








方案详情
文
理想情况下,在运行色谱方法时,样品稀释剂组分应尽可能接近方法起始条件。这样做的目的是最大程度减小由样品溶剂效应引起的谱带展宽和峰畸变,进而避免出现峰不对称性、峰分裂或数据不可用的情况。引起溶剂效应原因是稀释剂与流动相之间存在洗脱强度差异。当稀释剂的洗脱强度高于流动相时,峰展宽和峰形异常的情况通常会更加严重1-2。事实上,业内普遍认为,最好是将进样的样品溶解于起始流动相中。然而,给定样品的预处理常常需要将分析物溶解在与流动相组分相差很大的溶剂中。为了避免溶解度问题和峰形不佳的问题,许多方案都要求在预处理过程中挥干样品溶剂,然后将样品复溶于流动相中。然而,这个额外的步骤非常耗时,所需时间往往比HPLC分析的时间还要长1。一般而言,推荐的做法是避免使用比流动相更强的溶剂来溶解样品和标准品。这种做法基于如下假设:强于流动相的进样溶剂会干扰样品在柱头处的吸附,而采用大体积进样时尤其如此2。遗憾的是,这种做法在实践中可行性不佳,因为分析人员往往必须根据样品的溶解度来决定有机溶剂的含量,以确保样品能够完全溶解。对于扩散体积较大的传统LC系统,这种现象带来的问题较少,因为柱前样品/溶剂/流动相混合很充分,可以缓解溶剂效应造成的色谱峰问题。然而,对于现代的低分散U(H)PLC系统,如果以较大体积进样含有高有机相的样品,就会出现问题,并可能导致峰对称性变差或峰分裂。
方案详情

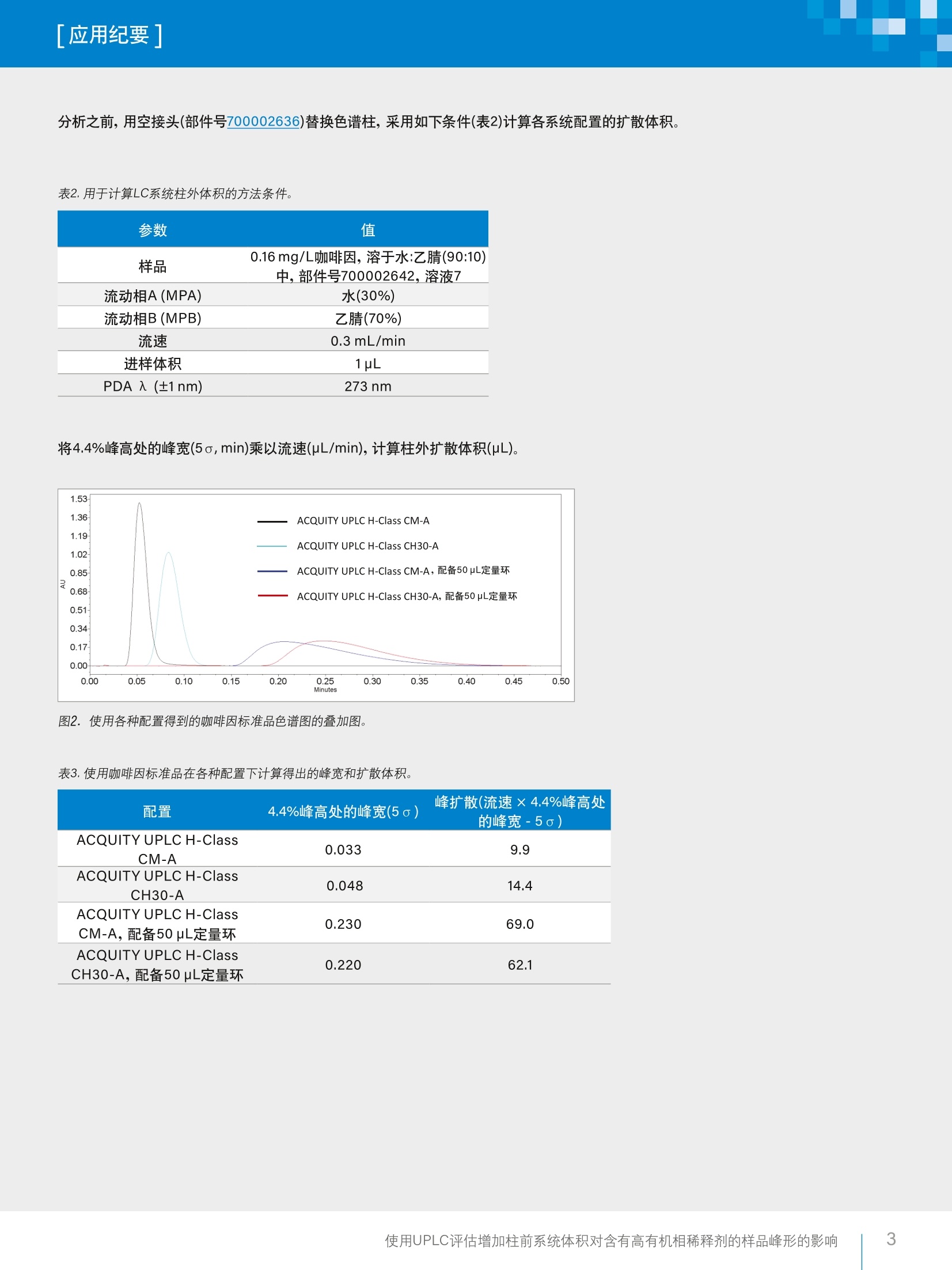
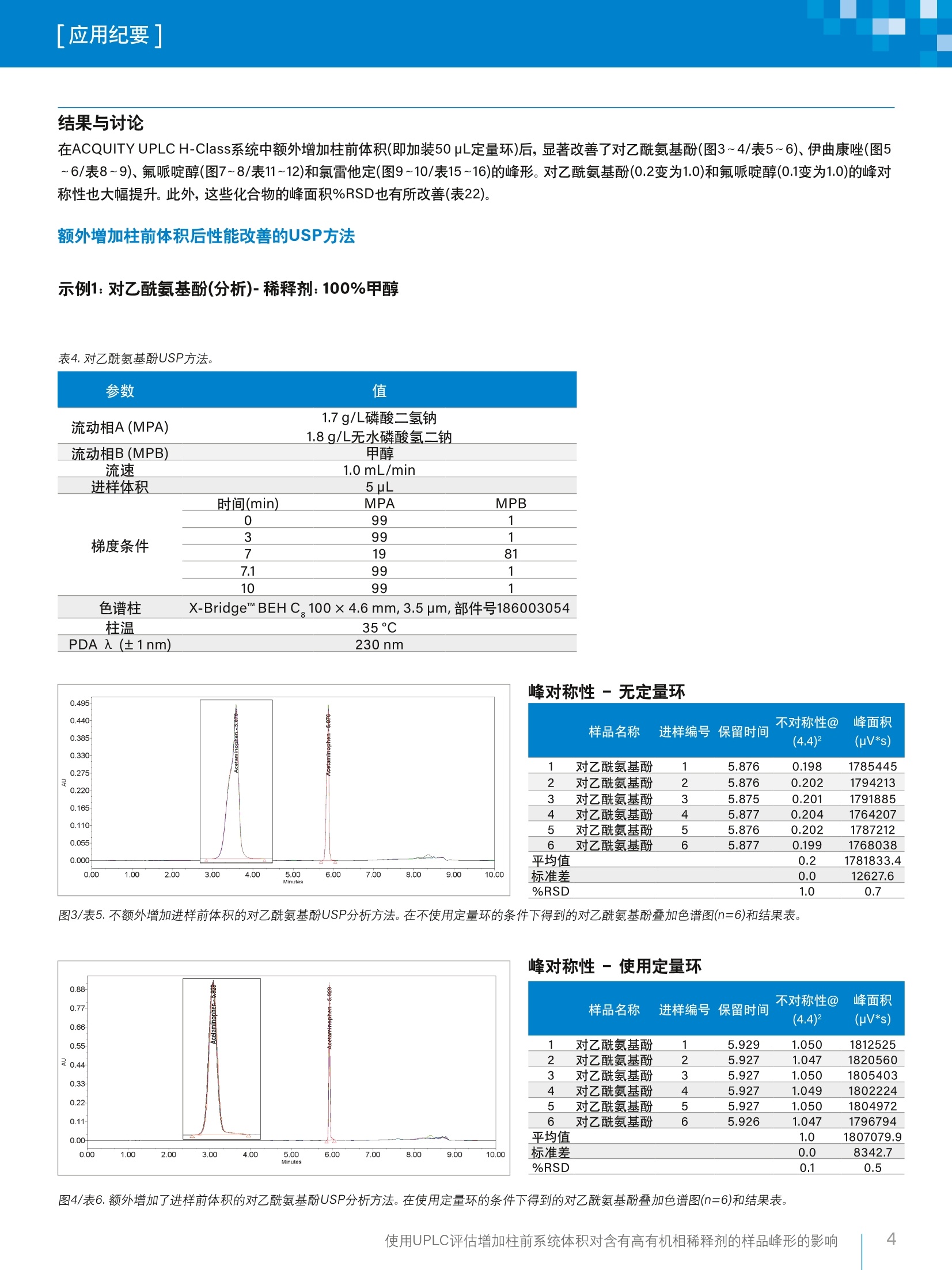
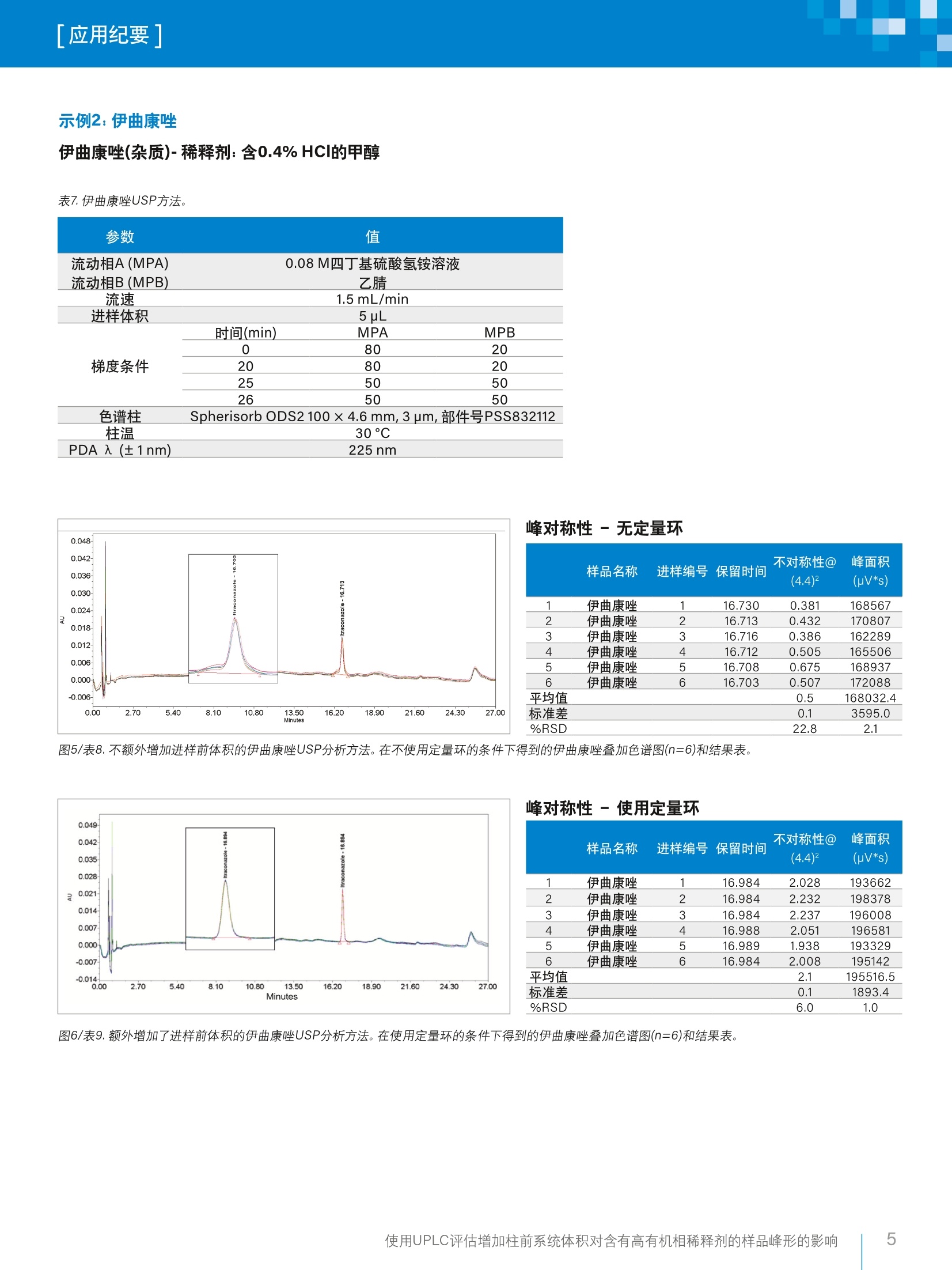
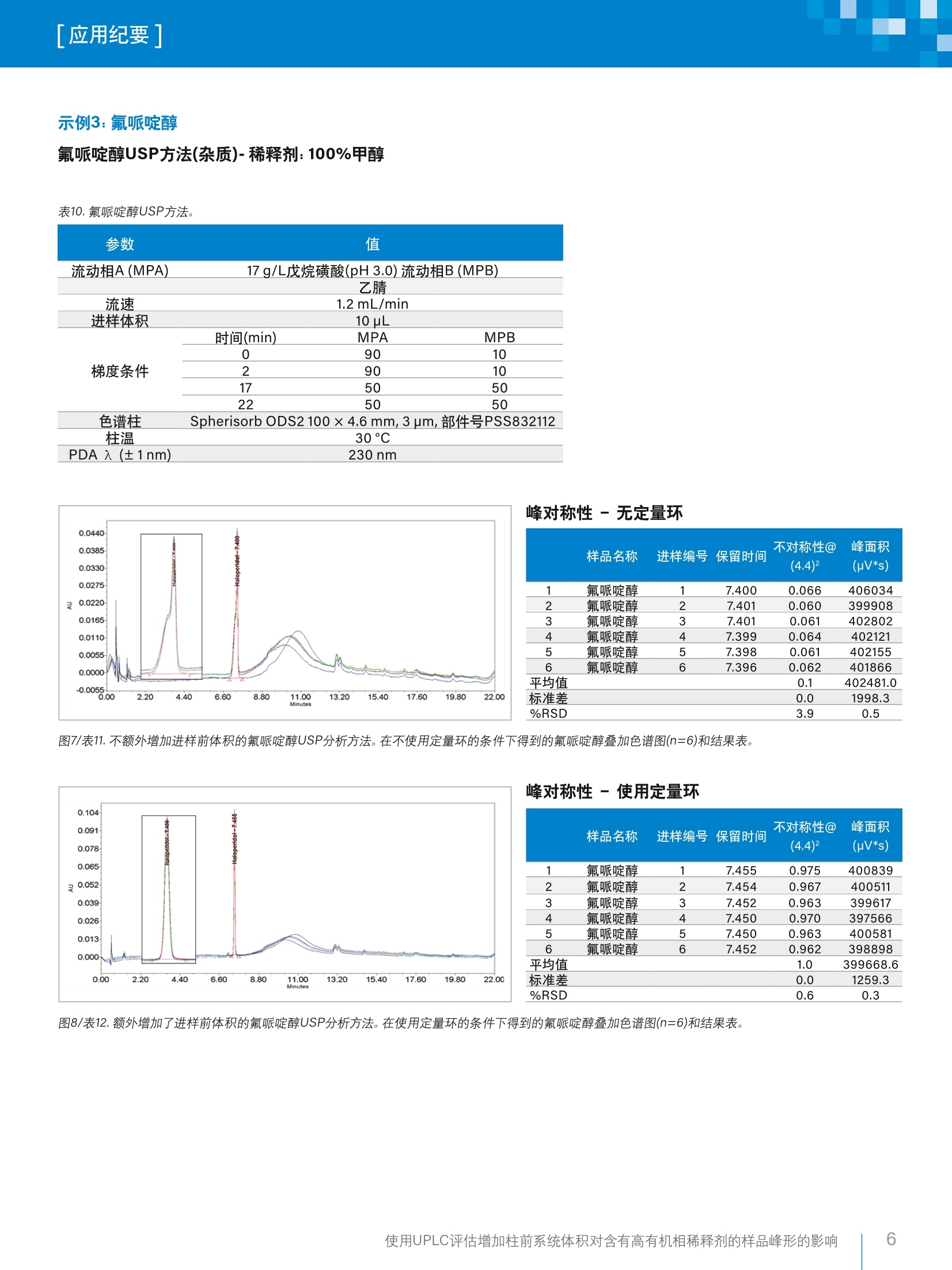
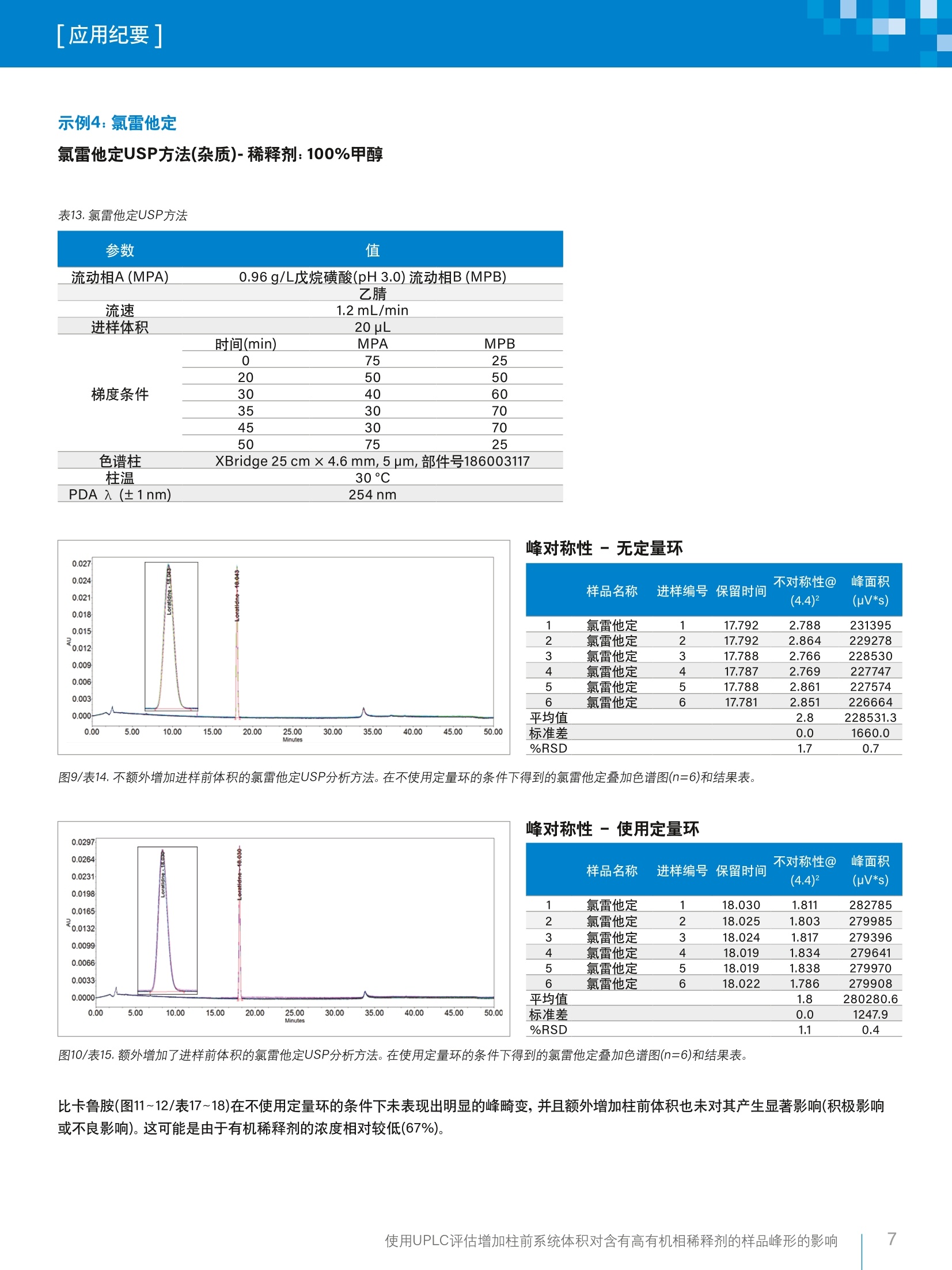
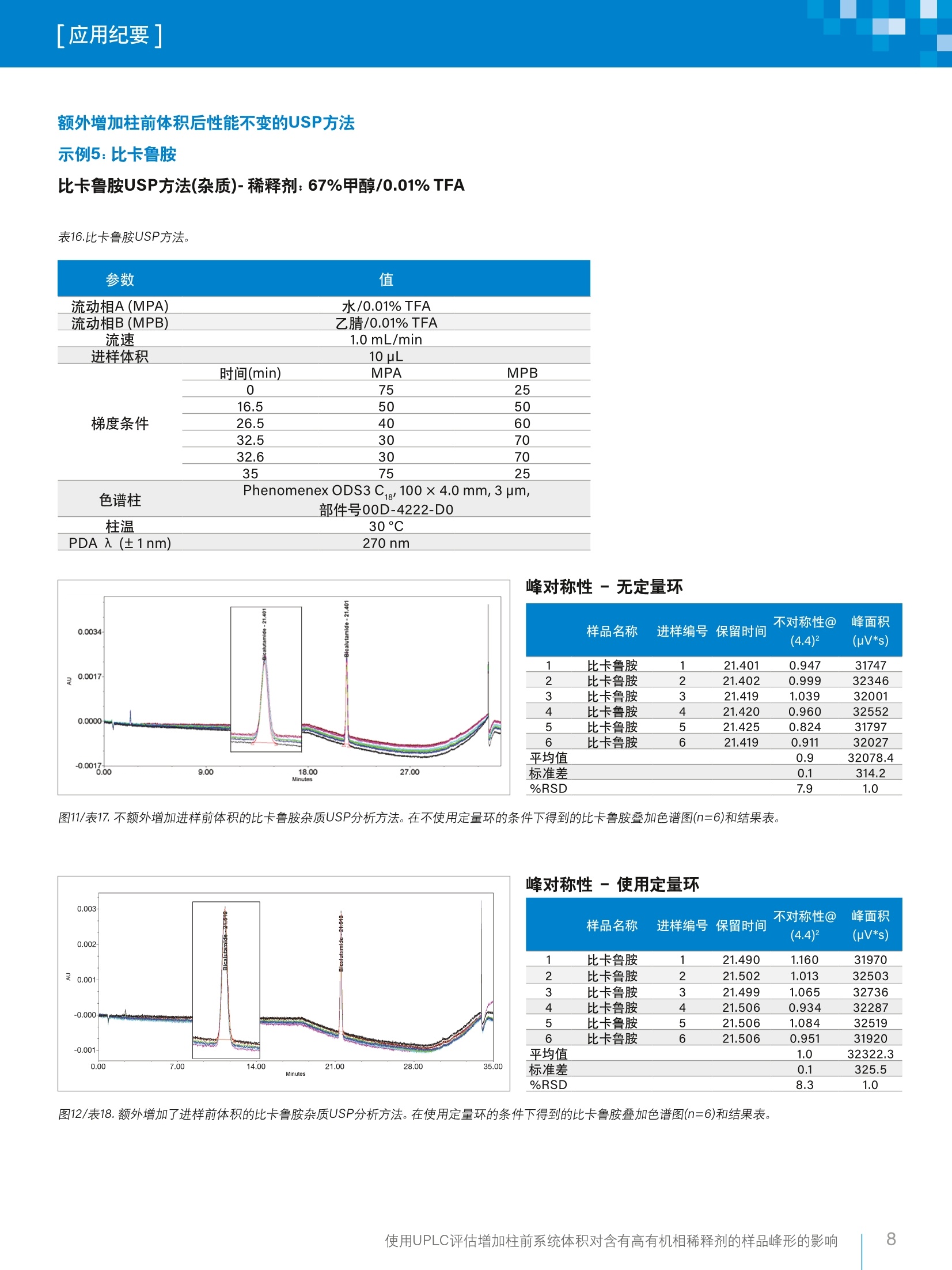
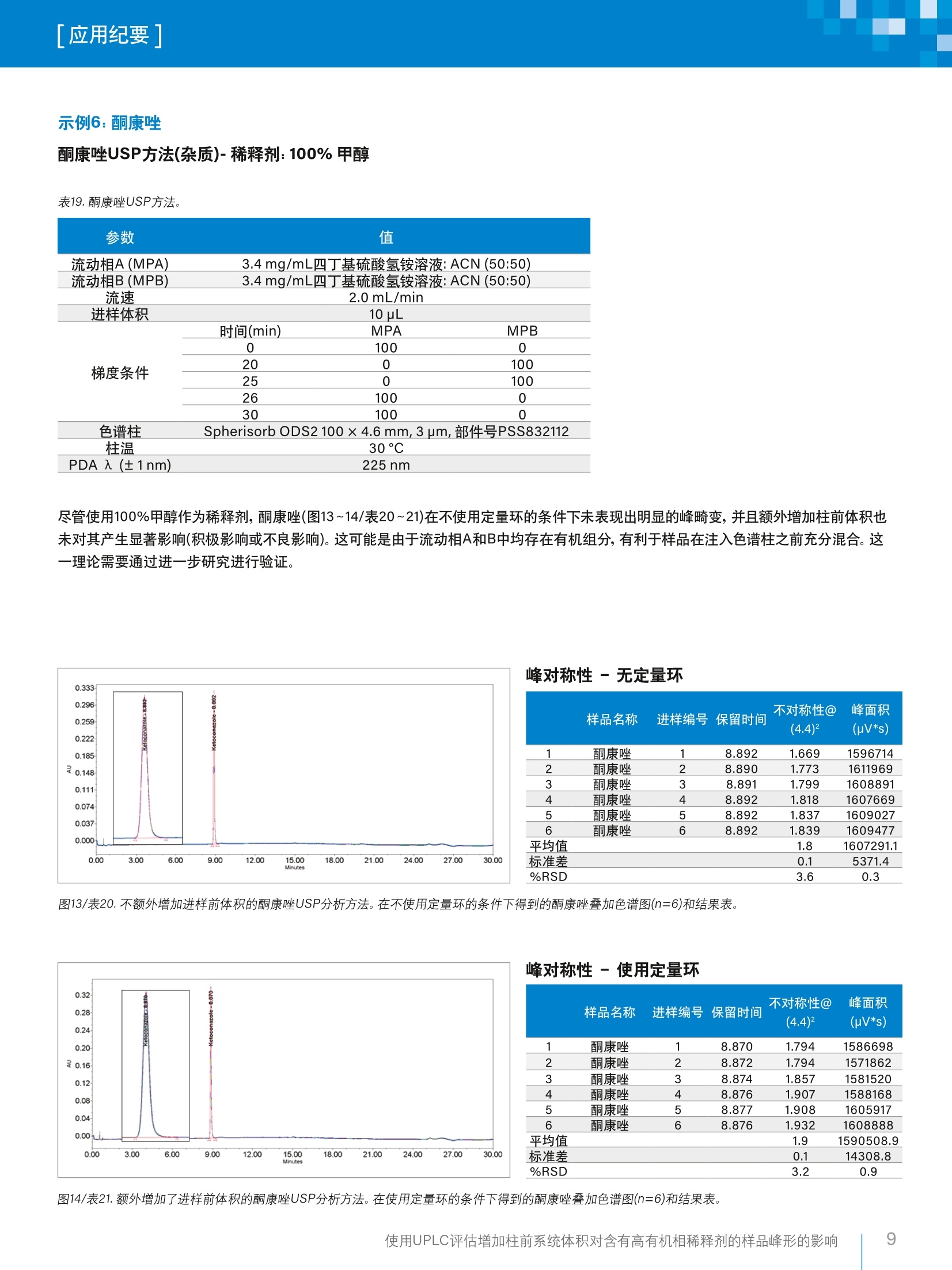
「应用纪要]WatersTHE SCIENCE OF WHAT'S POSSIBLE. 「应用纪要 使用UPLC评估增加柱前系统体积对含有高有机相稀释剂的样品峰形的影响 Chris Henry、Paul Rainville?、Pat McConville²和Paula Hong²沃特世公司(英国威姆斯洛)沃特世公司(美国马萨诸塞州米尔福德) 应用优势 在ACQUITYUPLC"H-Class系统上使用高有机相溶剂作为稀释剂成功运行了药典方法,且未发生峰畸变。 沃特世解决方案配备光电二极管阵列检测器的ACQUITYUPLC H-Class系统 Empower"3软件 关键词 USP,药典方法, UPLC, HPLC, Empower,监管,峰对称性 简介 理想情况下,在运行色谱方法时,样品稀释剂组分应尽可能接近方法起始条件。这样做的目的是最大程度减小由样品溶剂效应引起的谱带展宽和峰畸变,进而避免出现峰不对称性、峰分裂或数据不可用的情况。 引起溶剂效应的原因是稀释剂与流动相之间存在洗脱强度差异。当稀释剂的洗脱强度高于流动相时,峰展宽和峰形异常的情况通常会更加严重12。 