








方案详情
文
背景:埃博拉病毒(EBOV)主要攻击骨髓细胞,其致命感染会引起大量的T细胞死亡。研究已证明,受感染细胞所产生的外泌体中包含的埃博拉病毒蛋白VP40导致了受体免疫细胞的死亡。
方案详情

The Journal of Infectious DiseasesSUPPLEMENT ARTICLE BCycD1 Kinase Activity Ebola Virus VP40 Modulates Cell Cycle and Biogenesis ofExtracellular Vesicles Michelle L. Pleet, James Erickson, Catherine DeMarino, Robert A. Barclay, Maria Cowen, Benjamin Lepene, Janie Liang,Jens H. Kuhn, Laura Prugar, Spencer W. Stonier, John M. Dye,Weidong Zhou, Lance A. Liotta, M. Javad Aman, and Fatah Kashanchil Laboratory of Molecular Virology, School of Systems Biology, George Mason University, Manassas, Virginia;2Ceres Nanosciences, Inc., Manassas, Virginia; 3IntegratedResearch Facility at Fort Detrick, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Fort Detrick, Frederick, Maryland;Virology Division, USArmy Medical Research Institute of Infectious Diseases, Fort Detrick, Frederick,Maryland;5Center for Applied Proteomics and Molecular Medicine, George Mason University,Manassas, Virginia; Integrated BioTherapeutics, Inc.,Gaithersburg, Maryland Background. Ebola virus (EBOV) mainly targets myeloid cells; however, extensive death of T cells is often observed in lethalinfections. We have previously shown that EBOV VP40 in exosomes causes recipient immune cell death. Methods. Using VP40-producing clones, we analyzed donor cell cycle, extracellular vesicle (EV) biogenesis, and recipientimmune cell death. Transcription of cyclin D1 and nuclear localization of VP40 were examined via kinase and chromatin immu-noprecipitation assays. Extracellular vesicle contents were characterized by mass spectrometry, cytokine array, and western blot.Biosafety level-4facilities were used for wild-type Ebola virus infection studies. Results. VP40 EVs induced apoptosis in recipient T cells and monocytes. VP40 clones were accelerated in growth due to cyclinD1 upregulation, and nuclear VP40 was found bound to the cyclin D1 promoter. Accelerated cell cycling was related to EV bio-genesis, resulting in fewer but larger EVs. VP40 EV contents were enriched in ribonucleic acid-binding proteins and cytokines(interleukin-15, transforming growth factor-p1, and interferon-y). Finally, EBOV-infected cell and animal EVs contained VP40,nucleoprotein, and glycoprotein. Conclusions. Nuclear VP40 upregulates cyclin D1 levels, resulting in dysregulated cell cycle and EV biogenesis. Packaging ofcytokines and EBOV proteins into EVs from infected cells may be responsible for the decimation of immune cells during EBOVpathogenesis. Keywords.aapoptosis; cell cycle; Ebola virus; extracellular vesicles; VP40. Ebola virus (EBOV) is a negative sense, single-stranded ribonu-cleic acid (RNA) virus capable of causing severe hemorrhagicfever in humans and nonhuman primates (NHPs). Until 2013,EBOV was responsible for approximately 1600 deaths; how-ever, the most recent outbreak in Western Africa (Sierra Leone,Guinea, and Liberia), leading to eventual spread to Europeand the United States, resulted in over 28 600 cases and 11300deaths as of April 2016 [1, 2]. The progression of EBOV dis-ease (EVD) within infected individuals involves nonspecificinfluenza-like symptoms, cytokine storm, and potential hem-orrhagic manifestations including coagulopathy, leaky bloodvessels, and multiorgan failure. Ebola virus mainly targets host ( Presented i n part: American Society for Exosomes and Microvesicles, Asilomar ConferenceCenter, 2017, Pacific Beach, CA; 9th I nternational Symposium on Filoviruses, Welcome Hot e l M arburg, 2017 , M arburg, Germany; C e ll S y mposia on Em e rgi n g and Re-Emerging Vir u ses, S heraton Pen t agon City, 2017, A r lington, V A; Bio-Trac E xosomes: Principles, M ethods andApplications Workshop, 2018, Montgomery College Germantown Campus, Germa n town, MD ;2 nd annual Extracellular Vesicles and Infections Conference,2018, Potomac, MD. ) ( Correspondence: F . K ashanchi, P hD, Laboratory o f M o lecular V i rology, G eorge M a son U niversity, Discovery Hal l Room 182, 10900 University Blvd., Manassas, VA 20110 ( fk ashanc @ gmu .edu ) . ) ( The Journal of Infectious Diseases 2 018;218(S5):S365-87 ) ( C The Author(s) 2018. Pu b lished by Oxford Uni v e rsity Press for the Infectious Diseases Society o f A merica. A l l rights r e served. For p e rmissions, e- m ail: jo u rnals.permissions@oup.com . DOI: 1 0.1093/infdis/jiy472 ) cells ofthe myeloid lineage (monocytes, macrophages, and den-dritic cells), but it does not productively infect T cells. Despitethis targeting, lethal infections are accompanied by extensivecell death of CD4 and CD8 T-lymphocytes [3-10]. There areseveral proposed mechanisms for the induction of apoptosis inbystander lymphocytes during EBOV pathogenesis, includingthe following: Fas/Fas ligand and tumor necrosis factor (TNF)-TNF-related apoptosis-inducing ligand (TRAIL) interactions,impaired dendritic cell/T-cell contacts, and nitric oxide orviral glycoprotein (GP)-induced apoptosis [5, 9-11]. Likewise,we have recently proposed an additional potential mechanismfor the induction of bystander T-cell death in the form of theEBOV matrix protein transferred to recipient cells via exosomes[12, 13]. However, the extent to which each of these mecha-nisms may contribute to bystander lymphocyte apoptosis andEBOV pathogenesis is not well characterized. Nevertheless, theloss of circulating T-cell populations in infected individualsmay contribute to unchecked replication of the virus, poten-tially correlating with poor patient prognosis [13,14]. In recent years, it has become clear that exosomes-small(~50-120 nm in diameter), membrane-bound extracellularvesicles (EVs) produced from the late endosomal pathway-are important for intercellular communication, particularlyduring disease [15-18]. Various specific tetraspanin proteins such as CD63, CD81,and CD9 mark the surface of exosomesand can be used to distinguish them from other EVs [19]. Thecontent of exosomes can vary based upon cell type and envi-ronment; however, the selective packaging of cargo into exo-somes is largely regulated by the Endosomal Sorting ComplexesRequired for Transport (ESCRT) proteins. The ESCRT proteinscan be classified as ESCRT-I, -II, -III complex components andVPS4, which sequentially act to recognize cargo, package it intonascent vesicles, and pinch off the membrane while recyclingthe machinery back for future cargo packaging [20]. Duringinfection, various viral proteins and nucleic acids can becomeintegrated into exosomes, often by utilizing the ESCRT path-way [21-25]. Exosomes originating from infected cells can theninduce proviral effects in recipient cells [12, 13, 16, 17, 21-28].In addition to exosomes, other EVs such as exosome-like ves-icles, microvesicles, and apoptotic bodies are produced fromhost cells by various mechanisms. Microvesicles, sometimescalled ectosomes, are typically larger (100-1000 nm) in diame-ter and differ from exosomes mainly in that they arise from thedirect outward budding and fission of the plasma membrane,or ectocytosis [29-31].Microvesicles do not contain tetraspan-ins CD9 or CD81, and they have a similar lipid compositionas the plasma membrane from which they budd [28, 29, 32].Apoptotic bodies, unlike the other secreted vesicles, are formedonly during programmed cell death. They are generally larger(500-4000 nm in diameter) than microvesicles and may containdeoxyribonucleic acid (DNA) and histones [28, 33, 34]. The EBOV matrix protein VP40 is one of the most abundantviral proteins, and, therefore, it is commonly used as a diagnos-tic marker for patients [35]. Alone or in conjunction with theGP and nucleoprotein (NP) of EBOV, VP40 can independentlybud from cells to form virion-like particles (VLPs) that aremorphologically similar to infectious virions [36-41]. Recently,we found the first evidence that VP40 is also able to exit cellsby becoming packaged into exosomes [12, 13]. This finding isconsistent with previous studies that have shown filoviral VP40concentrating in late endosomal compartments and multivesic-ular exosomal bodies [42, 43]. Another study by Steele et al [44]showed that in EBOV-infected NHP and guinea pig models,VP40 antigen alone is concentrated in renal tubule epithelialcells, indicating that VP40 can exist outside of the virion (ie,potentially in exosomes) [45]. In the current study, we focused on (1) the mechanismsinvolved in the packaging of EBOV VP40 into exosomes andother EVs and, (2) the characterization of these vesicles. Wehave found that VP40 was able to affect cell cycle through cyclinD1 (which binds to cdk4 or cdk6) modulation to regulate thesynthesis of EVs in donor cells. This regulation was done bythe translocation of VP40 into the nucleus to stimulate over-transcription of cyclin D1 at its promoter, thus producing anactivated phenotype in VP40-producing cells. We tested theefficacy of cdk4/6 inhibitors for their ability to hinder donor cell EV production and found that Fascaplysin and Ribociclib(Ribociclib is US Food and Drug Administration [FDA]-approved) may impact the biogenesis of EVs. In previous stud-ies, we showed that VP40 was secreted in exosomes [12, 13],and here we additionally found that VP40 and other viral pro-teins (ie,NP and GP) were associated with EVs from wild-typeEBOV-infected cells and macaques. Finally, mass spectrometry(MS) and cytokine analysis showed that exosomes originatingfrom VP40-producing cells were enriched in several RNA-binding proteins and cytokines, which may play a pivotal rolein recipient cell outcome. MATERIALS AND METHODS Cell Culture and Reagents U937, CEM, and Jurkat cells were obtained from ATCC(Manassas, VA) and maintained in Roswell Park MemorialInstitute (RPMI) 1640 media containing 10% heat-inactivatedfetal bovine serum (FBS), 1% L-glutamine, and 1% streptomy-cin/penicillin (Quality Biological, Gaithersburg, MD). 293Tcells, VP40 clones, and HeLa cells were sustained in Dulbecco'smodified Eagle medium (DMEM) containing 10% heat-in-activated fetal bovine serum (FBS), 1% L-glutamine, and 1%streptomycin/penicillin (Quality Biological). Peripheral bloodmononuclear cells (PBMCs) were maintained in RPMI 1640media containing 20% FBS, 1% L-glutamine, and 1% strepto-mycin/penicillin. The PBMCs were also maintained with 50IU/mL human recombinant interleukin (IL)-2 (Sigma) beforetransfections, but they were incubated in the absence of exoge-nous growth factors once transfected. All cells were incubated at37°℃ with 5%CO..For antibiotic selection of transfected 293Tcells, hygromycin B (Invitrogen) was added to the culture thenext day. Cdk4/6 inhibitors Fascaplysin(0.1-1uM; Abcam) andRibociclib (LEEO11; 0.1-10.0 uM; MedChemExpress) wereused for the treatment of 293T and VP40 clone cell culturesfor the analysis of cyclin D1 inhibition, cell viability, and EVproduction. Plasmids, Transfections, and Generation of Resistant Clones EBOV Mayinga VP40 isolate was expressed from a plasmid(Invitrogen) with cytomegalovirus (CMV) promoter and specificantibiotic selection marker (pcDNA3.1/Hygro). Twenty micro-grams of Escherichia coli-purified DNA was transfected into293T cells using electroporation as previously described [46].Transfected cells were treated the next day with hygromycin B(200-400 ug/mL) for plasmid selection. To generate resistantclones, transfected cells were cultured for >3 weeks, followed byisolation of surviving colonies and multiple passages under spe-cific antibiotic selection. A total of 21 new resistant clones weregenerated, which produced varying amounts of VP40 (selectclones are shown in Supplementary Figure 1). Two clones, anintermediate and a high VP40-producing clone, were selectedto complement an existing low VP40-producing clone that we previously published (EVTR2C) [12]. For clarity, we renamedthis clone V2CL (VP40 293T Clone Low), whereas we desig-nated the medium and high clones V2CI (VP40 293T CloneIntermediate) and V2CH (VP40 293T Clone High), respec-tively. All resistant clones were kept under constant hygromycinB (200 ug/mL for V2CL; 400 ug/mL for V2CI and V2CH) anti-biotic selection. Transfections of U937 cells, HeLa cells, and PBMCs were per-formed with log-phase cells and attractene reagent (QIAGEN)according to the manufacturer's instructions. In brief, 1.65×10log-phase cells were incubated with transfection complexesformed from 1.5 ug of VP40-producing plasmid DNA and1.5 uL attractene reagent in serum-free media. Cells and com-plexes were incubated for 3 days in 100 uL, followed by analysisof cell viability by CellTiter-Glo assay. Capture of Extracellular Vesicles, Virus-Like Particles, and Proteins WithNanotrap Particles The Nanotrap (NT) particles we used here were provided byCeres Nanosciences, Inc. and have been described in detailpreviously [47]. In brief, NT particles are highly porous, mul-tifunctional hydrogel particles, approximately 700-800 nm indiameter and are composed of high-affinity aromatic dye com-pounds covalently linked to an interior hydrogel particle struc-ture. The 2 particles used frequently by our laboratory includeNT80 particles (Ceres no. CN1030), which contain a ReactiveRed 120 core bait, and NT82 particles (Ceres no. CN2010),which have Cibacron Blue F3GA. Both particle types have aNIPAm-Bis-AA matrix with a shell lacking vinyl sulfonic acid[47]. For the capture and isolation of EVs from cell culturesupernatants, a 30-uL slurry (30%) of 1:1 NT80 and NT82 par-ticles (Ceres Nanosciences) was incubated with 1 mL cell-free,filtered (0.22 um) supernatant. Samples were bound at 4℃ for24-72 hours. The NT pellets were isolated and washed with500 uL of sterile 1x phosphate-buffered saline (PBS) withoutcalcium and magnesium, followed by preparation for down-stream assays. For western blot analysis, washed NT pelletswere resuspended in 10 uL of Laemmli buffer and then loadedonto a 4-20% Tris-glycine sodium dodecyl sulfate (SDS) gel. For the analysis and characterization of VLP, VLPs containingEBOV VP40, NP, and GP produced in Sf9 insect cells throughrecombinant baculovirus infection were used (obtained fromIBT Bioservices). For analysis of VLP passage through 0.22-umfilters,5 g of VLP was spiked into 1 mL of sterile 1x PBS, fol-lowed by filtration (through 0.22 um) alongside a PBS control.The resulting filtrate was then incubated with 30 pL ofNT80/82particles (30% slurry) overnight at 4°C. NT pellets were used thenext day for SDS-polyacrylamide gel electrophoresis (PAGE)and western blot analysis for presence of EBOV proteins. Forthe analysis of VLP in comparison to EV distribution uponpassage through size exclusion fractionation before and afterfiltration, 10 ug of VLP was spiked into 10 mL of 5-day V2CH supernatants produced in exosome-free media. Supernatantswere either filtered (through 0.22 um filter) or directly incu-bated with ExoMAX Opti Enhancer (1:1 reagent/filtered super-natant; Systems