








方案详情
文
Over the last several years some focus has been placed on improving efficiency for the determination of
polychlorinated dibenzo-p- dioxins and polychlorinated dibenzo furans (D/Fs), as well as polychlorinated
biphenyl (PCBs) and the polybrominated diphenyl ethers (PBDEs). Most efforts in this area have been focused
on instrumentation; moving from expensive magnetic sector mass spectrometers 1 to triple quadrupoles 2,3 and
orbital trapping mass spectrometers 4 . Lesser efforts have been placed on sample preparation stages, which can
be divided into extraction, cleanup and concentration techniques. Although the manual preparation stage is
inherently inexpensive, compared to instrumentation costs, low level contamination or poor recoveries can result
in rework which negatively impacts the method, making this stage costly. Additionally, the extraction method
varies depending upon the matrix category, which adds to the complexity, thus multiple extraction methods may
be necessary depending upon the moisture and fat content of a matrix. The extraction of milk fat by an
automated acid hydrolysis and abbreviated Soxhlet extraction for determination of POPs have been previously
described 5 . By broadening the extraction method to include egg yolks, prepared cakes, prepared meats and raw
tissues, we are moving toward a single, matrix-independent, extraction method.
方案详情

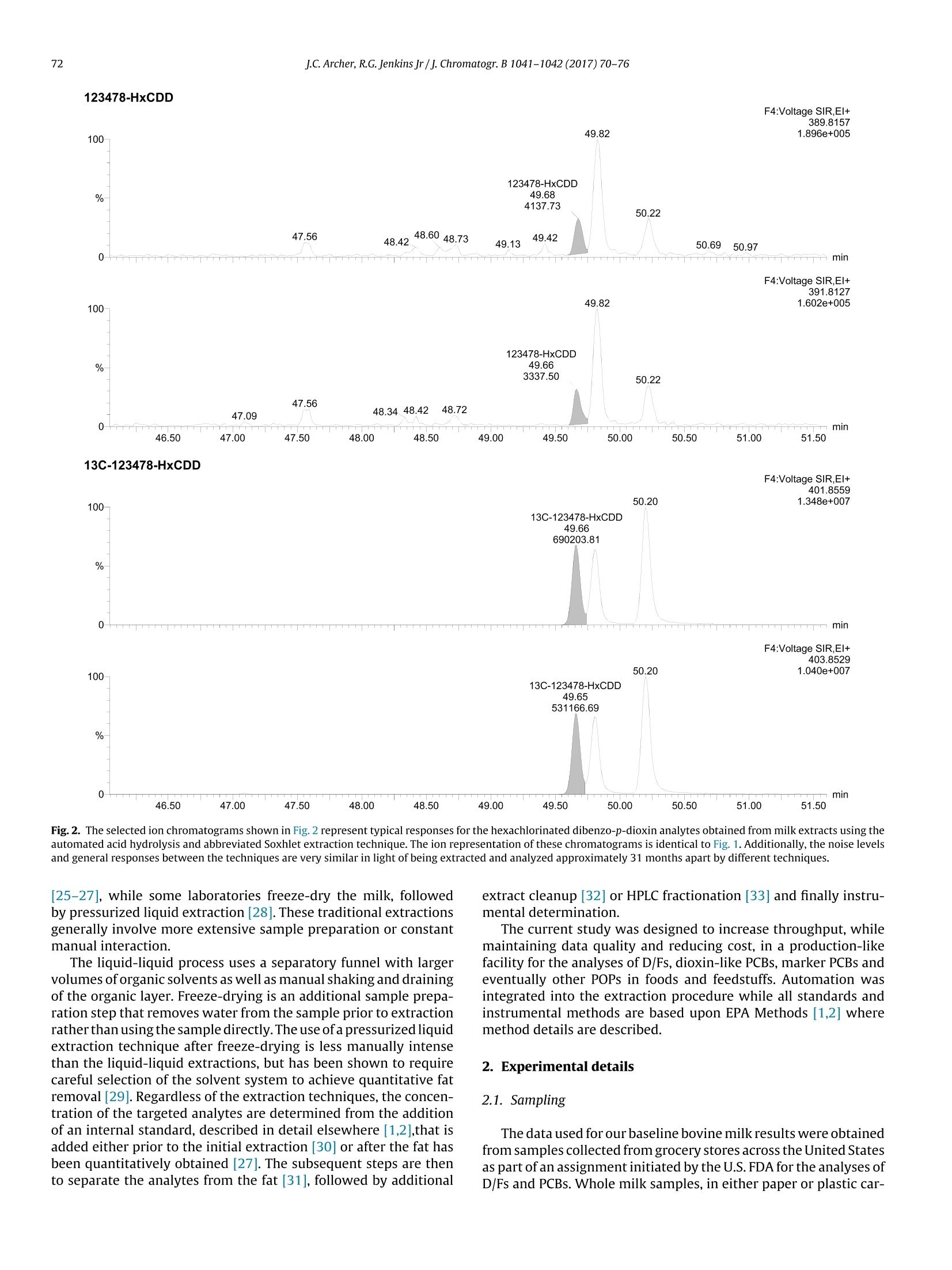
Journal of Chromatography B, 1041-1042(2017)70-76Contents lists available at ScienceDirect J.C. Archer, R.G. Jenkins Jr/J. Chromatogr. B 1041-1042 (2017)70-7671 Journal of Chromatography B ELSEVIER journal homepage: www.elsevier.com/locate/chromb 自动牛奶脂肪萃取分析POPs Automated milk fat extraction for the analyses of persistent organicpollutants Jeffrey C. Archer*, Roy G. Jenkins Jr U.S. Food & Drug Administration, Arkansas Regional Laboratory, 3900 NCTR Road, Building 26, Jefferson, AR 72079, United States ARTICLEIN F O ABSTRACT Article history: Received 14 July 2016Received in revised form 28 October 2016Accepted 3 December2016 Available online 6 December 2016 Keywords: POPsDioxinsExtractionAutomatedAcid hydrolysis We have utilized an automated acid hydrolysis technology, followed by an abbreviated Soxhlet extractiontechnique to obtain fat from whole milk for the determination of persistent organic pollutants, namelypolychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans and polychlorinated biphenyls. Theprocess simply involves (1) pouring the liquid milk into the hydrolysis beaker with reagents and stan-dards, (2) drying the obtained fat on a filter paper and (3)obtaining pure fat via the modified Soxhletextraction using 100 mL of hexane per sample. This technique is in contrast to traditional manually intenseliquid-liquid extractions and avoids the preparatory step of freeze-drying the samples for pressurizedliquid extractions. Along with these extraction improvements, analytical results closely agree betweenthe methods, thus no quality has been compromised. The native spike (n=12) and internal standard(n=24) precision and accuracy results are within EPA Methods 1613 and 1668 limits. While the median(n=6) Toxic Equivalency Quotient (TEQ) for polychlorinated dibenzo-p-dioxins/polychlorinated diben-zofurans and the concentration of the marker polychlorinated biphenyls show a percent difference of 1%and 12%,respectively, compared to 315 previously analyzed milk samples at the same laboratory usingliquid-liquid extraction. During our feasibility studies, both egg and fish tissue show substantial promiseusing this technique as well. 1. Introduction Polychlorinated dibenzo-p-dioxins and polychlorinated diben-zofurans (D/Fs) as well as polychlorinated biphenyls (PCBs) area group of compounds known as persistent organic pollutants(POPs). These environmental contaminants are typically monitoredby utilizing direct isotope dilution techniques as described in EPAMethods 1613 [1] and 1668 [2]. The D/Fs are commonly combinedas one family, although they are two classes of the 12 initial POPsslated for elimination under the Stockholm Convention