








方案详情
文
A group of novel taxoids, with modifications at C-7, C-10, C-30 and C-14 positions of paclitaxel, was synthesized in order to improve their biological profile by decreasing their affinity with P-glycoprotein (Pgp) and increasing cellular permeability. Most of the new taxoids demonstrated the similar potent cytotoxic activities in MCF-7 human tumor cell line as paclitaxel in vitro. In the permeability assay with monolayers of Caco-2 cells, most of the compounds demonstrated an increased trans-cellular transport in Ato-
B direction in comparison with paclitaxel. Among them the compounds T-13, T-15 and T-26 showed the highest permeability, and with efflux ratios better than that of ortataxel. The interaction of the compounds T-13 and T-26 with P-gp was evaluated using Madin-Darby canine kidney (MDCK)-multidrug resistance-1(MDR1) and MDCK-wild-type (WT). The results indicated that T-13 and T-26 were poor substrates for P-gp and possessed inhibiting effects of P-gp mediated efflux. It was thus clear that simultaneous modifications at the C-7, C-10 and C-30 positions of paclitaxel significantly impaired its interactions with P-gp and interfered with P-gp mediated efflux.
方案详情

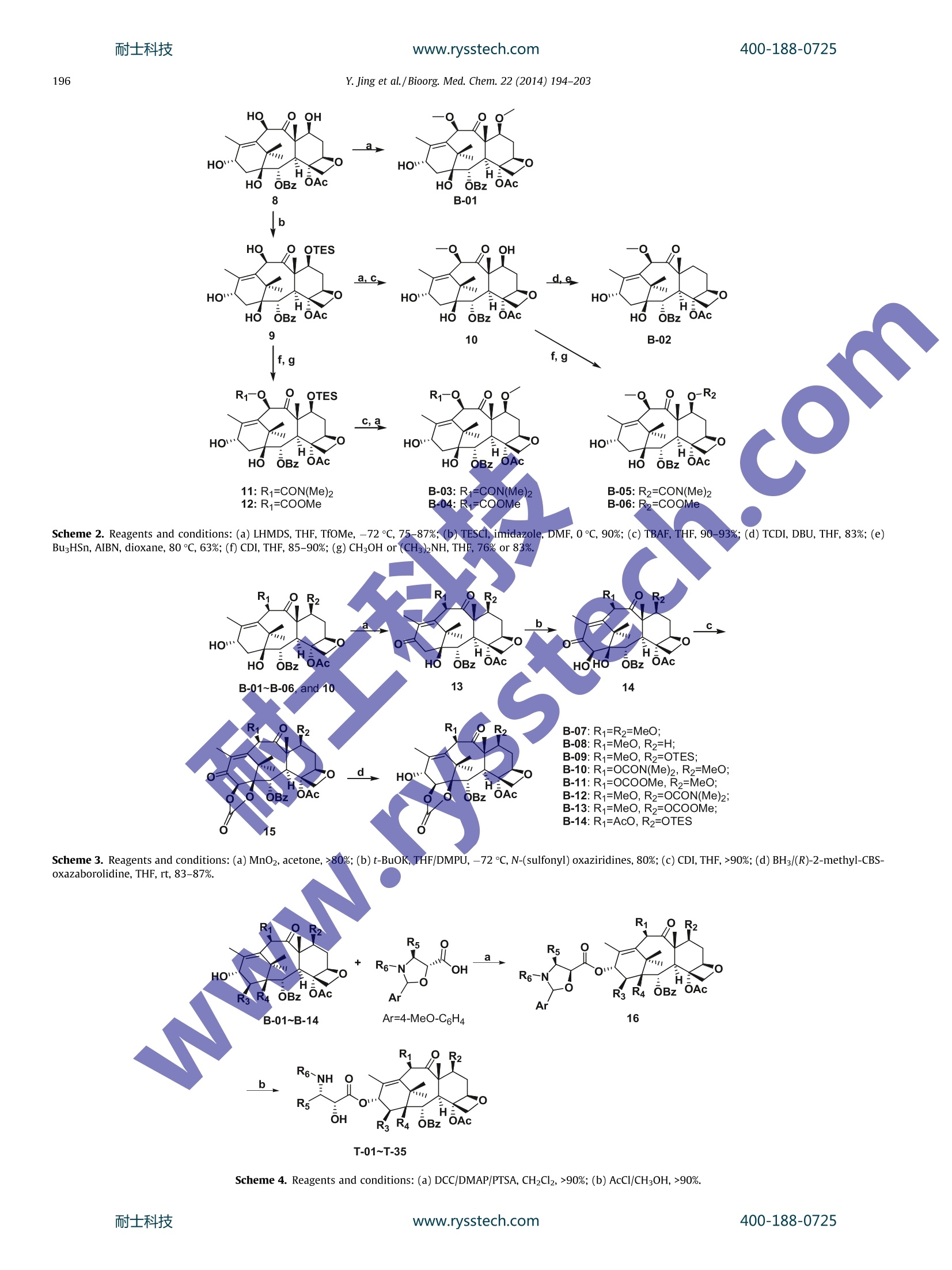
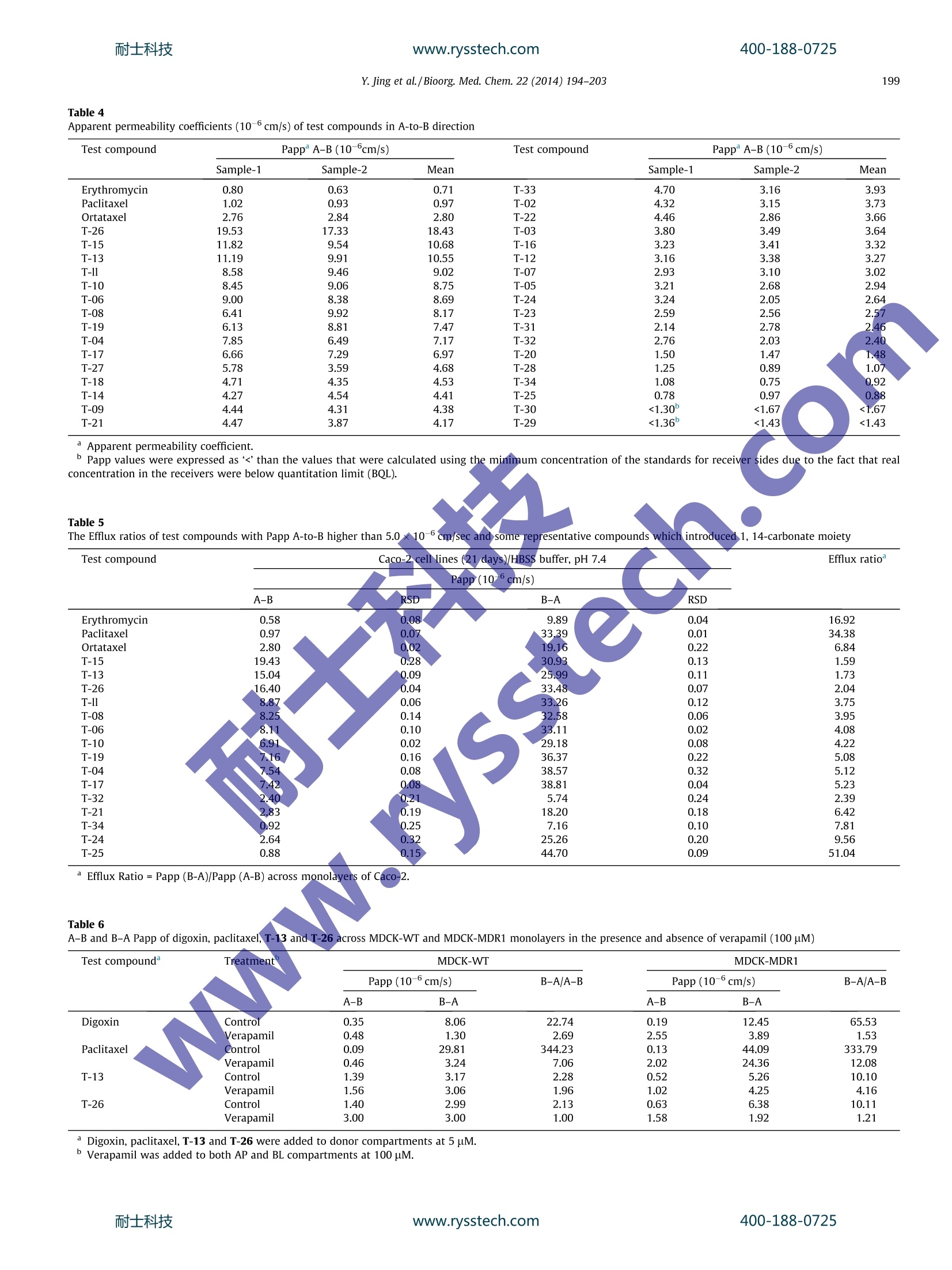
www.rysstech.comBioorganic & Medicinal Chemistry 22 (2014) 194-203耐士科技400-188-0725 耐士科技400-188-0725www.rysstech.comY. Jing et al./Bioorg. Med.Chem. 22 (2014)194-203195 Contents lists available at ScienceDirect Bioorganic & Medicinal Chemistry ELSEVIER journal homepage: www.elsevier.com/locate/bmc The synthesis of novel taxoids for oral administration CrossMark Yun-rong Jing, Wei Zhou , Wan-liang Li, Lin-xiang Zhao a*, Yong-feng Wangb,c,* Key Laboratory of Structure-Based Drug Design Discovery, Shenyang Pharmaceutical University, Shenyang,Liaoning 110016, PR ChinaD Chem-Pharm R&D Institute, Tianjin Tasly Group Co., Ltd, Tasly TCM Garden, No. 2, Pujihe East Road, Beichen District, Tianjin 300402, PR China‘Zhuhai Oxforston PharmTech Co., Ltd, Zhuhai, Guangdong 519085, China ARTICLEINFO A BSTRACT Article history: A group of novel taxoids, with modifications at C-7,C-10, C-3'and C-14 positions of paclitaxel, was syn- Received 19 July 2013 thesized in order to improve their biological profile by decreasing their affinity with P-glycoprotein (P- Revised 17 November 2013 gp) and increasing cellular permeability. Most of the new taxoids demonstrated the similar potent cyto- Accepted 18 November 2013toxic activities in MCF-7 human tumor ceelll line as paclitaxel in vitro. In the permeability assay with mon- Available online 27 November 2013olayers of Caco-2 cells, most of the compounds demonstrated an increased trans-cellular transport in A- to-B direction in comparison with paclitaxel. Among them the compounds T-13, T-15 and T-26 showed Keywords: the highest permeability,and with efflux ratios better than that of ortataxeI. The interaction of the com- Paclitaxel 1.14-Carbonate baccatin III pounds T-13i and T-26 with P-gp was evaluated using Madin-Darby canine kidney (MDCK)-multidrug Oral bioavailabilityresistance-1(MDR1) and MDCK-wild-type(WT). The results indicated that T-13 and T-26 were poor sub- P-gp substratestrates for P-gp and possessed inhibiting effects of P-gp mediated efflux. It was thus clear that simulta- Bi-directional Caco-2 cells permeability neous modifications at the C-7, C-10 and C-3' positions of paclitaxel significantly impaired its interactions MDCK-MDR1 monolayers with P-gp and interfered with P-gp mediated efflux. 2013 Elsevier Ltd. All rights reserved. 1. Introduction Taxoids have become one of the most potent and successfulanticancer drugs derived from nature. Paclitaxel, in particular,alone and combined with other drugs has been widely applied inchemotherapy for treatments of a range of tumors, such as ovarian,lung, bladder, cervix, and metastatic breast cancer (MBC),as wellas melanomas and glioma.However its application has been im-peded by its intrinsic physicochemical properties and enzymaticaffinities, such as poor aqueous solubility,and being a substrateof P-glycoprotein. As a result, its clinically available dosage formsare only injections, some of which contain a excipient with thepotented for causing allergic reactions. Development of a viableinjection formulation for taxoids and an oral taxoid have been achallenging task for pharmaceutical researchers. Paclitaxel is a highly substituted polyoxygenated diterpenoid. Inrecent years, a number of taxoids derived from 14-hydroxy-10-deacetylbaccatin III have been showed good oral bioavailability whilemaintaining anticancer activity.Ortataxel (Fig. 1), in particular, hasentered phase II clinical trials with oral administration. It was sug-gested that the better oral absorption of these new taxoids resultedfrom their impaired affinity with P-gp; therefore, they no longer ( * C orresponding a uthors. a t: Zhuhai O xforston PharmTech Co . , L t d, Z h uhai, Guangdong 51 9 085,China. Tel.: +8 6 24 23986420; fax: +86 24 2 3 98 6 420 (L.-x. Z . ); tel. : +86 22 26736391; fax: +86 2286342716(Y.-f.W.). ) ( E -mail a ddresses: linxia n g.zhao@vip. s ina.com ( L.-X. Zhao), y onken a fil @ ya h oo.co.uk (Y.-F. Wang). ) suffered efflux mediated by P-gp in the gastrointestinal tract. It wasobserved that modifications of paclitaxel at the C-7, C-10 and C-3'positions were crucial in weakening the interactions between taxanewith P-gp, for example, BMS-184476,milataxell and tesetaxelig. 1)11 in which modifications occurred at C-7 and C-3; while inthe case of simotaxel the modification of docetaxel was at the C-10and C-3'positions. In summary, modifications at the C-7,C-10, C-14 and C-3'positions of paclitaxel appear to disturb its interactionswith P-gp, as a result, its poor oral bioavailability and hydrophobic-ity-related drug resistance were improved. In order to explore the effects of modified C-7,C-10, C-14, andC-3' substituents on the affinity of paclitaxel analogs with P-gpand to find potent oral taxoid candidates, we decided to synthesizea group of new taxoids with modifications at the northern hemi-sphere of paclitaxel at C-7 (ester, amino ester, deoxy or methylether), C-10 (ester, amino ester, or methyl ether) and C-3'(3'N-Boc and 3'-aromatic ring or alkyl group) positions, as wellas a group of taxoids with modifications at the northern and thesouthern hemisphere of paclitaxel at C-7,C-10, C-14 (1,14-carbon-ate moiety)and C-3' positions. Herein, we report the design,syn-thesis, and biological evaluation of these novel taxoids. 