Biosciences) overnight at 4C to concentrateEVs. Extracellular vesicles were pelleted, resuspended in 0.5 mLof sterile 1× PBS, and loaded onto qEVoriginal size exclusioncolumns (IZON) according to the manufacturer’s instructions.Fraction numbers 2-11 (0.5 mL each) were collected and incu-bated with 30 uL of NT80/82 30% slurry overnight at 4℃. Theresulting NT pellets were then resuspended in Laemmli bufferfor SDS-PAGE and western blot analysis of EBOV protein andexosomal marker levels. Extracellular vesicles were obtained from EBOV-infectedcell cultures under biosafety level (BSL)-4 conditions. Humanumbilical vein endothelial cells (HUVECs) were infected withEBOV H.sapiens-tc/COD/1995/Kikwit (multiplicity of infec-tion [MOI] of 1) and incubated for 3 days. Two milliliters ofsupernatants were harvested, passed through a 0.22-um filter,and incubated with ExoMAX (1:1 reagent/filtered supernatant)reagent overnight at 4℃ to concentrate EVs. Extracellular vesi-cles were pelleted, resuspended in 0.5 mL of sterile 1× PBS, andloaded onto qEVoriginal size exclusion columns according tothe manufacturer's instructions. Purified vesicles were collectedas 0.5-mL fraction numbers 7-10. Each fraction was incubatedwith 30 uL of NT80/82 (30% slurry) at room temperature for 1hour. Extracellular vesicle-bound NTs were then washed oncewith 1x PBS and resuspended in 10 uL of 2x NuPAGE LDSsample buffer (Thermo Fisher) for SDS-PAGE and western blotanalysis of EBOV protein levels. Samples were heated at 95℃for 10 minutes twice before electrophoresis. For the capture and isolation of EVs from inactivated (gammairradiation) rhesus monkey (Macaca mulatta) serum samples(obtained from US Army Medical Research Institute of InfectiousDiseases), 100 uL of serum from each animal (n =3;2 EBOV+,1 EBOV-) were diluted in 400 uL of sterile 1× PBS without cal-cium or magnesium and passed through a 0.22-um filter. Sampleswere incubated with 25 uL of NT80/82 30% slurry for 48 hoursat 4°C. The NT pellets were washed with sterile 1× PBS withoutcalcium and magnesium and resuspended in 10 uL Laemmlibuffer for western blot analysis of exosomal markers and VP40protein. Nonhuman primate details are as follows: NHP 1(EBOV-)= day 0 prebleed serum; NHP 2 (EBOV+)= pooledserum from days 4 to 5 postinfection (pi), animal died on day 7;NHP 3 (EBOV+)= pooled serum from days 8 to 11 pi, animaldied on day 12. All experiments involving NHPs and inactivatedEBOV samples were carried out under the Institutional BiosafetyCommittee-approved institutional biosafety guidelines and wereperformed at BSL-2/2+level. Cell Cycle Analysis The 293T,V2CL, or V2CH cells were either plated in 0.1% FBSDMEM, blocked with hydroxyurea for 2-5 days, or preblocked Cell Treatment and Viability Assay Cells were seeded into 96-well plates at 50 000 cells per well infresh media followed by treatment. All wells were plated andtreated in triplicate. For treatment of CEM, Jurkat, U937, and293T cells with cell culture supernatants, control cells or resis-tant clones were cultured for 5 days in exosome-free media tomaximize EV concentration within the media, as previouslydescribed[26]. Media were then harvested from the cells,cen-trifuged for 10 minutes at 20 800 xg to remove cells and cellu-lar debris, and filtered (0.22 um). Seeded cells in fresh media(50 uL total) were then treated with 50 uL of supernatant. Alltreatments of CEM, Jurkat, U937, and 293T cells were incu-bated for 5 days, followed by measurement of cell viabilityusing CellTiter-Glo Cell Luminescence Viability kit (Promega,Madison, WI) as per manufacturer’s instructions. In brief,100 uL of CellTiter-Glo reagent were added to the wells (1:1reagent/cell suspension). The plate was shaken for 2 minutes,incubated at room temperature for 10 minutes, and followed bydetection of luminescence using the GloMax Explorer multide-tection system (Promega). For the viability analysis of cell cycleblocks, 293T,V2CL, and V2CH cells were seeded as describedabove. Starvation wells were plated in fresh 0.1% FBS DMEM,whereas all others were in 10% FBS DMEM. Cells were thentreated with either hydroxyurea (20mM) or nocodazole (50 ng/mL). Cells receiving nocodazole treatment were pretreated for18 hours with hydroxyurea (20 mM). It should be noted thathydroxyurea is normally used at 1-2 mM to block cells, but herewe have tested higher concentrations to block cells without celldeath. Cells were incubated for 5 days followed by measurementof cell viability using CellTiter-Glo assay as described above. Preparation of Whole-Cell Extracts and Western Blot Analysis Cell pellets were harvested, washed twice with 1x PBS with-out calcium and magnesium, and resuspended in lysis buffer(50 mM Tris-HCl [pH 7.5], 120 mM NaCl, 5 mM EDTA, 0.5%Nonidet P-40, 50 mM NaF, 0.2 mM Na, VO, 1 mM dithio-threitol [DTT], and 1 complete protease inhibitor mixturetable/50 mL [Roche Applied Science]). The suspension wasincubated on ice for 20 minutes with gentle vortexing every 5minutes, followed by centrifugation at 10000 xg at 4℃ for 10 minutes. Protein concentration from the lysate supernatant wasquantified using Bradford protein assay according to the manu-facturer's instructions (Bio-Rad). Preparation of EBOV-infected HUVEC lysates was per-formed as previously described [48]. Infected HUVECs werefixed in 10% neutral-buffer formalin at 4℃ in 6-well plates.Fixed HUVEC monolayers were washed in PBS, a solution of300 mM of Tris pH 8.0 containing 2% SDS was added, andcells were scraped off and transferred to polypropylene tubes.Samples were heated at 100℃ for 30 minutes, then 60℃ for 2hours. Excess SDS was dialyzed using spin filtration, and pro-tein content was determined by BCA assay. For western blot analysis, whole-cell extracts (10-30 ug)were resuspended in 10 uL of Laemmli buffer, heated at 95℃for 3 minutes, and loaded onto a 4-20% Tris-glycine SDS gel.NT particle pellets were resuspended in 10 uL of Laemmli buf-fer, heated at 95℃ for 3 minutes, and vortexed 3 times untilfully resuspended. The eluted material was then loaded ontoa 4-20% Tris-glycine gel. Gels were run at a maximum 150 Vand wet-transferred overnight at 50 mA onto polyvinylidenedifluoride (PVDF) membranes. Membranes were blocked in5% milk in 1× PBS containing 0.1% Tween 20 for 1 hour at 4C,then incubated overnight at 4℃ with appropriate primary anti-body: a-Caspase 3, a-poly(ADP-ribose)polymerase-1(PARP-1), a-Alix,a-VPS4,a-CHMP6,a-TSG101,a-EAP20,a-EAP45,a-cyclin D1, a-cyclin E, a-cyclin A, a-cdk2, a-cdk4, a-cdk6,a-cdc2 p34(a-cdk1), a- histone deacetylase 1 ([HDAC1] SantaCruz Biotechnology);a-B-actin (Abcam); a-CD63, a-CD81,a-CD9 (Systems Biosciences); a-cyclin B1 (Cell Signaling);and a-GP, a-NP, and a-VP40 (IBT Bioservices). Extracellularvesicles from EBOV-infected HUVECs and lysates from EBOV-infected cells were dry-transferred to PVDF membranes withthe iBlot system (Thermo Fisher). VP40 and NP were detected asdescribed above, whereas GP was detected with murine mono-clonal EBOV GP-specific antibody 6D8 [49]. Membranes werethen incubated with the appropriate horseradish peroxidase(HRP)-conjugated secondary antibody for 2 hours at 4℃ anddeveloped using Clarity or Clarity Max Western ECL Substrate(Bio-Rad). Luminescence was visualized on a ChemiDoc TouchImaging System (Bio-Rad). Isolation of Exosomes for Characterization and Functional Assays Our laboratory has substantial experience in the isolation andpreparation of exosomes and EVs for characterization and func-tional analysis, particularly in the context of viral infection [12,21, 22, 26, 27, 50-52]. For the experiments used here, 293T cellsand VP40 clones were grown in exosome-free DMEM contain-ing 10% heat-inactivated FBS (ultracentrifuged at 100 000 xgfor 90 minutes to remove vesicles), 1%L-glutamine, and 1%streptomycin/penicillin (Quality Biological). Exosome prepa-rations were made from exosome-free cell culture supernatant(filtered; 0.22 um) produced from fully confluent cells grown for 5 days. For isolation of exosomes for western blot analysisand AChE assay, NT80/82 particles were used as describedabove. For the characterization of exosomes by gradient sepa-ration and western blot, 10 mL of unfiltered cell culture super-natant was incubated with 10 mL of ExoMAX Opti Enhancer at4℃ overnight. The vesicle pellet was spun down at 1500 ×g for30 minutes and resuspended in 300 uL sterile 1× PBS withoutcalcium or magnesium. Resuspended vesicles were then placedon top of iodixanol (OptiPrep; Sigma) gradients prepared in 1xPBS in 1.2% increments ranging from 6% to 18%. The gradientwas ultracentrifuged for 90 minutes at 100000 xg in a SW41Ti rotor (Beckman). Gradient fractions were collected from thetop of the gradient in 1-mL increments and transferred to ster-ile 1.5-mL microcentrifuge tubes. Separated iodixanol fractionswere incubated with 30 uL of NT80/82 30% slurry at 4℃ over-night, followed by processing for western blot or MS analysis.Alternatively, select iodixanol fractions were pooled and resus-pended in sterile 1x PBS, followed by ultracentrifugation for 90minutes at 100000 xg in a Ti-70 rotor (Beckman) to pellet EVsfor the removal of iodixanol for downstream functional assays.Direct isolation of EVs from small volumes of cell-free super-natants from 293T and V2CH cells (grown in exosome-freemedia) was completed by ultracentrifugation for 90 minutesat 100 000 xg in a TL-100 rotor (Beckman). Resulting EV pel-lets were resuspended in 40 uL of sterile 1x PBS and used forZetaView analysis and subsequent functional assays. Acetylcholinesterase Assay ACHE protein levels of the exosomes were determined withtheFLUOROCET Ultrasensitive EExosome QuantitationAssay Kit (Systems Biosciences) following the manufacturer'sinstructions. In brief, a negative control containing buffer solu-tion (exosome negative) and standard curve were prepared.Nanoparticle pellet samples (from filtered cell culture media)were plated and treated. Fluorescence corresponding to AChEactivity was measured with a GloMax Explorer multidetectionsystem (Promega). The number of exosomes was calculatedusing the standard curve. ZetaView Nanoparticle Tracking Analysis Nanoparticle tracking analysis (NTA) was performed usingthe ZetaView Z-NTA (Particle Metrix) and its correspondingsoftware (ZetaView 8.04.02). One hundred nanometer poly-styrene nanostandard particles (Applied Microspheres) wereused to calibrate the instrument before sample readings at asensitivity of 65 and a minimum brightness of 20. Automatedquality control measurements including, but not limited to, cellquality check and instrument alignment and focus were alsoperformed before the use of the Zeta View for sample measure-ments. For each measurement, the instrument preacquisitionparameters were set to a temperature of 23℃, a sensitivity of85, a frame rate of 30 frames per second, and a shutter speed of 250. For each sample, 1 mL of the sample, diluted in deionizedwater, was loaded into the cell, and the instrument measuredeach sample at 11 different positions throughout the cell, with3 cycles of readings at each position. After automated analysisand removal of any outliers from the 11 positions, the mean,median, and mode (indicated as diameter) sizes and the con-centration of the sample were calculated by the machine soft-ware. Measurement data from the ZetaView were analyzedusing the corresponding software, ZetaView 8.04.02, andMicrosoft Excel 2016. We selected the mode, defined as the sizeof the most abundant particles, as the measurement for size inour analysis. Cytoplasmic Versus Nuclear Isolation Cytoplasmic and nuclear compartments were isolated andextractedl utsingttheNE-PERRNuclear and CytoplasmicExtraction Reagent kit (Thermo Fisher Scientific) according tothe manufacturer's instructions. In brief, log-phase 293T andV2CH cells (1×10) were harvested, washed once in 1× PBS,and incubated with ice cold CER I (100 uL). Samples were vor-texed for 15 seconds and incubated on ice for 10 minutes. Next,ice cold CER II was added (5.5 pL) to each sample, vortexed for5 seconds, and incubated on ice for 1 minute. Tubes were thenvortexed, spun for 5 minutes at 16000 xg, followed by trans-fer of cytoplasmic fractions to clean prechilled tubes. Insolublenuclei pellets were suspended in ice cold NER (50 uL), vortexedfor 15 seconds, and incubated on ice for 40 minutes with addi-tional vortexing every 10 minutes. Samples were centrifuged for10 minutes at 16000 xg, and resulting nuclear fractions weretransferred to clean prechilled tubes. Resulting fractions wereanalyzed for protein concentration by Bradford assay (Bio-Rad)and used for SDS-PAGE and subsequent western blot analysis. Kinase Assay Peptide kinase assays were performed as describedpre-viously [12]. In brief, immunoprecipitation (IP) was per-formed by incubation of 500 rg of 293T or V2CH whole-cellextracts with 10 ug of appropriate primary antibody (a-cyclinD1, a-normal rabbit immnunoglobulin [Ig]G; Santa CruzBiotechnology) and 1 mL of TNE50 + 0.1% NP-40 for 24hours at 4C. The next day complexes were precipitated with50 uL of a 30% slurry of A/G beads (Calbiochem) for 2 hoursat 4°C and washed twice with TNE50 + 0.1% NP-40 and oncewith kinase buffer. Phosphorylation reactions were performedwith IPed material (15 pL), 50 ug of appropriate peptide (780S[775RPPTLSPIPHIPR7871, 780A 75RPPTLAPIPHIPR7871[ABclonal Science]), and [y-32P] ATP (2 uL) as substratesin TTK kinase buffer containing 50 mM HEPES (pH 7.9),10 mM MgCl, 6 mM EGTA, and 2.5 mM DTT. Some sam-ples were also incubated with cdk4/6 inhibitor Fascaplysin(1 uM; Abcam). The reaction mixtures contained the follow-ing final concentrations: 40 mM β-glycerophosphate (pH 7.4), 7.5 mM MgCl,, 7.5mM EGTA, 5% glycerol, [y-32P] adenosinetriphosphate (ATP)(0.4 mM, 1 uCi), 50 mM NaF,1 mM ortho-vanadate, and 0.1% (v/v) β-mercaptoethanol. Reactions wereincubated at 37℃ for 1 hour, and samples (10 uL) were dottedonto Whatman glass microfiber filters, dried for 30 minutes,and subsequently submerged in 1x TE buffer with gentle agita-tion for 48 hours. Samples were quantified using a scintillationcounter (QuantaSmart CPM assay count). Chromatin Immunoprecipitation and Quantitative Polymearse ChainReaction for Cyclin D1 Promoter DNA Log-phase 293T and V2CH cells (3×10 cells) were harvestedand processed for chromatin IP (ChIP) and cyclin D1 pro-moter PCR using the Imprint Chromatin ImmunoprecipitationKit (Sigma) according to the manufacturer’s instructions. Inbrief, samples were cross-linked using 1% formaldehyde andincubated for 1 hour, followed by quenching with 1.25 M gly-cine (9:1 cell suspension/glycine). Samples were sonicated andused for IP with 1 ug of a-RNA PolII, a-p300 (Santa Cruz), ora-VP40 (IBT Bioservices) at 4C overnight. The next day, a 30%slurry of A/G beads (Calbiochem) was added and incubatedfor 2 hours at 4C. The