








方案详情
文
天然和合成阿片类药物一直是法医毒理学研究的一个重要方面。有比例相当高的犯罪和/或死亡事件都是由误用或滥用麻醉镇痛药(如羟考酮和氢可酮),以及非法阿片类药物海洛因所引起。法医实验室通常需要分析全血样本中的不同药物以确定确切的死亡原因,或是判断是否在药物作用下驾驶,或者出于其它犯罪或研究目的。
过去,通常用酸或酶使样品水解并释放出葡糖苷酸代谢物之后再进行GC/MS分析。通过酶水解将葡糖苷酸代谢物转化为其游离形式的处理步骤增加了分析时间和成本,而且无法确保水解程度始终如一。随着现代UPLC/MS/MS技术的出现,如今可直接对葡糖苷酸代谢物进行分析。
方案详情

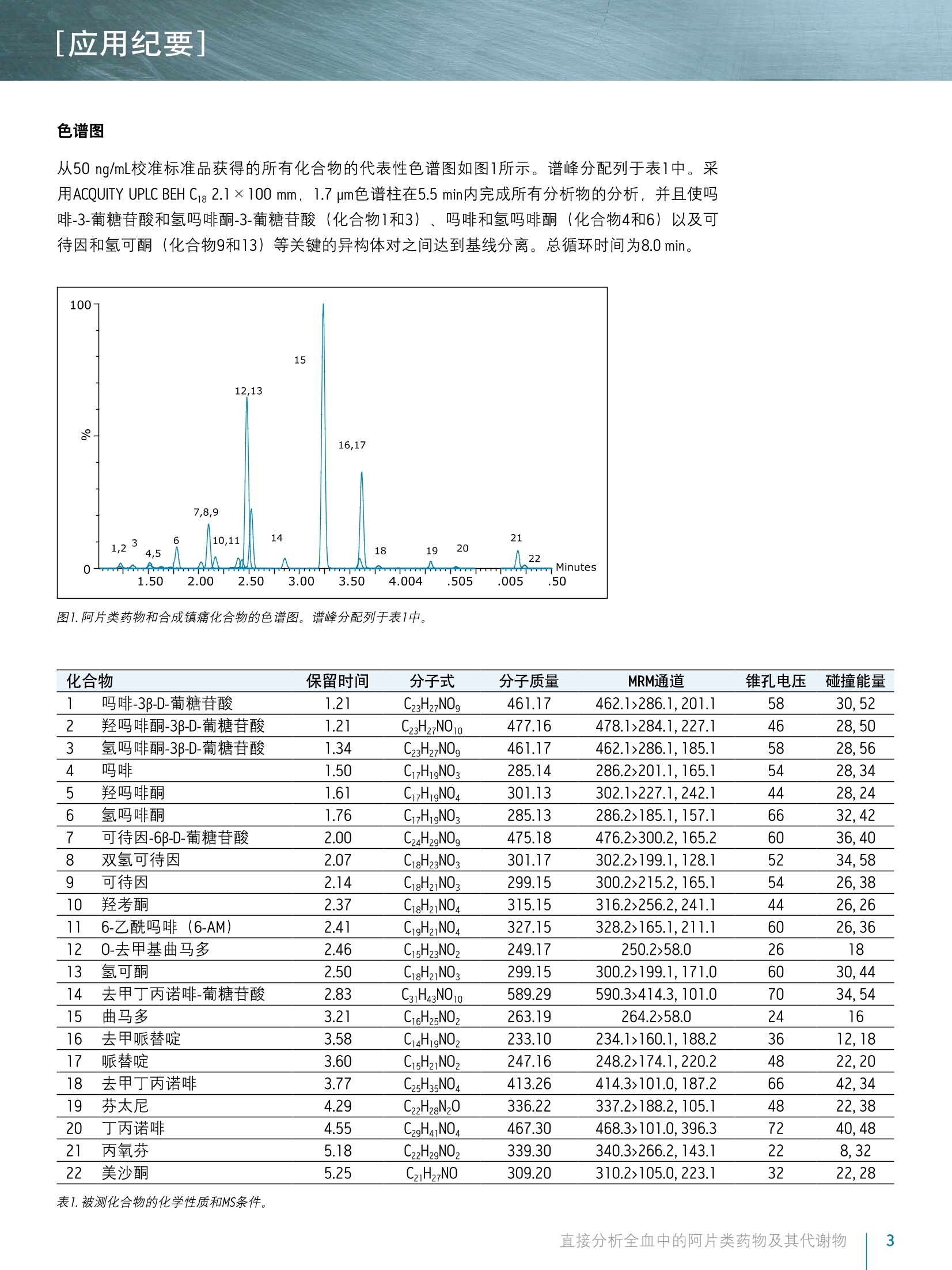
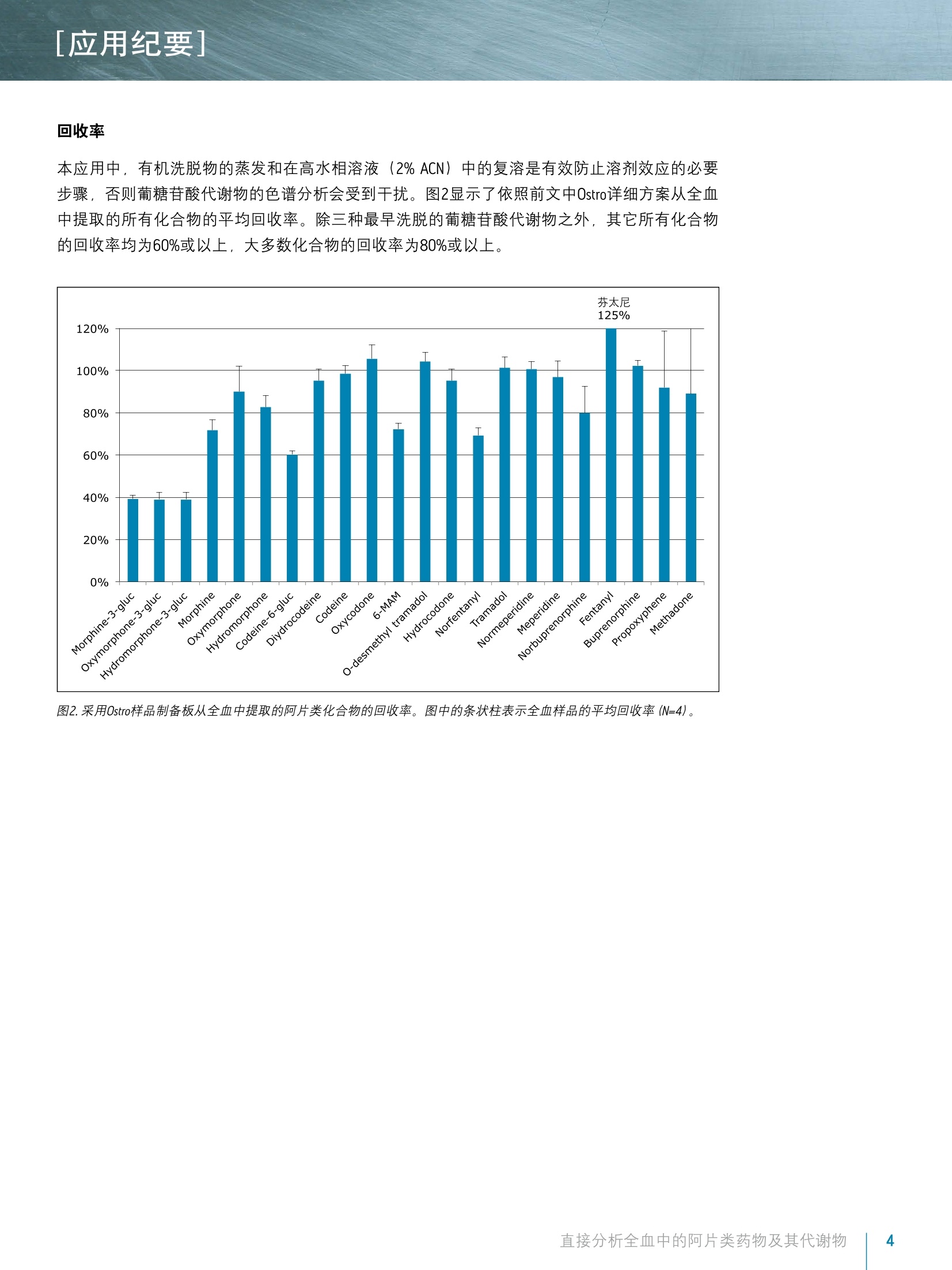
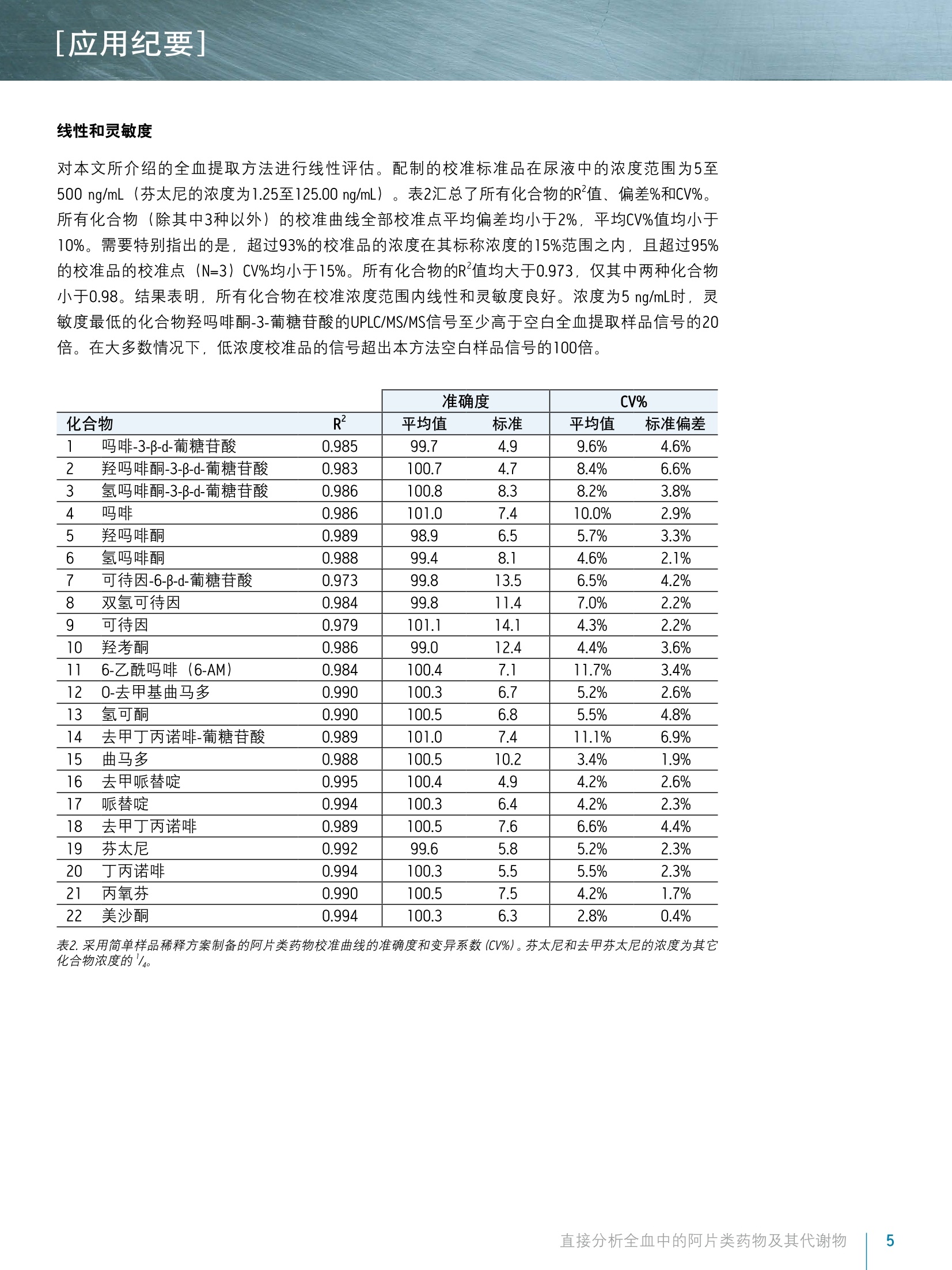
[应用纪要]THE SCIENCE OF WHAT'S POSSIBLE, [应用纪要] ■ ■ Waters 使用Ostro样品前处理板结合UPLC/MS/MS对全血中的阿片类药物及其代谢物进行法医毒理学直接分析 Jonathan P. Danaceau. Erin E. Chambers and Kenneth J. Fountain 沃特世公司(美国马萨诸塞州米尔福德) 应用优势 无需酶水解即可分析葡糖苷酸代谢物 适用于由22种阿片类药物和阿片类镇痛化合物构成的综合性药物组 ■与传统的LLE、SPE或蛋白质沉淀法相比,样品制备快速简单 去除了内源性磷脂质 对所有分析物和代谢物具有线性响应 沃特世解决方案 Ostro"96孔板 ACQUITY UPLCBEH色谱柱ACQUITY UPLC系统 XevoTQD质谱仪 关键词 阿片类药物,阿片类, UPLC,法医毒理学,全血 简介 天然和合成阿片类药物一直是法医毒理学研究的一个重要方面。有比例相当高的犯罪和/或死亡事件都是由误用或滥用麻醉镇痛药(如羟考酮和氢可酮),以及非法阿片类药物海洛因所引起。法医实验室通常需要分析全血样本中的不同药物以确定确切的死亡原因,或是判断是否在药物作用下驾驶,或者出于其它犯罪或研究目的。过去,通常用酸或酶使样品水解并释放出葡糖苷酸代谢物之后再进行GC/MS分析。