2. Results and discussion 2.1. Chemistry Following literature methods, a group of oxazolidine carboxylicacid derivatives 7a-d were prepared, ready for acylation with the R1 R2 R6~NH 11 R5 OH H: R3R4 Ortataxel: R1=AcO, R2=OH,R3,R4=1,14-carbonate Tesetaxel R5=CH2CH(CH3)2, R6=t-butyloxycarbonyl; BMS-184476:R1=AcO,R2=OCH2SCH3,R3=H, R4=OH, R5=C(CH3)3, R6=t-butyloxycarbonyl; Milataxel: R1=OH,R2=OCOCH2CH3, R3=H, Simotaxel: R4=OH,R5=2-furanyl, R6=t-butyloxycarbonyl; Ri=cyclopentylcarbonyloxy, R2=OH, R3=H,R4=OH, R5=2-thiophene R6=isopropyloxycarbonyl Figure 1. Taxanes bearing modifications at the key positions C-7, C-10, C-14 and C-3' endowed with cytotoxic activity against MDR cell lines. designed baccatins. First, O-benzyl, Boc-o-hydroxyacetate benzylester 1 was converted to its enolate with lithium, followed by addi-tion of (SR)-tert-butanesulfinamide 2 to give enantiopure 3-substi-tuted isoserine benzyl esters 3 in excellent yield and highdiastereoselectivity. After exchange of the amine protectivegroup and condensation with 1-dimethoxymethyl-4-methoxy-benzene, the oxazolidine carboxylic acid derivatives 7a-dwere ob-tained in high yields (Scheme 1). The designed baccatins were synthesized starting with 10-deacetylbaccatin ⅢI (10-DAB, 8), an abundant taxoid from renew-able parts of Taxus spp. plants.13 The first baccatin 7,10-di-methyl-baccatin ⅢI B-01 was prepared by methylation of 8 withmethyl trifluorosulfonate (TfOMe) in quantitative yield (Scheme 2). The 7-hydroxyl group of 8 was selectively protected by usingtriethylchlorosilane in quantitative yield.14 In the presence ofLHMDS, 9 was treated with TfOMe to introduce the methyl groupselectively at the 10-hydroxyl group. After the silyl group was re-moved, the 7-hydroxyl group of 10 underwent a selective methyl-thiocarbonylation withthiocarbonyldiimidazole, ready forradical deoxygenation. Treatment of the resulting compound withtributyltin hydride in the presence of AIBN at 80c14 gave the 7-deoxy derivative B-02 (Scheme 2 10-Carbonate-7-methyl-baccatin B-03 and 10-carbonate deri-vate B-04 were prepared through activation of the 10-hydroxyl group in 9 with carbonyldiimidazole, followed by acylation at C-10and methylation at C-7. Their reverse counterparts B-05 andB-06 were synthesized by selective acylation of the 7-hydroxyl in10 (Scheme 2). 1,14-Carbonate baccatins III B-07-B-14 also were prepared(Scheme 3). MnO2 oxidation of the C13-hydroxy group in B-01-B-06 and 10 gave 13, which underwent an enolation with N-(sulfo-nyl) oxaziridines followed by a diastereoselective 14β-hydroxyl-ation to give 14.15Treatment of 14 with carbonyldiimidazoleafforded the 1,14-carbonate derivatives 15. Diastereoselectivereduction of the C13-oxo group of 15 with BH.THF in the presenceof (R)-2-methyl-CBS-oxazaborolidine afforded the 13a-OH epimeronly, yield 85-90% with 99% ee. The target taxoids T-01-T-35 were synthesized by acylations ofB-01-14 with 7a-d, followed by deprotection with acetyl chlo-ride in MeOH (Scheme 4 and Table 1). 2.2. Biological evaluation 2.2.1. In vitro growth-inhibition assay The synthesized target compounds T-01-T-35 (Table 1) demon-strated almost the same inhibitory activity as paclitaxel (Table 2)in a test against eight selected human tumor cell lines at 1 uMconcentration. In order to substantiate the potency of the new Scheme 1. Reagents and conditions: (a) LHMDS, THF, -70C, 70%; (b) HCl/EtOAc, 83%; (c) Boc20 or BzCl, DMAP, CH2Cl2, 90%;(d) 1-dimethoxymethyl-4-methoxy-benzene,PPTS, toluene, 100 C, 80%; (e) H2,Pd(OH)2, MeOH, 87%. MOC Scheme 2. Reagents and conditions: (a) LHMDS, THF, TfOMe, -72℃,75-87%; (b) TESCl, imidazole, DMF, 0℃, 90%; (c) TBAF, THF, 90-93%;(d) TCDI, DBU, THF, 83%;(e)BuzHSn, AIBN, dioxane, 80 C, 63%; (f) CDI, THF,85-90%; (g) CHgOH or (CH3)2NH, THF, 76% or 83%. Scheme 3. Reagents and conditions:(a)MnO2, acetone,>80%;(b) t-BuOK,THF/DMPU, -72 C,N-(sulfonyl) oxaziridines, 80%; (c) CDI, THF,>90%;(d) BHs/(R)-2-methyl-CBS-oxazaborolidine, THF, rt, 83-87%. Scheme 4. Reagents and conditions: (a) DCC/DMAP/PTSA,CH2Cl2,>90%; (b) AcCl/CH;OH,>90%. R3 R4 T-02 H OH MeO T-03 compounds against paclitaxel sensitive cancer cell lines, the ICsovalues of the new compounds against the MCF-7 cell line weredetermined (Table 3). Most of new compounds demonstrated acomparable potency to paclitaxel against MCF-7 cells, with ICsovalues under 10 nM, except for T-12-T-15 bearing 10-methyl-7- dimethylaminocarbonyl and T-30-T-32 bearing 1,14-carbonate-10-methyl-7-dimethylaminocarbonyl groups. These compoundsshowed a decreased potency against the MCF-7 cell line, however,they are still cytotoxic agents with IC5o values under 65 nM. There-fore, it can be concluded that modifications at the C-7, C-10, C-14or C-3'position of taxane at one or two, three, or four positionsdid not reduce cytotoxicity significantly as compared to paclitaxel. 