事实上,业内普遍认为,最好是将进样的样品溶解于起始流动相中。然而,给定样品的预处理常常需要将分析物溶解在与流动相组分相差很大的溶剂中。为了避免溶解度问题和峰形不佳的问题,许多方案都要求在预处理过程中挥干样品溶剂,然后将样品复溶于流动相中。然而,这个额外的步骤非常耗时,所需时间往往比HPLC分析的时间还要长1。 一般而言,推荐的做法是避免使用比流动相更强的溶剂来溶解样品和标准品。这种做法基于如下假设:强于流动相的进样溶剂会干扰样品在柱头处的吸附,而采用大体积进样时尤其如此?。遗憾的是,这种做法在实践中可行性不佳,因为分析人员往往必须根据样品的溶解度来决定有机溶剂的含量,以确保样品能够完全溶解。 对于扩散体积较大的传统LC系统,这种现象带来的问题较少,因为柱前样品/溶剂/流动相混合很充分,可以缓解溶剂效应造成的色谱峰问题。 然而,对于现代的低分散U(H)PLC系统,如果以较大体积进样含有高有机相的样品,就会出现问题,并可能导致峰对称性变差或峰分裂。 为了研究这种现象并开发一种简单的解决方案来克服样品稀释剂中强溶剂的影响,本研究选择了要求样品稀释剂中有机溶剂含量在67%~100%之间的五种美国药典(USP)方法(即,对乙酰氨基酚、伊曲康唑、酮康唑、氯雷他定和比卡鲁胺)。 在ACQUITY UPLC H-Class系统上运行上述方法,并计算各种配置的扩散体积。然后,将这些方法与为了增加柱前体积而进行的结构改进和其它改进相结合,评估额外增加柱前体积对高有机相稀释剂所引起的峰对称性问题的影响。 实验 方法选自USP,标准品重复分析六次(n=6)。按照USP方法制备标准品(表1)。 在ACQUITY UPLC H-Class系统上采用两种配置运行USP方法,第一种配置不使用定量环,第二种配置是在样品管理器中进样器座上的位置6与色谱柱入口管路之间安装50pL定量环(如图1所示)。本研究目视评估了这两种条件下的色谱分析结果,以4.4%峰高处的峰不对称性平均值和峰面积%RSD作为色谱性能的指标。 除氯雷他定方法之外,其它所有方法都使用色谱柱管理器CM-A。氯雷他定方法使用CH30-AACQUITY柱温箱,以容纳25 cm HPLC色谱柱,详细信息请参阅USP方法。 化合物 稀释剂 对乙酰氨基酚 100%甲醇 伊曲康唑 含0.4%HCI的甲醇 氟哌啶醇 100%甲醇 氯雷他定 100%甲醇 比卡鲁胺 67%甲醇(含0.01%TFA) 酮康唑 100%甲醇 图1.加配的50 pL柱前定量环。 分析中用到的材料包括(分析标准品以粗体显示): 酮康唑: Sigma-Aldrich, 批号SLBR1290V 伊曲康唑:EEP参比标准品,Y0001100,批次2.0 比卡鲁胺:安:USP参比标准品-批号G01298 ( 氯雷他定:S Sigma-Aldrich-批号LRAA9165 ) ( 氟哌啶醇:Si g ma-Aldrich-批号LRAA7399 ) 对乙酰氨基基:: Sigma-Aldrich-批号SLBM5923V 水:EELGA Purelab ( :乙腈: Fisher Chemicals Optima, LC-MS级-批号1731013 ) ( 甲醇: Honeywell LC-MS Chromasolv-批号SZBG246C ) 三氟乙酸: Sigma-Aldrich-批号6942V 磷酸二氢钠:Sigma-Aldrich-批号BCBV1183 磷酸氢二钠(无水):Fisher Chemicals - 批号164693 四丁基硫酸氢铵:ACROS Organics-批号A0375G05 戊烷磺酸: ACROS Organics-批号A0378319 分析之前,用空接头(部件号700002636)替换色谱柱,采用如下条件(表2)计算各系统配置的扩散体积。 