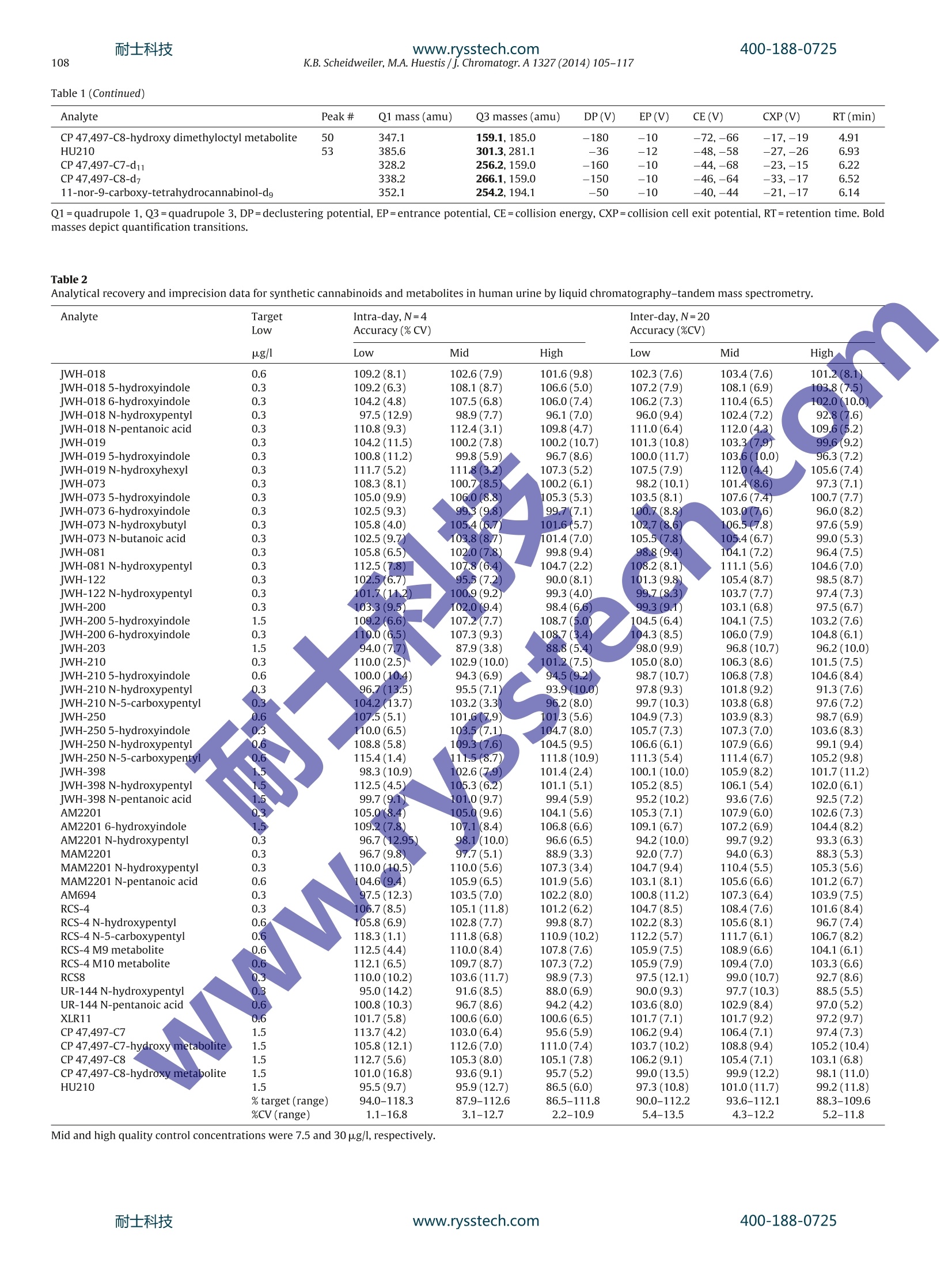
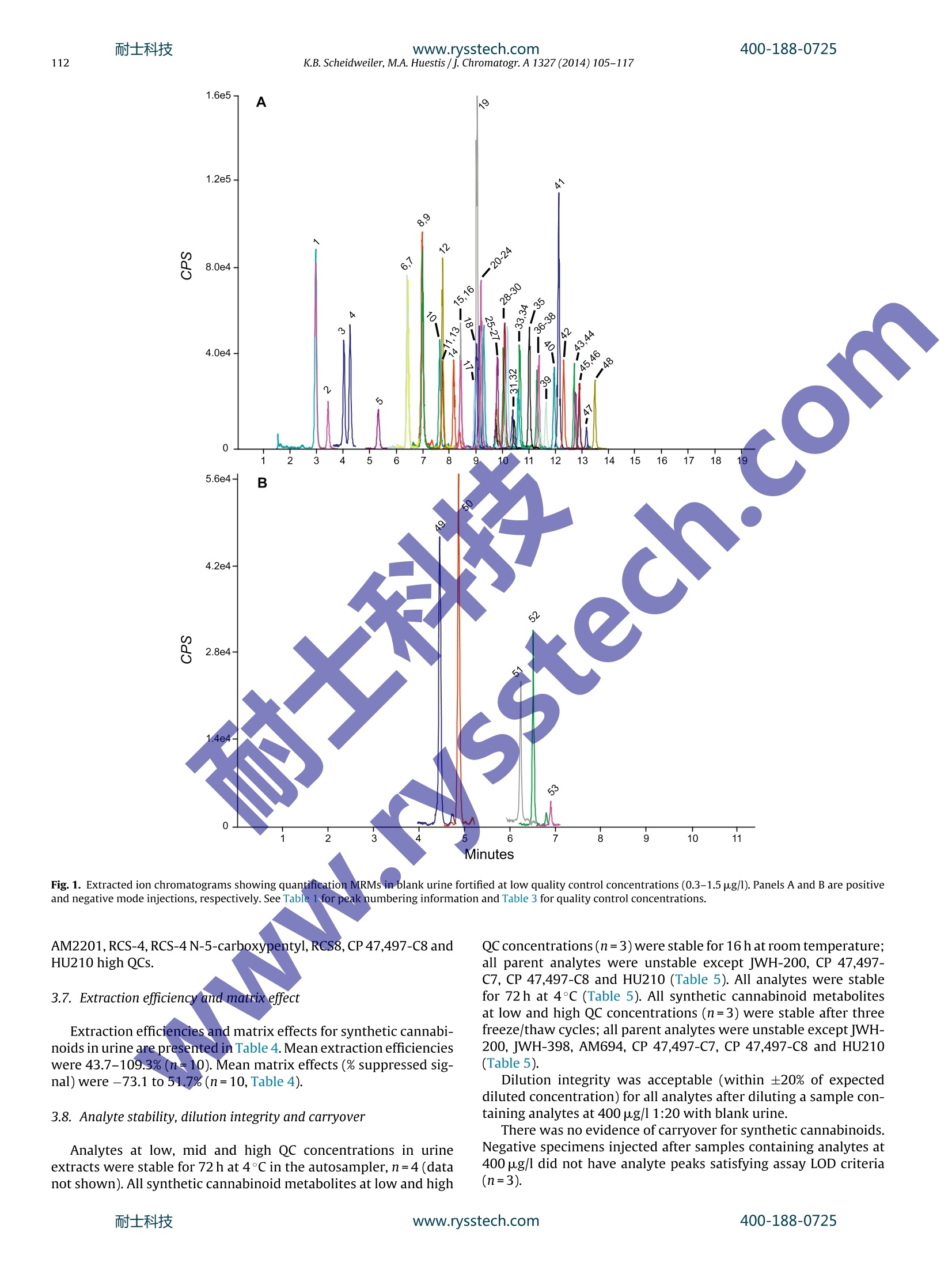
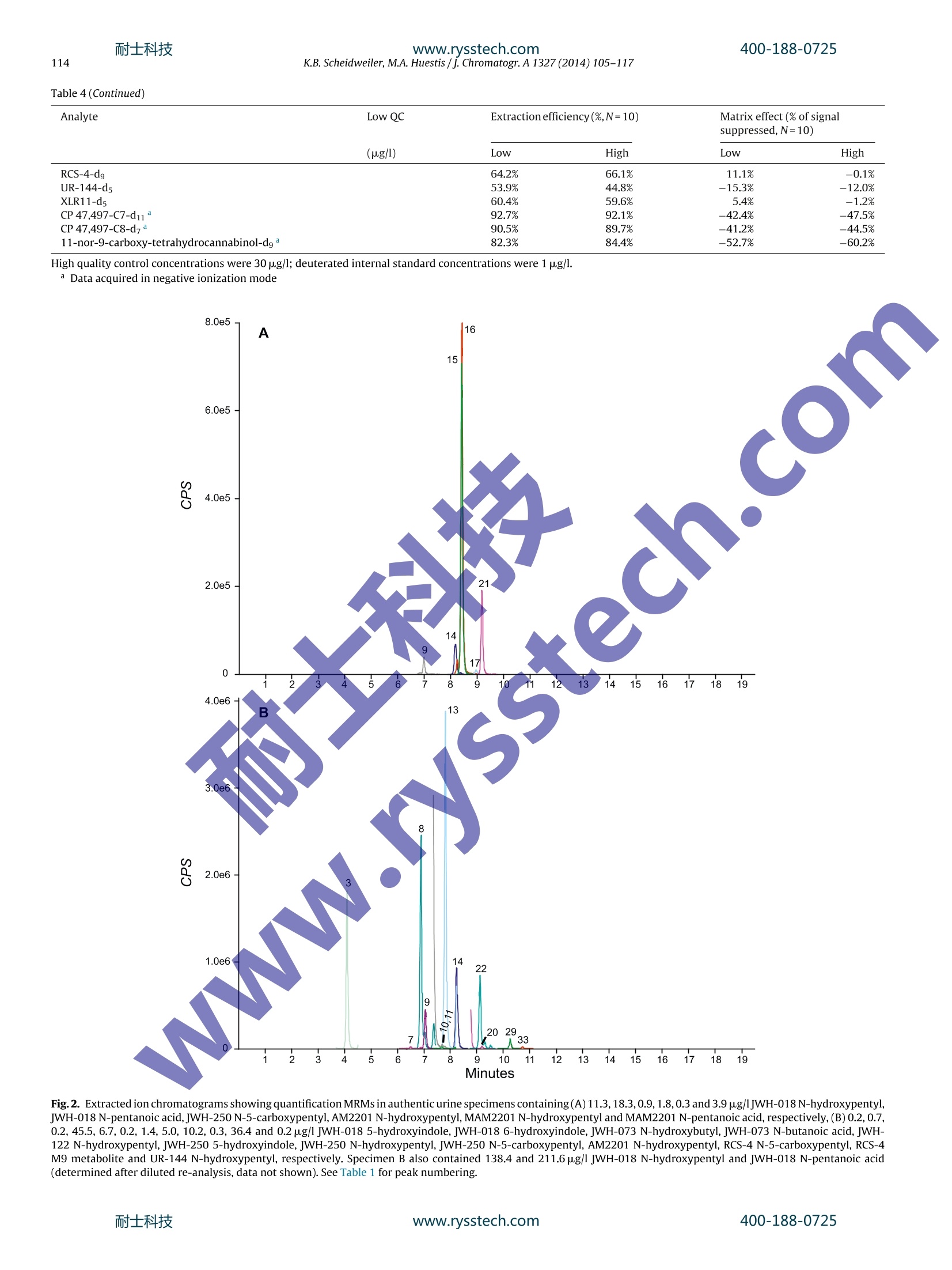
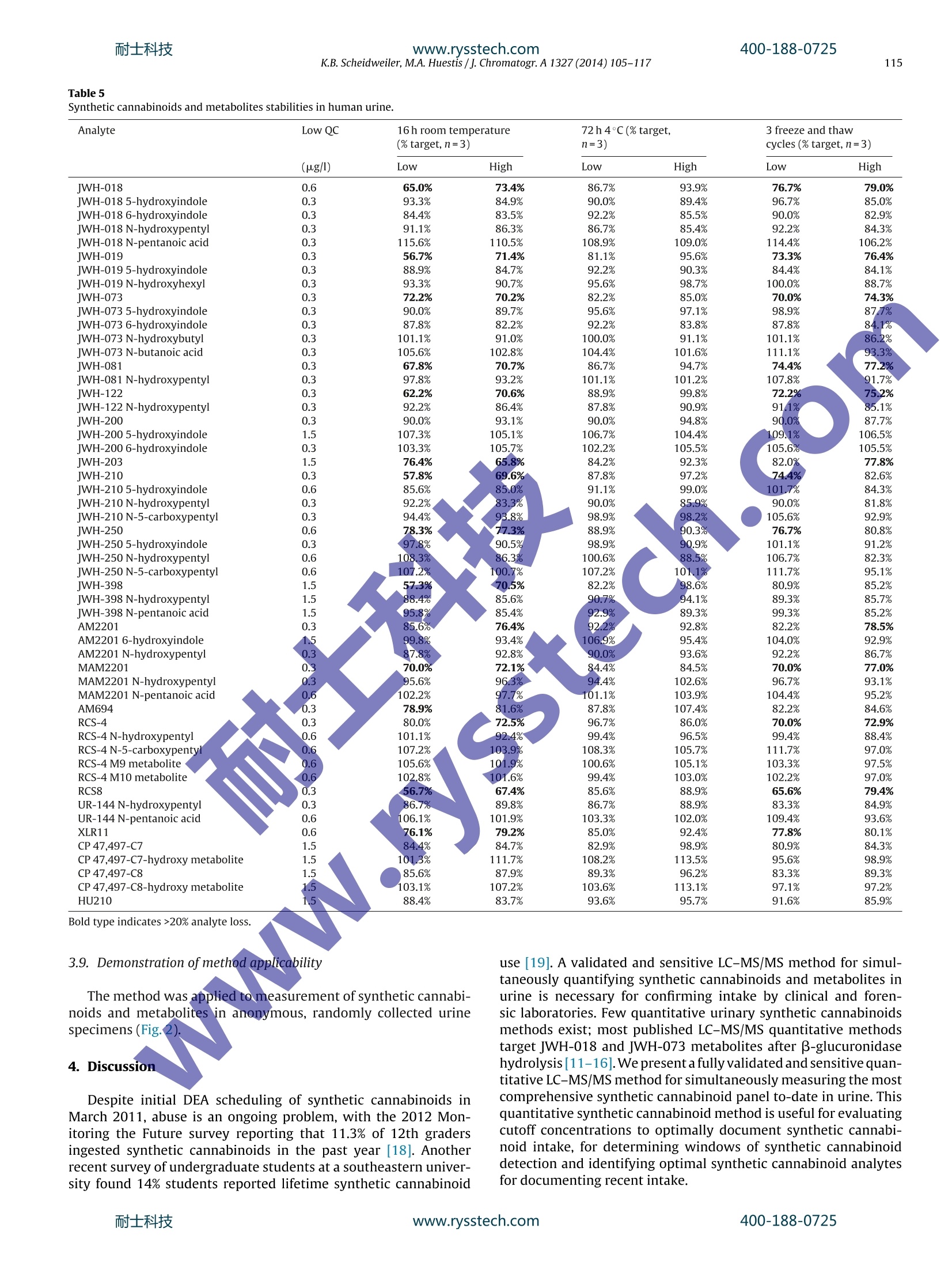
耐士科技400-188-0725www.rysstech.com 耐士科技400-188-0725www.rysstech.com106K.B. Scheidweiler, M.A. Huestis / J. Chromatogr. A1327 (2014) 105-117 Contents lists available at ScienceDirect Journal of Chromatography A journal homepage: www.elsevier.com/locate/chroma Simultaneous quantification of 20 synthetic cannabinoids and 21metabolites, and semi-quantification of 12 alkyl hydroxy metabolitesin human urine by liquid chromatography-tandem massspectrometry Karl B. Scheidweiler, Marilyn A. Huestis* Chemistry and Drug Metabolism, Intramural Research Program, National Institute on Drug Abuse, National Institutes of Health,Baltimore, MD 21224, USA Article history:Received 5 August 2013Received in revised form21 November 2013Accepted 20 December 2013Available online 30 December 2013 A BSTRACT Clandestine laboratories constantly produce new synthetic cannabinoids to circumvent legislative efforts,complicating toxicological analysis. No extensive synthetic cannabinoid quantitative urinary methodsare reported in the literature. We developed and validated a liquid chromatography-tandem mass spec-trometric (LC-MS/MS) method for simultaneously quantifyingJWH-018,JWH-019,JWH-073,JWH-081,JWH-122,JWH-200,JWH-210,JWH-250,JWH-398,RCS-4,AM-2201,MAM-2201, UR-144, CP 47,497-C7,CP 47,497-C8 and their metabolites, and JWH-203,AM-694, RCS-8, XLR-11 and HU-210 parent com-pounds in urine. Non-chromatographically resolved alkyl hydroxy metabolite isomers were consideredKeywords:Synthetic cannabinoidssemi-quantitative. B-Glucuronidase hydrolyzed urine was extracted with 1 ml Biotage SLE+ columns.UrineSpecimens were reconstituted in 150 uL mobile phase consisting of 50%A (0.01% formic acid in water)Metabolites and 50% B (0.01% formic acid in 50:50 methanol:acetonitrile). 4 and 25 pL injections were performed toAnalytical method acquire data in positive and negativeionization modes, respectively.The LC-MS/MS instrument consistedLC-MS/MS of a Shimadzu UFLCxr system and an ABSciex 5500 Qtrap mass spectrometer with an electrospray source.Gradient chromatographic separation was achieved utilizing a Restek Ultra Biphenyl column with a0.5 ml/min flow rate and an overall run time of 19.5 and 11.4 min for positive and negative mode methods,respectively. Quantification was by multiple reaction monitoring with CP 47,497 compounds and HU-210ionized via negative polarity; all other analytes were acquired in positive mode. Lower and upper limitsof linearity were 0.1-1.0 and 50-100 pg/l (r²>0.994). Validation parameters were evaluated at threeconcentrations spanning linear dynamic ranges. Inter-day analytical recovery (bias) and imprecision(N=20) were 88.3-112.2%and 4.3-13.5% coefficient of variation,respectively. Extraction efficiencies andmatrix effect (N=10)were 44-110 and -73 to 52%, respectively. We present a novelLC-MS/MS methodfor simultaneously quantifying 20 synthetic cannabinoids and 21 metabolites, and semi-quantifying 12alkyl hydroxy metabolites in urine. Published by Elsevier B.V. 1. Introduction Synthetic cannabinoids bind CB1 and/or CB2 receptors andwere originally developed for studying endocannabinoid phar-macology; however, now areabused drugs smoked or inhaledfor psychoactive effects,butdeceptively marketed as herbalincenses and air fresheners. Synthetic cannabinoid abuse resultedin increases in emergency room visits and occasional deaths [1-3]. ( * C orresponding author a t : Chemistry and Dru g Metabolism, IRP, N ational Insti-tute on Drug Abuse, National Institutes of Health, Biomedical Research Center, 251Bayview Boulevard, Suite 200, Room 05A-721,Baltimore, MD 21224, USA. Tel.: +1 443 740 2524; fax: +1 443 7402823. ) ( E-mail a ddress: mhuestis@intr a. nida.nih.gov (M .A. H u estis). ) Synthetic cannabinoid subclasses include napthoylindoles (JWH-015,JWH-018,JWH-019,JWH-073,JWH-081,JWH-122,JWH-200,JWH-210 andJwH-398),phenylacetylindoles (JWH-203,JWH-250,JWH-251, and RCS-8), benzoylindoles (RCS-4 and AM694), cyclo-hexylphenols (CP 47,497 C7 and C8analogs) and dibenzopyrans(HU-210). In July 2012 the United States Drug Enforcement Agency classi-fied JWH-018,JWH-019,JWH-073,JWH-081,JWH-122,JWH-200,JWH-203,JWH-250,JWH-398,AM694,AM2201, RCS-4,RCS-8,HU-210, CP 47,497-C7, CP 47,497-C8 and their analogs as schedule Icontrolled substances [4,5].Recently, UR-144, XLR11 and AKB48were temporarily added to the Schedule I controlled substancelist [6]. Most countries enacted similar legislation. Clandestine lab-oratories constantly synthesize new compounds in response tolegislative efforts, complicating drug testing. New synthetic cannabinoid structures may not cross-react inantibody-based techniques,leading laboratorians to consider massspectrometric screening [7-10]. Mass spectrometry is flexible,allowing incorporation of new analytes as rapidly as referencestandards become available. We recently published a liquidchromatography-tandem mass spectrometric (LC-MS/MS) qual-itative screening method employing spectral library searchingsimultaneously targeting 9 synthetic cannabinoids and 20 metabo-lites in urine [8]. Urinary