








方案详情
文
当静脉注射时,各种颗粒和纳米药物会激活补体,可能导致输液反应和其他药物不良反应。颗粒在治疗蛋白的配方中,由于运输、处理和给病人的治疗过程中产生的应力形成。在本研究中,IVIg溶液被储存在多种类型的小瓶和预填充的注射器中,并暴露在搅拌和冻融应力下产生颗粒。将应激样本加入人血清中,以确定这些颗粒是否激活补体。大小在2到10微米的亚可见IVIg颗粒其激活补体的方式与颗粒数量呈现出线性关系,而在较大粒子(>10微米)的剂量和补体激活之间几乎没有相关性。通过亚可见颗粒IVIg激活补体是另一种途径,如补体级联因子Bb的释放和无C4a生成的过敏性毒素C3a和C5a。亚可见颗粒的数量和形态取决于所施加的应力、配方和容器材料。但2- 10微米大小的微粒激活人血清补体的能力仅取决于微粒浓度。
点击下载
方案详情

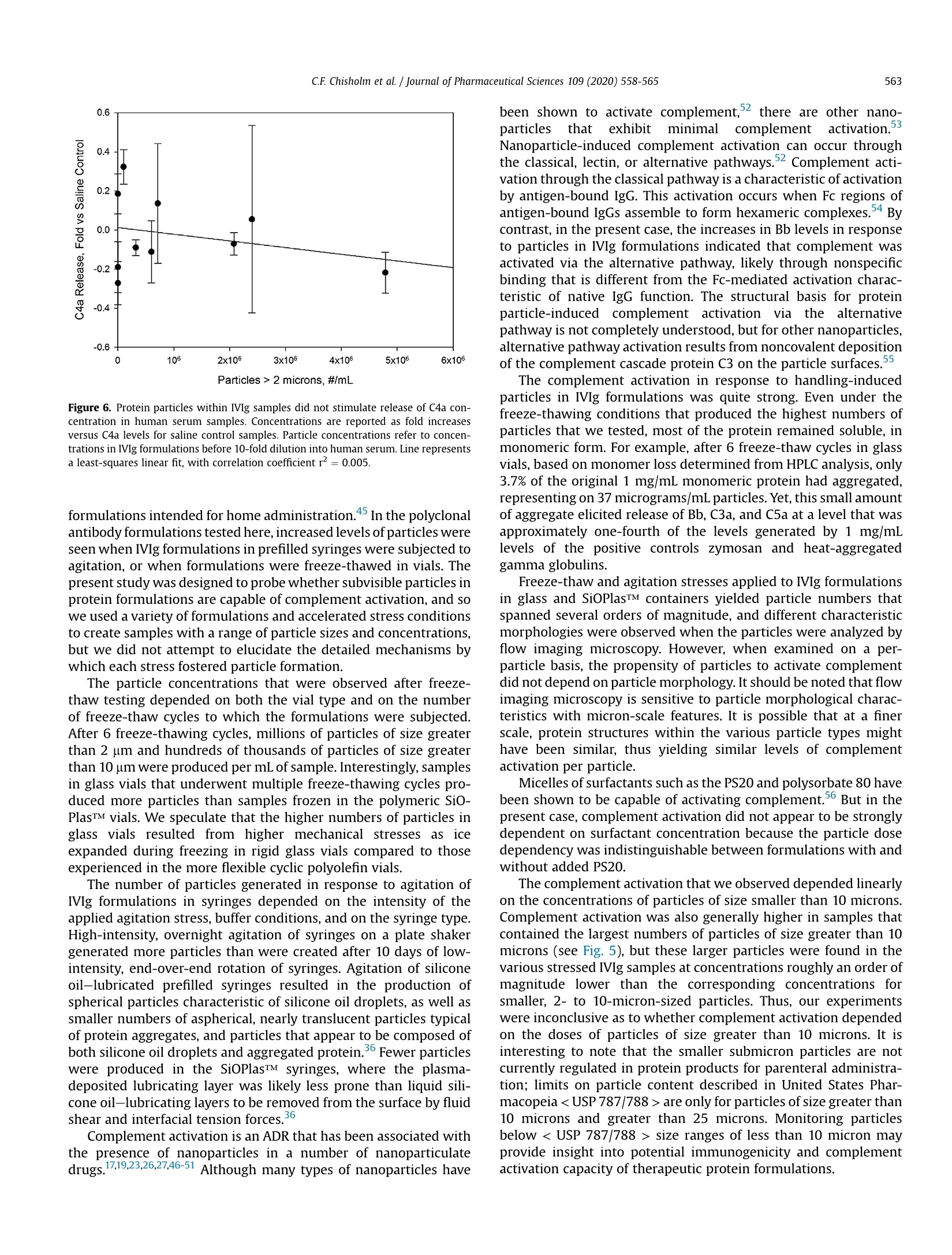
Journal of Pharmaceutical Sciences 109 (2020)558-565 C.F. Chisholm et al./ Journal of Pharmaceutical Sciences 109 (2020) 558-565559Accelerated Stress Testing of IVIg Formulations Using Agitation https://doi.org/10.1016/j.xphs.2019.10.0410022-3549/O 2020 American Pharmacists Association. Published by Elsevier Inc. All rights reserved. Contents lists available at ScienceDirect Iournal of Pharmaceutical Sciences journal homepage: www.jpharmsci.org Pharmaceutical Biotechnology Subvisible Particles in IVIg Formulations Activate Complement inHuman Serum Check for updates Carly F. Chisholm , William Behnke , Yekaterina Pokhilchuk 1,Ashley A. Frazer-Abel, Theodore W. Randolph1,* ' Department Chemical and Biological Engineering, Center for Pharmaceutical Biotechnology, University of Colorado, Boulder, Colorado 80309Exsera Biolabs, Anschutz Medical Campus, Aurora, Colorado 80045 ARTICLEINFO ABSTRACT Article history:Received 9 August 2019Revised 21 October 2019Accepted 22 October 2019Available online 28 October 2019 Keywords:IgG antibody(s)protein aggregationstabilityparticle sizenanoparticle(s)immune response(s) When administered intravenously, various particles and nanomedicines activate complement, poten-tially leading to infusion reactions and other adverse drug reactions. Particles form within formulationsof therapeutic proteins due to stresses incurred during shipping, handling, and administration to pa-tients. In this study, IVIg solutions were stored in multiple types of vials and prefilled syringes andexposed to agitation and freeze-thaw stresses to generate particles.The stressed samples were added tohuman serum to determine whether these particles activated complement. Subvisible IVIg particlesranging in size between 2 and 10 microns activated complement in a fashion that was linearly dependenton the number of particles dosed, whereas little correlation was found between doses of larger particles(>10 microns) and complement activation. Activation of complement by subvisible particles of IVIgfollowed the alternative pathway, as shown by the release of complement cascade factor Bb and theproduction of the anaphylatoxins C3a and C5a without generation of C4a. The number and themorphology of subvisible particles formed depended on the applied stress, formulation, and on thecontainer material. But the capacity of the 2- to 10-micron-sized particles to activate complement inhuman serum appeared to depend only on particle concentration. Introduction Therapeutic proteins offer effective treatments for a wide rangeof human diseases. However, in a fraction of patients, the safety andefficacy of these medicines are compromised by adverse drug re-actions (ADRs) that include unwanted+ bi 1m-m1u3n ee responses, infusionreactions, anaphylaxis, and even death.-13 Some of these ADRs arequite common.For example, hypersensitivity reactions after thefirst exposure to rituximab are reported in about half of patientswith B-cell malignancies,14 14.5% of patients were reported toexperience severe infusion reactions in response to the firstadministration of cetuximab,15 and 12.3% of patients receiving in-fusions of the anti-TNF-a antibody infliximab experienced infusionreactions. 6 Anaphylaxis, hypersensitivity, and increased (unde-sired) antibody responses all can result from complement activa-tion. Complement is normally activated as a biological defensemechanism against viral and microbial challenges,S, but ( * Correspondence to: Theodore W. Randolph ( T elephone: (303) 492-4776).E-mail address: t h eodore.r an dolph@color a do.edu ( T.W. R andolph). ) nanoparticles in intravenously administered drug formulations canalso activate the complement cascade.17-23 Once the complementcascade is activated, it can have strong proinflammatory andanaphylactoid outcomes.22 The strength of these responses isdriven largely by the extents to which C3a and C5a, the 2 ana-phylatoxins of the complement system, are produced. These 2signaling molecules have the capacity to cause blood vessel dila-tion, modulate cytokine production, cause histamine release andoxidative burst, and act as signals for chemotaxis of innate immunecells.19,24,25 Although complement activation-driven actions can be veryhelpful for fighting localized microbial infections, they can bedetrimental if they occur systemically, as happens in complementactivation-related pseudoallergy (CARPA). CARPA is an infusionreaction that has been associated with intravenous administrationof various liposomal26.27 and micellar formulations of drugs, as wellas to administration of nanoparticles and therapeutic anti-bodies.22,28-30C ACRAPRAP Acan provoke severe cardiopulmonary re-sponses including anaphylactic cardiogenic shock and death.22,28 Therapeutic proteins readily aggregate to form soluble oligo-mers and subvisible particles during production, shipping, storage, and administration to patients. These aggregates are a risk factorfor unwanted immune responses, which can lead to loss of thera-peutic efficacy and other adverse reactions.13,32-34 Protein aggre-gation is often initiated by