along withPCBs as a third class [3]. All three (D/Fs and PCBs) have toxicityeffects, are lipophilic in nature, and have been detected in our foodsupply[4-19]. The major non-occupational exposure to D/Fs and PCBs comesthrough the diet by consuming animal fats in our foods. For examplethe Belgian PCB/Dioxin incident [4] while an isolated contamina-tion of PCB oil to the feed supply, significantly contaminated the ( * Corresponding author. ) Belgian animal food supply. Additional examples of unintentionalcontaminations in feeds include ball clays in the United States inpoultry [5] and catfish [6] feeds, bakery waste in Germany [7], andcholine chloride that crossed borders in Europe [8]. Each of thesecases illustrate where contaminated animal feeds have threatenedour food supply. In the case of dairy animals exposed to contam-inated feeds, these compounds are excreted into the milk andeventually appear in our daily diet [9]. Levels of POPs have been reported in milk from surveil-lance programs throughout the world. Tetra and penta-chlorinateddibenzofuran levels in milk fat from Iran [10] have recently beenreported while additional cow milk surveillance programs havebeen and continue to be reported [11-13], including a study of sea-sonal variations in the U.K. [14]. Other dairy surveillance studieshave also been completed including buffalo[15]goat[16]and sheep[17]. Some of the surveillance studies have resulted in unexpectedfindings leading to follow-up studies such as the citrus pulp inci-dent[18] and Italian buffalo milk levels [19]. The U.S. Food and DrugAdministration (U.S.FDA) completed a raw bovine milk surveil-lance study of about 320 samples during Fiscal Year 2013. A similarstudy was designed for the surveillance of “grocery-purchased" Table 1 Comparison of 315 Historical Milk Results Extracted by Liquid-Liquid Process to 6 Samples Extracted by the Automated Acid Hydrolysis Technique. TEQ n=315 n=6 n=315 n=6 n=315 n=6 Sum of Dioxins Sum of Dioxins Sum of Dioxins and Sum of Dioxins and Sum of PCB28. Sum of PCB28. (2005 (2005 Dioxin-Like PCBs Dioxin-Like PCBs PCB52.PCB101, PCB52,PCB101, WHO-PCDD/F-TEQ) WHO-PCDD/F-TEQ) (2005 (2005 PCB138, PCB 153 PCB138, PCB 153 WHO-PCDD/F-TEQ) WHO-PCDD/F-TEQ) andPCB180 and PCB181 Average 0.354 0.292 0.474 0.359 0.504 0.348 sd 0.207 0.069 0.245 0.058 0.454 0.041 Median 0.321 0.318 0.435 0.372 0.398 0.352 Fig. 1. The selected ion chromatograms shown in Fig. 1 represent typical responses for the hexachlorinated dibenzo-p-dioxin analytes obtained from milk extracts using thetraditional liquid-liquid extraction. The first 2 chromatograms represent the M* and(M+2)* ion responses, masses 389.8 and 391.8, respectively, for the native or incurredlevels analytes, while the following two chromatograms represent the M* and (M+2) ion responses, masses 401.9 and 403.9, respectively, for the C-13 labeled internalstandards, as described in EPA Method 1613 [1]. whole bovine milk (pasteurized) completed Fiscal Year 2014. Thiswas followed by a new, “grocery-purchased"study for Fiscal Year2016. Several extraction techniques for the analyses of POPs in milkhave been explored over the years. The goal for each is the same;first isolate the sample lipid material, followed by the isolation of the POPs from the lipid material. Recently concentrations haveroutinely been reported on a lipid weight basis. There are severalmethods for the determination of fat in milk that have been used.including Babcock [20], the Gerber Method [21],as well as othermore recently developed and published techniques [22-24]. Mostlaboratories extract using some variation of liquid-liquid technique F4:Voltage SIR,EI+ 46.50 47.00 47.50 48.00 48.50 49.00 49.50 50.00 50.50 51.00 51.50 Fig. 2. The selected ion chromatograms shown in Fig. 2 represent typical responses for the hexachlorinated dibenzo-p-dioxin analytes obtained from milk extracts using theautomated acid hydrolysis and abbreviated Soxhlet extraction technique. The ion representation of these chromatograms is identical to Fig. 1. Additionally, the noise levelsand general responses between the