








方案详情
文
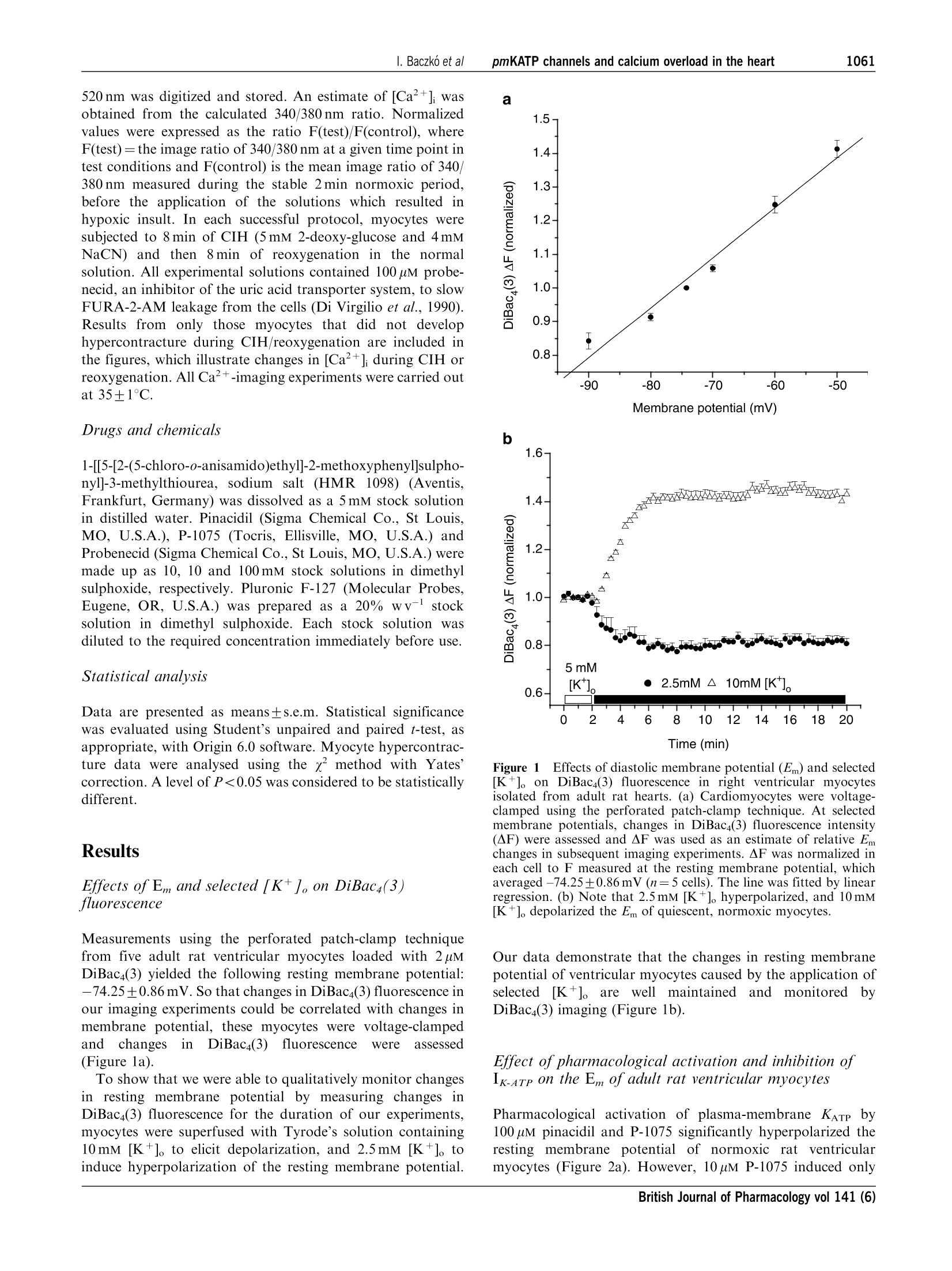
右心室的心肌细胞用2mM的生物膜电位荧光染料 