samples were washed once with eachTNE300 +0.1% NP40, TNE150 +0.1% NP40,TNE50 + 0.1%NP40, and IP Wash Buffer (Sigma) before the addition of pro-teinase K (800 units/mL). After a 15-minute incubation at 65℃,Reversing Solution (Sigma) was added, and samples were incu-bated at65℃ for an additional 90 minutes. The DNA was puri-fied, and quantitative PCR analysis was performed with 2 uLofundiluted aliquots of DNA using SYBR Green (Bio-Rad) withthe following pair of primers that amplify the cyclin D1 pro-moter region containing the RNA Pol II start site (+1): cyclinD1-Reverse (-180), (5'-CAC TTC GCA GCA CAG GAG-3,Tm = 56.3℃); and cyclin D1-Forward (+238), (5'-CGG ACTACA GGG GCA ACT-3,Tm=57.0C). Serial dilutions of DNAfrom 293T cells were used as quantitative standards. The PCRconditions were as follows: 1 cycle at 50℃ for 2 minutes, fol-lowed by 1 cycle at 95C for 2 minutes, and 42 cycles at 95℃for 15 seconds and 48.7C for 40 seconds. The absolute quan-tification of the samples was determined based on the cyclethreshold value relative to the standard curve. Quantitative PCRreactions were carried out in triplicate using the CFX96 RealTime System (Bio-Rad). Mass Spectrometry The EVs from NT80/82 NT particle-processed 10.8 and 12.0iodixanol fractions (as described above) were treated with 8 Mof urea with 0.00005% DTT to lyse the vesicles. The sampleswere reduced with 10 mM of DTT and alkylated with 50 mMof iodoacetamide. The samples were next diluted by a solu-tion of equal parts water and 500 mM of NH HCO,, followedby trypsin (Promega) digestion for 4 hours at 37℃. Sampleswere then centrifuged at 12000 xg at room temperature for 10 minutes, and supernatants were collected into a microcentri-fuge tube. ZipTip was then used to collect the peptide samples,which were dried and resuspended in 10 uL of 0.1% trifluo-roacetic acid solution before loading into an Orbitrap Fusionmass spectrometer. Bioinformatic searches from Swiss-Protwere used to identify peptides, and a label-free precursor iondetection method (Proteome Discoverer, version 1.3; ThermoFisher Scientific) was used for accurate mass measurements onproteins and peptides with specific retention times on precur-sors and fragments. Cytokine Analysis and Densitometry 293T and V2CH cells were cultured in exosome-free DMEM for5 days, followed by harvesting and filtering (0.22um) of super-natants. Ten milliliters of supernatant samples were incubated1:1 with ExoMAX at 4℃ overnight, spun down, resuspended,and separated on an OptiPrep iodixanol gradient, as describedabove. The 10.8 and 12.0 iodixanol fractions for each cell typewere combined and used side-by-side with unprocessed cellsupernatants (filtered; 0.22 um). For analysis of cytokine pro-files in whole supernatant versus in association with exosomes,human cytokine array membranes (Abcam) were used accord-ing to the manufacturer's instructions. In brief, membraneswere blocked at room temperature for 30 minutes, followed byincubation with sample overnight at 4℃ with gentle rocking.The next day, samples were aspirated, and membranes werewashed and then subsequently incubated overnight at 4°℃ withbiotin-conjugated a-cytokines. Membranes were then washed,incubated with HRP-conjugated streptavidin for 2 hours atroom temperature, and washed again.Detection of chemilum-inescence was accomplished via ChemiDoc Touch ImagingSystem (Bio-Rad). Raw densitometry counts were obtainedusing ImageJ software. Exposures were matched betweencytokine membranes according to positive control signals.Densitometry data were normalized by subtracting the back-ground represented by the negative control on each membrane. Statistical Analysis Standard deviation was calculated in all quantitative exper-iments done in triplicate. All Pvalues were calculated using2-tailed Student's t tests (Microsoft Excel) and were consideredstatistically significant when P<.05. RESULTS Cells Producing VP40 Are Dysregulated in Their Cell Cycle In our previous work, we used a stably transfected VP40-producing clone cell line that we named V2CL (EVTR2C [12];see Methods) to show that VP40 can be packaged into exosomesand thereby impact recipient cells. To further verify our find-ings, we transfected a new series of 293T cells with EBOV VP40plasmid (pcDNA3.1/Hygro) and treated them with hygromycinB for plasmid selection. After >3 weeks culture under constant antibiotic selection, surviving colonies (total of 21 clones) wereisolated and cultured for 5 days. EVs by NT80/82 particle incu-bation and western blot analysis for VP40 levels and exosomalmarkers. Results in Supplementary Figure l show a representa-tive panel of 6 of the resulting clones: V2C1-4,V2CI, and V2CH.Most of the V2C lines (19 of 21) produced similar levels of VP40in EV western blot analysis. However,2 lines, V2CI and V2CH,produced lower and higher levels of VP40, respectively, in com-parison to the rest of the clones, while still exhibiting higherlevels of exosomal marker Alix and Actin. This finding indicatesthat these 2 clones may produce higher levels of EVs with dif-ferential amounts of VP40 packaged. Compared side-by-side tothe pre-existing V2CL line, which produced very low levels ofextracellular VP40, V2CI produced intermediate levels, whereasV2CH released high levels of VP40 in EVs (data not shown).For this reason, we chose these 2 lines to utilize in future exper-iments to determine dose dependency of VP40 on donor andrecipient cell responses. In the process of generating these clones, we observed anotably accelerated growth in cells producing VP40 (data notshown). It is well documented for many viruses, includinghuman immunodeficiency virus type 1 (HIV-1), human cyto-megalovirus, human T-cell leukemia virus type 1 (HTLV-1),Epstein-Barr virus (EBV), human papillomaviruses (HPVs),Kaposi sarcoma herpesvirus, hepatitis B virus, hepatitis C virus,adenoviruses, influenza viruses, and infectious bronchitis virus,that infected cells are altered in their cell cycle to benefit viralreplication [53-56].Therefore, we asked whether a similar phe-notype occurred within VP40 clones. To address this question,we blocked control 293T,V2CL, and V2CH cells at the G, G,/S,and G,/M stages of the cell cycle for either 2 or 5 days to observechanges in cell morphology and viability (Figure 1). Data inFigure 1A show that after 2 days of cell cycle blocks, all cell typeshad the expected morphology with minimal cell death. Morespecifically, G cells appeared adherent and stretched, G,/S cellswere adherent and enlarged, whereas G,/M cells were smaller Figure1.Cell cycle analysis of VP40 clones. 293T, V2CL, and V2CH cells were seeded in a 96-well plate at 5×105 cells in 100 uL of fresh media. Cells were blocked at G(starvation; 0.1% fetal bovine serum DMEM), G,/S (20 mM of hydroxyurea), and G,/M (18-hour 20 mM of hydroxyurea pretreatment, followed by release for 1 hour, and sub-sequent treatment with 50 ng/mL of nocodazole) for 2 days. Blocked cells were then imaged (A) and assayed for cell viability with CellTiter-Glo (B). The same experiment wasrepeated; however, cells were allowed to incubate for 5 days after blocking. Cells were subsequently imaged (C) and assayed for cell viability via CellTiter-Glo (D). Statisticalanalysis by Student’s 2-tailed t test compares V2CL and V2CH cell cycle-blocked groups with corresponding 293T groups (t, P<.05; tt, P<.01; ttt, P<.001). AdditionalStudent’s 2-tailed ttest compares cell cycle-blocked groups with controls of their own cell type (**, P<.01; ***, P<.001). androunded. Results from the corresponding 2-day cell viabil-ity assay (Figure 1B) indicated that all blocked cells had reducedviability, as would also be expected with functioning cell cycleblocks. After 5 days, a different pattern between cell types wasobserved. Control 293T cells retained similar phenotypes at5 days compared with 2 days.However, it should be noted thatsignificant cell death was observed for 293T cells at both G,/Sand G,/M after 5 days (Figure 1C and D). In contrast, VP40donor cells expressed greater cell viability at G,/S and G,/M, inline with increasing amounts of VP40 produced (Figure 1D).Strikingly, with increased levels of VP40 produced, V2C cellsadhered less to the expected phenotype exhibited by 293T cells.At G after 5 days, V2C cells were more viable compared with293T cells and had clumped together to form structures thatclosely resembled viral synapses (Figure 1C), similar to thoseseen with viruses such as HIV-1, Japanese encephalitis virus,influenza A virus, and HTLV-1[57-61]. It is possible that theseclumped structures may represent a potential synaptic mecha-nism used by EBOV in epithelial cell infection when nutrientsare depleted and cell cycling is hindered. In addition, V2C cellsat G /S and G,/M continued to "slip through"cell cycle blocks,especially in V2CH cells, as demonstrated by many roundedcells (G,/M) present in G,/S-blocked cultures and many adher-ent cells (G,/S) in G,/M-blocked cultures (Figure 1C). This pat-tern was further demonstrated by an increase in cell viability ofV2C cells compared with 293T at G,/S and G,/M (Figure 1D).Finally, regardless of the length of incubation, there was aVP40 production level-dependent increase in the number andviability of V2C control cells compared with 293T, support-ing our previous observation of an increased growth rate inVP40-producing cells. Taken together, these results indicatethat cells producing VP40 may have accelerated rates of growthdirectly proportional to the amount of VP40 being produced.Furthermore, VP40-producing cells may potentially be ableto push past cell cycle blocks imposed by external or internalinfluences, indicating a dysregulation of cell cycle checkpointsand resulting in increased survival under various environmen-tal stresses. VP40 Increases Functional Cyclin D1 Levels in Donor Cells As V2C cells had been observed to have accelerated growth, wenext explored the effect of VP40 production on levels of cyclinand cdk proteins. Western blot analysis of cyclin and cdk lev-els in 293T, V2CL, V2CI, and V2CH cells showed that levels ofcdk1 increased in all V2C cells, whereas cdk2,-4,-6, and cyclinE showed only a small upregulation with increasing VP40 pro-duced (Figure 2A). Cyclins A and B1 were initially increasedwith low levels of VP40 but then decreased in a dose-dependentmanner with increasing levels of VP40 produced.On the otherhand, cyclin D1 increased in a VP40 dose-dependent manner.Increased levels of cyclin D1 have previously been shown toresult in accelerated cell cycling and many types of oncogenic phenotypes [62-64]. In addition, several viruses, includingHTLV-1, HPV, EBV, human neurotropic JC virus, and humanrespiratory syncytial virus, have previously been shown to influ-ence the levels of cyclin D in infected cells to aid in their virallife cycle[65-70]. Cyclin D1s primary role during cell cycle is to bind to itscdk partner(s), cdk4 or cdk6, to activate its kinase activity,and thereby phosphorylate its substrate Rb to induce cell cycleprogression from G to S phase [62]. Therefore, we investi-gated the ability of cyclin D1 from V2C cells to initiate phos-phorylation of its substrate Rb at the Serine-780 residue. Two13-mer peptides were generated: one 780S peptide mappingto a cyclin D1/cdk4 phosphorylation site of Rb, and one 780Apeptide (S780A point mutation) [71]. Control 293T and V2CHwhole-cell extracts were immunoprecipitated with a-cyclin D1or IgG. Precipitated complexes were incubated with either 780Sor 780A peptide, and some were incubated with Fascaplysin(cdk4/6 inhibitor). In vitro kinase assays were then performedusing [y-3?P] ATP. Results in Figure 2B show that in 293T cells,active cyclin D1 complexes phosphorylated the 780S peptideas expected, whereas the 780A peptide and the 780S peptideincubated with Fascaplysin had only minimal backgroundlevels of phosphorylation. It is interesting to note that V2CHcells had an increased level of phosphorylation of 780S by IPedcyclin D1 compared with 293T cells.Moreover, incubation withFascaplysin did not completely ameliorate the cyclin D1 activityon the 780S peptide. Taken together, these results indicate thatcyclin D1 has a higher level of activity in V2CH cells comparedwith 293T cells, and that inhibition of active cyclin D1/cdk4/6complexes in V2CH cells by Fascaplysin is less effective. Thisfinding could perhaps be due to an overall greater amount ofcyclin D1 present in V2CH cells, and therefore a higher concen-tration of Fascaplysin may be needed to prevent kinase activity. VP40 Is Present in the Nucleus and Binds the Cyclin D1 Promoter Based upon our observations of accelerated growth andincreased levels of cyclin D1 in cells producing VP40, we nextwished to investigate potential factors contributing to cyclinD1 transcription. Previous studies have shown the intriguingpresence of EBOV VP40 within the nucleus, especially at earlytime points in infection [72-74]. To confirm VP40 presencein the nuclear compartment of our VP40 clones, cytoplasmand nuclear fractions of 293T and V2CH cells were isolatedby NE-PER Nuclear and Cytoplasmic Extraction kit. CHMP6(cytoplasmic protein; ESCRT-III component) and HDAC1(nuclear protein involved in chromatin remodeling) were usedas controls for cytoplasmic and nuclear compartments, respec-tively. Actin was also used to measure even loading of samples,because Actin has been shown to be abundantly present in boththe cytoplasm and the nucleus [75, 76]. Results in Figure 2Cshow that VP40 protein was present in both the cytoplasm andthe nucleus of V2CH cells. We were surprised to find that the A C D Figure 2. Effect of nuclear VP40 on cyclin and cdk regulation and activity. (A) Log phase 293T, V2CL, V2Cl, and V2CH cells were harvested, lysed, and subjected to SDS/PAGE for western blot analysis of cyclin D1 (CycD1), cyclin E (CycE), cyclin A (CycA), cyclin B1 (CycB1), cdk4, cdk6, cdk2, cdk1, and actin levels. (B) Five hundred microgramsof 293T or V2CH whole-cell extracts were used for IP with 10 ug of either normal rabbit immunoglobulin G (lgG) or a-CycD1. IPed material was incubated with 50 uL 30%of Protein A/G for 2 hours, followed by two 1x PBS washes and 1 kinase buffer wash. Pellets were resuspended in kinase buffer, and 15 pL samples were incubated with50 ug of either no peptide, 780S, or 780A peptide, along with 2 pL of [y-32P] ATP. Some samples were also treated with Fascaplysin ([Fascap.]1 juM). All samples incubatedfor 1 hour, followed by dotting onto Whatman glass microfiber filters and drying for 30 minutes. Filters were incubated in 1x TE buffer with gentle agitation for 2 days, dried,and then quantified with a scintillation counter. Background levels of CycD1 kinase activity (o-CycD1 IP with 780A peptide substrate) are indicated by black bar. (C)Log phase293T and V2CH cells were harvested for