techniques are very similar in light of being extracted and analyzed approximately 31 months apart by different techniques. [25-27], while some laboratories freeze-dry the milk, followedby pressurized liquid extraction [28]. These traditional extractionsgenerally involve more extensive sample preparation or constantmanual interaction. The liquid-liquid process uses a separatory funnel with largervolumes oforganic solvents as well as manual shaking and drainingof the organic layer. Freeze-drying is an additional sample prepa-ration step that removes water from the sample prior to extractionrather than using the sample directly. The use ofa pressurized liquidextraction technique after freeze-drying is less manually intensethan the liquid-liquid extractions, but has been shown to requirecareful selection of the solvent system to achieve quantitative fatremoval [29]. Regardless of the extraction techniques,the concen-tration of the targeted analytes are determined from the additionof an internal standard, described in detail elsewhere [1,2],that isadded either prior to the initial extraction [30] or after the fat hasbeen quantitatively obtained [27]. The subsequent steps are thento separate the analytes from the fat [31], followed by additional extract cleanup [32] or HPLC fractionation [33] and finally instru-mental determination. The current study was designed to increase throughput, whilemaintaining data quality and reducing cost, in a production-likefacility for the analyses of D/Fs, dioxin-like PCBs, marker PCBs andeventually other POPs in foods and feedstuffs. Automation wasintegrated into the extraction procedure while all standards andinstrumental methods are based upon EPA Methods [1,2] wheremethod details are described. 2. Experimental details 2.1. Sampling The data used forour baseline bovine milk results were obtainedfrom samples collected from grocery stores across the United Statesas part of an assignment initiated by the U.S. FDA for the analyses ofD/Fs and PCBs. Whole milk samples, in either paper or plastic car- A Comparison of Native Spike Precision and Accuracy Results (n=12) to Established EPA Methods 1613 and 1618 Criteria. Precision and Accuracy results are based on percentrecoveries from two spiked levels. tons,were collected from November of 2013 through July of 2014.Atotal of 315 milk samples were analyzed under this assignment. Ifnot immediately analyzed upon receipt to the laboratory, sampleswere frozen at -20°C. Milk samples extracted using the new automated method, werepurchased from local grocery stores in central Arkansas duringOctober of 2015. Milk from three separate sources (brands) werecollected for this study, and maintained at 4°C. The incurred con-centrations measured in 3 sources (analyzed in duplicate) werecompared to the mean of 315 surveillance results from the previousU.S. FDA assignment. In addition to incurred level determinations,the milks were spiked at two levels, specified in section“4.2 NativeSpiked Level Recoveries," for a total of 18 milk extracts and 6 oilmethod blanks, which were used to verify that all solvents andreagents were D/F and PCB free, for 24 total data points extractedover 4 days. 2.2. Sample extraction of surveillance milk The surveillance samples were thawed (if frozen) and homog-enized. Approximately 100g of milk were accurately weighedand spiked with 13C-Labeled Internal Standards (Wellington Lab-oratories Inc., Guelph, Ontario, Canada and Cambridge IsotopeLaboratories, Inc. Tewksbury, MA) in Teflon bottles. An addi-tional 1g of sodium oxalate (Fisher Scientific, Fair Lawn, NJ)and 100 mL of anhydrous alcohol (JT Baker Center Valley, PA)were added to the samples prior to liquid-liquid extraction using100 mL 50:50 mixture of hexane (Optima 95%, Fisher Scientific)and ethyl ether (Honeywell, Burdick & Jackson,Muskegon,MI). Theresulting organic solvent was dried with sodium sulfate (Fisher Sci- entific) and then subjected to rotary-evaporation (Buchi, Flawil,Switzerland) to remove the solvent for fat determination. Thisprocess can take up to 12 h from start to fat determination for 6samples. Fat extracts were then diluted to approximately 10mL withdichloromethane(DCM)(Omnisolv Darmstadt,Germany)and sub-jected to Gel Permeation Chromatography (GPC) (J2 Scientific,Columbia, MO USA) for fat removal using a large column packedwith BioBeads (J2 Scientific). The collection of DCM was thenexchanged to hexane for an automated multi-column cleanupusing a PowerPrep (FMS, Inc. Watertown, MA). Various mix-tures of hexane, DCM and toluene (HPLC Grade, Fisher Scientific)with silica gel (acid, base, neutral), alumina and carbon columns(FMS, Inc.) were used for the cleanup and separation of D/Fs andPCBs. 