








方案详情
文
The [18F]fluorocyclobutyl group has the potential to be a metabolically stable prosthetic group for PET tracers. The synthesis of the radiolabeling precursor cis-cyclobutane-1,3-diyl bis(toluene-4-sulfonate) 8 was obtained from epibromohydrin in 7 steps (2% overall yield). The radiolabeling of this precursor 8 and its conjugation to L-tyrosine as a model system was successfully achieved to give the new nonnatural amino acid 3-[18F]fluorocyclobutyl-L-tyrosine (L-3-[18F]FCBT) [18F]17 in 8% decay-corrected yield
from the non-carrier-added [18F]fluoride. L-3-[18F]FCBT was investigated in vitro in different cancer cell lines to determine the uptake and stability. The tracer [18F]17 showed a time dependent uptake into different tumor cell lines (A549, NCI-H460, DU145) with the best uptake of 5.8% injected dose per 5 105 cells after 30 min in human lung carcinoma cells A549. The stability of L-3-[18F]FCBT in human and rat plasma and the stability of the non-radioactive L-3-FCBT in rat hepatocytes were both found to be excellent. These results show that the non-natural amino acid L-3-[18F]FCBT is a promising metabolically stable radiotracer for positron emission tomography.
方案详情

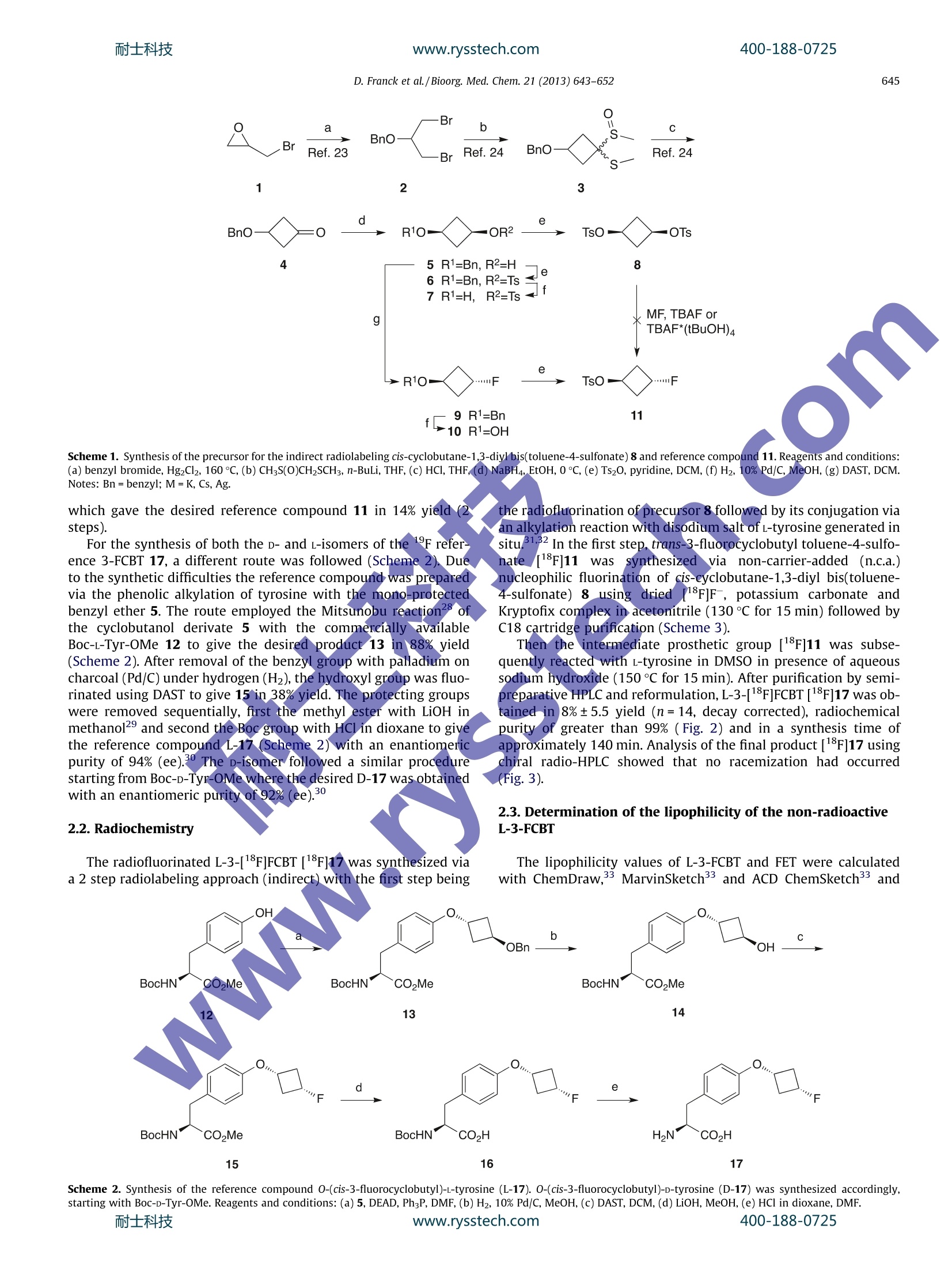
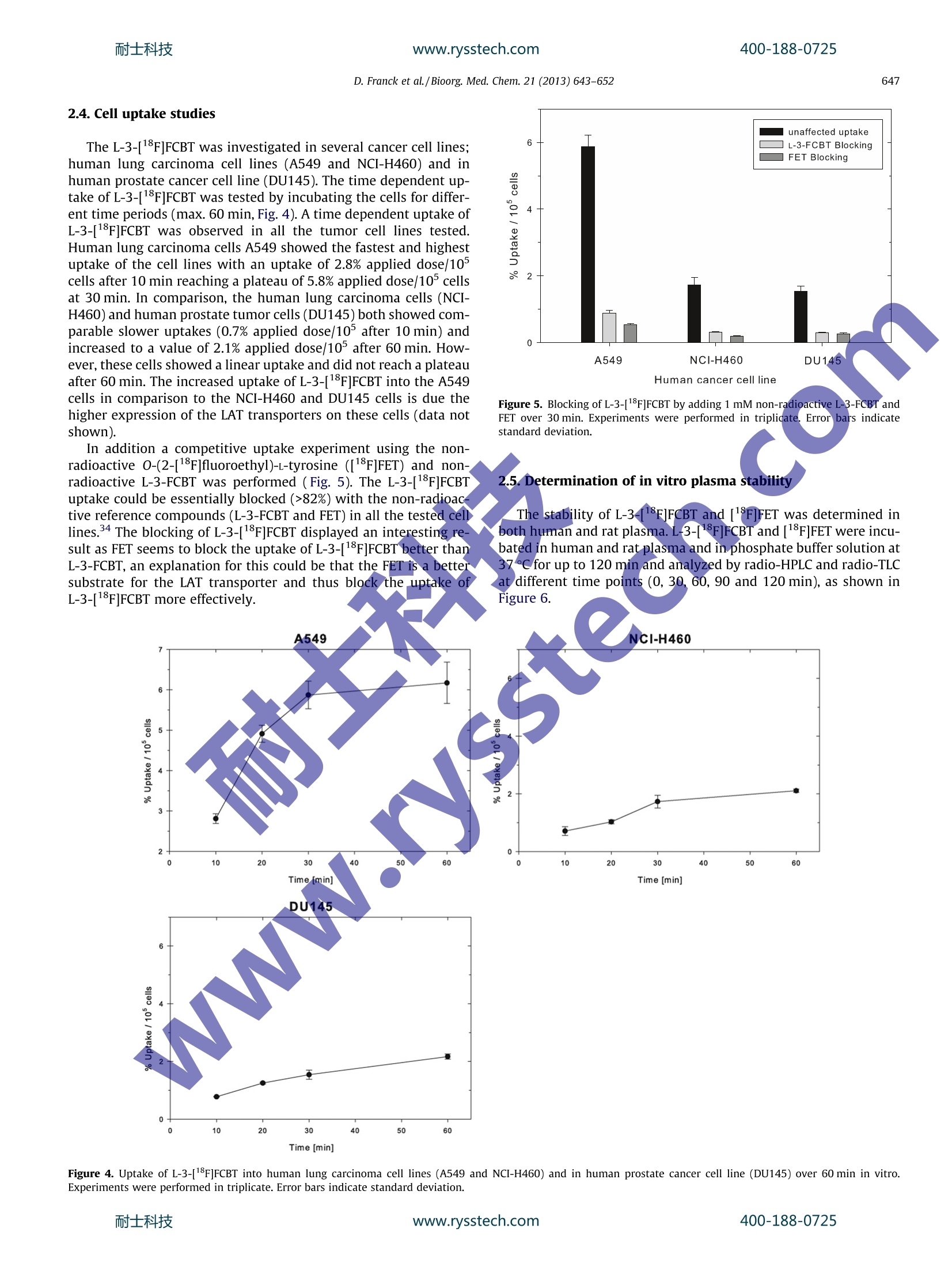
耐士科技www.rysstech.comBioorganic & Medicinal Chemistry 21 (2013)643-652400-188-0725 耐士科技400-188-0725www.rysstech.com644 Contents lists available at SciVerse ScienceDirect Bioorganic & Medicinal Chemistry ELSEVIER journal homepage: www.elsevier.com/locate/bmc Investigations into the synthesis, radiofluorination and conjugation of anew[F]fluorocyclobutyl prosthetic group and its in vitro stability using atyrosine model system Dominic Franck, Torsten Kniess,Jorg Steinbach, Sabine Zitzmann-Kolbe, Matthias Friebeb,Ludger M. Dinkelborg , Keith Grahamb,* Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, Dresden, GermanyGlobal Drug Discovery, Bayer Healthcare Pharmaceuticals, Berlin, Germany ARTICLE INFO Article history: Received 13 August 2012 Revised 12 November 2012 Accepted 29 November 2012 Available online 8 December 2012 Keywords: Tyrosine Cyclobutyl Stability Metabolism Fluorine-18 Positron emission tomography(PET) The [1F]fluorocyclobutyl group has the potential to be a metabolically stable prosthetic group for PETtracers. The synthesis of the radiolabeling precursor cis-cyclobutane-1,3-diyl bis(toluene-4-sulfonate) 8was obtained from epibromohydrin in 7 steps (2% overall yield). The radiolabeling of this precursor 8and its conjugation to l-tyrosine as a model system was successfully achieved to give the new non-natural amino acid 3-[18F]fluorocyclobutyl-l-tyrosine (L-3-[18F]FCBT)[18F]17 in 8% decay-corrected yieldfrom the non-carrier-added [18F]fluoride. L-3-[1F]FCBT was investigated in vitro in different cancer celllines to determine the uptake and stability. The tracer [1F]17 showed a time dependent uptake into dif-ferent tumor cell lines (A549, NCI-H460, DU145) with the best uptake of 5.8% injected dose per 5×10°cells after 30 min in human lung carcinoma cells A549. The stability of L-3-[F]FCBT in human and ratplasma and the stability of the non-radioactive L-3-FCBT in rat hepatocytes were both found to be excel-lent. These results show that the non-natural amino acid L-3-[18F]FCBT is a promising metabolically sta-ble radiotracer for positron emission tomography. Metabolism plays a huge role in the development of new drugsboth in the therapeutic and diagnostic field. With therapeutics,the metabolism of the drug can lead to biologically active metabo-lites which could cause numerous side-effects.For diagnostics, inparticular positron emission tomography (PET) radiopharmaceuti-cals, the images obtained from the PET camera are only visualizingthe radionuclide attached to the biologically active molecule. If thisradiolabeled molecule is metabolically unstable then the cameracannot distinguish between both the intact molecule and any ofits metabolites containing the PET radionuclide. This often leadsto undesirable accumulation in non-target organs increasing back-ground activity and giving misleading information. Therefore it isessential when synthesizing such radiolabeled biologically activemolecules that they remain metabolically stable to ensure thatthe image obtained is due to the injected PET tracer and not dueto one or more of its metabolites. In PET research when investigat-ing new biologically active molecules, it is attractive to synthesizean analog with a radiolabeled alkyl group attached to a hetero-atom, namely oxygen and nitrogen.However, it is reported in the ( * Corresponding author. ) literature that potential sites for metabolism are alkylated hetero-atoms.34 For 18F-labeled PET tracers, this type of metabolism hasbeen reported for different biological targets; for example. thedopamine transporter radiotracer [18F]FECNT; the vesicularacetylcholine transporter ligand [18F]FEOBV4 and the non-peptideendothelin type A receptor antagonist[18F]PD 156707(Fig.1). For these tracers the noticeable similarity is the [lF]fluor-oethylalkyl chain attached to either an oxygen or nitrogen hetero-atom. Radiotracers labeled with an N-or O-[18F]fluororethyl groupcan be prone to dealkylation through enzymatic reactions in thebody, particularly the monooxygenase cytochrome P450, to givepolar radiometabolites which can interfere in PET imaging. Allradiometabolites originating from N- or O-dealkylation are a con-sequence of the Phase I biotransformation and are believed to be[18F]fluoroethanol.3. The radiometabolite [18F]fluoroethanol canundergo an oxidative biotransformation to the aldehyde[18F]fluo-roethanol and can also be further oxidized to [18F]fluoroaceticacid.3.8 This radiometabolite can be further metabolized, mainlyin the liver, to the [F]fluoride ion which accumulates in the bone,skull and joints, thus leading altogether to a higher backgroundsignal and can influence the PET image quality. We hypothesized that replacing the [1F]fluoroalkyl chain witha [F]fluorocycloalkyl group could increase the metabolic stabilityof a PET tracer. In the literature numerous examples can be found ( 0968-0896/$- see front matter @ 20 1 2 Else v ier Ltd. All rights re s erved. http:// d x.doi. o rg/10 .1 016/j. b mc.2012. 