quantitative methods were only publishedfor single parent analytes and metabolites [11,12]or for metabolitesof JWH-018 and JWH-073[13-15]. The most comprehensive urinequantification method reported to-date targets 8 parent analytefamilies [16]. A comprehensive, up-to-date quantitative confirm-atory synthetic cannabinoid method is required for confirmingpresumptive positive and negative screening results, comparingscreening techniques and evaluating optimal cutoff concentrations.We present a fully validated LC-MS/MS method targeting 53 ana-lytes:JWH-018,JWH-019,JWH-073,JWH-081,JWH-122,JWH-200,JWH-210,JWH-250,JWH-398,RCS-4,AM2201,MAM2201,UR-144,CP 47,497-C7, CP 47,497-C8 and their metabolites, and JWH-203,AM694, RCS8, XLR11 and HU210 parent compounds in urine. Non-chromatographically resolved alkyl hydroxyl metabolite isomerswere semi-quantitative. 2. Methods 2.1. Reagents and supplies 2.2. Instrumentation An ABSciex API 5500 QTRAP triple quadrupole/linear ion trapmass spectrometer with a TurbolonSpray source operated in elec-trospray (ESI) mode (ABSciex, Foster City, CA) was coupled with anLC-20ADxr high performance liquid chromatography (HPLC) sys-tem (Shimadzu Corp., Columbia, MD). Analyst version 1.6.1 andMultiquant 2.1 were employed for data acquisition and analysis,respectively. 2.3. Calibrators, quality control and internalstandards Blank urine was evaluated to ensure absence of detectablesynthetic cannabinoids or metabolites prior to fortification withworking stock solutions to prepare calibrators and quality control samples. Primary stock solution containing 53 synthetic cannabi-noids and metabolites at 1000 pg/l was prepared in methanol(see analyte list in Table 1). Dilutions of the stock solution cre-ated calibrators at 0.1,0.2, 0.5,1.0,5.0, 10, 25, 50 and 100 pg/lwhen fortifying 20 pL standard solution into 200 pL blank humanurine. Quality control (QC) samples were prepared with different vialsof reference standard solutions than calibrators. Three mixed QCworking solutions containing the same analytes as present in cali-brators (see Table 1), ranging from 0.3 to 30 pg/l, were prepared inmethanol (see Table 2 for analyte QC concentrations). Deuterated internal standard (N=24, see Table 1) stock solu-tions were diluted in methanol producing a mixed internal standardsolution of 10 pg/l. 20 pL of 10 pg/l mixed internal standard solu-tion was added to blank urine,yielding 1 ug/l fortified urine internalstandard concentrations. All primary and working solutions were stored at-20°C inamber glass vials. 2.4. Specimen preparation approaches evaluated during methoddevelopment Preliminary synthetic cannabinoids recovery studies wereconducted in triplicate during method development to evalu-ate potential sample preparation approaches with Resprep C18columns, Strata C8 columns and SLE. Three sets of specimens wereprepared: urine fortified prior to extraction, urine fortified afterlextraction and neat. For Resprep C18 and Strata C8 sample preparations, 0.5mlblank urine containing 10pg/l JWH-018, RCS8, JWH-250 N-5-hydroxypentyl and1 JwH-250 N-5-carboxypentyl was dilutedwith 2.5ml 400mM! ammonium acetate buffer, pH 4.0 priorto addition of 50p.L glucuronidase solution (100,000 units glu-curonidase activity/ml). Screwtop glass tubes were capped andincubated at 55°C for 2 h, then centrifuged at 1600×g,4°C for5 min prior to application on conditioned columns. Resprep C18and Strata C8 columns were conditioned with acetonitrile and400mM ammonium acetate buffer, pH 4.0 and washed with3 ml water and 400 mM ammonium acetate buffer, pH 4.0:ace-tonitrile (80:20, v/v). Columns were dried via 40 psi positivepressure for 5min prior to elution with 3 ml 2% glacial aceticacid in acetonitrile followed by 3 ml hexane:ethyl acetate (90:10,v/v). Combined eluents were dried completely under nitrogen at40°C prior to reconstitution with 150 pL mobile phase A:B 50:50(v/v). For SLE preparation, 200 pL blank urine containing 10 pg/lJWH-018, RCS8,JWH-250 N-5-hydroxypentyl and JWH-250 N-5-carboxypentyl was diluted with 0.3 ml 400 mM ammonium acetatebuffer, pH 4.0 prior to addition of 40 pL glucuronidase solution(100,000 units glucuronidase activity/ml). Polypropylene micro-centrifuge tubes were capped and incubated at 55°C for 2h.Samples were centrifuged at 15,000×g, 4°C for 5 min after addi-tion of 0.5ml acetonitrile. Samples were transferred onto SLEcolumns and gently driven onto column phase with fine pres-sure control by slowly increasing pressure up to 1l/min (achieving21 ml/min through each column). After equilibration at ambientpressure for 5 min, analytes were eluted with 6ml ethyl acetateinto 16mmx100 mm conical polypropylene tubes. Positive pres-sure was gradually applied up to 51/min (100 ml/min through eachcolumn) with fine pressure control until elution was complete.Allsample extracts were completely dried at 45°C under nitrogen ina Zymark TurboVap. Samples were reconstituted in 150 pL mobilephase A:B 50:50 (v/v), vortexed 15 s prior to centrifugation at 4°C,4000 × g for 5 min and transferred to autosampler vials containing200pL glass inserts. Table1Liquid chromatography-tandem mass spectrometry parameters for synthetic cannabinoids and metabolites in human