2.3. Automated sample extraction ofmilk Milk samples were homogenized prior to adding approximately95g of sample into HydrothermIM(C. Gerhardt GmbH & Co. KG,Oberdollendorf,Germany) beakers. The samples were spiked with13 C-Labeled Internal Standards at least 1h prior to extractionfollowed by the addition of 1 g of sodium oxalate and 20 mLofanhy-drous alcohol. Methylene chloride sonicated and rinsed 240 mmdiameter pleated filters, M-N 715 -1/4, (Macherey-Nagel Duren,Germany) were then inserted in the automated acid hydrolysis sys-tem, HydrothermTM. Sulfuric acid (Acros Reagent ACS -NJUSA),2M solution, was used for the automated acid hydrolysis. The fatwas collected on the filters and rinsed with DI water during the 2 h Table 3A Comparison of Internal Standard Precision and Accuracy Results (n=24) to Established EPA Methods 1613 and 1618 Criteria. EPA Limits from EPA Method 1613 & 1668 process. After the standing liquid passed through the filters, theywere dried at 100°C for 1h. The dry filters were then placed into glass thimbles and thenloaded into SoxthermTM (C. Gerhardt GmbH & Co. KG)receivers. Thereceivers, containing boiling chips, were accurately weighed priorto the addition of the thimbles as a tare weight for fat determina-tion. Approximately 100 mL of hexane was added to each receiverprior to a 2h automated Soxhlet extraction using a SoxthermTMUpon the completion of the extraction, the thimble and filter wereremoved from the receiver. The receiver was then placed in theoven at 100°C for approximately 1.25 h to remove the remaininghexane for fat determination. The automated milk extraction to fatdetermination for 6 samples can be completed within 8h while anadditional batch of 6 samples can be subjected to the automatedSoxhlet extraction at the end of the day. These extracts were then subjected to GPC for fat removal. Thesample, exchanged from DCM to hexane, was then passed throughan acid/neutral silica mini-column and collected with hexane. Thehexane containing extract was then injected onto an 1100 AgilentHPLC (Agilent Technologies, Santa Clara, CA) using a pyrenylethylgroup bonded (5-PYE) 4.6 I.D. x250 mm column(Cosmosil,NacalaiUSA, San Diego, CA) for the separation of D/Fs and PCBs. 2.4. Instrumental analyses All D/F extracts, containing four non-ortho PCBs, were analyzedvia GC/HRMS using AutoSpec Ultima high resolution magnetic sec-tor instrument (Waters, Milford,MS). The gas chromatograph usedwas an Agilent 6890 Series (Agilent Technologies) with a 60m DB5-MS UI (Agilent, J&W, Santa Clara, CA) GC column, a 0.25 mm id.deactivated pre-column (Agilent Technologies), and a split split-less 0.4 mmx78.5 mm liner (Thermo Scientific, Waltham,MA). The columns and guard column were connected with a deactivatedpress-fit (Restek Universal). Direct isotope dilution was used forreporting of all analytes. Additionally,a13C-labeled Recovery stan-dard [1] containing 1,2,3,4-TCDD and 2,3,4,6,7,8-HxCDD was addedto each extract for a final volume of 10 uL with a 1.5 uL injection todetermine internal standard recoveries. The additional dioxin-like (DL) and marker PCBs were also ana-lyzed via direct isotope dilution with a Pegasus 4D-GCxGC-TOFMS(LECO,St.Joseph, MI) using an internal U.S. FDA method, describedelsewhere [34]. The injection liner, guard column and press-fitsused are identical to the above mentioned.A10 m PCB HT-8 primarycolumn (SGE Analytical Science, Austin, TX) with an additionalpress-fit connection to a 1 m DB-17MS secondary column (Agilent,J&W) was used. 3. Results and discussion 3.1. Incurred levels detected in milk A 2014 surveillance study for POPs in “grocery-purchased”whole milk across the United States was completed by the U.S.FDA. This study included the analyses of D/Fs,DL-PCBs, and markerPCBs for 315 samples, and was the largest consumer ready bovinemilk POPs study completed within the U.S. The results from the2014 study, summarized in Table 1, show the average and stan-dard deviation of the total Toxic Equivalency Quotient(TEQ),basedupon amount found per gram of fat, for the D/Fs, the D/Fs plus DL-PCBs, as well a the concentration sum of the marker PCBs. The 2014surveillancesamples were all extracted via liquid-liquid technique,rotary evaporated to constant weight for fat determination, andused as a baseline comparison for the results obtained with the newautomated method. Likewise, as a baseline comparison showing a typical set of chromatograms, representing ion responses form thehexachlorodibenzo-p-dioxins for the internal standard and nativeanalytes for the traditional extraction are shown in Fig. 1, These data for the automated extraction process from threesources are summarized in Table 1 for comparison to the histor-ical 2014 surveillance data. The D/F TEQ average for the automatedextraction was slightly lower than the 315 samples as was the PCBTEQ and marker PCB concentrations. The median values betweenthe methods were very close with a percent difference for the D/FTEQ of 1% while the marker PCB percent difference is just over12%. In addition to the close median results, the averages for theautomated method are within a single standard deviation of the315 surveillance results, suggesting that the extraction methodsare equivalent. Furthermore, the chromatograms shown in Fig. 2,which represent identical ions as Fig. 1 for the purpose of methodcomparison, also suggest that the methods are equivalent with nocompromised quality. 3.2. Native spiked level recoveries Three separate brands of milk were spiked at two separate con-centrations in duplicate for a total of 12 samples, and extracted over4 days. The incurred levels detected in the respective milks weresubtracted from the spiked amounts prior to calculating the pre-cision and accuracy data. The precision and accuracy data, shownin Table 2, were calculated based upon percent recoveries ratherthan the two spiked concentrations independently, thus show-ing precision and accuracy over the spiked range. The spike levelsfor 2,3,7,8-tetrachloro-dibenzo-p-dioxin and 2,3,7,8-tetrachloro-dibenzofuran were 0.8 pg/extract and 2.4pg/extract while the spikelevels for the penta-chlorinated through hepta-chlorinated diox-ins and furans and the non-ortho PCBs (77, 81,126 and 169)were 4 pg/extract and 12 pg/extract.The spike levels for octachloro-dibenzo-p-dioxin and octachloro-dibenzofuran were 8.0 pg/extractand 24 pg/extract while the additional D-L PCBs and marker PCBs(105, 114,118,123,156,157,167,189,28,52,101,138,153 and180) were spiked at 16.7 pg/extract and 50 pg/extract levels. Theaccuracy and precision data were compared to limits described inEPA Method 1613 [1] and EPA Method 1668 [2], shown in Table 2,and all results are within the established criteria. 3.3. Blank results To ensure that no data quality was compromised (from potentialcontaminations, to estimated detection limits) during the auto-mated extraction procedure, method blanks were compared tothe historical liquid-liquid extraction blanks. The average TEQ forthe 6 blank extracts obtained from the automated procedure was0.170 pg/g fat. The average plus 2 standard deviations (0.314pg/gfat) was equivalent to the average blank results plus 2 standarddeviations for historical liquid-liquid extractions of 0.316 pg/g fat. 3.4. Internal standard recoveries Internal standard recoveries were based upon 24 extracts thatinclude 6 oil method blanks, the 6 previously described incurredmilk samples, and the 12 previously described spiked milk sam-ples. All of the calculated precision and accuracy results, shown inTable 3, are within the established EPA Methods 1613[1] and 1668[2] limits, also shown in Table 3. 