








方案详情
文
随着标准将于2021年11月1日正式实施,为了帮助企业快速进行方法重现的工作,上海汉尧仪器设备有限公司作为YMC色谱柱中国区代理商,依托相关实验室进行了以下不同品种的指纹图谱和含量图谱的方法重现工作:
陈皮配方颗粒标准重现
甘草(甘草)配方颗粒标准重现
防风配方颗粒标准重现
知母配方颗粒标准重现
连翘(青翘)配方颗粒标准重现
厚朴(厚朴)配方颗粒标准重现
白芷(白芷)配方颗粒标准重现
虎杖配方颗粒标准重现
猫爪草配方颗粒标准重现
方案详情

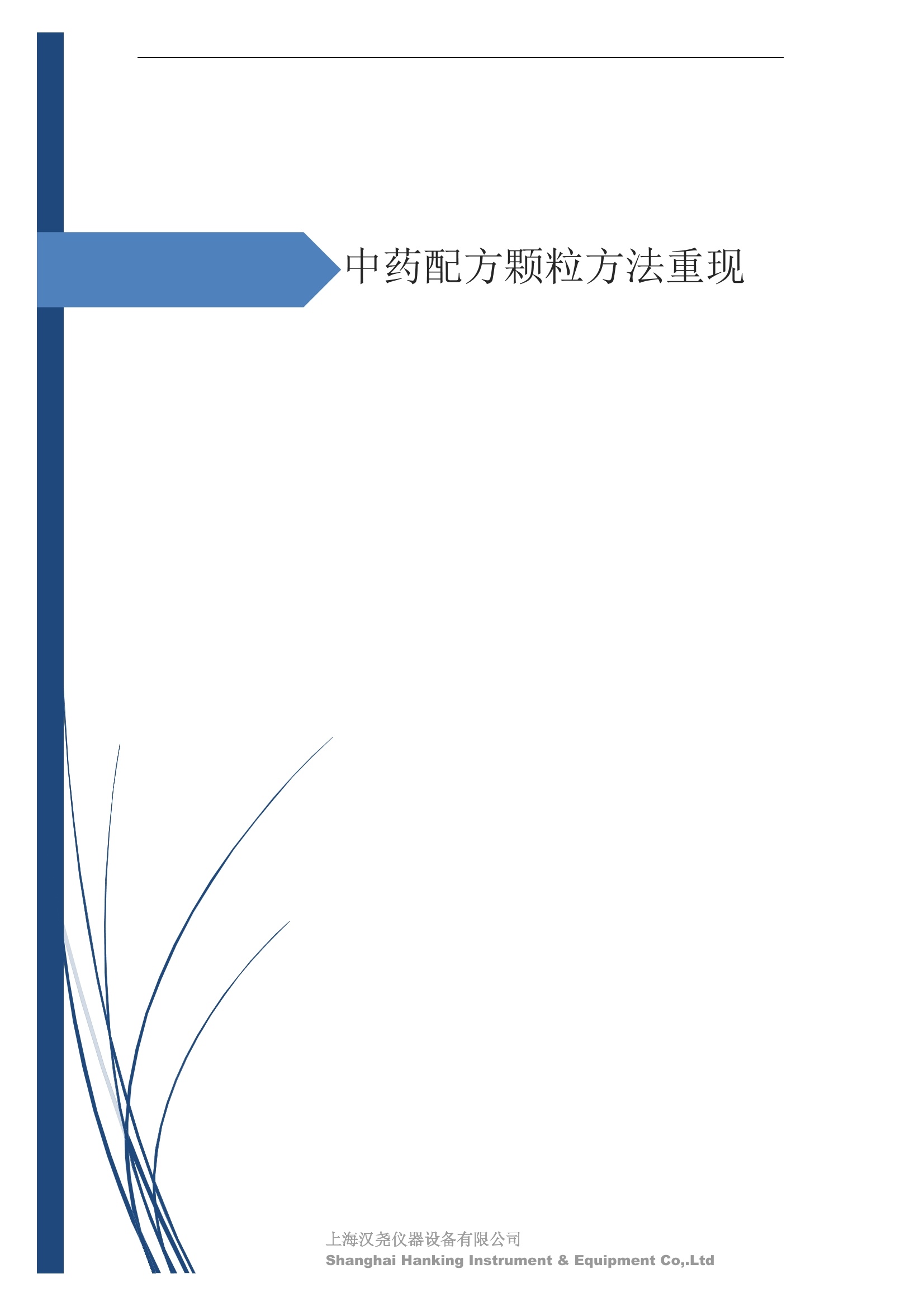
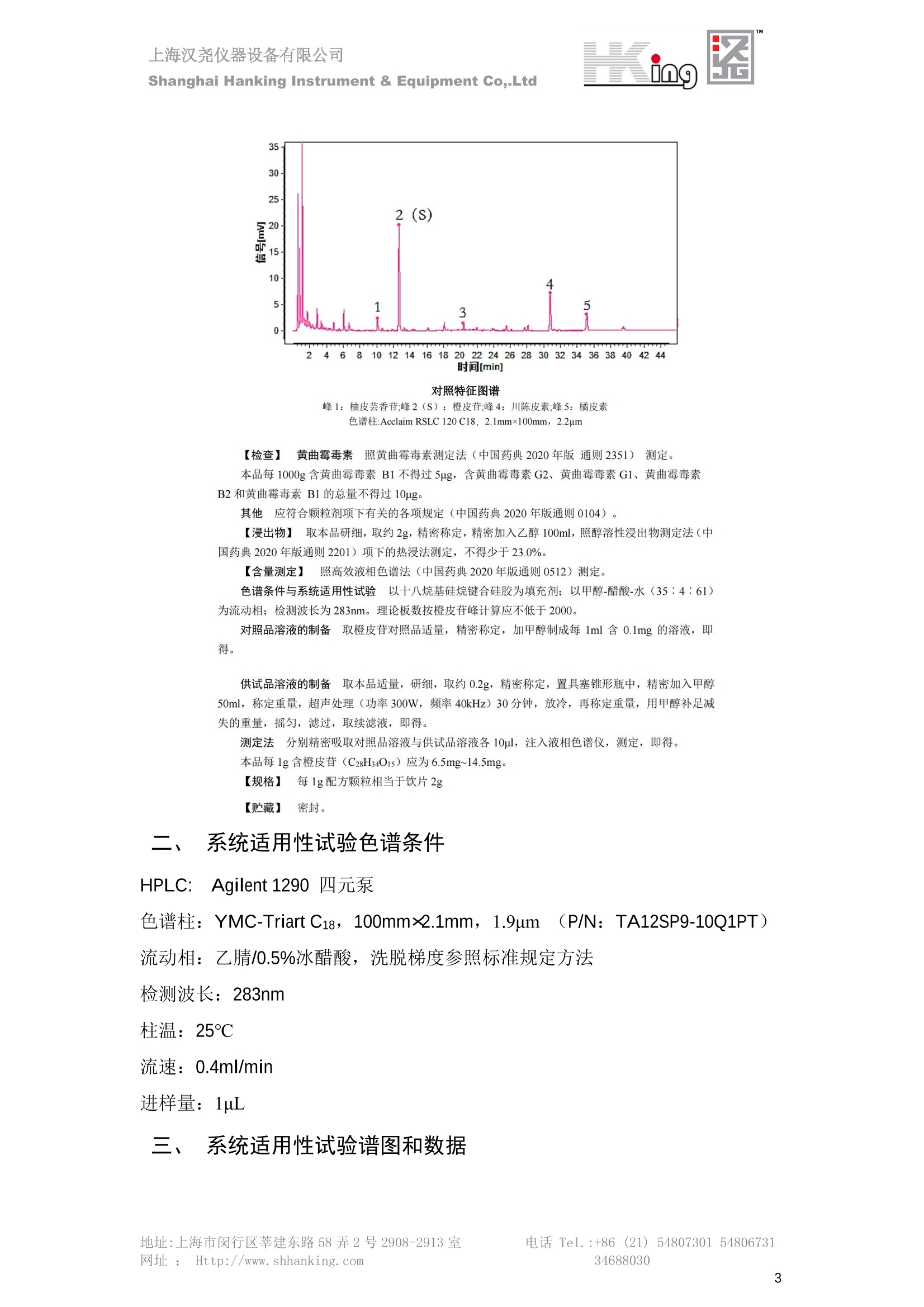
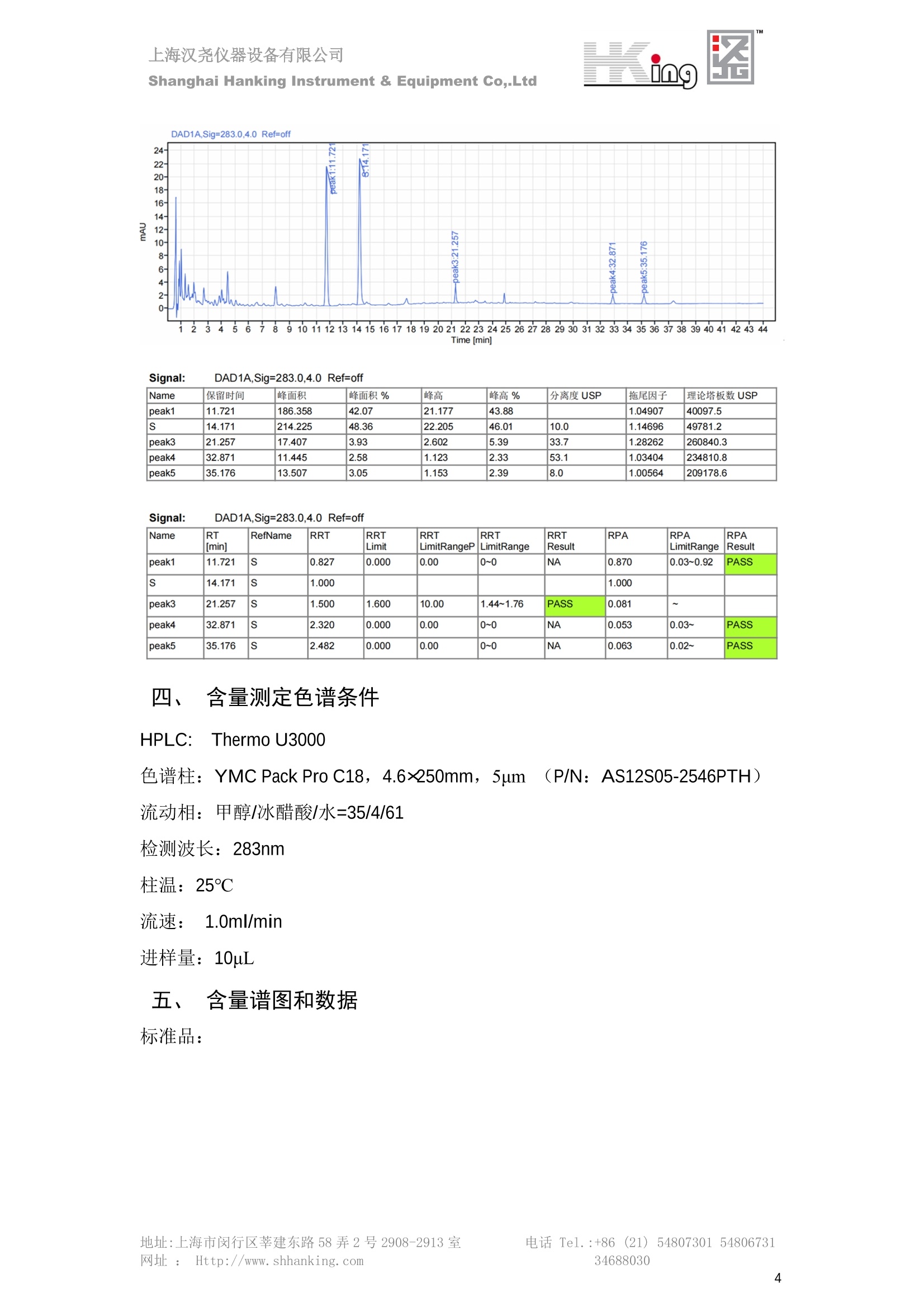
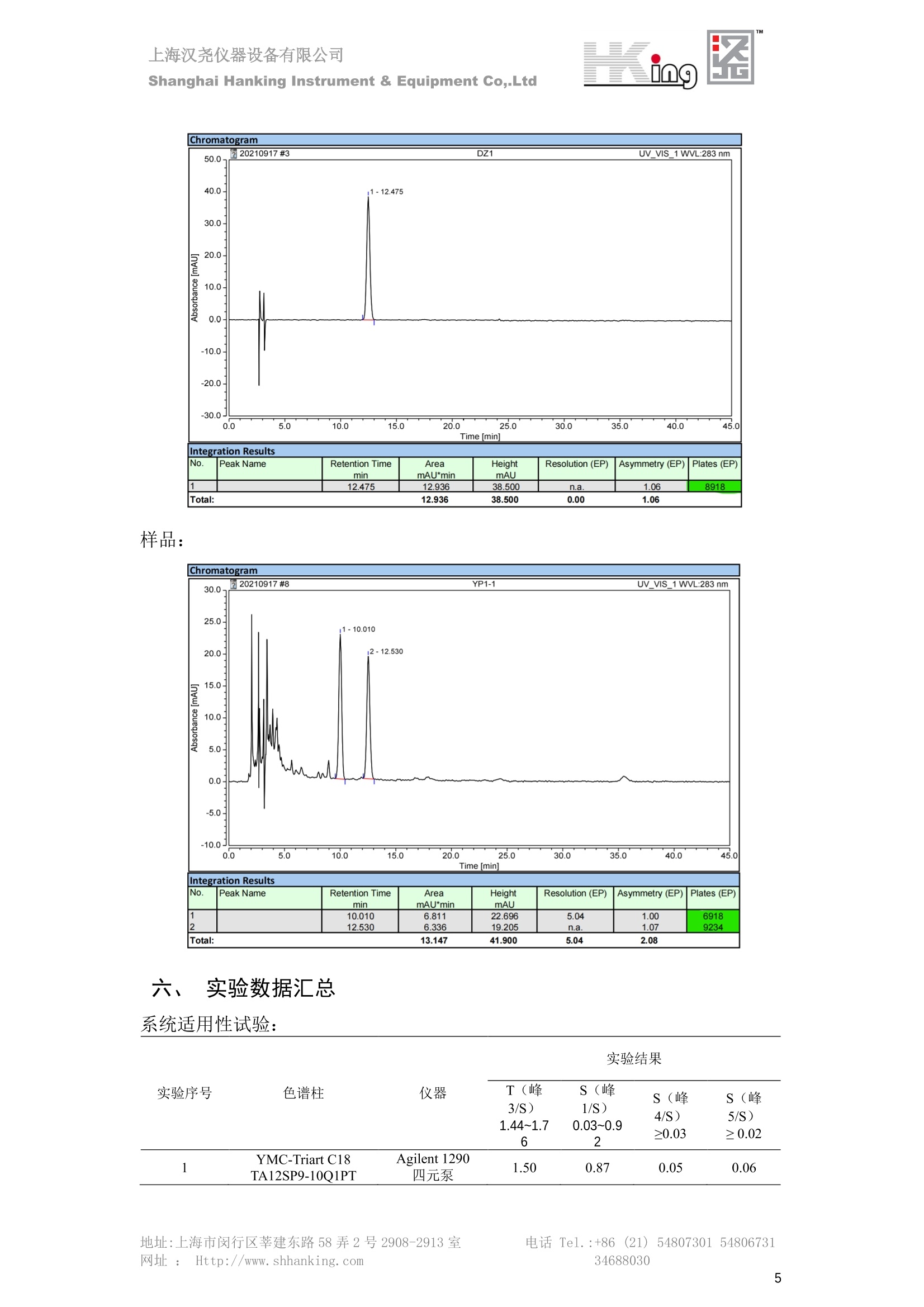
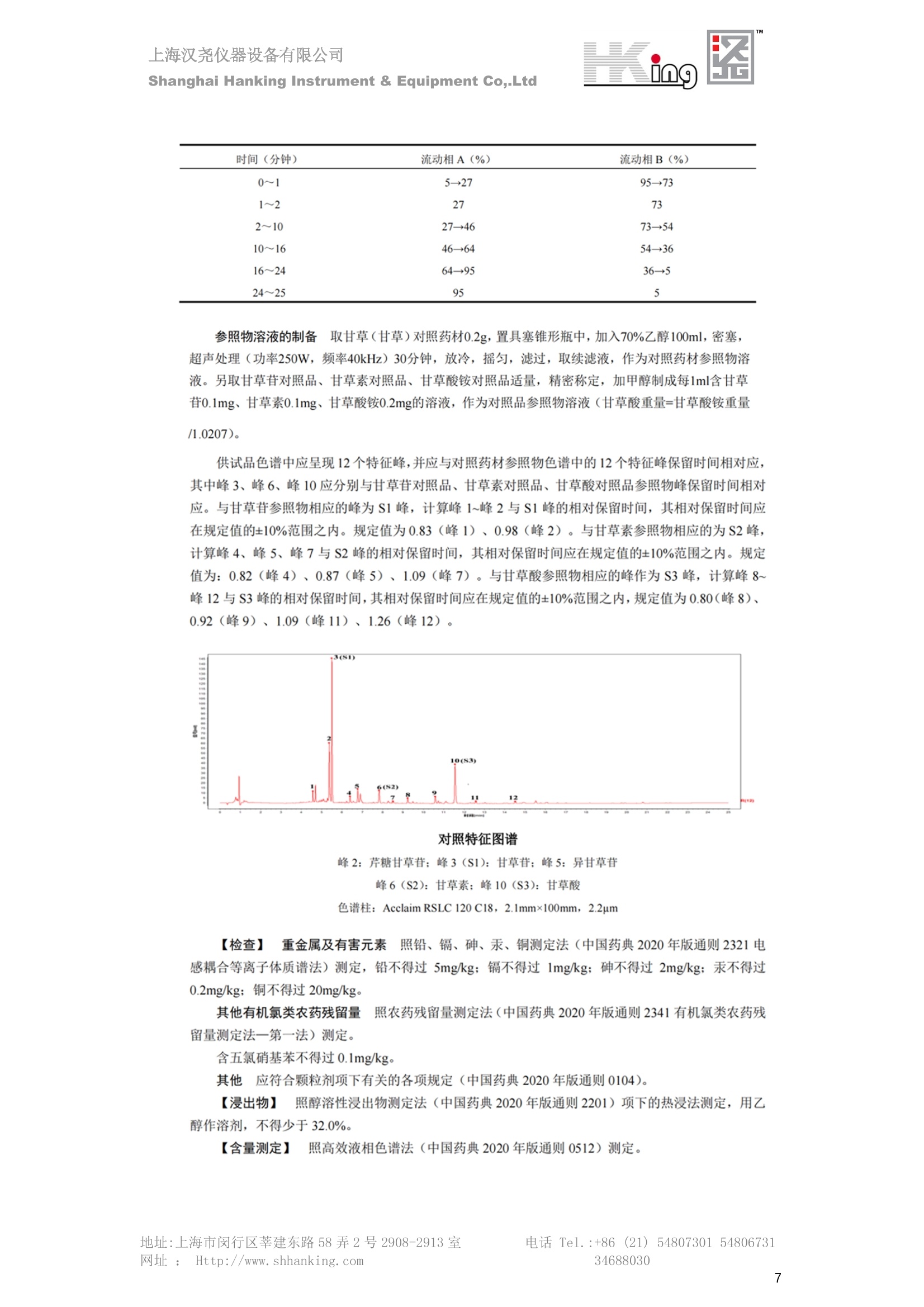
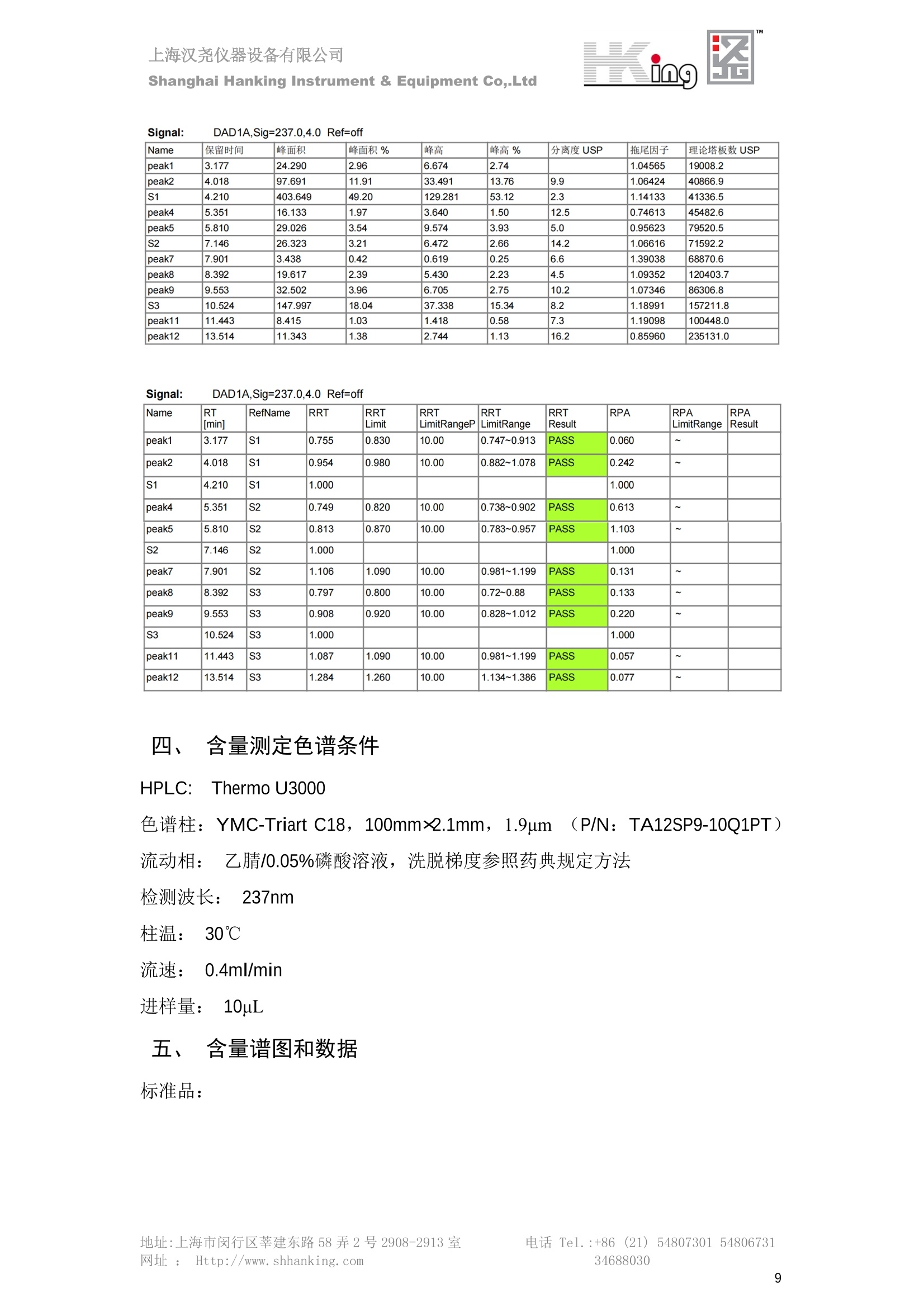
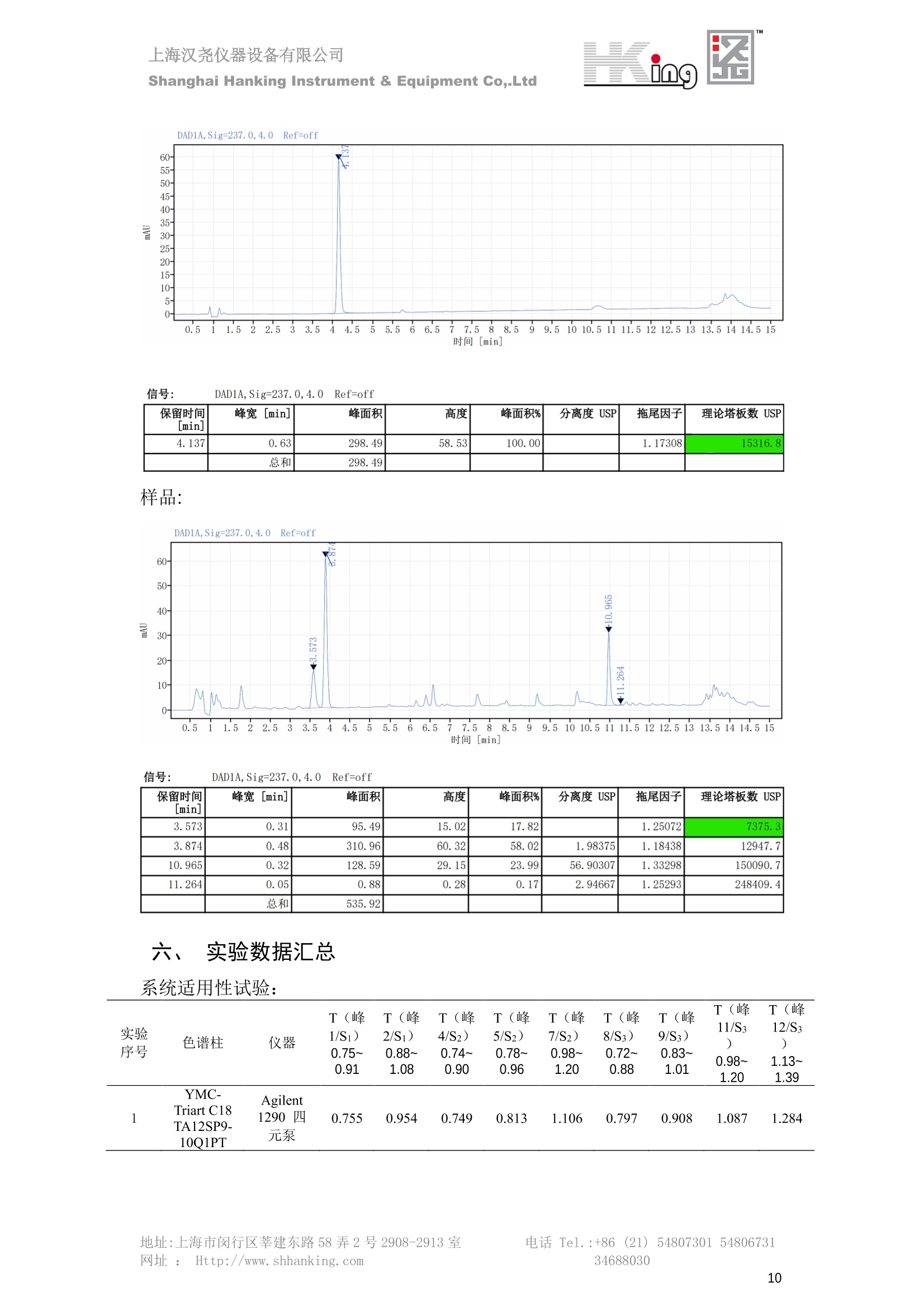
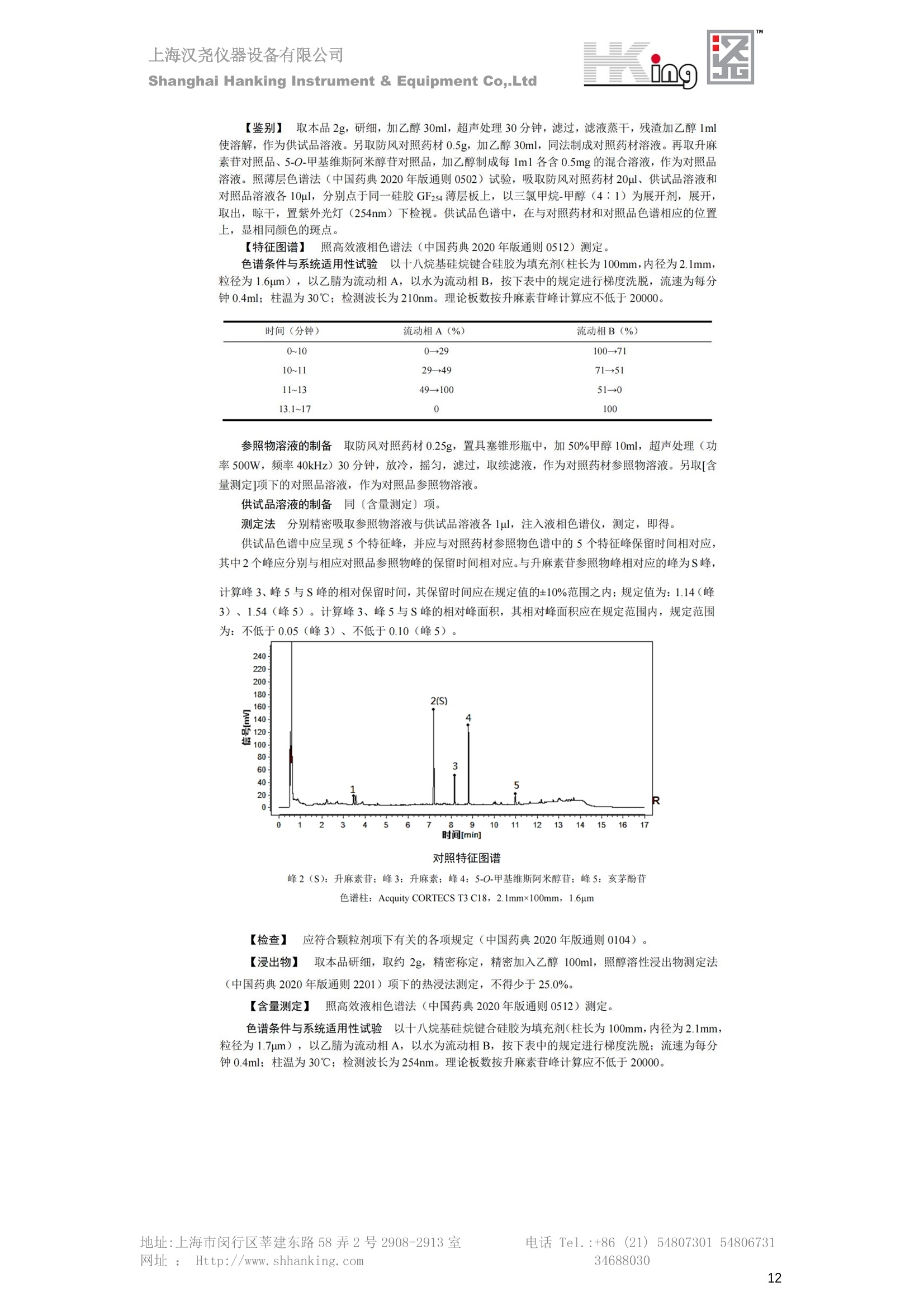
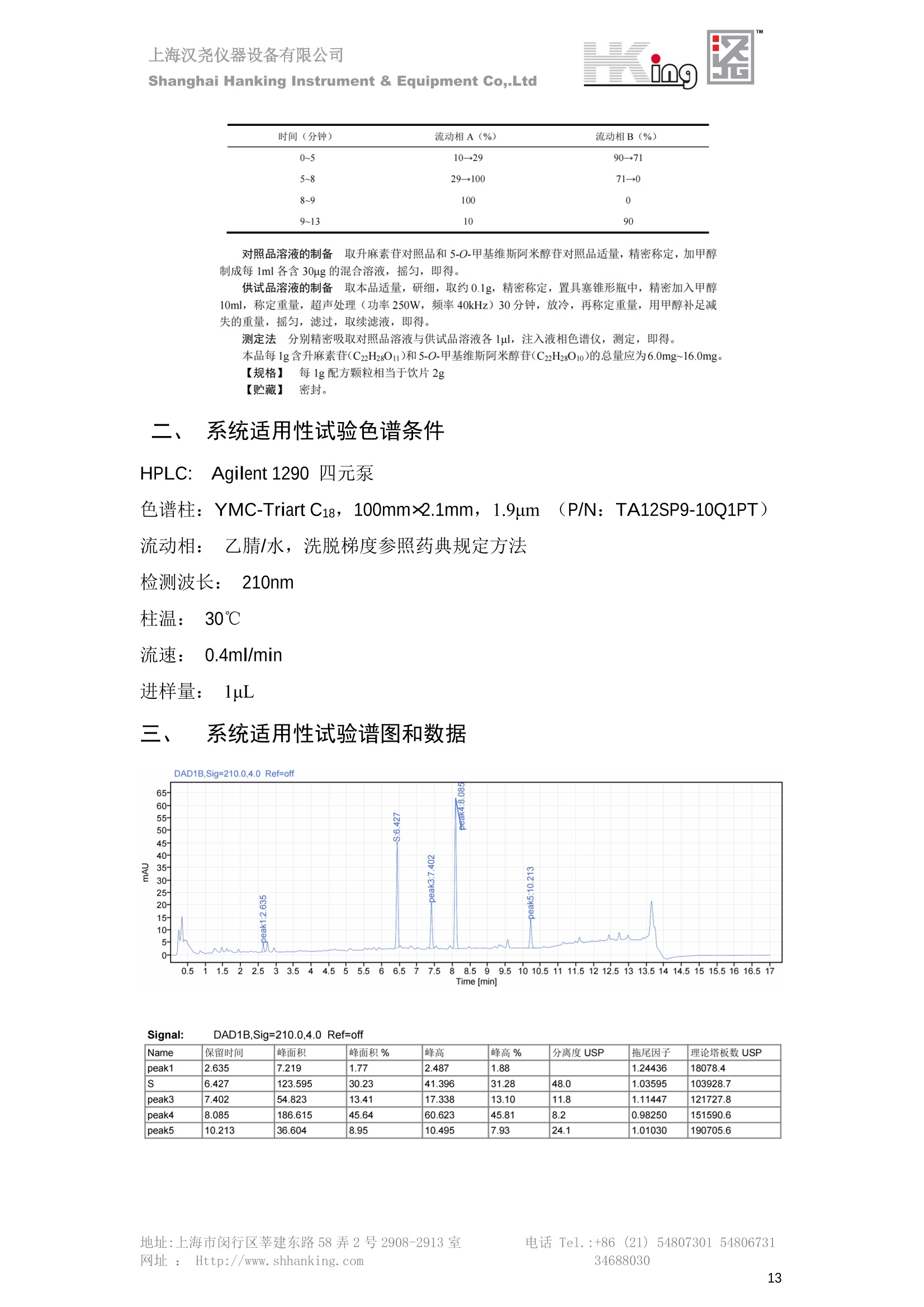
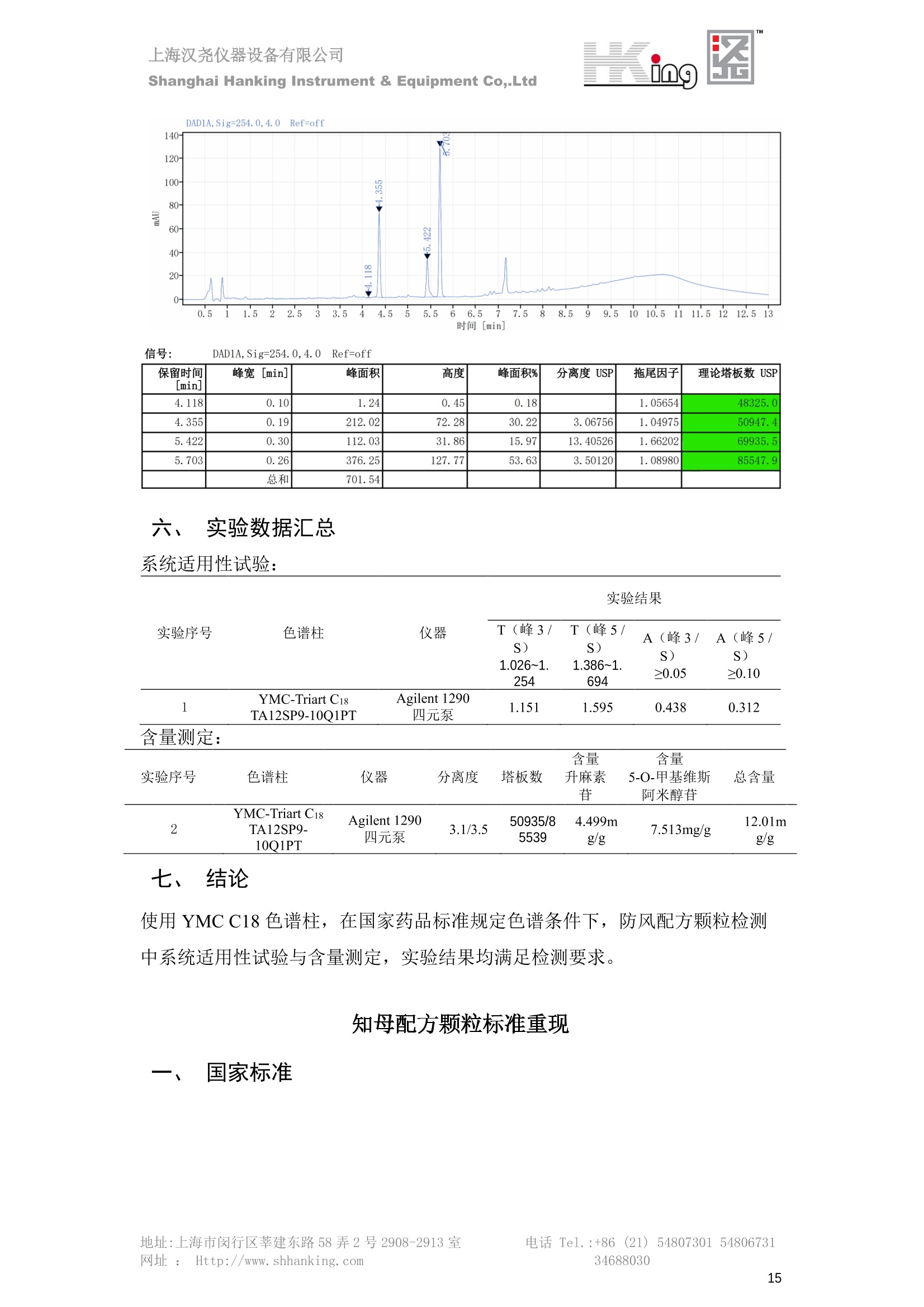
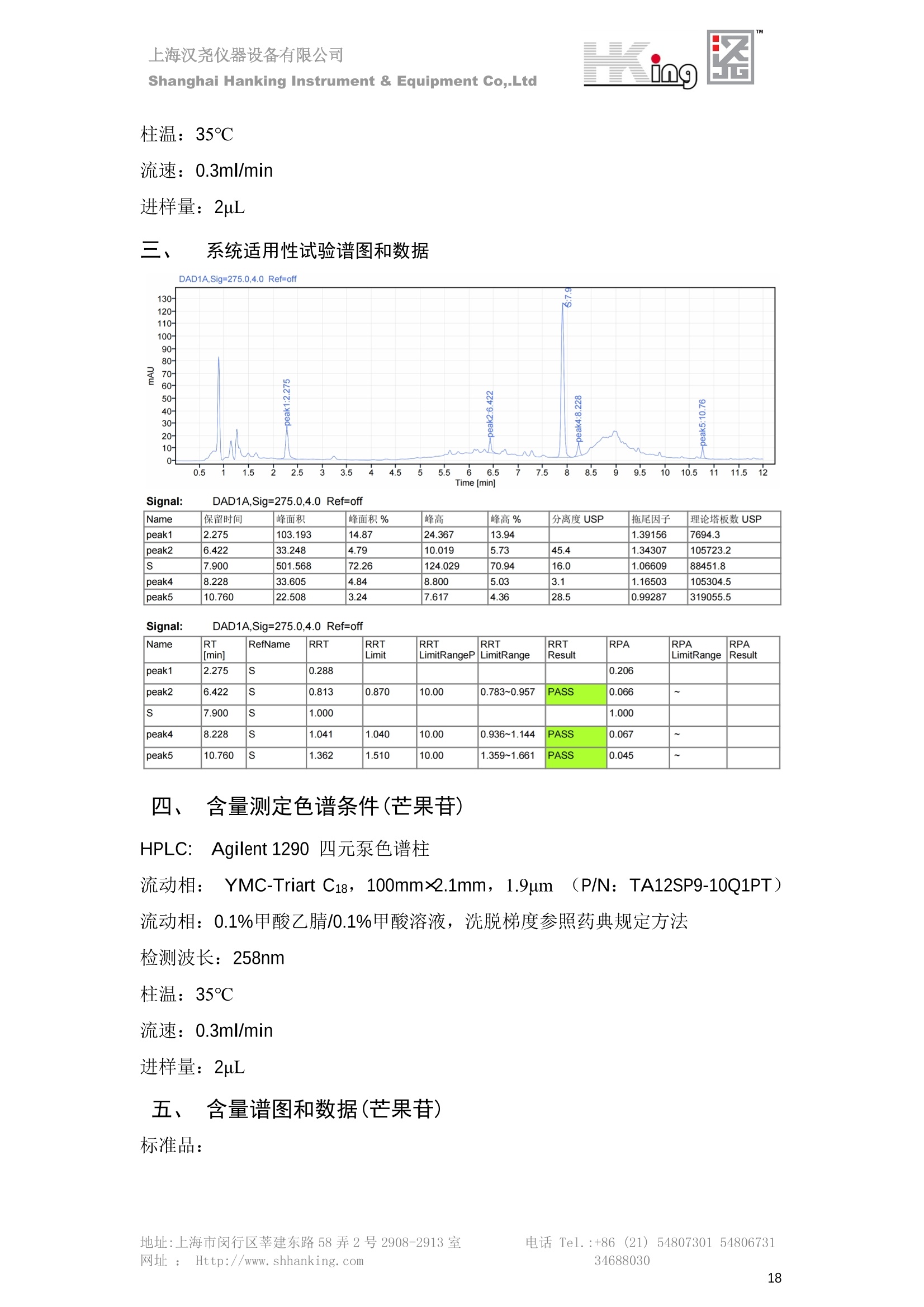
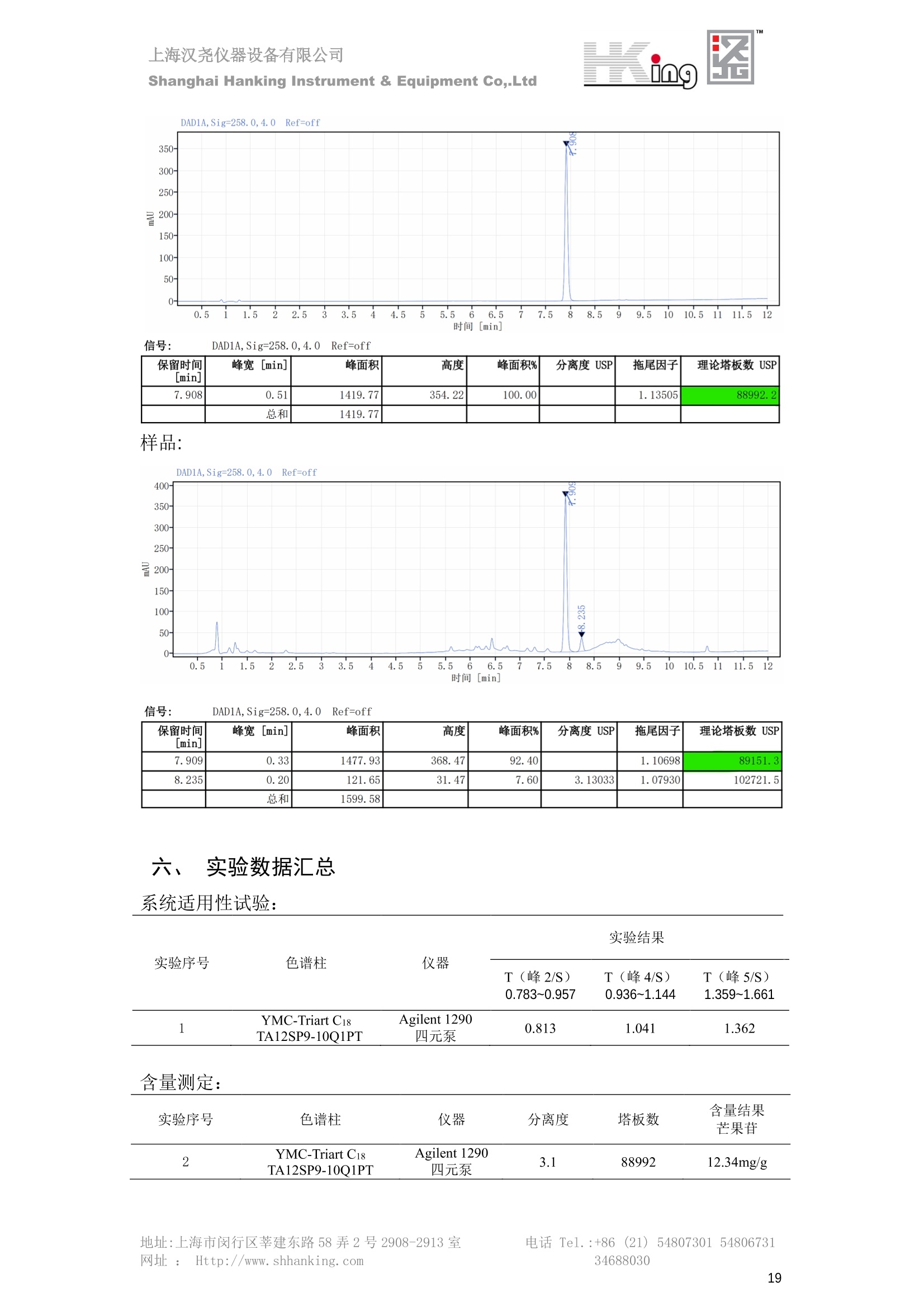
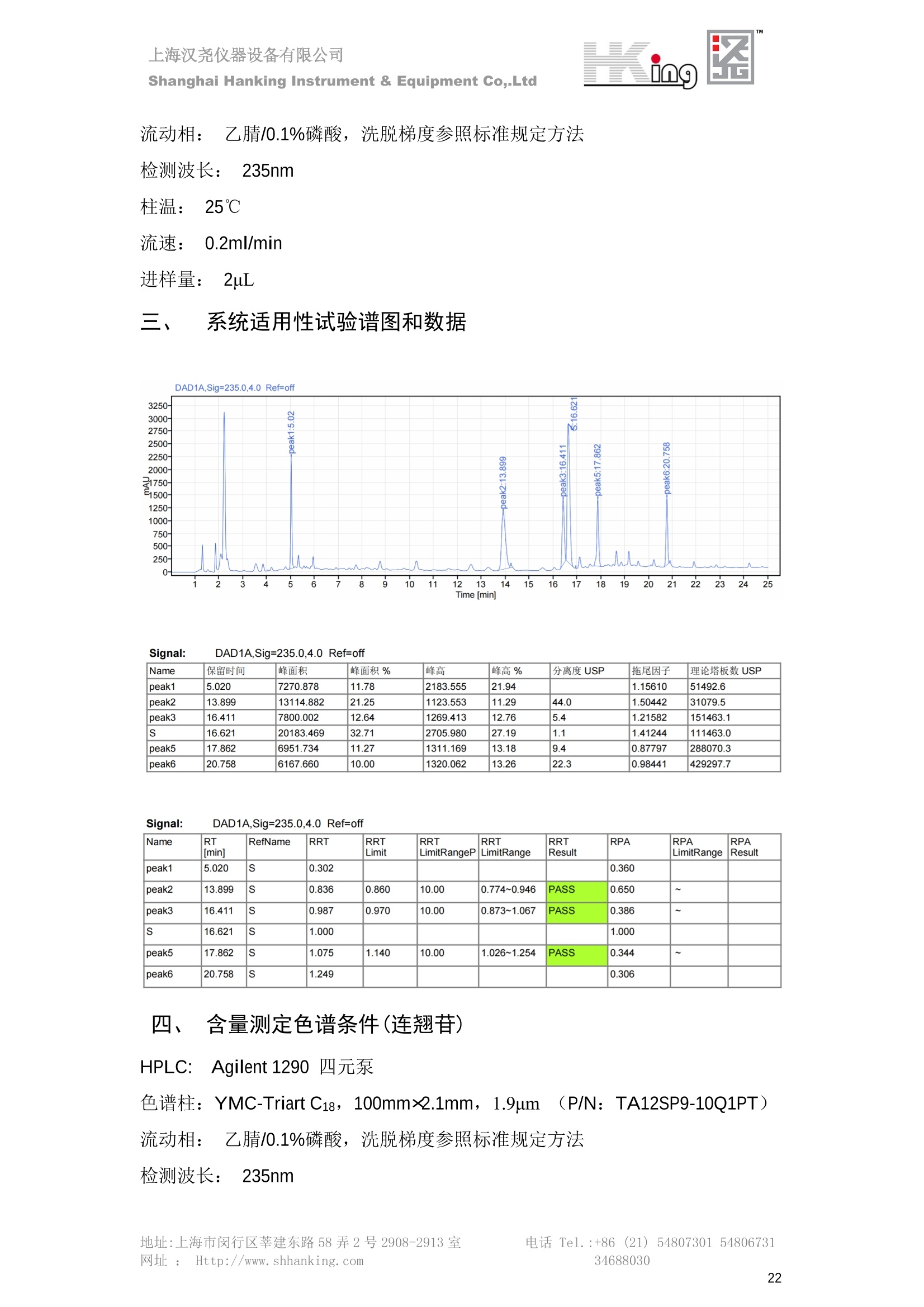
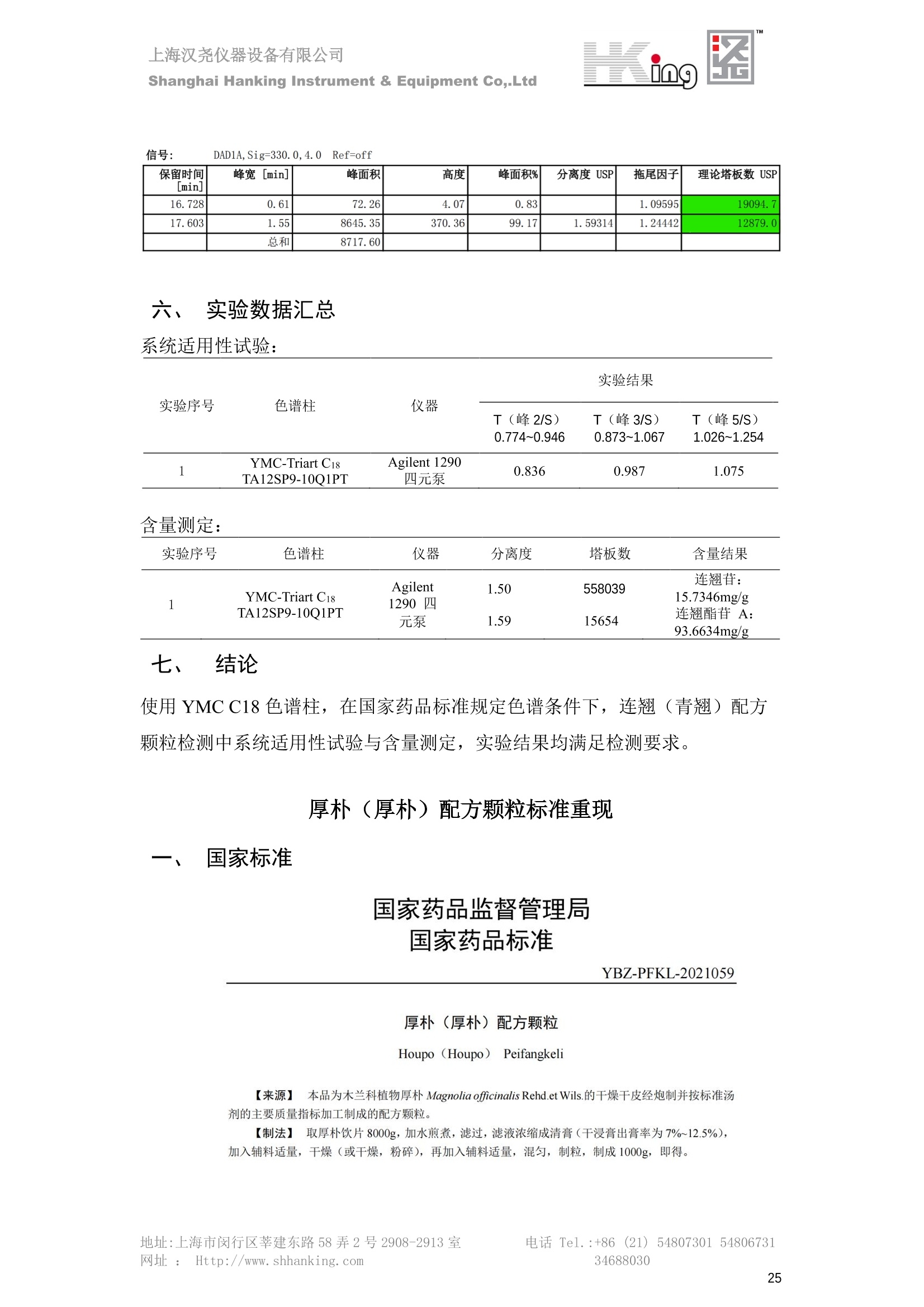
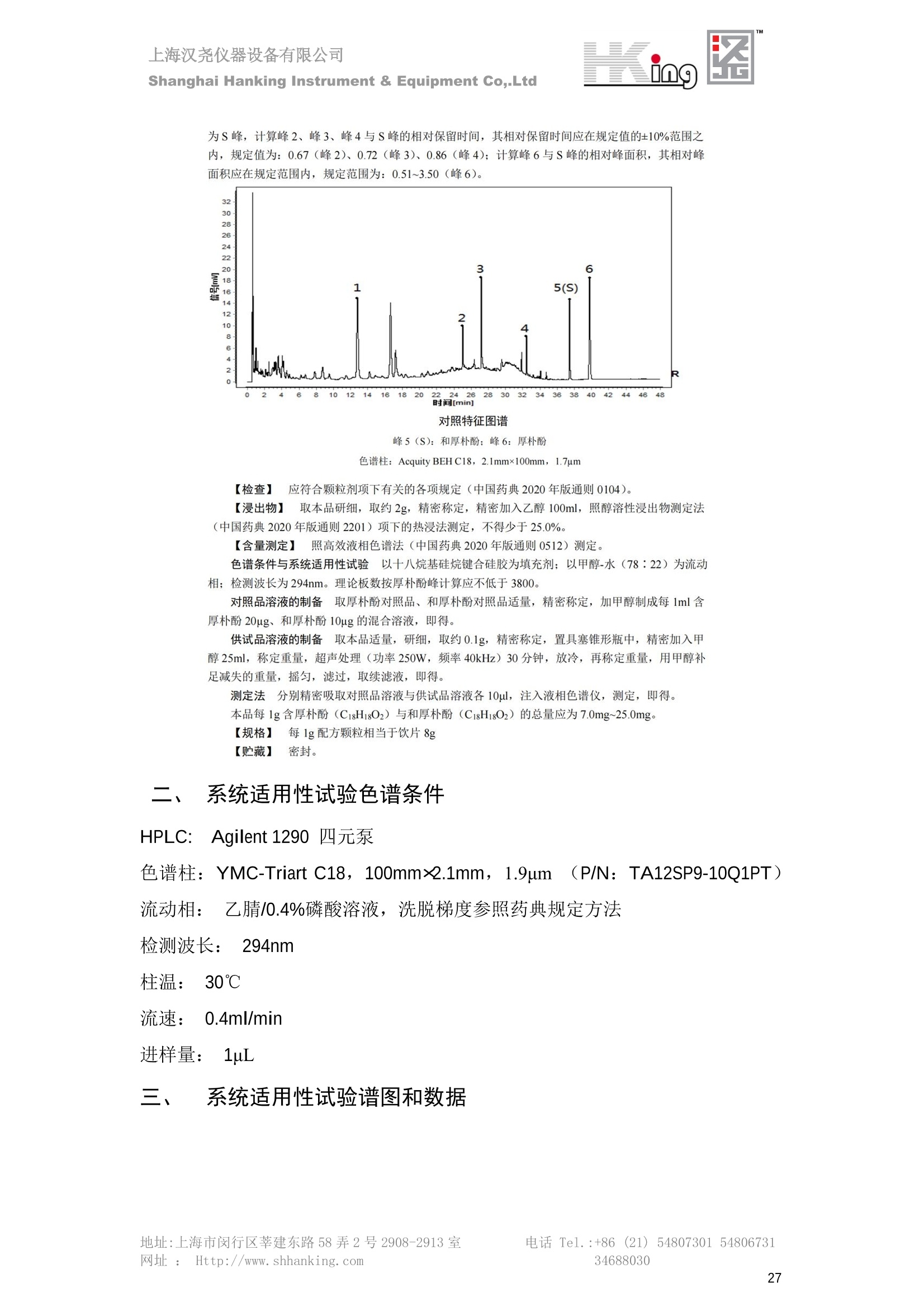
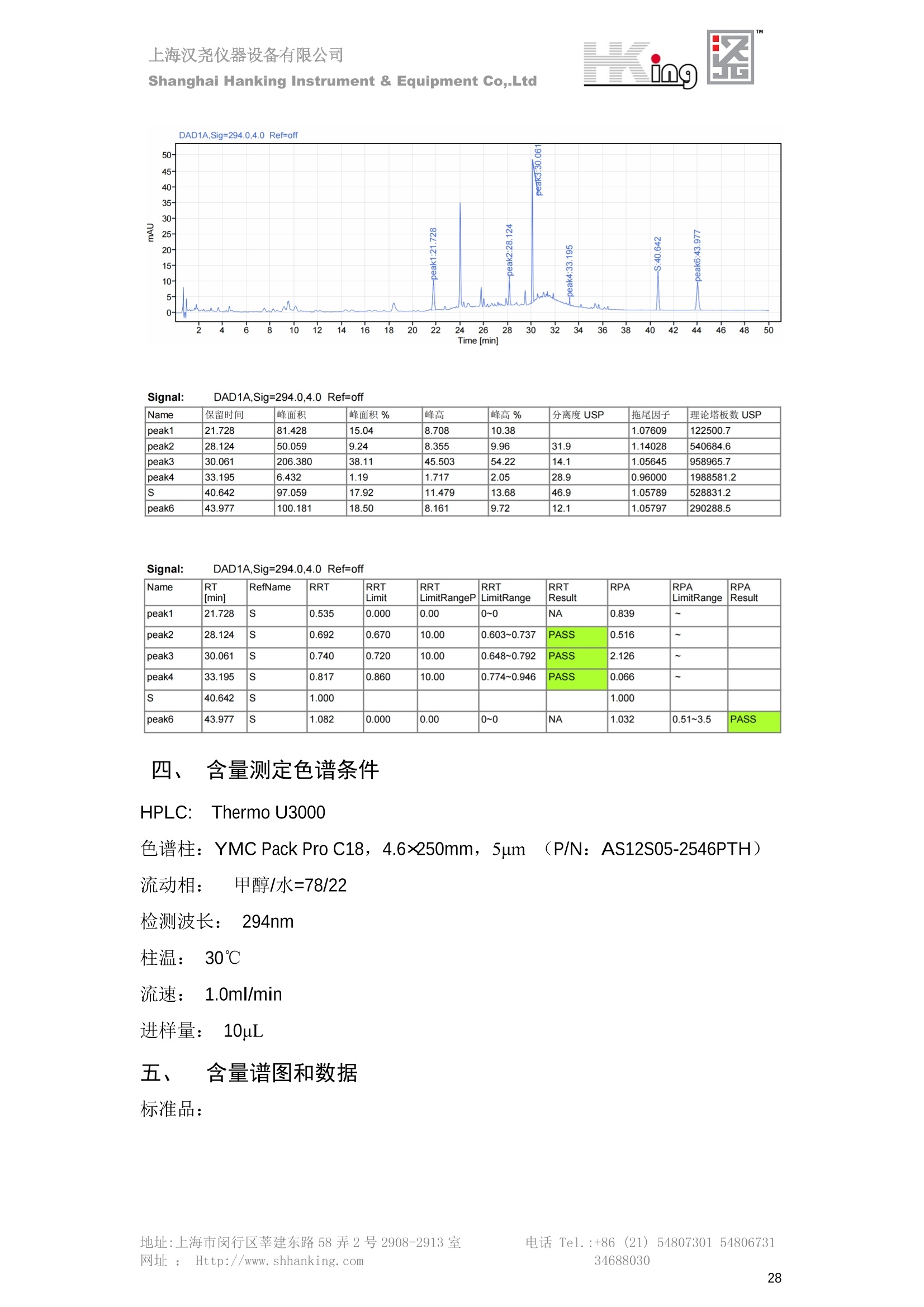
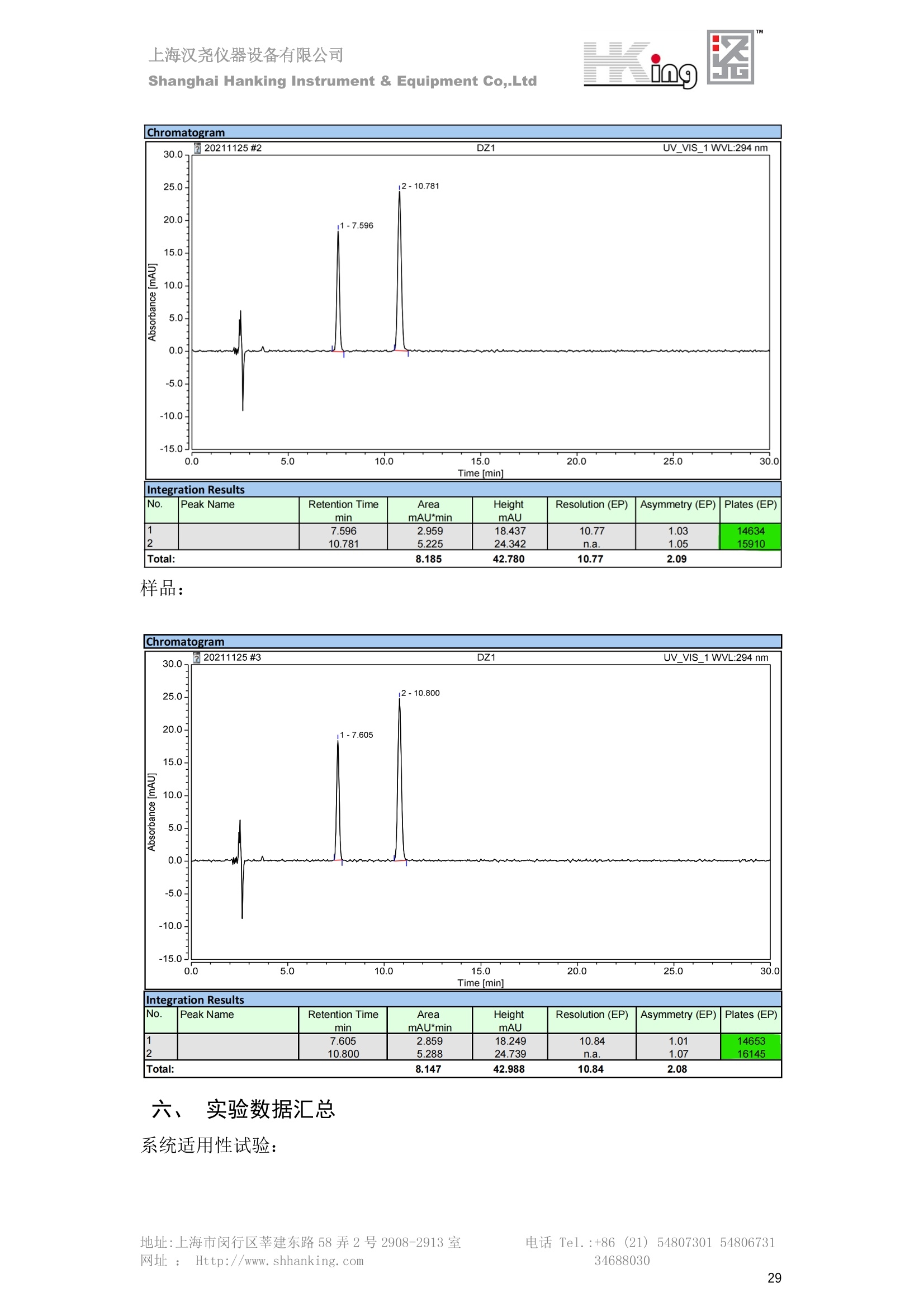
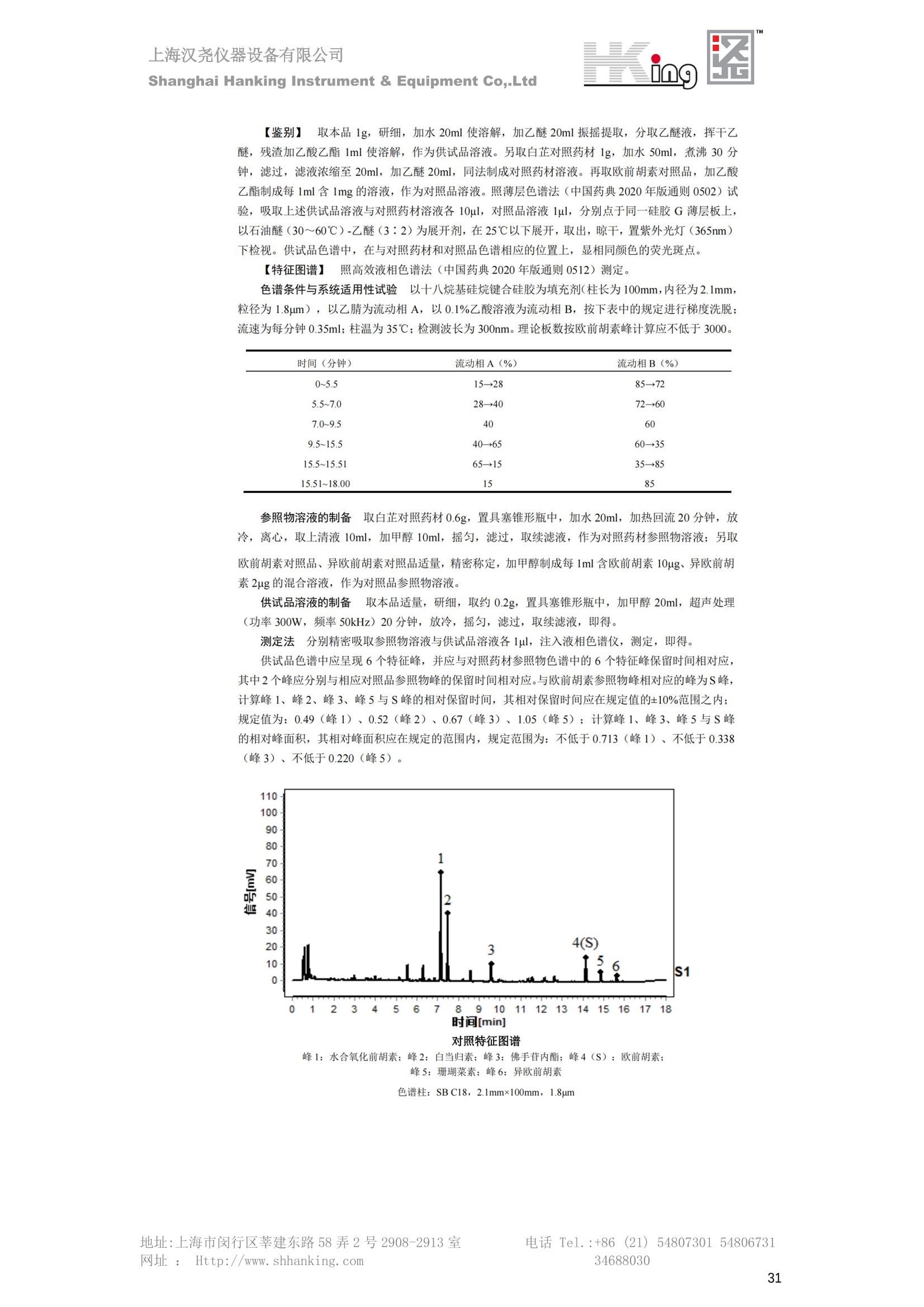
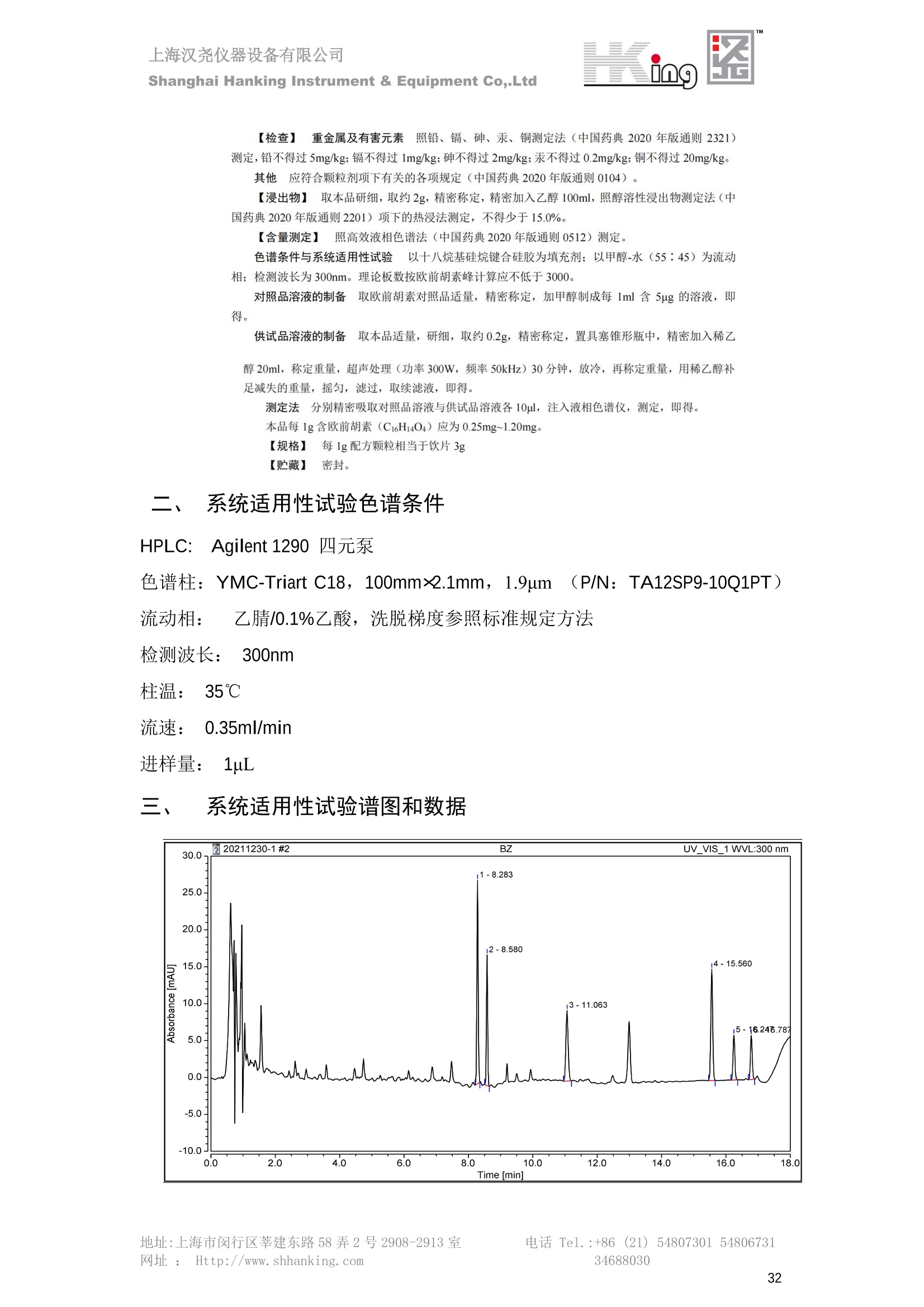
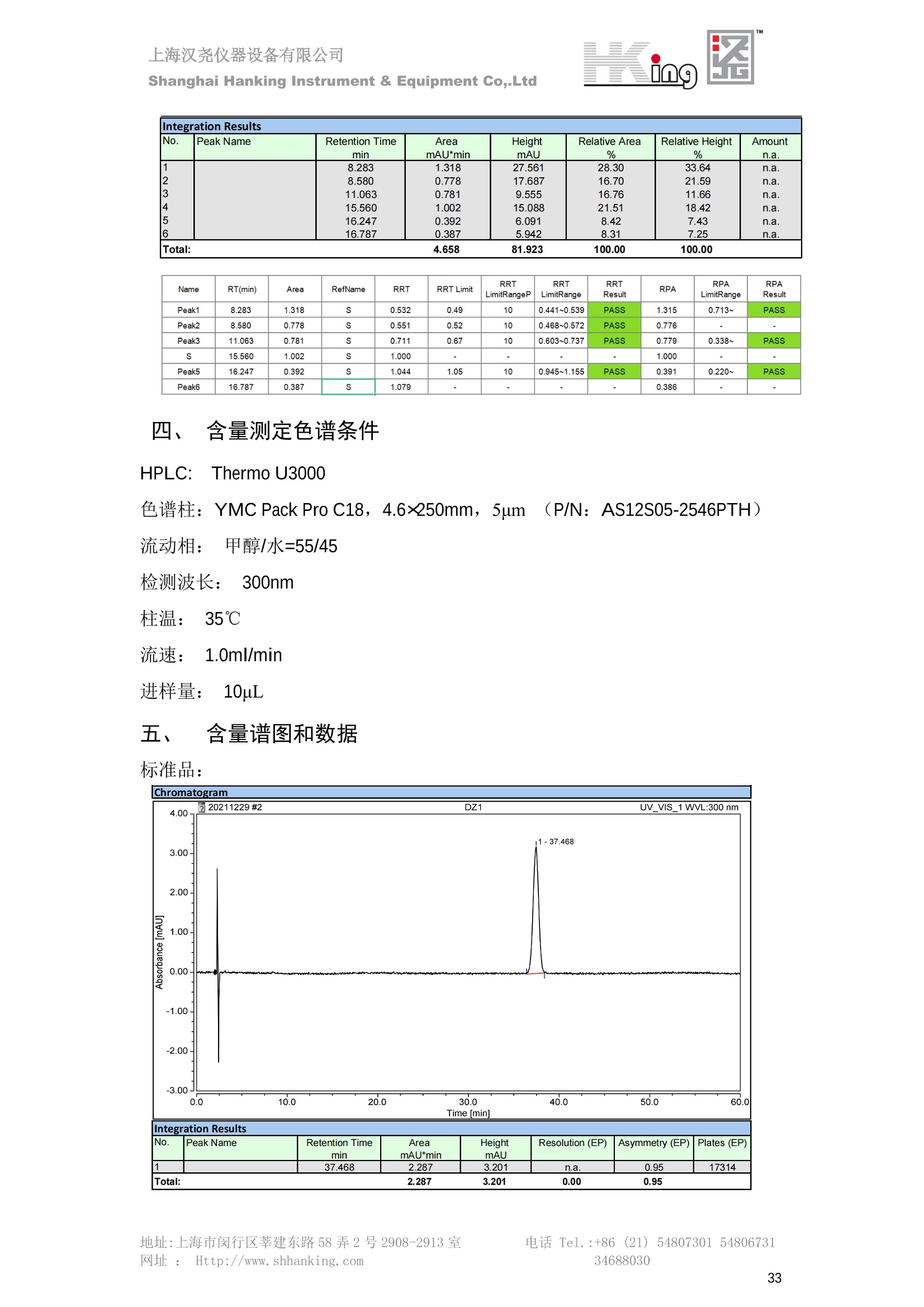
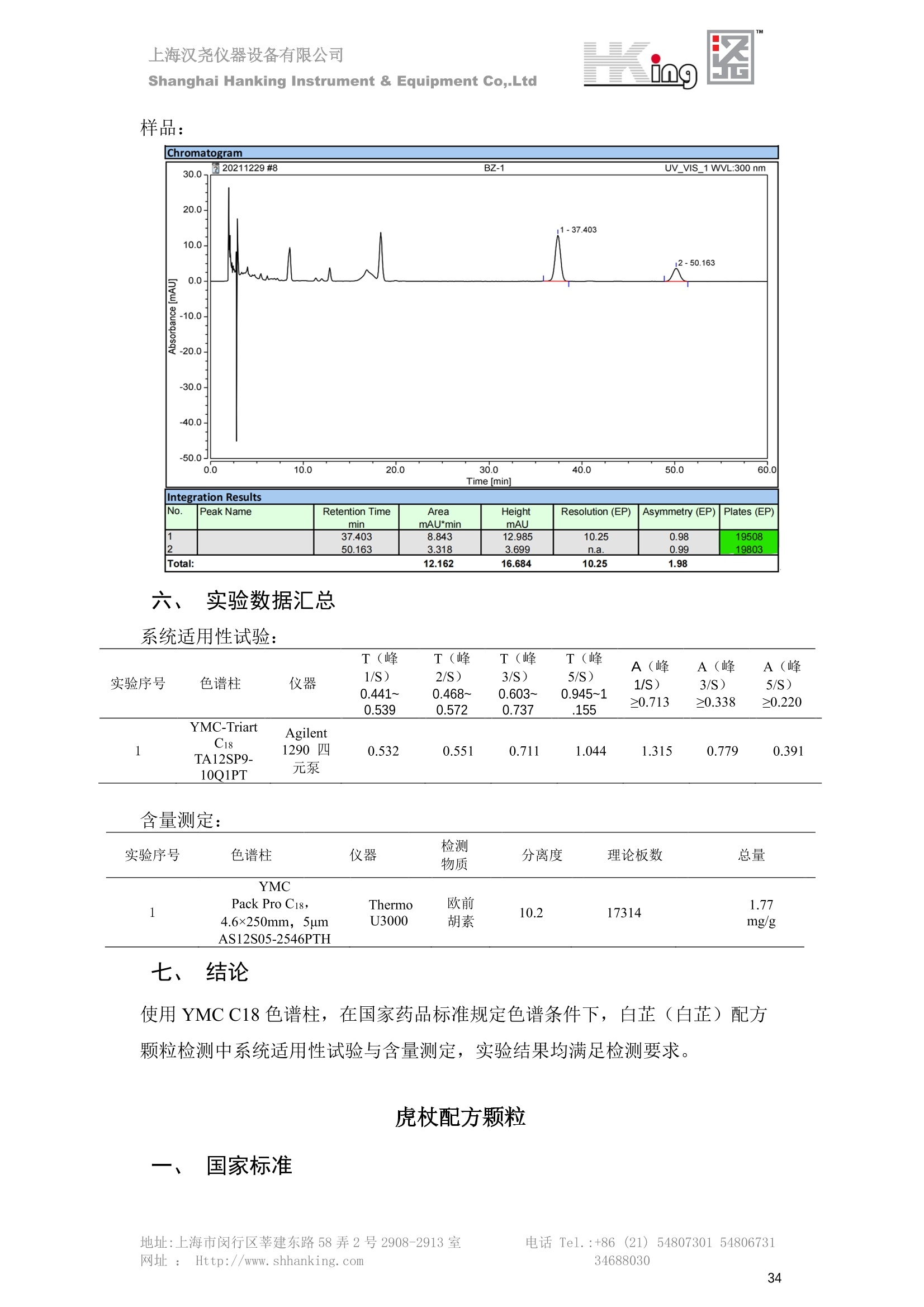
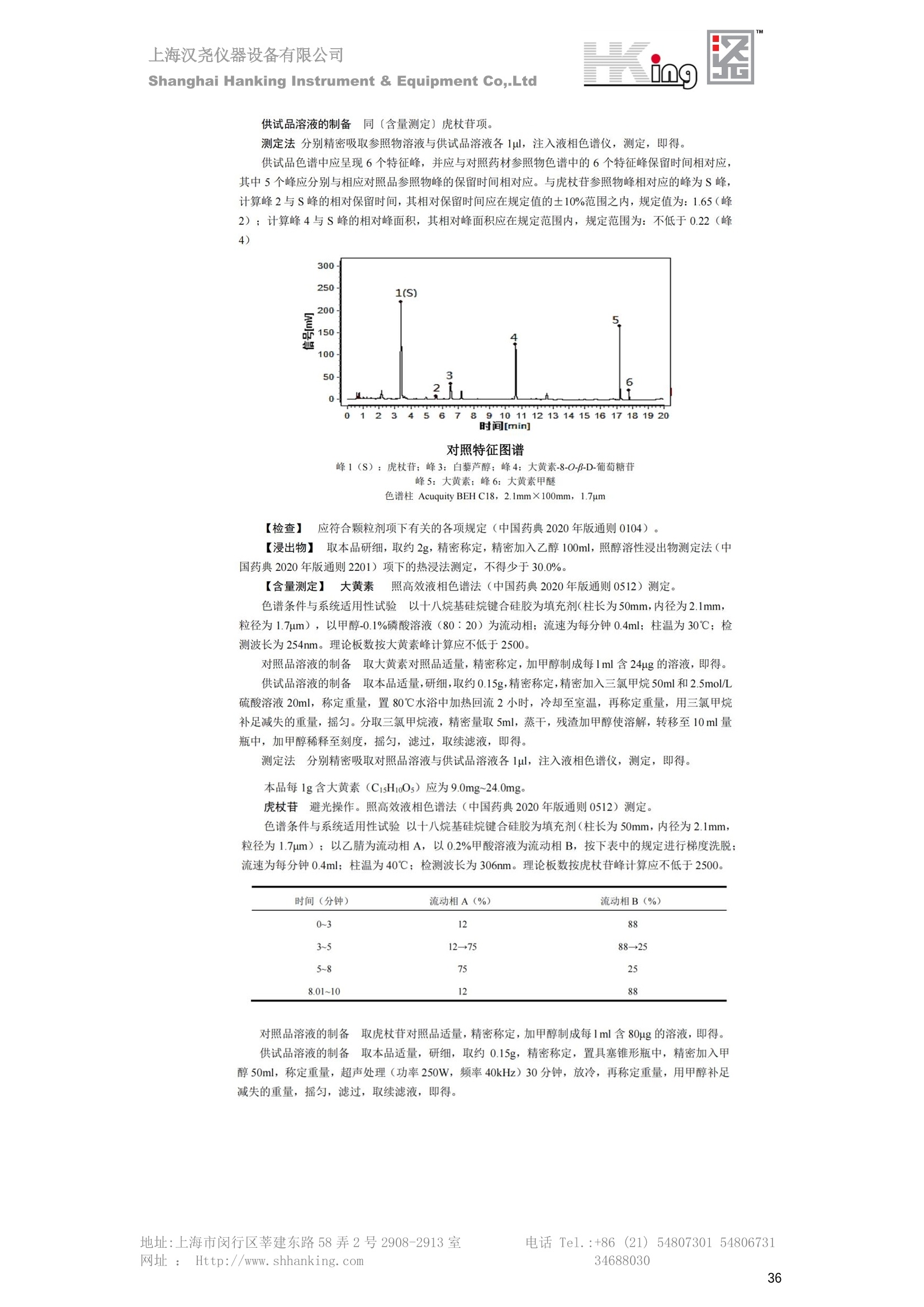
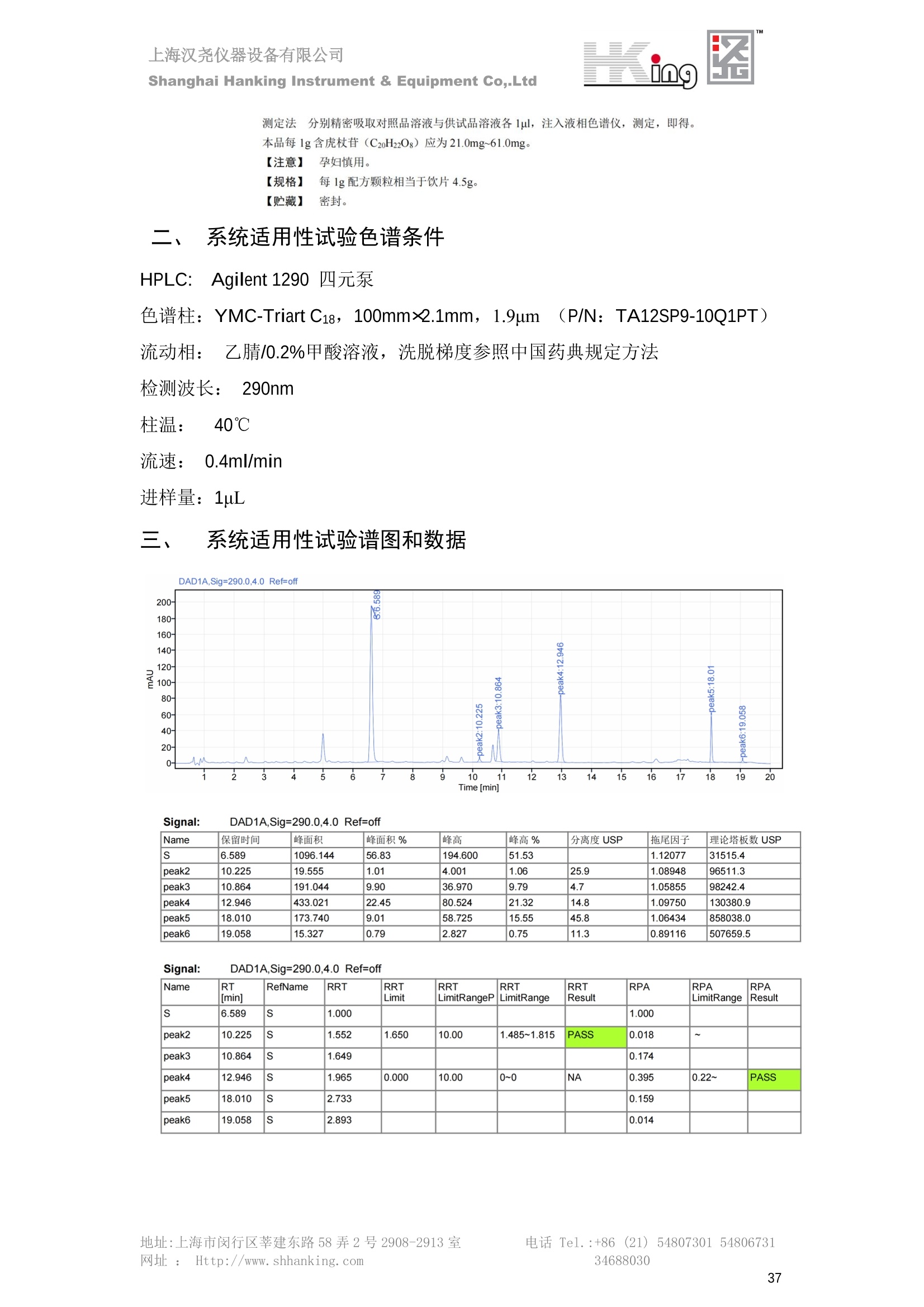
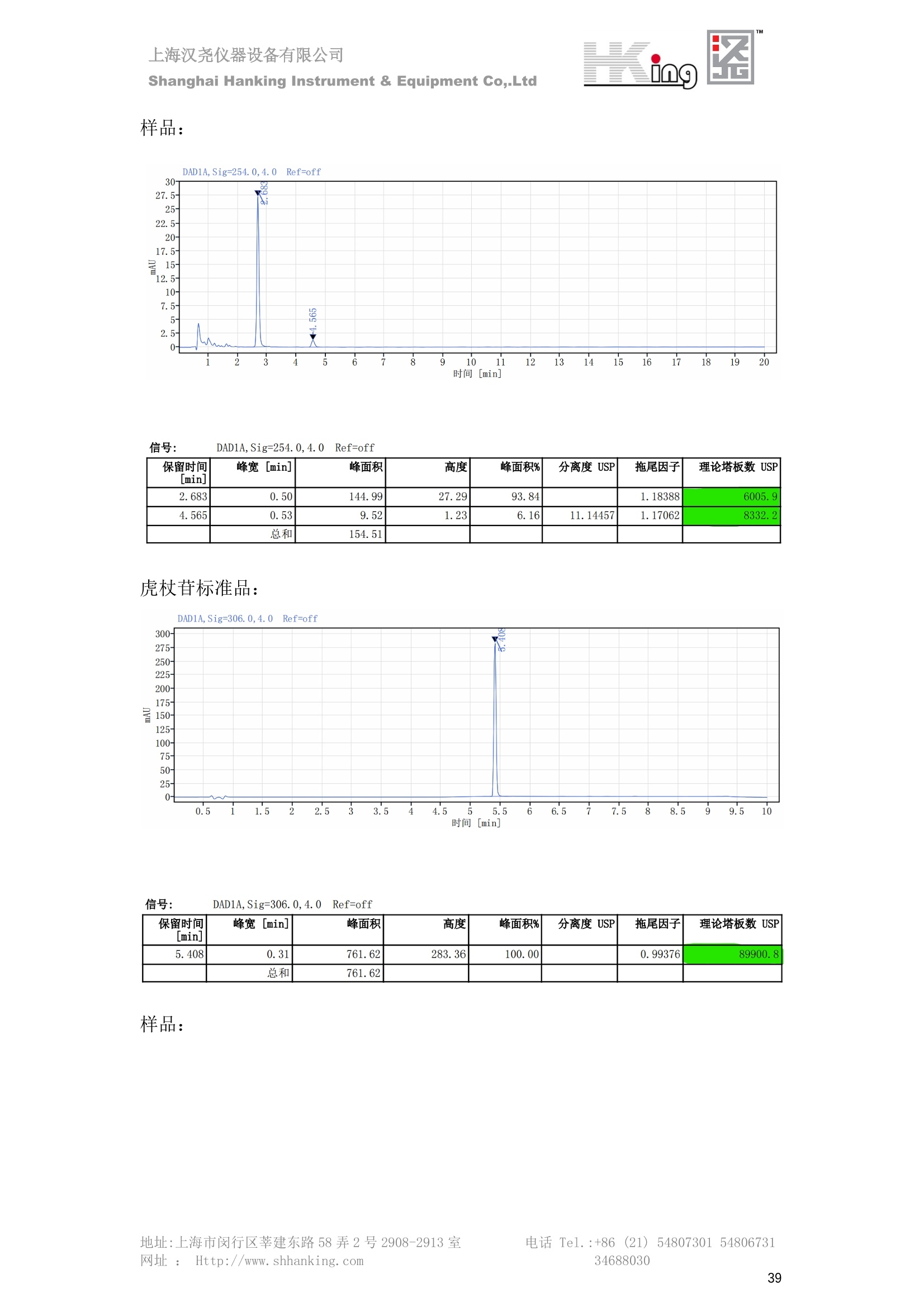
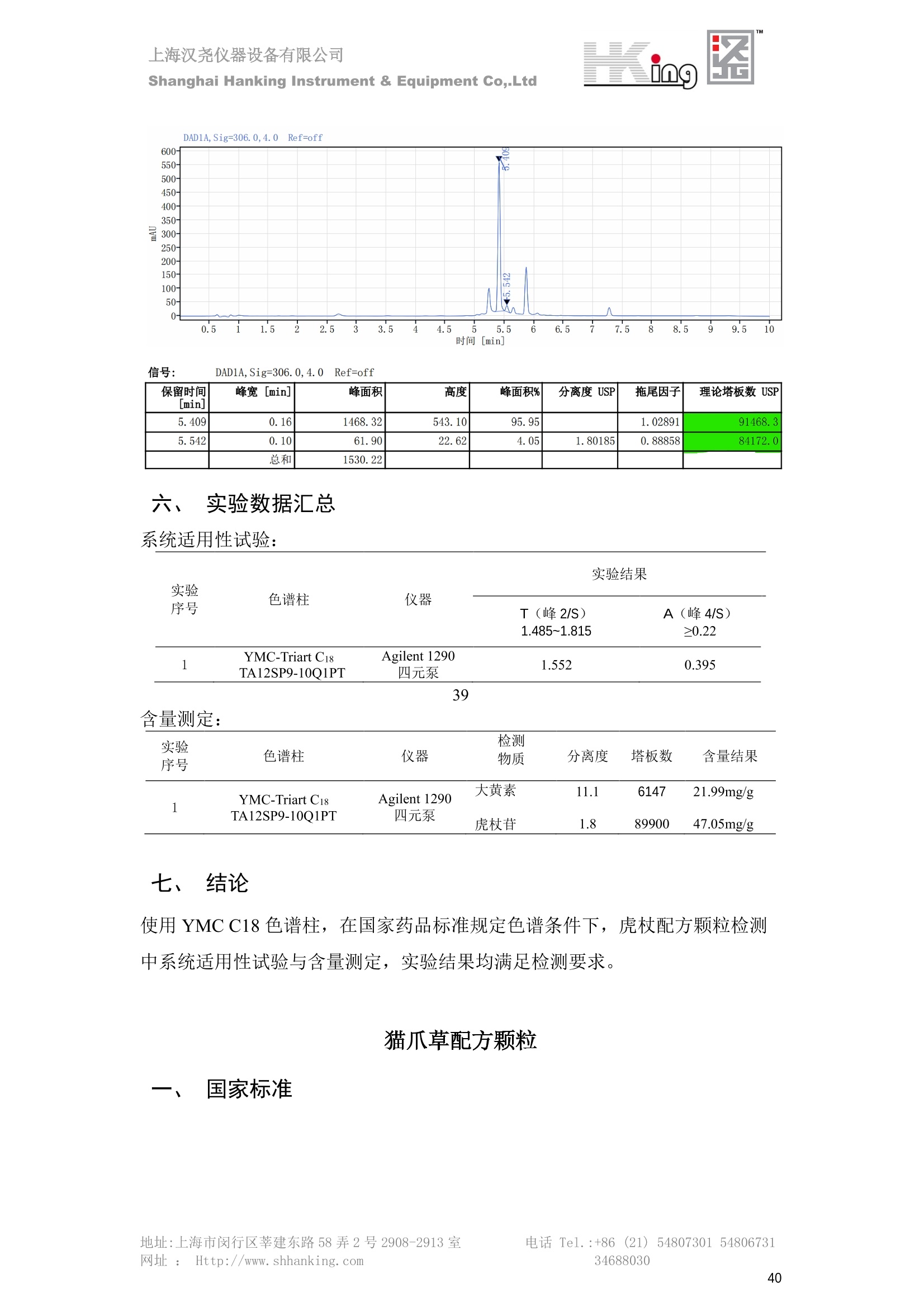
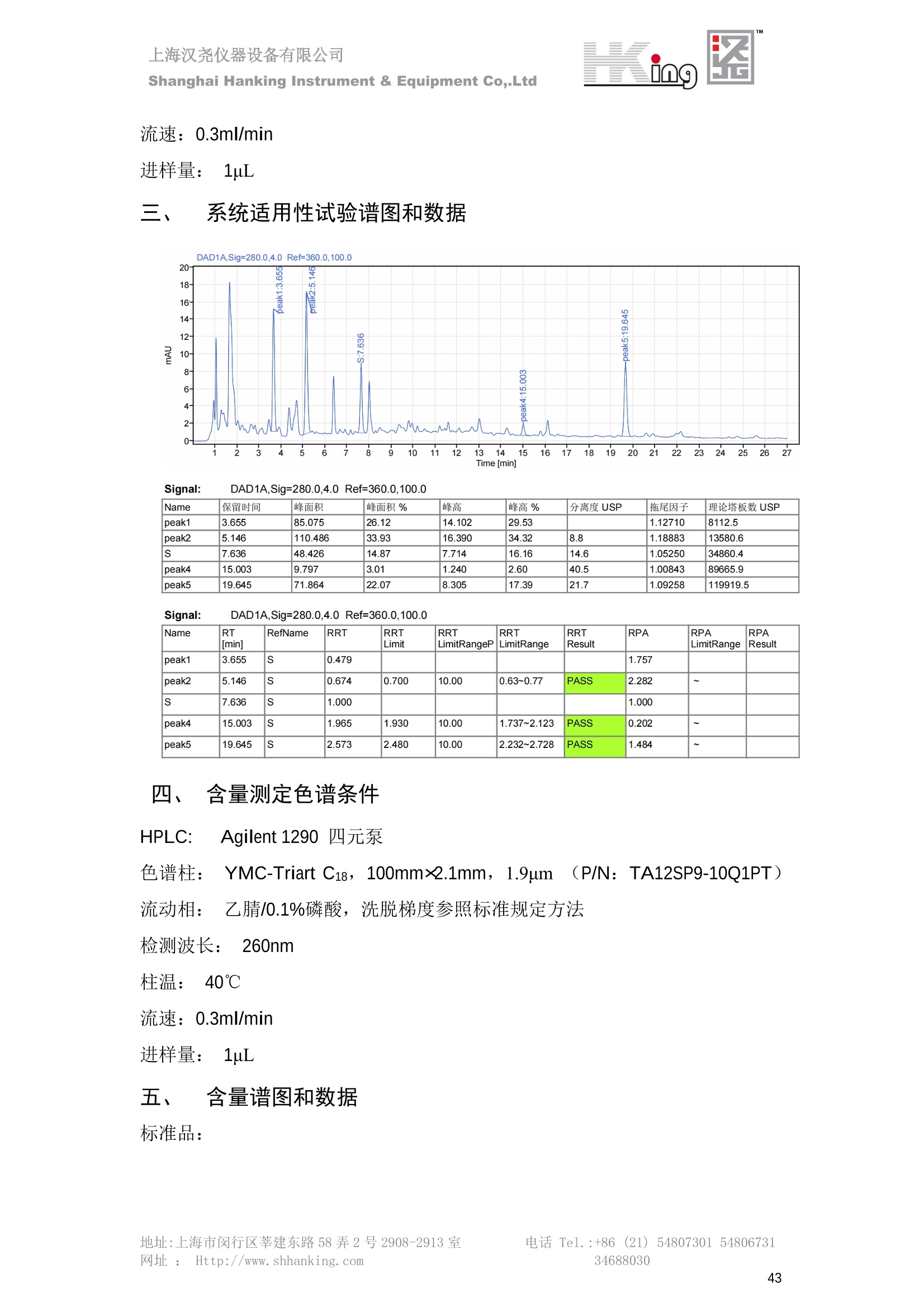
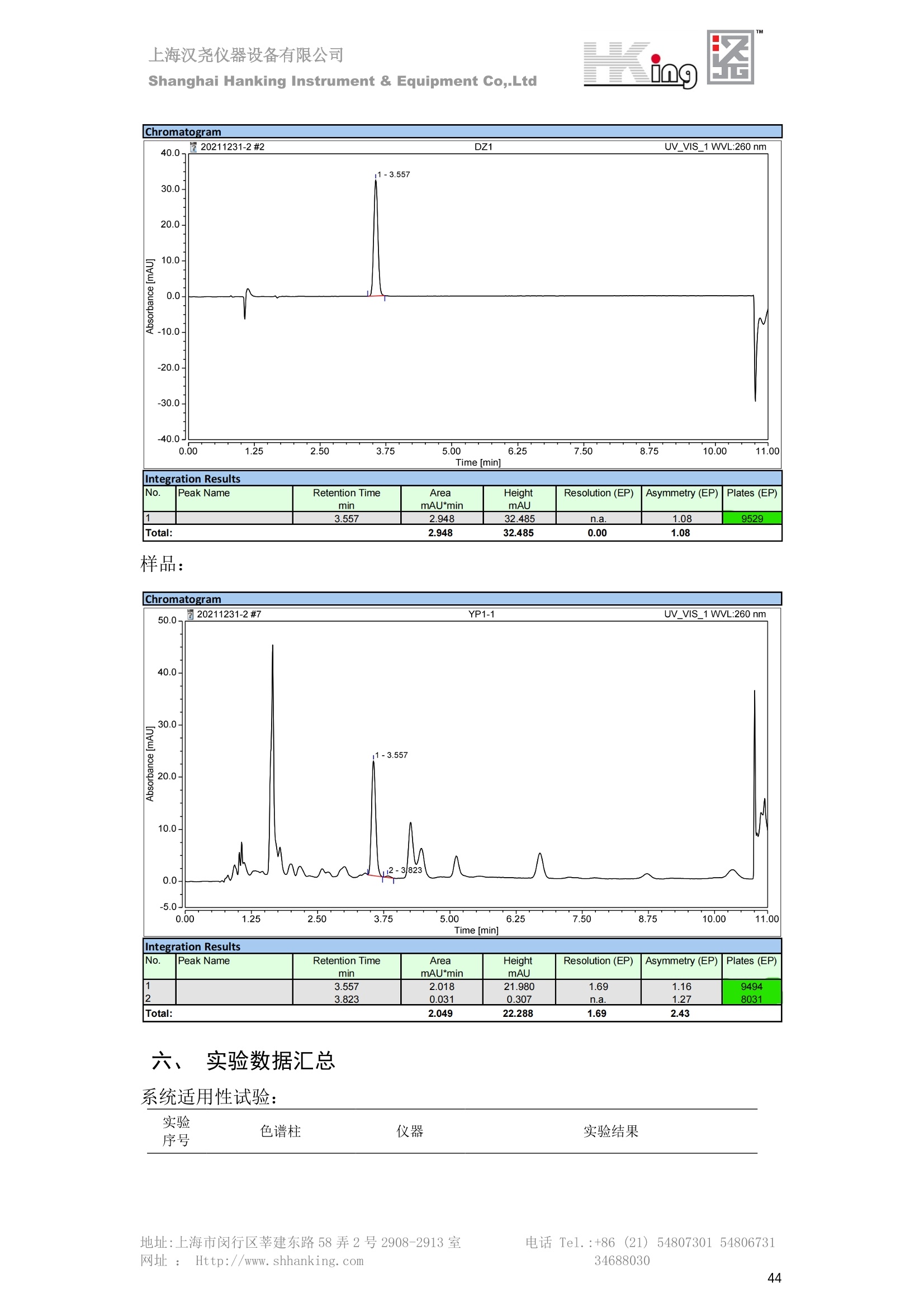
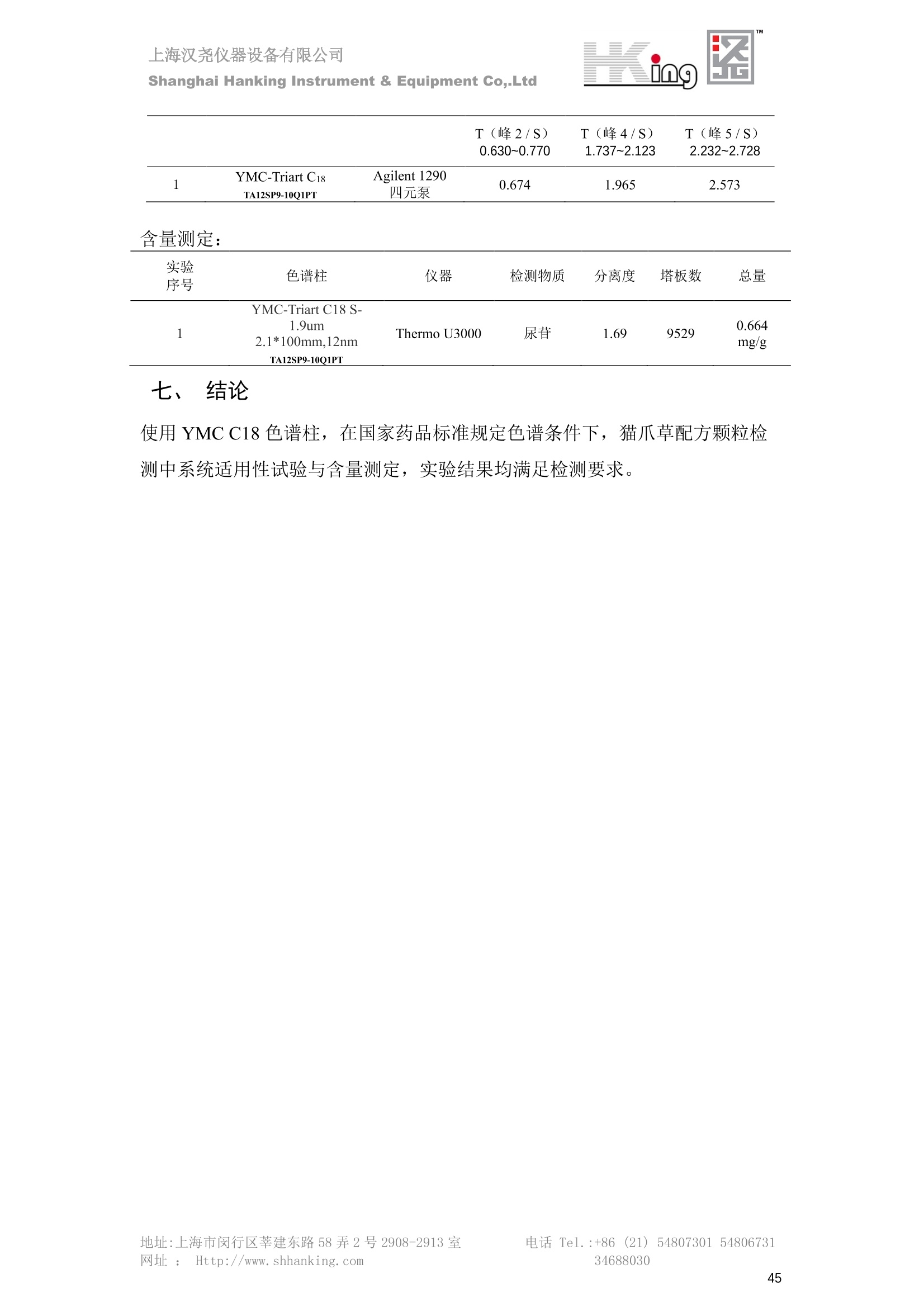
IN9上海汉尧仪器设备有限公司Shanghai Hanking Instrument & Equipment Co,.Ltd 中药配方颗粒方法重现 上海汉尧仪器设备有限公司 陈皮配方颗粒标准重现 .2 甘草(甘草)配方颗粒标准重现 .6 防风配方颗粒标准重现 .11 知母配方颗粒标准重现 .15 连翘(青翘)配方颗粒标准重现 20 厚朴(厚朴)配方颗粒标准重现. .25 白芷(白芷)配方颗粒 .30 虎杖配方颗粒 34 猫爪草配方颗粒. .40 陈皮配方颗粒标准重现 \ 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021025 陈皮配方颗粒 Chenpi Peifangkeli 【来源】 本品为芸香科植物橘 Citrus reticulate Blanco 及其栽培变种的干燥成熟果皮经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取陈皮饮片2000g,加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为25%~40%),加入辅料适量,干燥(或干燥,粉碎),再加入辅料适量,混匀,制粒,制成1000g, 即得。 【性状】 本品为棕黄色至棕色的颗粒;气香,味辛、苦。 【鉴别】 取本品1g,研细,加甲醇10ml,加热回流20分钟,滤过,取滤液5ml, 浓缩至约1ml,作为供试品溶液。另取陈皮对照药材1g,同法制成对照药材溶液。再取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。照薄层色谱法(中国药典2020年版通则0502)试验,吸取供试品溶液2~5ul、对照品溶液5ul、对照药材溶液3pl,分别点于同一用0.5%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯-甲醇-水(100:17:13)为展开剂,展至约3cm, 取出,晾干,再以甲苯-乙酸乙酯-甲酸-水(20:10:1:1)的上层溶液为展开剂,展至约8cm,取出,晾干,喷以三氯化铝试液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为2.2um);以乙腈为流动相A, 以0.5%冰醋酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为为每钟0.40ml;柱温为25℃;检测波长为283nm。理论板数按橙皮苷峰苷算应不低于15000。 时间(分钟) 流动相A(%) 流动相B(%) 0~15 13→20 87→80 15~25 20→34 80→66 25~44 34→42 66→58 参照物溶液的制备取陈皮对照药材 1g,置具塞锥形瓶中,加甲醇100ml,超声处理(功率300W,频率45kHz) 60分钟, 取出,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取柚皮芸香苷对照品、橙皮苷对照品、川陈皮素对照品、橘皮素对照品适量,精密称定,加甲醇制成成1ml各含100ug的混合溶液,作为对照品参照物溶液。 供试品溶液的制备同(含量测定)项。 测定法分别精密吸取取照物溶液与供试品溶液各 lul, 注入液相色谱仪,测定,即得。 供试品色谱中应呈现5个特征峰,并应与对照药材参照物色谱中的5个特征峰保留时间相对应,其中4个峰应分别与相应对照品参照物峰的保留时间相对应。与橙皮苷参照物峰相对应的峰为S峰,计算峰3与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:1.60(峰3);计算峰1、峰4、峰5与S峰的相对峰面积,其相对峰面积应在规定的范围内,规定范围为:0.03~0.92(峰1)、不低于0.03(峰4)、不低于0.02(峰5)。 对照特征图谱 峰1:柚皮芸香苷;峰2(S):橙皮苷;峰4:川陈皮素;峰5:橘皮素 色谱柱:Acclaim RSLC 120 C18, 2.1mmx100mm, 2.2um 【检查】 黄曲霉毒素 照黄曲霉毒素测定法(中国药典2020年版 通则2351)测定。 本品每1000g含黄曲霉毒素 B1 不得过 5ug, 含黄曲霉毒素 G2、黄曲霉毒素 G1、黄曲霉毒素B2和黄曲霉毒素 B1 的总量不得过 10pg。 其他 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 取本品研细,取约2g,精密密定,精密加入乙醇 100ml, 照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的热浸法测定,不得少于23.0%。 【含量测定】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-醋酸-水(35∶4:61)为流动相;检测波长为283nm。理论板数按橙皮苷峰计算应不低于2000。 对照品溶液的制备取橙皮苷对照品适量,精密称定,加甲醇制成醇 1ml 含 0.1mg的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.2g, 精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理(功率300W, 频率40kHz) 30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供与品溶液各 10ul, 注入液相色谱仪,测定,即得。 本品占 1g含橙皮苷(C28H34O15) 应为6.5mg~14.5mg。 【规格】 每1g配方颗粒相当于饮片2g 【贮藏】 密封。 二、 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18,1100mm×2.1mm,,1.9um (P/N:TA12SP9-10Q1PT) 流动相:乙腈/0.5%冰醋酸,洗脱梯度参照标准规定方法 检测波长:283nm 柱温:25C 流速:0.4ml/min 进样量::1uL 三、 系统适用性试验谱图和数据 Time [min] Signal: DAD1A,Sig=283.0,4.0 Ref=off Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度 USP 拖尾因子 理论塔板数 USP peak1 11.721 186.358 42.07 21.177 43.88 1.04907 40097.5 14.171 214.225 48.36 22.205 46.01 10.0 1.14696 49781.2 peak3 21.257 17.407 3.93 2.602 5.39 33.7 1.28262 260840.3 peak4 32.871 11.445 2.58 1.123 2.33 53.1 1.03404 234810.8 peak5 35.176 13.507 3.05 1.153 2.39 8.0 1.00564 209178.6 Signal: DAD1A,Sig=283.0,4.0 Ref=off Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRT LimitRange RRTResult RPA RPALimitRange RPAResult peak1 11.721 S 0.827 0.000 0.00 0~0 NA 0.870 0.03~0.92 PASS 14.171 S 1.000 1.000 peak3 21.257 S 1.500 1.600 10.00 1.44~1.76 PASS 0.081 ~ peak4 32.871 S 2.320 0.000 0.00 0~0 NA 0.053 0.03~ PASS peak5 35.176 S 2.482 0.000 0.00 0~0 NA 0.063 0.02~ PASS 四、 含量测定色谱条件 HPLC:Thermo U3000 色谱柱:YMC Pack Pro C18,, 4.6×250mm, 5um (P/N: AS12S05-2546PTH) 流动相:甲醇/冰醋酸/水=35/4/61 检测波长:283nm 柱温:25℃ 流速: 1.0ml/min 进样量:10uL 五、 含量谱图和数据 标准品: 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel. :+86 ((21)5480730154806731网址:Http://www. shhanking.com34688030 样品: 六、实验数据汇总 系统适用性试验: 实验序号 色谱柱 实验结果 仪器 T(峰 S(峰S(峰 S(峰 3/S) 1/S) 4/S) 5/S) 1.44~1.7 0.03~0.9 6 2 ≥0.03 ≥0.02 1 YMC-Triart C18 TA12SP9-10Q1PT Agilent 1290 四元泵 1.50 0.87 0.05 0.06 含量测定: 实验序号 色谱柱 仪器 分离度 塔板数 含量结果 YMC 2 Pack Pro C18, 4.6x250mm, 5um Thermo U3000 5.0 8918 9.85mg/g AS12S05-2546PTH 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,陈皮配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 甘草(甘草)配方颗粒标准重现 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021049 甘草(甘草)配方颗粒 Gancao (Gancao) Peifangkeli 【来源】 本品为豆科植物甘草 Glycyrrhiza uralensis Fisch. 