表2.用于计算LC系统柱外体积的方法条件。 参数 值 样品 0.16mg/L咖啡因,溶于水:乙腈(90:10) 中,部件号700002642,溶液7 流动相A(MPA) 水(30%) 流动相B(MPB) 乙腈(70%) 流速 0.3 mL/min 进样体积 1uL PDA 入 (±1nm) 273nm 将4.4%峰高处的峰宽(5o, min)乘以流速(uL/min),计算柱外扩散体积(pL)。 图2.使用各种配置得到的咖啡因标准品色谱图的叠加图。 表3.使用咖啡因标准品在各种配置下计算得出的峰宽和扩散体积。 配置 4.4%峰高处的峰宽(5o) 峰扩散(流速×4.4%峰高处的峰宽-5o) ACQUITY UPLC H-Class 0.033 9.9 CM-A ACQUITY UPLC H-Class 0.048 14.4 CH30-A ACQUITY UPLC H-Class CM-A,配备50uL定量环 0.230 69.0 ACQUITY UPLC H-Class 0.220 62.1 CH30-A,配备50pL定量环 结果与讨论 在ACQUITY UPLC H-Class系统中额外增加柱前体积(即加装50pL定量环)后,显著改善了对乙酰氨基酚(图3~4/表5~6)、伊曲康唑(图5~6/表8~9)、氟哌啶醇(图7~8/表11~12)和氯雷他定(图9~10/表15~16)的峰形。对乙酰氨基酚(0.2变为1.0)和氟哌啶醇(0.1变为1.0)的峰对称性也大幅提升。此外,这些化合物的峰面积%RSD也有所改善(表22)。 额外增加柱前体积后性能改善的USP方法 示例1:对乙酰氨基基(分析)-稀释剂:100%甲醇 表4.对乙酰氨基酚USP方法。 PDA入 (±1nm) 1 色谱柱 X-Bridge"BEH C, 100 ×4.6 mm,3.5 pm,部件号186003054 1.7g/L磷酸二氢钠 流动相A(MPA)1.8g/L无水磷酸氢二钠_ 流动相B(MPB) 甲醇 流速 1.0 mL/min 进样体积 5pL 时间(min) MPA MPB 0 99 1 3 99 1 峰对称性-无定量环 样品名称 进样编号保留时间 不对称性@ (4.4)2 峰面积 (pV*s) 1 对乙酰氨基酚 1 5.876 0.198 1785445 2 对乙酰氨基酚 2 5.876 0.202 1794213 3 对乙酰氨基酚 3 5.875 0.201 1791885 4 对乙酰氨基酚 4 5.877 0.204 1764207 5 对乙酰氨基酚 5 5.876 0.202 1787212 6 对乙酰氨基酚 6 5.877 0.199 1768038 平均值 0.2 1781833.4 标准差 0.0 12627.6 %RSD 1.0 0.7 图3/表5.