








Gyrase ATPase domain, the pharmaceutical underexploited segment of DNA gyrase, the sole Type II topoisomerase present in Mycobacterium tuberculosis represents an attractive target for anti-tubercular drug discovery. Here we report, the development of a novel series of MTB DNA gyraseB inhibitor identified through a medium throughput screening (MTS) of BITS in-house chemical library (3000 compounds). The MTS hit was further remodeled by chemical synthesis to identify the most potent analogue 27 exhibiting an in vitro gyrB inhibitory IC50 of 0.15 lM. The series also demonstrated well correlating gyrase super coiling activity and in vitro anti-mycobacterial potency against MTB H37Rv strain. Furthermore the compounds displayed good safety profile in their subsequent cytotoxicity and hERG toxicity
evaluations, to be worked out from a pharmaceutical point of view as potential anti-tubercular agents.
方案详情

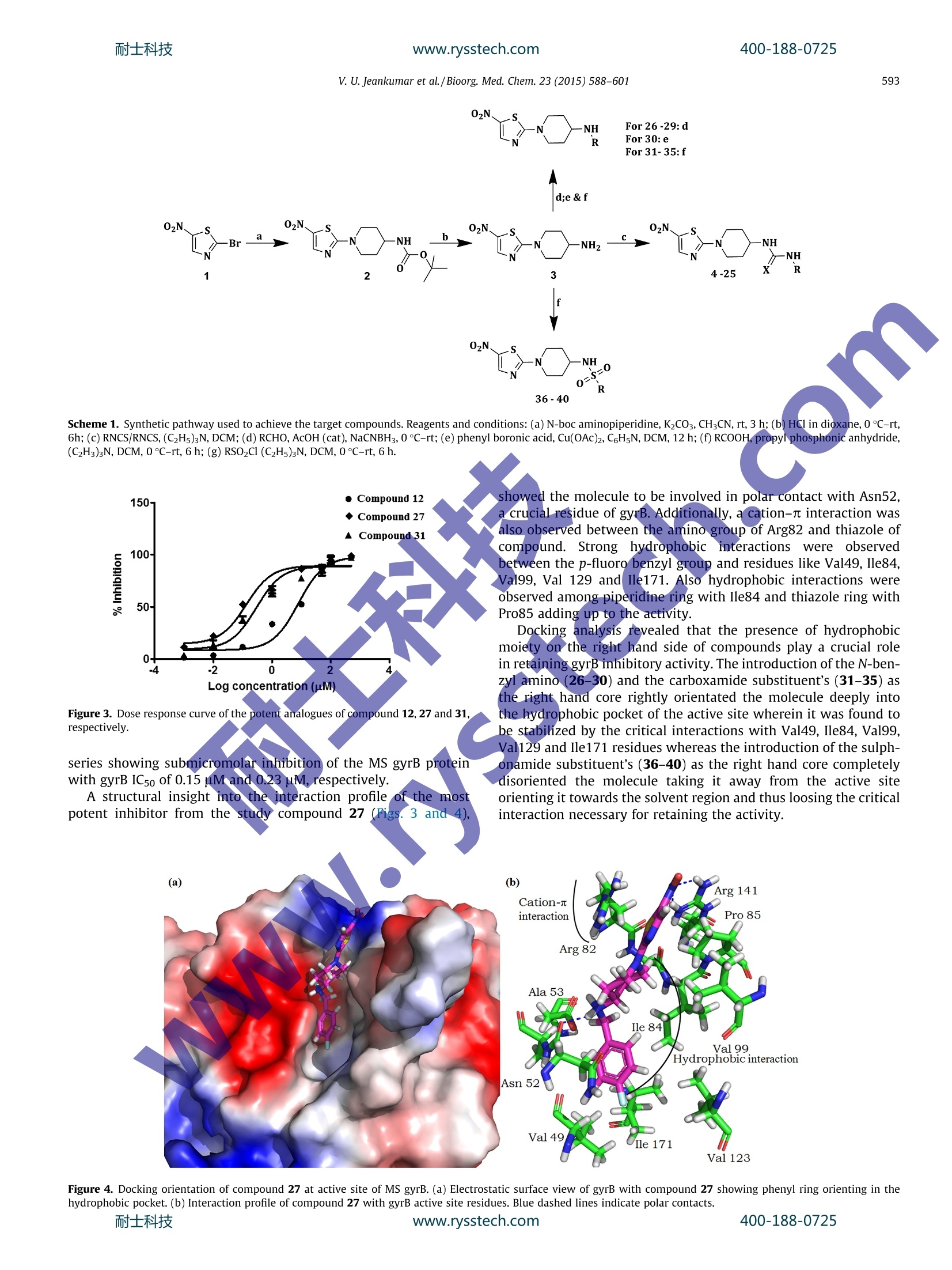
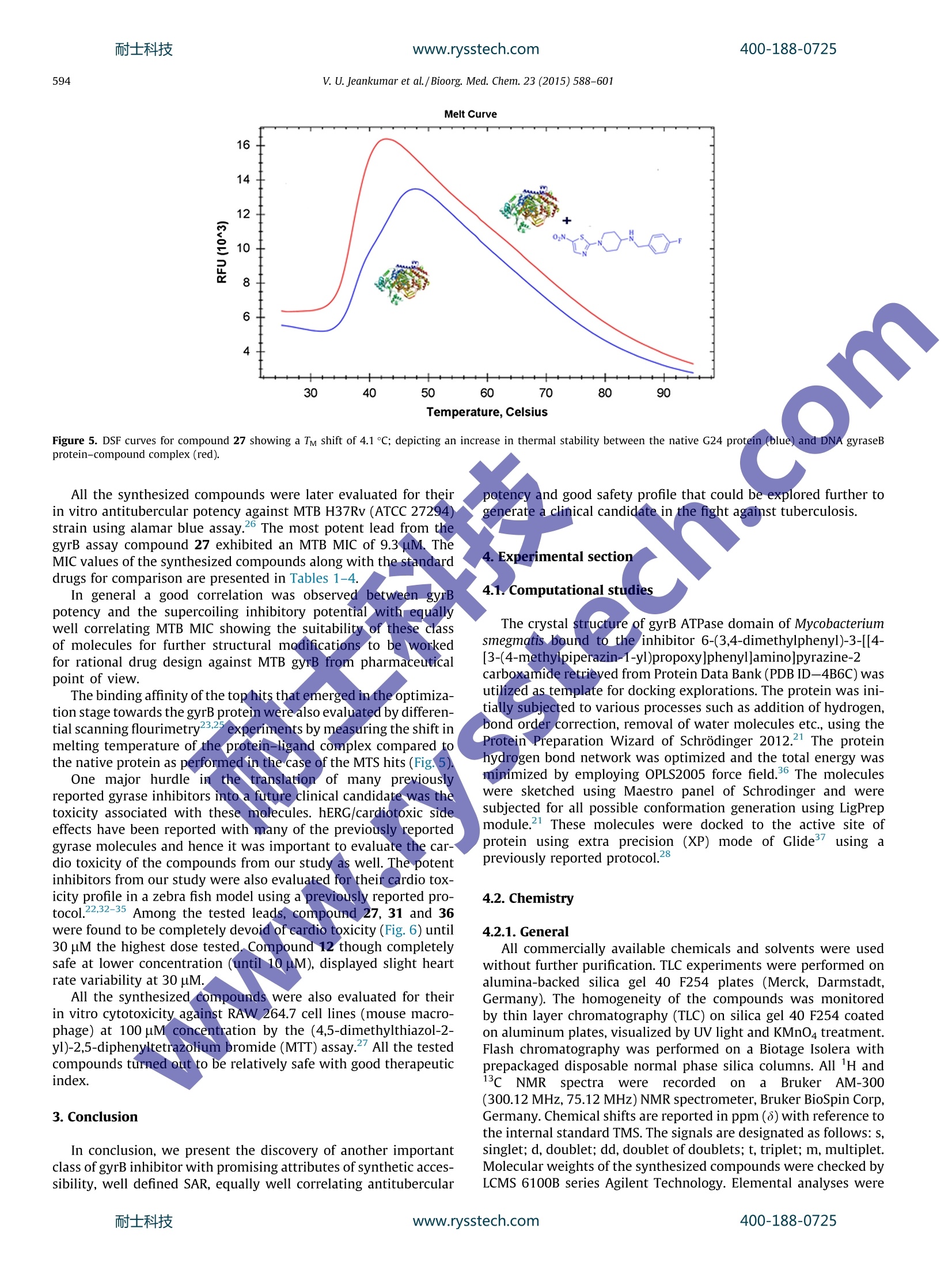
www.rysstech.comBioorganic & Medicinal Chemistry 23 (2015) 588-601耐士科技400-188-0725 耐士科技400-188-0725www.rysstech.com589V. U. Jeankumar et al./Bioorg. Med. Chem.23 (2015) 588-601 Contents lists available at ScienceDirect Bioorganic & Medicinal Chemistry ELSEVIER journal homepage: www.elsevier.com/locate/bmc Exploring the gyrase ATPase domain for tailoring newer CrossMarkanti-tubercular drugs: Hit to lead optimization of a novelclass of thiazole inhibitors ` Variam Ullas Jeankumar, Sonali Kotagiri ,Renuka Janupally, Priyanka Suryadevara ,Jonnalagadda Padma Sridevi, Raghavender Medishetti ·, Pushkar KulkarnibD,CPerumal Yogeeswari, Dharmarajan Sriram * Department of Pharmacy, Birla Institute of Technology 8r Science-Pilani, Hyderabad Campus, Shameerpet, R.R. District, Hyderabad 500078, Andhra Pradesh, India"Dr Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500046, India‘Zephase Therapeutics (An Incubated Company at the Dr Reddy’s Institute of Life Sciences), University of Hyderabad Campus, Gachibowli, Hyderabad 500046, India ARTICLEINFO Article history: Gyrase ATPase domain, the pharmaceutical underexploited segment of DNA gyrase, the sole Type IlReceived 9 September 2014 topoisomerase present in Mycobacterium tuberculosis represents an attractive target for anti-tubercularRevised 29 November 2014drug discovery. Herewe report, the development of a novel series of MTB DNA gyraseB inhibitorAccepted 1 December 2014identified through a medium throughput screening (MTS) of BITS in-house chemical library (3000 com-Available online 8 December 2014pounds). The MTS hit was further remodeled by chemical synthesis to identify the most potent analogue27 exhibiting an in vitro gyrB inhibitory ICso of 0.15 uM. The series also demonstrated well correlatingKeywords: gyrase super coiling activity and in vitro anti-mycobacterial potency against MTB H37Rv strain. Further-Mycobacterium tuberculosisGyrase ATPase domainmore t1he compounds displayed good safety profile in their subsequent cytotoxicity and hERG toxicityMedium throughput screeningevaluations, to be worked out from a pharmaceutical point of view as potential anti-tubercular agents.hERG◎ 2014 Elsevier Ltd. All rights reserved.Tuberculosis 1. Introduction The global burden of tuberculosis (TB) remains enormous;killing 1.3 million people in 2012, with an estimated 170,000deaths from MDR-TB.12 Furthermore, effective treatment of TB inpersons co-infected with HIV is complicated because of drug-druginteractions, which reduce patients compliance and thus lead tohigh rates of recurrence, appearance of extensively drug resistant(XDR) strains and mortality. Shorter and simpler regimens thatare safe, well tolerated, and effective against drug-susceptibleand drug-resistant TB, which are appropriate for joint HIV-TBtreatment and amenable to routine clinical settings, are neededurgently.3-9 The drug discoverypipeline for TB has been quite fruitful in thelast 10 years, with Bedaquiline (TMC 207) a novel adenosinetriphosphate synthase inhibitor being recently been granted licen-sure by the U.S. Food and Drug Administration under its acceler-ated approvalprocedure for treatment of MDR TB.1 Tencompounds have progressed through the clinical development * Corresponding author. Tel.: +91 40663030506; fax: +91 4066303998. ( E-mail a ddress: dsriram @ hyder ab ad.bits- p il a n i .ac.i n ( D. Sriram). ) pipeline, including six new compounds specifically developed fortuberculosis. Currently, two fluoroquinolones (MXF and GAT) arein Phase III clinical development for the treatment of drug-suscep-tible TB, and two novel compounds (OPC-67683 and PA-824) are inPhase II clinical trials for the treatment of both drug-susceptibleand drug-resistant diseases. Despite this progress, the global drugpipeline for tuberculosis is still insufficient to address the unmetneeds of treatment. 2 Also it is critical that a good supply ofnew chemical entities is forthcoming in order to overcome the highrates of attrition encountered during the drug discovery process.Phenotypic screening of compound libraries to identify novelleads is rapidly gathering momentum in tubercular drug discovery,with many potential leads in the current TB pipeline being identi-fied from whole cell based high-throughput screening strategy.However lack of knowledge about the specific biochemicaltarget(s) that these phenotypic hits inhibit and/or upregulate ham-per their lead progression potential and further development.4Atarget based screening could possibly offer an effective solutionto the limitation of phenotypic screening. In Mycobacterium tuberculosis (MTB), DNA gyrase is the soleType II topoisomerase that ensures the regulation of DNA topologyand has been genetically demonstrated to be a bactericidal drug target. The clinical success of fluoroquinolines have not only vali-dated the druggability of DNA gyrase as a potential anti-tuberculartarget but has also shown that inhibitors of this enzyme could alsobe active against non-replicating, persistent mycobacteria, andmight possibly reduce the duration of TB therapy. Howeverincreasing resistance to quinolone class of drugs calls for novelresearch in this field. Quinolones preferably targets gyrA subunit;the ATPase activity of bacterial DNA gyrase that resides in the gyrBsubunit remains pharmaceutically under-exploited even though itis an effective target of coumarins and related compounds.Inhibitor of gyrB subunit competitively inhibits the ATPase activityconferred by the DNA gyrB subunit and thereby abolishes theenergy-dependent reactions catalyzed by DNA gyrase. 5-20 In the present study, we explore a new class of DNA gyraseinhibitor derived from the medium throughput screening (MTS)of BITS-Pilani in house chemical library using the relatively under-exploited gyrase ATPase domain as the template. The hit obtainedwas further customized using medicinal chemistry strategies toobtain a lead molecule displaying considerable in vitro enzymeefficacy and anti-mycobacterial potency against MTB H37Rv strain. 2. Results and discussion The working outline utilized for identifying the inhibitors hasbeen depicted in Figure 1. In the preliminary stage of the study,BITS-Pilani in house database consisting of little more than 3000diverse compounds were first subjected to a series of in silico filter-ingfor compound redundancy, removing known anti-tubercularentities and finally for their physicochemical property clusteringusing the Lipinski rule of Five as the scoring filter. The basic goalof this strategy was to decrease the enormous virtual chemicalspace of small organic molecules to a manageable number of com-pounds that could inhibit the protein with the highest chance tolead to a drug candidate. 