1 1 .049 ) D. Franck et al./Bioorg. Med. Chem.21 (2013)643-652 moFigure 1. Selected examples of 18F-labeled radiotracers based on the fluoroalkyl group with radiometabolites reported.c where pharmaceuticals with cycloalkyl rings attached to heteroat.oms are more metabolically stable than their simple alkyl chaincounterparts.10-12Radiolabeledccyclobutyl groups are notunknown and the Goodman group have explored different cyclo-butyl unnatural amino acids for cancer imagingand interestinglyanti-1-amino-3-[1F]fluorocyclobutane-1-carboxylic: acid (anti-3-[F]FACBC), a potential imaging agent for prostate cancer,6their were no visible metabolism in vivo.17The model system cho-sen to test our hypothesis was a tyrosine based amino acid, as avariety of O-alkylated tyrosines are reported, for example, 0-(2-[18F]fluoroethyl)-L-tyrosine ([18F]FET)18 or DFMT 19.20These com-pounds are known to be transported into the cells via the wellcharacterized and extensively documented large amino acid trans-porter (LAT).21.22 Herein, we describe the preparation of the novel tyrosine deri-vateO-(cis-3-[1F]fluorocyclobutyl)L-tyrosine (L-3-[18F]FCBT)[1ǒF]17 via an indirect radiolabeling method and the synthesis ofthe corresponding reference compounds (11, L-171 and D-17).L-3-[18F]FCBT was tested in vitro for both its uptake in different tu-mor cell lines (A549, NCI-H460, DU145) and its metabolic stability. 2. Results and discussion 2.1. Chemistry The precursor cis-cyclobutane-1,3-diyl bis(toluene-4-sulfonate)8 for the indirect radiolabeling was prepared in a multistep synthe-sis starting from epibromohydrin 1 and benzyl bromide(Scheme 1).The first 3 steps are literature known and gave 3-(benzyloxy)cyclo-butanone 4.15.23.24 The reduction of 4 with sodium borohydride(NaBH4) gave the key intermediate cis-3-(benzyloxy)cyclobutanol5 in 86% yield. It was of interest to see if the basic reaction conditions of thereduction step would form another stereoisomer than the expectedcis-alcohol 5. The H NMR- and ES-mass spectrum confirmed thestructure of the alcohol 5. Since the NMR data do not give informa-tion about the cis/trans-stereochemistry, NOE NMR experimentwas performed and the cis-isomer 5 was confirmed as the majorcompound. Consequently, the observed stereoselective reductionsuggests an axial attack of the nucleophile NaBH4. Standard tosylation methods were tested to synthesize 6; usingtosyl chloride (TsCl) with or without the catalyst 4-dimethylami-nopyridine (DMAP) resulted in low yields (~30%). The use ofpara-toluenesulfonic anhydride (Ts20) in pyridine gave far superioryields (87%). The next step was the removal of the benzyl groupwith standard palIadium on charcoal catalyzed hydrogenation con-ditions. Repeating the tosylation of the deprotected compound 7with the Ts20 method afforded the precursor for the indirect label-ing cis-cyclobutane-1,3-diyl bis(toluene-4-sulfonate) 8 (65%, 3steps) The preparation of the 19F-reference compound trans-3-fluoro-cyclobutyl toluene-4-sulfonate 11 was initially investigated vianucleophilic fluorinations of the bis-tosylate 8 using variousdifferent known methods; these included potassium fluoride(KF),25 cesium fluoride (CsF),26 silver fluoride (AgF)7 or tetrabutyl-ammonium fluoride (TBAF)2 failed to give the desired referencecompound 11. Kim et al.26 recently described TBAF to be hygro-scopic which could have an influence on the TBAF reaction and theyreported the significantly less hygroscopic fluorination agentTBAF*(tBuOH)4 which was prepared simply by refluxing TBAF intBuOH/hexane and recrystallization at room temperature in 55%yield. The fluorination of the bis-tosylate 8 was tried with thefreshly prepared TBAF*(tBuOH)4 refluxing in acetonitrile (1 h).Unfortunately, all attempts under a variety of conditions rangingfrom low to high precursor 8 concentrations failed to give the de-sired fluorotosylate 11. Alternatively the fluorinated compound 9 was then preparedfrom cis-3-(benzyloxy)cyclobutanol 5 by treatment with diethyla-minosulfur trifluoride (DAST) at 0 °C in 37% yield. Fluorination wasalso successful with perfluoro-1-butanesulfonyl fluoride with amixture of triethylamine trihydrofluoride in presence of triethyl-amine (TEA) at room temperature albeit in lower yield (25%) thanthe DAST method. Removal of the benzyl group using catalytichydrogenation conditions with palladium on charcoal in methanol(Scheme 1) afforded the trans-3-fluorocyclobutanol 10, which wasconfirmed by 'H NMR spectroscopy. Unfortunately, the fluoroalco-hol 10 was found to be volatile. A high quantity of product was lostduring the solvent evaporation process and was avoided with care-ful evaporation of the solvent (40 C, 200 mbar). The fluoroalcohol10 was used without further purification in the tosylation step, Scheme 1. Synthesis of the precursor for the indirect radiolabeling cis-cyclobutane-1,3-diyl bis(toluene-4-sulfonate) 8 and reference compound 11. Reagents and conditions:(a) benzyl bromide, Hg2Cl2, 160 ℃, (b) CH;S(O)CH2SCHs,n-BuLi, THF, (c)HCl, THF, (d) NaBH4, EtOH, 0℃, (e) Ts2O0, pyridine,DCM, (f) H2, 10%Pd/C,MeOH, (g) DAST, DCM.Notes: Bn=benzyl; M=K, Cs, Ag. which gave the desired reference compound 11 in 14% yield (2steps). For the synthesis of both the D- and L-isomers of the 19F refer-ence 3-FCBT 17, a different route was followed (Scheme 2).).LDueto the synthetic difficulties the reference compound was preparedvia the phenolic alkylation of tyrosine with the mono-protectedbenzyl ether 5. The route employed the Mitsunobu reaction28ofthe cyclobutanol derivate 5 with the commercially/ availableBoc-L-Tyr-OMe 12 to give the desired product 13 in 88% yield(Scheme 