interactions with interfaces to whichproteins are exposed, such as air-water, container-water, and ice-water interfaces. These interfacial stresses can be magnified byhandling. For example, aggregation of proteins at the container-water interfaces found in silicone oil-lubricated syringes can begreatly accelerated by agitation35,36 and levels of particles withintherapeutic protein formulations may be dramatically increased byfreeze-thawing.37.38 We hypothesized that, like some nanoparticulate drugs, theparticles generated by handling of protein formulations may becapable of activating complement. To test this hypothesis, we firstconducted accelerated stability experiments to generate particleswithin a polyclonal antibody formulation (intravenous immuno-globulin, IVIg) by subjecting the antibody formulation to freeze-thaw stresses in multiple types of vials or to stresses caused byagitation in multiple types of prefilled syringes. We then testedwhether these handling-induced particles were capable of acti-vating complement when added to human serum samples. Com-plement activation Was measured usingenzyme-linkedimmunosorbent assays (ELISA) to quantify the levels of C3a andC5a, 2 key signaling molecules for the proinflammatory propertiesof complement. In addition, we measured levels of C4a and Bb toprobe which complement pathways might be activated. Productionof C4a is a marker of complement activation by the classical orlectin pathways, whereas production of Bb is indicative of com-plement activation via the alternative pathway. Materials and Methods Materials used were of USP grade or higher. Intravenousimmunoglobulin (IVIg; GAMMAGARD LIQUID@, Shire US Inc., Lex-ington, MA) was purchased from Wardenburg Pharmacy at theUniversity of Colorado at Boulder. Chemicals purchased from SigmaAldrich (St. Louis, MO) included sodium phosphate monobasic,sodium phosphate dibasic, and glycine. Chemicals purchased fromFisher Scientific (Waltham,MA) included polysorbate 20 (PS20,Tween 20TM, N.F., Multi-Compendial, J.T. Baker), 10X phosphate-buffered saline (PBS), and HyCloneTM water for injection. Silicon-ized glass syringes were BD Hypak SCF 1 mL long 27G1/2 (BDMedical-Pharmaceutical Systems, Franklin Lakes, NJ). SiOPlasTMsyringes (1 mL) (SiO2 Medical Products, Inc., Auburn, AL) werecomposed of cyclic olefin polymer (COP) syringe barrels whoseinterior surfaces were coated with a silica-based barrier coatingsystem and an organosiloxane lubricant, both applied by plasma-enhanced chemical vapor deposition.3 SiOPlasTM vials (6 mL)were composed of COP that was coated on the vial interior with asilica-based barrier coating system. 6 mL Ompi EZ-fill@ borosilicateglass vials (Schott, AG) were provided by SiO2 Medical Products, Inc.(Auburn, AL). Protein Formulation Before use, IVIg was formulated at a protein concentration of 1mg/mL in either PBS at pH 7.4, or in 250 mM glycine pH 4.25 with0.02%(v/v) PS20. To remove any pre-existing particulates, GAM-MAGARD LIQUIDQ samples containing 100 mg/mL immunoglob-ulin G in 250 mM glycine were centrifuged at 20,000× g for 20 minat 4°C and the supernatant was used as a stock solution. The IVIgstock solution was diluted 1:100 into either 0.22-micron-filteredPBS pH 7.4 or 250 mM glycine pH 4.25 with 0.02% (v/v) PS20 toyield a final concentration of 1 mg/mL IVIg. Siliconized glass and SiOPlasTM syringes were filled with 1 mg/mL IVIg in 250 mM glycine pH 4.25 with 0.02%(v/v) PS20. Syringeswere filled so that a uniform headspace gap of 4 mm was presentbetween the liquid and the stopper (Datwyler Pharma Packaging,Pennsauken,NJ). These syringes were rotated end over end at roomtemperature for 10 days, causing the air bubble in the headspacegap to move from one end of syringe to other end with each rota-tion. In a few of the syringes that were tested, the air bubblebecame lodged at one end of the syringe and failed to move as thesyringe was rotated. These syringes were excluded from furtheranalysis. After the 10 days of end-over-end rotation, each formu-lation was expelled from the syringe through the needle using anautomated syringe pump at 150 mm/min and sample was collectedin prerinsed polypropylene tubes and tested for subvisible particleconcentrations and for complement activation capacity. Another set of siliconized glass and SiOPlasTM syringes werefilled with 1 mg/mL IVIg in PBS. These syringes were placed hori-zontally on an orbital shaker and shaken overnight at room tem-perature. After shaking, the formulations were expelled from thesyringes through the needle using an automated syringe pump at150 mm/min, collected in prerinsed polypropylene tubes, andtested for subvisible particle concentrations and complementactivation capacity. Accelerated Stress Testing of IVIg Formulations Using Freeze- Thawing Borosilicate vials (6 mL) and SiOPlasTM vials (6 mL) were filledwith 4 mL of a formulation containing 1 mg/mL IVIg in PBS pH 7.4.The contents of the vials were subjected to 1 or 6 freeze-thaw cy-cles. In each freeze-thaw cycle, the vials were first dipped in liquidnitrogen for 2 min and then thawed in a water bath at 30°C for14.5 min. Each vial was swirled gently for mixing before nextfreeze-thaw cycle. Analysis of Subvisible Particle Concentrations Particle concentrations and images of particles in the variousstressed formulations were obtained using flow imaging micro-scopy (FlowCAMQ, Fluid Imaging Technologies,Scarborough, ME),as previously described. Samples that had been subjected to 6freeze-thaw