2.2.2. Caco-2 monolayers permeability assay Caco-2 monolayers have been widely used as an in vitro modelfor prediction of intestinal permeability and hence oral absorptionof therapeutic agents,especially during the early drug discoveryprocess for compounds screening and prioritizing. One of the char-acteristics affecting intestinal permeability of a compound is itssubstrate activity for P-glycoprotein (P-gp), an efflux transporterresponsible for multiple drug resistance expressed in many tissuesincluding epithelia cells of the intestinal mucosa.18 Therefore,mit-igating substrate activity for P-gp became one of the strategies indrug discovery for optimization of oral bioavailability. The trans-cellular permeability of the new taxoids T-01-T-34 were assessedand the results obtained are summarized in Table 4. Apparent per-meability coefficients (Papp) from A-to-B (apical to basolater of thecell monolayers) and B-to-A (basolateral to apical of the cell mon-olayers) were obtained by measuring the amount of the compoundtransported from the donor compartment at 10 uM to the receivercompartment after 90 min incubation. Quantification was done byLC-MS/MS. P-gp substrate activity wSfSSfas assessed by the efflux ratio(ER) (the ratio of Papp in the B-to-A direction over that in the A-to-B direction). Erythromycin, paclitaxel and ortataxel (T-35) were in-cluded as the parallel references in the same experiments. Thetrans-cellular transport of most of the new taxoids in the A-to-Bdirection. was increased compared with paclitaxel (Papp A-B=0.97), and over half the new taxoids demonstrated a similartrans-cellular efficacy to ortataxel (Papp A-B=2.80). It appearedthe compounds T-13, T-15 and T-26 were the most able com-pounds in the group to undergo the trans-cellular transport (Papp>10.0 ×10-6 cm/s). To further assess substrate activity for the P-gp, a group of com-pounds with the Papp in A-to-B higher than 5×10-6 cm/s waschosen for evaluation of efflux ratios in bi-directional Caco-2 cellspermeability studies and the results are summarized in Table 5.Paclitaxel exhibited asymmetric transport across Caco-2 monolay-ers with an efflux ratio higher than 34. Ortataxel1 also showedasymmetric transport but to a much less extent compared withpaclitaxel. A compound with an efflux ratio (Papp B-A/Papp A-B)greater than 2.0 is generally considered a substrate of an effluxtransporter. The compounds T-13 and T-15 showed efflux ratiosless than 2.0, indicating that they are probably not P-gp substrates.The compounds T-04, T-06, T-08, T-10, T-11, T-17 and T-19showed efflux ratios smaller than ortataxel (ER=6.84) suggestingthat modification at C-7, C-10 and C-3'positions could minimizesubstrate activity for P-gp. In addition to the modifications at the position C-7 and C-10, a1,14-carbonate moiety as seen in ortataxel was introduced incompounds T-20-T-34. Most of these compounds exhibited Pappsimilar to that of ortataxel. To further investigate the structure-transporter relationship for the new structure features, thecompounds T-21, T-24, T-25, T-32 and T-34 were examined by per-meability assessment bidirectionally in Caco-2 cell monolayers andtheir efflux ratios were determined (Table 5). Although some ofthem showed a decreased efflux ratios compared to that of paclit-axel, they still retained some substrate activity for P-gp. The resultssuggested that attempts to modify the C-7, C-10 and C-14 positionsof taxane simultaneously did not provide a synergistic effect onweakening its affinity for P-gp. Compound T-26 was an interestingcase which showed substantially reduced substrate activity for400-188-0725 Table 2In vitro growth-inhibition assay of T-01-T-35 Inhibition % at 1 uM Taxoid A2780 HeLa MCF-7 NCI-H460 A375 HT29 HL60 DU145 (ovarian) (cervix) (breast) (NSCL) (melanoma) (colon) (leukemia) (prostate) Paclitaxel 96.68 101.04 94.20 87.48 96.22 88.36 96.80 90.62 T-01 95.22 101.55 92.10 85.05 89.73 87.54 97.22 86.42 T-02 93.84 98.18 88.53 79.84 96.95 87.25 97.84 83.14 T-03 95.79 101.12 95.52 85.74 91.64 88.28 97.65 87.62 T-04 95.63 99.31 97.84 84.31 104.36 88.18 97.58 87.04 T-05 96.31 101.83 85.73 86.54 87.65 87.89 97.45 87.14 T-06 94.82 99.76 93.49 83.31 87.71 87.92 97.21 84.79 T-07 95.53 100.30 82.35 83.56 86.80 88.10 98.11 84.69 T-08 94.82 100.56 89.78 80.65 89.00 87.29 97.66 82.89 T-09 95.80 100.93 95.47 86.44 94.18 88.75 96.84 86.20 T-10 94.13 99.53 95.28 80.43 102.87 87.32 97.40 83.30 T-11 95.60 99.65 102.90 85.75 91.86 89.09 96.94 85.74 T-12 95.57 102.82 90.79 87.68 88.42 89.36 97.82 85.80 T-13 95.81 102.87 102.13 86.59 90.43 89.81 97.77 87.45 T-14 94.69 102.64 83.46 87.75 90.34 89.11 97.75 86.30 T-15 95.01 102.32 101.21 86.19 104.50 89.15 98.09 86.86 T-16 96.88 102.11 106.85 90.32 93.96 89.73 97.79 88.35 T-17 96.42 101.56 102.71 89.49 97.39 89.65 97.01 88.52 T-18 96.55 101.75 106.08 88.69 88.34 89.76 97.44 89.18 T-19 94.87 99.99 99.96 81.60 90.21 88.41 99.11 84.92 T-20 96.72 100.54 96.00 87.94 90.80 89.41 96.57 87.76 T-21 94.69 98.20 93.54 82.25 89.84 88.98 95.90 84.71 T-22 94.65 98.28 100.73 81.84 82.17 88.28 95.81 83.87 T-23 94.59 99.45 84.86 81.98 87.90 87.93 97.17 83.88 T-24 96.00 99.89 86.26 84.68 94.40 87.33 97.66 89.14 T-25 95.02 100.10 87.12 82.65 92.54 86.58 97.46 86.78 T-26 92.50 97.27 81.28 Table 3In vitro growth-inhibition assay of taxoids against MCF-7 tumor cell line Test compound MCF-7 (ICso, nM) Test compound MCF-7 (IC50,nM) Paclitaxel 7.05±0.05 2.10±0.08 T-03 3.33±0.06 3.40±0.08 T-04 3.06±0.21 4.04±0.14 T-06 1.58±0.04 T-24 4.49±0.18 T-08 1.70±0.06 T-26 7.93±0.08 T-10 1.40±0.02 T-27 14.31±0.12 T-11 2.90±0.08 T-28 6.44±0.25 T-12 64.67±0.18 T-29 3.24±0.12 T-13 9.63±0.12 T-30 15.40±0.16 T-14 46.63±0.21 T-31 18.21±0.21 T-15 13.26±0.12 21.17±0.24 T-17 4.97±0.05 3.84±0.07 T-19 2.81±0.02 T-434 2.94±0.11 T-20 6.93±0.16 All assays consisted of at least six replicate wells and were performed on threeseparate occasions. ICso values are presented as the mean+standard deviation.Cells were exposed to each agent for 72 h. P-gp as indicated by the efflux ratios (ER=2.04) compared to thatof ortataxel (ER=6.84). 2.2.3. P-gp-mediated BL-AP transport in MDCK-MDR1 andMDCK-WT monolayers The further investigating experiment of the interaction of thenew compounds T-13 and T-26 with P-gp were conducted. TheAP-BL and BL-AP Papp of paclitaxel, T-13, T-26, and digoxin were evaluated in MDCK-MDR1 and MDCK-WT cell monolayers in thepresence and absence of a known P-gp inbitor verapamil(100uM)(Table 6). As shown in Table 6, all the test compoundsexhibited efflux in the MDCK-MDR1 cells either similar or strongerthan those in the MDCK-WT cells. The efflux ratios of T-13 and T-26 were 6-fold and 30-fold less than that of digoxin and paclitaxel,respectively. The efflux of both compounds were reduced signifi-cantly in the presence of 100 uM verapamil suggesting that com-pounds T-13 and T-26 retained P-gp substrate activity althoughweaker than that of paclitaxel. 