urine. Analyte Peak# Q1 mass (amu) Q3 masses (amu) DP(V) EP(V) CE(V) CXP(V) RT(min) Positive mode method JWH-018 42 342.1 155.2,127.2 46 10 33,53 12,12 12.30 JWH-018 5-hydroxyindole 33 358.2 155.1,127.2 91 10 33,65 16,10 10.70 JWH-018 6-hydroxyindole 29 358.1 155.1,127.2 76 10 31,69 16,10 10.20 JWH-018 N-5-hydroxypentyl 16 358.2 155.1,127.2 56 10 29,65 12,14 8.48 JWH-018 N-pentanoic acid 15 372.2 155.0,126.9 106 10 31,71 14,14 8.47 IWH-019 45 356.0 154.9,127.0 26 10 33,65 16,14 12.90 JWH-019 5-hydroxyindole 36 372.0 155.0,127.0 11 10 33,71 12,12 11.40 JWH-019 N-6-hydroxyhexyl 23 372.0 154.9,127.0 86 10 29,73 14,12 9.34 IWH-073 39 328.0 155.2,127.1 51 10 31,63 12,14 11.70 JWH-073 5-hydroxyindole 27 344.2 155.0,127.0 51 10 33,65 12,18 9.88 JWH-073 6-hydroxyindole 24 344.2 155.1,127.0 46 10 31,67 10, 14 9.37 JWH-073 N-4-hydroxybutyl 10 344.1 155.0,127.0 36 10 29,51 12,18 7.68 JWH-073 N-butanoic acid 13 358.1 155.0,127.0 61 10 31,61 14,12 7.80 IWH-081 43 372.1 185.1,157.2 181 10 33,51 16,10 12.70 JWH-081 N-5-hydroxypentyl 18 388.2 185.1,113.9 56 10 29,99 16,14 9.09 IWH-122 46 356.1 169.0,115.0 51 10 33,91 16,12 12.90 JWH-122N-5-hydroxypentyl 22 372.1 169.0,115.0 51 10 29,85 12,14 9.29 WH-200 5 385.0 155.1,126.8 126 10 29,69 10,14 5.41 JWH-200 5-hydroxyindole 1 401.1 155.0,76.9 31 10 29,125 14,8 2.99 JWH-200 6-hydroxyindole 2 401.1 155.0,127.0 120 10 29,71 12,16 3.48 IWH-203 38 340.9 124.9,89.1 6 10 35,103 14,8 11.50 IWH-210 48 370.1 183.1,214.1 51 10 33,33 18,18 13.50 jWH-2105-hydroxyindole 41 386.2 183.0,155.1 51 10 35,49 14,14 12.20 JWH-210N-5-hydroxypentyl 30 386.2 183.1,155.1 30 10 31,47 16..12 10.20 JWH-210N-5-carboxypentyl 28 400.1 183.0,,155.0 40 10 33,49 14,18 10.10 IWH-250 35 336.1 121.2,.9.1 50 10 27,61 12,14 11.10 JWH-2505-hydroxyindole 20 352.1 121.0,91.0 66 10 27,65 www.rysstech.com Table 1 (Continued) Analyte Peak # Q1 mass (amu) Q3 masses (amu) DP(V) EP(V) CE (V) CXP(V) RT(min) CP 47,497-C8-hydroxy dimethyloctyl metabolite 50 347.1 159.1,185.0 -180 -10 -72,-66 -17,-19 4.91 HU210 53 385.6 301.3,281.1 -36 -12 -48,-58 -27,-26 6.93 CP47,497-C7-d11 328.2 256.2,159.0 -160 -10 -44,-68 -23,-15 6.22 CP 47.497-C8-d7 338.2 266.1,159.0 -150 -10 -46,-64 -33,-17 6.52 11-nor-9-carboxy-tetrahydrocannabinol-dg 352.1 254.2,194.1 -50 -10 -40,-44 -21,-17 6.14 Q1=quadrupole 1, Q3=quadrupole 3, DP=declustering potential, EP=entrance potential, CE=collision energy, CXP=collision cell exit potential, RT=retention time. Boldmasses depict quantification transitions. Table 2 Analytical recovery and imprecision data for synthetic cannabinoids and metabolites in human urine by liquid chromatography-tandem mass spectrometry. %CV (range) Mid and high quality control concentrations were 7.5 and 30 pg/l,respectively. Chromatographic separation was performed on an UltraBiphenyl column equipped with a guard column containing iden-tical packing material. Two LC-MS/MS methods were required, a19.5 min positive ionization mode method with 4 uL injection vol-ume and a 11.4 min negative ionization mode method with 25 uLinjection volume. For positive and negative mode methods, thecolumn oven and auto-sampler were maintained at 40 and 4°C,respectively. Gradient elution was performed for both methodswith (A) 0.01% formic acid in water and (B) 0.01% formic acid inacetonitrile:methanol (50:50, v/v) at a flow rate of 0.5 ml/min. Theinitial gradient conditions for the positive mode method were 40%B, held for 30 s, then increased to 90% B over 14.5 min, increased to98% B at 15.1 min held until 17.6 min, returnedto 40% B at 17.7 minand held until 19.5 min. Flow rate was ramped from 0.5 ml/minto 1.0ml/min at 15.2 min and returned to 0.5 ml/min at 18.0min.HPLC eluent was diverted to waste for the first 1.5 min and after16.5 min of analysis. Initial gradient conditions for the negativemode method were 40%B,held for 30 s,then increased to 90%B over6.5 min,increased to 98%B at 7.1 min held until 9.5 min,returned to40%B at 9.6 min and held until 11.4 min. Flow rate was ramped from0.5 ml/min to 1.0 ml/min at 7.3 min and returned to 0.5 ml/min at9.9 min. The divert valve was directed to waste for the first 3.0 minand after 7.1 min. 2.6. Hydrolysis optimization Hydrolysis conditions were optimized with blank urine fortifiedto contain 2500 pg/l JWH-018 N-5-hydroxypentyl-glucuronide.Amount of enzyme, pH, temperature and duration of incubationwere evaluated for optimally hydrolyzing glucuronides. 2.7. Data analysis Peak area ratios of analytes to corresponding internal standardswere calculated for each concentration to construct daily calibra-tion curves via linear least-squares regression with a 1/x²weightingfactor.See Table 3 for calibration linear ranges. 