separation of cytoplasmic (Cyt) and nuclear (Nuc)compartments with the NE-PER Nuclear and Cytoplasmic Extraction Reagent Kit(Thermo Fisher Scientific).Western blot analysis for levels of VP40, CHMP6, HDAC1, and Actin was performed. (D) Log phase 293T and V2CH cells (3×106) were harvestedand cross-linked with 1% formaldehyde for 1 hour followed by quenching with 1.25 glycine (9:1 cell suspension/glycine). Samples were sonicated, and 100 uL of each sample(corresponding to approximately 5×10 cells)was used for IP with 1 ug of anti-Pol ll, -p300, or o-VP40 at 4°C overnight. The next day, Protein A/G (30% slurry) was addedand incubated for 2 hours at 4°C. Complexes were washed once with each TNE300 +0.1% NP40, TNE150+0.1% NP40, TNE50 + 0.1% NP40, and IP wash buffer beforethe addition of proteinase K (800 units/mL). Samples were incubated for 15 minutes at 65C, reversing solution was added, and samples were incubated for an additional90 minutes at 65C. DNA was purified, and qPCR was performed with 2 uL of undiluted DNA with primers for the CycD1 promoter region (spanning-180 to +238 from themessenger RNA start site at+1). The absolute quantification of the samples was determined based on the cycle threshold value relative to the standard curve generated fromserial dilutions of DNA from 293T cells. Data are presented as percentage (%) of the input (DNA purified from sonicated samples before IP with specific antibodies). Student’s2-tailed t test compares V2CH ChlPed DNA with corresponding 293T ChIP samples (**, P<.01). levels of VP40 in the nuclear fraction were nearly equivalentwith those seen in the cytoplasmic compartment, representinga much higher proportion of nuclear VP40 than we had antic-ipated. Therefore, we hypothesized that VP40 may potentiallyact as a transcription factor for the cyclin D1 promoter to elicitincreased levels of cyclin D1, thereby accelerating the growth of host cells. RNA Polymerase II (Pol II) is found on the pro-moters of actively transcribed genes, and p300 has previouslybeen shown to be involved in the activation of transcriptionof cyclin D1 and other mitogen-induced genes involved in cellgrowth and differentiation [77, 78]. Along these lines, to ascer-tain whether Pol II, p300, or VP40 were potentially involved in the transcriptional upregulation of cyclin D1, we performedChIP analysis of the cyclin D1 promoter in both 293T andV2CH cells. Data in Figure 2D show that in 293T cells, back-ground levels of Pol II,p300, and VP40 were <3% of the DNAinput (2.8%,2.6%, and 2.7%, respectively). The background lev-els of these, particularly VP40, may be partially attributed tononspecific binding of proteins during the IP procedure duringChIP. Interestingly, Pol II on the cyclin D1 promoter more thandoubled to 5.9%, whereas p300 increased more than 4-fold to11.11% of DNA input in V2CH cells. Even more surprising,VP40 was present on the cyclin D1 promoter in V2CH cellsat an astounding 38.4% of DNA input. Collectively, these datasuggest that VP40, Pol II, and p300 are present at elevated lev-els on the cyclin D1 promoter in cells producing VP40, whichmay result in the upregulated transcription of cyclin D1. Thisin turn may be responsible for the accelerated cell cycling ofVP40-producing cells. Additional consequences ofthis abun-dance of nuclear VP40 are likely, which should be the focus offuture studies. Altered Growth Dynamics Occur in Other Cell Types Producing VP40 To determine whether production of VP40 resulted in upreg-ulated growth in other cell types, we transfected U937 mono-cytes and HeLa cells with VP40-producing plasmid usingattractene reagent alongside attractene-treated controls (noplasmid) for 3 days. Cells were then analyzed for differences inviability. Data in Figure 3A show that transfection with VP40had little effect on U937 cells, whereas HeLa cells were signifi-cantly increased in viability. Next, to explore the effect of VP40production in primary cells, PBMCs were transfected withCMV-VP40 for 3 days with attractene reagent, followed byanalysis of cell viability. Results in Figure 3B show one repre-sentative PBMC sample, as well as the pooled data for PBMCstested in biological triplicate (PBMC×3). For all PBMCs tested,an increase in cellular viability was observed upon transfec-tion with VP40 in comparison with control-treated cells.Collectively, these data point to an increased rate of growth insome cell types other than 293T, including primary cells, andthereby potentially indicating a cell type-dependent effect ofVP40 on growth regulation. VP40 Alters Exosome Biogenesis in a Cell Cycle-Dependent Manner We previously showed that exosomes from V2CL cells areenriched in specific markers CD63 and Alix, whereas donorcells contain higher levels of ESCRT proteins [12, 13]. Therefore,we asked whether the altered cell cycle dynamics in VP40 donorcells could impact EV production. Control 293T and V2CL cellswere blocked at G , G,/S, and G,/M stages of the cell cycle andanalyzed for levels of ESCRT proteins and exosomal markers(Figure 4A). Side-by-side analyses of EV contents and AChEactivity of the exosomes produced from cell cycle blocked cellswere likewise performed (Figure 4B and C). Data in Figure 4A Figure3. Alteration of cell viability by VP40 in multiple cell types. Cells including(A) U937, HeLa, and (B) 3 peripheral blood mononuclear cells (PBMCs) log-phasecultures (~1.65×105 cells) were transfected with attractene and 1.5 ug of cyto-megalovirus-VP40 plasmid. The PBMCs received a 1-time treatment of 50 IU/mLof interleukin-2 the day before transfection. Control cells received attractene treat-ment alone. Cell viability was assayed 3 days post-transfection. Statistical analysisby Student's 2-tailed t test compares control cells with transfected cells (*, P<.05;** P<.01). Intracellular Figure 4. Differential biogenesis of exosomes at different phases of the cell cycle. 293T and V2CL cells were blocked at G, (starvation; 0.1% fetal bovine serum DMEM),G,/S (20 mM of hydroxyurea), and G,/M (18-hour 20 mM of hydroxyurea pretreatment, followed by release for 1 hour and subsequent treatment with 50 ng/mL of nocodazole)for5 days. Control (unsynchronized) cells were also incubated for 5 days. Black arrows point to bands of noticeable difference between V2CL and 293T cells. (A) Blocked cellsand controls were harvested, washed twice in 1x PBS, and lysed. Samples were run on a 4-20% Tris-glycine gel and analyzed by western blot for the presence of ESCRTpathway proteins (VPS4, EAP45, TSG101,CHMP6,and EAP20), exosomal markers (Alix and CD63), and Actin. (B) Cell-free supernatants from blocked cells were harvestedand passed through a 0.22-um filter. One milliliter of filtered supernatant was incubated with 30 uL of NT80/82 particles overnight at 4°C. The next day, the NT pellet waswashed once in 1x PBS and resuspended in 10 uL Laemmli buffer, followed by SDS/PAGE and western blot analysis for VP40 protein, exosomal markers CD63 and Alix, andActin. (C) One milliliter of filtered supernatant from blocked and control cells was incubated with 30 pL of NT80/82 particles overnight at 4C. The next day,the NTpelletswere isolated, washed,and subjected to AChE assay for quantification of exosomes. Statistical analysis was completed by Student's 2-tailed ttest (*, P<.05;***,P<.001). Analysis of exosomal markers from cell cycle-blockedV2CL cell EVs revealed an increase in all forms of CD63at G compared with 293T cells (Figure 4B). When test-ing for AChE activity (indicative of exosomal abundance),the total number of exosomes from unsynchronized (con-trol) 293T vs V2CL cells was not significantly different(Figure 4C). However, it was interesting to note that at G,the number of exosomes was reduced by >1 log comparedwith unsynchronized control exosomes for both cell types.This finding was corroborated by the distinct reduction ofmost ESCRT (Figure 4A) and exosomal proteins at G,com-pared with levels at G,/S and G,/M phases (Figure 4A and B).Both cell types had similar levels of exosomes produced whenblocked at G,/S compared with unsynchronized (control) cells;however, V2CL cells produced significantly more exosomes atG,/M compared with control, whereas 293T cells did not. Inaddition, V2CL cells produced significantly fewer exosomes atG, and at G,/S phases compared with 293T cells at the same phases (Figure 4C). Together, these results suggest that exo-somes and EVs are produced in a cell cycle-dependent manner,with cells at G, phase producing significantly fewer vesicles.Moreover, cells producing VP40 may have an altered patternof cell cycle-driven exosomal production, as demonstrated bydifferential patterns of ESCRT and exosomal marker proteinexpression. Next, we wished to more closely examine the effects of cellcycle blocks on EV production in cells producing VP40 at lowand high levels. 293T, V2CL, and V2CH cells were blockedat G, G,/S, and G,/M phases for 5 days, followed by har-vesting of the cell culture supernatants, filtration (through0.22 um), and analysis of the resulting EVs by ZetaView.Data in Figure 5A display the peak diameter size (mode) ofvesicles released.V2CL cells had significantly larger vesiclesproduced at G,compared with 293T cells, whereas V2CHcells had larger EVs when not only blocked at G /S andG,/M phases, but also in untreated cultures when compared B Figure 5.EExtracellular vesicles released by VP40-producing cells at different phases of the cell cycle. 293T, V2CL, and V2CH cells were blocked at G, (starvation; 0.1%fetal bovine serum DMEM), G,/S (20mM of hydroxyurea),and G,/M (18-hour 20 mM of hydroxyurea pretreatment, followed by release for 1 hour and subsequent treatmentwith 50 ng/mL of nocodazole) for 5 days. Supernatants were harvested, filtered (0.22 um), and analyzed by ZetaView for size (peak [mode] diameter) (A) and concentration ofparticles (B). Statistical analysis by Student's 2-tailed ttest compares V2CL and V2CH cell cycle-blocked groups with corresponding 293T groups (t, P<.05; tt, P<.01; ttt,P<.001). Additional Student's 2-tailed ttest compares cell cycle-blocked groups with controls of their own cell type(*, P<.05; **, P<.01;***,P<.001). with 293T cells. In addition, larger vesicles were produced apoptotic bodies [16, 50, 79]. Second, in fractions 14.4-15.6,from all 3 cell types at G, in comparison with untreated con- CD63 was present in moderate amounts, whereas levels oftrols, although this difference was not consistently significant. CD9 and CD81 increased. This population was also consistentThe overall trend regarding EV concentration was that vesi- with an exosomal category containing viral particles demon-cles from V2C cells decreased in number in a VP40 level-de- strated in previous publications [16, 50, 79], although, in ourpendent manner when compared against 293T cell groups case, this group is likely to be exosomes that are larger in size(Figure 5B). Furthermore, amongst all cell types, the number or more dense than the smaller ones seen in fractions 8.4-12.0.of EVs present at G, phase were significantly reduced from Finally, the 16.8-18.0 fractions showed a distinct reduction ofunsynchronized (control) cultures. This finding supports CD63 but a drastic increase in CD9, CD81, and Actin. It isour earlier conclusions that exosomes may be produced at likely that these larger or denser vesicles are not true exosomeslower levels when cells are resting at G, (Figure 4B and C). but either microvesicles or apoptotic bodies. In contrast,It should be noted that ZetaView analysis detects the total V2CI cells had a noticeably different distribution of exoso-number of vesicles or large particles present, with exosomes mal markers, although 3 distinct populations were still pres-comprising only a fraction ofthese. Therefore, to determine the ent (Figure 6B). Fractions 8.4-9.6 were almost absent in CD9distribution of true exosomes containing VP40, we used our and CD81, although CD63 was still present in higher amountspreviously published strategy of exosome characterization by compared with background. In addition, a shift to the rightdensity gradient ultracentrifugation [50]. Through this meth- of the major CD63*CD9*CD81* population from fractionsodology we have shown that it is possible to clearly separate 10.8-12.0 in 293T EVs to fractions 12.0-14.4 was observed.out exosomes from potentially contaminating viral particles, Finally, a population with high levels of CD9 and CD81 andthus making more detailed characterizations of specific EV low levels of CD63 was seen from fractions 15.6-18.0, similarpopulations possible. Here, we grew control 293T and V2CI to 293T cells. VP40 protein, although present in all fractionscells for 5 days, harvested the supernatants, and incubated the to various extents, was seen to be enriched in each of the 3 ves-unfiltered supernatants in ExoMAX, followed by separation of icle populations, especially in fraction 18.0. This abundanceresulting extracellular material through iodixanol density gra- of VP40 in fraction 18.0 is likely to be due to the presencedient fractionation. Fractions were subjected to western blot of VLPs, beecause there was no filtration through 0.22 um ofanalysis for exosomal marker proteins and VP40. Results in the supernatants before vesicle enrichment and fractionationFigure 6A show that in 293T cells, CD63*CD9*CD81+ (exo- in this experiment. Altogether, these results suggest that cellssome) fractions fell into 3 distinct categories. First, fractions producing VP40 release fewer EVs that are slightly larger8.4-12.0 contained high levels of CD63 and moderate levels in size, and that these cells in general release significantlyof CD9 and CD81. Previous publications have classified this fewer (although larger) exosomes at G. Furthermore, out-group as true exosomes, distinct from viral particles and side of VLPs and virions, VP40 is released in EVs (primarily B V2CI Figure 6. lodixanol gradient separation of extracellular vesicles (EVs) from 293Tand VP40-producing cells. 