102 compounds that came out of thisexercise and those which were lead-like were tested for their inhi-bition of Mycobacterium smegmatis (MS) gyrB ATPase at single doseconcentration of 500 pM concentration in triplicates using a mala-chite green based assay adapted to 96 v-shaped well plate formatas described previously. The use of Mycobacterium smegmatis(MS) gyrB protein as a surrogate for gyrB protein from Mycobacte-rium tuberculosis (MTB) has been well demonstrated in litera-ture,17.22 as the activity of gyrBprotein from MTB was found bevery low when compared to that of MS." The MS gyrB was clonedinto pQE2 vector and was expressed in BL21 (DE3) pLysS cells, theexpressed protein was induced with a final concentration of0.2 mM IPTG (isopropyl--D thiogalactopyranoside) and was puri-fied using Ni-NTA column and were identified by SDS-PAGE.Novobiocin which has been previously demonstrated to be apotent inhibitor of DNA gyrB was used as a positive control inthe study.16 Compounds were also tested in the presence of Brij-35, a non-anionic detergent to ascertain whether these inhibitionswere an artifact due to sequestration of the enzyme by drug aggre-gates. The other artifacts like auto absorbance of the drug were alsoruled out by nullifying its absorbance during the reaction. The relative enzyme activity at 500 uM compound concentra-tion was used as a filter to score the compounds and select thestrongest inhibitors for further studies. Compounds inhibiting gyrBactivity ≥50% were retested at 100 uM concentration and the posi-tive hits from this study were further screened at 50 uM concen-tration. Twenty three compounds showed gyrB inhibition of≥50%, at 50 uM and these where further retested for gyrB inhibi-tory potency at 25 uM. Out of the seventeen compounds thatexhibited gyrB inhibition of 50% at 25 uM, twelve compounds werecherry picked and evaluated for their gyrB potency in a doseresponse in more detail. The binding affinity of the identified leads towards the gyrBprotein, was re-ascertained by differential scanning fluorimetryexperiments by measuring the fluorescence of the native proteinand protein-ligand complexes in presence of a fluorescent dyewhose fluorescence increases when exposed to non-polar residuesof the protein and reach the maximum when the protein dena-ture.23.25 A higher or positive shift of melting temperature (TM) ofprotein-ligand complex compared to the native protein TM signifya better stabilization of the protein-ligand complex, which in turnwould reflect on the inhibitor binding. The most potent analoguefrom the MTS screen displayed a TM shift of 2.6C confirming theiraffinity towards the gyrB protein. Further, the active hits were subjected to the DNA super coilinginhibitory assay23.24 using DNA gyrase protein from MTB (Inspiral-is, Norwich), to reconfirm their inhibitory potential; as any inhibi-tion of ATPase activity conferred by the DNA gyrB subunit shouldalso inhibit the supercoiling activity performed by both gyrA andgyrB domains.Novobiocin was used as a positive control in thestudy. As expected, the inhibitors showed goodcorrelationbetween the ATPase inhibitory activity and gyrase supercoilinginhibitory potency. The twelve potent hits retrieved from this study were thensubjected to a number of tertiary screens to assess their in vitroanti-tubercular potency and cell cytotoxicity. A major hurdle intarget based drug discovery is the lack oftranslation of the potencyand selectivity observed at the enzyme level into mycobacterialcidality. The compounds were first subjected to wholecell screen-ing against Mycobacterium tuberculosis H37Rv strain using alamarblue assay.26 with compound concentrations ranging from 50 ug/mL to 1.56 ug/mL in triplicates. The minimum inhibitory concen-tration (MIC99), defined as the minimum concentration of com-pound required tocompletely inhibit bacterial growth, wasdetermined for each compound. Isoniazid, rifampicin and ofloxacinwere used as reference compounds for comparison (Table 1). Since the predominant host cells for MTB are the lungmacrophages. These molecules were further interrogated for theirin vitro cytotoxicity against RAW 264.7 cell line (mouse macro-phage) at 100 uM concentration by the (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay." The hitsobtained were later clustered based on scaffold similarity. Among the small number of molecules identified,the subset ofnitrothaizolyl derivatives (represented inFFig.. 1, [HTS hits,compound 7, 8, and 12 in Table 1]) emerged as the lead seriesexhibiting promising in vitro potency and good correlation in thegyrB, supercoiling and MIC experiments. Compound 12 the mostpromising analogue from the assay, exhibited a gyrB inhibitoryIC50 of 8.07 uM. To provide a structural basis for further inhibitorimprovement, compound 12 was subjected to molecular dockingcalculations using the crystal structure of gyrB ATPase domain ofMycobacterium smegmatis retrieved from Protein Data Bank (PDBID-4B6C)22 as template; utilizing a protocol detailed in the exper-imental section. The compound exhibited good interaction profilein docking studies retaining critical polar and non polar interactioncomparable to that of (3,4-dichloro-5-methyl-N-[4-(5-nitro-1,3-thiazol-2-yl)cyclohexyl]-1H-pyrrole-2-carboxamide, a potent gyrBinhibitor [gyrB IC50=0.49 uM], identified from our earlier studywith an RMSD of less than 0.5 A as shown in Figure 2 suggestingthe suitability of the identified hit for further structure activityexplorations. The overall chemical structure topology of the identified MTShit incorporated a nitrothiazole left hand core that runs throughan aminopiperidine linker to the corresponding right handed ureaand thiourea phenyl units. Since a thiazole left hand core and theaminopiperidine linker wereaacommon1structural featureobserved in many of the previously reported gyrase inhibitors, itwas decided to retain these units in our initial structure activity profiling studies and introduce structural diversity by attemptingvarious explorations at the right hand core as shown in Table 1-4. Thus a library of 36 molecules with various urea, thiourea,amide, amino and sulphonamide substituent's as the right handcore were attempted in hit expansion step to have a clear under-standing of the role of these groups in activity determination. A retro synthetic evaluation of the identified MTS hits (7,8, and12) enabled us to design the reaction sequence depicted inScheme 1 for achieving the desired scaffold 1-(5-nitrothiazol-2-yl)piperidin-4-amine (3) for further optimizations in the ligandexpansion step. The synthesis of the target molecule was achievedin asimple and straight forwardmanner startingfromcommercially available 2-bromo-5-nitro thiazole. The constructionof target molecule began by condensing 2-bromo-5-nitro thiazolewith commercially available 4-N-Boc-aminopiperidine via a SnAr displacement to give nitrogen-linked analogue (3) in good yield.Subsequent Boc deprotection followed by coupling of the soobtained primary amine with various isocyanate afforded the cor-responding urea (4-10 and 18-21) derivatives in good yields asdepicted in Table 1. In a similar fashion, the thiourea (11-17 and22-25) and the sulphonamide derivatives (36-40) were assembledfrom the 1-(5-nitrothiazol-2-yl)piperidin-4-amine scaffold (3) byreacting with their corresponding isothiocyanate and sulphonylchloride, respectively. The amide derivatives (31-35) were devel-oped by coupling the scaffold derivative (3) with the respectiveacid using propyl phosphonic anhydride as the coupling agent.Finally reductive amination of 1-(5-nitrothiazol-2-yl)piperidin-4-amine with desired aldehydes gave the designed amino derivatives(26-29). The Chan-Lam methodology29-31was successful inachieving the N-phenyl analogue (30). Table 1In vitro biological evaluation of compounds 4-25 Compd X R gyrB assay (IC50) Supercoiling assay (ICso)" MIC99(uM) Cytotoxicity (% inhib:) 4 1.75 <1.56 9.40 32.3 6.37 17.83 33.1 9.8 12.5 17.04 30.9 COCH3 15.7 10.2 33.3 53.62 NO, 23.9 66.24 30.3 CH: 5.85 18.04 2.83 OCH3 3.56 9.25 34.5 34.5 S 9.8 12.5 17.93 18.1 S 8.07 11.2 17.11 25.1 S 8.707 10.12 16.3 14.2 S COCH, 23.31 22.36 64.02 S NO2 21.51 >25 120.1 CH 17.85 19.5 34.5 3.23 OCH3 4.538 12.5 33.02 14.70 2.136 8.24 9.02 34.1 111 17.15 36 11.9 16.41 49.1.3 OCH3 16.60 68.97 OCH: 16.4 17.9 S 13.05 31.5 31.3 S 7.6 .31.8 22.2 Novobiocin 180±3.9 nM nd Isoniazid 0.66 Rifampicin 0.23 Ofloxacin 2.16 nd MTB DNA gyrase super coiling activity. In vitro MTB activity. " At 100 uM against RAW 264.7 cells; nd: indicates not determined. Figure 2. Superimposed orientation of the docked conformations of compound 12(green) and most potent gyrB inhibitor from our earlier study compound 28 [gyrBIC50=0.49 uM] (blue). With respect to the structure-activity explorations; in