2.8. Method validation Specificity, sensitivity, linearity, imprecision, analytical recov-ery, extraction efficiency, matrix effect, stability, dilution integrityand carry-over were evaluated during method validation. Analyte peak identification criteria were relative retention timewithin ±0.1 min of the lowest calibrator and qualifier/quantifiertransition peak area ratios ±20% of mean calibrator transitionratios. We employed ±0.1 min as a peak identification retentiontime requirement based upon ourobservations of calibrator reten-tion time drift during method development. Retention times didnot drift by more than ±0.05 min, but we employed a wider±0.1 min retention time window requirement because largerretention time variation is expected with authentic specimen anal-ysis over time. Potential endogenous interferences were assessedby analyzing ten blank urine specimens from different individu-als. In addition, 83 potential interferences from commonly useddrugs were evaluated by fortifying drugs into low QC samples.Final interferent concentrations were 500 pg/l (see Supplemen-tary Table 1 for interferent list). Synthetic cannabinoids also wereindividually fortified into low QC samples at 200 pg/l (20pg/Ifor parent analytes and hydroxyindole minor metabolites).Nointerference was noted if all analytes in the low QCsamplequantified within ±20% of target concentrations withaccept-able qualifier/quantifier transition ratios. At least 25 scans wereacquired across each peak for accuratee peak area determina-tion. Limit of detection (LOD) was evaluated over three runs withduplicates from 3 different urine sources and defined as the lowestconcentration producing a peak eluting within ±0.1 min of ana-lyte retention time for the lowest calibrator with signal-to-noise≥3:1, Gaussian peak shape and qualifier/quantifier transition peakarea ratios ±20% of mean calibrator transition ratios for all repli-cates. Limit of quantification (LOQ) also was evaluated in the samemanner, and defined as the lowest concentration that met LODcriteria with signal-to-noise ≥10:1 and measured concentrationwithin ±20% of target. Performance at the LOQ was confirmed ineach batch of specimens and was each analytes’lowest limit oflinearity. Preliminary experiments with six sets of calibrators determinedthe most appropriate calibration model comparing goodness-of-fit via normalized residuals inspection for unweighted linear leastsquares,linear least squares employing 1/x and 1/x2 weighting. Cal-ibration curves were fit by linear least squares regression with atleast 6 concentrations across the linear dynamic range for eachana-lyte. Calibrators were required to quantify within ±20% and duringmethod validation correlation coefficients (R-) were required toexceed 0.99. 2.11. Analytical recovery and imprecision Intra- and inter-day analytical recovery (bias) and imprecisionwere determined from four replicates at three different QC con-centrations across the linear dynamic range of the assay. Analyticalrecovery was determined by comparing the mean result for all anal-yses to the nominal concentration value (i.e. mean % of expectedconcentration). Inter-day imprecision and analytical recovery wereevaluated on five different runs with four replicates in each run,analyzed on five separate days (n=20). Imprecision was expressedas % coefficient of variation (% CV) of calculated concentrations.One-way analysis of variance (ANOVA) was conducted on low,medium and high QCs to evaluate inter- and intra-day differencesin analyte concentrations. Table 3 Limit of quantification was the lower limit of linearity.V. Extraction efficiency and matrix effect were evaluated via threesets of samples as described by Matuszewski et al.(n=10 for eachset)[17]. In the first set, urine samples were fortified with ana-lytes and internal standards prior to SLE. In set 2, urine sampleswere fortified with analytes and internal standards after SLE, andthe third set contained analytes and internal standards in mobilephase. Extraction efficiency,expressed as a percentage, was cal-culated by dividing analyte mean peak areas of set 1 by set 2.Absolute matrix effect was calculated by dividing the mean peakarea of the analyte in set 2 by the mean analyte area in set 3.The value was converted to a percentage and subtracted from 100 to represent the amount of signal suppressed by the presence ofmatrix. 2.13. Analyte stability Analyte stability also was evaluated with blank human urine for-tified with analytes of interest at low and high QC concentrations(n=3). Analyte short-term temperature stability was evaluated forfortified human urine stored in the dark in polypropylene micro-centrifuge tubes for 16 h at room temperature, 72 h at 4°C, 72 hon the autosampler (4°C), and after three freeze-thaw cycles at-20°C. On the day of analysis, internal standard was added to eachspecimen and analyzed as described. Analyte autosampler stability was assessed by re-injecting QC specimens after 72 h, and com-paring calculated concentrations to values obtained against theoriginal calibration curve. 2.14. Dilution integrity Dilution integrity was evaluated by diluting a fortified urinesample(n=3)containing all analytes at 400 pg/11:20(v/v). Internalstandards were added and samples extracted as described. Dilutionintegrity was maintained if specimens quantified within ±20% of20 pg/l. 2.15. Carry-over Carry-over was investigated in triplicate by injecting extractedblank urine samples containing internal standards immediatelyafter samples containing target analytes at 400 pg/l. Blank urinespecimens could not meet LOD criteria to document absence ofcarryover. 2.16. Authentic specimens Anonymous, randomly collected authentic urine specimenswere analyzed to assess method utility. 