2). After removal of the benzyl group with palladium oncharcoal (Pd/C) under hydrogen (H2), the hydroxyl group was fluo-rinated using DAST to give 15 in 38% yield. The protecting groupswere removed sequentially, first the methyll ester with LiOH inmethanol2 and second the Boc group with HCl in dioxane to givethe reference compound L-17 (Scheme 2) with an enantiomericpurity of 94% (ee).30 TheD-isomer followed a similar procedurestarting from BoC-D-Tyr-OMe where the desired D-17 was obtainedwith an enantiomeric purity of 92%(ee).3 2.2. Radiochemistrv The radiofluorinated L-3-[18F]FCBT [18F]1 was synthesized viaa 2 step radiolabeling approach (indirect) with the first step being the radiofluorination of precursor 8 followed by its conjugation viaan alkylation reaction with disodium salt of L-tyrosine generated insitu.31.32 In the first step, trans-3-fluorocyclobutyl toluene-4-sulfo-nate[18F]11 was synthesizedvia non-carrier-added(n.c.a.)nucleophilic fluorination of cis-cyclobutane-1,3-diyl bis(toluene-4-sulfonate) 8 using dried [F]F, potassium carbonate andKryptofix complex in acetonitrile (130 ℃ for 15 min) followed byC18 cartridge purification (Scheme 3). Then the intermediate prosthetic group [18F]11 was subse-quently reacted with L-tyrosine in DMSO in presence of aqueoussodium hydroxide (150C for 15 min). After purification by semi-preparative HPLC and reformulation, L-3-[1F]FCBT[1F]17 was ob-tained in 8%±5.5 yield (n=14, decay corrected), radiochemicalpurity of greater than 99% (Fig. 2) and in a synthesis time ofapproximately 140 min. Analysis of the final product [1F]17 usingchiral radio-HPLC showed that no racemization had occurred(Fig. 3). 2.3. Determination of the lipophilicity of the non-radioactiveL-3-FCBT The lipophilicity values of L-3-FCBT and FET were calculatedwith ChemDraw,33 MarvinSketch33and ACD ChemSketch33 and Scheme 2. Synthesis of the reference compound O-(cis-3-fluorocyclobutyl)-l-tyrosine (L-17). O-(cis-3-fluorocyclobutyl)-D-tyrosine (D-17) was synthesized accordingly,starting with Boc-D-Tyr-OMe. Reagents and conditions: (a) 5, DEAD, Ph3P, DMF, (b) H2, 10% Pd/C, MeOH, (c) DAST, DCM, (d)LiOH,MeOH, (e) HCl in dioxane, DMF.耐士科技 www.rysstech.com 400-188-0725 D. Franck et al./Bioorg. Med. Chem. 21 (2013)643-652 8 [18F]11 [18F]17 Scheme 3. Radiosynthesis ofO-(cis-3-[18F]fluorocyclobutyl)-L-tyrosine [18F]17 via the intermediate trans-3-[18F]fluorocyclobutyl toluene-4-sulfonate [18F]11. Figure 2. C18 radio-HPLC analysis of the final product L-3-[F]FCBT[F]17 showed no impurities in the radio- as well as in the UV-trace. Time [min] Figure 3. Chiral radio-HPLC analysis of final product L-3-[18F]FCBT[18F]17 showed no impurities and only L-isomer to be present (top). Co-injection of the non-radioactive L-isomer of 3-FCBT L-17 with traces of p-isomer visible (bottom). experimentally determined by the extraction (Table 1). Thecalculated lipophilicity values of L-3-FCBT and FET are found tobe in the similar range and indicating very hydrophilic character.These calculated trends were confirmed by the experimentaldetermination of the logD of L-3-FCBT and was found to be-0.83. Comparing the experimental value of FET found in literature(-1.51), 18 L-3-FCBT showed a higher lipophilicity of -0.83 whichcould influence the pharmacokinetics of the tracer for futurein vivo experiments. Table 1Calculated and experimental partition coefficient log D of L-3-FCBT and FET Method L-3-FCBT FET logD logD ChemDraw 1.03 1.39 MarvinSketch -1.08 -1.14 ACD 1.9 -2.2 Experimental MarvinSketch -1.51 Experimental log D value of FET according Ref.18. The L-3-[18F]FCBT was investigated in several cancer cell lines;human lung carcinoma cell lines (A549 and NCI-H460) and inhuman prostate cancer cell line (DU145). The time dependent up-take of L-3-[18F]FCBT was tested by incubating the cells for differ-ent time periods (max. 60 min, Fig. 4). A time dependent uptake ofL-3-[18F]FCBT was observed in all the tumor cell lines tested.Human lung carcinoma cells A549 showed the fastest and highestuptake of thecell lines with an uptake of 2.8% applied dose/10cells after 10 min reaching a plateau of 5.8% applied dose/10cellsat 30 min. In comparison, the human lung carcinoma cells (NCI-H460) and human prostate tumor cells (DU145) both showed com-parable slower uptakes (0.7% applied dose/10 after 10 min) andincreased to a value of 2.1% applied dose/10after 60 min. How-ever, these cells showed a linear uptake and did not reach a plateauafter 60 min. The increased uptake of L-3-[18F]FCBT into the A549cells in comparison to the NCI-H460 and DU145 cells is due thehigher expression of the LAT transporters on these cells (data notshown). Figure 5. Blocking of L-3-[F]FCBT by adding 1 mM non-radioactive L-3-FCBT andFET over 30 min. Experiments were performed in triplicate.Error bars indicatestandard deviation. In addition a competitive uptake experiment using the non-radioactive O-(2-[18F]fluoroethyl)-l-tyrosine ([18F]FET) and non-radioactive L-3-FCBT was performed (Fig. 5). The L-3-[18F]FCBTuptake could be essentially blocked (>82%) with the non-radioactive reference compounds (L-3-FCBT and FET) in all the tested celllines.34 The blocking of L-3-[18F]FCBT displayed an interesting re-sult as FET seems to block the uptake of L-3-[18F]FCBTbetter thanL-3-FCBT, an explanation for this could be that the FET is a bettersubstrate for the LAT transporter and thus block the uptake ofL-3-[18F]FCBT more effectively. The stability of L-3-[18F]FCBT and [1F]FET was determined inboth human and rat plasma.L-3-[18F]FCBT and [18F]FET were incu-bated in human and rat plasma and in phosphate buffer solution at37°C for up to 120 min and analyzed by radio-HPLC and radio-TLCat cdifferent time points (0, 30, 60, 90 and 120 min), as shown inFigure 6. A549 6 0 3 2 0 10 20 30 40 50 60 Time [min] DU145 6 0 0 10 20 30 40 50 60 Time [min] Figure 4. Uptake of L-3-[18F]FCBT into human lung carcinoma cell lines (A549 and NCI-H460) and in human prostate cancer cell line (DU145) over 60 min in vitro.Experiments were performed in triplicate. Error bars indicate standard deviation. L-3-[18F]FCBT showed over 120 min an excellent stability(>99.9%) in human and rat plasma.