不额外增加进样前体积的对乙酰氨基酚USP分析方法。在不使用定量环的条件下得到的对乙酰氨基酚叠加色谱图(n=6)和结果表。 图4/表6.额外增加了进样前体积的对乙酰氨基酚USP分析方法。在使用定量环的条件下得到的对乙酰氨基酚叠加色谱图(n=6)和结果表。 使用UPLC评估增加柱前系统体积对含有高有机相稀释剂的样品峰形的影响 示例2:伊曲康唑 伊曲康唑(杂质)-稀释剂:含0.4%HCI的甲醇 表7.伊曲康唑USP方法。 参数 值 流动相A(MPA) 0.08M四丁基硫酸氢铵溶液 流动相B(MPB) 乙腈 流速 1.5 mL/min 进样体积 5pL 时间(min) MPA MPB 0 80 20 20 80 20 25 50 50 26 50 50 色谱柱 Spherisorb ODS2 100 × 4.6 mm, 3 um,部件号PSS832112 柱温 30°C 峰对称性-无定量环 样品名称 进样编号保留时间 不对称性@ 峰面积 (4.4)2 (uV*s) 1 伊曲康唑 1 16.730 0.381 168567 2 伊曲康唑 2 16.713 0.432 170807 3 伊曲康唑 3 16.716 0.386 162289 4 伊曲康唑 4 16.712 0.505 165506 5 伊曲康唑 5 16.708 0.675 168937 6 伊曲康唑 6 16.703 0.507 172088 平均值 0.5 168032.4 标准差 0.1 3595.0 %RSD 22.8 2.1 图5/表8.不额外增加进样前体积的伊曲康唑USP分析方法。在不使用定量环的条件下得到的伊曲康唑叠加色谱图(n=6)和结果表。 峰对称性-使用定量环 样品名称 进样编号保留时间 不对称性@ 峰面积 (4.4)2 (uV*s) 1 伊曲康唑 1 16.984 2.028 193662 2 伊曲康唑 2 16.984 2.232 198378 3 伊曲康唑 3 16.984 2.237 196008 4 伊曲康唑 4 16.988 2.051 196581 5 伊曲康唑 5 16.989 1.938 193329 6 伊曲康唑 6 16.984 2.008 195142 平均值 2.1 195516.5 标准差 0.1 1893.4 %RSD 6.0 1.0 图6/表9.外外增加了进样前体积的伊曲康唑USP分析方法。在使用定量环的条件下得到的伊曲康唑叠加色谱图(n=6)和结果表。 示例3:氟哌啶醇 氟哌啶醇USP方法(杂质)-稀释剂:100%甲醇 表10.氟哌啶醇USP方法。 参数 值 流动相A(MPA) 17 g/L戊烷磺酸(pH3.0)流动相B(MPB) 乙腈 流速 1.2 mL/min 进样体积 10 uL 时间(min) MPA MPB 0 90 10 梯度条件 2 90 10 17 50 50 22 50 50 色谱柱 Spherisorb ODS2 100 ×4.6 mm,3 um, 部件号PSS832112 柱温 30°C PDA 入(±1nm) 230 nm 峰对称性-无定量环 不对称性@ 峰面积 样品名称 进样编号保留时间 (4.4)² (uV*s) 1 氟哌啶醇 1 7.400 0.066 406034 2 氟哌啶醇 2 7.401 0.060 399908 3 氟哌啶醇 3 7.401 0.061 402802 4 氟哌啶醇 4 7.399 0.064 402121 5 氟哌啶醇 5 7.398 0.061 402155 6 氟哌啶醇 6 7.396 0.062 401866 平均值 0.1 402481.0 标准差 0.0 1998.3 %RSD 3.9 0.5 图7/表11.