通过酶水解将葡糖苷酸代谢物转化为其游离形式的处理步骤增加了分析时间和成本,而且无法确保水解程度始终如一。随着现代UPLC/MS/MS技术的出现,如今可直接对葡糖苷酸代谢物进行分析析。 全血分析可采用多种样品制备方法,包括液液萃取(LLE)和固相萃取(SPE)。最简单的一种方法就是在细胞溶解之后进行蛋白质沉淀。本研究中介绍了一种快速直接的样品制备方法,采用Ostro样品制备板对全血样品进行预处理以裂解细胞,然后用乙腈沉淀,再用96孔板洗脱。所有样品预处理均在Ostro板孔中进行,无需离心或从各个离心管中转移样品。 样品制备完成之后,对22种阿片类药物及其代谢物进行UPLC/MS/MS分析。葡糖苷酸代谢物可直接进行分析,无需酶水解或化学水解。校准曲线呈线性,并可轻松达到相应的检测限。 实验 LC条件 系统: ACQUITY UPLC 色谱柱: ACQUITY UPLC BEH C18 2.1×100 mm, 1.7 pm (部件号186002352) 柱温: 30℃ 进样体积: 10pL 流速: 0.4mL/min流动相A: 0.1%甲酸的MilliQ水溶液流动相B: 0.1%甲酸梯度: 初始条件为2%的流动相B。流动相B在6 min内增加至47.2%,然后在0.5 min内返回至2%。重新平衡系统1.5 min。整个循环时间为8.0min。MS条件 质谱仪: Xevo TQD电离模式: ESI+采集模式: MRM (离子通道参阅表1)毛细管电压: 1kV碰撞能量(eV): 针对具体化合物进行优化(参阅表1)锥孔电压(V): 针对具体化合物进行优化(参阅表1) ( 数据管理 ) ( WatersMassLynx软件4.1版 ) 样品描述 所有化合物和内标物(IS)均购自Cerilliant (德克萨斯州圆石))。另外,除氢吗啡酮-3-葡糖苷酸、可待因-6-葡糖苷酸和去甲丁丙诺啡-葡糖苷酸之外,所有化合物均有对应的气代内标。对于这些化合物,具有极为相似的响应的的代IS,被选择作为替代物。 所有化合物的混合储备液(10 pg/mL; 芬太尼浓度为2.5 pg/mL)用甲醇配制。工作溶液需每天以基质(血液)配制的高浓度标准品和QC样品制备,并进行连续稀释以得到所需浓度。所有分析物的校准浓度范围为5至500 ng/mL, 但芬太尼的浓度配制为其它分析物浓度的25%((1.25至125.00ng/mL)。所有内标混合储液(5 pg/mL;芬太尼为1.25 ug/mL)用乙腈配制。 