2.2.4. Effects of T-13 and T-26 on digoxin uptake The inhibitory interactions of compounds T-13 and T-26 on P-gp activity were investigated by assessing their effect on digoxinefflux in MDCK-MDR1 cells. As shown in Table 7, in the presenceof 10 uM ofT-13 and T-26, the efflux ratio of digoxin reduced from27 down to 8 and 6, respectively. In contrast under the same con-dition, paclitaxel had no effect on P-gp mediated digoxin efflux.The results suggested that the T-13 and T-26 could inhibit P-gpactivity. 3. Conclusions In summary, a group of novel taxoids was synthesized withmodifications at the C-7, C-10, C-14 and C-3' positions. Theiranticancer activities were evaluated against MCF-7 human breasttumor cell line in vitro by MTT assay and their affinities withP-gp were measured in the bi-directional Caco-2 cell permeability400-188-0725 Table 4Apparent permeability coefficients (10-6 cm/s) of test compounds in A-to-B direction Test compound Papp A-B (10-6cm/s) Test compound PappA-B (10-6cm/s) Sample-1 Sample-2 Mean Sample-1 Sample-2 Mean Erythromycin 0.80 0.63 0.71 T-33 4.70 3.16 3.93 Paclitaxel 1.02 0.93 0.97 T-02 4.32 3.15 3.73 Ortataxel 2.76 2.84 2.80 T-22 4.46 2.86 3.66 T-26 19.53 17.33 18.43 T-03 3.80 3.49 3.64 T-15 11.82 9.54 10.68 T-16 3.23 3.41 3.32 T-13 11.19 9.91 10.55 T-12 3.16 3.38 3.27 T-11 8.58 9.46 9.02 T-07 2.93 3.10 3.02 T-10 8.45 9.06 8.75 T-05 3.21 2.68 2.94 T-06 9.00 8.38 8.69 T-24 3.24 2.05 2.64 T-08 6.41 9.92 8.17 T-23 2.59 2.56 2.57 T-19 6.13 8.81 7.47 T-31 2.14 2.78 2.46 T-04 7.85 6.49 7.17 T-32 2.76 2.03 2.40 T-17 6.66 7.29 6.97 T-20 1.50 1.47 1.48 T-27 5.78 3.59 4.68 T-28 1.25 0.89 1.07 T-18 4.71 4.35 4.53 T-34 1.08 0.75 0.92 T-14 4.27 4.54 4.41 T-25 0.78 0.97 0.88 T-09 4.44 4.31 4.38 T-30 <1.30 <1.67 <1.67 T-21 4.47 3.87 4.17 T-29 <1.36 <1.43 <1.43 Apparent permeability coefficient. Table 5 The Efflux ratios of test compounds with Papp A-to-B higher than 5.0 ××10-6cm/sec and some representative compounds which introduced 1, 14-carbonate moiety Test compound Caco-2 cell lines (21 days)/HBSS buffer, pH 7.4 Efflux ratio Papp (10 cm/s) A-B RSD B-A RSD Erythromycin 0.58 0.08 9.89 0.04 16.92 Paclitaxel 0.97 0.07 33.39 0.01 34.38 Ortataxel 2.80 0.02 19.16 0.22 6.84 T-15 19.43 0.28 30.93 0.13 1.59 T-13 15.04 0.09 25.99 0.11 1.73 T-26 16.40 0.04 33.48 0.07 2.04 T-11 8.87 0.06 33.26 0.12 3.75 T-08 8.2 0.14 32.58 0.06 3.95 T-06 8.11 0.10 33.11 0.02 4.08 T-10 6.91 0.02 29.18 0.08 4.22 T-19 6 0.16 36.37 0.22 5.08 T-04 .54 0.08 38.57 0.32 5.12 T-17 .42 0.08 38.81 0.04 5.23 T-32 .40 0.21 5.74 0.24 2.39 T-21 2.83 0.19 18.20 0.18 6.42 T-34 0.92 0.25 7.16 0.10 7.81 T-24 2.64 0.32 25.26 0.20 9.56 T-25 0.88 0.15 44.70 0.09 51.04 Efflux Ratio=Papp(B-A)/Papp(A-B) across monolayers of Caco-2.Table 6A-B and B-A Papp of digoxin, paclitaxel, T-13 and T-26 across MDCK-WT and MDCK-MDR1 monolayers in the presence and absence of verapamil (100 uM) Test compound Treatment MDCK-WT Papp(10-6cm/s) B-A/A-B MDCK-MDR1 Papp (10-6cm/s) B-A/A-B A-B B-A A-B B-A Digoxin Control 0.35 8.06 22.74 0.19 12.45 65.53 Verapamil 0.48 1.30 2.69 2.55 3.89 1.53 Paclitaxel Control 0.09 29.81 344.23 0.13 44.09 333.79 Verapamil 0.46 3.24 7.06 2.02 24.36 12.08 T-13 Control 1.39 3.17 2.28 0.52 5.26 10.10 T-26 Verapamil 1.56 3.06 1.96 1.02 4.25 4.16 Control 1.40 2.99 2.13 0.63 6.38 10.11 Verapamil 3.00 3.00 1.00 1.58 1.92 1.21 Digoxin, paclitaxel, T-13 and T-26 were added to donor compartments at 5uM. Verapamil was added to both AP and BL compartments at 100 pM. Table 7 Permeability of digoxin in the presence and absence of test compounds Test compounds Treatment° Papp Efflux ratio° A-B B-A Sample-1 Sample-2 Mean Sample-1 Sample-2 Mean Digoxin (5 pM) Alone 0.63 0.68 0.65 17.22 18.02 17.62 26.92 Paclitaxel 0.60 0.69 0.64 14.73 19.76 17.25 26.88 T-13 1.70 1.62 1.66 13.27 13.78 13.53 8.15 T-26 1.37 2.21 1.79 11.34 11.21 11.27 6.30 Paclitaxel, T-13 and T-26 were added to donor compartments at 10 uM. ” Apparent permeability coefficient. ‘Effux ratio=Papp (B-A)/Papp (A-B) across monolayers of MDCK-MDR1. assay in comparison with paclitaxel and ortataxel. T-13, T-15 andT-26 were found could not to be substrates for P-gp, and demon-strated significant enhancement of transcellular transport in theA-to-B direction. Compounds T-04, T-06, T-08, T-10, T-11, T-17and T-19 appear to be poor substrates for P-gp. Furthermore T-13 and T-26 were found not only poor substrates for P-gp but alsopossessed inhibiting effects of P-gp mediated efflux in MDCK-MDR1 and MDCK-WT monolayers.S. The results indicated simultaneous modifications at C-7 (ester,amino ester, deoxy or methyl ether), C-10 (ester, amino ester ormethyl ether) and C-3' positions (3'N-Boc and 3'-aromatic ring oralkyl group) seem to play an important role in modulating P-gpmediated efflux. Moreover, the results from the new taxoids T-20-T-34 indicated the presence of the 1,14-carbonate moiety onthe taxanes seems not be a necessary prerequisite in weakeningthe affinity with P-gp. However, the role of the 1,14-carbonatemoiety on weakening taxoid affinity with P-gp was seen in the caseofT-26 and T-06, with efflux ratio improved to 4.08 from 2.04. Theimproved physicochemical and biological profiles of the com-pounds T-13, T-15 and T-26 warrant a continuing study to assesswhether or not they are qualified as oral administration drug can-didates of taxoids. The results will be reported in due time. 4. Experimental 4.1. Syntheses All anhydrous reactions were performed in oven-dried glass-ware under argon. Tetrahydrofuran was distilled over sodium/ben-zophenone. Chemicals were obtained from J&K Chemica Co. andwere used without further purification. Reactions were monitoredby thin-layer chromatography on 0.25 mm1 E. Merck silica gelplates (60 F254) using UV light as a visualizing agent. For silicagel chromatography, the flash chromatography technique (BiotageIsolera I) was used, with silica gel (300-400 mesh) and p.a. gradesolvents. Melting points were determined on a YRT-3 Thomas-Hoover capillary apparatus and were not corrected. H and 13cNMR and 2D NMR spectra were recorded on a Bruker Avance III400 spectrometer with Me4Si or CHCl (in CDCl3) as internal refer-ence. Mass spectra were obtained on an Agilent G6300 Trap MSDequipped with an ESI ionization source. 