的干燥根和根茎经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取甘草饮片3000g, 加水煎煮,滤过,滤液浓缩宿清膏(干浸膏出膏率为20%~33%),干燥(或干燥,粉碎),加辅料适量,混匀,制粒,制成1000g, 即得。 【性状】 本品为黄色至棕黄色的颗粒;气微,味甜而特异。 【鉴别】 取本品0.2g,研细,加水20ml使溶解,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇液,蒸干,残渣加甲醇 5ml 使溶解,作为供试品溶液。另取甘草(甘草)对照药材 0.5g,加水50ml,煮沸30分钟,滤过,滤液浓缩至20ml, 同法制成对照药材溶液。再取甘草酸铵对照品,加甲醇制成每 lml 含 2mg 的溶液,作为对照品溶液。照薄层色谱法(中国药典2020年版通则0502)试验,吸取上述三种溶液夜 2ul,别点点于同一用1%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15:1:1:2)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱和对照品品谱相应的位置上,显相同颜色的荧光斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为2.2um);以乙腈为流动相A, 以0.1%磷酸溶液为流动相B, 按下表中的规定进行梯度洗脱;流速为每分钟0.3ml;柱温为30℃;检测波长为 237nm。理论板数按甘草酸峰计算应不低于5000。 时间(分钟) 流动相A(%) 流动相B(%) 0~1 5→27 95→73 1~2 27 73 2~10 27→46 73→54 10~16 46→64 54→36 16~24 64→95 36→5 24~25 95 5 参照物溶液的制备取甘草(甘草)对照药材0.2g,置具塞锥形瓶中,加入70%乙醇100ml,密塞,超声处理(功率250W,频率40kHz) 30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取甘草苷对照品、甘草素对照品、甘草酸铵对照品适量,精密密定,加甲醇制成每1ml含甘草苷0.1mg、甘草素0.1mg、甘草酸铵0.2mg的溶液,作为对照品参照物溶液(甘草酸重量=甘草酸铵重量/1.0207)。 供试品色谱中应呈现12个特征峰,并应与对照药材参照物色谱中的12个特征峰保留时间相对应,其中峰3、峰6、峰10应分别与甘草苷对照品、甘草素对照品、甘草酸对照品参照物峰保留时间相对应。与甘草苷参照物相应的峰为S1 峰,计算峰1~峰2与S1峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内。规定值为0.83(峰1)、0.98(峰2)。与甘草素参照物相应的为 S2 峰,计算峰4、峰5、峰7与S2峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内。规定值为:0.82(峰4)、0.87(峰5)、1.09(峰7)。与甘草酸参照物相应的峰作为S3峰,计算峰8~峰12与S3 峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为0.80(峰8)、0.92(峰9)、1.09(峰11)、1.26(峰12)。 对照特征图谱 峰2:芹糖甘草苷;峰3(SI):甘草苷;峰5:异甘草苷峰6(S2):甘草素;峰10(S3):甘草酸 色谱柱: Acclaim RSLC 120 C18, 2.1mmx100mm, 2.2um 【检查】 重金属及有害元素 照铅、镉、砷、汞、铜测定法(中国药典2020年年版则2321电感耦合等离子体质谱法)测定,铅不得过 5mg/kg;镉不得过 1mg/kg;砷不得过 2mg/kg;汞不得过0.2mg/kg;铜不得过 20mg/kg。 其他有机氯类农药残留量 照农药残留量测定法(中国药典2020年版通则2341有机氯类农药残留量测定法一第一法)测定。 含五氯硝基苯不得过 0.1mg/kg。 其他 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的热浸法测定,用乙醇作溶剂,不得少于32.0%。 【含量测定】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为2.2um);以乙腈为流动相A, 以 0.05%磷酸溶液为流动相B, 按下表中规定进行梯度洗脱;流速为每分钟0.4ml;检测波长为237nm。理论板数按甘草苷峰计算应不低于5000。 时间(分钟) 流动相A(%) 流动相B(%) 0~2 19 81 2~12.5 19→50 81→50 12.5~13 50→100 50→0 13~15 100→19 0→81 对照品溶液的制备取甘草苷对照品、甘草酸铵对照品适量,精密称定,加70%乙醇制成每 1ml含甘草苷60ug、甘草酸铵0.1mg 的溶液,即得(甘草酸重量=甘草酸铵重量/1.0207)。 供试品溶液的制备取本品适量,研细,取约0.1g,精密称定,置具塞锥形瓶中,精密加入70%乙醇50ml,密塞,称定重量,超声处理(功率250W, 频率40kHz) 30分钟,放冷,再称定重量,用70%乙醇补足减失的重量,摇匀、滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供试品溶液各lul, 注入液相色谱仪,测定,即得。 本品每1g含甘草苷(C21H22O)应为15.0mg~35.0mg、含甘草酸 (C42H62016)应为29.0mg~80.0mg。【注意】 不宜与海藻、京大戟、红大戟、甘遂、芫花同用。 【规格】 每1g配方颗粒相当于饮片3g 【贮藏】 密封。 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱: YMC-Triart C18, 100mm×2.1mm, 1.9um((P/N: TA12SP9-10Q1PT) 流动相:Z乙腈/0.1%磷酸溶液,洗脱梯度参照药典规定方法 检测波长:237nm 流速:0.3ml/min 进样量: 1uL 系统适用性试验谱图和数据 地址:上海市闵行区莘建东路58弄2号2908-2913 室 电话 Tel.:+86(21)5480730154806731网址:Http://www.shhanking. com 34688030 Signal: DAD1A,Sig=237.0,4.0 Ref=off Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度 USP 拖尾因子 理论塔板数 USP peak1 3.177 24.290 2.96 6.674 2.74 1.04565 19008.2 peak2 4.018 97.691 11.91 33.491 13.76 9.9 1.06424 40866.9 S1 4.210 403.649 49.20 129.281 53.12 2.3 1.14133 41336.5 peak4 5.351 16.133 1.97 3.640 1.50 12.5 0.74613 45482.6 peak5 5.810 29.026 3.54 9.574 3.93 5.0 0.95623 79520.5 S2 7.146 26.323 3.21 6.472 2.66 14.2 1.06616 71592.2 peak7 7.901 3.438 0.42 0.619 0.25 6.6 1.39038 68870.6 peak8 8.392 19.617 2.39 5.430 2.23 4.5 1.09352 120403.7 peak9 9.553 32.502 3.96 6.705 2.75 10.2 1.07346 86306.8 S3 10.524 147.997 18.04 37.338 15.34 8.2 1.18991 157211.8 peak11 11.443 8.415 1.03 1.418 0.58 7.3 1.19098 100448.0 peak12 13.514 11.343 1.38 2.744 1.13 16.2 0.85960 235131.0 Signal: DAD1A,Sig=237.0,4.0 Ref=off Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRTLimitRange RRTResult RPA RPALimitRange RPAResult peak1 3.177 S1 0.755 0.830 10.00 0.747~0.913 PASS 0.060 ~ peak2 4.018 S1 0.954 0.980 10.00 0.882~1.078 PASS 0.242 ~ S1 4.210 S1 1.000 1.000 peak4 5.351 S2 0.749 0.820 10.00 0.738~0.902 PASS 0.613 ~ peak5 5.810 S2 0.813 0.870 10.00 0.783~0.957 PASS 1.103 ~ S2 7.146 S2 1.000 1.000 peak7 7.901 S2 1.106 1.090 10.00 0.981~1.199 PASS 0.131 ~ peak8 8.392 S3 0.797 0.800 10.00 0.72~0.88 PASS 0.133 ~ peak9 9.553 S3 0.908 0.920 10.00 0.828~1.012 PASS 0.220 ~ S3 10.524 S3 1.000 1.000 peak11 11.443 S3 1.087 1.090 10.00 0.981~1.199 PASS 0.057 ~ peak12 13.514 S3 1.284 1.260 10.00 1.134~1.386 PASS 0.077 ~ 四、 含量测定色谱条件 HPLC:Thermo U3000 色谱柱:YMC-Triart C18,100mmx2.1mm, 1.9um(P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.05%磷酸溶液,洗脱梯度参照药典规定方法 检测波长: 237nm 柱温: 30℃ 流速: 0.4ml/min 进样量: 10pL 五、 含量谱图和数据 标准品: 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86((21)54807301 54806731网址:Http://www.shhanking. com34688030 时间 [min] 信号: DAD1A,Sig=237.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 4.137 0.63 298.49 58.53 100.00 1.17308 15316.8 总和 298.49 样品: 0.5 1.522.533.544.5555.566.5 77.588.599.51010.5 11 11.51212.51313.51414.515时间 [min] 信号: DAD1A,Sig=237.