样品制备 在Ostro样品制备板的孔中加入150 uL 0.1 M ZnSO/0.1 M NHCH,COOH的水溶液,制备全血样品。向ZnSO_/NHCH,COOH溶液中加入50uL全血样品,并简单混合(5s),使细胞裂解。然后,在制备的样品中加入含有内标的乙腈600uL。涡旋混合3 min之后,将样品洗脱至96孔收集板,在N,下蒸干,之后复溶于50uL含0.1%甲酸的2%乙腈溶液中。取10uL注入LC/MS/MS系统。 结果与讨论 表1中列出了22种筛选化合物和代谢物,它们构成一组全面的化合物组,包括天然阿片类药物、半合成阿片片及合成麻醉性镇痛化合物。这些化合物多数为弱碱性,其pKa值大约为8至9。它们的极性范围较广,LogP值的范围为-3.48(吗啡-3β-d-葡糖苷酸)至5.0(美沙酮))。所用MRM通道同见表1。除曲马朵和0-去甲基曲马朵之外,均列出了主要MRM通道和确证MRM通道及其碰撞能量。 色谱图 从50 ng/mL校准标准品获得的所有化合物的代表性色谱图如图1所示。谱峰分配列于表1中。采用ACQUITY UPLC BEH C1g 2.1×100 mm, 1.7 um色谱柱在5.5 min内完成所有分析物的分析,并且使吗啡-3-葡糖苷酸和氢吗啡酮-3-葡糖苷酸(化合物1和3)、吗啡和氢吗啡酮(化合物4和6)以及可待因和氢可可(化合物9和13)等关键的异构体对之间达到基线分离。总循环时间为8.0 min。 8- 22 mmm Minutes 12,13 7,8,9 143 6 10,111,24,5 图1.阿片类药物和合成镇痛化合物的色谱图。谱峰分配列于表1中。 化合物 保留时间 分子式 分子质量 MRM通道 锥孔电压 碰撞能量 1 吗啡-3B-D-葡糖苷酸 1.21 C23H27NO。 461.17 462.1>286.1,201.1 58 30,52 2 羟吗啡酮-3B-D-葡糖苷酸 1.21 C23H27NO10 477.16 478.1>284.1,227.1 46 28.50 3 氢吗啡酮-3B-D-葡糖苷酸 1.34 C23H27N0。 