4.1.1.(SR,2R,3S)-N-tert-Butanesulfinyl-O-methoxycarbonyl-3-substituted isoserine benzyl ester 3 A typical procedure is given for the preparation of (SR, 2R, 3S)-N-tert-Butane-sulfinyl-O-methoxy carbonyl-3-isobutyl isoserine benzyl ester: To a solution ofcompound 1(141.00 g,0.53 mol) in THF (1.5 L) was added LiHMDS(530 mL, 0.53 mol) at -70 C dropwise over 1 h. The reaction mix-ture was stirred at -70 ℃ for 30 min. Subsequently, a solution ofcompound 2c (20.00 g, 0.11 mol) in THF (250 mL) was added at -70℃ dropwise over 30 min. The reaction was stirred at -70℃for 4 h. The resulted mixture was poured into sat. NH4Cl solution(2L) and extracted with EtOAc (1.20 L×3). The combined organiclayer was dried over NazSO4, filtered and concentrated. The residuewas purified by column chromatography (Petroleum ether, Petro-leum ether/EtOAc=10:1) to give a mixture of compound 3c (solid)with a small amount of compound 2c (oil), which was filtered andwashed with Petroleum ether to give compound 3c (30g, yield62%6)) asa white solid. H NMR (400 MHz, CDCl3) 8 7.41 (dd,J=7.9, 1.3 Hz, 2H), 7.38-7.27 (m, 3H), 5.30 (d,J=12.2Hz, 1H),25 (s, 1H), 5.05 (d,J=12.2Hz, 1H), 4.32 (dd,J=11.2, 5.6 Hz,1H), 3.97 (ddd,J=10.9, 5.3, 4.0 Hz[,z ,1 H1)H),, 2.55 (t,J=10.8 Hz, 1H),1.72-1.60 (m, 1H), 1.49 (s, 9H), 1.39-1.30(m, 1H), 1.22 (s, 9H),0.90 (dd,J=11.2,6.6Hz,6H). 4.1.2.(2R,3S)-3-Substituted isoserine benzyl ester 4 A typical procedure is given for the preparation of (2R,3S)-3-isobutyl isoserine benzyl ester: A mixture of compound 3c(18.00 g, 39.13 mmol) in 4 N HCl/EtOAc (500 mL) was stirred atroom temperature for 10 h. The resulted mixture was concen-trated, and the residue was partitioned between DCM (250mL)and water (200 mL). The separated aqueous layer was washed withDCM (150mL×2). The aqueous layer was adjusted to pH 9-10with NH3H20(28%), and then extracted with DCM (200 mL×3).The combined organic layers were dried over NazSO4, filteredand concentrated to give compound 4c (9.50 g, yield 95.7%) as awhite solid. H NMR (400 MHz, CDCl3) 8 7.49-7.30 (m, 5H), 5.24(s, 2H), 4.06 (d,J=2.3 Hz, 1H), 3.21-3.09 (m, 1H), 1.77-1.59 (m,1H), 1.43-1.25 (m, 2H), 0.92 (d,J=6.6 Hz, 3H), 0.89 (d,J=6.6 Hz,3H). 4.1.3. (2R,3S)-N-Substituted-3-substituted isoserine benzyl ester5 A typical procedure is given for the preparation of (2R,3S)-N-tert-butyloxycarboryl-3-isobutyl isoserine benzyl ester: A mixtureof compound 4c (9.03 g, 35.8 mmol),(Boc)20 (9.38 g, 43.00 mmol)and TEA (9.04 g, 89.50 mol) in DCM (380mL) was stirred at roomtemperature overnight. The resulted mixture was concentrated,and the residue was purified by column chromatography (Petro-leum ether, Petroleum ether/EtOAc=10:1,5:1) to give compound5d (11.82 g, yield94.4%) as a yellow oil. H NMR (400 MHz, CDCl3)8 7.41 (m, 5H), 5.30 (d,J=12.0Hz, 1H), 5.12 (d,J=12.0 Hz,1H),4.63 (d,J=10.0Hz, 1H), 4.22-4.14 (m, 2H), 3.11 (d, J=5.1 Hz,1H), 1.67 (m, 1H), 1.56-1.46 (m, 1H), 1.44 (s, 9H), 1.42-1.35 (m,1H), 0.96(d,J=6.6Hz, 3H), 0.95 (d,J=6.6 Hz, 3H). 4.1.4. (4S,5R)-N-Substituted-2-(4-anisyl)-4-substituted-5-oxazolidinecarboxylic benzyl ester 6 A typical procedure is given for the preparation of (4S,5R)-N-butyloxycarboryl-2-(4-anisyl)-4-isobutyl-5-oxazolidinecarboxylicbenzyl ester: To a solution of compound 5d(11.00 g, 31.32 mmol) and PPTS (0.74 g, 2.96 mmol) in toluene (160 mL) was added a solu-tion1-dimethoxymethyl-4-methoxy-benzene(6.84 g, 37.54 mmol)dropwise at 90°℃ over 20 min. The reaction was stirred at 90-100 ℃ for 2 h. 1-Dimethoxymethyl-4-methoxy-benzene (2.45 g)added to the reaction mixture additionally. The reaction was stirredfor another 2 h. The resulting mixture was concentrated, and theresidue was purified by column chromatography (Petroleumether/EtOAc=30:1,20:1,10:1) to give compound 6d(13.83g, yield92%) as a yellow oil mixture with 4-methoxybenzaldehyde.HNMR(400 MHz, CDCl3) 8 7.42-7.23 (m,7H), 6.89-6.76 (m, 2H), 6.22 (s,1H),5.16 (S,2H), 4.43 (d,J=1.3Hz, 1H), 4.24 (s, 1H), 3.73 (s, 3H),1.73-1.56 (m, 2H), 1.51 (d,J=6.1Hz, 1H), 1.24(s,9H), 0.91 (d,J=5.4 Hz, 3H), 0.86(d,J=5.4Hz,3H). 4.1.5. (4S,5R)-N-Substituted-2-(4-anisyl)-4-substituted- 5-oxazolidinecarboxylic acid 7a-d A typical procedure is given for the preparation of (4S,5R)-N-butyloxycarboryl-2-(4-anisyl)-4-isobutyl-5-oxazolidinecarboxylicacid 7d: To a mixture of compound 6 (15.60 g, 33.20 mmol) inMeOH (400 mL) was added Pd(OH)2(7.80g). The reaction was stir-red at room temperature under H2 atmosphere (20 psi) for 1 h. TLCshowed the reaction was complete. The resulting mixture was fil-tered, and the filtrated was concentrated. The residue was purifiedby column chromatography (Petroleum ether/EtOAc=10:1,5:1)togive 7d (10.98 g, yield 87.2%) as a white solid. 7a: mp: 127~128℃; MS (m|z) ESI: 402.0 (M-H);‘HNMR(400 MHz, CDCl3) 8 7.53-7.19 (m, 12H), 6.89 (s, 1H), 6.85 (d,J=8.0Hz, 2H), 5.48(s, 1H), 4.91(s, 1H),3.82(S, 3H).13C NMR(101MHz, CDCl3)8 172.74, 159.99, 135.30,,130.73, 129.53,128.78,128.70,128.30, 128.16,127.06,113.56,55.30. 7b: mp: 129~130℃; MS (m/z) ESI: 398.0 (M-H);HNMR(400 MHz, DMSO) 8 7.48-7.23 (m, 7H), 6.96 (d.J=8.3Hz, 2H),6.28 (s, 1H), 5.15 (s, 1H), 4.53 (s, 1H), 3.78 (s, 3H), 0.95 (s,9H).3c NMR (101 MHz, DMSO) 8 170.83,160.12,129.17,128.92,128.08,127.19,113.81,91.49,79.80,55.64,27.90. 7c: mp: 125~126℃; MS (m/z) ESI: 399.0(M-H); H NMR(400 MHz, CDCl3); 8.599(d, J-4.6Hz, 1H),7.85 (td, J=7.8,1.5 Hz, 1H), 7.45 (m, 3H),), 7.34 (dd, ]=7.1,5.3 Hz, 1H), 6.93 (d,J=8.6Hz, 2H), 6.23 (s,11H),.5.58(d, J=6.6Hz, 1H), 4.64i (d,J=6.6Hz,1H), 3.82(s,3H), 1.07(s,9HH)).13CNMR(101MHz, CDCl3);169.83,160.50,151.95,148.21,138.29,130.48, 128.87,123.34,121.49, 113.87,92.39,81.08,63.56,55.34,27.80. 