3. Results 3.1. Chromatography Baseline chromatographic resolution between all analytes wasnot possible; all isobaric compounds were baseline resolved exceptfor isomeric hydroxypentyl compounds and JWH-019,JWH-122,JWH-019 hydroxyhexyl and JWH-122 N-hydroxypentyl. Although,JWH-019,JWH-122 andjWH-019 hydroxyhexyl,JWH-122 hydrox-ypentyl isobaric pairs were not chromatographically baselineresolved, specific product ions differentiated the isobars. Weadded JWH-018 N-5-hydroxypentyl,JWH-019 N-6-hydroxyhexyl,JWH-073 N-4-hydroxybutyl, JWH-081 N-5-hydroxypentyl, JwH-122 N-5-hydroxypentyl, JWH-210 N-5-hydroxypentyl, JWH-250N-5-hydroxypentyl,JWH-398 N-5-hydroxypentyl, AM2201N-4-hydroxypentyl, MAM22011 N-4-hydroxypentyl, RCS-4N-5-hydroxypentyl and UR-144 N-5-hydroxypentyl alkyl hydroxymetabolite standards, but since isomeric baseline separation wasnot possible, we can only identify these peaks as alkyl hydroxymetabolites without assigning hydroxy position on the pentylchain. We employed ±0.1 min as a peak identification retentiontime requirement based upon our observations of calibrator reten-tion time drift during method development. Retention times didnot drift by more than ±0.05 min, but weemployed a wider±0.1 min retention time window requirement because largerretention time variation is expected with authentic specimen anal-ysis over time. 3.2. Evaluation ofpotential sample preparation approaches For simplicity sake, we randomly selected two parent com-pounds and two metabolites for screening sample preparationapproaches before method validation for all analytes. Resprep C18and Strata C8 columns provided efficient recoveries for syntheticcannabinoid metabolites (83.1-98.7%recovery), but less than 27.3%synthetic cannabinoid parent analyte recovery from urine (Supple-mentary Table 2). SLE achieved 61.3-103.3% extraction efficienciesfor all our urinary synthetic cannabinoid analytes of interest (Sup-plementary Table 2). Matrix effect was -14.0 to 6.0 for all samplepreparations. We previously evaluated synthetic cannabinoid hydrolysiscontaining JWH-018, JWH-073, JWH-122, JWH-210, JWH-250,AM2201 and RCS-4 metabolites [8], but we wanted to verifyoptimal hydrolysis conditions for the larger urine specimen vol-ume required in this method. We did not have fresh authenticspecimens during hydrolysis optimization, therefore, we con-firmed equivalent JWH-018N-5-hydroxypentyl-glucuronidehydrolysis efficiency as observed during hydrolysis optimizationfor our previous qualitative method. We found that addition of300 pL 400 mM ammonium acetate, pH 4.0, 40 pL 100,000 unitsbeta-glucuronidase/ml and hydrolysis at 55°C for 2h achievedoptimalJWH-018N-5-hydroxypentyl-glucuronide hydrolysis(>95.8% conversion to un-conjugated JWH-018 N-hydroxypentylmetabolite). 3.4. Specificity Urine samples from ten synthetic cannabinoid-abstinent indi-viduals contained no peaks fulfilling LOD criteria. None of the 83potential exogenous interferences fortified at 500 pg/l into low QCsamples produced transition ratio or quantification criteria failure.MRM ion chromatograms from a blank urine specimen, a blankurine specimen fortified at low QCconcentrations and anonymous,random urine specimens are depicted in Figs.1 and 2. No syntheticcannabinoid parent analytes or hydroxyindole metabolites fortifiedinto low QCs at 20 pg/l interfered with any other analytes.JWH-250N-5-carboxypentyl interfered with JWH-210 5-hydroxyindolewhen fortified into low QCs at>100 pg/l; no other hydoxypentylorpentanoic metabolites interfered with other analyte low QC quan-tification when fortified at 200 pg/l. 3.5. Sensitivity and linearity Initial experiments were conducted with six sets of calibra-tion curves fit via unweighted linear least squares and linear leastsquares with 1/x and 1/x² weighting factor to identify the mostappropriate calibration model. Inspection of residuals indicated lin-ear least squares with 1/x²weighting factor produced the best fitfor the calibration data. All correlation coefficients exceeded 0.994(Table 3). Table 3 details LOD, LOQ, linearity and mean calibration results.LOD were between 0.05 and 1.0 pg/l; LOQ were between 0.1and 1.0 pug/l.Assays were linear to 50 pg/l for all analytes except100 pg/l for JWH-2005-hydroxyindole,JWH-398 N-pentanoic acidand AM2201 6-hydroxyindole. 3.6. Analytical recovery and imprecision Analytical recovery and imprecision were evaluated at threeconcentrations across the linear dynamic range.Analytical recoveryin urine ranged from 83.3 to 118.3% of expected concentra-tions for intra-day and inter-day analytical recoveries (Table 2).Intra-day and inter-day imprecision were 0.8-9.1 and 4.3-13.5%CV, respectively (Table 2). There were few significant effectsof day on most analyte QC concentrations (F4,15=0.03-3.02,p>0.05); however, in 25 cases day did have an influence(F4.15=3.12-9.98, p<0.05) for JWH-250 carboxypentyl, AM694and RCS-4 carboxypentyl low QCs; JWH-019 5-hydroxyindole,JWH-203, JWH-210 5-hydroxyindole, JWH-398, RCS8, UR-144N-hydroxypentyl, UR-1444 N-pentanoic acid, XLR11 and CP47,497-C8 mid Qcs and JWH-018 6-hydroxyindole, JWH-019N-hydroxyhexyl, jwH-203, JWH-210 5-hydroxyindole, JwH-250 N-hydroxypentyl, JWH-250 N-5-carboxypentyl, JWH-398, AM2201, RCS-4,RCS-4N-5-carboxypentyl,RCS8, CP47,497-C8 andHU210 high QCs. 3.7. Extraction efficiency and matrix effect Extraction efficiencies and matrix effects for synthetic cannabi-noids in urine are presented in Table 4. Mean extraction efficiencieswere 43.7-109.3%(n=10). Mean matrix effects (% suppressed sig-nal) were -73.1 to 51.7%(n=10, Table 4). 