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 3.573 0.31 95.49 15.02 17.82 1.25072 7375.3 3.874 0.48 310.96 60.32 58.02 1.98375 1.18438 12947.7 10.965 0.32 128.59 29.15 23.99 56.90307 1.33298 150090.7 11.264 0.05 0.88 0.28 0.17 2.94667 1.25293 248409.4 总和 535.92 六、实验数据汇总 系统适用性试验: T(峰 T(峰 T(峰 T(峰 T(峰 T(峰 T(峰 T(峰 11/S3 T(峰 12/S3 实验 仪器 1/S1) 2/S1) 4/S2) 5/S2) 7/S2) 8/S3) 9/S3) 色谱柱 ) ) 序号 0.75~ 0.88~ 0.74~ 0.78~ 0.98~ 0.72~ 0.83~ 0.98~ 1.13~ 0.91 1.08 0.90 0.96 1.20 0.88 1.01 1.20 1.39 YMC- Agilent 1290 四 1 Triart C18 TA12SP9- 0.755 0.954 0.749 0.813 1.106 0.797 0.908 1.087 1.284 10Q1PT 元泵 含量测定: 实验序号 色谱柱 仪器 分离度 塔板数 含量结果 甘草苷 甘草酸 1 YMC-Triart C18 TA12SP9- 10Q1PT Agilent 1290 二元泵 2.0/2.9 15316/1745 67 30.56mg/g 45.04mg/g 七、 结论 使用 YMC C18色谱在,在国家药品标准规定色谱条件下,甘草配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 防风配方颗粒标准重现 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021041 防风配方颗粒 Fangfeng Peifangkeli 【来源】 本品为伞形科植物防风 Saposhnikovia divaricata (Turcz.) Schischk.的干燥根经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取防风饮片2000g,加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为27%~43%),加入辅料适量,干燥(或干燥,粉碎),再加入辅料适量,混匀,制粒,制成 1000g, 即得。 【性状】 本品为浅棕黄色至棕褐色的颗粒;气微,味微苦。 【鉴别】 取本品2g,研细,加乙醇30ml,超声处理30分钟,滤过,滤液蒸干,残渣加乙醇1ml使溶解,作为供试品溶液。另取防风对照药材 0.5g, 加乙醇30ml, 同法制成对照药材溶液。再取升麻素苷对照品、5-0-甲基维斯阿米醇苷对照品,加乙醇制成每 lml 各含0.5mg 的混合溶液,作为对照品溶液。照薄层层谱法(中国药典2020年版通则0502)试验,吸取防风对照药材20ul、供试品溶液和对照品溶液各 10ul, 分别点于同一硅胶 GF254 薄层板上,以三氯甲烷-甲醇(4:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下视视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为1.6um),以乙腈为流动相A, 以水为流动相B,按下表中的规定进行梯度洗脱,流速为每分钟0.4ml;柱温为30℃;检测波长为210nm。理论板数按升麻素苷峰计算应不低于20000。 时间(分钟) 流动相A(%) 流动相B(%) 0~10 0→29 100→71 10~11 29→49 71→51 11~13 49→100 510 13.1~17 0 100 参照物溶液的制备取防风对照药材 0.25g, 置具塞锥形瓶中,加50%甲醇10ml, 超声处理(功率500W,频率40kHz) 30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取[含量测定]项下的对照品溶液,作为对照品参照物溶液。 供试品溶液的制备同(含量测定)项。 测定法分别精密吸取参照物溶液与供试品溶液溶 lul,注入液相色谱仪,测定,即得。 供试品色谱中应呈现5个特征峰,并应与对照药材参照物色谱中的5个特征峰保留时间相对应,其中2个峰应分别与相应对照品参照物峰的保留时间相对应。与升麻素苷参照物峰相对应的峰为S峰, 计算峰3、峰5与S峰的相对保留时间,其保留时间应在规定值的±10%范围之内;规定值为:1.14(峰3)、1.54(峰5)。计算峰3、峰5与S峰的相对峰面积,其相对峰面积应在规定范围内,规定范围为:不低于0.05(峰3)、不低于0.10(峰5)。 对照特征图谱 峰2(S):升麻素苷;峰3:升麻素;峰4:5-0-甲基维斯阿米醇苷;峰5:亥茅酚苷 色谱柱:Acquity CORTECS T3 C18, 2.1mmx100mm, 1.6um 【检查】 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 取本品研细,取约2g,精密称定,精密加入乙醇 100ml,照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的热浸法测定,不得少于25.0%。 【含量测定】 照高效液相色谱法(中国药典2020年版通则0512)测定。 ( 色谱条件与系统适用性试验 以十八 烷 基硅烷键合硅胶 为 填充 剂 ( 柱 长为 1 00 mm,内径 为 2. 1mm, 粒径为1 .7um), 以 乙腈 为流 动相A,以 水 为 流动 相B,按下 表中的规定进 行 梯 度洗脱 ;流 速 为 每 分钟 0.4m l;柱 温 为 30℃; 检测波 长 为 254n m 。理 论板 数 按 升 麻 素苷峰计算 应 不低于 2 0 000。 ) 时间(分钟) 流动相A(%) 流动相B(%) 0~5 10→29 90→71 5~8 29→100 71→0 8~9 100 0 9~13 10 90 对照品溶液的制备r取升麻素苷对照品品5-0-甲基维斯阿米醇苷对照品适量,精密称定,加甲醇制成每lml各含 30ug 的混合溶液,摇匀,即得。 供试品溶液的制备取本品适量,研细,取约0.1g, 精密称定,置具塞锥形瓶中,精密加入甲醇10ml,称定重量,超声处理(功率250W, 频率40kHz) 30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供试品溶液溶1ul, 注入液相色谱仪,测定,即得。 本品每1g含升麻素苷(C22H28Om)和5-0-甲基维斯阿米醇苷(C22H28O10)的总量应为6.0mg~16.0mg。 【规格】 每 1g配方颗粒相当于饮片2g 【贮藏】 密封。 二、系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱: YMC-Triart C18, 100mm×2.1mm, 1.9um((P/N: TA12SP9-10Q1PT) 流动相::乙腈/水,洗脱梯度参照药典规定方法 检测波长:210nm 柱温: 30℃ 流速: 0.4ml/min 进样量:11uL 三、 系统适用性试验谱图和数据 Time [min] Signal: DAD1B,Sig=210.0,4.0 Ref=off Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度 USP 拖尾因子 理论塔板数 USP peak1 2.635 7.219 1.77 2.487 1.88 1.24436 18078.4 S 6.427 123.595 30.23 41.396 31.28 48.0 1.03595 103928.7 peak3 7.402 54.823 13.41 17.338 13.10 11.8 1.11447 121727.8 peak4 8.085 186.615 45.64 60.623 45.81 8.2 0.98250 151590.6 peak5 10.213 36.604 8.95 10.495 7.93 24.1 1.01030 190705.6 Signal: DAD1B,Sig=210.0,4.0 Ref=off Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRT LimitRange RRTResult RPA RPALimitRange RPAResult peak1 2.635 0.410 0.000 10.00 0~0 NA 0.058 ~ S 6.427 S 1.000 1.000 peak3 7.402 S 1.152 1.140 10.00 1.026~1.254 PASS 0.444 0.05~ PASS peak4 8.085 S 1.258 0.000 10.00 0~0 NA 1.510 ~ peak5 10.213 1.589 1.540 10.00 1.386~1.694 PASS 0.296 0.1~ PASS 四、含量测定色谱条件 HPLC:Agilent 1290 四元泵 色谱柱: YMC-Triart C18, 100mm×2.1mm, 1.9pm((P/N: TA12SP9-10Q1PT) 流动相:乙腈/水,洗脱梯度参照药典规定方法 检测波长:254nm 柱温:330℃ 流速:0.4ml/min 进样量:11uL 五、 含量谱图和数据 标准品: 时间 [min] 信号: DAD1A, Sig=254.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 4.359 0.27 137.10 46.90 47.45 1.06979 50935.6 5.706 0.25 151.82 51.47 52.55 17.34617 1.07302 85539.5 总和 288.92 样品: 地址:上海市闵行区莘建东路58弄2号2908-2913室 电话 Tel.:+86(21) 54807301 54806731网址:Http://www.shhanking. com 34688030 DAD1A,Sig=254.0,4.0Ref=off 时间 [min] 信号: DAD1A, Sig=254.0,4.0Ref=off 保留时间[min] 峰宽[min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 4.118 0.10 1.24 0.45 0.18 1.05654 48325.0 4.355 0.19 212.02 72.28 30.22 3.06756 1.04975 50947.4 5.422 0.30 112.03 31.86 15.97 13.40526 1.66202 69935.5 5.703 0.26 376.25 127.77 53.63 3.50120 1.08980 85547.9 总和 701.54 六、S实验数据汇总 系统适用性试验: 实验序号 色谱柱 仪器 分离度 塔板数 含量 升麻素 苷 含量 5-0-甲基维斯 阿米醇苷 总含量 实验序号 色谱柱 仪器 T(峰3/ S) T (峰5/ S) A(峰3/ A(峰5/ 2 3.1/3.5 S) S) YMC-Triart C18 TA12SP9- Agilent 1290 1.026~1. 50935/8 4.499m 12.01m 10Q1PT 四元泵 254 5539 g/g 7.513mg/g g/g 1 YMC-Triart C18 TA12SP9-10Q1PT Agilent 1290 四元泵 1.151 1.595 0.438 0.312 七、 结论 使用 YMC C18色谱在,在国家药品标准规定色谱条件下,防风配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 知母配方颗粒标准重现 一、国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021149 知母配方颗粒Zhimu Peifangkeli 【来源】 本品为百合科植物知母 Anemarrhena asphodeloides Bge.的干燥根茎经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取知母饮片1800g, 加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为28%~43%),加辅料适量,干燥(或干燥、粉碎),加辅料适量,混匀,制粒,制成 1000g,即得。 【性状】 本品为黄色至棕黄色的颗粒;气微,味微苦。 【鉴别】 取本品0.2g,研细,加稀乙醇10ml, 超声处理20钟钟,取上清液作为供试品溶液。另取芒果苷对照品,加稀乙醇制成每1ml 含 0.5mg 的溶液,取知母皂苷BⅡ对照品,加30%丙酮制成每 1ml 含 lmg的溶液,分别作为对照品溶液。照薄层色谱法(中国药典2020年版通则0502)试验, 吸取上述三种溶液各 4ul, 分别点于同一硅胶 GF254 薄层板上,以正丁醇-冰醋酸-水(4:1:5)的上层溶液为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与芒果苷对照品色谱相应的位置上,显相同颜色的斑点。