7d: mp: 137~138 ℃; MS (m/z) ESI: 378.0 (M-H); 1H NMR(400 MHz, CDCl3) 9.08 (s, 1H), 7.39 (d,J=8.1 Hz, 2H), 6.92 (d,J=8.5 Hz, 2H), 6.31 (s, 1H), 4.54 (s, 1H), 4.41 (s, 1H), 3.84 (s, 3H),1.69 (m, 3H), 1.36 (s, 9H), 1.04 (d, J=5.9Hz, 6H).C NMR(101 MHz, CDCl3)8 175.66,159.93,153.59,128.16,113.63,90.64,81.29,58.94,55.30, 43.97,28.24,25.57, 23.13,21.87. 7,10-Dimethyl-baccatin II B-01 To a solution of compound 8 (0.52 g, 0.96 mmol) in THF (6 mL)wasadded1.0M LiHMDS(3.82 mL, 3.82 mmol) at -72℃dropwise. The reaction mixture was stirred at --72 °C for 40 min.Subsequently, a solution of TfOMe (0.33 mL, 2.87 mmol) in THF(2mL) was added at -72C dropwise over 5 min. The reactionwas stirred at -72 C for 4 h. The resulting mixture was pouredinto sat. NH4Cl solution (20 mL) and extracted with EtOAc(20 mL×3). The combined organic layer was dried over NazSO4,filtered and concentrated. The residue was purified by columnchromatography (n-hexane/EtOAc=1:1) gave B-01 (0.48g,0.84 mmol, 87%) as a white solid. Compound B-02 was prepared according to Ref. 14, gave awhite solid in 63%yield. MS (m/z) ESI: 565.2 (M+Na); 1H NMR(400MHz, CDCl3)8 8.15-8.09 (m, 2H), 7.61 (m, 1H), 7.49 (m,2H), 5.62 (d,J=7.4Hz, 1H, H-2), 5.01-4.95 (m, 2H, H-10, H-10,H-5), 4.95-4.87 (m, 1H, H-13), 4.32 (d,J=8.4Hz, 1H, H-20), 4.20 (d,J=8.5 Hz, 1H,H-20), 3.87 (d,J=7.3 Hz, 1H, H-3), 3.45 (s, 3H),2.32 (d,J=7.0Hz, 1H, H-14), 2.29 (s, 4H, H-14), 2.26-2.22(m,2H, H-6) 2.08 (t,J=5.2 Hz, 3H), 1.97-1.91 (m, 1H, H-7), 1.74 (s,3H), 1.60-1.51 (m, 1H, H-7), 1.13 (s, 3H), 1.08 (s, 3H). 13c NMR(101 MHz, CDCl3)8209.67,170.62,167.21,142.99,133.54,130.11,129.61,128.64,128.57, 86.21,84.43,82.65, 82.00, 79.07,75.85,67.84,57.77,56.44,53.18,45.61,42.55,42.15,38.78),35.55,29.69,27.24,26.35,22.65,19.99,19.75,16.56,15.37,14.84,14.75. 10-Methyl-7-dimethylaminocarbonyl-baccatin III B-05.n1 To a solution of compound 10 (0.73g, 1.31 mmol) in THF(10mL) was added carbonyldiimidazole (0.44g, 2.62 mmol) atroom temperature. The reaction was stirred for 4 h. The resultingmixture was poured into water (20 mL) and extracted with EtOAc(10mL×3). The combined organic layer was dried over NazSO4,filtered and concentrated. The crude product was dissolved inTHF (10mL), and then 33% aqueous dimethylamine (0.50mL) solu-tion was added. stirred for 2 h. The reaction solution was concen-trated and then purified by column chromatography (n-hexane/EtOAc=3:4) gave B-05 (0.46 g, 0.96 mmol, 73%) as a white solid. 13-Oxo-7,10-substituted-1,14-carbonate-baccatin IH 15. Synthesis of compound 15 following the procedures of Ref. 157,10-Substituted-1,14-carbonate-baccatin II B-07-B-14. A typical procedure is given for the preparation of 10-methyl-7-dimethylaminocarbonyl-1,14-carbonate baccatin II B-12: A solu-tion of 13-oxo-10-methyl-7-dimethylaminocarbonyl-1,14-carbon-ate baccatin ⅢI (0.78 g, 1.17 mmol) in anhydrous THF (5 mL) wasadded to (R)-2-methyl-CBS-oxazaborolidine (0.07 mL, 0.23 mmol)and 1.0 M BH3/THF(5.85 mL, 5.85 mmol) at room temperature, un-der nitrogen and stirring. After 5 h, the reaction was quenched byaddition of water. The reaction mixture was extracted with EtOAcand the organic phase was dried, filtered and evaporated under re-duced pressure. Chromatography of the residue (SiO2, n-hexane/EtOAc=1:1) gave B-12 (0.68 g, 1.02 mmol,87.3%) as a white solid.MS (m/z) ESI: 694.2 (M+Na)*; HNMR (400 MHz, CDCl;)8 8.03 (d,J=7.3 Hz, 2H), 7.62 (t,J=7.4Hz, 1H), 7.49 (t, J=7.7 Hz, 2H), 6.08(d,J=7.2 Hz, 1H, H-2), 5.49 (dd,J=10.7, 7.2 Hz, 1H, H-7), 5.23 (s,1H,H-10), 5.04 (t, 1H,H-13), 4.97 (d,J=8.3Hz, 1H, H-5), 4.80 (d,J=5.8 Hz, 1H, H-14), 4.32 (d, J=8.4Hz, 1H, H-20), 4.22(d,J=8.4Hz, 1H, H-20), 3.87 (d, J=7.2Hz, 1H, H-3), 3.677 (d,J=5.3 Hz, 1H, OH-13), 3.40 (s, 3H), 2.87 (s, 6H), 2.54 (m,J=14.5,9.5, 7.3 Hz, 1H, H-6), 2.31 (s, 3H), 2.17 (s, 3H), 1.92 (ddd,J=14.2,8.1, 2.6 Hz, 1H, H-6), 1.81 (s, 3H), 1.24 (s, 3H), 1.14 (s, 3H). 1’cNMR (101 MHz, CDCl3)8205.73,171.28,170.21,164.84, 155.01 ,152.98,141.50,134.17, 133.99,129.83,128.91,128.14,88.54,84.21, 83.86,82.88,80.06,75.99,72.28,71.79,69.45,57.11,57.00,46.25, 41.32,36.53,35.89,33.73,25.62,22.21,21.33,14.91,10.85. General procedure for the syntheses of protected taxoids 16through the coupling of side-chain precursor 7a~d with baccatinderivatives B-01-B-14. A solution of 4 equiv 7a in dry CH2Cl2, was added with 1equivbaccatin derivative, 2 equiv DCC, 0.5 equiv DMAP and 0.2 equivPTSA, at room temperature with stirring overnight. The reactionwas evaporated under reduced pressure. Chromatography of thecrude mixture(n-hexane/EtOAc=2:1) afforded 16 as a white solid. A general procedure for the syntheses of the target taxoids T-01-T-35 through deprotection of the fully protected taxoids 16 isgiven below. The fully protected taxoids 16 were dissolved in THF and added at0°C to a0.2 M solution of acetyl chloride in MeOH. After 2 h the reac-tion was quenched by addition of saturated aqueous NH4Cl solution.The organic phase was dried, filtered, and evaporated under reducedpressure. Chromatography(SiO2,n-hexane/EtOAc=5:6) gave the tar-get taxoids as white solids, in greater than 90% yield. The analyses of T-01-T-35 were listed in the supportinginformation. 