293T and V2CI cells were grown in exosome-free mediafor 5 days, followed by harvesting of the supernatant and incubation with ExoMAX(1:1 reagent/filtered supernatant) reagent overnight at 4°C. The EVs were pelleted,resuspended in 300 uL of sterile 1x PBS, and loaded onto a 6-18% iodixanol den-sity gradient (1.2% increments). Samples were ultracentrifuged for 90 minutes at100000 xg, followed by harvesting and isolation of each fraction, and incubationwith 30 pL of NT80/82 particles overnight at 4℃. The NT pellets were washed in1x PBS, resuspended in 12 uL of Laemmli buffer, and loaded onto a 4-20% Tris-glycine gel. Western blot of 293T (A) and V2CI (B) fractions were analyzed for levelsof VP40, CD63, CD81, CD9, and Actin. Major groups of EVs or exosome type areindicated by black boxes. exosomes) consistent with our previous observations [12, 13],but it may also be released in other larger vesicles and poten-tially as free protein or within protein complexes. Filtration Effectively Removes Virion Particles From Samples, LeavingViral Protein-Containing Extracellular Vesicles Our rationale for utilizing 0.22-um filters in our experimentswas based upon the knowledge that Ebola virions and VLPsare commonly 1-2 um in size or larger, are rarely straight,and therefore should not be able to effectively pass througha 0.22-um filter [36, 37, 39, 42, 80]. In fact, Beniac et al [80]previously showed that approximately 53% of infectious viri-ons were 982 ± 79 nm in length, with all other virions occur-ring in multiples of that size. No smaller virions containing complete genomes were observed in their investigation of over2000 virions by electron microscopy. On the other hand, EVsand exosomes below 220 nm in diameter will be able to passthrough 0.22-um filtration for downstream analysis. To test thisconcept, we used VLPs containing VP40, NP,and GP EBOVproteins. A simple filtration (0.22 um) of VLPs spiked into 1 mLPBS is shown in Supplementary Figure 2A, alongside filteredPBS alone (mock) and unfiltered VLP controls. Filtered sam-ples were incubated with NT80/82 particles and processed forwestern blot analysis of EBOV proteins. The results indicate thatfiltration of VLPs in small volumes drastically reduced the levelsof EBOV proteins present to undetectable levels. Next, to further characterize the distribution of VLPs incomparison to EVs by size, we spiked 10 ug of VLPs into10 mL of V2CH supernatants. VLP-spiked supernatants werethen either filtered (0.22 um) or left unfiltered and incubatedwith a vesicle-enrichment agent (ExoMAX). The resultingmaterial was then loaded on top of qEV (IZON) size exclu-sion columns, and fractions were collected corresponding tothe column void volume (fraction nos. 2-6) and those knownto contain vesicles larger than 70 nm in diameter (fractionnos. 7-11). Fractions were next incubated with NT80/82 par-ticles to concentrate material, followed by western blot analy-sis for viral proteins, exosomal markers, and Actin. Results inSupplementary Figure 2B show that, unsurprisingly, proteinsoriginating from unfiltered VLPs mostly appeared in the firstfraction after the void volume of the column (fraction no. 7),with some overlap into the next 1-2 fractions. This was shownby the strong presence of NP and GP, particularly the largerglycosylated form of GP, and the low levels of Actin, whichwe have not observed to be present in our control VLP butis present in EVs (Supplementary Figure 2B, compare lane1 to lanes 9-12). However, upon filtration through 0.22 um,approximately all EBOV proteins vanished from fractionnumber 7, and there was a sizeable reduction in the levels ofGP and NP in fraction numbers 8 and 9. Notably, not all NPsand GPs(originating from VLPs) were eliminated from thefractions after filtration. Another interesting observation centered around the visi-ble forms of GP. Specifically, in the unfiltered fractions, withthe higher fraction numbers (corresponding to smaller vesi-cles), the larger glycosylated form of GP decreased while thelevels of lower molecular weight forms of GP increased. In thefiltered fractions, the glycosylated larger form of GP was pres-ent only at very low levels, whereas lower molecular weightGP levels were relatively high. In comparison to the VLP con-trol (lane1), the predominant form of GP present was theglycosylated form. Together, these results suggest that glyco-sylated GPs may be the principal form within VLPs, whereasthe lower molecular weight forms of GP may be more likely tobe associated with EVs. Collectively, these data may indicate that VLPs appear mainly in the larger fraction number 7 andto a lesser extent in fraction numbers 8 and 9, whereas EVsand true exosomes will be present in fraction numbers 8-11. VP40 Is Present at Varying Levels in Extracellular Vesicles In Vitro andIn Vivo We have previously shown that exosomes from VP40-producingtransfected cells contain VP40 in vitro [12, 13], but, to ourknowledge, VP40 has never been shown to be present in EVsin the context of full viral infection. To address this, we infectedHUVECs with EBOV/Kik (MOI of 1) under BSL-4 contain-ment for 3 days, followed by harvesting of the supernatants,filtration through 0.22 um, and incubation with ExoMAX.Enriched vesicles were then loaded onto qEV size exclusion col-umns. Fraction numbers 7-10 were then collected, incubatedwith NT80/82 particles, and analyzed by western blot for EBOVproteins VP40, NP, and GP, and Actin. Results in Figure 7Ademonstrate that VP40, NP, and GP were all present in theEVs concentrated from EBOV-infected cells. In addition, all 3EBOV proteins were observed to be most highly concentratedin fraction number 8, followed closely by fraction number 9,which we had shown to be the fractions where most EVs andexosomes appear (Supplementary Figure 2B). VP40 and NPwere also present in fraction number 7, whereas GP was presentat very low levels in this fraction. It is possible that this fraction Figure 7. The presence of VP40 in exosomes in in vitro and in vivo EBOV-infectedcells. (A) HUVECs were cultured and infected with EBOV (MOI of 1) and incubatedfor 3 days under BSL-4 containment. Two milliliters supernatant were harvested,passed through a 0.22-um filter, and incubated with ExoMAX (1:1 reagent/filteredsupernatant) reagent overnight at 4°C. EVs were pelleted, resuspended in 0.5 mL1x PBS, and loaded on qEV columns. Fraction numbers 7-10 (0.5 mL each) werecollected and separately incubated with 30 pL NT80/82 at room temperature for1 hour. The EV-bound NTs were washed with 1x PBS, followed by resuspension in10 uL 2x NuPAGE LDS sample buffer, heating at 95℃ for 10 minutes, and loadingonto a 4-12% Tris-glycine gel for subsequent western blot analysis for VP40, GP,NP, and Actin levels. Negative control (Null) samples consisted of purified exosomesfrom uninfected HUVECs.(B) Gamma-irradiated and inactivated NHP (rhesus mon-key) serum samples were obtained. NHP 1: day 0 prebleed sample. NHP 2: pool ofday 4 and day 5 postinfection (pi); NHP 2 died on day 7 post-EBOV infection. NHP3: pool of day 8-11 pi; NHP 3 died on day 12 post-EBOV infection. One hundredmicroliters of serum were diluted with 400 uL sterile 1x PBS and filtered (0.22 um).Twenty-five microliters NT80/82 particles were incubated with the filtered samplesat 4°C overnight. The next day, NT pellets were washed once in 1x PBS, resus-pended in 12 uL Laemmli buffer, run on 4-20% SDS/PAGE, and analyzed by west-ern blot for VP40 protein and exosomal markers CD81 and CD9. may represent slightly larger vesicles that do not incorporateGP. All 3 proteins were much less abundant in fraction number10, which contained smaller vesicles. Actin followed a similartrend, with the most Actin present in fraction numbers 8-9,and little to no detectable protein present in fraction numbers7 or 10. Collectively, these results indicate that not only VP40but GP and NP are also packaged into vesicles in cells activelyinfected with EBOV. To take this one step further, we next used inactivated NHPserum samples from 2 infected macaques and 1 uninfectedprebleed animal. Inactivated samples were filtered (0.22 um),EVs were concentrated with NT80/82 particles, and sampleswere analyzed by western blot for levels of VP40 and exoso-mal proteins. Data in Figure 7B show that exosomes from all3 NHP serum samples were concentrated, as demonstratedby the presence of CD81 and CD9 tetraspanins. In addition,VP40 appeared at fairly high levels in the 2 serum samplesfrom infected animals. Quantitative densitometry was usedto calculate the approximate amounts of VP40 present in ourV2CH cell EVs and how much was released from infectedNHP samples. V2CH cells released approximately 13.7 ng ofEV-associated VP40/mL, whereas NHP 2 and NHP 3 released~319 and ~206.5 ng of EV-associated VP40/mL serum,respectively. In addition, we determined that VP40 composedon average approximately 55.8% of purified VLPs (containingGP, NP, and VP40; data not shown). It is interesting to notethat more VP40 was present from NHP 2 serum, which suc-cumbed to infection sooner (day 7 vs day 12) and whose serumwas collected at earlier time points in infection in comparisonwith NHP 3 (days 4-5 vs days 8-11). Therefore, these resultsindicate that VP40 is present in EVs from in vivo EBOV infec-tion, with higher levels potentially being prevalent duringearlier time points or with more robust infection. Althoughwestern blot confirmed the presence of captured exosomes, wecannot be certain that we did not capture other EVs or freeproteins in our analysis of NHP samples. Therefore, it cannotbe ruled out that VP40 may be present in not only exosomes,but also a combination of EVs and/or as a free protein in vivo.Taken together, these results indicate that relatively high levelsof VP40, NP, and GP, are present in EVs from cells infectedwith EBOV both in vitro and in vivo. VP40 in Extracellular Vesicles Induces Apoptosis in Recipient T Cells andMonocytes During EVD, bystander T cells mainly die by apoptosis,whereas monocytes/macrophages appear to die by necrosis[5, 7, 9, 10, 81-83]. We have previously shown that EVs fromV2CL cells can induce death in recipient immune cells [12, 13].Therefore, to test the ability of recipient cells to withstand lowversus high levels of EV-associated VP40, we incubated CEM,Jurkat, U937, and 293T cells with 5-day filtered supernatantsfrom 293T,V2CL, and V2CH cells, followed by measurement Figure 8. Recipient monocyte and T-cell apoptosis. (A) Supernatants from 293T (Control), V2CL, and V2CH cells (5 days, 0.22 um filtered, in exosome-free media) were usedto treat CEM, Jurkat, U937, and 293T recipient cells. Recipient cells were seeded in a 96-well plate at 5×10 cells in 50 uL of fresh media, treated with 50 uL of filteredsupernatants, and incubated 5 days, followed by analysis by CellTiter-Glo for cell viability. Statistical analysis by Student’s 2-tailed ttest compares supernatant-treated groupswith control cells of the same type (*, P<.05; **, P<.01;***,P<.001).293T, V2CL, CEM, Jurkat, and U937 cells were grown in exosome-free media for 5 days, followedby harvesting and filtering (0.22 um) of the supernatant. Log-phase CEM, Jurkat, and U937 cells were plated at a density of 1×106 cells/mL and treated with increasingconcentrations (100, 250, and 500 uL)of supernatant from either 293T, V2CL, or their own cell type. Cells were incubated for 5 days, followed by harvesting of the cells andlysis. Lysates of CEM (B), Jurkat (C), and U937 (D) cells were then run on a 4-20%Tris-glycine gel and subjected to western blot analysis for apoptotic markers procaspase3 and PARP-1 and their cleaved forms, and Actin. of cell viability 5 days later.Results in Figure 8A show that CEMand Jurkat T cells had a similar reduction in cellular viabilityfrom treatment with supernatants from both V2CL and V2CHcells. However, treatment of U937 monocytes with supernatantsfrom these 2 lines showed a dose-dependent reduction in cel-lular viability. These data suggest that the T cells were equallysensitive to both low and high levels of extracellular VP40,whereas monocytes were more sensitive to higher doses ofEV-associated VP40. It is interesting to note that when 293Tcells were incubated with supernatants from VP40-expressingcells, 293T cells showed a significant increase in viability, poten-tially indicating a cell-type dependent mechanism of cell deathinduction by EV-associated VP40. The mechanism of cell death under these conditions hasnot been previously explored. Along these lines, we incubatedrecipient CEM and Jurkat T cells and U937 monocytes withincreasing concentrations of EVs from V2CL cells, EVs fromcontrol 293T cells, and EVs from their own cell type that wereproduced in the absence of VP40. Western blots were performedfor cell death markers including Caspase 3 and PARP-1. Data inFigure 8B-D show that treatment with increasing concentra-tions of EVs from V2CL cells induced cell death by apoptosis, asshown by cleavage of Caspase 3 and PARP-1, in recipient T cells and monocytes. This phenotype was not observed with 293Tcell EV treatment or EVs from the same cell type. It should benoted that very high levels of EVs from 293T cells stimulatedsome low levels of PARP-1 cleavage in all cell types; however,there was no cleavage of Caspase 3 detected. Together, thesedata indicate that EVs from VP40-producing cells cause deathby apoptosis in recipient T cells and monocytes. Because our V2C cells were kept under constant anti-biotic selection, we wished to verify that induction of celldeath in recipient cells was not due to residual hygromycinB within the 5-day supernatants. Therefore, 293T and V2CHcells were grown for 5 days in exosome-free media, followedby harvesting of the supernatants, filtration (0.22 um), andultracentrifugation. The resulting EV pellets were then resus-pended in sterile PBS and used to treat recipient CEM cellsin increasing concentrations. Cells were treated for 3 daysfollowed by analysis of cell viability. Data in Figure 9A showthat in comparison with CEM cells treated with the highestconcentration of 293T EVs (75 000 particles/cell),there wasa significant dose-dependent decrease in recipient cell via-bility when treated with purified EVs from V2CH cells. Thisfinding further verifies that EVs from V2C cells negativelyimpact recipient immune cells. Figure 9. Induction of recipient T-cell death by purified VP40 EVs.(A) 293T andV2CH cells were grown in exosome-free media for 5 days, followed by harvest-ing of cell-free supernatants and filtration through 0.22 um. Supernatants werethen spun at 100 000 xg for 90 minutes to pellet EVs, followed by resuspension insterile 1x PBS. Concentrations of resulting ultracentrifuged EVs were determinedwith ZetaView analysis, followed by treatment of CEM cells with increasing con-centrations of EVs (10000, 25000, or 75000 particles/cell) from V2CH cell type.Controls included CEM cells that were left untreated and CEM cells that receiveda treatment of the highest concentration of 293T cells (75000 particles/cell). Cellswere incubated for 3 days followed by analysis of cell viability by CellTiter-Glo.