cycles contained concentrations of particles thatapproached or exceeded the upper limit for the flow imaging mi-croscopy instrument, and so these formulations were diluted 100-fold with PBS, pH 7.4, before analysis. Particle counts in the stressedformulations were measured in aliquots that were sampled fromthe same individual syringes and vials that were tested for com-plement activation. Each sample contained a distribution of particlesizes; no effort was made to fractionate the particles before testingthe stressed formulation for their ability to activate complement. Analysis of Soluble Protein Fractions by Size-ExclusionChromatography Size exclusion chromatography was used to monitor the reten-tion of monomeric protein and the appearance of any soluble ag-gregates in IVIg samples after application agitation or freeze-thawstresses. A TSKgel G3000SWxL column (TOSOH Biosciences,Montgomeryville PA) was used with an Agilent 1100 series system(Santa Clara, CA). The eluent was monitored at an absorbance of280 nm in the Agilent ChemStation software. The mobile phase of100 mM sodium sulfate, 100 mM sodium phosphate, and 0.05% (w/v) sodium azide at pH 6.7 was passed through the system at 0.6 mL/ Table 1. Concentrations of Subvisible Particles in Stressed Samples Submitted for Complement Activation Testing Applied Stress Container Type/Buffer Condition Particles of Size >2 Particles of Size >10 Micron, #/mL Micron, #/mL Unstressed control Polypropylene/250 mM glycine pH 4.25, 0.02%(v/v) polysorbate 20 300 10 Single freeze-thaw cycle SiOPlasTM Vial/250 mM glycine pH 4.25,0.02%(v/v) polysorbate 20 7.1×10 5.1×104 Single freeze-thaw cycle Borosilicate Glass Vial/250 mM glycine pH 4.25, 0.02%(v/v) polysorbate 20 3.2×10 8.0×10+ Six freeze-thaw cycles SiOPlasTM Vial/250 mM glycine pH 4.25, 0.02%(v/v) polysorbate 20 2.1×10° 3.6×10 Six freeze-thaw cycles Borosilicate Glass Vial/250 mM glycine pH 4.25,0.02%(v/v) polysorbate 20 4.8×106 2.3×10 Orbital shaker, overnight SiOPlasTM Syringe/PBS pH 7.4 2.4×10° 1.7×10 Orbital shaker, overnight Siliconized Glass Syringe/PBS pH 7.4 6.0×10 1.1×10 End-over-end rotation, 10 d SiOPlasTM Syringe/250 mM glycine pH 4.25, 0.02%(v/v) polysorbate 20 3×103 90 End-over-end rotation, 10 d Siliconized Glass Syringe/250mM glycine pH 4.25, 0.02%(v/v) polysorbate 20 1.0×10' 2×10 Samples were pooled from 3 samples from each stress/container combination. Container-to-container variation in particle concentrations in samples before pooling was <15% min. Peak areas in chromatograms were quantified in GRAMS/AIsoftware version 9.1 (Thermo Fisher Scientific Inc., Waltham, MA).After application of accelerated stress conditions, the fractional lossof soluble protein from the (essentially) particle-free, centrifuged,and filtered starting formulations was quantified using the ratio ofthe peak areas for stressed versus unstressed formulations. Complement Activation in Human Serum Samples Samples pooled from 3 vials or syringes of the various stressedIVIg formulations as well as unstressed control samples of eachformulation were delivered to Exsera BioLabs (Denver, CO) foranalysis of their capacity to activate complement. Complementactivation was measured in normal human serum pooled from 3individual donors who had been previously screened for normalcomplement function. Test samples of stressed IVIg formulationswere diluted 10-fold into pooled human serum, mixed, and allowedto incubate at 37°C for 30 min. After incubation, samples werestored at-80°C until further testing was conducted. For analysis ofcomplement activation, concentrations of four complementcascade activation fragments,C3a, Bb, C4a,and C5a, were measuredby ELISA using kits purchased from Quidel Corporation (San Diego,CA). C4a was chosen because it is a marker of activation of theclassical or lectin pathway, Bb as a distinct marker of the alternativepathway for complement activation, C3a as the central point ofcomplement activation, and C5a as a marker of terminal comple-ment pathway activation.4 Triplicate ELISA measurements wereconducted for each of the four complement cascade proteins. Inaddition to testing of stressed IVIg samples, several controls wereanalyzed. These control samples contained pooled serum alone orsamples of serum to which saline solution PBS with zymosan or PBSwith heat-aggregated gamma globulins were added in a 1:9 ratio.The average of the measurements was plotted versus particlesample counts as a fold increase compared to concentrationsmeasured in the saline control. Results Particle Formation During Accelerated Stress Testing ofIVIgFormulations As expected, each of the accelerated stress testing methodsgenerated microparticles within the tested formulations (Table 1).Freeze-thawing of IVIg in PBS pH 7.4 resulted in the highest numberof subvisible particles. A single freeze-thaw cycle produced 3.2 ×10 and 7.1 ×105 particles of size greater than 2 micron in boro-silicate and SiOPlasTM vials, respectively. Particles larger than 10microns were also produced, with 8.0×104 particles detected inborosilicate vials and 5.1 x104 particles found in SiOPlasTM vials. Application of multiple freeze-thaw cycles further increasedparticle numbers by about an order of magnitude (Table 1). After 6cycles, particle concentrations in borosilicate vials and SiOPlasTMvials had increased to 4.8 ×10 and 2.1×106 particles/mL of sizegreater than 2 microns, respectively. Particles larger than 10 micronwere also generated, with 2.3×10 and 3.6×10 in