461.17 462.1>286.1,185.1 58 28.56 4 吗啡 1.50 C7H1gNO3 285.14 286.2>201.1,165.1 54 28,34 5 羟吗啡酮 1.61 C17H1gNO 301.13 302.1>227.1.242.1 44 28,24 6 氢吗啡酮 1.76 C17H1gNO 285.13 286.2>185.1,157.1 66 32,42 7 可待因-6B-D-葡糖苷酸 2.00 CzAH2NO。 475.18 476.2>300.2,165.2 60 36.40 8 双氢可待因 2.07 C18H23NO: 301.17 302.2>199.1,128.1 52 34,58 9 可待因 2.14 C18H2NO: 299.15 300.2>215.2,165.1 54 26.38 10 羟考酮 2.37 C18H2NO 315.15 316.2>256.2,241.1 44 26.26 11 6-乙酰吗啡 :(6-AM) 2.41 CgHNO 327.15 328.2>165.1,211.1 60 26,36 12 0-去甲基曲马多 2.46 C15H23NO, 249.17 250.258.0 26 18 13 氢可酮 2.50 C18H2N03 299.15 300.2>199.1,171.0 60 30,44 14 去甲丁丙诺啡-葡糖苷酸 2.83 C31H43NO1o 589.29 590.3>414.3,101.0 70 34,54 15 曲马多 3.21 C16H25NO2 263.19 264.2>58.0 24 16 16 去甲哌替啶 3.58 C4H19NO, 233.10 234.1>160.1,188.2 36 12,18 17 哌替啶 3.60 CsHNO, 247.16 248.2>174.1,220.2 48 22,20 18 去甲丁丙诺啡 3.77 C25H35NO4 413.26 414.3>101.0,187.2 66 42,34 19 芬太尼 4.29 C22H28N20 336.22 337.2>188.2,105.1 48 22,38 20 丁丙诺啡 4.55 C29H41NO4 467.30 468.3>101.0,396.3 72 40,48 21 丙氧芬 5.18 C22H29NO2 339.30 340.3>266.2,143.1 22 8,32 22 美沙酮 5.25 C21H27NO 309.20 310.2>105.0.223.1 32 22,28 ( 表1.被测化合物的化学性质和MS条件。 ) 回收率 线性和灵敏度 对本文所介绍的全血提取方法进行线性评估。配制的校准标准品在尿液中的浓度范围为5至500 ng/mL (芬太尼的浓度为1.25至125.00 ng/mL)。表2汇总了所有化合物的R值、偏差%和CV%。所有化合物(除其中3种以外)的校准曲线全部校准点平均偏差均小于2%,平均CV%值均小于10%。需要特别指出的是,超过93%的校准品的浓度在其标称浓度的15%范围之内,且超过95%的校准品的校准点(N=3) CV%均小于15%。所有化合物的R值均大于0.973,仅其中两种化合物小于0.98。结果表明,所有化合物在校准浓度范围内线性和灵敏度良好。浓度为5 ng/mL时, 灵敏度最低的化合物羟吗啡酮-3-葡糖苷酸的UPLC/MS/MS信号至少高于空白全血提取样品信号的20倍。在大多数情况下,低浓度校准品的信号超出本方法空白样品信号的100倍。 准确度 CV% 化合物 R2 平均值 标准 平均值 标准偏差 1 吗啡-3-B-d-葡糖苷酸 0.985 99.7 4.9 9.6% 4.6% 2 羟吗啡酮-3-B-d-葡糖苷酸 0.983 100.7 4.7 8.4% 6.6% 3 氢吗啡酮-3-B-d-葡糖苷酸 0.986 100.8 8.3 8.2% 3.8% 4 吗啡 0.986 101.0 7.4 10.0% 2.9% 5 羟吗啡酮 0.989 98.9 6.5 5.7% 3.3% 6 氢吗啡酮 0.988 99.4 8.1 4.6% 2.1% 7 可待因-6-B-d-葡糖苷酸 0.973 99.8 13.5 6.5% 4.2% 8 双氢可待因 0.984 99.8 11.4 7.0% 2.2% 9 可待因 0.979 101.1 14.1 4.3% 2.2% 10 羟考酮 0.986 99.0 12.4 4.4% 3.6% 11 6-乙酰吗啡(6-AM) 0.984 100.4 7.1 11.7% 3.4% 12 0-去甲基曲马多 0.990 100.3 6.7 5.2% 2.6% 13 氢可酮 0.990 100.5 6.8 5.5% 4.8% 14 去甲丁丙诺啡-葡糖苷酸 0.989 101.0 7.4 11.1% 6.9% 15 曲马多 0.988 100.5 10.2 3.4% 1.9% 16 去甲哌替啶 0.995 100.4 4.9 4.2% 2.6% 17 哌替啶 0.994 