the firststage of lead expansion step, various substituted phenyl urea (4-10) and thiourea derivatives (11-17); analogues to the lead mole-cules were attempted and evaluated for the biological potency asshown in Table 1.The entire set of molecules tested showed goodto potent gyrase inhibitory potential and as is evident from theTable 1, the urea analogues (4-10) were the preferred right handpartners than their thiourea counterparts (11-17). With respectto the various substituents attempted on the phenyl ring; onlysmaller substituent were accommodated; as the unsubstituted, flu-oro, methoxy and chloro substituted derivatives showed morepotent inhibition of the gyrB protein than the derivatives withbulkier substituents like acetyl and nitro groups. Encouraged bythese results, it was further decided to attempt various benzylicurea and thiourea derivatives as the right hand partner in the nextstage of study. Only the more potent right hand substitutions fromthe earlier study viz.; the unsubstituted, fluoro, methoxy andchloro substituted rings were attempted in this stage. This modifi-cation was found to be suitable as it led to identification of manypotent inhibitors of gyrase protein with good in vitro antitubercu-lar potency. The docking studies of these compounds revealed some of theimportant differences among their binding patterns at the gyrBactive site. A deeper look into the interaction profile diagram ofthe active molecules showed the phenyl/benzyl ring to be deeplyoriented into the hydrophobic region where it was found to be sta-bilized by the hydrophobic interaction with residues Val49, le84,Val99, Val123 and Val128, previously reported to be critical inretaining the activity;22.24 the left hand nitrothiazole core was Table 2In vitro biological evaluation of compounds 26-30 O2N S 02N S -R -N -N 26-29 30 R Compd R gyrB assay (IC5o) Supercoiling assay (ICso) MIC99(uM) Cytotoxicity (%inhib:) 26 H 9.25 11.31 39.26 48 27 F 0.1496 0.52 9.3 39.9 28 CI 19.9 20.12 35.42 47.9 29 OCH: 17.42 16.4 17.93 47.9 30 H 7.33 8.4 41.06 45 MS gyrB ATPase activity. Cytotoxicity (%inhib:) 39 OCH: 14.93 31.3 23.5 40 H 52.9 >25 65.4 38.9 dMS gyrB ATPase activity. MTB DNA gyrase supercoiling activity.in vitro MTB activity." At 100 uM against RAW 264.7 cells, nd: indicates not determined. found to be oriented towards the solvent accessible surface of pro-tein. The inactive molecules oriented in a completely differentdirection thereby taking it away from the active site thus losingcritical non-polar interaction with the amino acid residues presentin the vicinity of the protein essential for retaining the activity. Theimproved activity of the urea analogues compared to the thioureaanalogues was found to be due to the better electrostatic interac-tion that the urea derivatives (oxygen being more electronegative[EN-3.44]) retained with the amino acids residues in the active sitevicinity like Arg82 than their thiourea counterparts, the sulfur group in thiourea analogues being less electronegative [EN-2.58]lacked these interactions, thus resulting in their reduced activity. Later, various N-benzyl/phenyl amino (26-30), carboxamide(31-35) and sulphonamide derivatives (36-40) were also exploredas the right hand partner to have a clear profiling of the structureactivity relationship. All these modification resulted in very potentmolecules as shown in Tables 2-4. The N-benzyl amino (26-30)and carboxamide analogues (31-35) were more preferred as theright hand partner than their corresponding sulphonamideanalogues (36-40), with two molecules (27 and 31) one from each Scheme 1. Synthetic pathway used to achieve the target compounds. Reagents and conditions: (a) N-boc aminopiperidine, K2CO3,CH;CN, rt, 3 h;(b) HCl in dioxane,0 °C-rt,6h; (c) RNCS/RNCS,(C2Hs)3N,DCM;(d) RCHO, AcOH (cat), NaCNBH3,0 C-rt;(e) phenyl boronic acid, Cu(OAc)2, C6HsN, DCM, 12 h;(f) RCOOH, propyl phosphonic anhydride,(C2H3)3N, DCM, 0 C-rt, 6 h; (g) RSO2Cl (C2H5)3N, DCM,0°C-rt, 6 h. showed the molecule to be involved in polar contact with Asn52,a crucial residue of gyrB. Additionally, a cation-n interaction wasalso observed between the amino group of Arg82 and thiazole ofcompound. Stronghydrophobic interactionswere observedbetween the p-fluoro benzyl group and residues like Val49,Ile84,Val99, Val 129 and Ile171. Also hydrophobic interactions wereobserved among piperidine ring with Ile84 and thiazole ring withPro85 adding up to the activity. Figure 3. Dose response curve of the potent analogues of compound 12, 27 and 31,respectively. series showing submicromolar inhibition of the MS gyrB proteinwith gyrB IC5o of 0.15 pM and 0.23 pM, respectively. A structural insight into the interaction profile of the mostpotent inhibitor from the studycompound 27 (Figs.3 and 4), Docking analysis revealed that the presence of hydrophobicmoiety on the right hand side of compounds play a crucial rolein retaining gyrB inhibitory activity. The introduction of the N-ben-zyl amino (26-30) and the carboxamide substituent's (31-35) asthe right hand core rightly orientated the molecule deeply intothe hydrophobic pocket of the active site wherein it was found tobe stabilized by the critical interactions with Val49, Ile84,Val99,Val129 and Ile171 residues whereas the introduction of the sulph-onamide substituent's (36-40) as the right hand core completelydisoriented the molecule taking it away from the active siteorienting it towards the solvent region and thus loosing the criticalinteraction necessary for retaining the activity. Figure 4. Docking orientation of compound 27 at active site of MS gyrB. (a) Electrostatic surface view of gyrB with compound 27 showing phenyl ring orienting in thehydrophobic pocket. (b) Interaction profile of compound 27 with gyrB active site residues. Blue dashed lines indicate polar contacts.www.rysstech.com 400-188-0725 Temperature, Celsius Figure 5. DSF curves for compound 27 showing a TM shift of 4.1℃; depicting an increase in thermal stability between the native G24 protein (blue) and DNA gyraseB All the synthesized compounds were later evaluated for theirin vitro antitubercular potency against MTB H37Rv (ATCC 27294)strain using alamar blue assay.2 The most potent lead from thegyrB assay compound 27 exhibited an MTB MIC of 9.3 uM. TheMIC values of the synthesized compounds along with the standarddrugs for comparison are presented in Tables 1-4. All the synthesized compounds were also evaluated for theirin vitro cytotoxicity against RAW 264.7 cell lines (mouse macro-phage) at 100 pM concentration by the (4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. All the testedcompounds turned out to be relatively safe with good therapeuticindex. 3.Conclusion In conclusion, we present the discovery of another importantclass of gyrB inhibitor with promising attributes of synthetic acces-sibility, well defined SAR, equally well correlating antitubercular 4.1.Computational studies The crystal structure of gyrB ATPase domain of Mycobacterium smegmatis bound to the inhibitor 6-(3,4-dimethylphenyl)-3-[[4-[3-(4-methylpiperazin-1-yl)propoxy]phenyl]amino]pyrazine-2carboxamide retrieved from Protein Data Bank (PDB ID-4B6C) wasutilized as template for docking explorations. The protein was ini-tially subjected to various processes such as addition of hydrogen,bond order correction, removal of water molecules etc., using theProtein Preparation Wizard of Schrodinger 2012.21 The proteinhydrogen bond network was optimized and the total energy wasminimized by employing OPLS2005 force field.36 The moleculeswere sketched using Maestro panel of Schrodinger and weresubjected for all possible conformation generation using LigPrepmodule.1 These molecules were docked to the active site ofprotein using extra precision (XP) mode of Glide using apreviously reported protocol.28 4.2. Chemistry 4.2.1.General All commercially available chemicals and solvents were usedwithout further purification. TLC experiments were performed onalumina-backed silica gel 40 F254 plates (Merck, Darmstadt,Germany). The homogeneity of the compounds was monitoredby thin layer chromatography (TLC) on silica gel 40 F254 coatedon aluminum plates, visualized by UV light and KMnO4 treatment.Flash chromatography was performed on a Biotage Isolera withprepackaged disposable normal phase silica columns. All H and1'c NMRspectrawere recorded onaBruker AM-300(300.12 MHz, 75.12 MHz) NMR spectrometer, Bruker BioSpin Corp,Germany. Chemical shifts are reported in ppm (8) with reference tothe internal standard TMS. The signals are designated as follows: s,singlet; d, doublet; dd, doublet of doublets;t, triplet; m, multiplet.Molecular weights of the synthesized compounds were checked byLCMS 6100B series Agilent Technology. Elemental analyses were Mean (±S.E.M.) of the heart rates of atria and ventricles of compounds 12, 27, 31, 36treatment groups. (*p<0.05, **p<0.01 and ***p<0.001). Statistical significance wasanalyzed comparing control group Vs treated groups. carried out on an automatic Flash EA 1112 Series, CHN Analyzer(Thermo). The synthesis of the target molecule was achieved in asimple and straight forward manner as depicted in Scheme 1 start-ing from commercially available2-bromo-5-nitro thiazole, whichin turn can also be prepared by diazotization of relatively cheaper2-amino-5-nitro thiazole. 