不额外增加进样前体积的氟哌啶醇USP分析方法。在不使用定量环的条件下得到的氟哌啶醇叠加色谱图(n=6)和结果表。 峰对称性-使用定量环 样品名称 进样编号保留时间 不对称性@ 峰面积 (4.4)2 (uV*s) 1 氟哌啶醇 1 7.455 0.975 400839 2 氟哌啶醇 2 7.454 0.967 400511 3 氟哌啶醇 3 7.452 0.963 399617 4 氟哌啶醇 4 7.450 0.970 397566 5 氟哌啶醇 5 7.450 0.963 400581 6 氟哌啶醇 6 7.452 0.962 398898 平均值 1.0 399668.6 标准差 0.0 1259.3 %RSD 0.6 0.3 图8/表12.额外增加了进样前体积的氟哌啶醇USP分析方法。在使用定量环的条件下得到的氟哌啶醇叠加色谱图(n=6)和结果表。 示例4:氯雷他定 氯雷他定USP方法(杂质)-稀释剂:100%甲醇 表13.氯雷他定USP方法 参数 值 流动相A(MPA) 0.96 g/L戊烷磺酸(pH3.0)流动相B(MPB) 乙腈 流速 1.2mL/min 进样体积 20pL 时间(min) MPA MPB 0 75 25 20 50 50 梯度条件 30 40 60 35 30 70 45 30 70 50 75 25 色谱柱 XBridge 25 cm ×4.6 mm,5 um,部件号186003117 柱温 30°C PDA 入(±1nm) 254nm 峰对称性-无定量环 样品名称 进样编号保留时间 不对称性@ 峰面积 (4.4)² (uV*s) 1 氯雷他定 1 17.792 2.788 231395 2 氯雷他定 2 17.792 2.864 229278 3 氯雷他定 3 17.788 2.766 228530 4 氯雷他定 4 17.787 2.769 227747 5 氯雷他定 5 17.788 2.861 227574 6 氯雷他定 6 17.781 2.851 226664 平均值 2.8 228531.3 标准差 0.0 1660.0 %RSD 1.7 0.7 图9/表14.不额外增加进样前体积的氯雷他定USP分析方法。在不使用定量环的条件下得到的氯雷他定叠加色谱图(n=6)和结果表。 峰对称性-使用定量环 样品名称尔 进样编号保留时间 不对称性@ (4.4)2 峰面积 (uV*s) 1 氯雷他定 1 18.030 1.811 282785 2 氯雷他定 2 18.025 1.803 279985 3 氯雷他定 3 18.024 1.817 279396 4 氯雷他定 4 18.019 1.834 279641 5 氯雷他定 5 18.019 1.838 279970 6 氯雷他定 6 18.022 1.786 279908 平均值 1.8 280280.6 标准差 0.0 1247.9 %RSD 1.1 0.4 图10/表15.