3.8. Analyte stability, dilution integrity and carryover Analytes at low, mid and high QC concentrations in urineextracts were stable for 72 h at 4°C in the autosampler, n=4(datanot shown). All synthetic cannabinoid metabolites at low and high QC concentrations (n=3) were stable for 16 h at room temperature;all parent analytes were unstable except JWH-200, CP 47,497-C7, CP 47,497-C8 and HU210 (Table 5). All analytes were stablefor 72 h at 4°C (Table 5). All synthetic cannabinoid metabolitesat low and high QC concentrations (n=3) were stable after threefreeze/thaw cycles; all parent analytes were unstable except JWH-200, JWH-398, AM694, CP 47,497-C7, CP 47,497-C8 and HU210(Table5). Dilution integrity was acceptable (within ±20% of expecteddiluted concentration) for all analytes after diluting a sample con-taining analytes at 400 pg/l 1:20 with blank urine. There was no evidence of carryover for synthetic cannabinoids.Negative specimens injected after samples containing analytes at400 pug/l did not have analyte peaks satisfying assay LOD criteria(n=3). Table 4Mean extraction efficiencies and matrix effects for synthetic cannabinoids and metabolites extracted from urine by supported-liquid extraction. Analyte Low QC Extraction efficiency(%,N=10) Matrix effect (%of signal suppressed,N=10) (ug/l) Low High Low High RCS-4-do 64.2% 66.1% 11.1% -0.1% UR-144-d5 53.9% 44.8% -15.3% -12.0% XLR11-d5 60.4% 59.6% 5.4% -1.2% CP47,497-C7-d11 92.7% 92.1% -42.4% -47.5% CP47,497-C8-d7 90.5% 89.7% -41.2% -44.5% 11-nor-9-carboxy-tetrahydrocannabinol-dg 82.3% 84.4% -52.7% -60.2% High quality control concentrations were 30 pg/l; deuterated internal standard concentrations were 1 ug/l. Data acquired in negative ionization mode Minutes Fig. 2. Extractedion chromatograms showing quantification MRMs in authentic urine specimens containing(A)11.3,18.3,0.9,1.8,0.3 and 3.9 pg/1JWH-018N-hydroxypentyl,JWH-018 N-pentanoic acid,JWH-250N-5-carboxypentyl,AM2201 N-hydroxypentyl,MAM2201 N-hydroxypentyl and MAM2201 N-pentanoic acid, respectively,(B)0.2,0.7,0.2, 45.5, 6.7,0.2, 1.4, 5.0,10.2,0.3,36.4 and 0.2 ug/l JWH-018 5-hydroxyindole,JWH-018 6-hydroxyindole, JWH-073 N-hydroxybutyl, JWH-073 N-butanoic acid, JWH-122 N-hydroxypentyl, jwH-250 5-hydroxyindole, JWH-250 N-hydroxypentyl, JWH-250 N-5-carboxypentyl, AM2201 N-hydroxypentyl, RCS-4 N-5-carboxypentyl, RCS-4M9 metabolite and UR-144 N-hydroxypentyl,respectively. Specimen B also contained 138.4 and 211.6pg/l JWH-018 N-hydroxypentyl and JWH-018 N-pentanoic acid(determined after diluted re-analysis, data not shown). See Table 1 for peak numbering. Table 5 Synthetic cannabinoids and metabolites stabilities in human urine. Bold type indicates >20% analyte loss. The method was applied to measurement of synthetic cannabi-noids and metabolites in anonymous, randomly collected urinespecimens (Fig.2). 4. Discussion Despite initial DEA scheduling of synthetic cannabinoids inMarch 2011, abuse is an ongoing problem, with the 2012 Mon-itoring the Future survey reporting that 11.3% of 12th gradersingested synthetic cannabinoids in the past year [18]. Anotherrecent survey of undergraduate students at a southeastern univer-sity found 14% students reported lifetime synthetic cannabinoid use [19]. A validated and sensitive LC-MS/MS method for simul-taneously quantifying synthetic cannabinoids and metabolites inurine is necessary for confirming intake by clinical and foren-sic laboratories. Few quantitative urinary synthetic cannabinoidsmethods exist; most published LC-MS/MS quantitative methodstarget JWH-018 and JWH-073 metabolites after B-glucuronidasehydrolysis[11-16].We present a fully validated and sensitive quan-titative LC-MS/MS method for simultaneously measuring the mostcomprehensive synthetic cannabinoid panel to-date in urine. Thisquantitative synthetic cannabinoid method is useful for evaluatingcutoff concentrations to optimally document synthetic cannabi-noid intake, for determining windows of synthetic cannabinoiddetection and identifying optimal synthetic cannabinoid analytesfor documenting recent intake. Our goal during validation of the current method was toassess whether more B-glucuronidase enzyme was required toachieve similarly optimal hydrolysis as this method requires alarger 200 pL urine volume compared to 100 pL urine for theprevious validated synthetic cannabinoid screening method [8].We evaluated JWH-018 N-5-hydroxypentyl-glucuronide, the onlysynthetic cannabinoid glucuronide metabolite reference standardcommercially available at the time of method development, forevaluating the effect of increasing urine volume on hydrolysisefficiency, as we did not have fresh authentic specimens. Usingthe current method's hydrolysis conditions achieved similar JWH-018 N-5-hydroxypentyl-glucuronide hydrolysis as observed duringdevelopment of the previous qualitative synthetic cannabinoidassay inferring