borosilicateand SiOPlasTM vials,respectively. The number of particles produced as a result of agitationstresses depended on the container type and also on the type ofagitation that was applied.41 Overnight agitation of IIg formula-tions in PBS pH 7.4 using an orbital shaker generated 6.0 ×10particles of size greater than 2 microns in siliconized glass syringes,and 2.4× 10 particles/mL in corresponding SiOPlasTM syringes(Table 1). As was the case with the freeze-thaw studies, roughly anorder of magnitude fewer large particles (>10 micron) were formedin syringes of both types. End-over-end rotation for 10 days was the gentlest acceleratedstability test that was applied.After IVIg formulations in glycine pH4.25 underwent end-over-end rotation for 10 days, 1.0×10 and3 × 10°particles/mL in the particle size range greater than 2 mi-crons were detected in siliconized glass syringes and SiOPlasTMsyringes, respectively. Correspondingly few particles larger than 10micron were also generated, with 2.0 ×10’ and 90 particles/mLdetected in siliconized glass and SiOPlasTM syringes, respectively. Collections of typical flow imaging microscopy images of par-ticles produced by the various accelerated stress methods arepresented in Figure 1. Samples stressed by freeze-thawing in vialspredominately contained aspherical microparticles characteristic ofproteinaceous aggregates, whereas samples stressed by agitation insiliconized glass syringes contained numerous spherical particlescharacteristic of droplets of lubricants (Fig. 1a,c,e and f). Agitation inthe SiOPlasTM syringes produced particles of various morphologiesthat may have comprised both irregularly shaped protein aggre-gates and spherical droplets of silicone oil (Fig. 1b and d). For all of the accelerated stress conditions tested, size-exclusionchromatographic analysis showed that only minimal (<5%)amounts of insoluble protein were formed. No soluble aggregateswere detected under any condition. Application of 6 freeze-thawcycles to IVIg in borosilicate glass vials produced the highestnumbers of particles per mL-nearly 5 million particles greaterthan 2 microns-but this represented a loss of only 3.7% of theoriginal monomeric protein. Complement Activation in Human Serum in Response to ParticlesProduced Under Accelerated Stress Conditions Particles of size greater than 2 microns within IVIg formulationsthat had been subjected to accelerated stress conditions activatedcomplement in a linear, dose-dependent fashion when the IVIgformulations were diluted 10-fold into human serum (Figs. 2-4). e C o ovennght shakingaannglass syringes on an orbital shaker; (b) overnight shaking in SioPlasw syringes on an orbital shaker: (c) 10 days of end-over-end rotationagm vias.overnight shakaneinges: (d) 10 days of end-over-end rotation in SiOPlasa syringes; (e) freeze-thawing 6 times in glass vials; (f) freeze-thawing 6 times in SiOPlasu vials. tgure 1. Flow imaging microscopy of stressed Mg samples. colages are randomly selected set of images of partidles present in lVg sanmle.tar ered e rotatoe slicone Figure 2. Effect of sample particle levels on Bb concentration in human serum sam-ples. Concentrations are reported as fold increases versus Bb levels for saline controlsamples. Particle concentrations refer to concentrations in IVIg formulations before 10-fold dilution into human serum. Line represents a least-squares linear fit, with cor-relation coefficient r²=0.94. Figure 4. Effect of sample particle levels on C5a concentration in human serumsamples. Concentrations are reported as fold increases versus C5a levels for salinecontrol samples. Particle concentrations refer to concentrations in IVIg formulationsbefore 10-fold dilution into human serum. Line represents a least-squares linear fit,with correlation coefficientr²=0.99. Levels of complement activation were generally higher in sampleswith larger numbers of particles of size greater than 10 microns, butthese larger particles were present at low levels that did not allowany clear dose-dependence to be resolved (see Fig.5). and C5a concentrations that were 2.4- and 8.9-fold higher thanthose observed in saline controls. The observed complement activation was consistent with acti-vation through the alternative pathway. No increase over salinecontrol levels was seen for C4a, which is a marker for the classicalor lectin pathways (Fig. 6). By contrast, Bb, a marker of the alter-native pathway for complement activation, increased linearly (r-=0.94) as the concentrations of particles in the size range greaterthan 2 microns increased (Fig.2). For comparison, positive controls containing 1 mg/mL zymosanor 1 mg/L heat-aggregated gamma globulins stimulated ca. 11-foldincreases (vs. saline) of C3a, Bb, and C4a, and ca 32-fold increases(vs. saline) in C5a levels. Discussion Increases in concentrations of the anaphylatoxins C3a and C5awere also observed when particles were diluted into serum (Fig. 3and 4). As with the Bb response, the fold increases versus salinecontrol were linearly dependent on particle dose, with correlationcoefficients r²of 0.85 and 0.99 for C3a and C5a, respectively. Re-sponses to the IVIg formulations that were subjected to 6 freeze-thaw cycles in borosilicate glass vials produced increases in C3a Subvisible particles are ubiquitous in formulations of therapeuticproteins, but their levels can be greatly increased by stressesencountered during shipping, handling, and storage.42-44 Agitationstresses are incurred as a result of