[13F]FET showed similar stabil-ity for 120 min (data not shown). The compounds were also stablein phosphate buffer solution over 120 min. With [F]FET, a veryslight polar radio-peak was observed after 60 min in human andrat plasma, but was deemed to be insignificant as the amountwas below 1%. In humans, [18F]FET was reported to be relativemetabolically stable with 95% and 87% intact tracer at 5 and120 min, respectively. 2.6. Determination of in vitro stability of L-3-FCBT in rathepatocytes The stability of the non-radioactive L-3-FCBT was tested in anin vitro experiment using hepatocytes isolated from Han Wisterrats. This assay is important in the early preclinical phase of drugdevelopment as hepatocytes play a major role in a variety ofmetabolic processes in particular the cytochrome P450 (CP450)system. The metabolic stability was examined by incubating L-3-FCBT with the hepatocytes and analyzing at different time-points(0, 2,8, 16,30,45 and 60 min) by HPLC-MS and the results areshown in Table 2. The results clearly show that the stability ofL-3-FCBT in this assay is excellent and warrants further investiga-tions in in vivo models. 3. Conclusion Figure 6. In vitro stability of L-3-[18F]FCBT in human plasma over 120 min; radioactivity signal of radio-HPLC (left) and radio-TLC (right). Rat data identical-not shown.耐士科技 www.rysstech.com 400-188-0725 Determination of in vitro metabolic stability in rat hepatocytes of non-radioactiveL-3-FCBT Incubation time (min) (%) Intact FCBT 0 100 2 100 8 99 16 98 30 97 45 95 60 94 4. Experimental 4.1. Chemistry All solvents and chemicals unless otherwise stated where pur-chased from Aldrich (Germany), Fluka (Switzerland), Acros Organ-ics (Belgium), ABCR (Germany) or Merck (Germany)andusedwithout further purification. The O-(2-fluoroethyl)-L-tyrosine(FET) reference compound was purchased from ABX GmbH (Ger-many). Silica chromatography was performed on Biotage IsoleraFlash Purification System from Biotage"(Sweden) with the corre-spondingSNAP cartridges (10-340 g silica gel); solvent gradientswere generated automatically, entering TLC values and were runingeneral from 5% of the weak solvent to 100% of the strong sol-ventin10 column volumes. Thin-layer chromatography (TLC)wascarried out with ssilica gel plates (TLC Silica gel 60 F254,20 ×20mm) from Merck (Germany) visualized under UV at254nm or withKMnostaining. HPLC system used an Agilent1100 and 1200HPLC system with a binary pump, autosamplerand diode array detector (DAD) and an attached gamma-detector(NaI) GABI from Raytest (Germany), using the Agilent ‘ChemSta-tion'software. Analytical HPLC column ACE C18 (50×4.6 mm)was obtained from ACE (Great Britain), the Astec Chirobiotic T Chi-ral HPLC ccolumn (250x4.6 mm) from Sigma-Aldrich (Germany).Semi-preparative Knauer HPLC system consisted of a Knauerpump,, UV detector and an attached gamma-detector (NaI) GABItromRaytest using the Knauer ‘ChromGate’software. Semi-preparative HPLC was performed with Phenomenex SynergiHydro-RP 4p(250x10 mm) from Phenomonex (USA). Massspectra (MS) were obtained on a ZQ 4000 LC and LCT TOF massspectrometer from Waters/Micromass (USA) for electrosprayionization (ESI) and a DSQ CTC from Thermo Scientific (USA) for chemical ionization (CI). 1H and 19F nuclear magnetic resonance(NMR) spectra were recorded with the Bruker AVANCE-300 and400 MHz and the Bruker DRX-600 NMR spectrometer. 4.2. cis-3-(Benzyloxy)cyclobutanol (5) 3-(Benzyloxy)cyclobutanone 4 was prepared according to themethod described earlier.15.23,24 To a suspension of 4 (10.4g;59.0 mmol) in ethanol (100 mL) was added sodium borohydride(2.45 g; 64.9 mmol) in portions over 30 min. The mixture was stir-red at 0 ℃ for 3 h and monitored by TLC upon reaction completion(ethyl acetate/hexane 1:1). The pale yellow mixture was passedthrough a pad of Celite@, concentrated and the residue was re-dissolved in methanol (CARE: gas evolution), then neutralized with3 M HCl and concentrated in vacuo.To the residue was added water(80 mL) and extracted with diethyl ether (80 mL). The combined or-ganic phases were dried with anhydrous sodium sulfate (Na2SO4),filtered and concentrated in vacuo. The crude product was purifiedby silica chromatography with a gradient of ethyl acetate and hex-ane. Product fractions were combined and concentrated to give 5(8.97 g, 86%) as a clear colorless oil. TLC (ethyl acetate/hexane 1:1,Rr~0.28). H NMR (400 MHz, CDCl3): 8 ppm 1.87 (d,J=6.32 Hz,OH) 1.90-2.00 (m, 2H, CH) 2.68-2.79 (m, 2H, CH) 3.64 (quin,J=6.95 Hz, 1H, CH)3.92 (sxt,J=6.97 Hz, 1H, CH) 4.43 (S,2H, CH2)7.28-7.40 (m,5H,CHarom). 13cNMR(101MHz, CDCl3): 8 ppm 39.6(CH2) 41.4 (CH2) 59.8 (CH) 64.4 (CH) 70.4(CH2)127.7 (CH) 127.9(CH) 127.9(CH) 128.5 (CH) 138.1 (C).ESI-MS: m/e 179[M+H]*. 4.3. cis-3-(Benzyloxy)cyclobutyl toluene-4-sulfonate (6) 4.4. cis-3-Hydroxycyclobutyl toluene-4-sulfonate (7) A solution of cis-3-(benzyloxy)cyclobutyl toluene-4-sulfonate 6(1.8 g; 5.42 mmol) in absolute ethanol (35 mL) was stirred at roomtemperature with palladium on charcoal 10% (1.02g; 0.96 mmol)and a positive pressure of hydrogen (balloon) for 4 h. After reactioncompletion, the catalyst was removed by filtration over Celiteandconcentrated in vacuo to give the desired product 7 (1.3 g, 96%) asa yellow oil which was used without further purification. TLC(ethyl acetate/hexane 1:2, Rr~0.11).H NMR (300 MHz, CDCl3): 8ppm 1.73 (br s, QH) 2.06-2.19 (m, 2H, CH) 2.46 (s, 3H, CH3)2.64-2.78 (m,2H, CH) 3.92 (quin, J=7.02 Hz, 1H, CH) 4.42 (quin,J=7.06 Hz, 1H, CH) 7.35 (d, J=7.91 Hz, 2H, CHarom) 7.79 (d,J=8.10 Hz,2H, CHarom). 