再喷以香草醛硫酸酸液,在105℃加热至斑点显色清晰,在日光下检视。供试品色谱中,在与知母皂苷BⅡ对照品色谱相应的位置上,显相同颜色的斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验检测波长为275nm,其他同(含量测定)芒果苷项下。 参照物溶液的制备 取知母对照药材 2g, 加水30ml, 加热煮沸30分钟,滤过,滤液减压蒸干,残渣加30%甲醇100ml, 超声处理(功率250W,频率40kHz) 30 分钟, 放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取芒果苷对照品适量,精密称定,加30%甲醇制成每 1ml 含50ug的溶液,作为对照品参照物溶液。 供试品溶液的制备 同(含量测定)芒果苷项下。 测定法分别精密吸取参照物溶液与供试品溶液溶2ul,注入液相色谱仪,测定,即得。 供试品色谱中应呈现5个特征峰,并应与对照药材参照物色谱中的5个特征峰保留时间相对应,与芒果苷参照物峰相对应的峰为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内。规定值为::00.87(峰2)、1.04(峰4)、1.51(峰5)。 对照特征图谱 峰2:新芒果苷;峰3(S):芒果苷;峰4:异芒果苷 色谱柱: BEH C18, 2.1 mmx100mm, 1.7um 【检查】 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104) 【浸出物】 照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的热浸法法定,用乙醇作溶剂,不得少于17.0%。 【含量测定】 芒果苷照高效液相色谱法(中国药典2020年版通则0512)测定。色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm, 内径为2.1mm,粒径为1.7um);以0.1%甲酸乙腈溶液为流动相A, 0.1%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为每分钟0.3ml;柱温为35℃;检测波长为258nm。理论板数按芒果苷峰计算应不低于20000。 时间(分钟) 流动相A(%) 流动相B(%) 0~2 5 95 2~4 5→15 95→85 4~6 15 85 6~9 15→80 85→20 9~9.1 80→100 20→0 9.1~10.6 100 0 10.6~10.7 100→5 0→95 10.7~12 5 95 对照品溶液的制备 取芒果苷对照品适量,精密称定,加30%甲醇制成每 lml 含50ug的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.1g, 精密称定,置具塞锥形瓶中,精密加入30%甲醇25ml, 密塞,称定重量,超声处理(功率250W, 频率40kHz) 30分钟, 取出,放冷,再称定重量,用30%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供试品溶液各 2ul,注入液相色谱仪,测定,即得。本品每lg含芒果苷(C19HisOn)应为4.5mg~14.5mg。 知母皂苷BⅡ 照高效效相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以辛烷基硅烷键合硅胶为填充剂(柱长为250mm,内径为4.6mm, 粒径为5.0um);以乙青-水(25:75)为流动相;流速为每分钟1.0ml, 柱温为30℃;蒸发光散射检测器检测。理论板数按知母皂苷BⅡ峰计算应不低于10000。 对照品溶液的制备 取知母皂皂BⅡ对照品适量,精密称定,加30%丙酮制成每 1ml 含0.50mg的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.1g, 精密称定,置具塞锥形瓶中,精密加入30%丙酮15ml, 密塞,称定重量,超声处理(功率250W,频率40kHz) 30分分,取出,放冷,再称定重量,用30%丙酮补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液5ul、10ul, 供共品溶液5~10ul, 注入液相相谱仪,测定,用外标两点法对数方程计算,即得。 本品占lg含知母皂苷BⅡ (C45H76019) 应为 11.0mg~41.0mg。 【规格】 每1g配方颗粒相当于饮片 1.8g 【贮藏】 密封。 二、 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18,100mm×2.1mm, 1.9um(P/N:TA12SP9-10Q1PT) 流动相:0.1%甲酸乙腈/0.1%甲酸溶液,洗脱梯度参照药典规定方法 检测波长:275nm 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86((21)54807301 54806731网址:Http://www.shhanking. com34688030 柱温:35℃ 流速:0.3ml/min 进样量:2uL 三 系统适用性试验谱图和数据 Time [min] Signal: DAD1A,Sig=275.0,4.0 Ref=off Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度 USP 拖尾因子 理论塔板数 USP peak1 2.275 103.193 14.87 24.367 13.94 1.39156 7694.3 peak2 6.422 33.248 4.79 10.019 5.73 45.4 1.34307 105723.2 7.900 501.568 72.26 124.029 70.94 16.0 1.06609 88451.8 peak4 8.228 33.605 4.84 8.800 5.03 3.1 1.16503 105304.5 peak5 10.760 22.508 3.24 7.617 4.36 28.5 0.99287 319055.5 Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRTLimitRange RRTResult RPA RPALimitRange RPAResult peak1 2.275 0.288 0.206 peak2 6.422 0.813 0.870 10.00 0.783~0.957 PASS 0.066 ~ 7.900 S 1.000 1.000 peak4 8.228 1.041 1.040 10.00 0.936~1.144 PASS 0.067 ~ peak5 10.760 1.362 1.510 10.00 1.359~1.661 PASS 0.045 ~ 四、 含量测定色谱条件(芒果苷) HPLC:Agilent 1290 四元泵色谱柱 流动相: YMC-Triart C18, 100mm×2.1mm,1.9pm(P/N:TA12SP9-10Q1PT) 流动相:0.1%甲酸乙腈/0.1%甲酸溶液,洗脱梯度参照药典规定方法 检测波长:258nm 柱温:35°C 流速: 0.3ml/min 进样量:2uL 五、 含量谱图和数据(芒果苷) 标准品: 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86(21)5480730154806731网址:Http://www.shhanking.com34688030 时间 [min] 信号: DAD1A,Sig=258.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 7.908 0.51 1419.77 354.22 100.00 1.13505 88992.2 总和 1419.77 样品: 保留时间[min] 峰宽[min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 7.909 0.33 1477.93 368.47 92.40 1.10698 89151.3 8.235 0.20 121.65 31.47 7.60 3.13033 1.07930 102721.5 总和 1599.58 六、实验数据汇总 系统适用性试验: 实验序号 色谱柱 仪器 实验结果 T(峰2/S) T(峰4/S) T (峰5/S) 0.783~0.957 0.936~1.144 1.359~1.661 1 YMC-Triart C18 TA12SP9-10Q1PT Agilent 1290 四元泵 0.813 1.041 1.362 含量测定: 实验序号 色谱柱 仪器 分离度 塔板数 含量结果 芒果苷 2 YMC-Triart C18 TA12SP9-10Q1PT Agilent 1290四元泵 3.1 88992 12.34mg/g 电话 Tel.:+86(21) 54807301 54806731 34688030 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,知母配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 连翘(青翘)配方颗粒标准重现 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021178 连翘(青翘)配方颗粒 Lianqiao (Qingqiao))Peifangkeli 【来源】 本品为木犀科植物连翘 Forsythia suspensa (Thunb.) Vahl 的干燥初熟果实经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取连翘(青翘)饮片3300g,加水煎煮,同时提取挥发油适量(以β-环糊精包合,备用),滤过,滤液浓缩成清膏(干浸膏出膏率为 19%~30%),干燥(或干燥,粉碎),加入连翘挥发油包合物(相当于0.7ml连翘挥发油)及辅料适量,混匀,制粒,制成1000g, 即得。 【性状】 本品为浅棕黄色至黄棕色的颗粒;气微香,味苦。 【鉴别】 取本品 1g,研细,加甲醇25ml,超声处理20分钟,滤过,取续滤液,作为供试品溶液。另取连翘对照药材1g, 加石油醚(30~60℃)20ml,密塞,超声处理15分钟,滤过,弃去石油醚液,残渣挥干石油醚,加甲醇20ml, 超声处理20分钟,滤过,滤液蒸干,残渣加甲醇5ml使溶解,作为对照药材溶液。再取连翘苷对照品,加甲醇制成每 1ml含0.25mg的溶液,作为对照品溶液。照薄层色谱法(中国药典2020年版通则0502)试验,吸取上述三种溶液液 3ul,分别点于同一硅胶G 薄层板上,以三氯甲烷-甲醇(8:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置日光下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验同(含量测定)连翘苷项。 参照物溶液的制备 取连翘对照药材 0.3g, 加水20ml, 煎煮30分钟,放冷,离心,取上清液,蒸干,加50%甲醇25ml, 超声处理40分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取(含量测定)连翘苷、连翘酯苷A项页对照品溶液,作为对照品参照物溶液。 供试品溶液的制备 同(含量测定)连翘苷项。 测定法 分别精密吸取参照物溶液与供试品溶液溶2ul, 注入液相色谱仪,测定,即得。 供试品色谱中应呈现6个特征峰,并应与对照药材参照物勿谱中的6个特征峰保留时间相对应,其中2个峰应分别与对照品参照物峰的保留时间相对应。与连翘酯苷A参照物峰相对应的峰为S峰,计算峰2、峰3、峰5与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:0.86(峰2)、0.97(峰3)、1.14(峰5)。 对照特征图谱 峰2:连翘酯苷I;峰3:连翘酯苷H;峰4(S):连翘酯苷A;峰6:连翘苷 色谱柱: ZORBAX Eclipse plus C18, 100mm×2.1mm, 1.8um 【检查】 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的冷浸法测定,用乙醇作溶剂,不得少于32.0%。 