vibration during shipping andduring routine handling of liquids. Although liquid formulations oftherapeutic proteins are typically intended to remain in liquid format controlled temperature, accidental freezing can occur, especiallyduring shipping in containers cooled with gel packs,or in Particles > 10 microns,#/mL Particles > 2 micron,#/mL Figure 3. Effect of sample particle levels on C3a concentration in human serumsamples. Concentrations are reported as fold increases versus C3a levels for salinecontrol samples. Particle concentrations refer to concentrations in IVIg formulationsbefore 10-fold dilution into human serum. Line represents a least-squares linear fit,with correlation coefficient r²=0.85. Figure 5. Effect of concentrations of particles of sizes larger than 10 microns on C5a(filled circles) and C3a (open circles) concentrations in human serum samples. Con-centrations are reported as fold increases versus C5a and C3a levels for saline controlsamples. Particle concentrations refer to concentrations in IVIg formulations before 10-fold dilution into human serum. Line represents a least-squares linear fit, with cor-relation coefficients r²=0.60 and r²=0.47 for C5a and C3a, respectively. Figure 6. Protein particles within IVIg samples did not stimulate release of C4a con-centration in human serum samples. Concentrations are reported as fold increasesversus C4a levels for saline control samples. Particle concentrations refer to concen-trations in IVIg formulations before 10-fold dilution into human serum. Line representsa least-squares linear fit, with correlation coefficient r²=0.005. formulations intended for home administration.45 In the polyclonalantibody formulations tested here, increased levels of particles wereseen when IVIg formulations in prefilled syringes were subjected toagitation, or when formulations were freeze-thawed in vials. Thepresent study was designed to probe whether subvisible particles inprotein formulations are capable of complement activation, and sowe used a variety of formulations and accelerated stress conditionsto create samples with a range of particle sizes and concentrations,but we did not attempt to elucidate the detailed mechanisms bywhich each stress fostered particle formation. The particle concentrations that were observed after freeze-thaw testing depended on both the vial type and on the numberof freeze-thaw cycles to which the formulations were subjected.After 6 freeze-thawing cycles, millions of particles of size greaterthan 2 um and hundreds of thousands of particles of size greaterthan 10 um were produced per mL of sample. Interestingly,samplesin glass vials that underwent multiple freeze-thawing cycles pro-duced more particles than samples frozen in the polymeric SiO-PlasTM vials. We speculate that the higher numbers of particles inglass vials resulted from higher mechanical stresses as iceexpanded during freezing in rigid glass vials compared to thoseexperienced in the more flexible cyclic polyolefin vials. The number of particles generated in response to agitation ofIVIg formulations in syringes depended on the intensity of theapplied agitation stress, buffer conditions, and on the syringe type.High-intensity, overnight agitation of syringes on a plate shakergenerated more particles than were created after 10 days of low-intensity, end-over-end rotation of syringes. Agitation of siliconeoil-lubricated prefilled syringes resulted in the production ofspherical particles characteristic of silicone oil droplets, as well assmaller numbers of aspherical, nearly translucent particles typicalof protein aggregates, and particles that appear to be composed ofboth silicone oil droplets and aggregated protein.Fewer particleswere produced in the SiOPlasTM syringes, where the plasma-deposited lubricating layer was likely less prone than liquid sili-cone oil-lubricating layers to be removed from the surface by fluidshear and interfacial tension forces. Complement activation is an ADR that has been associated withthe presence of nanoparticles in a number of nanoparticulatedriugs17,19,23,26,27,46-51 AAlltthehough many types of nanoparticles have been shown to activate complement,2 there are other nano-particlestthatt6exhibititminimal complement activation.3Nanoparticle-induced complement activation can occur throughthe classical, lectin, or alternative pathways.Complement acti-vation through the classical pathway is a characteristic of activationby antigen-bound IgG. This activation occurs when Fc regions ofantigen-bound IgGs assemble to form hexameric complexes.4 Bycontrast, in the present case, the increases in Bb levels in responseto particles in IVIg formulations indicated that complement wasactivated via the alternative pathway, likely through nonspecificbinding that is different from the Fc-mediated activation charac-teristic of native IgG function. The structural basis for proteinparticle-induced complement activation)nviathe alternativepathway is not completely understood, but for other nanoparticles,alternative pathway activation results from noncovalent depositionof the complement cascade protein C3 on the particle surfaces. The complement activation in response to handling-inducedparticles in IVIg formulations was quite strong. Even under thefreeze-thawing conditions that produced the highest numbers ofparticles that we tested, most of the protein remained soluble, inmonomeric form. For example, after 6 freeze-thaw cycles in glassvials,based on monomer loss