3.5. Fat determination The automated acid hydrolysis and Soxhlet extraction methodsfor the fat determination was used to calculate 6 replicates of threedifferent whole milk samples and 6 replicates of oil blanks. Each of the three milk brands averaged 3.06%, 3.12% and 2.97% withrelative standard deviations of 5.9%, 2.5% and 3.4%, respectively.Additionally, the oil recovery was 95.9% with a 2.3%RSD. 4. Conclusion Statistical evaluation of 315 previously extracted whole milksamples by liquid-liquid technique provided baseline data to com-pare the results using an automated acid hydrolysis process. Theresults of the extraction techniques are statistically equivalentwhen evaluating detection limits and internal standard recoveries.The automated acid hydrolysis process surpasses the other tech-niques by a reduction in time and manual intervention with thesample, as well as a reduction in the quantity of organic solventsand reagents used. The automated process allows the milk to be poured into theacid hydrolysis beaker then spiked with internal standard andallowed to sit for at least one hour. Just prior to extraction, thesample is mixed with the anhydrous alcohol and sodium oxalate.No additional preparation is needed for the acid hydrolysis step.A batch of 6 samples can be prepared and completed through fatdetermination within a single 8 h workday. Meanwhile, an addi-tional batch of 6 can be started the same day, allowing 12 samplesto reach fat determination on the second day. Using this processthroughout a workweek, 54 fat determinations can be completed.Per sample, only 100 mL of 2M sulfuric acid, 100 mL of hexane,1 gof sodium oxalate, and 20 mL of anhydrous alcohol as solvents andreagents are used for the fat determination. This automated process is currently being evaluated for the fatextraction of other matrices including egg, fish tissue and animalfeeds. Similarly, results obtained will be compared to that of his-torical data from samples collected in the U.S. Acknowledgements The authors would like to acknowledge each of the U.S. FDAPOPs team members at the Arkansas Regional Laboratory for theirinvolvement in this project - it would not have been accomplishedwithout everyone's help. We would also like to thank our DeputyDirector Paul Wynne and Branch Director Russell Fairchild for theircontinued support during this project as well as our supervisorBill Nelson for his detailed review and suggestions to strengthenthis manuscript. Finally, we would like to thank Marcus Kranz ofGerhardt for the analytical discussions as the project proceeded. Mention of trade names or commercial products in this presen-tation is solely for the purpose of providing specific information anddoes not imply recommendation or endorsement by the U.S. Food& Drug Administration. The views expressed in this manuscript arethose of the authors and do not necessarily reflect the views orpolicies of the U.S. Food & Drug Administration. References ( [1 ] EPA Method 1613 Tetra- through Octa-Chlorinated Dioxins and Furans by I sotope Dilution H RGC/HRMS US E PA Office 1 1 of Water, Washington D.C., 1994. [2] EPA M ethod 1 6 68, R e vision A: Chlorinated Biphenyl Congeners in Water, Soil,Sediment, and Tissue by HRGC/HRMS US EPA Office of Water, WashingtonD.C., 1999. ) [3] http://chm.pops.int/TheConvention/ThePOPs/The12InitialPOPs/tabid/296/-Default.aspx DOI. [4] A. Bernard, F. Broeckaert, G. De Poorter, A. De Cock, C. Hermans, C. Saegerman,G. Houins, The Belgian PCB/dioxin incident: analysis of the food chaincontamination and health risk evaluation,Environ. Res. 88 (2002)1-18. [5] D.G. Hayward, D. Nortrup, A. Gardner, M. Clower Jr., Elevated TCDD in chickeneggs and farm-raised catfish fed a diet with ball clay from a Southern UnitedStates mine, Environ.Res.81 (1999)248-256. [6] J.K. Huwe,J.C. Archer, Dioxin congener patterns in commercial catfish fromthe United States and the indication of mineral clays as the potential source,Food Addit. Contam. A 30 (2013)331-338. ( [ 7] R . Hoogenboo m , T. Bovee, L. P o r tier, G . B or, G . van d