【含量测定】 连翘苷照高效液相色谱法(中国药典2020年版通则0512)项测定。色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(柱长为100mm, 内径为2.1mm, 粒径为1.8um);以乙腈为流动相A, 以 0.1%磷酸溶液为流动相B, 按下表中的规定进行梯度洗脱;流速为每分钟0.2ml;柱温为25℃;检测波长为 235nm。理论板数按连翘苷峰计算应不低于10000。 时间(分钟) 流动相A(%) 流动相B(%) 0~2 6→16 94→84 2~12 16 84 12~28 16→48 84→52 对照品溶液的制备 取连翘苷对照品照量,精密称定,加50%甲醇制成每 1ml 含 0.05mg的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.2g,精密称定,置具塞锥形瓶中,精密加入50%甲醇25ml,密塞,称定重量,超声处理(功率250W, 频率40kHz) 40分钟,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取吸照品溶液与供试品溶液品2ul, 注入液相色谱仪,测定,即得。本品每1g含连翘苷(C27H34On)应为10.0mg~30.0mg。 连翘酯苷A 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(柱长为 100mm, 内径为2.1mm,粒径为1.8um);以乙腈-0.4%醋酸溶液(14:86)为流动相;流速为每分钟0.3ml;检测波长为 330nm。理论板数按连翘酯苷A峰计算应不低于8000。 对照品溶液的制备 取连翘酯苷A对照品适量,精密青定,加50%甲醇制成每lml 含0.05mg 的溶液,即得(临用配制)。 供试品溶液的制备 取本品适量,研细,取约0.2g, 精密称定,置具塞锥形瓶中,精密加入50%甲醇25ml,密塞,称定重量,超声处理(功率250W,频率40kHz) 40分钟,放冷,再称定重量,用50%甲醇补足减失的重量,摇匀,滤过,精密量取续滤液1ml, 置 10ml量瓶中,加50%甲醇至刻度,摇匀,即得。 测定法分别精密吸取对照品溶液与供试品溶液各2pl,注入液相色谱仪,测定,即得。本品每1g含连翘酯苷 A (C29H36Oi5)应为 55.0mg~121.0mg。 【规格】 每lg配方颗粒相当于饮片3.3g 【贮藏】 密封。 二、 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱::1YMC-Triart C18,100mmx2.1mm,1.9um (P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.1%磷酸,洗脱梯度参照标准规定方法 检测波长: 235nm 柱温: 25℃ 流速: 0.2ml/min 进样量: 2uL 三 系统适用性试验谱图和数据 Time [min] Signal: DAD1A,Sig=235.0,4.0 Ref=off Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度 USP 拖尾因子 理论塔板数 USP peak1 5.020 7270.878 11.78 2183.555 21.94 1.15610 51492.6 peak2 13.899 13114.882 21.25 1123.553 11.29 44.0 1.50442 31079.5 peak3 16.411 7800.002 12.64 1269.413 12.76 5.4 1.21582 151463.1 S 16.621 20183.469 32.71 2705.980 27.19 1.1 1.41244 111463.0 peak5 17.862 6951.734 11.27 1311.169 13.18 9.4 0.87797 288070.3 peak6 20.758 6167.660 10.00 1320.062 13.26 22.3 0.98441 429297.7 Signal: DAD1A,Sig=235.0,4.0 Ref=off Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRTLimitRange RRTResult RPA RPALimitRange RPAResult peak1 5.020 S 0.302 0.360 peak2 13.899 S 0.836 0.860 10.00 0.774~0.946 PASS 0.650 ~ peak3 16.411 S 0.987 0.970 10.00 0.873~1.067 PASS 0.386 ~ S 16.621 S 1.000 1.000 peak5 17.862 S 1.075 1.140 10.00 1.026~1.254 PASS 0.344 ~ peak6 20.758 S 1.249 0.306 四、 含量测定色谱条件(连翘苷) HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18,100mm×2.1mm,1.9um (P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.1%磷酸,洗脱梯度参照标准规定方法 检测波长: 235nm 柱温: 25℃ 流速: 0.2ml/min 进样量: 2uL 含量测定色谱条件(连翘酯苷A) HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18,,1100mm×2.1mm,1.9um (P/N:TA12SP9-10Q1PT)流动相: 乙腈/0.4%醋酸=14/86 检测波长: 330nm 柱温: 25℃ 流速: 0.3ml/min 进样量: 2uL 五、 含量谱图和数据 连翘苷标准品: 信号: Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 20.758 0.41 782.89 185.40 100.00 1.08962 558039.7 总和 782.89 样品: 地址:上海市闵行区莘建东路58弄2号2908-2913室 电话 Tel.:+86((21)54807301 54806731网址:Http://www.shhanking. com 34688030 信号: DAD1A,Sig=235.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 20.773 0.20 2002.01 506.97 93.43 1.07734 488170.8 20.928 0.17 140.79 36.24 6.57 1.50990 1.06052 677501.1 总和 2142.80 连翘酯苷A标准品: DAD1A,Sig=330.0,4.0Ref=off 信号: DAD1A,Sig=330.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 17.967 1.61 563.58 26.00 100.00 1.03306 15654.2 总和 563.58 样品: DAD1A,Sig=330.0,4.0Ref=off 地址:上海市闵行区莘建东路58弄2号2908-2913室 电话 Tel.:+86(21) 54807301 54806731网址:Http://www.shhanking. com 34688030 信号: DAD1A,Sig=330.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 16.728 0.61 72.26 4.07 0.83 1.09595 19094.7 17.603 1.55 8645.35 370.36 99.17 1.59314 1.24442 12879.0 总和 8717.60 六、实验数据汇总 系统适用性试验: 实验序号 色谱柱 仪器 实验结果 T(峰2/S) T(峰3/S) T(峰5/S) 0.774~0.946 0.873~1.067 1.026~1.254 1 YMC-Triart C18 TA12SP9-10Q1PT Agilent 1290 四元泵 0.836 0.987 1.075 含量测定: 实验序号 色谱柱 仪器 分离度 塔板数 含量结果 1 Agilent 1290四 元泵 1.50 558039 连翘苷: YMC-Triart C18 TA12SP9-10Q1PT 15.7346mg/g 1.59 15654 连翘酯苷A: 93.6634mg/g 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,连翘(青翘)配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 厚朴(厚朴)配方颗粒标准重现 一\ 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021059 厚朴(厚朴)配方颗粒 Houpo (Houpo)Peifangkeli 【来源】 本品为木兰科植物厚朴 Magnolia officinalis Rehd.et Wils.的干燥干皮经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取厚朴饮片8000g,加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为7%~12.5%),加入辅料适量,干燥(或干燥,粉碎),再加入辅料适量,混匀,制粒,制成1000g, 即得。 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86 ((21) 54807301 54806731网址:Http://www.shhanking. com34688030 【性状】 本品为黄棕色至棕色的颗粒;气微,味苦。 【鉴别】 取本品1g,研细,加加醇 5ml, 超声处理30分钟,滤过,滤液作为供试品溶液。另取厚朴(厚朴)对照药材1g,同法制成对照药材溶液。再取厚朴酚对照品、和厚朴酚对照品,加甲醇制成每 1ml 各含 1mg 的混合溶液,作为对照品溶液。照薄层色谱法(中国药典2020年版年则0502)试验,吸取供试品溶液和对照药材溶液溶2ul,对照品溶液5ul,分别点于同一硅胶G 薄层板上,以甲苯-甲醇(17:1)为展开剂,展开,取出,晾干,喷以1%香草醛硫酸溶液,在100℃加热至斑点显色清晰。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm, 内径为2.1mm,粒径为1.7um),以乙腈为流动相A, 以0.4%磷夜溶液为流动相B, 按下表中的规定进行梯度洗脱;流速为每分钟0.4ml;柱温为30℃;检测波长为 294nm。理论板数按和厚朴酚峰计算应不低于50000。 时间(分钟) 流动相A(%) 流动相B(%) 0~7 8 92 7~14 8→10 92→90 14~19 10→11 90→89 19~28 11→20 89→80 28~34 20→48 80→52 34~48 48 52 48~50 48→8 52→92 参照物溶液的制备 取厚朴(厚朴)对照药材lg,置具塞锥形瓶中,加水20ml,加热回流30分钟,放冷,离心,取上清液,蒸干,加甲醇25ml, 超声处理(功率250W, 频率40kHz) 30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取(含量测定)项下对照品溶液,作为对照品参照物溶液。 供试品溶液的制备取本品适量,研细,取约0.2g, 置具塞锥形瓶中,加甲醇25ml, 超声处理(功率250W, 频率40kHz) 30分钟, 放冷,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取取照物溶液与供试品溶液各1pl, 注入液相色谱仪,测定,即得。 供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的6个特征峰保留时间相对应,其中2个峰应分别与相应对照品参照物峰的保留时间相对应。与和厚朴酚参照物峰相对应的峰 为S峰,计算峰2、峰3、峰4与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:0.67(峰2)、0.72(峰3)、0.86(峰4);计算峰6与S峰的相对峰面积,其相对峰面积应在规定范围内,规定范围为:0.51~3.50(峰6)。 