4.2.2. tert-Butyl (1-(5-nitrothiazol-2-yl)piperidin-4-yl)carbamate (2) 4-N-Boc-aminopiperidine (0.96 g, 4.78 mmol) was added lotwise to a well stirred suspension of 2-bromo-5-nitro thiazole(1 g, 4.78 mmol) and potassium carbonate (0.79 g 5.74 mmol)in acetonitrile (15 mL). The reaction mixture was then stirredat room temperature for about 4 h (monitored by TLC & LCMSfor completion), and solvent evaporated under reduced pressure.The residue was further diluted with water (10mL) and ethylacetate (20 mL) and the layers separated. The aqueous layerWas re-extracted with ethyl acetate (2×15mL) and thcombined organic layer was washed with brine (15 mL), driedover anhydrous sodium sulfate and evaporated under reduced pressure and the residue purified by column chromatographyusing hexane/ethylacetate as eluent to give the desired product2 (1.32 g, 84%) as yellow solid. H NMR (DMSO-d6): 8H 1.39 (s,9H), 1.44-4.03 (m, 9H), 8.38 (s, 1H), 13C NMR (DMSO-d6): 8c171.9,154.8,147.6,135.7,77.8,47.1,46.1,30.7,28.2. ESI-MSm/z 329.1 (M+H). Anal. Calcd for C13H20N404S: C, 47.55; H,6.14; N,17.06. Found: C, 47.52; H, 6.11; N, 17.09. 4.2.3.1-(5-Nitrothiazol-2-yl)piperidin-4-amine (3) HCl in dioxane (2 M solution,15 mL) was added drop wise to asolution of tert-butyl (1-(5-nitrothiazol-2-yl)piperidin-4-yl)carba-mate (1 g, 3.05 mmol) in dry dioxane (20 mL) at 0 °Cover a periodof 10 min. The reaction was slowly warmed to room temperatureand stirred at that temperature for 6 h (monitored by TLC andLCMS for completion). The solvent was then removed underreduced pressure, and the residue diluted with water (5 mL). Thesolution was adjusted to pH 8 with solid sodium bicarbonate.The aqueous phase was back extracted with dichloromethane(3×25 mL) and dried over sodium sulfate. The combined organicphases were concentrated under reduced pressure to give the deprotected product (0.58 g, 83.4%) as yellow solid, which wasimmediately taken to the next step. 4.2.4. General procedure for the synthesis of urea/thioureaderivatives (4-25) Toa ssolution of 1-(5-nitrothiazol-2-yl)piperidin-4-amine(1 mmol) in dry dimethylformamide (3 mL) was added triethyl-amine (2 mmol) and corresponding isocyanate/isothiocyanate(1.1 mmol) at O℃. The reaction mixture was slowly warmed tort and stirred at rt for 6 h (monitored by TLC & LCMS for comple-tion). The reaction mixture was washed with water (2mL), brine(2 mL), dried over anhydrous sodium sulfate and evaporated invacuo. The residue obtained was then either recrystallised fromdiethylether or purified by column chromatography. 4.2.4.1.1-(1-(5-Nitrothiazol-2-yl)piperidin-4-yl)-3-phenylurea(4). The compound was synthesized according to the generalprocedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g,0.438 mmol), triethylamine (0.089 g, 0.88 mmol), and phenyl iso-cyanate (0.057 g, 0.482 mmol) to afford 4 (0.12 g, 79%) as pale yel-low solid. Mp: 180-182 C. 1H NMR(CDCl3): 8H 1.43-4.01(m,9H),7.23-7.61(m, 5H), 8.39 (s, 1H). 1’C NMR (CDCl3): 8c 171.8, 153.9,147.6, 139.5,135.4,128.7,127.9,121.6,47.6,45.5, 31.2. ESI-MSm/z 348.2 (M+H). Anal. Calcd for C15H17N503S: C, 51.86; H, 4.93;N, 20.16. Found: C, 51.89; H, 4.91; N,20.15. (0.1 g, 0.438 mmol), triethylamine (0.089 g,,0.88mmol), and4-nitro phenyl isocyanate (0.079g, 0.482 mmol) to afford 8(0.134g, 77.9%) as yellowish-orange solid. Mp: 194-196℃.1HNMR (CDCl3): 8H 1.46-3.93 (m, 9H), 7.84-8.39 (m, 5H). 13c NMR(CDCl3):8c172.1,154.2,147.6,145.6,143.4,135.6,123.8,120.1,47.6,45.7,31.3. ESI-MS m|z 393.1(M+H). Anal. Calcd forC15H16N60sS: C, 45.91; H, 4.11; N, 21.42. Found: C, 45.88; H,4.10;N, 21.41. 4.2.4.6. 1-(4-Tolyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)urea(9). The compound was synthesized according to the generalprocedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g,0.438 mmol), triethylamine (0.089g, 0.88mmol), and 4-tolylisocyanate (0.064 g, 0.482 mmol) to afford 9 (0.133 g,84.2%) as yel-lowish-orange solid. Mp: 182-184C.H NMR (CDCl3): 8H 1.38-1.96 (m, 4H), 2.31 (s, 3H), 2.79-3.96 (m, 5H),7.23-7.51 (m, 4H)8.4 (s,1H). 13C NMR (CDCl3): 8171.9,154.3,147.6,137.2,136.3,135.5,129.3,121.7,47.6,45.6,31.1,21.6. ESI-MS mjz: 362.2.3(M+H). Anal. Calcd for C16H19N503S: C, 53.17; H, 5.30; N, 19.38.Found: C, 53.16; H, 5.32; N, 19.36. 4.2.4.3. 1-(4-Chlorophenyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)urea (6)i) The compound was synthesized according to thegeneral procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1g, 0.438 mmol), triethylamine(0.089g, 0.88 mmol), and4- chloro phenyl isocyanate (0.074g, 0.482 mmol) to afford 6(0.12g, 71.9%) as red solid. Mp: 189-191C. 1H NMR (CDCl):8H1.46-3.92(m,9H), 7.63-7.84(m,4H),8.39(s,1H)13CNMR(CDCl3):8c172.1,154.2,147.6,137.6,135.5,132.9,129.1,120.6,47.5,45.7,31.3. ESI-MS m/z 382.4 (M+H)*. Anal. Calcd for C15H16CIN503S: C,47.18; H, 4.22; N, 18.34.Found: C,47.22; H, 4.19;N, 18.32. 4.2.4.4.1-(4-Acetylphenyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)urea (7). The compound was synthesized according tothe general procedure using,1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438mmol), triethylamine (0.089g, 0.88 mmol),and 4-acetyl phenylisocyanate (0.078 g, 0.482 mmol) to afford 7(0.119g, 70%) as yellowish-orange solid. Mp: 176-178℃. 1HNMR (CDCl3): on 1.37-1.45(m, 2H), 1.88-1.95 (m, 2H), 2.48 (s,3H), 2.78-3.89 (m, 5H),7.69-7.87(m, 4H), 8.39 (s, 1H). "’C NMR(CDClg): 8189.2,172,154.1,147.7,144.1,136.8,135.6,128.8,121.6,47.7, 45.7, 31.2, 26.8. ESI-MS m/z 390.4(M+H). Anal. Calcdfor C17H19N504S: C, 52.43; H, 4.92; N, 17.98. Found: C,52.47; H,4.9; N, 18.01. ( 4.2.4.5. 1-(4-Nitrophenyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4- yl)urea ( 8). The compound was synthesized according to the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine ) 4.2.4.7..l1-(4-Methoxyphenyl)-3-(1-(5-nitrothiazol-2-yl)piperi-din-4-yl)urea.(10) The compound was synthesized according tothe general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1g, 0.438 mmol),triethylamine (0.089g, 0.88 mmol),and 4-methoxy isocyanate(0.072g,0.482 mmol) to afford 10(0.141g, 85.5%) as yellowish-orange solid. Mp: 165-167C. 1HNMR(CDCl3): 8H 1.36-3.81 (m, 9H), 3.84 (s, 3H), 7.01-7.39 (m,4H), 8.37 (s, 1H). 13C NMR (CDCl): 8172,158.6,154.2,147.5,135.6, 131.8,120,114.6,56.1,47.5,45.3,31.1. ESI-MS m|zB78.2(M+H)t. Anal. Calcd for C16H19N504S: C, 50.92; H, 5.07; N,18.56. Found:C, 50.88; H, 5.03; N,18.54. 4.2.4.8.1-(1-(5-Nitrothiazol-2-yl)piperidin-4-yl)-3-phenylthio-urea.(11) The compound was synthesized according to the gen-eral procedure using1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1 g, 0.438 mmol), triethylamine (0.089 g, 0.88 mmol),and phe-nyl isothiocyanate (0.065 g, 0.482 mmol) to afford 11 (0.137 g,85.6%) as pale yellow solid. Mp: 180-182 C. 'H NMR (CDCl;): 8n1.39-3.14 (m, 9H), 6.98-7.64 (m, 5H), 8.39 (s, 1H). 13C NMR(CDCl3):8c176.5, 172,147.6,138.6,135.4,128.7,128.2, 126.7,50.6, 46.5, 31.9. ESI-MS m/z 364.1 (M+H) . Anal. Calcd for C15H17N5O2S2: C, 49.57;H,4.71;N,19.27.Found: C, 49.55; H, 4.69;N,19.28. 4.2.4.9. 