额外增加了进样前体积的氯雷他定USP分析方法。在使用定量环的条件下得到的氯雷他定叠加色谱图(n=6)和结果表。 比卡鲁胺(图11~12/表17~18)在不使用定量环的条件下未表现出明显的峰畸变,并且额外增加柱前体积也未对其产生显著影响(积极影响或不良影响)。这可能是由于有机稀释剂的浓度相对较低(67%)。 额外增加柱前体积后性能不变的USP方法 示例5:比卡鲁胺 比卡鲁胺USP方法(杂质)-稀释剂:67%甲醇/0.01% TFA 表16.比卡鲁胺USP方法。 参数 值 流动相A(MPA) 水/0.01% TFA 流动相B(MPB) 乙腈/0.01%TFA 流速 进样体积 1.0 mL/min 10pL 时间(min) MPA MPB 0 75 25 梯度条件 16.5 50 50 26.5 40 60 32.5 30 70 32.6 30 70 35 75 25 色谱柱 Phenomenex ODS3 Cg100×4.0mm, 3 pm, 部件号00D-4222-D0 柱温 30°C PDA 入(±1nm) 270nm 峰对称性-无定量环 样品名称 进样编号保留时间 不对称性@ 峰面积 (4.4)2 (uV*s) 1 比卡鲁胺 1 21.401 0.947 31747 2 比卡鲁胺 2 21.402 0.999 32346 3 比卡鲁胺 3 21.419 1.039 32001 4 比卡鲁 4 21.420 0.960 32552 5 比卡鲁胺 5 21.425 0.824 31797 6 比卡鲁胺 6 21.419 0.911 32027 平均值 0.9 32078.4 标准差 0.1 314.2 %RSD 7.9 1.0 图11/表17.不额外增加进样前体积的比卡鲁胺杂质USP分析方法。在不使用定量环的条件下得到的比卡鲁胺叠加色谱图(n=6)和结果表。 峰对称性-使用定量环 样品名称 进样编号保留时间 不对称性@ 峰面积 (4.4)2 (pV*s) 1 比卡鲁胺 1 21.490 1.160 31970 2 比卡鲁胺 2 21.502 1.013 32503 3 比卡鲁胺 3 21.499 1.065 32736 4 比卡鲁按 4 21.506 0.934 32287 5 比卡鲁胺 5 21.506 1.084 32519 6 比卡鲁按 6 21.506 0.951 31920 平均值 1.0 32322.3 标准差 0.1 325.5 %RSD 8.3 1.0 图12/表18.额外增加了进样前体积的比卡鲁胺杂质USP分析方法。在使用定量环的条件下得到的比卡鲁胺叠加色谱图(n=6)和结果表。 表19.酮康唑USP方法。 参数 值 流动相A(MPA) 3.4 mg/mL四丁基硫酸氢铵溶液: ACN(50:50) 流动相B(MPB) 3.4mg/mL四丁基硫酸氢铵溶液:ACN (50:50) 流速 进样体积 2.0 mL/min 10 pL 时间(min) MPA MPB 0 100 0 梯度条件 20 0 100 25 0 100 26 100 0 30 100 0 色谱柱 Spherisorb ODS2 100 × 4.6 mm, 3 um, 部件号PSS832112 柱温 30°C PDA 入 (±1nm) 225nm 尽管使用100%甲醇作为稀释剂,酮康唑(图13~14/表20~21)在不使用定量环的条件下未表现出明显的峰畸变,并且额外增加柱前体积也未对其产生显著影响(积极影响或不良影响)。