峰5(S):和厚朴酚;峰6:厚朴酚 色谱柱: Acquity BEH C18, 2.1mmx100mm, 1.7um 【检查】 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 取本品研细,取约2g, 精密称定,精密加入乙醇100ml,照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的热浸法测定,不得少于25.0%。 【含量测定】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(78:22)为流动相;检测波长为294nm。理论板数按厚朴酚峰计算应不低于3800。 对照品溶液的制备 取厚朴酚对照品、和厚朴酚对照品适量,精密称定,加甲醇制成每1ml含厚朴酚20ug、和厚朴酚 10ug 的混合溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.1g, 精密称定,置具塞锥形瓶中,精密加入甲醇25ml,称定重量,超声处理(功率250W,频率40kHz) 30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液与供试品溶液 10pl, 注入液相色谱仪,测定,即得。 本品品1g含厚朴酚(C18H18O2)与和厚朴酚(Ci8H18O2)的总量应为7.0mg~25.0mg。 【规格】 每lg配方颗粒相当于饮片8g 【贮藏】 密封。 二、 系统适用性试验色谱条件 HPLC: Agilent 1290 四元泵 色谱柱:YMC-Triart C18l,, 1100mmx2.1mm, 1.9pm(P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.4%磷酸溶液,洗脱梯度参照药典规定方法 检测波长: 294nm 柱温: 30℃ 流速: 0.4ml/min 进样量: 1uL 三、 系统适用性试验谱图和数据 Time [min] Signal: DAD1A,Sig=294.0,4.0 Ref=off Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度 USP 拖尾因子 理论塔板数 USP peak1 21.728 81.428 15.04 8.708 10.38 1.07609 122500.7 peak2 28.124 50.059 9.24 8.355 9.96 31.9 1.14028 540684.6 peak3 30.061 206.380 38.11 45.503 54.22 14.1 1.05645 958965.7 peak4 33.195 6.432 1.19 1.717 2.05 28.9 0.96000 1988581.2 S 40.642 97.059 17.92 11.479 13.68 46.9 1.05789 528831.2 peak6 43.977 100.181 18.50 8.161 9.72 12.1 1.05797 290288.5 Signal: DAD1A,Sig=294.0,4.0 Ref=off Name RT[min1 RefName RRT RRTLimit RRTLimitRangeP RRTLimitRange RRTResult RPA RPALimitRange RPAResult peak1 21.728 S 0.535 0.000 0.00 0~0 NA 0.839 ~ peak2 28.124 S 0.692 0.670 10.00 0.603~0.737 PASS 0.516 ~ peak3 30.061 S 0.740 0.720 10.00 0.648~0.792 PASS 2.126 ~ peak4 33.195 0.817 0.860 10.00 0.774~0.946 PASS 0.066 ~ S 40.642 S 1.000 1.000 peak6 43.977 S 1.082 0.000 0.00 0~0 NA 1.032 0.51~3.5 PASS 四、 含量测定色谱条件 HPLC:Thermo U3000 色谱柱:YMC Pack Pro C18,,4.6×250mm, 5pm(P/N:AS12S05-2546PTH) 流动相: 甲醇/水=78/22 检测波长: 294nm 柱温: 30℃ 流速: 1.0ml/min 进样量: 10uL 五、 含量谱图和数据 标准品: 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86((21)5480730154806731网址:Http://www. shhanking. com34688030 Total: 8.185 42.780 10.77 2.09 样品: 六、 实验数据汇总 系统适用性试验: 地址:上海市闵行区莘建东路58弄2号2908-2913室 电话 Tel. :+86 ((21) 54807301 54806731网址:Http://www.shhanking. com 34688030 实验 序号 色谱柱 仪器 T(峰2/S) T(峰3/S) T(峰4/S) A(峰6/S) 0.603~0.737 0.648~0.792 0.774~0.946 0.51~3.50 仪器 分离度 塔板数 厚朴酚 和厚朴酚 总量 序号 YMC 1 Pack Pro 1 C18 TA12SP9-10Q1PT C18, 1290 四元 泵 Thermo 0.692 14634/ 3.3531 1.032 11.8198 15.1729 4.6×250mm, 5um U3000 10.8 15910 mg/g mg/g mg/g AS12S05- 2546PTH 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,厚朴配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 白芷(白芷)配方颗粒 \ 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021004 白芷(白芷)配方颗粒 Baizhi (Baizhi) FPeifangkeli 【来源】 本品为伞形科植物白芷 Angelica dahurica (Fisch. ex Hoffm.) Benth.et Hook.f.的干燥根经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取白芷芷片3000g,加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为20%~33%),加入辅料适量,干燥(或干燥,粉碎),再加入辅料适量,混匀,制粒,制成 1000g, 即得。 【性状】 本品为浅黄色至棕黄色的颗粒;气芳香,味辛,微苦。 【鉴别】 取本品 1g,研细,加水20ml使溶解,加乙醚20ml振摇提取,分取乙醚液,挥干乙醚,残渣加乙酸乙酯1ml使溶解,作为供试品溶液。另取白芷对照药材 1g, 加水 50ml, 煮沸30分钟,滤过,滤液浓缩至20ml, 加乙醚20ml, 同法制成对照药材溶液。再取欧前胡素对照品,加乙酸乙酯制成 1ml含lmg的溶液,作为对照品溶液。照薄层色谱法(中国药典2020年版通则0502)试验,吸取上述供试品溶液与对照药材溶液材10pl, 对照品溶液 lul,分别点于同一硅胶G 薄层板上,以石油醚(30~60℃)-乙醚(3:2)为展开剂,在25℃以下展开,取出,晾干,置紫置光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。 【特征图谱】 照高效液相相谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为1.8um),以乙腈为流动相A, 以0.1%乙酸溶液为流动相B, 按下表中的规定进行梯度洗脱;流速为每分钟0.35ml;柱温为35℃;检测波长为300nm。理论板数按欧前胡素峰计算应不低于3000。 时间(分钟) 流动相A(%) 流动相B(%) 0~5.5 15→28 85→72 5.5~7.0 28→40 72→60 7.0~9.5 40 60 9.5~15.5 40→65 60→35 15.5~15.51 65→15 35→85 15.51~18.00 15 85 参照物溶液的制备取白芷对照药材 0.6g, 置具塞锥形瓶中,加水20ml, 加热回流20分钟,放冷,离心,取上清液10ml,加甲醇10ml,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另另欧前胡素对照品、异欧前胡素对照品适量,精密称定,加甲醇制成醇1ml 含欧前胡素 10pg、异欧前胡素2ug的混合溶液,作为对照品参照物溶液。 供试品溶液的制备 取本品适量,研细,取约0.2g, 置具塞锥形瓶中,加甲醇 20ml, 超声处理(功率300W, 频率50kHz) 20 放钟,放冷,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取参照物溶液与供试品溶液各1pl,注入液相色谱仪,测定,即得。 供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的6个特征峰保留时间相对应,其中2个峰应分别与相应对照品参照物峰的保留时间相对应。与欧前胡素参照物峰相对应的峰为S峰,计算峰1、峰2、峰3、峰5与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内;规定值为:0.49(峰1)、0.52(峰2)、0.67(峰3)、1.05(峰5);计算峰1、峰3、峰5与S峰的相对峰面积,其相对峰面积应在规定的范围内,规定范围为:不低于0.713(峰1)、不低于0.338(峰3)、不低于0.220(峰5)。 对照特征图谱 峰1:水合氧化前胡素;峰2:白当归素;峰3:佛手苷内酯;峰4(S):欧前胡素; 峰5:珊瑚菜素;峰6:异欧前胡素 色谱柱: SB C18, 2.1mmx100mm, 1.8um 【检查】 重金属及有害元素 照铅、镉、砷、汞、铜测定法(法国药典2020年版通则2321)测定,铅不得过 5mg/kg;镉不得过 1mg/kg;砷不得过 2mg/kg;汞不得过 0.2mg/kg;铜不得过 20mg/kg。 其他应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 取本品研细,取约2g,精密称定,精密加入乙醇100ml,照醇溶性浸出物勿定法(中国药典2020年版通则2201)项下的热浸法测定,不得少于15.0%。 【含量测定】 照高效液相色谱法(中国药典2020年版通则0512)测定。色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(55:45)为流动相;检测波长为300nm。理论板数按欧前胡素峰计算应不低于3000。对照品溶液的制备 取欧前胡素对照品适量,精密称定,加甲醇制成每 lml 含 5ug的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.2g, 精密称定,置具塞锥形瓶中,精密加入稀乙 醇20ml, 定定重量,超声处理(功率300W, 频率50kHz) 30分钟,放冷,再称定重量,用稀乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供试品溶液各10ul, 注入液相色谱仪,测定,即得。 本品每1g含欧前胡素(C16H1404) 应为 0.25mg~1.20mg。 【规格】 每1g配方颗粒相当于饮片3g 【贮藏】 密封。 