1-(4-Fluorophenyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thiourea (12).The compound was synthesized accordingto the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol),and 4-fluoro phenyl isothiocyanate (0.074 g, 0.482 mmol) to afford12 (0.167 g, 67.7%) as pale yellow solid. Mp: 169-171C.1H NMR(CDCl;): 81.41-3.13(m,9H), 6.54-7.01(m,4H).13CNMR(CDCl;):8_176.8,172.1,161,147.7,135.6,133.9,130.8,115.6,50.7, 46.6, 32.ESI-MS m/z 382.3(M+H). Anal. Calcd for C15H16FN502S2: C, 47.23;H, 4.23;N, 18.36. Found: C, 47.23; H, 4.23; N, 18.36. 4.2.4.10. 1-(4-Chlorophenyl)-3-(1-(5-nitrothiazol-2-yl)piperi-din-4-yl)thiourea (13) The compound was synthesized accord-ing to tthe generalprocedureusing1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1g, 0.438 mmol), triethylamine (0.089 g,0.88 mmol),and 4-chloro phenylisothiocyanate(0.081 g,0.482 mmol) to afford 13 (0.136 g, 78.2%) as red solid. Mp: 189-191℃. 1H NMR (CDCl3): 8q 1.43-3.18 (m, 9H), 6.76-7.21 (m,4H), 8.38 (s, 1H). C NMR (CDCl3): 8c177,172.1,147.7, 136.8,135.8,133.7,130.8,129.2,50.8,46.7,32.1. ESI-MS m/z 398.3(M+H). Anal. Calcd for C15H16ClN5O2S2: C, 45.28; H, 4.05; N,17.60. Found: C, 45.27; H, 4.03; N, 17.59. 4.2.4.11. 1-(4-Acetylphenyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thiourea (14). The compound was synthesized accord-ing to the general procedure using 1-(5-nitrothiazol-2-yl)piperi-din-4-amine (0.1g, 0.438 mmol), triethylamine (0.089g,0.88 mmol), and14-acetylphenyl isothiocyanate (0.085 g,0.482 mmol) to afford 14 (0.114 g, 64%) as yellowish-orange solid.Mp: 176-178C. 1H NMR (CDCl3): 8H 1.41-3.12 (m, 12H), 6.76-7.21 (m, 4H), 8.38 (s, 1H).13C NMR (CDCl3): 8189.6,176.8,172,147.8,143.2,137.1,135.4,129.7,126.6,50.6,46.5, 32, 26.9. ESI-MS m|z 406.1 (M+H). Anal. Calcd for C17H19N503S2: C, 50.35; H,4.72; N, 17.27. Found: C, 50.33; H, 4.71; N, 17.25. 4.2.4.12. 1-(4-Nitrophenyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thiourea (15). The compound was synthesized accordingto the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol),and 4-nitro phenyl isothiocyanate (0.087 g, 0.482 mmol) to afford15 (0.146g, 81.6%) as yellowish-orange solid. Mp: 194-196℃.H NMR (CDCl3): 8H 1.43-3.16 (m, 9H), 7.02-8.38 (s, 5H). 13cNMR (CDCl3): 8c176.9,172,147.3,144.7,144.1,135.4,124.6,123.9,50.8,46.7,32. ESI-MS m|z 409.1(M+H). Anal. Calcd forC15H16N604S2: C, 44.11; H, 3.95; N, 20.58. Found: C, 44.12; H,3.97; N, 20.56. 4.2.4.13. 1-(4-Tolyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thiourea (16). The compound was synthesized accordingto the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol)and 4-tolyl isothiocyanate (0.072g, 0.482 mmol)to afford 16(0.137g, 83%) as yellowish-orange solid. Mp:1182--184C.1HNMR (CDCl3):8H 1.37-3.21(m, 12H), 6.46-6.89 (m,4H),8.37 (S,1H). 1’C NMR (CDCl3): 8c 176.6,171.9,147.3,136.8,135.6, 135.3,129.1,126.2,50.6,46.4,31.8,21.7.ESI-MS mjz 378.1(M+H).Anal.Calcd for C16H19N502S2: C, 50.91; H, 5.07; N,18.55. Found: C,50.93; H, 5.06; N, 18.53. 4.2.4.14. 1-(4-Methoxyphenyl)-3-(1-(5-nitrothiazol-2-yl)piperi-din-4-yl)thiourea (17). The compound was synthesized accord-ing othegeneralprocedure using1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1g,0.438 mmol), triethylamine (0.089 g,0.88 mmol), and 4-methoxy isothiocyanate (0.08g, 0.482 mmol)to afford 17 (0.159 g, 92.4%) as yellowish-orange solid. Mp: 165-167C.1HNMR (CDCl3): 8H11.35-3.19(m,9H), 3.83(s,3H), 6.42-6.76 (m, 4H), 8.39 (s, 1H). 13C NMR (CDCl3): 8176.8,171.8,158.9,147.2,135.4,130.4,127.2,114.7,56.3,50.6,46.5,31.7.ESHMS m/z 394 (M+H)*. Anal. Calcd for C16H19N503S2: C,48.84; H,4.87; N, 17.80. Found: C, 48.87; H, 4.85,N, 17.79. 4.2.4.15. 1-Benzyl-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)urea(18). The compound was synthesized according to the generalprocedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1g,0.438 mmol), triethylamine (0.089 g, 0.88 mmol), and benzyl iso-cyanate (0.064 g, 0.482 mmol) to afford 18 (0.131 g, 82.9%) as paleyellow solid. Mp: 196-198C.H NMR (CDCl3): 8H 1.38-4.21(m,11H), 7.21-7.39 (m, 5H), 8.38 (s, 1H). 13C NMR (CDCl3): 8 172,157.3,147.7,140.8,135.7,128.2,126.9,126.5,47.4,45.5,42.8,31.3. ESI-MS m|z 362.1 (M+H). Anal. Calcd for C16H19N5O3S: C,53.17; H, 5.30; N, 19.38. Found: C, 53.15; H, 5.29; N, 19.39. ( 4.2.4.16.1-(4-Fluorobenzyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)urea (19). The compound w a s synthesized according to the general pr ocedure using 1- ( 5-nitrothiazol-2-yl)piperidin-4- amine ( 0.1g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol), and4-fluorobenzyl isocyanate (0 . 073 g, 0.482 mmol) to afford 19 (0.127 g , 76.5% ) as pale yellow solid. M p : 180-182℃. H N MR (CDCl3):8H1.42-4.21( m , 11H), 7.16-7.38(m, 4H), 8 . 40 (s, 1 H). ) 13C NMR (CDCl3): 8c 172.1,160.7,157.4,147.8,135.6,133.3,128.2,115.6,47.6,45.5,42.8, 31.4. ESI-MS m|z 380.1(M+H)*.Anal.Calcd for C16H18FN5O3S: C, 50.65; H,4.78; N, 18.46. Found: C,50.63; H,4.77;N, 18.48. 4.2.4.17.1-(4-Chlorobenzyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)urea (20). The compound was synthesized according tothe general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol),and 4-chlorobenzyl isocyanate (0.081 g, 0.482 mmol) to afford 20(0.156g, 90.2%) as pale yellow solid. Mp: 195-197℃. H NMR(CDCl3): 8H1.44-4.24(m, 11H), 7.32-7.39 (m, 4H), 8.39 (s, 1H).13C NMR (CDCl): 8 172,157.6,147.7,136.2,135.7,134.3,132.6,128.8,47.5,45.6,42.7, 31.3. ESI-MS m/z 396.1 (M+H). Anal. Calcdfor C16H18ClN503S: C, 48.54; H, 4.58;N, 17.69. Found: C48.56; H,4.54; N, 17.7. 4.2.4.18B..1-(4-Methoxybenzyl)-3-(1-(5-nitrothiazol-2-yl)piperi-din-4-yl)urea (21). The compound was synthesized accord-ingto the generalprocedure usingi-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1g, 0.438 mmol), triethylamine (0.089 g,0.88mmol), and 4-methoxybenzyl isocyanate (0.079 g,0.482 mmol) to afford 21 (0.146 g, 85.9%) as pale yellow solid.Mp: 198-200C.1H NMR (CDCl): 8H 1.36-3.80 (m, 9H), 3.86(s, 3H), 4.16 (s, 2H), 6.98-7.21 (m, 4H),8.38 (s, 1H).C NMR(CDCl3): 8c171.9,159,157.6,147.6,135.5,130.6,130.3,113.9,56.1,47.6, 45.4, 42.6,31.2. ESI-MS m/z 392.1 (M+H)*. Anal. Calcdfor C17H21N5O4S: C, 52.16; H,5.41; N, 17.89. Found: C, 52.17; H,5.4; N, 17.87. 4.2.4.19. 1-Benzyl-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thio-urea (22). The compound was synthesized according to thegeneral procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1 g, 0.438 mmol), triethylamine (0.089 g, 0.88 mmol), and ben-zyl isothiocyanate (0.072 g, 0.482 mmol) to afford 22 (0.132 g,80%) as pale yellow solid. Mp: 195-197°C. 1H NMR (CDCl3): 81.38-3.21(m, 9H), 4.21 (s, 2H), 7.22-7.33 (m, 5H), 8.39 (s, 1H).13C NMR (CDCl3): 8c180.9,171.8,147.6,138.1,135.6,128.6,27.1,126.9,51.4, 50.6, 46.9, 32.1. ESI-MS m/z 378.2 (M+H)*. Anal.Calcd for C16H19N502S2: C, 50.91; H,5.07; N, 18.55. Found: C,50.98; H, 5.05; N,18.53. 4.2.4.20.1-(4-Fluorobenzyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thiourea (23). The compound was synthesized accordingto the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol),and 4-fluorobenzyl isothiocyanate (0.073 g, 0.482 mmol) to afford23 (0.143 g, 82.66%) as pale yellow solid. Mp: 177-179℃. 1HNMR (CDCl3): 8H 1.38-3.16 (m, 9H), 4.21 (s,2H), 7.16-7.38(m,4H), 8.42 (s, 1H). 13C NMR (CDCl3): 8c 181, 172,160.7, 147.6,135.7, 133.5,128.7,115.4,51.6, 50.7, 47, 32. ESI-MS m/z 396.1(M+H)*. Anal. Calcd for C16H18FN502S2: C, 48.59; H, 4.59; N,17.71. Found: C,48.62; H, 4.56; N, 17.74. 4.2.4.21.1-(4-Chlorobenzyl)-3-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)thiourea (24). The compound was synthesized accordingto the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), triethylamine (0.089g, 0.88 mmol),and 4-chlorobenzyl isothiocyanate (0.081 g, 0.482 mmol) to afford24 (0.167 g, 92.8%) as pale yellow solid. Mp: 147-149 C. 1H NMR(CDCl3): 8H 1.40-3.19(m, 9H), 4.29 (s, 2H), 7.33-7.39 (m, 4H),8.44 (s, 1H). 13c NMR (CDCl;): 8c181.2, 171.9, 147.7,135.8,135.6,134.7,132.5,128.6,51.5,50.6,46.9,32.1. ESI-MS mz412.4 (M+H). Anal. Calcd for C16H18ClN502S2: C, 46.65; H, 4.40;N, 17.Found: C, 46.63; H, 4.38; N, 17.1. 4.2.4.22. 1-(4-Methoxybenzyl)-3-(1-(5-nitrothiazol-2-yl)piperi-din-4-yl)thiourea (25). The compoundwas synthesizedaccording to the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), triethylamine (0.089 g,0.88 mmol), andd44-methoxybenzylisothiocyanate(0.086g,0.482mmol) to afford 25 (0.156g, 87.1%) as pale yellow solid.Mp: 167-169C.1HNMR (CDCl3): 8q 1.33-3.19 (m, 9H), 3.82 (s,3H)4.24(s,2H), 6.89-7.19 (m, 4H), 8.38 (s,1H).113C NMR (CDCl3):8。