er W eg,C. O n stenk, W . T ra a g , The G e rm a n ba k ery w a ste incident ; use of a combine d a p p roac h of s creening and confirm a ti o n for d i o x ins i n feed and f o od, T a la n ta 63 (2004) 1249-1253. ) ( [8] J . J . Llerena, E . Abad, J . Caixach, J . Rivera, An episode of dioxin contamination i n f eedi ng stuff: t he cholin e chloride case, C h emosphe r e 53 (2003)67 9 - 6 83. ) ( [9] J.K . Huwe, D . J . S mith, Laboratory a nd on-fa r m stu d ies on the bioaccumula t ion and e limination of dioxins from a co n taminated m in eral sup p lement f e d todairy cows, J . Agric. Fo o d C hem. 53 (2005)2362 - 2370. ) ( [ 10] A . N akisa, Z.N. Khorasgani , M. Rezaei, N. I mani, S. Rezaee, D etermination of t he m ost toxic polych l orodib e n z ofuran s in fr e sh milk f r om so u thwest Ira n , Food N u t r. Sci . 06 (20 1 5)955-963. ) ( [111 B . D urand,B. D u f our,D. F ra i sse,S. Defour, K . Duhem, K. Le-Barillec , Lev e ls of 0 W P CDDs, PCDFs and d ioxin-like PCBs in r a w cow’s milk collected in Fr a nce in 2006, C h emosphere 70 (2008) 689-693. ) ( [12] ] S . Aslan,M. Kemal Ko r u c u, A . Karadem ir ,E . Durm u soglu, Levels of PCDD/Fs in l ocal a n d n on-local f ood s amples c o llected fro m a h i ghly p o lluted area in T ur k ey , Chem o sphere 80 (2010)1 2 13 - 12 1 9 . ) ( [13] H .D. H ess, M . Geinoz, A farm survey on t h e p r esence of d i o x ins and dl-P C B in b eef p rodu c tion s y stem s in S w itz e rl a n d, Biotechno l. A g ro n . Soc.15 ( 2 0 11)31 - 37. ) ( [14] I .R. Lake, C .D. Foxall, A . Fernandes , M. L e wis, M. R o se, O . White, A. Dowding, 1p S easonal varia t ions in the levels of PCDD/Fs , P CB s and P B DEs in c o ws’mil k , Chemosphere 90 (2013)72-7 9 . ) ( [ 15] S .P. D e Filippis, C . Ch i rollo, G. Br a mbilla , A. A nastasio, P. Sarnelli, E. D e Felip, A.di Domenico, A.L . Iamiceli, M.L. Cortesi,Polychlorodibenzodioxin and - furan a nd dioxin-like polychl o robiphen y l di stribution in t i s sues and da i ry prod u cts of dairy Buffaloes,J. Agr i c. Food Chem. 61 ( 2 013)655 2 -6561 . ) ( [16] A . C ostera,C. F eidt , P. March a nd , B. L e Bizec, G. Ryc h en,PCDD / F a n d PCB t ransfer to mil k in goats e xposed t o a long-term i ntake of con t aminate d hay, Chemosphere 6 4 (2006)650-657. ) ( [17] E .G. H errera Nunez, M . Perugini, M. Esposito,L. B ald i, M. Amorena,Sheep milk as a poten t ial i n d icator of envir o nmental exposure to d i oxin-like polychlorin a ted b iphenyls (dl - P CBs), Sm a ll R umin. Res. 1 06 (Suppl)( 2 012) S49-S53. ) ( [18] R. Malisch , Increase of the PCDD/F-contam i nat i on of m i lk, butter and meatsampl e s by use of contaminat e d citrus pulp,Chemosp h ere 40 (2000 ) 1041-10 5 3. ) ( [19]] 1M. E sposito, F .P. Serpe, F . Neugebauer, S . Cavallo, P. G allo, G. Col a russo, L. B ald i , G. Iovane, L . Serpe, Cont a mination levels and congener distribution of P CDDs, P C DFs and di o xin-like PCBs in bu f fa lo 's m i l k fro m Caserta pr o vinc e (Italy), Chemosphere 79 (2010) 341 - 3 48. ) ( [20] D .E. B AILEY , Stu d y o f Babcock te s t for bu t terfat in m i l k,J. D a i ry S c i. 2(19 18 ) 331-373. ) ( [21] D .H. Kleyn,J. M . Lync h , D.M.Bar b ano, M.J. Bloom , M .W. M itchell, D etermination of fat in r a w and processed milks by the gerber . method : c ollabor a tive st u dy, J. AOCA I nt. 8 4 (20 0 1)1499. ) ( [ 22] Q . Xin, H . Z. L i ng, T . J . Long, Y . Zhu, The r a pid d e termination of fat a nd proteincontent i n f resh raw milk using the l a ser light scattering technology,O p t. Laser.En g . 44(2006)858-869. ) ( [23] P I . Manir a kiz a , A.C o vaci, P . Sch e pens,Compara t iv e s t ud y o n total lipid determ i nation u s ing Soxhlet, R o ese-Gottlieb, B l igh & Dyer, and m o dified Bl i gh ) ( &D y er extracti o n methods, J . Food Com p os. Anal . 