二、 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18,100mm×2.1mm, 1.9um (P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.1%乙酸,洗脱梯度参照标准规定方法 检测波长: 300nm 柱温: 35℃ 流速: 0.35m1/min 进样量: 1uL 三、 系统适用性试验谱图和数据 电话 Tel.:+86((21)5480730154806731 Integration Results No. Peak Name Retention Timemin AreamAU*min Height mAU Relative Area% Relative Height % Amountn.a. 123456 8.2838.58011.06315.56016.24716.787 1.3180.7780.7811.0020.392 0.387 27.56117.6879.55515.0886.0915.942 28.3016.7016.7621.518.42 8.31 33.6421.5911.6618.427.437.25 n.a.n.a.1.n.a.n.a.n.a.n.a. Total: 4.658 81.923 100.00 100.00 RRT RRT RRT RPA RRT RRT Limit RPA RPA Name RT(min) Area RefName LimitRangeP LimitRange Result LimitRange Result Peak1 8.283 1.318 0.532 0.49 10 0.441~0.539 PASS 1.315 0.713~ PASS Peak2 8.580 0.778 0.551 0.52 10 0.468~0.572 PASS 0.776 - Peak3 11.063 0.781 0.711 0.67 10 0.603~0.737 PASS 0.779 0.338~ PASS 15.560 1.002 1.000 - - 1.000 Peak5 16.247 0.392 1.044 1.05 10 0.945~1.155 PASS 0.391 0.220~ PASS Peak6 16.787 0.387 1.079 - - - 0.386 四、含量测定色谱条件 HPLC:TThermo U3000 色谱柱: YMC Pack Pro C18, 4.6×250mm, 5pm ((P/N: AS12S05-2546PTH) 流动相: 甲醇/水=55/45 检测波长:3300nm 柱温: 35℃ 流速: 1.0ml/min 进样量:110uL 五、 含量谱图和数据 标准品: Total: 2.287 3.201 0.00 0.95 样品: 六、实验数据汇总 系统适用性试验: T(峰 T(峰 T(峰 T(峰 A(峰 A(峰 A(峰 实验序号 色谱柱 仪器 1/S) 2/S) 3/S) 5/S) 1/S) 3/S) 5/S) 0.441~ 0.539 0.468~ 0.572 0.603~ 0.737 0.945~1 .155 >0.713 ≥0.338 ≥0.220 YMC-Triart Agilent 1 C1: TA12SP9- 1290 四 元泵 0.532 0.551 0.711 1.044 1.315 0.779 0.391 10Q1PT 含量测定: 实验序号 色谱柱 仪器 检测 分离度 理论板数 总量 物质 1 10.2 17314 YMC Pack Pro C18, Thermo 欧前 1.77 4.6×250mm, 5um U3000 胡素 mg/g AS12S05-2546PTH 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,白芷(白芷)配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 虎杖配方颗粒 国家标准 国家药品监督管理局国家药品标准 YBZ-PFKL-2021060 虎杖配方颗粒Huzhang Peifangkeli 【来源】 本品为蓼科植物虎杖 Polygonum cuspidatum Sieb.et Zucc.的干燥根茎及根经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取虎杖饮片4500g, 加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为12%~22%),干燥(或干燥,粉碎),加入辅料适量,混匀,制粒,制成1000g, 即得。 【性状】 本品为黄棕色至棕褐色的颗粒;气微,味微苦、涩。 【鉴别】 取本品0.15g,研细,加甲醇10ml,超声处理30分钟,滤过,滤液蒸干,残渣加甲醇2ml 使溶解,作为供试品溶液。另取虎杖对照药材0.1g, 同法制成对照药材溶液。再取大黄素对照品、虎杖苷对照品适量,加甲醇制成每 1ml各含 lmg 的溶液,作为对照品溶液。照薄层色谱法(中国药典2020年版通则0502)试验,吸取上述供试品溶液与对照药材溶液各 2ul, 虎杖苷对照品溶液2pl,大黄素对照品溶液1pl, 分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(4:1:0.1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。 【特征图谱】 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为1.7um);以乙腈为流动相A, 以0.2%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为每分钟 0.4ml;柱温为40℃;检测波长为290nm。理论板数按虎杖苷峰计算应不低于3000。 时间(分钟) 流动相A(%) 流动相B(%) 0~7 12→20 88→80 7~10 20→28 80→72 10~12 28 72 12~15 28→30 72→70 15~16 30→80 70→20 16~18 80 20 18~18.01 80→12 20→88 18.01~20 12 88 参照物溶液的制备 取虎杖对照药材 0.4g,加水40ml,煮沸30分钟,滤过,取滤液蒸干,残渣加甲醇50ml, 超声处理(功率250W, 频率40kHz) 30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液。另取虎杖苷对照品、白藜芦醇对照品、大黄素对照品、大黄素甲醚对照品和大黄素-8-0-B-D-葡萄糖苷对照品适量,精密称定,加甲醇制成每 lml各含50ug 的混合溶液,作为对照品参照物溶液。 供试品溶液的制备 同(含量测定)虎杖苷项。 测定法分别精密吸取参照物溶液与供试品溶液各 1ul,注入液相色谱仪,测定,即得。 供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的6个特征峰保留时间相对应,其中5个峰应分别与相应对照品参照物峰的保留时间相对应。与虎杖苷参照物峰相对应的峰为S峰,计算峰2与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:1.65(峰2);计算峰4与S峰的相对峰面积,其相对峰面积应在规定范围内,规定范围为:不低于0.22(峰4) 对照特征图谱 峰1(S):虎杖苷:峰3:白藜芦醇:峰4:大黄素-8-O-B-D-葡萄糖苷 峰5:大黄素;峰6:大黄素甲醚 色谱柱 Acuquity BEH C18, 2.1mm×100mm, 1.7um 【检查】 应符合颗粒剂项下有关的各项规定(中国药典2020年版通则0104)。 【浸出物】 取本品研细,取约2g,精密称定,精密加入乙醇100ml, 照醇溶性浸出物测定法(中国药典2020年版通则2201)项下的热浸法测定,不得少于30.0%。 【含量测定】 大黄素 照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(充长为50mm,内径为 2.1mm,粒径为1.7um), 以甲醇-0.1%磷酸夜液(80:20)为流动相;流速为每分钟0.4ml;柱温为30℃;检测波长为 254nm。理论板数按大黄素峰计算应不低于2500。 对照品溶液的制备 取大黄素对照品适量,精密称定,加甲醇制成每1ml 含24pg的溶液,即得。 供试品溶液的制备取本品适量,研细,取约0.15g,精密称定,精密加入三氯甲烷50ml 和2.5mol/L硫酸溶液20ml,定定重量,置80℃水浴中加热回流2小时,冷却至室温,再称定重量,用三氯甲烷补足减失的重量,摇匀。分取三氯甲烷液,精密量取 5ml, 蒸干,残渣加甲醇使溶解,转移至10ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供与品溶液各1pl,注入液相色谱仪,测定,即得。 本品每 1g 含大黄素 (CisH1oOs) 应为9.0mg~24.0mg。 虎杖苷避光操作。照高效液相色谱法(中国药典2020年版通则0512)测定。 色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂(柱长为50mm,内径为2.1mm,粒径为1.7um);以乙腈为流动相A, 以0.2%甲酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为每分钟0.4ml;柱温为40℃;检测波长为306nm。理论板数按虎杖苷峰计算应不低于2500。 时间(分钟) 流动相A(%) 流动相B(%) 0~3 12 88 3~5 12→75 88→25 5~8 75 25 8.01~10 12 88 对照品溶液的制备 取虎杖苷对照品适量,精密精定,加甲醇制成醇1ml 含 80ug的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.15g, 精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理(功率250W, 频率40kHz) 30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法分别精密吸取对照品溶液与供与品溶液各 1ul, 注入液相色谱仪,测定,即得。本品每lg含虎杖苷(C20H22Og)应为21.0mg~61.0mg。【注意】 孕妇慎用。【规格】 每1g配方颗粒相当于饮片4.5g。【贮藏】 密封 二、 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18, 100mmx2.1mm,1.9um (P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.2%甲酸溶液,洗脱梯度参照中国药典规定方法 检测波长: 290nm 柱温: 40℃ 流速: 0.4m1/min 进样量::11pL 三、 系统适用性试验谱图和数据 AM 08 Time [min] Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度USP 拖尾因子 理论塔板数 USP 6.589 1096.144 56.83 194.600 51.53 1.12077 31515.4 peak2 10.225 19.555 1.01 4.001 1.06 25.9 1.08948 96511.3 peak3 10.864 191.044 9.90 36.970 9.79 4.7 1.05855 98242.4 peak4 12.946 433.021 22.45 80.524 21.32 14.8 1.09750 130380.9 peak5 18.010 173.740 9.01 58.725 15.55 45.8 1.06434 858038.0 peak6 19.058 15.327 I0.79 2.827 0.75 11.3 0.89116 507659.5 Signal: DAD1A,Sig=290.0,4.0 Ref=off Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRT LimitRange RRTResult RPA RPALimitRange RPAResult S 6.589 S 1.000 1.000 peak2 10.225 S 1.552 1.650 10.00 1.485~1.815 PASS 0.018 ~ peak3 10.864 S 1.649 0.174 peak4 12.946 S 1.965 0.000 10.00 0~0 NA 0.395 0.22~ PASS peak5 18.010 S 2.733 0.159 peak6 19.058 2.893 0.014 四、 含量测定色谱条件(大黄素) HPLC: Agilent 1290 四元泵 色谱柱: YMC-Triart C18, 100mm×2.1mm, 1.9um(P/N:TA12SP9-10Q1PT)流动相: 甲醇/0.1%磷酸=80/20 检测波长:254nm 柱温: 30℃ 流速: 0.4m1/min 进样量: 1pL 含量测定色谱条件(虎杖苷) HPLC: Agilent 1290 四元泵 色谱柱: YMC-Triart C18, 100mmx2.1mm, 1.9um(P/N: TA12SP9-10Q1PT) 流动相: 乙腈/0.2%甲酸,洗脱梯度参照标准方法 检测波长:306nm 柱温: 40℃ 流速: 0.4ml/min 进样量::11pL 五、 含量谱图和数据 大黄素标准品: 信号: DAD1A,Sig=254.