180.9,172,158.8,147.5,135.6,130.6,130.2,113.9,56.1,51.4,50.4,47,31.9. ESI-MS m/z 408.2(M+H). Anal. Calcd for C17H21N5O3S2: C, 50.10; H, 5.19; N, 17.19. Found: C, 50.09; H, 5.17; N, 17.21. 4.2.5. General procedure for the synthesis of sub: N-benzylderivatives (26-30) ToaSsolutionof 1-(5-nitrothiazol-2-yl)piperidin-4-amine(1 mmol) in dry methanol (3mL) was added the correspondingaldehyde (1.1 mmol) and catalytic amount of acetic acid. The reac-tion mixture was stirred at rt for 3 h. Sodiumtriacetoxy borohy-dride resin was subsequently added and the stirring continued atrt for another 6 h (monitored by TLC & LCMS for completion).The reaction mixture was filtered and the filtrate evaporated undervacuo. The residue was further diluted with dichloromethane,washed with water (2mL), brine (2 mL), dried over anhydroussodium sulfate and evaporated in vacuo. The residue obtainedwas then either recrystallised from diethylether or purified by col-umn chromatography. 4.2.5.2.N-(4-Fluorobenzyl)-1-(5-nitrothiazol-2-yl)piperidin-4-amine (27). The compound was synthesized according tothe general procedure using1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), and 4-fluoro benzaldehyde(0.06g,0.482 mmol) to afford 27 (0.123 g, 883.7%) as pale yellow solid.Mp: 119-121C. H NMR (CDCl3): 8H 1.36-3.94 (m, 11H), 7.09-7.15 (m, 2H), 7.36-7.41 (m,2H), 8.37 (s,1H). C NMR (CDC): 8.171.9,162.6,147.7,137.1,135.5,129.7,129.6,114.8,114.5,51.7,48.8, 46.7, 30.7. ESI-MS m/z 337.1 (M+H)*. Anal. Calcd for C15H17-FN402S: C, 53.56; H, 5.09; N, 16.66. Found: C, 53.55; H, 5.07; N,16.68. 4.2.5.3.. Nl-(4-Chlorobenzyl)-1-(5-nitrothiazol-2-yl)piperidin-4-amine (28). The compound1 was synthesized according tothe general procedure using1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), and 4-chloro benzaldehyde (0.068 g,0.482 mmol) to afford 28(0.119g,76.7%) as pale yellow solid.Mp: 126-128C. H NMR (CDCl3): 8H 1.36-3.75 (m, 11H),7.33-7.39 (m, 4H), 8.4(S, 1H).1C NMR (CDCl3): 8c172,147.7,138.3,135.3, 132.8,130.1,128.7,51.7,48.6,46.6,30.8 .ESI-MS m/z353.3 (M+H). Anal. Calcd for C15H17ClN402S: C, 51.06; H, 4.86;N, 15.88. Found: C, 51.07; H, 4.85; N, 15.89. 4.2.5.4.N-(4-Methoxybenzyl)-1-(5-nitrothiazol-2-yl)piperidin-4-amine (29). The compound was synthesized according tothe general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), and 4-methoxybenzaldehyde (0.066 g,0.482 mmol) to afford 29 (0.136 g, 88.9%) as pale yellow solid. Mp: 143-145C. 1H NMR (CDCl3):8g 1.28-3.18 (m, 9H), 3.71 (s,2H), 3.87 (s, 3H), 6.95-7.29(m, 4H), 8.38 (s, 1H). 13CNMR (CDCl3):8c 171.8, 159.1,147.6,135.6,132.4,131.6,113.8, 56, 51.6,48.7,46.6, 30.6. ESI-MS m/z 349.1 (M+H). Anal. Calcd for C16H20N403S:C, 55.16; H, 5.79; N, 16.08. Found: C, 55.17; H, 5.8; N, 16.09. 4.2.6. Procedure for the synthesis of 1-(5-nitrothiazol-2-yl)-N-phenylpiperidin-4-amine (30) A suspension of 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g,0.438 mmol), phenylboronic acid (0.134g, 1.1 mmol), anhydrousCu(OAc)2 (0.174 g, 0.96 mmol), pyridine (0.087, 1.1mmol) indichloromethane (10 mL) was stirred at room temperature for24 h. The reaction was stirred at room temperature and the pro-gress monitored by TLC and LCMS. The reaction mixture was fur-ther quenched with water and the layers separated. The aqueouslayer was again extracted with dichloromethane (15 mL). The com-bined organic layer was washed with brine (10 mL), dried overanhydrous sodium sulfate, evaporated in vacuo and residue puri-fied by column chromatography using hexane/ethylacetate as elu-ent to afford 30(0.07 g, 52.6%) as orange solid. Mp:183-185C. 1HNMR(CDCl3): 81.59-3.91(m,9H),6.86-7.29(m,5H), 8.39 (s, 1H).13Cc NMR (CDCl3): 8171.9, 147.7,147.4,135.8,129.6,120.6,113.7,55.9, 49, 31.6. ESI-MS m/z 305.1(M+H)*. Anal. Caled for C14H16N4OS:C,55.25; H, 5.30; N,18.41. Found: C, 55.23; H, 5.29; N,18.39. 4.2.7. General procedure for the synthesis of amide derivatives(31-35) Toasolution of 1-(5-nitrothiazol-2-yl)piperidin-4-amine(1 mmol) in dry dichloromethane (3 mL) was added triethylamine(2 mmol) and corresponding acid (1 mmol) at 0°C. Propylphos-phonic anhydride solution (≥50 wt % in ethyl acetate; 2.5 mmol)was then added drop wise to the reaction mixture and was stirredat rt for 6 h (monitored by TLC & LCMS for completion). The reac-tion mixture was then washed with water (2 mL), brine(2 mL), anddried over anhydrous sodium sulfate and evaporated in vacuo. Theresidue obtained was then recrystallised from diethylether. 4.2.7.1. N-(1-(5-Nitrothiazol-2-yl)piperidin-4-yl)benzamide(31) The compound was synthesized according to the generalprocedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g,0.438 mmol), and benzoic acid (0.054 g, 0.438 mmol) to afford 31(0.123 g, 84.3%) as pale yellow solid. Mp:249-251 C. lH NMR(CDCl3): 8H 1.62-4.12(m, 9H), 7.45-8.37(m, 5H), 8.41 (s, 1H).c NMR (CDCl3):8c172,165.7,147.6,134.4,131.2,131.1,128.2,127.2, 47.4, 45.5, 30.4. ESI-MS m|z 333.1 (M+H). Anal. Calcd forC15H16N403S: C, 54.20; H, 4.85; N, 16.86. Found: C, 54.18; H,4.86; N, 16.83. 4.2.7.2. 4-Fluoro-N-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)benz-amide (32). The compound was synthesized according tothe general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), and 4-fluorobenzoic acid (0.061g,0.438 mmol) to afford 32 (0.126g, 82.4%) as pale yellow solid.Mp: 175-177℃.H NMR (CDCl3): 8 1.40-3.91 (m, 9H), 7.46-8.4(m, 5H), "’c NMR (CDCl3): 8c172.1,166.9,166.2,147.6,134.6,129.6,128.9,115.5,47.5,45.6, 30.5. ESI-MS m|z 351 (M+H)*. Anal.Calcd for C15H15FN403S: C, 51.42; H, 4.32; N, 15.99. Found: C,51.44; H, 4.31; N, 16. 4.2.7.3. 4-Chloro-N-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)benz-amide (33). The compound was synthesized according to thegeneral procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1g, 0.438 mmol), and 4-chorobenzoicacid (0.069g,0.438 mmol) to afford 33 (0.14g, 87.5%) as pale yellow solid. Mp:210-212℃. 'H NMR (CDCl3): 8 1.42-3.94 (m, 9H), 7.73-7.92 400-188-0725 (m, 4H), 8.39 (s,1H).1CNMR(CDCl3): 8 172.1,167.1,147.7,137.8,134.5,132.4,130.3,129.1,47.6,45.5, 30.5. ESI-MS m/z 366(M+H)*.Anal. Calcd for C15H15CIN403S: C, 49.11; H, 4.12; N, 15.27. Found: C,49.11; H, 4.12; N,15.27. 4.2.7.4. 4-Methoxy-N-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)benzamide (34). The compound was synthesized accord-ing to the general procedure using 1-(5-nitrothiazol-2-yl)piperi-din-4-amine (0.1 g, 0.438 mmol), and 4-methoxybenzoic acid(0.067 g, 0.438 mmol) to afford 34 (0.136 g, 86%) as pale yellowsolid. Mp: 177-179C.1H NMR (CDCl3): 8H 1.36-3.71 (m, 9H),3.86 (s, 3H), 7.21-8.38 (m,5H) 1cNMR (CDCl3): 8 171.9, 166.9,164.2,147.6,134.6,132.8,126.5,114.6,56.2,47.4,45.4, 30.3.ESI-MS m/z 363.1 (M+H)*. Anal. Calcd for C16H18N404S: C, 53.03;H, 5.01; N, 15.46. Found: C, 53.01;H, 5.03; N, 15.43. 4.2.7.5. N-(1-(5-Nitrothiazol-2-yl)piperidin-4-yl)-2-phenylacet-amide (35). The compound was synthesized according tothe general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1g,C0.438 mmol), andphenylacetic acid (0.06g,0.438 mmol) to afford 35 (0.12 g, 79%) as pale yellow solid. Mp:190-192C.H NMR (CDCl3): 8H 1.45-3.69(m, 9H), 3.87 (s,2H),7.20-7.30 (m, 5H), 8.39 (s, 1H). 13c NMR (CDCl3): 8172,169.5,147.6, 136.4,135.8,128.8,128.1,126.2,47.11,44.8,42.3,30.4.ESI-MS m/z 347 (M+H). Anal. Calcd for C16H18N403S: C, 55.48;H,5.24; N, 16.17. Found: C, 55.46; H, 5.27; N, 16.15. 4.2.8. General procedure for the synthesis of sulphonamidederivatives (36-40) Toasolutionof1-(5-nitrothiazol-2-yl)piperidin-4-amine(1 mmol) in dry dimethylformamide (3 mL) was added triethyl-amine(2 mmol) and corresponding sulphonyl chloride (1.1 mmol)at 0 C. The reaction mixture was slowly warmed to rt and stirredat rt for 6h (monitored by TLC & LCMS for completion). The reac-tion mixture was washed with water (2 mL), brine (2 mL), driedover anhydrous sodium sulfate and evaporated in vacuo. The resi-due obtained was then either recrystallised from diethylether orpurified by column chromatography. 4.2.8.1.N-(1-(5-Nitrothiazol-2-yl)piperidin-4-yl)benzenesulfon-amide (36). The compound was synthesized according to thegeneral procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine(0.1g,0.438 mmol), andbenzene.Ssulfonylchlorid:e (0.085 g,0.482 mmol) to afford 36 (0.136 g, 84.5%) as pale yellow solid.Mp: 184-186℃.H NMR (CDCl;):8g 1.33-3.84 (m, 9H), 7.68-7.84 (m, 5H), 8.39 (s, 1H). 