determined from HPLC analysis, only3.7% of the original 1 mg/mL monomeric protein had aggregated,representing on 37 micrograms/mL particles. Yet, this small amountof aggregate elicited release of Bb, C3a, and C5a at a level that wasapproximately one-fourth of the levels generated by 1 mg/mLlevels of the positive controls zymosan and heat-aggregatedgamma globulins. Freeze-thaw and agitation stresses applied to IVIg formulationsin glass and SiOPlasTM containers yielded particle numbers thatspanned several orders of magnitude, and different characteristicmorphologies were observed when the particles were analyzed byflow imaging microscopy. However, when examined on a per-particle basis, the propensity of particles to activate complementdid not depend on particle morphology. It should be noted that flowimaging microscopy is sensitive to particle morphological charac-teristics with micron-scale features. It is possible that at a finerscale, protein structures within the various particle types mighthave been similar, thus yielding similar levels of complementactivation per particle. Micelles of surfactants such as the PS20 and polysorbate 80 havebeen shown to be capable of activating complement.56 But in thepresent case, complement activation did not appear to be stronglydependent on surfactant concentration because the particle dosedependency was indistinguishable between formulations with andwithout added PS20. The complement activation that we observed depended linearlyon the concentrations of particles of size smaller than 10 microns.Complement activation was also generally higher in samples thatcontained the largest numbers of particles of size greater than 10microns (see Fig. 5), but these larger particles were found in thevarious stressed IVIg samples at concentrations roughly an order ofmagnitude lower than the corresponding concentrations forsmaller, 2- to 10-micron-sized particles. Thus, our experimentswere inconclusive as to whether complement activation dependedon the doses of particles of size greater than 10 microns. It isinteresting to note that the smaller submicron particles are notcurrently regulated in protein products for parenteral administra-tion; limits on particle content described in United States Phar-macopeia < USP 787/788> are only for particles of size greater than10 microns and greater than 25 microns. Monitoring particlesbelow < USP 787/788> size ranges of less than 10 micron mayprovide insight into potential immunogenicity and complementactivation capacity of therapeutic protein formulations. Caution should be used in extending these results to the pre-diction of complement activation-related ADRs in clinical settings.Thresholds and extents of complement activation may vary frompatient to patient because of genetic and acquired factors,21,57-59making direct prediction of in vivo responses to particles prob-lematic.60 Furthermore, the use of cell-free serum in our in vitro testmay underestimate the capacity for particles to activate comple-ment; in whole blood, the presence of B cells increases sensitivity tocomplement activation by rituximab in patients. Finally, com-plement activation by nanoparticles can be highly material-specific, and particles formed from other protein drugs, forexample, monoclonal antibodies, may not behave in a fashionsimilar to particles found in the polyclonal mixture of antibodiespresent in IVIg. Conclusions We observed that as little as a single unintended freeze-thawingof an IVIg formulation creates a sufficient number of particles toactivate complement in an in vitro serum-based assay. Complementactivation was directly proportional to particle concentration for 2-to 10-micron-sized particles in stressed samples, whereas no clearcorrelation was observed for >10-micron particles in the samesamples. The level of activated complement was not dependent onthe specific particle morphologies generated by multiple types ofstresses, or on the presence of nonionic surfactants in the formu-lations. Although these results may not be easily extrapolated toquantitatively predict risk of ADRs in patients infused with othermonoclonal antibody drug products, it is clearly prudent to takesteps in handling, storage, and administration of therapeutic pro-tein products to minimize generation of particles in infused ther-apeutic protein products, and it may be important to monitorparticles below 10 microns in size for insight into immunogenicity. Acknowledgments Funding for this work was provided by the NIH under RO1EB006006, and by SiO2 Medical Products, Inc. References 1. Bertolotto A, Malucchi S, Sala A. et al.Differential effects of three interferonbetas on neutralising antibodies in patients with multiple sclerosis: a follow upstudy in an independent laboratory. j Neurol Neurosurg Psychiatry.2002;73(2):148-153. 2.1Deisenhammer F, Schellekens H, Bertolotto A. Measurement of neutralizingantibodies to interferon beta in patients with multiple sclerosis. JNeurol.2004:251:31-39. 3.Goodin DS, Frohman EM, Hurwitz B, et al. Neutralizing antibodies to interferonbeta: assessment of their clinical and radiographic impact: an evidence report:report of the therapeutics and technology assessment subcommittee of theAmerican Academy of Neurology. Neurology. 2007;68(13):977-984. 4.Hartung HP, Schellekens H, Munschauer FE. Neutralizing antibodies to inter-feron beta in patients with multiple sclerosis: scientific background and clinicalimplications-introduction. J Neurol. 2004;251:1-3. 