1 4 (2001)93- 1 0 0. [ 2 4] 1 . Stefanov, B . Vla e m i nck , V. F i evez, A novel pr o cedure for rou t ine milk fat ) ( extraction b a s ed on dichloromethane , J. Food Com p os. Anal. 23 (2 0 10) 852-855. ) ( [ 2 5] L .R a mos, E. E l jarrat,L. M . H e rnandez, L. Alonso,J. R i v e ra, M .J . Gonzalez, L eve l s of PCDDs and P C DFs in far m cow's milk located near potential contam i na nt s ources in A sturias ( Spain). Comparison with levels found i n co n tr o l, rural farms and comm e rcial pasteurized cow's milks, Chemosphere 35 (1997) 2167-2179. ) ( [26] U 1 .R u off,H. Kar, H . - G.Wa l te, Dioxins, diox i n-like PCBs a nd non-dio x in-like PCBs in dairy products on the German m arket a nd t he temporal t endency in Schl e swig-H o lstein, J . f u r Verbr a uch e rschutz und Lebensmittelsicherheit 7 (2011)11 - 17. ) ( [27] E . E l j arrat, A. Monjonell, J . Caixach , J . R iver a , Tox i c potency of poly c hlorinateddibenzo- p -dioxi n s,polychlorinated dibenzofurans, and polychlorinated b iphenyls in food sam p les from Catalonia (Spain), J.A g ric . F ood Chem.50 (2002)1161 - 1 167. ) ( [28] R I . Liza k ,S. M aszewski,J. P i skorska-Pliszczynska,Occurance and profiles o f p olychlorinated d benzo-p -d ioxin s , debenzofurans, a n d dioxin - l i ke p olychlorina t ed bip h enyls in p o lish f a rm m i lk, B Vet. I Pulawy 53 (2009)833-838. ) [291D.G. Hayward, T.S. Pisano, W.J.W.R.J. Scudder, Multi-residue method forPesticides and POPs in milk and cream using comprehensive two dimensionalgas chromatography and time of flight mass spectrometry,OrganohalogenCompd.72(2010)100-103. ( [30] L I . B ertocchi,S . Ghidini, G. F e d r i zzi, V. Loren z i, Case-st u dy a n d ri s kmanagement of dioxins and P C Bs bovine milk contaminations in a high i ndustrialized a rea i n N orth e rn Italy, Environ . Sci. Poll u t . Res.22(2015)9775-9785. ) ( [31] I D .Z a cs, V. B artkevics, A. Viks n a, Content of polychlor i nateddiben z o-p-dioxins, d ibenzof u rans and d ioxin- l i k e polychlorinated bi p h e nyls in fish from Latvian lakes , Chemosphere 9 1 (20 1 3)179-186. ) ( [32] J .F . Foca n t , C . Pirard, E . D e P auw , Autom a ted sample prepa r ation- f ractionation f or the measurement o f dioxins a nd r elated comp o unds in b i ologi ca l matrices: a review, Talanta 63 (2 0 04) 1 1 01 - 1 11 3. ) ( [33] X . O rtiz, R. Mar t i, M. J . Monta n a, M. G a ss e r, L. Margarit, F. B ro t o , J. D iaz-Ferrero, F racti o nati o n of p ersistent organic pollut a nts in fish oil b y high-pe r forma n ce l iq u id chrom a to g raph y on a 2-(1-pyrenyl)ethyl s il i ca column, Anal. Bio a nal. Chem. 398 (2010)985-994. ) ( [34] A. Adeuya, V.E., Litman, J.C. Archer, GCxGC-TOFMS method development for r outine analysis of PCB and PBDE in food. , Organohalogen Compounds, 7 2 1125-1128. ) Over the last several years some focus has been placed on improving efficiency for the determination ofpolychlorinated dibenzo-p- dioxins and polychlorinated dibenzo furans (D/Fs), as well as polychlorinatedbiphenyl (PCBs) and the polybrominated diphenyl ethers (PBDEs). Most efforts in this area have been focusedon instrumentation; moving from expensive magnetic sector mass spectrometers 1 to triple quadrupoles 2,3 and orbital trapping mass spectrometers 4 . Lesser efforts have been placed on sample preparation stages, which can be divided into extraction, cleanup and concentration techniques. Although the manual preparation stage isinherently inexpensive, compared to instrumentation costs, low level contamination or poor recoveries can result in rework which negatively impacts the method, making this stage costly. Additionally, the extraction method varies depending upon the matrix category, which adds to the complexity, thus multiple extraction methods may be necessary depending upon the moisture and fat content of a matrix. The extraction of milk fat by an automated acid hydrolysis and abbreviated Soxhlet extraction for determination of POPs have been previously described 5 . By broadening the extraction method to include egg yolks, prepared cakes, prepared meats and rawtissues, we are moving toward a single, matrix-independent, extraction method.
确定






还剩5页未读,是否继续阅读?

产品配置单



中国格哈特为您提供《巧克力蛋糕、熟鸡蛋、鱼等中萃取前处理检测方案(抽提萃取)》,该方案主要用于水产品中前处理检测,参考标准--,《巧克力蛋糕、熟鸡蛋、鱼等中萃取前处理检测方案(抽提萃取)》用到的仪器有格哈特全自动超级水解脂肪测定系统HT6、格哈特全自动快速索氏提取SOXTHERM、德国加液器MM
推荐专场



该厂商其他方案
更多