4.2.1. Growth-inhibition assay in vitro The inhibitory effects of test compounds on proliferation rateswere determined by a modified MTT assay (Amresco),whichindirectly determines cell number by measuring membrane-asso-ciated proteins. Breast carcinoma cell1 1lines MCF-77(ATCC),non-small cell lung carcinoma cell lines NCI-H 460 (ATCC), ovariancarcinoma cell line A2780 (ECACC), melanoma carcinoma cell linesA-375 (ATCC), colorectal carcinoma cell lines HT-29 (ATCC), cervi-cal carcinoma cell line HeLa (ATCC), leukemia carcinoma cell linesHL-60 (ATCC), and prostate carcinoma cell lines DU145 (ATCC)were grown in DMEM or IMDM with 10% fetal calf serum. The cellswere plated at a density of 2000-5000 cells/well/100 ul on 96-wellmicrotiter plates in complete growth medium and incubated at37 °C overnight to allow the cells to attach to the substrate beforedrugs exposure. The test compounds were suspended in DMSO at100 mM and stored at -20C. Serial dilutions in media were pre-pared to make the final desired concentrations (1 uM). Duplicatewells were exposed to various treatments. After 72 h at 37°C, 5%CO2 incubation, the wells were added for 4 h with 10 pl of 5 mg/mL MTT in phosphate-buffered saline and incubated at 37℃. Atthe end of the incubation period, the cells were fixed with 100 ulmodified lysis reagent (10% SDS, 5% isopropanol with 0.012 N HClin distilled water), dispersed by pipetting until homogeneous.Optical density was measured at 580 to 680 nm by microplatereader, within 15 min. 4.2.2. Caco-2 cells permeability assay The Caco-2 cells were harvested and seeded on the apical side ofa Millicell-24 well plate. After 21 days culture, take cell cultureplate out of incubator, pre-incubated with HBSS, pH 7.4 at 37℃,and then measure TEER values at room temperature. The trans-monolayer electrical resistance of Caco-2 monolayers from ran-domly selected wells was measured by Millicell-ERSvoltohmmeterreading,TTEER value was 359±12Q cm(Mean±SD) higher than 250 Q cm . Prewarm apical and basolat-eral plates at 37℃ for about 5 min. The assay was initiated bythe addition of the test compounds (10 uM) from the apical(0.8 mL) incubation at 37C with 5% Co2 and 95% air. The apicalplate was separated from the basolateral plate after 90-min incu-bation, and 100 ul samples were transferred from basolateral plateto an opaque plate. The samples were prepared for LC-MS/MS anal-ysis: donor samples (1:10 diluted): 6 ul of donor sample +54 ul0.4% DMSO HBSS + 60 ul ACN with IS (Osalmid or Imipramine); re-ceiver sample: 60 ul of receiver sample+60 pl ACN with IS (Osal-mid or Imipramine). Erythromycin, metoprolol, atenolol asreference compounds, paclitaxel and ortataxel as positive control. Drug permeability : Papp Where V is the volume in the acceptor well, the area is the surfacearea of the membrane and time is the total transport time in sec-onds. [drug]donor, 0-min is the donor drug concentration count mea-suredat O-min;druglacceptor, 90-mi,n isSthe acceptor drugconcentration count measured at 90-min; The dilution factor isthe fold of dilution of the donor sample before loaded to LC-MS/MS. For Millicell-24 cell culture plates: surface area of the mem-brane=0.7 cm²,VA=0.8 mL (A-to-B) or 0.4 mL (B-to-A). 4.2.3.MDCK-MDR1 cells and MDCK-WT cells transport MDCK-MDR1 cells were seeded onto polycarbonate filter Trans-wells, and confluent MDCK-MDR1 monolayers expressing P-gpwere obtained 5 days postseeding. The transmonolayer electrical resistance of MDCK-MDR1 monolayers from randomly selectedwells was 2870±210Q cm² (Mean±SD) higher than 1000Q cm² before use in transport experiments. MDCK-WT cells wereseeded at a density of 5.0×104 cells/well, and matured exhibitinga TEER 280±12 Q cm higher than 200 Q cm before use in trans-port experiments after 6 days. Cell monolayer integrity was evalu-ated by measurement of Lucifer yellow permeability by monitoringthe change in TEER over the course of the experiment. Transportexperiments of the test compounds were performed at 37C inHBSS at pH 7.4. Separate the apical plate from the basolateral plateafter 90 min incubation. A 100 ul sample was taken from the donorcompartment to determine the concentration of the compoundremaining in the donor chamber at the end of the experiment. Acknowledgments We grateully acknowledge Tianjin Tasly Group Co., Ltd for thefinancial support for the project and the scientist of ShanghaiChemPartner Co., Ltd for performing pharmacological experiments.We acknowledge Dr. W.Q. Chen for his many helpful suggestions. SuppIementary data Supplementary data associated with this article can be found, inhe online version, at http://dx.doi.org/10.1016/j.bmc.2013.11.037. References and notes ( (a ) Stephenson, F .; A T ale o f T axol, F SU Research i n R e view, Vo l . XII, No. 3 , T allahassee, FL, 2002, p . 1 2.; ( S aros y, G.; K ohn, E.; Stone, D. A.; Rothenberg , M. ; Jac o b, J ; Ad a mo,D.O; ( O g n ibene, F . P .; Cunni on , R. E . ; R ee d , E. J . Cl i n. Oncol.1992, 10,1165; ((C )H ol mes , F. A . ; W alters, R . S . ; T heriault, R . L .; For m an, A . D .; N ewton ,L . K.; R aber, M . N N . ; ; Bu z da r ,A. U.; F r y e, D . K.; H o r tob a gyi, G. N . J . N a t l. Canc e r I ns t . 1 1 991 ,83, 1 7 9 7 ; ( d) Rowinsky , E. K.; C azenave, L . A .; D on e hower, R. C . J . . Natl . Cance r Ins t . 199 0 , 8 2 ,1247 ; (e ) M c Gui r e, W. P .; Rowinsky, E . K.; R osenhe i m , N .B. : Gr u m b i ne , F. C.; Ett i nger, D. S .; Ar m str o ng, D. K. A n n. Intern. le d . 1 989. 1 1 1, 273 ; ( f ) W ani, M. C . ; T a y lor, H . L.; W a ll, M . E. ; C o ggon, P. ; c Pha i l, A. T . J . Am. Ch e m. So c . 1971 , 93,2325. ) 2.(a) Chow,L W.; Xu, B.; Gupta,S.; Freyman, A.; Zhao,Y.; Abbas, R.; Vo Van, M.L.;Bondarenko, I. Br. J. Cancer. 2013,108,1985; (b) Rugo, H. S.; Campone, M.;Am1aadori, D.; Aldrighetti, D.; Conte, P.; Wardley, A.; Villanueva, C.; Melisko,M.;McHenry, M. B.; Liu, D.; Lee, F.; Pivot, X. Breast Cancer Res. Treat. 2013,139,411;) Esteva, F. J; Franco, S. X.; Hagan, M. K.; Brewster, A. M.; Somer, R. A.; Williams, W.; Florance, A. M.; Turner, S.; Stein,S.; Perez, A. Oncologist 2013; (d)Henderson,I. C.; Berry, D. A.; Demetri, G. D.; Cirrincione, C. T.; Goldstein, L. J.;Martino,S.; Ingle, J. N.; Cooper, M. R.; Hayes, D. F.; Tkaczuk, K. H.; Fleming, G.;Holland,J.F.; Duggan,D. B.; Carpenter,J. T.; Frei,E.,II; Schilsky, R. L.; Wood, W.C.; Muss, H. B.; Norton, L. J. Clin. Oncol. 2003, 21, 976. ( 3. ( a )Jeyap a lan, S.; Bo x erman , J . ; D o na h u e ,J; G o l dman, M.; K i n s e l la,T. ; Di p et r illo, T .; Ev a ns, D .; Elin z ano,H.; C o nstantinou , M.; Sto p a , E.; P ut h a w ala, Y.; Ciel o , D . ; Santanie l lo, A .;O y elese, A. ; Mant r ip r ag a da, K.; Rosa t i, K.; I s d ale,D.;Safra n , H. J.Clin . Oncol . 