4.5.cis-Cyclobutane-1,3-diyl bis(toluene-4-sulfonate) (8) ( A solution o f c is-3-hydroxycyclobutyl toluene-4-sulfonate 7 (4.29g; 17.7 mmol) in dichloromethane was c ooled to 0 C. ) Pyridine (2.9 mL) was added, followed by the addition of p-tolu-enesulfonic anhydride (8.67g; 26.6mmol). The mixture wasstirred 72 h at room temperature. The reaction mixture was con-centrated to dryness and re-suspended in diethyl ether (75 mL).The suspension was washed with 0.5 M HCl, saturated sodiumbicarbonate solution and brine. The mixture was dried over sodiumsulfate, filtered and concentrated in vacuo. The crude product waspurified by silica chromatography eluting with ethyl acetate/hex-ane (1:6) to give pure 8 (5.35 g, 64%) as a white solid. H NMR(600 MHz, CDCl3): 8 ppm 2.29-2.37 (m, 2H, CH), 2.45 (s, 6H,CH3), 2.60-2.68 (m, 2H, CH), 4.40 (quin, 2H, CH), 7.33 (d, 4H,CHarom), 7.71-7.75(m, 4H, CHarom). 13c NMR (151MHz, CDCl3): 8ppm 21.7 (CH3), 39.1 (CH2), 39.1 (CH), 127.8 (CH),130.0(CH),133.4 (C)145.2 (C). CI-MS:m/e 396 [M]*. 4.6. trans-3-(Fluorocyclobutyl) benzyl ether (9) To an ice-cooled solution of cis-3-(benzyloxy)cyclobutanol 5(0.9g; 5.54mmol) in dry dichloromethane (25 mL)) was addeddiethylaminosulfur trifluoride (0.86 mL; 6.54 mmol). The mixturewas stirred for 2 h at O Cunder nitrogen and was allowed to warmto room temperature overnight. The yellow-brown reaction mix-ture was washed with water (20 mL), the organic layer was sepa-rated and the aqueous layer was extracted with dichloromethane(2x20mL). The organic layers were combined, dried with sodiumsulfate, filtered and concentrated in vacuo. The residue was puri-fied by silica chromatography with a gradient of ethyl acetate/hex-ane to give pure 9 (306 mg, 37%) as a clear colorless oil. TLC (ethylacetate/hexane1:2, Rr~0.67). H NMR (300 MHz, CDCl3): 8 ppm2.34-2.63 (m, 4H, CH), 4.31-4.41 (m, 1H, CH), 4.43 (s, 1H, CH),5.14-5.19 (m,0.5H, CH-F), 5.28-5.38 (m, 0.5H, CH-F), 7.28-7.58(m, 5H, CHarom). 19F'F NMR (400 MHz, CDCl3): 8 ppm -176.4. ESI-MS: m/e 254 M+H 4.7. ttrans-3-Fluorocyclobutyl toluene-4-sulfonate (11) A solution of trans-3-(fluorocyclobutyl)benzyl ether 9 (152 mg;0.84mmol) in methanol (10 mL) was stirred with 10% palladiumon charcoal (140 mg) and with a positive pressure of hydrogen(balloon) at room temperature. The mixture was filtered and thesolvent was partly evaporated in vacuo (40℃/200 mbar, volatile!)to give 10 as a clear colorless oil with traces of methanol. The prod-uct was used without further purification due to the low boilingpoint. TLC (ethyl acetate/hexane 1:2, R ~0.26; stained withKMnO4). 1H NMR (400 MHz, CDCl3): 8 ppm 2.22-2.39 (m, 2H,CH), 2.48-2.63 (m, 2H, CH), 4.59-4.72 (m, 1H, CH), 5.15-5.39(m,1H, CH), OH not visible; 19F NMR(376 MHz, CDCl3): 8 ppm-178.3. A solution of trans-3-fluorocyclobutanol 10 (50 mg;0.56 mmol)in dry dichloromethane (5 mL) was cooled to 0℃ and pyridine(82pl; 1.0 mmol) was added, followed by p-toluenesulfonic anhy-dride (201 mg; 0.62 mmol). The mixture was stirred for 5 h at 0℃under nitrogen and was allowed to warm to room temperatureovernight. The yellow solution was concentrated in vacuo. Theresulting residue was diluted with HC1 0.5 M (5 mL), extracted withdiethyl ether (10 mL) and the organic layer was washed with satu-rated sodium bicarbonate and brine. The mixture was dried with so-dium sulfate, filtered and concentrated in vacuo. The crude oil waspurified by silica chromatography, eluting with a gradient of ethylacetate/hexane to give 11 (59 mg, 14%, over 2 steps) as a clear color-less oil. TLC(ethyl acetate/hexane 1:2,Rr~0.52).HNMR(400MHz,CDCl3): 8 ppm 2.47 (s, CH3), 2.48-2.62 (m, 4H, CH), 5.03-5.08(m,1H, CH), 5.10-5.19 (m, 0.5H, CH-F), 5.24-5.33 (m, 0.5H, CH-F),7.36 (d,J=8.10 Hz, 2H, CHarom), 7.79 (d,J=8.29 Hz, 2H, CHarom).3c NMR (100 MHz, CDCl3): 8 ppm 21.7 (CH3), 38.8 (CH2), 72.3(CH), 85.7 (CH), 127.9 (CH), 129.9 (CH), 133.5 (C), 145.1 (C).19FNMR(376 MHz, CDCl3): 8 ppm-178.8.ESI-MS:m/e 245 [M+H]t. 4.8. Methyl O-[trans-3-(benzyloxy)cyclobutyl]-N-(tert-butoxycarbonyl)-L-tyrosine (13) To a solution of Boc-Tyr-OMe L-12 (1.02 g; 3.35 mmol) and cis-3-(benzyloxy)cyclobutanol 5 (1.33 g; 7.37 mmol) in dry DMF(25mL) was added diethyl diazocarboxylate (1.20 mL; 7.37 mmol).The yellow solution was stirred under nitrogen and triphenylphos-phine (1.98 g; 7.37 mmol) was added. The mixture was stirred atroom temperature for 23 h and concentrated in vacuo.The crudeoil was dissolved in chloroform (50 mL) and washed with water(3×30mL) to remove dimethylformamide. The organic layerwas dried with sodium sulfate, filtered and concentrated in vacuoto give a brown oil. The crude product was purified by silica chro-matography, eluting with a gradient of ethyl acetate/hexane to givethe L-13 (1.35 g, 88%) as a clear colorless oil. TLC (ethyl acetate/hexane 1:2, Rr~0.46). 'H NMR (400 MHz, CDCl3):8 ppm 1.43(s, 9H, CH3), 2.38-2.56 (m, 2H, CH), 2.94-3.11 (m, 2H, CH), 3.72(s, 3H, CH3), 4.30-4.39 (m, 1H, CH), 4.46 (s, 2H, CH2), 4.50-4.60(m, 1H, CH), 4.78-4.88 (m, 1H, CH), 4.90-5.00 (m, 1H, NH), 6.71(d, 2H, CHarom), 7.02 (d, 2H, CHarom), 7.29-7.42 (m, 5H, CHarom).CI-MS: m/e 356 [M-Boc]t. 4.9. Methyl N-(tert-butoxycarbonyl)-0-(trans-3-hydroxycyclobutyl)-L-tyrosine (14) A solution of methyl O-[trans-3-(benzyloxy)cyclobutyl]-N-(tert-butoxycarbonyl)-L-tyrosine 13 (1.35g; 2.96 mmol) in methanol(20 mL) was stirred with 10% palladium on charcoal(500 mg,50% wet). The mixture was stirred under a positiveeppressure ofhydrogen (balloon) at room temperature for 2 h..TThe mixturewas filtered and the solvent concentrated in vacuo. The crude oilwas ddissolved in dichloromethane, filtered through Celite",washed with dichloromethane and concentrated in vacuo to give14(1.02 g, 94%) as a clear colorless oil which was used without fur-ther purification. TLC (ethyl acetate,Rf~0.54).HNMR (400 MHz,CDCl3): 8 ppm 1.42 (s, 9H, CH3), 1.80(br s,1H, OH), 2.34-2.59 (m,2H, CH), 3.02 (m, 2H, CH), 3.72 (s,3H, CH3), 4.47-4.59 (m, 1H, CH),4.60-4.70(m, 1H, CH), 4.79-4.90 (m, 1H, CH), 4.96 (d,J=7.91 Hz,1H, NH), 6.71 (d, 2H, CHarom), 77.02 (d, 2H, CHarom). ESI-MS: m/e366 [M+H]*. 