100.3 6.4 4.2% 2.3% 18 去甲丁丙诺啡 0.989 100.5 7.6 6.6% 4.4% 19 芬太尼 0.992 99.6 5.8 5.2% 2.3% 20 丁丙诺啡 0.994 100.3 5.5 5.5% 2.3% 21 丙氧芬 0.990 100.5 7.5 4.2% 1.7% 22 美沙酮 0.994 100.3 6.3 2.8% 0.4% 表2.采用简单样品稀释方案制备的阿片类药物校准曲线的准确度和变异系数(CV%)。芬太尼和去甲芬太尼的浓度为其它化合物浓度的'/. 本研究介绍的方法展示了采用Ostro样品制备板结合UPLC/MS/MS对全血样品中的22种目标阿片类化合物及其代谢物进行分析。所有化合物的分析在5.5 min内完成,而且所有同位素化合物对达到完全分离。Ostro样品制备板的使用实现了快速的孔内细胞溶解和蛋白质沉淀,以及随后洗脱至96孔收集板。与在单个试管中进行蛋白质沉淀相比,此方法提高了样品通量,并且有效地消除了内源性磷脂质化合物。所有分析物在整个校准浓度范围内显示出良好的线性,而且本方法可在最低校准点浓度下实现灵敏可靠的检测。 ( 参考文献 ) ( 1 . Goldberger B A , Cone EJ . Co n firmatory tes t s for dr u gs in th e wo r kplace by g aschromatography-mass spectrometry. Journal of Chromatography A. 1 994; 674(1-2): 73-86. ) ( 2 . W a ng P, et al. I n complete re c overy of prescription op i oids in u r i ne using enzymatichydrolysis of g lucuronide metabolites. Journal o f Analytical T o xicology. 2 006;30(8): 570-575. ) ( 3. G ustavsson E, e t al. Validation of direct injection electrospray LC-MS/MS for confirmation ofopiates in u r ine drug testing. Journal of Ma s s Spectrometry. 