13c NMR (CDCl;): 8171.9, 147.4,140.8,135.9,132,130.4,129.2,48.9, 46.5,31.2.ESI-MS m/z 369.2(M+H) . Anal. Calcd for C14H16N404S2: C,45.64; H, 4.38; N, 15.21.Found:C, 45.66; H, 4.39; N, 15.2. 4.2.8.2. 4-Fluoro-N-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)ben-zenesulfonamide:(37). The compound was synthesizedaccording to the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1g, 0.438 mmol), and 4-fluorobenzenesulfonylchloride (0.094g, 0.482 mmol) to afford 37 (0.126g,74.6%) as pale yellow solid.Mp: 177-179C. 1H NMR (CDCl3): 81.36-3.88(m,9H), 7.4-7.89(m,4H), 8.41(s,1H).CNMR(CDCl3):8c172,166.2,147.4,140.2,135.9,130.6,116.1,49,46.6, 31.3. ESI-MS m|z 387.1 (M+H). Anal. Calcd for C14H15FN404S2: C, 43.51; H,3.91; N, 14.50. Found: C, 43.53; H, 3.89; N,14.48. 4.2.8.3.5.4-Chloro-N-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)ben-zenesulfonamide (38). The compound was synthesizedaccording to the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), and 4-chlorobenzenesulfonylchloride (0.1 g, 0.482 mmol) to afford 38 (0.152 g, 86.4%) as pale yellow solid. Mp: 201-203C. 1HNMR (CDCl;): 8q 1.37-3.84 (m, 9H), 7.67-8.05 (m, 4H), 8.36 (s, 1H).13C NMR (CDCl3):8171.8,147.5,140.6,137.2,135.9,129.4,128.2,48.9,46.6,31.3.ESI-MS m|z 403.4 (M+H). Anal. Calcd for C14H15CIN404S2: C,41.74; H, 3.75; N, 13.91. Found: C, 41.76; H, 3.77; N, 13.93. 4.2.8.4. 4-Methoxy-N-(1-(5-nitrothiazol-2-yl)piperidin-4-yl)benzenesulfonamide (39). The compound was synthesizedaccording to the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol), and 4-methoxybenzenesulfonylchloride (0.1 g, 0.482 mmol) to afford 39 (0.147 g, 84.5%)as pale yellow solid. Mp: 186-188C.H NMR (CDCl3): 8n 1.34-3.8 (m, 9H), 3.83 (m, 3H), 7.21-7.61(m, 4H), 8.38(s, 1H)..13cNMR (CDCl): 8c171.7,163.8,147.3,136.8,135.9,126.2,114.7,56.1,48.9, 46.5, 31.2. ESI-MS m/z399.1(M+H).Anal.Calcd for C15.H18N405S2: C,45.21; H, 4.55; N, 14.06. Found: C, 45.23; H,4.54;N14.07. 4.2.8.5. N-(1-(5-Nitrothiazol-2-yl)piperidin-4-yl)-1-phenylme-thanesulfonamide (40). The compound1 was synthesizedaccording to the general procedure using 1-(5-nitrothiazol-2-yl)piperidin-4-amine (0.1 g, 0.438 mmol),and phenylmethane sul-fonylchloride (0.092g, 0.482 mmol)to afford 40 (0.118 g,70.2%) aspale yellow solid. Mp: 141-143C. 1H NMR (CDCl3): 8g1.38-3.84(m, 9H), 4.29 (s, 2H),7.23-7.51 (m, 5H), 8.39 (s, 1H). 13c NMR(CDCl3): oc171.8,147.4,136,133.4,130.7,128.8,125.5,66.7,49,46.7,31.3.ESI-MS m|z 383.1(M+H).Anal. Calcd for C15H18N404S2:cl, 47.11; H, 4.74; N, 14.65. Found: C, 47.13; H, 4.71; N, 14.67. 4.3. Biological evaluation 4.3.1. Protein cloning,expression and purification Cloning of M. smegmatis gyraseB gene was done separately byamplifying the gyrB gene from mc²155 genomic DNA using thespecific forward and reverse primers using specific restrictionenzymes5'CACCCATATGGTGGCTGCCCAGAAGAACAA 3' (Ndel),and5AGCTAAGCTTTTAAACATCCAGGAAGCGAA 3'(Hind III),respectively. The final PCR amplicons obtained were cloned inexpression vector pQE2 (Qiagen) with a 6-His tag and was thentransformed into host BL21 (DE3) pLysS cells, specifically becauseof the compatibility of the host system for gyrB protein expressionand structurally similar protein folding. The vector transformedcells were later grown at 37℃ in LB broth containing 100 ug/mLampicillin upto an optical density (OD) of 0.4 to 0.6 at (A595). Fur-ther for the increased production of protein the bacterial cells,were induced with isopropyl-B-p-thiogalactopyranoside (IPTG) ata final concentration of 0.1 mM while the cells were in exponentialgrowth phase, and the cell growth was continued for another 12 hat 18C, as low temperature promotes the slow release of thedesired protein. The bacterial cells were then centrifuged at7000 rpm, 4℃ for 15 min. The cell pellets were re-suspended inPBSG buffer (PBS containing 5% glycerol), further lysed using soni-cator (20 s pulse and 45 s halt) and centrifuged the crude lysate at8000 rpm, 4℃ for 10 min, subsequently centrifugation wasrepeated for the supernatant of previous step at 10,000 rpm at4°℃ for 35 min for a clearer supernatant. The cell extract was laterapplied to pre-equilibrated Ni-NTA column (GE), and the columnwas washed with wash buffer (5% glycerol in PBS and 500 mMNaCl) and the protein was eluted using different concentration ofimidazole ranging from 10 mM to 200 mM in the elution buffer(5% Glycerol, 140 mM NaCl in 25 mM Tris-Cl (pH 8.0)). Fractionscontaining the desired gyrB enzyme was confirmed by runningan SDS-PAGE(sodium dodecyl sulfate-polyacrylamide gel electro-phoresis), subsequently pooled the 100 mM and 200 mM fractionsand dialyzed against the dialyses buffer (15% Glycerol, 140 mMNaCl in 25 mM Tris-HCl (pH7.4)), aliquoted and stored at -80 ℃. 4.3.2. MS gyrB ATPase assay The ATPase assay was performed similar to the previously pub-lished method using Mycobacterium smegmatis gyraseB domain,it was performed in 30 uL reaction volume consisting of assay buf-fer (60 mM HEPES-KOH pH 7.7, 250 mM potassium glutamate,200 mM KCl,2 mM magnesium chloride, 1 mM DTT, 2% Glycerol,4% DMSO, 0.001% BriJ) incubated with 90 nM gyrB, 0.65 mM ATPand inhibitors for 120 min at 25C. The reaction was initiated bythe addition of 16 uL of MgCl2 solution, as metal ion triggers theenzymatic function. Later the reaction was quenched by theaddition of 20 uL of malachite green (Bioassay systems, USA) andincubated for 20 min, absorbance was measured at 635 nm wave-length against the blank. Throughout the assay novobiocin wasconsidered as positive control. Dose-response curve was plottedfor the potent leads using GraphPad Prism software (GraphPadSoftware Inc., La Jolla, CA) by taking log (inhibitor concentration)on the X-axis and response (%inhibition) on the Y-axis. 4.3.3. MTB DNA Super coiling assay The binding affinities of the most potent ligands were evaluatedby measuring fluorescence of the native protein and the protein-ligand complexes in the presence of a fluorescent dye whosefluorescence increases when exposed to non-polar residues of theprotein and reaches a maximum when the protein denatures. Inbrief, the native protein (7.5 pL(1.5 mg/mL)of protein +3.5 pL ofbuffer [50 mM Tris (pH 7.4), 1 mM ethylene diamine tetra-aceticacid (EDTA), 5 mM DTT]) was subjected to stepwise heating in agradient PCR instrument (Bio-Rad) from 25℃ to 100℃ with anincrement of 0.6℃/min in the presence of the fluorescent dyeSYPRO Orange [2.5 pL (1:100); Sigma]. As the temperatureincreases, the stability of the protein decreases and becomes zeroat equilibrium, where the concentrations of folded and unfoldedprotein become stable. This temperature was noted as the meltingtemperature (Tm). The dye exhibited maximum fluorescence at thispoint as it was exposed to the hydrophobic portion of the proteinas a result of protein denaturing. A higher or positive shift in Tmof the protein-ligand complex compared with the native proteinsignifies better stabilisation of the protein-ligand complex, whichin turn is a reflection of inhibitor binding. 4.3.5. In vitro Mycobacterium tuberculosis microplatealamarBlue@ assay (MABA) Briefly, the inoculum was prepared from fresh LJ medium re-suspended in 7H9-S medium (7H9 broth, 0.1% casitone, 0.5% glyc-erol, supplemented oleic acid, albumin, dextrose, and catalase[OADC]), adjusted to a McFarland tube No. 1, and diluted 1:20;100 uL was used as inoculum. Each drug stock solution wasthawed and diluted in 7H9-S at four-fold the final highest concen-tration tested. Serial two-fold dilutions of each drug were prepareddirectly in a sterile 96-well microtiter plate using 100 uL 7H9-S. Agrowth control containing no antibiotic and a sterile control werealso prepared on each plate. Sterile water was added to all perim-eter wells to avoid evaporation during the incubation. The platewas covered, sealed in plastic bags and incubated at 37℃ in nor-mal atmosphere. After 7 days incubation, 30 pL of alamar bluesolution was added to each well, and the plate was re-incubatedovernight. A change in colour from blue (oxidised state) to pink(reduced) indicated the growth of bacteria, and the MIC wasdefined as the lowest concentration of drug that prevented thischange in colour. 