4.10. Methyl N-(tert-butoxycarbonyl)-O-(cis-3-fluorocyclobutyl)-L-tyrosine (15) To an ice-cooled solution of methyl N-(tert-butoxycarbonyl)-0-(trans-3-hydroxycyclobutyl)-l-tyrosine 14 (658 mg; 1.80 mmol) indry dichloromethane((25mL) wass added DAST (358pL;2.70 mmol). The mixture was stirred at 0°C for 3 h and was al-lowed to reach room temperature overnight. The crude productwas concentrated in vacuo and purified by column chromatogra-phy with a gradient of ethyl acetate/hexane to give 15 (257 mg,38%) as white crystals. TLC (ethyl acetate/hexane 1:2, Rf ~0.62).'H NMR (300 MHz, CDCl): S ppm 1.42 (s, 9H, CHg), 2.36-2.53(m, 2H, CH), 2.95-3.08 (m, 2H, CH), 3.72 (S, CH3), 4.17-4.27 (m,1H, CH), 4.50-4.60 (m, 1H,CH), 4.81 (quin,0.5H, CH-F), 4.94 (quin,0.5H, CH-F), 4.96 (d,J=7.91Hz, 1H, NH), 6.73 (d,J=8.59 Hz, 2H,CHarom), 7.03 (d,J=8.59 Hz, 2H, CHarom). 19F NMR (376 MHz,CDCl3): 8 ppm1-169.3. ESI-MS: m/e 368 [M+H]*. 4.11. N-(tert-Butoxycarbonyl)-O-(cis-3-fluorocyclobutyl)-L-tyrosine (16) To a solution of methyl N-(tert-butoxycarbonyl)-O-(cis-3-flu-orocyclobutyl)-L-tyrosine 15 (85mg;23 umol) in methanol(3.5mL) was added 1 M lithium hydroxide (650pL; 65umol).The clear mixture was stirred for 6 h at room temperature. After TLC showed full conversion, the pH of the mixture was adjustedto pH 5 with 1 M HCl and concentrated in vacuo. The resultingoil was dissolved in ethyl acetate, washed with brine and evapo-rated to dryness, was then re-dissolved in ethyl acetate, dried withsodium sulfate, filtered and concentrated in vacuo to give 16(80 mg, 97%) as a white solid. TLC (ethyl acetate/hexane 1:1, spotat start). H NMR (400 MHz, CDCl3): 8 ppm 1.41 (s, 9H, 3 CH3),2.33-2.51(m, 2H, CH), 2.75 (d, 1H, CH), 2.90 (dd, 2H, CH), 3.12(d, 2H, CH), 4.16-4.27 (m, 1H, CH), 4.52 (d,J=5.56 Hz, 1H, CH),4.76 (quin, 0.5H, CH-F), 4.90 (quin, 0.6H, CH-F), 4.98 (d,J=7.58Hz, 1H, NH), 6.05 (br s, COOH), 6.74 (d,J=8.34Hz, 2H,CHarom), 7.09 (d, J=8.34 Hz, 2H, CHarom). 19F NMR (376 MHz,CDCl3): 8 ppm -169.2. ESI-MS: m/e 354 [M+H]*. 4.12. 0-(cis-3-Fluorocyclobutyl)-L-tyrosine hydrochloride salt(L-17) A solution of N-(tert-butoxycarbonyl)-O-(cis-3-fluorocyclobu-tyl)-L-tyrosine 16 (79 mg; 0.22 mmol) in DMF (0.6 mL) was treatedwith 4 M HCl in dioxane (0.5 mL; 2.24 mmol) for 1 h at room tem-perature. The mixture was concentrated in vacuo, re-suspended inethanol/water (1:1) and extracted with a small amount of dichlo-romethane. To reduce the organic solvent, the aqueous phasewas concentrated and gave after freeze-drying the product 1725mg, 44%) as a white solid. TLC (DCM/MeOH 9:1, spot at start).HNMR (300 MHz, CD30D): ppm 2.19-2.36(m, 2H,CH), 2.95-3.12 (m, 2H, CH), 3.24-4.16 (m, 1H, CH), 4.27-4.37 (m, 1H, CH),4.84 (quin, 1H,CH-F), 4.92 (quin, 1H, CH-F), 6.85 (d, 2H, CHarom),7.20(d, 2H, CHarom), COOHH and NH2 not visible. 13c NMR(151MHz, CD30D):8 ppm 36.7 (CH2), 40.3(CH2), 40.5 (CH2),55.8(CH), 63.6 (CH), 63.8 (CH), 81.0 (CH), 82.4 (CH), 116.6 (CH),128.1(CH), 131.7(CH), 158.3 (C), 171.8 (C). ESI-MS: m/e 254[M+H]*. HPLC (chiral): ratio D/L 3%: 97%, rt=3.84 min. 4.13. 0-(cis-3-Fluorocyclobutyl)-D-tyrosine hydrochloride salt(D-17) O-(cis-3-Fluorocyclobutyl)-p-tyrosine hydrochloride salt D-17was synthesized according to the procedure described for its L-iso-mer L-17. 4.13.1. Methyl O-[trans-3-(benzyloxy)cyclobutyl]-N-(tert-butoxycarbonyl)-D-tyrosine (D-13) Clear oil, 1.29 g(3.81 mmol) of D-12 gave D-13 (1.16 g, 72%).TLC (ethyl acetate/hexane 1:2, Rr ~0.39). 1H NMR (300MHz,CDCl3): 8 ppm 1.43 (s, 9H, CHz), 2.37-2.58 (m, 4H, CH), 2.92-3.11(m,3H, CH3), 3.72 (s, 3H, CH3), 4.30-4.39 (m, 1H, CH), 46 (s, 2H,CH2), 4.54 (m,J=7.16 Hz, 1H, CH), 4.79-4.88 (m, 1H, CH), 4.96(d, J=7.91 Hz, 1H, NH), 6.71(d,J=8.48 Hz, 2H, CHarom), 7.02 (d,J=8.48 Hz, 2H, CHarom), 7.28-7.41 (m, 5H, CHarom). ESI-MS: m/e456[M+H]*. 4.13.2. Methyl N-(tert-butoxycarbonyl)-O-(trans-3- hydroxycyclobutyl)-D-tyrosine (D-14) Clear oil, 1.52 g (2.96 mmol) of D-13 gave D-14 (1.35 g, 97%).TLC (DCM/MeOH 9:1, Rf ~0.59). H NMR (300MHz, CDCl3): 8ppm 1.42 (s, 9H, CH3), 1.63 (br s, 1H, OH), 2.34-2.57 (m, 4H, CH),2.93-3.11 (m, 2H, CH2), 3.72 (s, 3H, CH3), 4.48-4.59 (m, 1H, CH),4.59-4.70 (m, 1H, CH), 4.80-4.89 (m, 1H, CH), 4.97 (d,J=8.10 Hz,1H, NH), 6.71(d,J=8.67 Hz, 2H, CHarom) 7.02 (d,J=8.48 Hz, 2H,CHarom). ESI-MS: m/e 366 [M+H]*. 4.13.3.Methyl N-(tert-butoxycarbonyl)-O-(cis-3- fluorocyclobutyl)-D-tyrosine (D-15) White solid, 147 mg (4.16 mmol) of D-14 gave ofD-15 (1.52 mg,8%). TLC (ethyl acetate/hexane 1:2, Rr~0.62). H NMR (300 MHz, 400-188-0725 CDCl3): 8 ppm 1.43 (S, 9H, CH3) 2.37-2.52 (m, 4H, CH) 2.94-3.10(m, 2H, CH2) 3.72 (s, 3H, CH3) 4.17-4.26 (m, 1H, CH) 4.51-4.59(m, 1H, CH) 4.72-4.93 (m, 1H, CH) 4.94-4.99 (m, 1H, NH) 6.73(d, J=8.59 Hz, 2H, CHarom) 7.03 (d,J=8.34Hz, 2H, CHarom). FNMR (376 MHz, CDCl3): 8 ppm -169.3. ESI-MS: m/e 368 [M+H]*. White solid, 19 mg (56 umol) of D-15 gave D-16 (21 mg, 95%).TLC (ethyl acetate/hexane 1:1, spot at start).H NMR (300 MHz,CD30D): 8 ppm 1.38 (s, 9H, CH3), 2.17-2.37 (m, 2H, CH), 2.76-2.88 (m, 1H, CH), 2.93-3.04 (m, 1H, CH), 3.04-3.12 (m, 1H,CH),4.28 (d,J=4.04Hz, 2H, CH2), 4.71-4.81 (m, 1H, NH), 6.76 (d,J=8.59 Hz, 2H, CHarom), 7.13 (d,J=8.59 Hz, 2H, CHarom). 19FNMR(376 MHz, CD3OD):8 ppm-170.5. 4.13.5.O-(cis-3-Fluorocyclobutyl)-D-tyrosine hydrochloride salt(D-17) White solid, 14 mg(48 umol) of D-16 gave D-17 (17 mg, 92%).TLC (DCM/MeOH 9:1, spot at start). H NMR (400 MHz, CD30D):ǒ ppm 2.17-2.36 (m, 2H, CH), 2.94-3.15 (m, 2H, CH), 3.19-3.28(m, 1H, CH), 4.17 (dd,J=7.58,5.31 Hz, 1H, CH), 4.32 (m, 1H, CH),4.77 (quin, J=6.57 Hz, 1H, CH), 6.85 (d,J=8.59 Hz, 2H, CHarom),7.20 (d,J=8.59 Hz, 2H, CHarom). 