2013 ; (b) Tha k ur, A. ; Joshi, N . ;S h anmu g am, T . ; B a nerjee, R . Cance r L ett. 2013 , 334,27 4 ;(c) Giussa ni , P.; Bass i , R.; A nelli, V.; Bri o s chi, L . ; D e Z en, F.; R iccitelli, E. ; Carol i , M. ; Ca m pane l la, R . ; Gaini, S. M .; Viani, P. ; R iboni, L. C ancer Invest.2012, 3 0.27. ) ( 4. W enk, M. R . ; F a hr, A .; R e szka, R.; S e eli g ,J. J. P h arm. Sci.1996,85 , 22 8 . 5. ( ( a ) Bardelmeijer, H . A .; O uwehand, M. ; Bei j ne n , J. H . ; Sc h ellens, J. H . ; v a n T el l ingen,O. I nvest. New Drugs 2 0 04, 22, 2 1 9;(b ) B a r delmei j er, H. A.; Be i jnen,J. H .; B rouwer, K . R . ; Rosin g , H. ; N o o ijen , W . J .; S c h e llens, J. H.; v an Tell i ngen , O . Clin . Cancer Res. 2000,6,4416; ( c) Sparr e boom, A.; v an Asperen,J ; M a yer, U.; S chinkel, A . H . ; Smit, J . W.; Meijer, D. K . F. ; Borst , P . ; Noojij e n, W. J; Beijne n , J . H . ; va n T ell i ngen, O. Proc. N at l . Ac a d. Sc i . U.S.A . 1997, 9 4,2031. ) ( 6. (a) Singla, A. K . ; Garg, A . ; Aggarwa l ,D. Int. J. Pharm. 2002,235, 1 79;( b )Adams,J.D . ; F lora, K . P .; G ol d spiel, B . R . ; W i lson, J. W. ; Arb u ck, S. G.; F in l e y , R. J. Na t l . Cancer Inst . Monogr . 1993, 15,14 1 ; ( c) Weiss, R. B . ; Donehower,R. C. ; Wiernik, P . H.; Ohnuma,T.; Gral l a, R . J.; T r u m p, D. L. ; B ake r jr,J. R . ; Van E c ho, D. A . ; V o n H off, D. D . ; L eyland-Jones, B. J. C lin. Oncol. 1 9 90, 8 ,1 2 63. ) 7.. (a)Vredenburg, M. R.;Ojima, I.; Veith, J; Pera, P.; Kee, F. C.; Sharma, A.; Kanter,P.; Greco, W. R.; Bernacki, R. J. J.Natl. Cancer Inst. 2001, 93, 1234; (b) Polizzi, D.;Pratesi, G.; Monestiroli, S.; Tortoreto, M.; Zunino,F.; Bombardelli,E.; Riva, A.;Morazzoni, P.; Colombo,T.; D'Incalci, M.; Zucchetti, M. Clin. Cancer Res. 2000, 6,2070; (c) Nicoletti, M. I.; Colombo, T.; Rossi, C.; Monardo, C.; Stura, S.;Zucchetti,M.; Riva, A.; Morazzoni,P.; Donati,M. B.; BombOarIdDelli, E.; D'Incalci,M.; Giavazzi, R. Cancer Res. 2000, 60, 842; (d) Ferlini, C.; Distefano, M.;Pignatelli, F.; Lin, S.; Riva, A.; Bombardelli,E.; Mancuso,S.; Ojima, I.; Scambia, G.Br. J. Cancer 2000, 83, 1762;(e) Ojima, I.; Kuduk, S. D.; Pera, P.; Veith, J. M.;Bernacki, R. J. J. Med.Chem. 1997,40,279;(f)Ojima, I.; Slater,J. C.; Kuduk, S. D.; Takeuchi, C. S.; Gimi, R.H.; Sun, C.-M.; Park,Y. H.;Pera, P.; Bernacki, R. J.J. Med.Chem. 1996, 39,3889;(g)Ojima, I.; Duclos,O.; Kuduk, S.D.; Sun, C.-M.; Slater,J.C.; Lavelle, F.; Veith, J. M.; Bernacki, R. J. Bioorg. Med. Chem. Lett. 1994, 4, 2631. 8. (a) Wu,Q.; Bounaud,P.-Y.;Kuduk, S. D.; Yang, C.-P. H.;Ojima, I.; Horwitz, S. B.;Orr, G. A. Biochemistry 1998, 37, 11272; (b) Ojima, I.; Slater,J.C.; Michaud,E.;Kuduk, S. D.; Bounaud, P.-Y.; Vrignaud, P.; Bissery, M.-C.; Veith, J. M.; Pera, P.;Bernacki,R. J.J. Med.Chem. 1996,39,3889;(c)Ojima, I.; Duclos, O.; Dorman,G.;Simonot, B.; Prestwich, G. D.; Rao, S.; Lerro, K. A.; Horwitz, S. B. J. Med. Chem.1995,38,3891. 9. Altstadt,T.J.; Fairchild, C. R.; Golik,J.; Johnston, K. A.; Kadow,J. F.; Lee, F. Y.;Long, B. H.; Rose, W. C.; Vyas,D. M.; Wong, H.; Wu, M.J.; Wittman, M. D.J.Med.Chem. 2001,44,4577. 10.Sampath, D.; Discafani,C. M.; Loganzo, F.; Beyer,C.; Liu, H.; Tan, X.; Musto, S.;Annable, T.; Gallagher, P.; Rios, C.; Greenberger,L.M. Mol. Cancer Ther. 2003,2,873. 11.S:hionoya,M.; Jimbo, T.; Kitagawa, M.; Soga, T.; Tohgo, A. Cancer Sci. 2003, 94,459. 12.Wang, Y.; He, Q. F.; Wang, H. W.; Qin, Y. j. Org. Chem. 2006, 71,1588. 13.1Blechert, S.; Guenard, D. The Alkaloids In Brossi, A.,Ed.; Academic Press: SanDiego,1990; p 39, 195. 144.(a)Ishiyama, T.; Iimura, S.; Yoshino, T.; Chiba,J.; Uoto, K.; Terasawa,H.; Soga, T.Bioorg. Med. Chem. Lett. 2002, 12, 2815;(b) Chen, S.-H.; Huang, S.; Gao, Q.;Golik, J; Farina, V. J. Org.Chem. 1994,59,1475;(c) Chaudhary, A. G.; Rimoldi, J.M.; Kingston, D. G. I. J.Org. Chem. 1993, 58,3798. 15.Baldelli, E.; Battaglia, A.; Bombardelli,E.; Carenzi, G.; Fontana, G.; Gambini, A.;Gelmi, M. L.; Guerrini, A.; Pocar, D. J. Org. Chem. 2003, 68,9773. ( 16. (a )Battaglia , A . ; B a ld e l l i, E.;B o mbardelli, E . ; Ca r enzi, G., et al Tetrahedron 200 5 , 6 1 , 7727; ( b ) Lacca b u e, D.; Pr a tesi, G. D r ugs Future 2001, 26, 53 3 . ) ( 17. (a ) Hubatsch, I . ; Ragn a rsson , E. G .; Artursson, P . N at. Protoc 2 . 0 20 0 07 . ,2, 4 21 6 11 2 ; ( b : ) A 03 rtursson, P .; P alm, K .; L uthman, K . A d v. D r ug De l iv. R e v. 2 0 01, 46 , 2 7 ; ( c ) H ilgers, A. R. ; Conradi,R. A. ; Burton, P . S. Pharm Res 1990, 7 , 902; ( d) H i dalgo, I. J ; R au b , T. J . ; Borchardt, R. T. Gastroenterology 1 989, 96,736. ) ( 18. ( a ) Sugano, K.; K ansy, M.; Artursson, P.; Avdeef, A . ; Bendels, S . ; D i, L .; Ecker, G. F .; Falle r, B. ; Fischer, H . ; Gere b tzof f , G. ; Lenner n aes,H . ; S enne r, F. N a t. R e v.Drug D isc. 2010,9 , 5 9 7; ( b ) Ballato r e, C . ; Hy de, E.; Dei c hes, R. F .; L e e Vir gi n ia , M .-Y. ; T r ojanowski, J . Q ; Hu r yn, D.; Smith, A. B., I I I B i oorg. M e d. Chem. Le t t. 2007,17, 3 642;( c) Ric e , A.; Li u , Y.; M i ch a elis, M. L.; Him e s,R. H . ; Georg,G. I .; A u dus, K. L. J.Med. Chem. 2005. 48.832. ) ( 19. V redenburg , M. R. ; Oj i ma, I.; Veith , J; P era, P.; K e e, K . ; Cab r al, F.; S h a rma, A.; K anter, P . ; G reco, W . R. ; Bernacki, R. J .J. Natl. C a ncer Inst. 20 0 1, 93 , 123 4 . ) 20.F1DA, 2006. Drug Interaction Studies-Study Design, Data Analysis, andImplications for Dosing and Labeling. http://www.fda.gov/OHRMS/DOCKETS/98fr/06d-0344-gdl0001.pdf. 21.(a) Kim, R. B. Drug Metab. Rev. 2002, 34, 47; (b) Litman, T.; Druley, T. E.; Stein,W. D.; Bates, S. E. Cell. Mol. Life Sci.2001, 58,931; (c) Wacher, V. J.; Wu, C. Y.;Benet, L. Z. Mol.Carcinog. 1995, 13, 129. 22.Sakehan, P.; Streng, R.; Scudierok, D.; Monks, A.; McMahon, J; Vistica,Warren,J. T.; Bokesch, H.; Kenney, S.; Boyd, M.R. J. Natl. Cancer Inst. 1990, 81107. 23.Taub, M. E.; Podila, L.; Ely, D.; Almeida, L. Drug Metab. Dispos. 2005, 33,1679. $ -see front matter @ Elsevier Ltd. All rights reserved.http://dx.doi.org/j.bmc.ww.rysstech.com耐士科技 士科技
确定








还剩8页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中主要物质含量分析检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多