that the current conditions would also achieve opti-mal hydrolysis ofJWH-073,JWH-122,JWH-210,JWH-250,AM2201and RCS-4 metabolites. It is unknown whether these hydrolysisconditions are optimal for other synthetic cannabinoid glucuronideconjugates. Previously, de Jager et al. presented a synthetic cannabi-noid urinary method targeting JWH-018 N-5-hydroxypentyl,JWH-018 N-pentanoic acid, JWH-019 5-hydroxyindole, JWH-073 N-4-hydroxybutyl, JWH-073N-butanoic acid, JwH-122N-5-hydroxypentyl,JWH-200 5-hydroxyindole,JJWH-2505-hydroxyindole, JWH-250N-5-carboxypentyl, JWH-398N-hydroxypentyl and RCS-4 N-5-hydroxypentyl metabolites [16].500 pL urine was hydrolyzed with B-glucuronidase priortoliquid-liquid extraction and analysis on an ABSciex 5500 QTRAPLCMSMS achieving linear ranges of 0.1-10 pg/l. Our currentLCMSMS method achieves similar LLOQ with increased linearityto 50 or 100 pg/l with only a 200 pL urine sample. Other methodstargeting only JWH-018 and JWH-073 metabolites achieved LLOQof 0.1-1.8 pg/l, similar or lower LLOQ than our current methodbut only 2-14 analytes [11-15].Two of the previous methodsemployed 1 and 2 ml sample volumes,5-10 fold larger than ourmethod [11,12], with Chimalakonda et al. and Yanes et al. onlyrequiring 40 and 100 uL urine, respectively [13,15]. Our method was unable to achieve baseline chromatographicresolution for alkyl hydroxy metabolite isomers (i.e. JWH-018N-2, N-3, N-4, and N-5-hydroxypentyl compounds have sim-ilar retention times) providingsemi-quantitative total alkyBhydroxy metabolite concentrationsforr any synthetic cannabi-noid. This is a potential limitation for distinguishing the syntheticcannabinoid ingested, as AM2201 metabolically forms JWH-018N-5-hydroxypentyl with minimal JwH-018 N-4-hydroxypentyl,and JWH-018 forms less JWH-018 N-5-hydroxypentyl than JWH-018 N-4-hydroxypentyl. Our quantitative method includes the'AM2201 specific metabolites AM2201 N-4-hydroxypentyl andAM2201 6-hydroxyindole, but these compounds are less abun-dant than JWH-018 N-5-hydroxypentyl after AM2201 intake. Weadded jWH-018 N-5-hydroxypentyl,JWH-019 N-6-hydroxyhexyl,JWH-073 N-4-hydroxybutyl,JWH-081 N-5-hydroxypentyl,JWH-122 N-5-hydroxypentyl, JWH-210 N-5-hydroxypentyl, JWH-250N-5-hydroxypentyl, JWH-398N-5-hydroxypentyl, AM2201 N-4-hydroxypentyl, MAM22011N-4-hydroxypentyl, RCS-4 N-5-hydroxypentyl and UR-1444 N-5-hydroxypentyl alkyl hydroxymetabolite standards, but since isomeric separation was notpossible, we can only identify these peaks as alkyl hydroxymetabolites without assigning hydroxy position on the sidechain. We opted to include parent analytes, even though metabolitespredominate in urine [20], to evaluate if parent analytes assistdetermining recent intake. This complicated sample preparationduring method development, observing low recoveries of nonpolarparent analytes (<27%) with traditional C8 and C18 reverse phasesolid phase extraction columns; SLE+achieved>44% parent analyterecoveries. Poor availability of analytical reference standards impedesmethod development for synthetic cannabinoids and other emerg-ing drugs of abuse. Deuterated analogs compensate for variableanalyte recovery or matrix effect enabling accurate quantification.Commercially available deuterated internal standards were uti-lized when available; deuterated analytes with similar functionalgroups and nearest retention time were utilized when matcheddeuterated internal standards were unavailable. There was oneexception for this internal standard selection scheme; we selecteddeuterated JWH-018 parent analyte as an internal standard forJWH-210 5-hydroxyindole after observing poor QC performancewith JWH-018 5-hydroxyindole-dg as internal standard.JWH-018-dg co-elutes with JWH-210 5-hydroxyindole better accounting formatrix effect. ANOVA revealed 25 of 159 cases where statistically signif-icant between-batch differences in measured QC concentrationoccurred. The explanation for these differences is unknown, pos-sibly due to variable matrix effect, extraction efficiencies, orday-to-day variability in fortifying calibrators and QCs. Matcheddeuterated internal standards were not available for 16 of 25cases that may explain between-day variability for these analytes.Although statistically significant, these effects are not likely clini-cally relevant since all QC concentrations were within 80-120% oftarget and maximum differences between days was 22.8%. We are uncertain why >100 ug/1JWH-250 N-5-carboxypentylconcentrations interfered withJWH-2105-hydroxyindole quantifi-cation at 0.6ug/1.JWH-2105-hydroxyindole ion ratio criteria werewithin 80-120% of target, but the low QC quantified at 193% of tar-get. Interference appears unlikely because retention times differby 5.2 min and parent masses by 20 amu for these two analytes.It also seems unlikely that JWH-250 N-5-carboxypentyl would beconverted to JWH-210 5-hydroxyindole during sample prepara-tion or analysis. jWH-250 N-5-carboxypentyl standard may havea trace JWH-210 5-hydroxyindole impurity. False positive resultscould occur when both JWH-250N-5-carboxypentyl and JWH-2105-hydroxyindole co-occur in specimens, presence of JWH-210 N-hydroxypentyl and/orJWH-210N-5-carboxypentyl is required fordocumentingJWH-210 