4.3.6. Screening for hERG channel inhibition The potent compounds were further examined for hERG chan-nel inhibition by assessing their arrhythmogenic potential on thezERG gene, which is orthologous to the hERG gene. Zebrafish(Danio rerio) were acquired commercially and were maintainedaspreviously described by Banote et al. All experiments wereperformed as per the guidelines published by the US National Insti-tutes of Health for care and use of zebrafish. In brief, zebrafish weremaintained in the re-circulatory system with 14 h light and 10 hdark cycles, with 28°C as optimum temperature.34 Breeding wascarried out using 2 females: 3 males in a separate breeding cage.Embryos were collected into petridishes containing E3 mediumat incubated at 28°C temperature. Three day post fertilized embryos are used for screening of testdrugs and the embryos were segregated and washed and distrib-uted in 24 well plate probably six embryos with 250 uL of 0.1%DMSO per well. Stock solution of the drug was prepared in 100%DMSO and the 2X working concentrations were prepared accord-ingly by serial dilutions. Each well was then added with 250 uLof 2X working concentration and embryos were allowed toincubate at the optimum temperature for 4 h. The respective drugconcentration exposed embryos were treated with tricaine and theheart rate were measured, the time taken for 30 atrial and ventric-ular beats were measured for each embryo, from which number ofheart beats per minute was calculated by using the for-mula=1800/X=beats/minute (where X=time in seconds). Allthe statistical analysis was done using GraphPad Prism softwareapplying ONEWAY-ANOVA and Dunnett's post-test. 4.3.7. In vitro cytotoxicity screening All the test compounds were further examined for toxicity in aRAW 264.7 mouse macrophage cell line at a concentration of50 uM. The assay was performed in RPMI media with 5% FBS and1% antibiotic. After 72 h of drug exposure, viability of the cellswas assessed on the basis of cellular conversion of MTT into a for-mazan product using the Promega Cell Titer 96 non-radioactivecell proliferation assay." Acknowledgement D.S. acknowledges the University Grant Commission, India for aU.G.C. research award. ( 1. 1 h t tp ://www.who.int/ m e d i ac e ntr e /factsheets/fs10 4 / en/28/ 04/2014. ) ( 2. W \ HO Global Tuberculosis Report 2013 http : //www.who.int/tb/publications/ global _ r eport/en/ . ) 3Dye, C.; Williams, B. G. Science 2010, 328,856.45 ( Siddiqi, M. L. ; Kuma r , A. Expert Opi n Drug Disco v . 2009,4 , 1005. ) ( L i e nhardt, C.; V ernon, A.; Raviglione, M. C . C urr. O pin. P ulm. Med. 2010, 16, 1 8 6 . ) ( 6. P ayne, D. J. ; Gwynn, M.N. ; H olmes,D. J . ; Po m pliano, D. L. N at. Re v . Drug Disc. 2007,6,29. ) ( 7. 1 K oul, A . ; Ar n oult, E.; Lo u nis, N. ; Guillemon t , J ; Andries, K. Nature 2011,4 6 9, 483. ) ( 8 R avi g li on e , M. C . Bull. World Health Organ. 2007, 85, 3 27. 9 ) ( G ill,S. K.; Xu, H.; Kirchhoff, P . D.; C ierpicki,T.; Turb i ak, A. J; W a n, B.; Zhang, N. ; P eng, Kuan-Wei; Fr a nzblau, S.G. ; Ga r cia, G. A.; Showalter,H. D. H. J. Med. C h e m. 2012, 5 5,3814. ) ( 10. A ndr i es, K .; V erh a sselt, P . ; G uillemont, J ; G o hlman n , H. W . ; Neef s , J . M . ; W inkle r , H . ; V en Gestel, J .; T i mmerman, P.; Zhu, M . ; L e e , E.; W illiams, P . ; de C h a ffoy, D .; H u i tric, E.; H offne r , S .; Ca m bau, E . ; Tr uff o t-Pernot, C . ; Lounis, N.; J arlier, V . A . Science 2005 , 307,223. ) 11.Ma, Z.; Lienhardt, C.; Mcllleron, H.; Nunn, A.; Wang, X. Lancet 2010, 375,2100.12.Lienhardt, C.; Raviglione, M.; Spigelman, M.; Hafner, R.; Jaramillo, E.; Hoelscher, M.; Zumla, A.; Gheuens,J. J. Infect. Dis. 2012,205,S241. ( 3 . D o ver, L. G. ; Coxon, G. D. J . Med.Chem. 2011 , 5 4 ,6157 . ) Cooper, C. B. J. Med. Chem. 2013,56,7755. ( O bla k , M . ; Kot ni k, M. ; Sol m aj e r, T . Curr. Med. Chem. 2007, 14,2033. ) ( b. C hopra, S . ; M atsuyama, K.; Tra n , T.; Maleric h , J . P .; Wan, B.; Fra n zblau, S. G.; L un, S .; G uo, H .; M aiga, M . C . ; B i s h a i, W. R. ; Ma d rid, P. B . J . A n t i microb. Chemother. 2012, 67,415. ) ( 17. K al e , M . G . ; R aichur k ar, A.; P, S . H . ; Waterson , D. ; McKinney, D . ; Manjunath a, M . R .; K ranthi , U . ; K o ushik, K.; Je n a, L . K. ; S h in d e, V. ; R u drapatna,S.; Barde, S . Humnaba d kar, V.; M adhavapeddi, P . ; Basavarajappa,H. ; Ghosh, A . ; R amya , V.; Gup t ha, S .; Shar m a, S.; Vac anguys.: A i has p ati, P .; Kumar, K. N .; G iridhar, J ; Redd y, P and u ga,V.; G a ngu l y, S . ; A huja, V. ; Gaonkar,S.; Kumar, C . N.;Ogg , D ; T ucke r A.; B oriack-Sjodi n , P. A . ; de Sousa, S. M.; Sambandamu rt hy, V . K .; Gho r pade, S . R . J . Med.Chem. 2013,56,8834 . ) ( 18 5 . . 1 M axwell, A. Trends M icrobiol. 1997, 5 , 1 02. ) ( 19. M a xw ell, A.; L awso n , D. M. Cu rr . T op. Med. Che m . 2 003, 3 , 2 2 83. ) 20.D.Cole,S.T.; Brosch,R.; Parkhill,J.; Garnier, T.; Churcher (C, Harriss. D.; Gordon, S.V.; Eiglmeier, K.; Gas,S.; Barry, C. E.; Tekaia, F.; Badcock, K., Basham,D.; Brown,D.; Chillingworth,T.; Connor, R.; Davies, R.; Devlin, K.; Feltwell, T.; Gentles, S.;Hamlin, N.; Holroyd, S.; Hornsby, T.; Jagels. K.; Krogh, A.: McLean Moule, S.;Murphy,L.; Oliver, K.; Osborne,J.; Quail, M. A.; Rajandream,M.A . Rogers,J; ( R utter, S .; Seeger , K.; S k elton,J; S qu a res, R . ; Squares,S. ; Sulston, J . E . ; T a ylor, K .;Whi t ehe a d,S.; B a rrell , B. G. Nature 1998, 3 93,537. ) 21. Schrodinger Suite 2012 Protein Preparation Wizard; Epik version 2.2,Schrodinger, LLC, New York, NY, 2012; Impact version 5.7, Schrodinger, LLC,New York, NY, 2012; Prime version 2.3, Schrodinger, LLC, New York, NY, 2012 ( 22. S hi r ude, P . S .;M a d h avaped d i, P. ; Tu c k er, J . A . ; Mu r ugan , K.; Pat i l, V . ;B asavarajappa, H.; R aich u rkar, A. V .; H umnaba d kar, V.; Hu s sein, S. ; S harma, S.; Ra m ya, V . K .; Narayan , C. B .; Ba l ganesh , T . S.; Sambandamurt h y, V. K. ACS Chem. Biol. 2013,8 , 519 . ) ( 23. J eankum a r, V. U. ; Janupally, R . ; Pul l a , V. K.; Son i , V.; Sri d evi, J. P.; Su r yadevara, P .; Shr a van, M .; Medish e tti, R . ; K u lka r ni , P . ; Yogeeswari, P . ; S r iram, D. I n t . J . A ntimicrob. Ag e nts 2013, 4 3, 269. ) ( 24. f. Jeankum a r, V . U . ; R e n uka, J; Sa n tosh, P.; S r i d evi, J . P . ; S u ryadevara, P. ; Y ogeeeswari, P . ; Sriram, D. Eur. J . Med. Chem. 2013, 7 0 , 1 4 0. ) ( 25. N iesen, F. H .; B erglund, H.; V eda d i, M. Nat. P rotoc. 2007,2, 2212. ) 26. ( Franzbalu, G. S.; Wi t zig, S. R. ; McLaughlin, C . J; Torres, P. ; Madico,Hernandez, A .; Degnan, T. M.; Cook, B . M . ;Quenzer, K. V .; Ferguson, M . .R. G ilman, H . R . J . C l in. Microbiol. 1998, 3 6, 362. ) ( 27. G e r l i er, D.; Th omasset , N. Im m unol. Methods. 1986,94,5 7 . ) ( 28. Je ankumar, V. U. ; Renuka, J.; K o t agiri, S . ; S axena,S. ; K a kkan, S . S : Sr i d e v i ,J. P. Y e l lanki, S.; K u lkarni, P. ; Yogeesw a r i , P. ; Sriram, D . C hemMedCh em: 2 01 4 , 9. 1850. ) ( 29. Ch a n, D . M . T . ; Monaco, K. L .; Wan g , R. P .; Winter, M . P.T e t rahedr o n L e tt. 1 998, 39.2933. ) ( 30. L am, P. Y . S.; Cl a rk, C . G .; Sa u b ern , S . ; Adams,J ; W i nter s, M . P .; C han , D . M. T . ; Combs , A. T etrahedron L ett. 1998, 39 , 94 1 . ) ( 31. J eankum ar , V . U. ; A l okam, R . ; S ridevi,J . P . ; Surya devar a, P.; M at i kond a , S. S. ; P eddi , S.; Sahithi , S .; Alvala , M.; Yoge e swari, P. ; Sr iram, D . Ch em. B iol. Drug Des. 2014,83,498. ) ( 3 2 2 . C ha u dha r i, G . H . ; C he n nubhotla , K . S . ; C ha t ti K. ; Ku l kar ni, P . j . Phar m acol. T oxi c ol . Methods 2013, 67 , 115 . ) ( 33. B a n ote , R . K .; K o uta r apu, S . ; Ch ennubh ot la, K . S. C h att i, K.; Kulkarni,P. Epilepsy B e hav. 201 3,27,212. ) ( 3 4 . Ku lkar ni , P . ; C haud h ari, G . H . ; Srip u r a m , V .B an o te, R . K.; Kirla, K. T . ; S ultana, R ao, P.; Oruganti, S. ; Ch atti , K. . P h Pharma co l. Re p .2014,66,179. ) http://dx.doi.org/j.bmc.@Elsevier Ltd. All rights reserved.www.rysstech.com士科技 www.rysstech.com耐士科技
确定












还剩12页未读,是否继续阅读?

上海鑫欣生物科技有限公司为您提供《化学药中主要物质含量分析检测方案 》,该方案主要用于化药新药研发中其他检测,参考标准--,《化学药中主要物质含量分析检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多