19F NMR (376 MHz, CD30D):ppm -170.5. ESI-MS: m/e 254 [M+H]*. HPLC (chiral): ratio D/L96%: 4%, rt=5.43 min.30 4.14. Radiosynthesis The aqueous n.c.a. [18F]fluoride from the cyclotron target (Eck-ert & Ziegler in Adlershof, Berlin, Germany) was separated from the[180]water with a QMA Sep-Pak" cartridge and collected into a5 mL Wheaton glass V-vial (Wheaton Industries Inc., Millville,USA) containing a solution of Kryptofix" 2.2.2 (5 mg; 13 umol)and K2CO3 (1 mg; 15 umol) in acetonitrile (0.95 mL) and water(0.05mL). The solvents were evaporated and the residue dried at110°C under a light nitrogen stream, more anhydrous acetonitrile(ACN) was added, and the drying process was repeated. The pre-cursor cis-cyclobutane-1,3-diylbis(toluene-4-sulfonate)8 (5mg;13 umol) in anhydrous ACN(0.5 mL) was added to the reaction vialcontaining the dry [18F]fluoride-cryptate complex and the reactionmixture was stirred at 130 C for 15 min in a sealed vial. The crudeintermediate was purified by passing through a Waters C18 lightSep-Pak"(equilibrated with 5 mL ethanol followed by 5 mL waterand dried by passing 20 mL air through), washing with water(5 mL) and eluted with ACN (0.8 mL) and the solvent was evapo-rated at 110℃ with a nitrogen stream. The trans-3-[18F]fluorocyc-lobutyl toluene-4-sulfonate [18F]11 was diluted with DMSO (1 mL)and added to L-tyrosine (~5 mg;28 umol) with aqueous 10% so-dium hydroxide (2 equiv, 22.1 uL). The reaction was heated at150℃ for 10 min, cooled to 501C and quenched by addition ofwater acidified with 1 M HCl to pH 2 (4mL). The crude productwas purified by semi-preparative HPLC (Synergi Hydro RP 4p,250×10 mm; 15% acetonitrile in water acidified with 1 M HCl topH2; flow 3 mL/min). The product peak was collected, diluted withwater acidified with 1 M HCl to pH 2 (25 mL) and trapped on aWaters C18 environmental cartridge (equilibrated with 10 mL eth-anol, 10 mL acidified water and the cartridge dried by passing20 mL air through). The cartridge was washed with water acidifiedwith 1 M HCl to pH2 (5 mL) and the product was eluted with frac-tions (0.5 ml) of 1:1 mixture of ethanol and phosphate bufferedsaline (total volume: 1.5 mL). Starting from 881 MBq[F]fluoride,44 MBq (12%, decay corrected) of the desired product ([F]17) wasobtained in excellent radiochemical purity (>99%) and within144 min. Specific activity was found to be 25 GBq/umol. Radio-chemical and chemical purity were determined by analytical radio-HPLC (UV at 280nm))usinga ACEC18 column(50x4.6 mm) at a flow rate of 2 mL/min using a gradient(0-7 min) from ACN +0.1% TFA/water +0.1% TFA 5:95 (v/v) to 95:5(v/v). The retention time of [18F]17 was 2.4-2.7 min. [F]FET was synthesized via a 2 step reaction. In the first step,2-bromoethyl 4-nitrobenzenesulfonate(10 mg; 49 umol))ino-dichlorobenzene (0.5 mL) was reacted with the dry[lF]fluoride-cryptate complex (preparation mentioned above) for 10 min at120 °C in a sealed 5 mL Wheaton glass V-vial followed by distilla-tion of the product 1-bromo-2-[1F]fluoroethane at 120℃ forabout 20 min into a receiver vial containing DMSO (0.5 mL).36The 2-bromoethyl 4-nitrobenzenesulfonate was synthesizedaccording to literature3using 4-nitrobenzenesulfonyl chloride inpresence of 1,2,2,6,6-pentamethylpiperidine. L-Tyrosine(5mg;28 umol) dissolved in anhydrous DMSO (0.2 mL) and 2M NaOH(27.6uL; 55 umol) were added to the receiver vial and heated at120°C for 15 min in a sealed vial. The reaction mixture was dilutedwith water (4mL) and purified by semi-preparative HPLC (ACE 5C18, 250×4.6 mm; 15% acetonitrile +0.1% TFA in water +0.1%TFA; flow 3 mL/min). The product peak (retention time ~15 min)was collected, diluted with water acidified with 1 M HCl to pH 2(35mL) and trapped on a Waters C18 plus cartridge (equilibratedwith 5 mL ethanol, 10 mL acidified water and the cartridge driedby passing 20 mL air through). The cartridge was washed withwater acidified with 1 M HCl to pH 2 (5 mL) and the product waseluted with fractions (0.5 ml) of 1:1 mixture of ethanol and wateracidified with 1 M HCl to pH 2. Starting from 2889 MBq[F]fluo-ride, 136 MBq (12%, decay corrected) of the desired product wasobtained in excellent radiochemical purity (>99%) and within160 min. 4.15.Determination of the lipophilicity of the non-radioactiveL-3-FCBT The logD assay was performed manually in two single vialsL-3-FCBT (0.1 mg) was dissolved in 600 uL octanol saturated with10 mM phosphate buffer pH 7.4 (with the help of sonication for3 min) and equilibrated by shaking for 4 min at a constant temper-ature (20C). Phases were separated by centrifugation at 10 min(20℃,3000 rpm). The sample was analyzed by a Waters Alliance2695 HPLC with a Xterra MS C18 2.5 um column (4.6×30mm)with a gradient of 5-65% ACN +0.01% TFA in water +0.01% TFA in3 min, hold for 2 min and a flow rate of 1.5 mL/min. The sampleswere quantified at 254nm by area integration. 4.16. Cell uptake studies The A549 and DU145 cells were cultured in Dulbecco's ModifiedEagle’s Medium (DMEM) (invitrogen) supplemented with 10% fetalbovine serum (FBS) and the NCI-H460 were cultured in DMEM/F-12 (1:1) (invitrogen) supplemented with 10% FBS. The cells wereseeded 1-2 days prior to the assay and grown until sub-confluencyin 48 well plates.Prior to the assay, the cell culture medium wasremoved and the cells were washed with 500 pL phosphate buf-fered saline (PBS). After adding 250 uL of PBS +0.1% Bovine serumalbumin (BSA) containing 0.2 MBq of L-3-[1F]FCBT in 8% EtOH perwell, the cells were incubated at 37℃ in a humidified atmosphere(5% CO2) for 10, 20, 30 and 60 min. For the blocking experiment,the cells were co-incubated with non-radioactive FET (1 mM) andnon-radioactive L-3-FCBT (1 mM) for 30 min to monitor radioactiv-ity uptake. To stop tracer uptake after the time point,the incuba-tion buffer was removed, the cells were washed with 500 uL PBSand lysed with 500 pL 1 M NaOH. Subsequently, the amount ofradioactivity in the cell lysate was determined in a Perkin Elmer1480 Wizard 3" Automated Gamma Counter. Aliquots of theapplied tracer amount were measured in a gamma counter to determine the total amount in counts per minute (cpm) togetherwith the samples to correct for radioactivity decay. Cell numbersper well were determined after detaching cells by trypsinizationin 3 wells prior to start of the assay and counted with the Invitro-gen Automated Cell Counter. The mean number ofcells was calcu-lated and normalized to 105 cells. The assay was performed intriplicates. 