intake. All analytes were stable under all evaluated conditions exceptmost parent analytes showed >20% loss after 16 h at room temper-ature or after three freeze/thaw cycles in fortified urine. Similarly,Dresen et al. showed >15%JWH-073,JWH-081 and methanan-damide decreases in fortified serum after 72h[21] and wepreviously showed THC instability in urine after 16 h at room tem-perature [22]. Instability appears to be related to analyte polaritywith more polar JWH-200, CP 47,497-C7, CP 47,497-C8 and HU210being stable while other parent analytes were unstable. Parent ana-lyte instability may partially explain not detecting parent syntheticcannabinoids in urine [20]. We present a fully validated quantitative LC-MS/MS methodfor the most comprehensive synthetic cannabinoid urine methodto-date targeting 53 analytes: JwH-018, JWH-019, JWH-073,JWH-081,JWH-122,JWH-200,JWH-210,JWH-250,JWH-398,RCS-4, AM2201, MAM2201, UR-144, CP 47,497-C7, CP 47,497-C8and their metabolites, and JWH-203, AM694,RCS8, XLR11 andHU210 parent compounds. 200pL urine was hydrolyzed with β-glucuronidase before SLE extraction. Two injections were requiredto acquire data for 53 analytes; 4 uL positive mode injection witha 19.5 min runtime for 48 analytes and 25 pL negative modeinjection with a 11.4 min runtime for CP 47,497-C7, CP 47,497-C7 hydroxy metabolite, CP 47,497-C8, CP 47,497-C8 hydroxymetabolite and HU210. This method should accommodate newsynthetic cannabinoids as certified reference standards becomeavailable. Inclusion of new analytes requires method re-validationfor the new analytes; specificity, sensitivity, linearity, imprecision,analytical recovery, extraction efficiency, matrix effect, stability, dilution integrity and carry-over would need to be evaluatedA7for new analytes. However, use of broad spectrum SLE+ samplepreparation and scheduled MRM methodologies should elimi-nate re-optimization of sample preparation and most instrumentsettings. Acknowledgments The authors would like to recognize Hua-Fen Liu, Xiaohong Chenand Sumandeep Rana's advice during method development. Thisresearch was supported by the Intramural Research Program of theNational Institute on Drug Abuse,National Institutes of Health. Appendix A. Supplementary data Supplementary material related to this article can be found,in the online version, at http://dx.doi.org/10.1016/j.chroma.2013.12.067. References ( [1] Substance Abuse and M ental Health S ervices Ad m inistration , Drug Abu s e Warning N e twork, 2 0 11: N a t ional E s t i mates of Drug- r elated E m ergency D ep a rtment Visits, Rock v ille, MD, 2 01 3 . ) ( [2] M . Wikstr om , G . Theland e r, M. Da h lgr e n, R. Kr o nstrand, J. A n al. Toxic o l. 3 7 (2013) 4 3 . ) ( [3] A.C. Y o ung, E . S chw a rz, G . M e dina, A. Obaf e m i, S. Y . Fe n g, C. Ka n e, K. K l e i n schmidt,Am. J. Emerg.Med. 30 (2012). ) ( [4] D rug E n f o rcement Agency, United States D e p artme n t of Justice, Fed. Reg i st.76 (2011) 1 1075. ) [6] Drug Enforcement Agency, United States Department of Justice, Fed. Regist. 78(2013)28735. [7] F. Guale,S. Shahreza,J.P. Walterscheid, H.H. Chen, C. Arndt, A.T.Kelly,A. Moza-yani,J. Anal. Toxicol. 37 (2013)17. [8]A. Wohlfarth, K.B.Scheidweiler, X.H. Chen, H.F. Liu, M.A. Huestis, Anal.Chem.85(2013)3730. [9] A.H. Wu, R. Gerona, P. Armenian,D. French,M. Petrie, K.L. Lynch, Clin. Toxicol.(Phila)50(2012)733. [10]D.K.Wissenbach, M.R. Meyer, D.Remane,A.A. Philipp,A.A. Weber, H.H. Maurer,Anal. Bioanal. Chem. 400 (2011)3481. [11] S. Beuck, I. Moller, A. Thomas, A. Klose, N. Schlorer, W. Schanzer, M. Thevis,Anal. Bioanal. Chem.401 (2011)493. [12]M.A. ElSohly, W. Gul, K.M. Elsohly, T.P. Murphy, V.L. Madgula,S.I.Khan, J.Anal.Toxicol. 35 (2011)487. [13] K.C. Chimalakonda, C.L. Moran, P.D. Kennedy, G.W. Endres, A. Uzieblo, PJ.Dobrowolski, E.K. Fifer,J. Lapoint, L.S. Nelson, R.S. Hoffman, L.P. James, A.Radominska-Pandya,J.H. Moran, Anal. Chem. 83 (2011)6381. [14]C.L. Moran, V.H. Le, K.C. Chimalakonda, A.L. Smedley, F.D. Lackey, S.N. Owen,P.D. Kennedy, G.W. Endres, F.L. Ciske,J.B. Kramer, A.M. Kornilov, L.D. Bratton,P.J.Dobrowolski,W.D. Wessinger, W.E. Fantegrossi, P.L. Prather, L.P. James, AA.Radominska-Pandya,J.H. Moran, Anal. Chem.83 (2011)4228. [15]E.G. Yanes, D.P. Lovett, J. Chromatogr.B: Analyt. Technol. Biomed. Life Sci. 909(2012)42. ( [16] A.D . de J ager , J.V. W a rner, M . Henman, W. F e rguson, A. H al l,j. C hr oma t ogr. B: A nal yt . T echnol. B iomed. Life S c i. 8 97 (2012)22 . ) [17]B.K. Matuszewski, M.L.Constanzer, C.M. Chavez-Eng, Anal. Chem. 75 (2003).3019. [18]L.D. Johnston, P.M. O'Malley, J.G. Bachman, JE. Schulenberg, Monitoring theFuture National Results on Drug Use: 2012 Overview, Key Findings on Adoles-cent Drug Use, Institute for Social Research, The University of Michigan, AnnArbor,Ann Arbor,2013. ( [19]J.M . St ogner, B . L. M i ller, J . S ubst. U se ( 2 013) , h t tp : //dx.doi.org/10.3109/ 1465 9891 . 2013.770571, A dv ance on l ine pu b licat i on. ) [20] M.R. Meyer, F.T. Peters, Ther.Drug Monit. 34 0112)615 士科技www.rysstech.com 耐士科技www.rysstech.com
确定








还剩11页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案(制备液相色谱)》,该方案主要用于化药新药研发中临床前研究检测,参考标准--,《化学药中主要物质含量分析检测方案(制备液相色谱)》用到的仪器有
相关方案
更多
该厂商其他方案
更多