2007;42(7): 881-889. ) ( 4. Murphy CM, H u estis M A . LC-ESI-MS/MS analysis for the quantification of morphine,codeine, morphine-3-B-D-glucuronide, morphine-6-B-D-glucuronide, and codeine-6-B-D- g lucuronide in human u r ine. J ournal of Mass S pectrometry.2 0 05;40(11): 1412-1416. ) ( 5. Edinboro L , B a cker RC , Poklis A. D ir e ct ana l ysis of opiates in u r in e by li q ui d chromatography-tandem ma s s spectrometry. Jou r nal of A nalytical Toxicology. 20 0 5;29(7):704-710. ) ( 6. French D , Wu A , Lynch K. Hydrophilic in t eraction LC-MS/MS analysis of opioids in ur i ne:significance of glucuronide metabolites. Bioanalysis. 2011; 3(23):2603-2612. ) THE SCIENCE OF WHAT'S POSSIBLE. ( 免费售后服务热线:800(400)820 2676 Www.waters.com ) 直接分析全血中的阿片类药物及其代谢物
确定



还剩4页未读,是否继续阅读?

产品配置单


沃特世科技(上海)有限公司(Waters)为您提供《全血中的阿片类药物及其代谢物进行法医毒理学直接分析法》,该方案主要用于全血/血清/血浆中--检测,参考标准--,《全血中的阿片类药物及其代谢物进行法医毒理学直接分析法》用到的仪器有Waters Xevo TQD 三重四极杆质谱、ACQUITY UPLC 超高效液相色谱
推荐专场






相关方案
更多
该厂商其他方案
更多