(B) 293T and V2Cl cells were grown in exosome-free media for 5 days. Cell-freesupernatants were harvested and incubated with equal volumes of ExoMAX over-night at 4°C. The EVs were pelleted, resuspended in 400 uL sterile 1x PBS, andloaded onto a 6-18% iodixanol density gradient (1.2% increments). Samples wereultracentrifuged for 90 minutes at 100 000 ×g, followed by harvesting and isolationof each fraction. Select fractions (13.2+ 14.4 and 16.8+ 18.0) were pooled andsubjected to a second ultracentrifuge spin for 90 minutes at 100000 xg dilutedin 1x PBS to pellet the EVs away from residual iodixanol. Resulting EV pelletswere resuspended in 100 uL sterile 1x PBS and used to treat recipient CEM cellsat a concentration of 10000 particles per cell (concentrations determined byZetaView analysis). Cells were incubated for 5 days followed by analysis of viabil-ity by CellTiter-Glo assay. Statistical analysis by Student's 2-tailed ttest comparesgroups treated with EVs from V2C cells to those treated with EVs from 293T cells(*,P<.05; ***,P<.001). Given our previous characterization of EVs from V2CI cellsin comparison with 293T cells (Figure 6), we next wished todetermine which EV fractions were responsible for the induc-tion of cell death in recipient cells. Supernatants from 293T and V2CI cells were harvested and separated through an iodixanolgradient, followed by pooling of select fractions from each celltype. Pooling of fractions 13.2+ 14.4 and 16.8+18.0 were cho-sen because, in V2CI cells, these were the 2 groups with thehighest levels of VP40 and exosomal markers. Pooled fractionswere then diluted in sterile PBS and subjected to a second ultra-centrifuge spin to isolate the EVs away from residual iodixa-nol. The resulting EV pellets were then resuspended in PBS andused to treat recipient CEM cells at a concentration of 10000particles/cell for 5 days, followed by analysis of cell viability.Treatment with EVs from the 13.2 + 14.4 fractions from V2CIcells resulted in a significant decrease in viability in CEM cellsin comparison with those treated with 293T EVs from the samefractions (Figure 9B). On the other hand, T cells treated withthe 16.8 + 18.0 fractions from either cell type did not differ intheir viability. Collectively, these data may signify that the EVswithin the 13.2 and 14.4 fractions from V2C cells can inducecell death in recipient immune cells. Cdk4/6 Inhibitor Treatment of VP40-Producing Cells Affects ExtracellularVesicle Release Our data showed that EVs from VP40-producing cells werecapable of causing apoptosis in recipient immune cells (Figure 8)and that the biogenesis of these EVs was potentially tied to reg-ulation of the cell cycle (Figures 4 and 5). Furthermore, wedemonstrated that accelerated cell cycle was potentially drivenby the upregulation of cyclin D1 by nuclear VP40 (Figure 2).Therefore, we next investigated the utility of cdk4/6 inhibitorsFascaplysin and Ribociclib (LEE011; FDA-approved) in reduc-ing EV production from 293T andV2CH cells. Cells were treatedfor 5 days with increasing concentrations of Fascaplysin (0.1,0.5,and 1.0 uM) and Ribociclib (0.1, 1.0, and 10.0 uM), followed byanalysis of cell viability and EV concentration. When 293T cellswere treated with low levels of either inhibitor, the number ofEVs released significantly decreased. However, with increasingconcentrations of drugs, the production of vesicles increasedin a dose-dependent manner (Figure 10A). These results maybe due to the toxicity of the drugs at high levels, as the viabilityof both 293T and V2CH cells were significantly decreased withincreased doses of either drug (Figure 10B). More specifically,low levels of cdk4/6 inhibitors in 293T cells may result in theblocking of cell cycle at G,, and thereby result in lower levels ofEVs released, whereas higher levels of the drugs may becometoxic, and result in the increased release of vesicles. In contrast,V2CH cells showed significantly decreased levels of EV pro-duction with all doses of Fascaplysin, although with the highestdose EV production decreased less significantly in comparisonto the lowest dose tested (Figure 10A). These EV reductionsoccurred despite significant decreases in cell viability in V2CHcells (Figure 10B). Ribociclib treatment of V2CH cells showedless significant results compared with Fascaplysin treatment,with only the highest dose resulting in any significant reduction B Drug-Treated Cell Viability Control □DMSO Low Medium Fascaplysin Ribociclib Figure 10.Effect of cdk4/6 inhibitors on VP40-producing cell viability and extracellular vesicle biogenesis.293T and V2CH cells were treated with low, medium, and highconcentrations of Fascaplysin (0.1, 0.5, 1.0 uM) or Ribociclib (0.1, 1.0, 10.0 pM) for 5 days. DMSO was also used at a final concentration of 1%. (A) Supernatants wereharvested, filtered (0.22 um), and analyzed by ZetaView for concentration of particles. (B) Cell viability was also measured by CellTiter-Glo. Statistical analysis by Student’s2-tailed ttest compares drug-treated groups to untreated controls of their own cell type (*,P<.05;**, P<.01;***P<.001). ofEV numbers or cellular viability (Figure 10A and B). Together,these results suggest that Fascaplysin treatment results in fewerEVs from V2CH cells compared with 293T cells with higherdoses, although high doses also result in significant decreases inviability in both cell types. In addition,Ribociclib treatment mayimpact the cell viability and EV biogenesis of VP40-producingcells less than 293T cells. The indication that 293T cells react toboth cdk4/6 inhibitors in a similar way, but V2CH cells do not,hints that these drugs may impact cell cycle regulation and EVbiogenesis pathways in distinctly different ways. Although themechanism of action for Fascaplysin is currently uncharacter-ized, Ribociclib is known to interact directly with cdk4 and cdk6molecules at their ATP pocket [84, 85]. Future study into the divergent mechanisms used by these drugs and how they mayinteract with pathways modulated by VP40 may potentially beadvantageous to determine the potential therapeutic utility ofcdk4/6 inhibitors in infection. Extracellular Vesicles From VP40 Donor Cells Contain CytokinesAssociated With Ebola Virus Disease Pathogenesis We previously showed that exosomes from VP40-producing cellsare capable of inducing cell death in recipient T cells and mono-cytes [12, 13]. We also confirmed here that the mechanism of celldeath is by apoptosis (Figure 8). Along these lines, we attemptedto gain insight into proteins contained in VP40 exosomes thatmay potentially point to a mechanism for the induction of cell death in recipient cells. 293T, V2CL, and V2CH cells werecultured for 5 days in exosome-free media. Supernatants wereharvested, filtered (0.22 um), and incubated with ExoMAXto enrich for EVs. Samples were then loaded onto a 6%-18%OptiPrep iodixanol gradient, and 10.8 and 12.0 fractions wereharvested for use. We chose the 10.8 and 12.0 fractions becauseprevious publications have shown that these fractions are spe-cifically enriched for exosomes, with fewer contaminating ves-icles or viral particles [16,79]. Fractions were then incubatedwith NT80/82 particles and processed for analysis of tandemmass spectra. The data presented in Supplementary Table 1 showthe proteins significantly upregulated in V2C EVs, with proteinscores from 10.8 and 12.0 fractions for each cell type summated.Overall, there was an interesting trend gleaned from the numberof proteins identified from each sample. The 10.8 fractions from293T, V2CL, and V2CH cells had 826, 537, and 261 detectedproteins, respectively, potentially showing an overall decreasein the number of proteins present with increase in VP40 pro-duction levels (data not shown). However, for the 12.0 fraction,the opposite trend was seen: 293T, V2CL, and V2CH EVs had387, 1393, and 1680 proteins identified, respectively (data notshown). These results could potentially indicate that the totalnumber of EVs had shifted from the 10.8 fraction more towardsthe 12.0 fraction for V2C cells, whereas 293T cells possibly pos-sessed more EVs in the 10.8 fraction in comparison to the 12.0. These data support our previous conclusions that the EVsfrom V2C cells are shifted more towards the right (ie, are moredense; Figure 6), and therefore they may contain a greater andmore diverse quantity of cargo. The presence of heat shockprotein (HSP)90, HSP70 and heat shock cognate 71 seen inabundance across all 3 cell types was indicative of all samplescontaining exosomes. A large increase in the number of pro-teins related to RNA binding was seen with increasing levels ofVP40. Specifically, numerous RNA binding proteins, includingheterogeneous nuclear ribonucleoproteins, were found in EVsfrom V2C cells, whereas only 1 was detected in 293T cell EVs.In addition, several DEAD-box protein (DDX)-type helicaseswere shown to be upregulated in EVs from VP40-producingcells. Other categories of interest that were upregulated with thepresence of VP40 included additional vesicle-associated, trans-lation-associated, nuclear or transcription-associated, protea-some or cell death-related, protein transport, and RNA splicingproteins. Of particular interest, 3 cytokines (vascular endothe-lial growth factor [VEGF], transforming growth factor beta-1[TGF-β1] precursor, and insulin-like growth factor II [IGF-II])and 2 corresponding binding proteins (latent-transforminggrowth factor beta-binding protein 1 precursor and insulin-likegrowth factor-binding protein 4 precursor) were found to bepresent in the EVs. It has been well documented that an eruption of cyto-kines, or a “cytokine storm,often accompanies fatal casesof EBOV infection1 [6, 8, 86, 87]. Specifically, cytokines including granulocyte-macrophage colony-stimulating fac-tor (GM-CSF), growth-regulated oncogene (GRO)-a, IL-1B,IL-2, IL-6,IL-8, IL-10, IL-15, IL-18, interferon (IFN)-a, -B,and -y, IFN-inducible protein-10 (IP10), Eotaxin, monocytechemoattractant protein (MCP)-1, macrophage-inflammatoryprotein (MIP)-1a and -1, macrophage colony-stimulatingfactor (MCSF), RANTES, TGF-B1, TNF-a, and VEGF have allbeen implicated as differentially up- or downregulated duringEBOV pathogenesis [8, 86-89]. This led us to question whetheradditional cytokines could potentially be present or upregulatedin VP40 exosomes or EVs. To this end, filtered 5-day superna-tants and pooled 10.8-12.0 exosome fractions from 293T andV2CH cells were run side-by-side on human cytokine arrays.Filtered exosome-free DMEM media was also used as control.Densitometry was then performed, and cytokine counts abovethe DMEM background levels were graphed in SupplementaryFigure 3. In the supernatants, some cytokines (ENA-78, IL-12,IL-15, RANTES, TNF-a, TNF-B, epidermal growth factor[EGF], IGF-1, and Oncostatin M) were present at higher lev-els in V2CH cells compared with 293T cells (SupplementaryFigure 3A). In contrast, 293T cell supernatants were slightlymore enriched in several different cytokines (GRO, GRO-a,I-309, IL-1a, IL-1B, and IFN-y). However, when cytokines fromthe enriched exosomal fractions were analyzed, an increase inalmost all cytokine levels was observed compared with thoseseen in whole supernatants (Supplementary Figure 3B vs A). Inparticular, V2CH exosomes possessed higher levels of ENA-78,GRO,IL-12,IL-15, IFN-y,MCP-1, MCSF, MIG, TARC, TGF-B1,TNF-B,EGF, and Oncostatin M and lower amounts of MIP-1 8and leptin compared with 293T exosomes. Additional cytokinesthat were undetectable in whole supernatants that were foundin exosomal samples included IL-2, IL-3, IL-4, IL-5, MCP-1, -2,and -3, MCSF, macrophage-derived chemokine (MDC), stemcell factor (SCF), angiogenin, platelet-derived growth factor(PDGF) BB, and leptin. This finding could potentially be due toour selective enrichment workflow, such that concentrations ofcytokine-associated exosomes were much higher than those inwhole supernatant samples and therefore more easily detectedby the assay. Cytokines such as granulocyte colony-stimulatingfactor (GCSF), GM-CSF, GRO-a,I-309,IL-1a,IL-1B,IL-2,IL-3,IL-4, IL-5,IL-8,IL-10,IL-13,MCP-2,MCP-3,MDC,RANTES,SCF, SDF-1,TNF-a, IGF-1, angiogenin, thrombopoietin, VEGF,and PDGF BB were present in both 293T and V2CH exosomesat similar levels. It is interesting to note that exosomes fromboth cell types were shown to possess VEGF,TGF-B1, and IGF-1, further supporting our MS results (Supplementary Table 1).Collectively, these data indicate that EVs, specifically exosomes,from VP40-producing cells may contain a diverse set of uniqueor upregulated proteins. In addition, a plethora of cytokinesare associated with vesicles released from virally affected andhealthy host cells. Moreover, specific cytokines may be con- centrated within EVs from cells producing VP40, which may potentially play a role in the cytokine storm associated withthe pathogenesis of EBOV, induction of cell death in recipientimmune cells, and poor outcome of infected individuals. DISCUSSION We previously showed that EBOV VP40 can become incorpo-rated into exosomes and thereby negatively impact recipient Tcells and monocytes [12, 13]. In line with this, we have addi-tionally shown here that VP40-containing vesicles can inducecell death by apoptosis in these recipient cells (Figure 8). Thiscell death was more exacerbated in monocytes treated with EVsfrom cells that produced higher levels of VP40. On the otherhand, EVs from cells that produced both low and high levelsof VP40 were equally destructive to T cells. Bystander T-celldeath by apoptosis is one of the hallmarks of EVD, especiallyfatal cases [5, 7, 9, 10, 81-83]. Furthermore, we determinedthat VP40 EVs that sediment within the 13.2 and 14.4 fractionsof iodixanol density gradients are able to significantly reducerecipient T-cell death, but EVs that fall in the 16.8 and 18.0fractions do not (Figure 9B). This finding may be noteworthy,because we believe the 16.8 + 18.0 fractions mainly containapoptotic bodies, large microvesicles, and VLPs in the case ofV2C cells, thus potentially indicating that these other extracel-lular particles may not be a significant contributor to the overalldecreased health of recipient immune cells. Therefore, VP40EVs of intermediate density may comprise a potential mecha-nism of bystander immune cell apoptosis in EVD. Interestingly,in contrast to immune cells, 293T (human embryonic kidneyepithelial) cells that were treated with EVs from both low andhigh V2C cells showed an increased cell viability. This couldpotentially be due to a cell type-dependent mechanism ofactionon recipient cells by VP40-associated vesicles. In addition,293T cells may be altogether resistant to VP40 EV componentsresponsible for cell death in immune cells and may converselybe positively stimulated by the vesicles. Further study into thespecific components responsible for these phenotypes in recip-ient cells is needed, but the resistance of certain cell types couldbe quite significant in the context of infection. Monocytes ormacrophages and dendritic cells are the first sustained targetof EBOV, andT cells die relatively early in infection; however,epithelial and endothelial cells are also productively infected bythe virus, albeit at later time points in infection [3-10]. If theseuninfected nonimmune cells (endothelial and epithelial) arebombarded with vesicles containing viral components