这可能是由于流动相A和B中均存在有机组分,有利于样品在注入色谱柱之前充分混合。这一理论需要通过进一步研究进行验证。 峰对称性-无定量环 6 (4.4)2 (uV*s) 1 8.892 1.669 1596714 8.890 1.773 1611969 2 酮康唑 2 3 酮康唑 3 8.891 1.799 4 1608891 5 1.818 1607669 8.892 1.837 1609027 1609477 6 8.892 1.839 1607291.1 1.8 0.1 5371.4 3.6 0.3 图13/表20.不额外增加进样前体积的酮康唑USP分析方法。在不使用定量环的条件下得到的酮康唑叠加色谱图(n=6)和结果表。 峰对称性-使用定量环 样品名称送进样编号保留时间 不对称性@(4.4) 峰面积(uV*s) 1 酮康唑 1 8.870 1.794 1586698 2 酮康唑 2 8.872 1.794 1571862 3 酮康唑 3 8.874 1.857 1581520 4 酮康唑 4 8.876 1.907 1588168 5 酮康唑 5 8.877 1.908 1605917 6 酮康唑 6 8.876 1.932 1608888 平均值 1.9 1590508.9 标准差 0.1 14308.8 %RSD 3.2 0.9 图14/表21.额外增加了进样前体积的酮康唑USP分析方法。在使用定量环的条件下得到的酮康唑叠加色谱图(n=6)和结果表。 使用UPLC评估增加柱前系统体积对含有高有机相稀释剂的样品峰形的影响 表22.在增加或不增加50pL柱前体积的条件下得到的所有化合物的分析结果汇总。 化合物 H-Class H-Class (加装50 uL定量环) 平均值 峰面积 平均 峰面积 不对称性 %RSD 不对称性 @ 4.4(n=6) (n=6) @ 4.4(n=6) %RSD (n=6) 对乙酰氨基酚 0.2 0.7 1.0 0.5 伊曲康唑 0.5 2.1 2.1 1.0 氟哌啶醇 0.1 0.5 1.0 0.3 氯雷他定 2.8 0.7 1.8 0.4 比卡鲁胺 0.9 1.0 1.0 1.0 酮康唑 1.8 0.3 1.9 0.9 结果与讨论 利用50 uL定量环增加柱前体积可显著影响对乙酰氨基酚、伊曲康唑、氟哌啶醇以及氯雷他定的峰形和峰面积%RSD,说明这是在低扩散LC系统上使用高有机相稀释剂时解决峰畸变问题的一个有效策略。 采用该解决方案,需要遵循药典方法参数的客户只需进行合规实验室环境下完全受支持的简单配置更改,就能解决所得数据不理想/不可用的问题。 ( 参考文献 ) ( 1:C E ric Loeser, Patrick Drum:Using strong injectionsolvents with 100% aqueous mobile phase inRP-LC.J.Sep.Sci 2006,29,2847-2852. ) ( 2. Sonia Keunchkarian, Mario Reta,Lilian Romero, Cecilia Castells: E ffect of sample solvent on thechromatographic peak shape of analytes elutedunder reversed-phase liquid chromatographic conditions. Journal of Chromatography A. ) 扫一扫,关注沃特世微信 沃特斯中国有限公司沃特世科技(上海)有限公司 北京:010-52093866 上海:021-61562666 广州:020-28295999 成都:028-67653588 香港:852-29641800 免费售后服务热线:800(400)820 2676www.waters.com 使用UPLC评估增加柱前系统体积对含有高有机相稀释剂的样品峰形的影响 使用UPLC评估增加柱前系统体积对含有高有机相稀释剂的样品峰形的影响 简介理想情况下,在运行色谱方法时,样品稀释剂组分应尽可能接近方法起始条件。这样做的目的是最大程度减小由样品溶剂效应引起的谱带展宽和峰畸变,进而免出现峰不对称性、峰分裂或数据不可用的情况。引起溶剂效应的原因是稀释剂与流动相之间存在洗脱强度差异。当稀释剂的洗脱强度高于流动相时,峰展宽和峰形异常的情况通常会更加严重1-2。事实上,业内普遍认为,最好是将进样的样品溶解于起始流动相中。然而,给定样品的预处理常常需要将分析物溶解在与流动相组分相差很大的溶剂中。为了避免溶解度问题和峰形不佳的问题,许多方案都要求在预处理过程中挥干样品溶剂,然后将样品复溶于流动相中。然而,这个额外的步骤非常耗时,所需时间往往比HPLC分析的时间还要长1。一般而言,推荐的做法是避免使用比流动相更强的溶剂来溶解样品和标准品。这种做法基于如下假设:强于流动相的进样溶剂会干扰样品在柱头处的吸附,而采用大体积进样时尤其如此2。遗憾的是,这种做法在实践中可行性不佳,因为分析人员往往必须根据样品的溶解度来决定有机溶剂的含量,以确保样品能够完全溶解。对于扩散体积较大的传统LC系统,这种现象带来的问题较少,因为柱前样品/溶剂/流动相混合很充分,可以缓解溶剂效应造成的色谱峰问题。然而,对于现代的低分散U(H)PLC系统,如果以较大体积进样含有高有机相的样品,就会出现问题,并可能导致峰对称性变差或峰分裂。结果与讨论利用50 μL定量环增加柱前体积可显著影响对乙酰氨基酚、伊曲康唑、氟哌啶醇以及氯雷他定的峰形和峰面积%RSD,说明这是在低扩散LC系统上使用高有机相稀释剂时解决峰畸变问题的一个有效策略。采用该解决方案,需要遵循药典方法参数的客户只需进行合规实验室环境下完全受支持的简单配置更改,就能解决所得数据不理想/不可用的问题。
确定



还剩8页未读,是否继续阅读?

产品配置单

沃特世科技(上海)有限公司(Waters)为您提供《高有机相稀释剂中样品峰形检测方案(液相色谱仪)》,该方案主要用于其他中样品峰形检测,参考标准--,《高有机相稀释剂中样品峰形检测方案(液相色谱仪)》用到的仪器有二维ACQUITY UPLC系统
推荐专场






相关方案
更多
该厂商其他方案
更多