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 2.682 0.63 209.64 40.05 100.00 1.13846 6147.5 总和 209.64 样品: 信号: DAD1A,Sig=254.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 2.683 0.50 144.99 27.29 93.84 1.18388 6005.9 4.565 0.53 9.52 1.23 6.16 11.14457 1.17062 8332.2 总和 154.51 虎杖苷标准品: 保留时间[min 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 5.408 0.31 761.62 283.36 100.00 0.99376 89900.8 总和 761.62 样品: 地址:上海市闵行区莘建东路58弄2号2908-2913室 电话 Tel.:+86 ((21) 54807301 54806731网址:Http://www.shhanking. com 34688030 时间[min] 信号: DAD1A,Sig=306.0,4.0Ref=off 保留时间[min] 峰宽 [min] 峰面积 高度 峰面积% 分离度 USP 拖尾因子 理论塔板数 USP 5.409 0.16 1468.32 543.10 95.95 1.02891 91468.3 5.542 0.10 61.90 22.62 4.05 1.80185 0.88858 84172.0 总和 1530.22 六、实验数据汇总 系统适用性试验: 实验 色谱柱 仪器 实验结果 序号 T(峰2/S) A(峰4/S) 1.485~1.815 >0.22 1 YMC-Triart C18 TA12SP9-10Q1PT Agilent 1290 四元泵 1.552 0.395 含量测定: 实验 色谱柱 仪器 检测 物质 分离度 塔板数 含量结果 序号 1 YMC-Triart C18 Agilent 1290 大黄素 11.1 6147 21.99mg/g TA12SP9-10Q1PT 四元泵 虎杖苷 1.8 89900 47.05mg/g 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,虎杖配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 猫爪草配方颗粒 国家标准 地址:上海市闵行区莘建东路58弄2号2908-2913室 电话 Tel.:+86 ((21) 54807301 54806731网址:Http://www. shhanking. com 34688030 山东省药品监督管理局中药配方颗粒标准 SDPFKL-2021077 猫爪草配方颗粒 Maozhuacao Peifangkeli 【来源】 本品为毛莨科植物小毛莨 Ranunculus ternatus Thunb. 的干燥块根经炮制并按标准汤剂的主要质量指标加工制成的配方颗粒。 【制法】 取猫爪草饮片2200g,加水煎煮,滤过,滤液浓缩成清膏(干浸膏出膏率为22.5%~34.0%),加入辅料量量,干燥(或干燥,粉碎),再加入辅料适量,混匀,制粒,制成 1000g, 即得。 【性状】 本品为浅黄色至黄棕色的颗粒;气微,味微甘、微苦。 【鉴别】 取本品 1g,研细,加稀乙醇10ml,超声处理30分钟,滤虑,取滤液作为供试品溶液。另取猫爪草对照药材 1g,同法制成对照药材溶液。照薄层色谱法(《中国药典》2020年版通则0502)试验,吸取上述两两溶液各2~5ul,分别点于同一硅胶G 薄层板上,以正丁醇-无水乙醇-冰醋酸-水(8:2:2:3)为展开剂,展开,取出,晾干,喷以茚三酮试液,热风吹至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的主斑点。 【特征图谱】 照高效液相色谱法(《中国药典》2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm, 内径为2.1mm, 粒径为1.8um);以乙腈为流动相A, 以0.1%磷酸溶液为流动相B,按下表中的规定进行梯度洗脱;流速为每分钟 0.3ml;柱温为40℃; 检测波长为280nm。理论板数按尿苷峰计算应不低于1000。 时间(分钟) 流动相A(%) 流动相B(%) 0~2 0 100 2~27 0→15 100→85 40 参照物溶液的制备 取猫爪草对照药材1g, 加水25ml,加热回流30分钟,滤过,取滤液作为对照药材参照物溶液。另取尿苷对照品、5-羟甲基糠醛对照品,加30%甲醇制成每 1ml 各含 30ug的溶液,作为对照品参照物溶液。 供试品溶液的制备 同(含量测定)项。 测定法 分别精密吸取参照物溶液与供与品溶液各 1ul, 注入液相色谱仪,测定,即得。 供试品色谱中应呈现与对照药材参照物色谱相对应的5个特征峰,其中2个峰应分别与相应对照品参照物峰的保留时间相对应,以5-羟甲基糠醛参照物峰相应的峰为S峰,计算峰2、峰4、峰5与S峰的相对保留时间,应在规定值的±10%范围之内,规定值为:0.70(峰2)、1.93(峰4)、2.48(峰5)。 对照品溶液的制备 取尿苷对照品适量,精密称定,加30%甲醇制成每1ml含 30ug 的溶液,即得。 对照特征图谱 峰1:尿苷;峰3(S):5-羟甲基糠醛 色谱柱 HSS T3 Cis, 2.1mmx100mm, 1.8um 【检查】 应符合颗粒剂项下有关的各项规定(《中国药典》2020年版通则0104)。 【浸出物】 照醇溶性浸出物测定法(《中国药典》2020年版通则2201)项下的热浸法测定,用乙醇作溶剂,不得少于20.0%。 【含量测定】 照高效液相色谱法(《中国药典》2020年版通则0512)测定。 色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为100mm,内径为2.1mm,粒径为1.8um);以乙腈为流动相A, 以0.1%磷酸溶液为流动相B,按下表中的规定进行梯度洗度;流速为每分钟0.3ml;柱温为40℃; 检测波长为 260nm。理论板数按尿苷峰计算应不低于1000。 时间(分钟) 流动相A(%) 流动相B(%) 0~8 0 100 8~8.1 0→20 100→80 8.1~11 20 80 对照品溶液的制备 取尿苷对照品适量,精密称定,加30%甲醇制成每 1ml含30ug的溶液,即得。 供试品溶液的制备 取本品适量,研细,取约0.5g, 精密称定,置具塞锥形瓶中,精密加入30%甲醇15ml,密塞,称定重量,超声处理(功率250W,频率40kHz)30分钟,放冷,再称定重量,用30%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。 测定法 分别精密吸取对照品溶液与供试品溶液溶1ul, 注入液相色谱仪,测定,即得。 本品每lg含尿苷(C)H12N2O6)应为0.10mg~1.2mg。 【规格】 每lg配方颗粒相当于饮片2.2g 【贮藏】 密封。 二、.\ 系统适用性试验色谱条件 HPLC:Agilent 1290 四元泵 色谱柱:YMC-Triart C18, 100mm×2.1mm, 1.9um(P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.1%磷酸,洗脱梯度参照规定方法 检测波长: 280nm 柱温: 40℃ 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86((21)5480730154806731网址:Http://www. shhanking. com34688030 流速:0.3ml/min 进样量: 1uL 三 系统适用性试验谱图和数据 Signal: DAD1A,Sig=280.0,4.0 Ref=360.0,100.0 Name 保留时间 峰面积 峰面积% 峰高 峰高% 分离度USP 拖尾因子 理论塔板数 USP peak1 3.655 85.075 26.12 14.102 29.53 1.12710 8112.5 peak2 5.146 110.486 33.93 16.390 34.32 8.8 1.18883 13580.6 7.636 48.426 14.87 7.714 16.16 14.6 1.05250 34860.4 peak4 15.003 9.797 3.01 1.240 2.60 40.5 1.00843 89665.9 peak5 19.645 71.864 22.07 8.305 17.39 21.7 1.09258 119919.5 Signal: DAD1A,Sig=280.0,4.0 Ref=360.0,100.0 Name RT[min] RefName RRT RRTLimit RRTLimitRangeP RRTLimitRange RRTResult RPA RPALimitRange RPAResult peak1 3.655 S 0.479 1.757 peak2 5.146 0.674 0.700 10.00 0.63~0.77 PASS 2.282 ~ 7.636 S 1.000 1.000 peak4 15.003 S 1.965 1.930 10.00 1.737~2.123 PASS 0.202 ~ peak5 19.645 S 2.573 2.480 10.00 2.232~2.728 PASS 1.484 ~ 四、 含量测定色谱条件 HPLC: Agilent 1290 四元泵 色谱柱: YMC-Triart C18, 100mm×2.1mm, 1.9um0((P/N:TA12SP9-10Q1PT) 流动相: 乙腈/0.1%磷酸,洗脱梯度参照标准规定方法 检测波长: 260nm 柱温: 40℃ 流速: 0.3ml/min 进样量: 1uL 五、 含量谱图和数据 标准品: 地址:上海市闵行区莘建东路58弄2号2908-2913室电话 Tel.:+86((21)5480730154806731网址:Http://www. shhanking. com34688030 样品: 六、 实验数据汇总 系统适用性试验: 实验 序号 色谱柱 仪器 实验结果 T(峰2/S) T(峰4/S) T(峰5/S) 0.630~0.770 1.737~2.123 2.232~2.728 1 YMC-Triart C18 Agilent 1290 四元泵 0.674 1.965 2.573 TA12SP9-10Q1PT 含量测定: 实验 序号 色谱柱 仪器 检测物质 分离度 塔板数 总量 YMC-Triart C18 S- 1 1.9um 2.1*100mm,12nm Thermo U3000 尿苷 1.69 9529 0.664 mg/g TA12SP9-10Q1PT 七、 结论 使用 YMC C18色谱柱,在国家药品标准规定色谱条件下,猫爪草配方颗粒检测中系统适用性试验与含量测定,实验结果均满足检测要求。 Shanghai Hanking Instrument & Equipment Co,.Ltd 地址:上海市闵行区莘建东路网址:Http://www.shhanking.com.电话 Tel.:+ 中药是指基于传统中医理论指导下在临床使用的植物、动物等天然物质及其复方。中药配方颗粒标准的公布将帮助改善大家印象中传统中药品质良莠不齐的刻板印象,标准中规定的多组分有效成分相比于传统中药饮片的单成分含量更全面。 随着标准将于2021年11月1日正式实施,为了帮助企业快速进行方法重现的工作,上海汉尧仪器设备有限公司作为YMC色谱柱中国区代理商,依托相关实验室进行了不同品种的指纹图谱和含量图谱的方法重现工作。中药配方颗粒方法重现报告如下:
确定


















还剩44页未读,是否继续阅读?

上海汉尧仪器设备有限公司为您提供《中药配方颗粒中指纹图谱和含量图谱检测方案 》,该方案主要用于中药配方颗粒中特征图谱检测,参考标准--,《中药配方颗粒中指纹图谱和含量图谱检测方案 》用到的仪器有
相关方案
更多
该厂商其他方案
更多