such asVP40 during early infection, it is possible that the recipient cellsmay be stimulated and primed for future infection. This type ofmechanism has been seen before in the case of HIV-1 TAR, inwhich exosomes containing this viral RNA predisposed recipi-ent cells to be more receptive to infection [26]. Should this typeof pathogenic process take place, inhibition of vesicle biogenesis from infected cells should be further explored to determinepotential benefits in recipient cells. To determine pathways contributing to EV biogenesis indonor cells, we investigated the cell cycle of VP40-producingcells. Our rationale for doing these experiments was that whencells contained VP40, they replicated faster than control cells,leading us to ask whether cell cycle patterns were affected.While exploring differences in cell cycle dynamics with theVP40-producing clones, it is important to note that althoughour vectors have SV40 origin of replication and cells containT-antigen, it is possible that over time few copies of the plasmidcan be integrated into the host genome because these cells areRec+ and allow recombination. However, we have not observedany differences between these clones that cannot be attributedto a dose response of VP40 production and cell cycle effects.Here, V2C cells were observed to be altered in a cell cycle-de-pendent manner in not only their exosomal biogenesis pathwaycomponents (ESCRT proteins), but also in their morphology,growth patterns, and EV content and abundance (Figures 1,4,5, and 6). Moreover, V2C cells exhibited increased levels ofcyclin D1 protein and activity (Figure 2). Cyclin D is a welldescribed cell cycle regulator and a potential oncogene knownto be upregulated in several cancers [62-64]. In addition, cyclinD1 was previously shown to be produced at elevated levels as aresult of many viral infections, especially those known to causecancers[65-70]. Although EBOV has not been shown to resultin higher incidences of cancer in survivors, it is possible thatthis phenotype may be a transient one and only in effect fora brief time before other virulence factors and the replicationof the virus results in the infected cells'death. Because EBOVhas been shown to best thrive in actively growing cells [90],upregulation of cyclin D1 may simply be a strategy used by thevirus to aid its own replication. However, larger proportionsof recurrent, persistent, and asymptomatic EBOV infectionshave come to light with study of the increasing numbers ofEBOV survivors, which is a significant topic of concern, as thiscan potentially lead to transmission of virus and future out-breaks [91-100]. How the virus manages to remain long termwithin human and NHP hosts has not yet been conclusivelydetermined; however, viral persistence and replication withinCD68* monocyte or macrophage cell types has been detectedwithin the brain, eyes, and testes of NHP survivors of infection[101]. If these central nervous system-resident myeloid cellsare also dysregulated in cell cycle, this could potentially repre-sent a method used by EBOV to counteract its own cytotoxicityto support prolonged viral replication or latency. Previous studies have shown VP40 to be transiently presentin the nucleus of infected cells at early time points [72-74].Here, we have shown that VP40 is present at relatively highlevels within the nucleus of stably transfected cells, and thatVP40 is found on a large proportion of cyclin D1 promoters (Figure 2C and D). It may be possible that early in infection,VP40 translocation into the nucleus may serve a role in tran-scriptional regulation of host genes to best benefit the virus. Inall likelihood, nuclear VP40 action as a transcriptional regulatoris not limited to the cyclin D1 gene. Investigation into the directinteractions and far-reaching effects of VP40 in the nucleuscould be vitally important for elucidating the molecular basis ofEBOV pathogenesis in host cells. Cell cycle dysregulation of VP40-producing cells alsoresulted in an altered pattern of EV biogenesis (Figures 4 and 5).It is interesting that all cells tested at G produced significantlyfewer EVs that were also larger than unsynchronized con-trols. Because many eukaryotic cells are quiescent in normal,healthy, in vivo environments, this could represent an import-ant trend that inherently differentiates EVs from cells that arenot frequently dividing versus actively replicating or cancercells. Our results also showed that EVs from VP40-producingcells were generally fewer in number but denser and larger insize (Figures 5 and 6). Larger vesicles of higher density wouldimply an enrichment of cargo within VP40 EVs, which wassupported by a greater abundance of proteins detected by MSanalysis (Supplementary Table 1). The increased numbers anddiversity of proteins found in EVs from VP40-producing cellscould influence the outcomes of recipient cells, particularlyif those proteins were associated with nucleic acids. A collec-tion of RNA-binding proteins was found from MS analysisof EVs from V2C cells, and although we do not yet know theRNAs associated with these proteins, they may be importantfor activities in recipient cells. An assortment of cytokines wasalso found, with an upregulation of IFN-y, IL-15, MCP-1, andTGF-B1 seen in VP40 EVs (Supplementary Figure 3). That wewere able to detect these cytokines at elevated levels in exoso-mal preparations from VP40-producing cells may indicate thatVP40 stimulates the production of some of these, potentially asother direct or indirect targets of nuclear VP40 transcriptionalregulation. Moreover, the fact that we were able to detect cyto-kines in exosomal preps in all cell types is a significant findingin and of itself. In our study, the majority of cytokines testedwere enriched in exosomes compared with whole supernatants.If this is the case in vivo, vesicles heavily laden with cytokinesoriginating from infected cells could act as a “cytokine bomb"on recipient immune cells. In this way, a few EVs from a sin-gle diseased cell would be able to enact devastating effects ona recipient cell. This may also be of substantial importance formany other viral or inflammatory diseases in which cytokinesplay an inflammatory or damaging role. Due to the observed upregulation of cyclin D1 proteinand activity, we also explored the ability of Ribociclib andFascaplysin to inhibit cdk4/6 activity and thereby slow therelease of vesicles in a cell cycle-driven manner from VP40 cells(Figure 10). Results showed that Fascaplysin was able to sig-nificantly decrease the number of vesicles produced from both 293T and V2CH cells with low, nontoxic doses. In contrast,Ribociclib was only able to decrease levels of EVs producedfrom 293T cells at doses that did not affect cell viability. The dif-ferences between 293T and V2CH cell responses to the inhibi-tors suggest that VP40-producing cells may be less susceptibleto the effects of Ribociclib. Future mechanistic studies into themolecular action of these drugs in connection with VP40 mayelucidate potential therapeutic utility or other more directeddrug targets. We previously showed that EBOV VP40 is packaged intoexosomes in VP40-transfected cells [12, 13]. Here, we showthat VP40 is found within EVs isolated from both in vitroand in vivo wild-type EBOV infection (Figure 7A and B).Furthermore, EVs from EBOV-infected cells contained bothNP and GP proteins of the virus. NP is responsible for encapsi-dating the RNA genome of the virus as soon as it is synthesizedand for formation of the nucleocapsid (in conjunction withother viral proteins) [102]. Because NP is the main viral proteinresponsible for binding of the RNA genome (each NP is boundto 6 bases of RNA [103]), the presence of NP within EVs couldalso potentially hint at the presence of bound viral RNA withinEVs as well. On the other hand, GP is the sole surface proteinof EBOV and is important for both viral entry and egress [104].GP has also been implicated in pathogenesis, because GP itselfhas shown cytotoxic effects in infected cells. Furthermore, sol-uble GP is found in large quantities in the blood of infectedpatients, and it has been proposed to potentially play a rolein immune evasion by absorption of neutralizing antibodies[105, 106]. Our experiments involving filtration of VLPs fol-lowed by size exclusion separation of extracellular materialsshowed that VLPs were able to be removed from samples byfiltration, whereas smaller vesicles in the VLP preparations thatcontained EBOV proteins were able to pass through 0.22-umfiltration (Supplementary Figure 2B). These results also inter-estingly hinted towards a potential role for GP in EVs, as unfil-tered VLPs showed enrichment of primarily glycosylated GPin the fractions corresponding to larger vesicles (fraction num-ber 7), whereas filtered VLPs showed mainly lower molecularweight forms of GP, which colocalized with exosomal markers.Therefore, if EVs are studded with GP proteins, this could forman additional previously unknown source of immune subver-sion and viral masking used by EBOV. Presence of other viralproteins or RNAs in EVs were not explored here and will beanalyzed in future studies. CONCLUSIONS In conclusion, we show that VP40 dysregulated the cell cyclein donor cells through translocation to the nucleus and upreg-ulation of functional cyclin D1 protein. In turn, cell cycle wasshown to be a regulator of EV biogenesis. Extracellular vesi-cles from VP40 cells were also altered in their content,whichincluded RNA-binding proteins and cytokines important for EBOV pathogenesis. In addition, NP and GP proteins weredetected within EVs from EBOV-infected cells. Collectively,the cell cycle-related effects of VP40 within donor cells mayhave repercussions on the virulence and replication efficiencyof EBOV, the upregulation and packaging of proteins intoEVs, and the overall outcome of EV-recipient cells in infectedindividuals. Supplementary Data Supplementary materials are available at The Journal of Infectious Diseasesonline. Consisting of data provided by the authors to benefit the reader, theposted materials are not copyedited and are the sole responsibility of theauthors, so questions or comments should be addressed to the correspond-ing author. Notes Acknowledgments.is.We thank all members of the Kashanchi laboratory,especially Gwen Cox, Angela Schwab, and Yao Akpamagbo. We also thankLaura Bollinger for thorough grammatical editing of the manuscript. Disclaimer. The views and conclusions contained in this document arethose of the authors and should not be interpreted as necessarily represent-ing the official policies, either expressed or implied, of the US Departmentof the Army, the US Department of Defense, the US Department of Healthand Human Services, or of the institutions and companies affiliated with.the authors. Financial support. This work was funded by National Institutesof Health Grants AI078859, AI074410, AI127351-01,AI043894, andNS099029 (to F. K.) and R33 CA206937 and R01AR068436 (to L. A. L.).This work was supported in part through Battelle Memorial Institute'sprime contract with the US National Institute of Allergy and InfectiousDiseases under Contract No. HHSN272200700016I (to J. L., J. H. K.).Support was also kindly provided by the Defense Threat ReductionAgency (Grant CB4088; to J. M. D.). Potential conflicts offinterest. B. L.isemployed by CeresNanosciences, Inc. M. J. A. is the President and Chief Scientific Officerof Integrated BioTherapeutics, Inc. All authors have submitted the ICMJEForm for Disclosure of Potential Conflicts of Interest. Conflicts that theeditors consider relevant to the content of the manuscript have beendisclosed. References 1. Centers of Disease Control. Outbreaks chronology: Ebola virus disease| Ebolahemorrhagic fever. Available at: https://www.cdc.gov/vhf/ebola/outbreaks/his-tory/chronology.html#modalIdString_outbreaks. Accessed 9 March 2017. 2. World Health Organization. Ebola virus disease. Available at:http://www.who.int/mediacentre/factsheets/fs103/en/.Accessed 9 March 2017. 3. Feldmann H, Geisbert TW. Ebola haemorrhagic fever. Lancet 2011; 377:849-62 4. Martines RB, Ng DL, Greer PW, Rollin PE, Zaki SR. Tissue and cellular tro-pism, pathology and pathogenesis of Ebola and Marburg viruses. J Pathol 2015;235:153-74 5. Wauquier N, Becquart P, Padilla C, Baize S, Leroy EM. Human fatal Zaire Ebolavirus infection is associated with an aberrant innate immunity and with massivelymphocyte apoptosis. PLoS Negl Trop Dis 2010; 4:pii: e837. 6. MIessaoudi I, Basler CF. Immunological features underlying viral hemorrhagicfevers. Curr Opin Immunol 2015;36:38-46. 7. Bradfute SB, Braun DR, Shamblin JD, et al. Lymphocyte death in a mouse modelof Ebola virus infection. J Infect Dis 2007; 196(Suppl2):S296-304. 8. Rougeron V, Feldmann H, Grard G, Becker S, Leroy EM. Ebola and Marburg hae-morrhagic fever. J Clin Virol 2015; 64:111-9. 9. Falasca L, Agrati C, Petrosillo N, et al. Molecular mechanisms of Ebola viruspathogenesis: focus on cell death. Cell Death Differ 2015; 22:1250-9. 10. Gupta M, Spiropoulou C, Rollin PE. Ebola virus infection of human PBMCscauses massive death of macrophages, CD4 and CD8 T cell sub-populations invitro. Virology 2007;364:45-54. 11. Yaddanapudi K, Palacios G, Towner JS, et al. Implication of a retrovirus-like glyco-protein peptide in the immunopathogenesis of Ebola and Marburg viruses. FASEBJ2006;20:2519-30. 12. Pleet ML, Mathiesen A, DeMarino C, et al. Ebola VP40 in exosomes can causeimmune cell dysfunction. Front Microbiol 2016; 7:1765. 13. Pleet ML, DeMarino C, Lepene B, Aman MJ, Kashanchi F. The role of exosomalVP40 in Ebola virus disease. DNA Cell Biol 2017;36:243-8. 14. Bradfute SB, Warfield KL, Bavari S. Functional CD8+ T cell responses in lethalEbola virus infection. JImmunol 2008;180:4058-66. 15. Fleming A, Sampey G, Chung MC, et al. The carrying pigeons of the cell: exo-somes and their role in infectious diseases caused by human pathogens. PathogDis 2014;71:109-20. 16. Schwab A, Meyering SS, Lepene B, et al. Extracellular vesicles from infected cells:potential for direct pathogenesis. Front Microbiol 2015; 6:1132. 17. Anderson MR, Kashanchi F, Jacobson S.Exosomes inviral disease.Neurotherapeutics 2016; 13:535-46. 18. Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis andsecretion to biological function. Immunol Lett 2006;107:102-8. 19. Andreu Z, Yanez-M6 M. Tetraspanins in extracellular vesicle formation and func-tion. Front Immunol 2014;5:442. 20. Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell 2011;21:77-91. 21. Ahsan NA, Sampey GC, Lepene B, et al. Presence of viral RNA and proteins inexosomes from cellular clones resistant to rift valley fever virus infection. FrontMicrobiol 2016; 7:139. 22.. Jaworski E, Narayanan A, Van Duyne R, et al. Human T-lymphotropic virus type1-infected cells secrete exosomes that contain Tax protein. J Biol Chem 2014;289:22284-305. 