5.Hartung HP, Munschauer F, Schellekens H. Significance of neutralizing anti-bodies to interferon beta during treatment of multiple sclerosis: expert opin-ions based on the Proceedings of an International Consensus Conference. Eur JNeurol. 2005;12(8):588-601. 6.1Hemmer B, Stuve O, Kieseier B, Schellekens H, Hartung HP. Immune responseto immunotherapy: the role of neutralising antibodies to interferon beta in thetreatment of multiple sclerosis. Lancet Neurol. 2005;4(7):403-412. 7.M1alucchi S, Sala A, Gilli F, et al. Neutralizing antibodies reduce the efficacy ofbeta IFN during treatment of multiple sclerosis. Neurology.2004;62(11):2031-2037. 8.Yanai H, Hanauer SB. Assessing response and loss of response to biologicaltherapies in IBD. AmJ Gastroenterol. 2011;106(4):685-698. 9.N1anda KS, Cheifetz AS, Moss AC. Impact of antibodies to infliximab on clinicaloutcomes and serum infliximab levels in patients with inflammatory boweldisease (IBD): a meta-analysis. Am J Gastroenterol. 2013;108(1):40-47. quiz 48. 10. Ordas I, Feagan BG, Sandborn WJ. Therapeutic drug monitoring of tumor ne-crosis factor antagonists in inflammatory bowel disease. Clin GastroenterolHepatol. 2012;10(10):1079-1087.quiz e85-e86. 11. O'Meara S, Nanda KS, Moss AC. Antibodies to infliximab and risk of infusionreactions in patients with inflammatory bowel disease: a systematic reviewand meta-analysis. Inflamm Bowel Dis. 2014;20(1):1-6. 12.Vennegoor A, Rispens T, Strijbis EM, et al. Clinical relevance of serum natali-zumab concentration and anti-natalizumab antibodies in multiple sclerosis.Mult Scler.2013;19(5):593-600. 13. Kotarek J, Stuart C, De Paoli SH, et al. Subvisible particle content, formulation,and dose of an erythropoietin peptide mimetic product are associated withsevere adverse postmarketing events. J Pharm Sci. 2016;105(3):1023-1027. 14. Picard M, Galvao VR. Current knowledge and management of hypersensitivityreactions to monoclonal antibodies. J Allergy Clin Immunol Pract. 2017;5(3):600-609. 15. Atwal D, Safar AM, Govindarajan R, Makhoul I. Severe first infusion reactionrelated to cetuximab in cancer patients in Arkansas. J Oncol Pharm Pract.2019;25(5):1130-1134. 16C.hoquette D, Faraawi R, Chow A, Rodrigues J, Bensen WJ, Nantel F. Incidenceand management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42(7):1105-1111. 17.Szebeni J, Alving CR, Savay S, et al. Formation of complement-activating par-ticles in aqueous solutions of Taxol: possible role in hypersensitivity reactions.Int Immunopharmacol. 2001;1(4):721-735. 18. Szebeni J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology. 2005;216(2-3):106-121. 19.Andersen AJ, Hashemi SH, Andresen TL, Hunter AC, Moghimi SM. Complement:alive and kicking nanomedicines. J Biomed Nanotechnol. 2009;5(4):364-372. 20. Moghimi SM, Andersen AJ, Ahmadvand D, Wibroe PP, Andresen TL, Hunter AC.Material properties in complement activation. Adv Drug Deliv Rev.2011;63(12):1000-1007. 21. Moghimi SM, Farhangrazi ZS.Nanomedicine and the complement paradigm.Nanomedicine. 2013;9(4):458-460. 22. Szebeni J. Complement activation-related pseudoallergy: a stress reaction inblood triggered by nanomedicines and biologicals. Mol Immunol. 2014;61(2):163-173. 23.Boraschi D, Italiani P, Palomba R, et al. Nanoparticles and innate immunity:new perspectives on host defence. Semin Immunol.2017;34:33-51. 24.1Sacks SH. Complement fragments C3a and C5a: the salt and pepper of theimmune response. Eur J Immunol. 2010;40(3):668-670. 25. Klos A,Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the ana-phylatoxins in health and disease. Mol Immunol. 2009;46(14):2753-2766. 26S.zebeni J, Baranyi L, Savay S, et al. Role of complement activation in hyper-sensitivity reactions to doxil and hynic PEG liposomes: experimental andclinical studies. J Liposome Res. 2002;12(1-2):165-172. 27. Chanan-Khan A, Szebeni J, Savay S, et al. Complement activation following firstexposure to pegylated liposomal doxorubicin (Doxil): possible role in hyper-sensitivity reactions. Ann Oncol. 2003;14(9):1430-1437. 28.Meszaros T, Csincsi AI, Uzonyi B, et al. Factor H inhibits complement activationinduced by liposomal and micellar drugs and the therapeutic antibody ritux-imab in vitro.Nanomedicine. 2016;12(4):1023-1031. 29.Fulop T, Meszaros T, Kozma GT, Szebeni J. Jozsi M. Infusion reactions associatedwith the medical application of monoclonal antibodies: the role of comple-ment activation and possibility of inhibition by factor H. Antibodies (Basel).2018;7(1):14. 30.van der Kolk LE, Grillo-Lopez AJ, Baars jW,Hack CE, van Oers MH. Complementactivation plays a key role in the side-effects of rituximab treatment. Br JHaematol. 2001;115(4):807-811. 31. Randolph TW, Carpenter JF. Engineering challenges of protein formulations.AIChE J. 2007;53(8):1902-1907. 32. Rosenberg AS, Worobec A. A risk-based approach to immunogenicity concernsof therapeutic protein products, part 1: Considering consequences of the im-mune response to a protein. BioPharm International. 2004;17:22-26.500 33.Rosenberg AS. Immunogenicity of biological therapeutics: a hierarchy of con-cerns. Dev Biol (Basel). 2003;112:15-21. 34.Rosenberg AS. Effects of protein aggregates: an immunologic perspective.AAPSJ. 2006;8(3):E501-E507. 35. Thirumangalathu R. Krishnan S, Ricci MS, Brems DN, Randolph TW,Carpenter JF. Silicone oil- and agitation-induced aggregation of a monoclonalantibody in aqueous solution. J Pharm Sci. 2009;98(9):3167-3181. 