4.17. Determination of in vitro plasma stability The tracers L-3-[18F]FCBT[18F]17 and [18F]FET were incubatedin plasma of human and rats at 37℃ for different time periods(0, 30, 60,90 and 120 min) at a concentration of 22 MBq/mL. Sam-ples were precipitated with MeOH and subsequently centrifugedfor 15 min at 3000 rpm and the supernatant was analyzed withan Agilent 1200 HPLC-system with a Raytest radio-detection withthe column Phenomenex Luna 5 um C18100 A LC column(250×4.6 mm) with a gradient of 5-95% ACN in water in 10 minand a flow rate of 1 mL/min and by TLC using the solvent Buta-nol/AcOH/H20 12:3:5 for developing the TLC plate.The stabilityof the test compound was quantified by comparison of the remain-ing amount at the different time points with the amount of the0 min sample (control) and is expressed in% of initialconcentration. 4.18.Determination of in vitro metabolic stability in rathepatocytes Acknowledgments We are grateful to Sylvia Zacharias (Bayer Healthcare Pharma-ceuticals) for her assistance with the biological studies and UrsulaMoenning (Bayer Healthcare Pharmaceuticals) for the in vitrohepatocytes stability study. ( 1 . Kumar, G . N.; S urapaneni, S. Med. R es. Rev. 2001, 21, 397. ) 2.Smith, D. A.; van de Waterbeemd, H.; Walker, D. K. In Pharmacokinetics andMetabolism in Drug Design; Mannhold, R., Kubinyi, H., Folker, G., Eds.; Wiley-VCH: Weinheim, 2001; Vol. 31, pp 99-122. ( 3. Zoghbi, S.; S hetty, H .; I chise, M.; F ujita, M.; I maizumi, M.; Liow, J; Sh a h, J.;Musachio, J.; P ike, V.; Innis, R. J. Nucl. Med . 2006,4 7 ,52 0 . ) ( 4. . M M ulholland, G.; W ieland, D.; Kilbourn, M.; F rey, K.; Sherman, P.; C arey,J; Kuhl,D. Synapse 1998, 30,263. ) ( 5. Holtke, C.; L aw, M.; Wagner, S.; K o pka, K.;Fa u st, A.; Breyholz, H. ; Schober, O. ; ) Bremer, C.; Riemann, B.; Schafers, M. Bioorg. Med. Chem. 2009, 17,7197. 6.Guengerich, P. F. Chem. Res. Toxicol. 2001, 14,611. 7. Dedeurwaerdere,S.; Gregoire, M.-C.; Vicash, L.; Roselt, P.; Binns, D.; Fookes, C.;Greguric, I.; Pham, T.; Loc’h, C.; Katsifis, A.; Hicks, R. J.; O'Brien, T.; Myers, D. E.Eur. J. Nucl. Med.Mol. Imaging 2009, 36, 958. 8.Luurtsema, G.; Schuit, R.; Takkenkamp, K.; Lubberink, M.; Hendrikse, NN.Windhorst, A.; Molthoff, C.; Tolboom, N.; van Berckel, B.; Lammertsma, A. NuclMed. Biol. 2008,35,869. 9. Sykes, T.; Ruth, T.; Adam, M. Nucl. Med. Biol. 1986,13,497. ( 10. D I ounay, A . ; Barta, N. ; Bikker, J; Borosky, S.; Campbell, B.; Crawford , T.;Denny, L .; Evans, L .; Gray, D.; Lee, P. Bioorg. Med. C hem. L e tt. 2009, 19,1 1 5 9 . ) 11. Manoury, P. M.; Binet, J; Rousseau, J.; Lefevre-Borg, F.; Cavero,I. J. Med. Chem.1987,30,1003. 12. Sorensen, B.; Rohde,J.; Wang,j; Fung, S.; Monzon, K.; Chiou, W.; Pan, L.; Deng,X.; Stolarik, D. A.; Frevert, E. U. Bioorg. Med. Chem. Lett.2006, 16,5958. 13. McConathy,J.; Goodman, M. M. Cancer Metastasis Rev.2008,27,555. Shoup, T. M.; Goodman, M. M. J. Labelled Compd. Radiopharm. 1999,42, 215.14McConathy,J.; Voll, R. J.; Yu, W.; Crowe, R. J.; Goodman, M. M. Appl. Radiat. Isot.2003,58,657. Schuster, D. M.; Votaw, J. R.; Nieh, P.T.; Yu, W.;Nye, J. A.; Master, V.; Bowman,F. D.; Issa, M. M.;Goodman, M. M. J. Nucl. Med. 2007, 48,56. Yu, W.; Williams, L.; Camp,V. M.; Malveaux, E.; OIson, J.J; Goodman, M. M.Bioorg. Med. Chem. 2009, 17,1982. 18.Wester, H. J.; Herz, M.; Weber, W.; Heiss, P.; Senekowitsch-Schmidtke, R.;Schwaiger, M.; Stocklin, G. J. Nucl. Med. 1999,40,205. 19.Tsukada, H.; Sato, K.; Fukumoto, D.; Kakiuchi, T. Eur. J. Nuc. Med. Mol. Imaging2006, 33,1017. 20. ULrakami, T.; Sakai, K.; Asai, T.; Fukumoto, D.; Tsukada, H.; Oku, N. Nucl. Med.Biol. 2009.36,295. 21. Fraga, S.; Pinho, M., Soares-Da-Silva, P. Amino Acids 2005, 29, 229. 22. Kanai, Y., Endou, H. Curr. Drug Metab.2001,2,339. 23. Avram, M., Nenitzescu, C.; Maxim, M. Ber. 1957,90,1424. 24. OCgura, K.; Yamashita, M.; Suzuki, M.; Furukawa, S.; Tsuchihashi, G. Bull. Chem.Soc. Jpn. 1984, 571637. 25.i. Pan, D.; Gambhir, S. S.; Toyokuni, T.; Iyer, M. R.; Acharya, N.; Phelps, M. E.;Barrio, J. R. Bioorg. Med. Chem. Lett. 1998,8,1317. 26.Kim, D. W.; Jeong, H.J.; Lim, S. T.; Sohn, M.-H. Angew. Chem., Int. Ed.2008, 47,8404. ( 2 7. W ilkinson, J. A. Chem. Rev. 1992, 92, 5 05. ) 28.James, M. L.; Fulton,R. R.; Vercoullie,J.; Henderson, D. J.; Garreau,L.; Chalon, S.;Dolle, F.; Costa, B.; Selleri, S.; Guilloteau, D.; Kassiou, M. J. Nucl. Med. 2008, 49,814. 29. Lampe,J.; Floor, S.; Gross,J.;Nishida,C.; Jiang, Y.; Trnka,M.; de Montellano, P.J.Am. Chem. Soc. 2008, 130,16168. 30. (Chiral HPLC was performed on an Astec Chirobiotic T Chiral HPLC column(250×4.6 mm, Sigma-Aldrich, Germany) using isocratic solvent 70% EtOH/30% water with a flow of 1 mL/min. Retention time of l-isomer: 3.84 min; D-isomer 5.43 min. 31. Tang, G.; Tang, X.; Wang, M.; Luo, L.; Gan, M.; Huang, Z. J. Labelled Compd.Radiopharm. 2003, 46, 661. 32. Wagner, S.; Law, M.; Riemann, B.; Pike, V.; Breyholz, H.; Holtke, C.; Faust, A.;Renner, C.; Schober, O.; Schafers,M.J. Labelled Compd. Radiopharm. 2006,49,177. 33. ChemDraw, version 12, CambridgeSoft; ACD ChemSketch, version 12.0,Advanced Chemistry Development Inc.; MarvinSketch, version 5.10.0,ChemAxon. 34. The tumor cells (A549, NCI-H460 and DU145) were co-incubated with 1 mM ofnon-radioactive FET and with 1mM non-radioactive L-3-FCBT at 37°C for30 min. 35.PFauleit, D.;Floeth, F.; Herzog, H.; Hamacher, K.; Tellmann, L.; Muller, H.-W.;Coenen, H. H.; Langen, K.-J. Eur.J. Nucl. Med. Mol. Imaging 2003, 30,519. 36. Zuhayra, M.; Alfteimi, A.; Papp, L.; Lutzen, U.; Lutzen, A.; van Forstner, C.;Meller, B.; Henze, E. Bioorg. Med. Chem. 2008, 16,9121. ( 37. T ada, M.; Shijima, H.; Nakamura, M. Org. Biomol . Chem. 2003 , 1,2499. ) ww.rysstech.com耐士科技 www.rysstech.com耐士科技
确定








还剩8页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中主要物质含量分析检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多