23. Pegtel DM, van de Garde MD, Middeldorp JM. Viral miRNAs exploiting theendosomal-exosomal pathway for intercellular cross-talk and immune evasion.Biochim Biophys Acta 2011;1809:715-21. 24. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, et al. Functional delivery ofviral miRNAs via exosomes. Proc Natl Acad Sci U S A 2010;107:6328-33. 25. Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell HostMicrobe 2013;14:232-41. 26. Narayanan A, Iordanskiy S, Das R, et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J Biol Chem 2013;288:20014-33 27. Sampey GC, Saifuddin M, Schwab A, et al. Exosomes from HIV-1-infected cellsstimulate production of pro-inflammatory cytokines through trans-activatingresponse (TAR) RNA. J Biol Chem 2016;291:1251-66. 28. Sampey GC, Meyering SS, Zadeh MA, Saifuddin M, Hakami RM, Kashanchi FExosomes and their role in CNS viral infections. J Neurovirol 2014; 20:199-208. 29. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, andfriends. J Cell Biol 2013; 200:373-83. 30. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interac-tions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014;30:255-89. 31. van der Pol E,Coumans FA, Grootemaat AE, et al. Particle size distribution ofexo-somes and microvesicles determined by transmission electron microscopy, flowcytometry, nanoparticle tracking analysis, and resistive pulse sensing. J ThrombHaemost 2014;12:1182-92. 32. Kastelowitz N, Yin H. Exosomes and microvesicles: identification and targeting byparticle size and lipid chemical probes. Chembiochem 2014;15:923-8. 33. Vader P, Breakefield XO, Wood MJ. Extracellular vesicles: emerging targets forcancer therapy. Trends Mol Med 2014; 20:385-93. 34. Akers JC, Gonda D, Kim R, Carter BS, Chen CC. Biogenesis of extracellular vesi-cles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies.J Neurooncol 2013;113:1-11. 35. Broadhurst MJ, Brooks TJ, Pollock NR. Diagnosis of Ebola virus disease: past,present, and future. Clin Microbiol Rev 2016;29:773-93. 36.T1immins J, Scianimanico S, Schoehn G, Weissenhorn W. Vesicular release ofEbola virus matrix protein VP40. Virology 2001; 283:1-6. 37. Kallstrom G, Warfield KL, Swenson DL, et al. Analysis of Ebola virus and VLPrelease using an immunocapture assay. J Virol Methods 2005; 127:1-9. 38. Licata JM, Johnson RF, Han Z, Harty RN. Contribution of Ebola virus glycopro-tein, nucleoprotein, and VP24 to budding of VP40 virus-like particles. J Virol2004;78:7344-51. 39. Bavari S, Bosio CM, Wiegand E, et al. Lipid raft microdomains. J Exp Med 2002;195:593-602. 40. Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer.J Virol 2001;75:5205-14. 41. Noda T, Sagara H, Suzuki E, Takada A, Kida H, Kawaoka Y. Ebola virus VP40drives the formation of virus-like filamentous particles along with GP. J Virol2002;76:4855-65. 42. Kolesnikova L, Bamberg S, Berghofer B, Becker S. The matrix protein of Marburgvirus is transported to the plasma membrane along cellular membranes: exploitingthe retrograde late endosomal pathway. JVirol 2004; 78:2382-93. 43. Kolesnikova L, Bugany H, Klenk HD, Becker S. VP40, the matrix protein ofMarburg virus, is associated with membranes of the late endosomal compartment.JVirol 2002;76:1825-38 44. Steele K, Crise B, Kuehne A, Kell W. Ebola virus glycoprotein demonstrates differ-ential cellular localization in infected cell types of nonhuman primates and Guineapigs. Arch Pathol Lab Med 2001;125:625-30. 45. Reynard O, Reid SP, Page A, et al. Unconventional secretion of Ebola virus matrixprotein VP40. J Infect Dis 2011; 204(Suppl3):S833-9. 46. Kashanchi F, Duvall JF, Brady JN. Electroporation of viral transactivator proteinsinto lymphocyte suspension cells. Nucleic Acids Res 1992; 20:4673-4. 47. Jaworski E, Saifuddin M, Sampey G, et al. The use of Nanotrap particles technol-ogy in capturing HIV-1 virions and viral proteins from infected cells. PLoS One2014;9:e96778. 48. Sadick JS, Boutin ME, Hoffman-Kim D, Darling EM. Protein characterizationof intracellular target-sorted, formalin-fixed cell subpopulations. Sci Rep 2016;6:33999. 49. Wilson JA, Hevey M, Bakken R, et al. Epitopes involved in antibody-mediated. protection from Ebola virus. Science 2000;287:1664-6. 50. DeMarino C, Pleet ML, Cowen M, et al. Antiretroviral drugs alter the content ofextracellular vesicles from HIV-1-infected cells. Sci Rep 2018; 8:7653. 51. Barclay RA, Pleet ML, Akpamagbo Y, Noor K, Mathiesen A, Kashanchi F.Isolation of exosomes from HTLV-infected cells. Methods Mol Biol 2017;1582:57-75. 52. Barclay RA, Schwab A, DeMarino C, et al. Exosomes from uninfected cells activatetranscription of latent HIV-1. J Biol Chem 2017; 292:11682-701. 53.Nascimento R, Costa H, Parkhouse RM. Virus manipulation of cell cycle.Protoplasma 2012; 249:519-28. 54. Fehr AR, Yu D. Control the host cell cycle: viral regulation of the anaphase-pro-moting complex. J Virol 2013;87:8818-25. 55.DIove B, Brooks G, Bicknell K, Wurm T, Hiscox JA. Cell cycle perturbationsinduced by infection with the coronavirus infectious bronchitis virus and theireffect on virus replication. J Virol 2006;80:4147-56. 56. Chaurushiya MS, Weitzman MD. Viral manipulation of DNA repair and cell cyclecheckpoints. DNA Repair (Amst) 2009;8:1166-76. 57. Bracq L, Xie M, Benichou S, Bouchet J. Mechanisms for cell-to-cell transmissionof HIV-1. Front Immunol 2018;9:260 58. Sylwester A, Wessels D, Anderson SA, et al. HIV-induced syncytia of a T cell line. form single giant pseudopods and are motile. J Cell Sci 1993; 106(Pt 3):941-53. 59. Wang P, Li M, Lu W, Zhang D, Hu Q, Liu Y. DC-SIGN promotes Japanese enceph-alitis virus transmission from dendritic cells to T cells via virological synapses.Virol Sin 2017; 32:495-502. 60. Ceccaldi PE, Delebecque F, Prevost MC, et al. DC-SIGN facilitates fusion of den-dritic cells with human T-cell leukemia virus type 1-infected cells. J Virol 2006;80:4771-80. 61. Kononova AA, Cheresiz SV, Chechushkov AV, Razumova YV, Pokrovskii AG.Comparative study of fusogenic activity of H1 and H5 subtypes influenza virushemagglutinins. Bull Exp Biol Med 2017;164:85-9. 62. Casimiro MC, Velasco-Velazquez M, Aguirre-Alvarado C, Pestell RG. Overviewof cyclins D1 function in cancer and the CDK inhibitor landscape: past and pres-ent. Expert Opin Investig Drugs 2014;23:295-304. 63. Pestell RG. New roles of cyclin D1. Am J Pathol 2013;183:3-9. 64. Klein EA, Assoian RK. Transcriptional regulation ofthe cyclin D1 gene at a glance.J Cell Sci 2008; 121:3853-7. 65. Santiago F, Clark E, Chong S, et al. Transcriptional up-regulation of the cyclin D2gene and acquisition ofnew cyclin-dependent kinase partners in human T-cellleukemia virus type 1-infected cells. J Virol 1999; 73:9917-27. ( 66. K ehn K , D e ng L, de la F u ente C , e t al. The r ole of cyclin D2 and p21/wafl i nhuman T-cell leukemia virus type 1 in f ected cells. R e trovirology 2004;1:6. ) 67. Zhou L, Kang D, Xu C, Zhao W, Tian B, Chen L. Expression of cyclin D1 andcyclin E significantly associates with human papillomavirus subtypes in Bowenoidpapulosis. Acta Histochem 2013; 115:339-43. 68. Ripple MJ, Parker Struckhoff A, Trillo-Tinoco J, et al. Activation of c-Myc andcyclin D1 by JCV T-antigen and B-catenin in colon cancer. PLoS One 2014;9:e106257. ( 69. X iang Z, L i ang Z, Yanfeng H, Leitao K. Persistence of RSV promotes proli f eration a nd epithelial- m esenc h ymal tra n sition of b ronchial epithelial cells through Nodal signaling. J Med Microbiol 2017; 66:1499- 5 05. ) 70. Xu Y, Shi Y, Yuan Q, et al.Epstein-Barr virus encoded LMP1 regulates cyclin D1promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J Exp Clin CancerRes 2013;32:90. 71. Kitagawa M, Higashi H, Jung HK, et al. The consensus motif for phosphorylationby cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2.EMBO J 1996; 15:7060-9. 72. Bjorndal AS, Szekely L, Elgh F. Ebola virus infection inversely correlates with theoverall expression levels of promyelocytic leukaemia (PML) protein in cultured.cells. BMC Microbiol 2003; 3:6. 73. Nanbo A, Watanabe S, Halfmann P, Kawaoka Y. The spatio-temporal distributiondynamics of Ebola virus proteins and RNA in infected cells. Sci Rep 2013; 3: doi:.10.1038/srep01206. 74. Del Vecchio K, Frick CT, Gc JB, et al. A cationic, C-terminal patch and structuralrearrangements in Ebola virus matrix VP40 protein control its interactions withphosphatidylserine. J Biol Chem 2018;293:3335-49. 75. Moore HM, Vartiainen MK. F-actin organizes the nucleus. Nat Cell Biol 2017;19:1386-8. 76. Falahzadeh K,Banaei-Esfahani A, Shahhoseini M. The potential roles of actin inthe nucleus. Cell J 2015;17:7-14. 77. Albanese C, DAmico M, Reutens AT, et al. Activation of the cyclin D1 gene bythe E1A-associated protein p300 through AP-1 inhibits cellular apoptosis. J BiolChem 1999;274:34186-95. 78. Byun JS, Wong MM, Cui W, et al. Dynamic bookmarking of primary responsegenes by p300 and RNA polymerase II complexes. Proc Natl Acad Sci U S A 2009;106:19286-91. 79. Cantin R, Diou J, Belanger D, Tremblay AM, Gilbert C. Discrimination betweenexosomes and HIV-1: purification of both vesicles from cell-free supernatants. J.Immunol Methods 2008; 338:21-30. 80. BFeniac DR, Melito PL, Devarennes SL, et al. The organisation of Ebola virusreveals a capacity for extensive, modular polyploidy. PLoS One 2012; 7:e29608. 81. Olejnik J, Alonso J, Schmidt KM, et al. Ebola virus does not block apoptotic signal-ing pathways.J Virol 2013;87:5384-96. 82. Geisbert TW, Hensley LE, Larsen T, et al. Pathogenesis of Ebola hemorrhagic feverin cynomolgus macaques: evidence that dendritic cells are early and sustained tar-gets of infection. Am J Pathol 2003; 163:2347-70. 83. Bradfute SB, Swanson PE, Smith MA, et al. Mechanisms and consequences of ebo-lavirus-induced lymphocyte apoptosis. JImmunol 2010;184:327-35. 84. Chen P, Lee NV, Hu W, et al. Spectrum and degree of CDK drug interactions pre-dicts clinical performance. Mol Cancer Ther 2016;15:2273-81. 85. Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): mechanism of action andclinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in varioussolid tumors. Clin Cancer Res 2017;23:3251-62. 86. Messaoudi I, Amarasinghe GK, Basler CF. Filovirus pathogenesis and immuneevasion: insights from Ebola virus and Marburg virus. Nat Rev Microbiol 2015;13:663-76. 87. Villinger F, Rollin PE, Brar SS, et al. Markedly elevated levels of interferon (IFN)-. gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alphaassociated with fatal Ebola virus infection. J Infect Dis 1999; 179(Suppl 1):S188-91 88.BIixler SL, Goff AJ. The role of cytokines and chemokines in Filovirus infection.Viruses 2015;7:5489-507. 89. Baize S, Leroy EM, Georges AJ, et al. Inflammatory responses in Ebola virus-in-fected patients. Clin Exp Immunol 2002; 128:163-8. 90. Kota KP, Benko JG, Mudhasani R, et al. High content image based analysis iden-tifies cell cycle inhibitors as regulators of Ebola virus infection. Viruses 2012;4:1865-77. 91. Jacobs M, Rodger A, Bell DJ, et al. Late Ebola virus relapse causing meningoen-cephalitis: a case report. Lancet 2016; 388:498-503. 92. Varkey JB, Shantha JG, Crozier I, et al. Persistence of Ebola virus in ocular fluidduring convalescence. N Engl J Med 2015;372:2423-7. 93. Deen GF, Broutet N, Xu W,et al. Ebola RNA persistence in semen of Ebola virusdisease survivors-final report. N Engl J Med 2017;377:1428-37. 94. Uyeki TM, Erickson BR, Brown S, et al. Ebola virus persistence in semen of malesurvivors. Clin Infect Dis 2016;62:1552-5. 95. Diallo B, Sissoko D, Loman NJ, et al. Resurgence of Ebola virus disease in guinealinked to a survivor with virus persistence in seminal fluid for more than 500 daysClin Infect Dis 2016; 63:1353-6 96. Christie A, Davies-Wayne GJ, Cordier-Lassalle T, et al. Possible sexual trans-mission of Ebola virus-Liberia, 2015. MMWR Morb Mortal Wkly Rep 2015;64:479-81. 97. Mate SE, Kugelman JR, Nyenswah TG, et al. Molecular evidence of sexual trans-mission of Ebola virus. NEngl JMed 2015;373:2448-54 ( 98. B lackley D J, Wiley MR, Ladner JT, et al. Reduced evolutionary rate in reemerged E b ola virus transmission c hains. Sci Adv 2016;2:e1600378. ) 99. Arias A, Watson SJ, Asogun D, et al. Rapid outbreak sequencing of Ebola virus inSierra Leone identifies transmission chains linked to sporadic cases. Virus Evol2016;2:vew016. 100. Rodriguez LL, Roo AD, Guimard Y, et al. Persistence and genetic stability ofEbola virus during the outbreak in Kikwit, Democratic Republic of the Congo,1995. J Infect Dis 1999; 179(Supp1):S170-6. 101. Zeng X, Blancett CD, Koistinen KA, et al. Identification and pathological char-acterization of persistent asymptomatic Ebola virus infection in rhesus monkeys.Nat Microbiol 2017;2:17113. 102. Martin B, Canard B, Decroly E. Filovirus proteins for antiviral drug discovery:Structure/function bases of the replication cycle. Antiviral Res 2017;141:48-61. 103. Bharat TA, Noda T, Riches JD, et al. Structural dissection of Ebola virus and itsassembly determinants using cryo-electron tomography. Proc Natl Acad Sci U SA 2012;109:4275-80. 104. Martin B, Reynard O, Volchkov V, Decroly E. Filovirus proteins for antiviral drugdiscovery: structure/function of proteins involved in assembly and budding.Antiviral Res 2018;150:183-92. 105. Lee JE, Saphire EO. Ebolavirus glycoprotein structure and mechanism of entry.Future Virol 2009;4:621-35. 106. Martin B, Hoenen T, Canard B, Decroly E. Filovirus proteins for antiviral drugdiscovery: a structure/function analysis of surface glycoproteins and virus entry.Antiviral Res 2016; 135:1-14. Ebola Virus VPModulates Cell Cycle and Biogenesis of Extracellular Vesicles · JID (Suppl)· S S·JID (Suppl)· Pleet et al 背景:埃博拉病毒(EBOV)主要攻击骨髓细胞,其致命感染会引起大量的T细胞死亡。研究已证明,受感染细胞所产生的外泌体中包含的埃博拉病毒蛋白VP40导致了受体免疫细胞的死亡。方法:通过克隆技术表达VP40,研究者分析了供体细胞循环、细胞外囊泡的生物发生过程和受体免疫细胞的死亡。通过激酶和染色质免疫沉淀实验,研究者研究了细胞周期蛋白D1的转录和VP40的核定位。通过质谱、细胞因子和蛋白质免疫印迹实验,研究者检测了细胞外囊泡的内含物。野生型埃博拉病毒感染实验在生物安全等级为4级的实验室平台进行。结果:含有VP40的细胞外囊泡诱导了受体T细胞和单核细胞的凋亡。细胞周期蛋白D1的表达水平上调,导致了表达VP40的克隆加速生长,而核内的VP40蛋白会结合到细胞周期蛋白D1的启动子上。细胞循环被加快,会影响细胞外囊泡的生物发生过程,最终产生了数量较少、但粒径更大的细胞外囊泡。含有VP40的细胞外囊泡的内含物富含能结合核糖核酸的蛋白和细胞因子(白细胞介素-15、转化生长因子-β1和干扰素-γ)。最终,受埃博拉病毒感染的细胞及其细胞外囊泡中包含了VP40、核蛋白质和糖蛋白。结论:细胞核内的VP40上调了细胞周期蛋白D1的表达水平,导致异常的细胞周期和异常的细胞外囊泡生物发生过程。被埃博拉病毒感染的细胞所产生的细胞外囊泡中包含的细胞因子和埃博拉病毒蛋白引起了免疫细胞的大量死亡。
确定






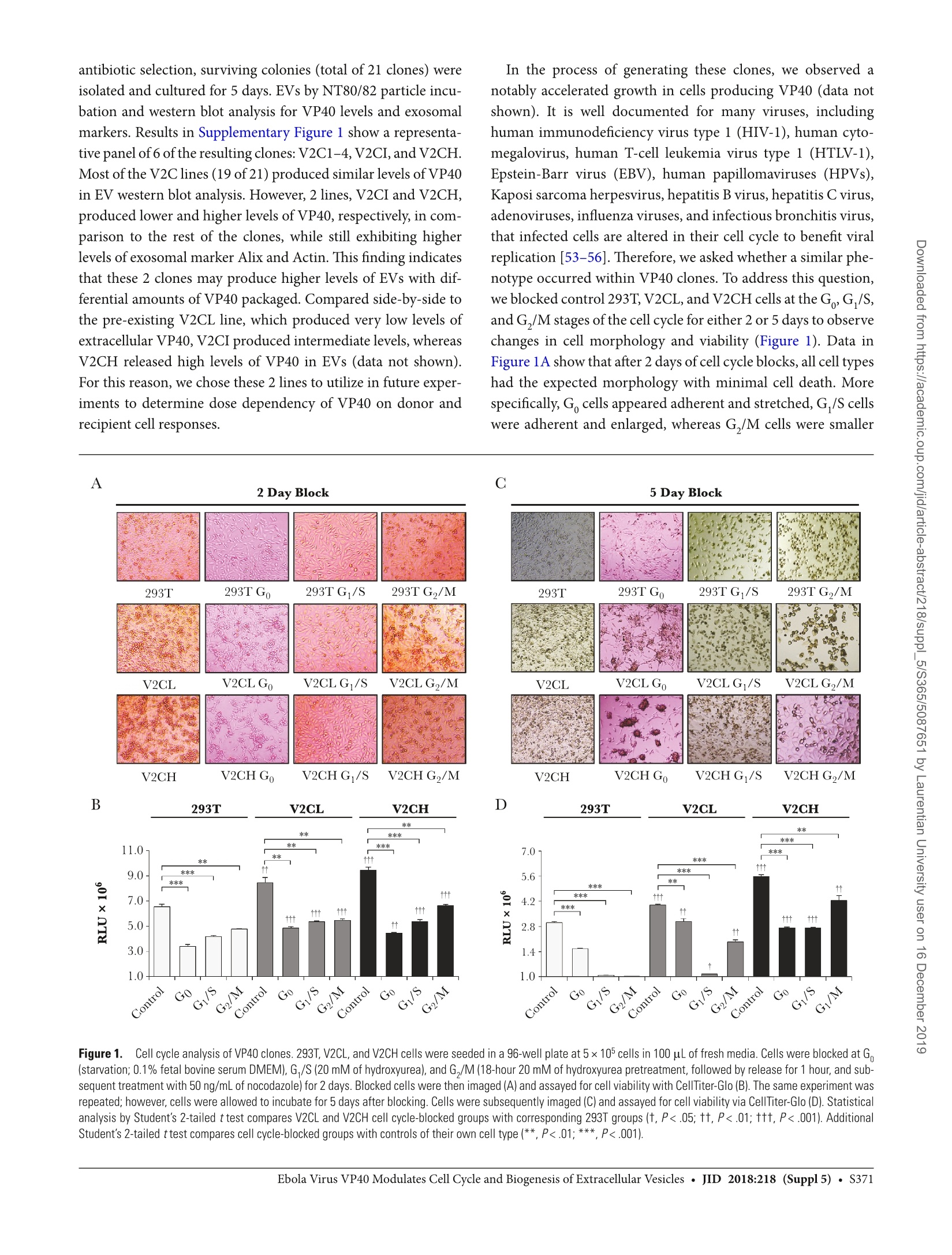

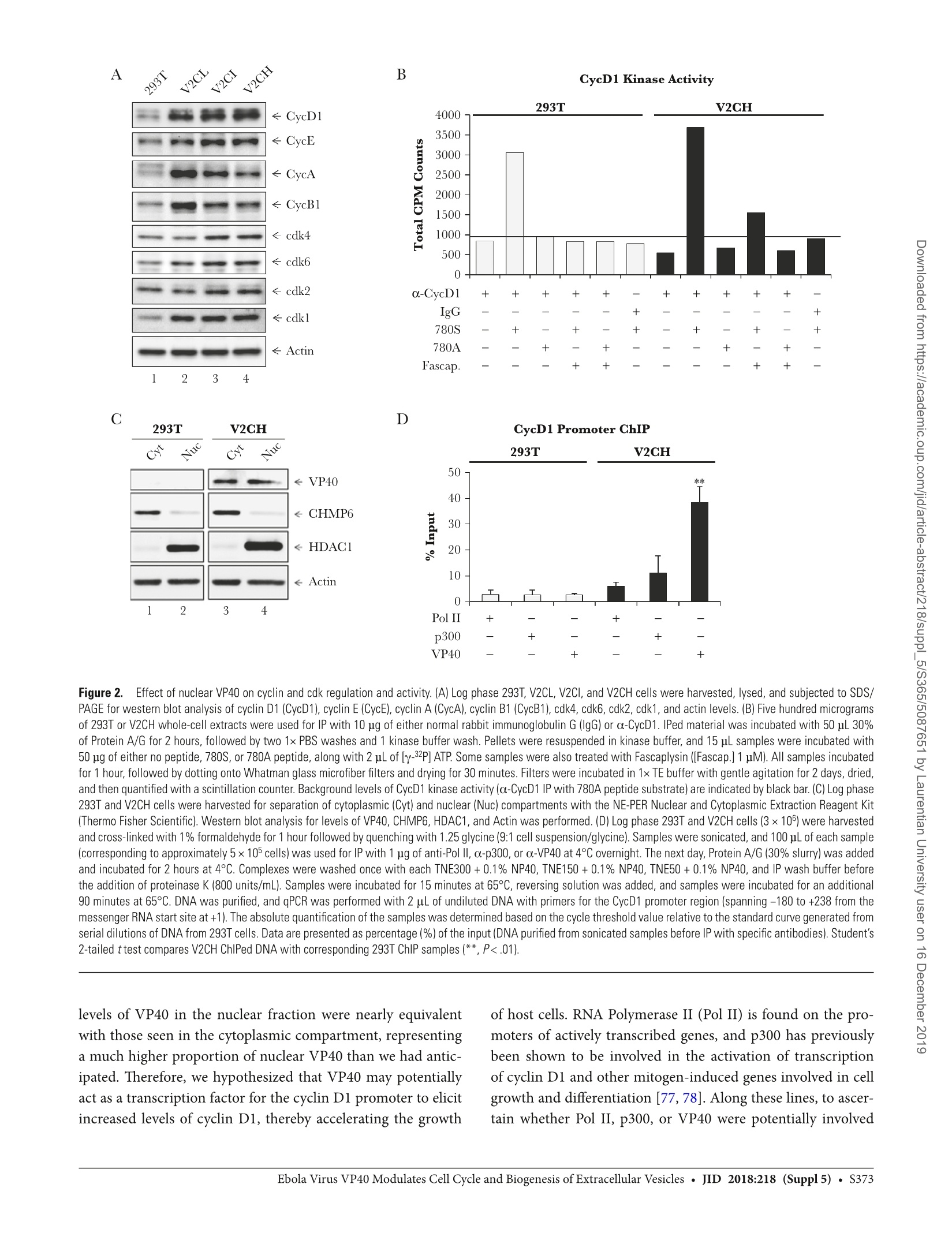
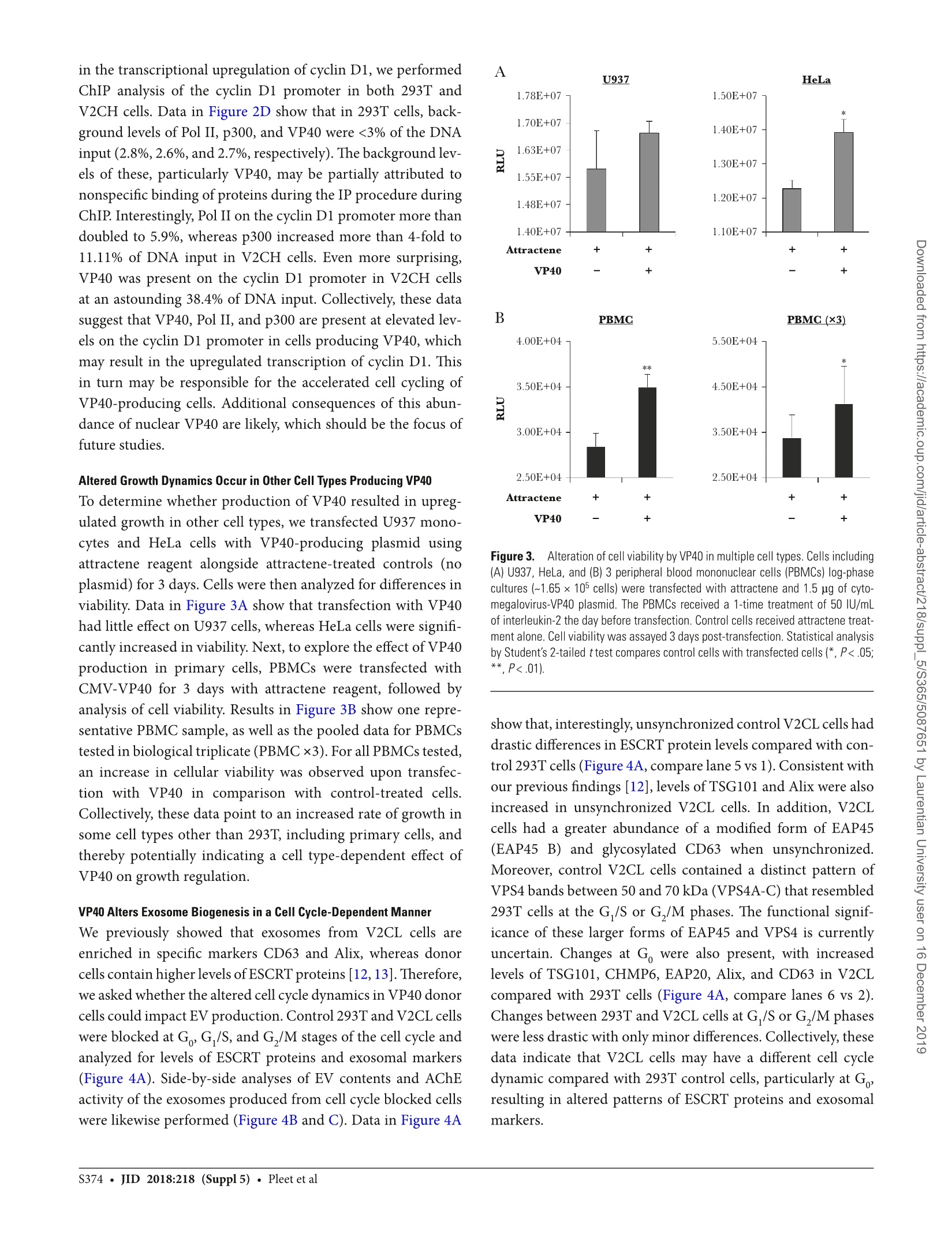
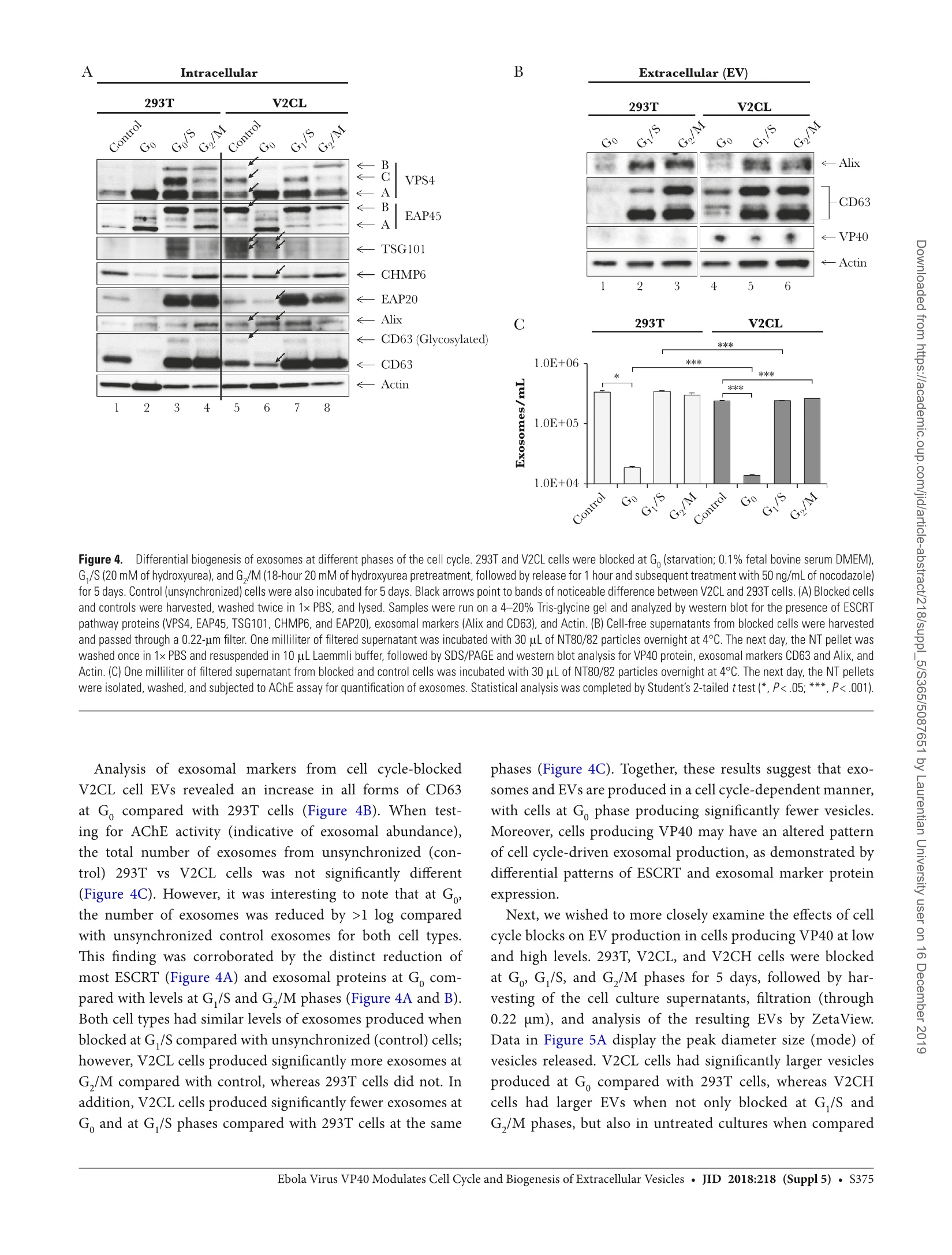
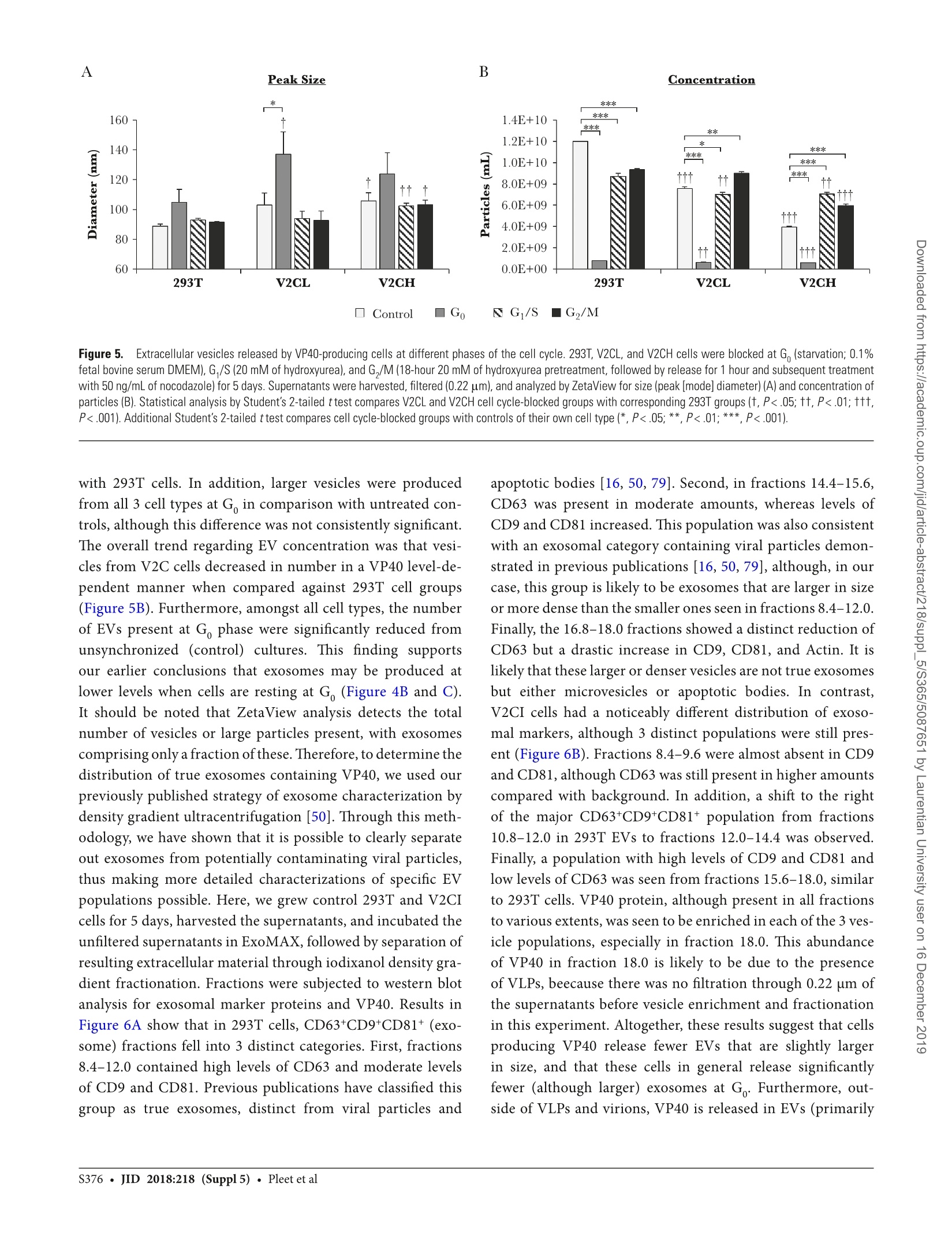
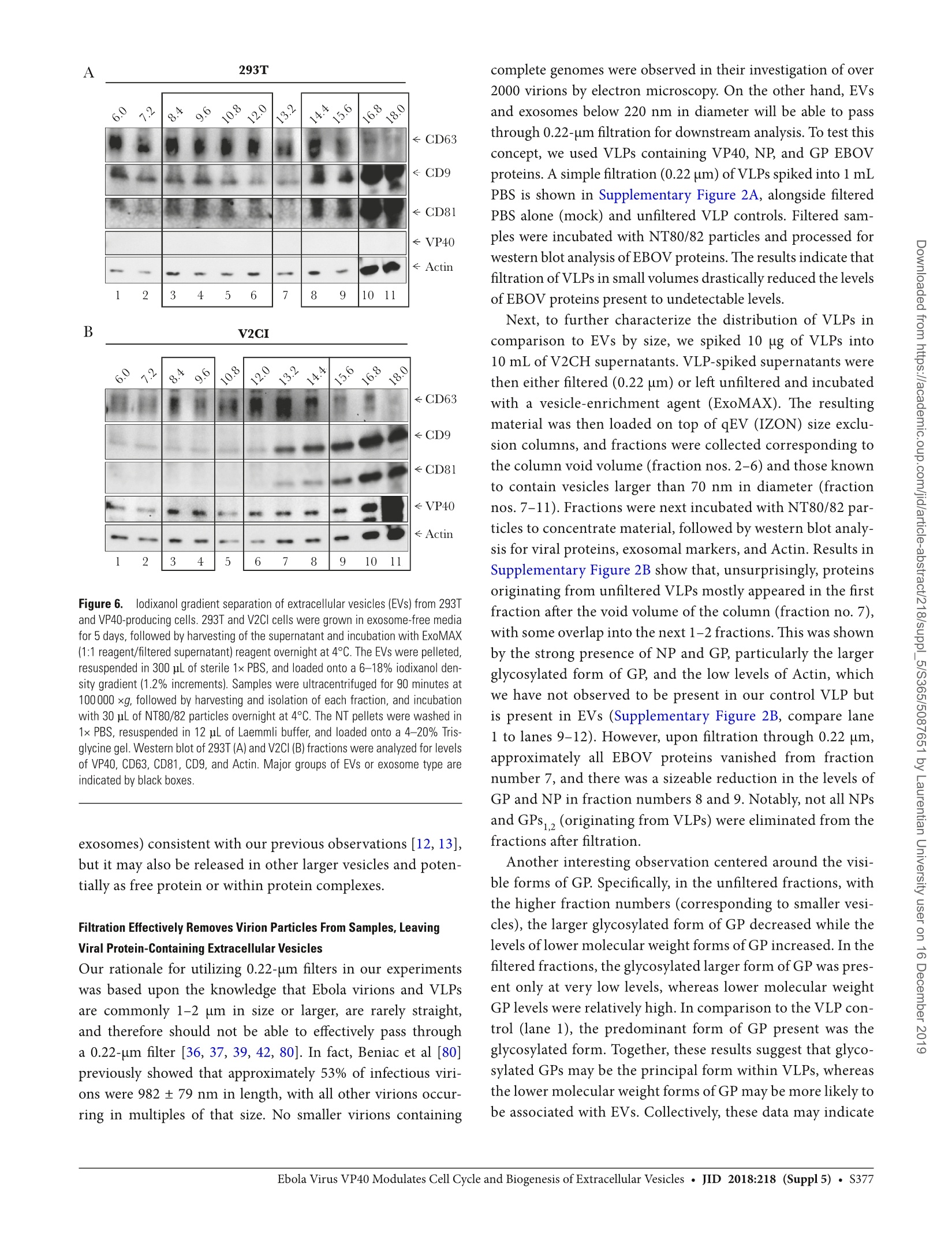

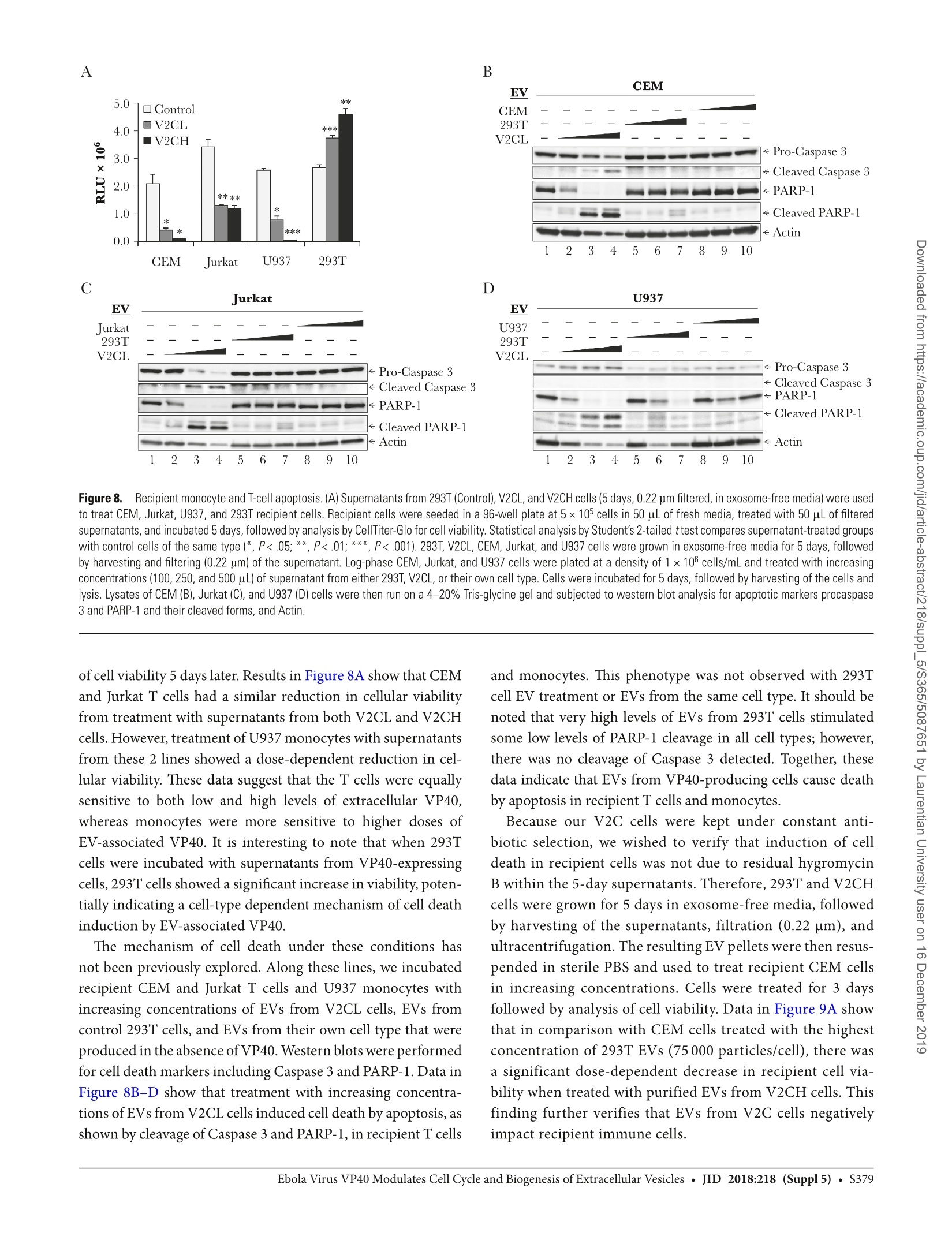






还剩21页未读,是否继续阅读?

产品配置单

大昌华嘉科学仪器为您提供《埃博拉病毒VP40中影响细胞循环和细胞外囊泡的生物发生过程检测方案(Zeta电位仪)》,该方案主要用于其他中影响细胞循环和细胞外囊泡的生物发生过程检测,参考标准--,《埃博拉病毒VP40中影响细胞循环和细胞外囊泡的生物发生过程检测方案(Zeta电位仪)》用到的仪器有Microtrac纳米粒度及Zeta电位分析仪
推荐专场






相关方案
更多
该厂商其他方案
更多