36.Gerhardt A, McGraw NR, Schwartz DK, Bee JS, Carpenter JF, Randolph TW.Protein aggregation and particle formation in prefilled glass syringes. J PharmSci. 2014;103(6):1601-1612. 37. Pardeshi NN, Zhou C, Randolph TW, Carpenter JF. Protein nanoparticles pro-mote microparticle formation in intravenous immunoglobulin solutions duringfreeze-thawing and agitation stresses. J Pharm Sci. 2018;107:1852-1857. 38.Liu L, Ammar DA, Ross LA, Mandava N, Kahook MY, Carpenter JF. Silicone oilmicrodroplets and protein aggregates in repackaged bevacizumab and rani-bizumab: effects of long-term storage and product mishandling. Invest Oph-thalmol Vis Sci. 2011;52(2):1023-1034. 39. WeikartCM, Pantano CG,Shallenberger JR. Performance stability of silicone oxide-coated plastic parenteral vials. PDA J Pharm Sci Technol. 2017;71(4):317-327. 40.Janeway CA, Travers P, Walport M, Schlomchik M. Immunobiology: The ImmuneSystem in Health. 6 ed. New York, NY: Garland Science Publishing; 2005:37-101 41. Kiese S, Papppenberger A, Friess W, Mahler HC. Shaken, not stirred: mechanicalstress testing of an IgG1 antibody.J Pharm Sci. 2008;97(10):4347-4366. 42.Nejadnik MR, Randolph TW, Volkin DB, et al. Postproduction handling andadministration of protein pharmaceuticals and potential instability issues.I Pharm Sci.2018;107:2013-2019. 43. Jiskoot W, Nejadnik MR, Sediq AS. Potential issues with the handling of bi-ologicals in a hospital. J Pharm Sci. 2017;106(6):1688-1689. 44.Vlieland ND, Nejadnik MR, Gardarsdottir H, et al. The impact of inadequatetemperature storage conditions on aggregate and particle formation in drugscontaining tumor necrosis factor-alpha inhibitors. Pharm Res.2018;35(2):42. 45.Vlieland ND, Gardarsdottir H, Bouvy ML, Egberts TCG, van den Bemt BJF. Themajority of patients do not store their biologic disease-modifying antirheu-matic drugs within the recommended temperature range. Rheumatology.2016;55(4):704-709. 46...Savay S, Szebeni J, Baranyi L, Alving CR. Potentiation of liposome-inducedcomplement activation by surface-bound albumin. Biochim Biophys Acta.2002;1559(1):79-86. 47. Szebeni J. Complement activation-related pseudoallergy caused by amphiphilicdrug carriers: the role of lipoproteins. Curr Drug Deliv. 2005;2(4):443-449. 48.Moghimi SM, Andersen AJ, Hashemi SH, et al. Complement activation cascadetriggered by PEG-PL engineered nanomedicines and carbon nanotubes: thechallenges ahead. J Control Release. 2010;146(2):175-181. 49. MIoghimi SM, Hunter AC. Complement monitoring of nanomedicines and im-plants. Adv Drug Deliv Rev. 2011;63(12):963-964. 50.1Boraschi D, Costantino L, Italiani P. Interaction of nanoparticles with immu-nocompetent cells: nanosafetyyconsiderations. Nanomedicine(Lond).2012;7(1):121-131. 51. Szebeni J, Storm G. Complement activation as a bioequivalence issue relevantto the development of generic liposomes and other nanoparticulate drugs.Biochem Biophys Res Commun. 2015;468(3):490-497. 52. Moghimi SM, Simberg D, Skotland T, Yaghmur A, Hunter C. The interplay be-tween blood proteins, complement, and macrophages on nanomedicine per-formance and responses. J Pharmacol Exp Ther. 2019;370:581-592. 53.Thomas DG, Chikkagoudar S, Heredia-Langer A, et al. Physicochemical signa-tures of nanoparticle-dependent complement activation. Comput Sci Discov.2014;7(1):015003. 54.Diebolder CA, Beurskens FJ, de Jong RN, et al. Complement is activated by IgGhexamers assembled at the cell surface. Science. 2014;343(6176):1260-1263. 55. K1lapper Y,Hamad OA, Teramura Y, et al. Mediation of a non-proteolytic acti-vation of complement component C3 by phospholipid vesicles. Biomaterials.2014;35(11):3688-3696. 56. Weiszhar Z, Czúcz j, Revesz C, Rosivall L, Szebeni J, Rozsnyay Z. Complementactivation by polyethoxylated pharmaceutical surfactants: cremophor-EL,Tween-80 and Tween-20.Eur J Pharm Sci. 2012;45(4):492-498. 57. Agbeko RS, Fidler KJ, Allen ML, Wilson P, Klein NJ, Peters MJ. Genetic variabilityin complement activation modulates the systemic inflammatory responsesyndrome in children. Pediatr Crit Care Med. 2010;11(5):561-567. 58.Heurich M, Martinez-Barricarte R, Francis NJ, et al. Common polymorphisms inC3, factor B, and factor H collaborate to determine systemic complement ac-tivity and disease risk. Proc Natl Acad Sci U S A. 2011;108(21):8761-8766. 59.Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype:dictating risk for inflammation and infection. Trends Immunol. 2012;33(10):513-521. 60. Szebeni J, Muggia F, Gabizon A, Barenholz Y. Activation of complement bytherapeutic liposomes and other lipid excipient-based therapeutic products:prediction and prevention. Adv Drug Deliv Rev. 2011;63(12):1020-1030. 61.1ilPKozma GT, Meszarosa T, Weiszhar Z, et al. Variable association of complementactivation by rituximab and paclitaxel in cancer patients in vivo and in theirscreening serum in vitro with clinical manifestations of hypersensitivity: apilot study.Eur J Nanomed. 2015;7(4):289-301. 当静脉注射时,各种颗粒和纳米药物会激活补体,可能导致输液反应和其他药物不良反应。颗粒在治疗蛋白的配方中,由于运输、处理和给病人的治疗过程中产生的应力形成。在本研究中,IVIg溶液被储存在多种类型的小瓶和预填充的注射器中,并暴露在搅拌和冻融应力下产生颗粒。将应激样本加入人血清中,以确定这些颗粒是否激活补体。大小在2到10微米的亚可见IVIg颗粒其激活补体的方式与颗粒数量呈现出线性关系,而在较大粒子(>10微米)的剂量和补体激活之间几乎没有相关性。通过亚可见颗粒IVIg激活补体是另一种途径,如补体级联因子Bb的释放和无C4a生成的过敏性毒素C3a和C5a。亚可见颗粒的数量和形态取决于所施加的应力、配方和容器材料。但2- 10微米大小的微粒激活人血清补体的能力仅取决于微粒浓度。点击下载
确定







还剩6页未读,是否继续阅读?

产品配置单



大昌华嘉科学仪器为您提供《血清中IVIg配方中的亚可见颗粒可活化人血清中补体检测方案(图像粒度粒形)》,该方案主要用于全血/血清/血浆中生化检验检测,参考标准--,《血清中IVIg配方中的亚可见颗粒可活化人血清中补体检测方案(图像粒度粒形)》用到的仪器有流式颗粒成像分析系统FlowCam®8100、FlowCam® 5000颗粒分析仪、颗粒成像法+光阻法分析系统 FlowCam® + LO
推荐专场






相关方案
更多
该厂商其他方案
更多