DiBac4(3)孵育20分钟,从而使心肌细胞肌纤维膜上显示均相荧光检测探针的分布。
方案详情

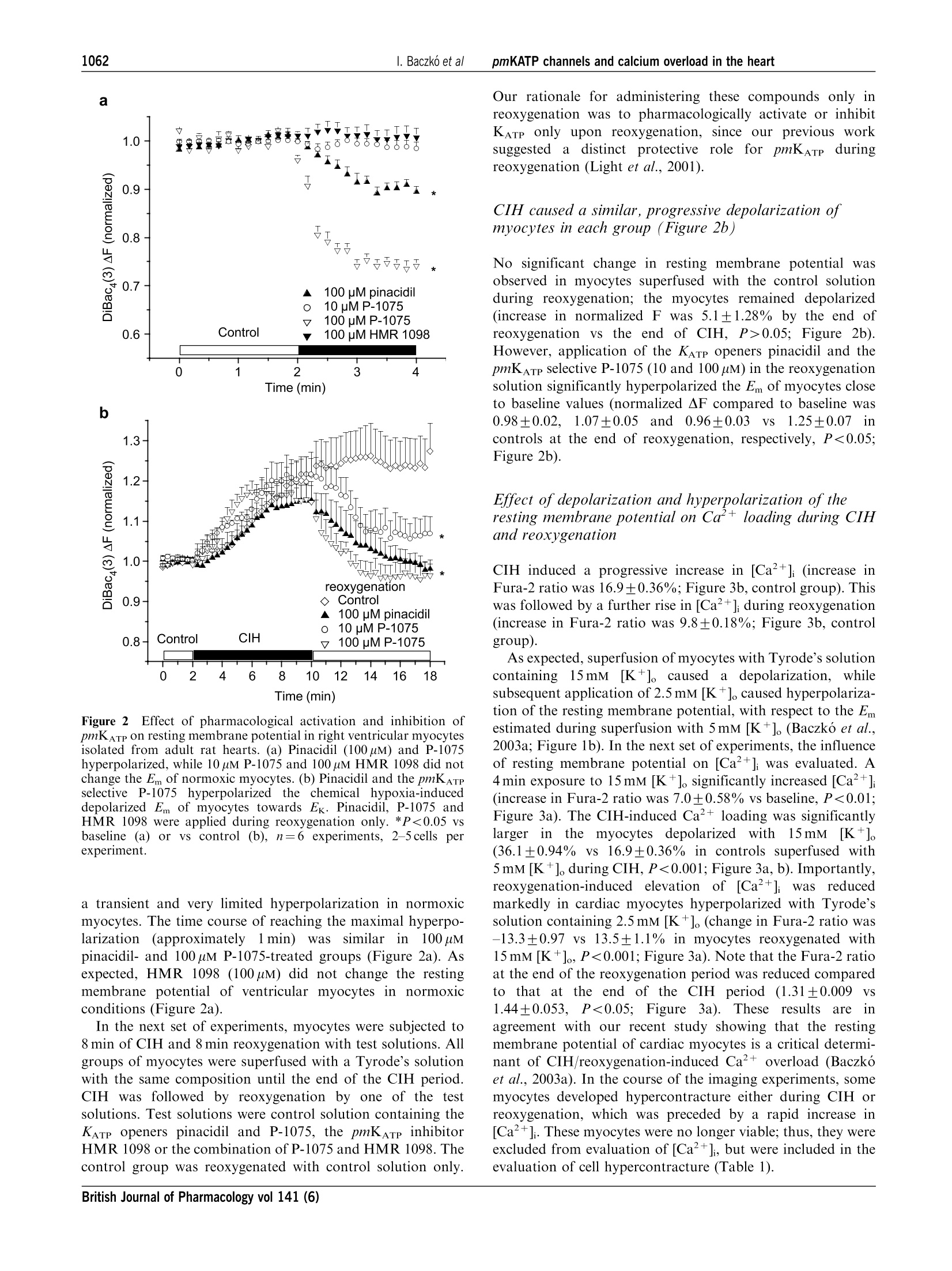
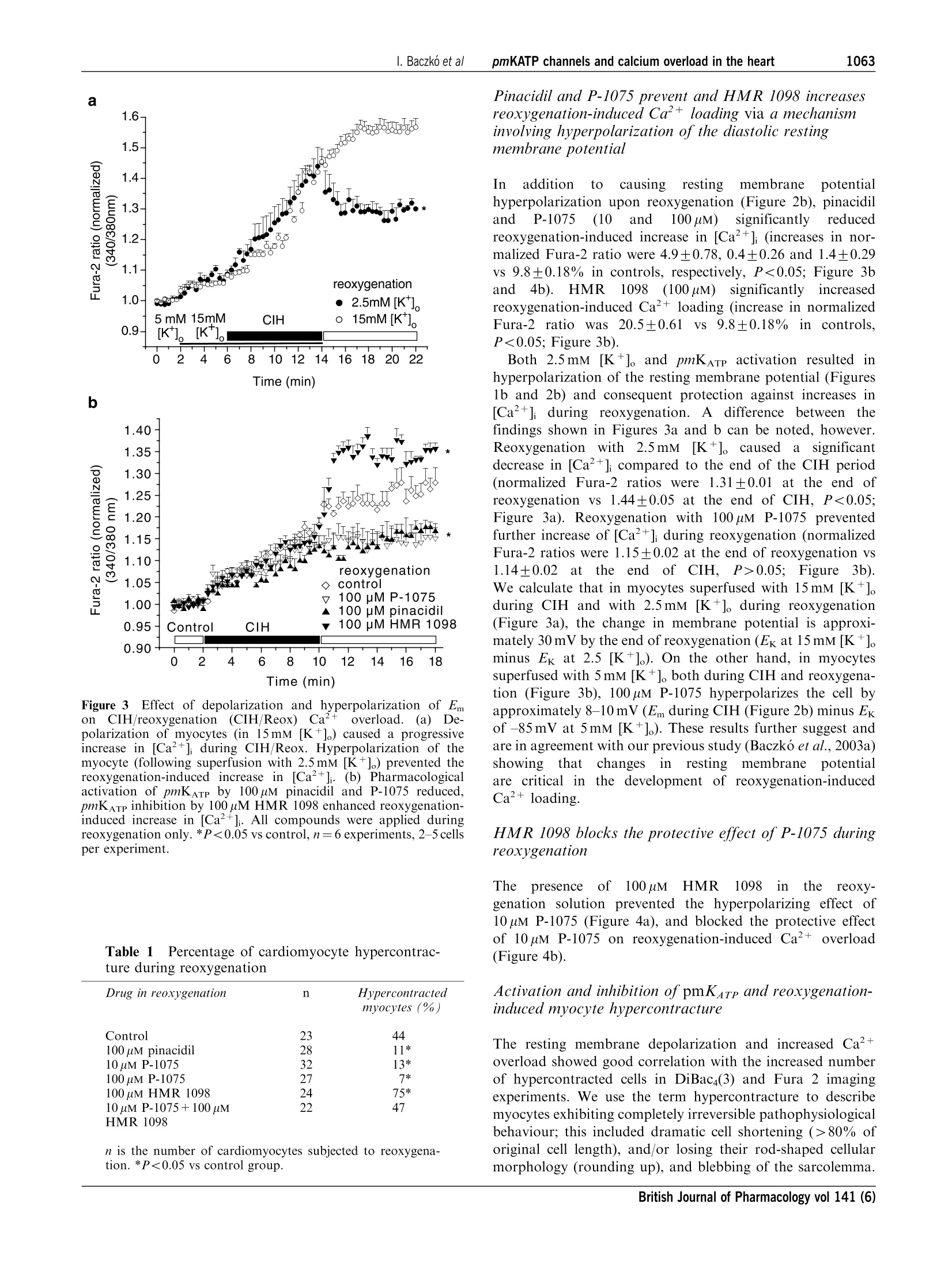
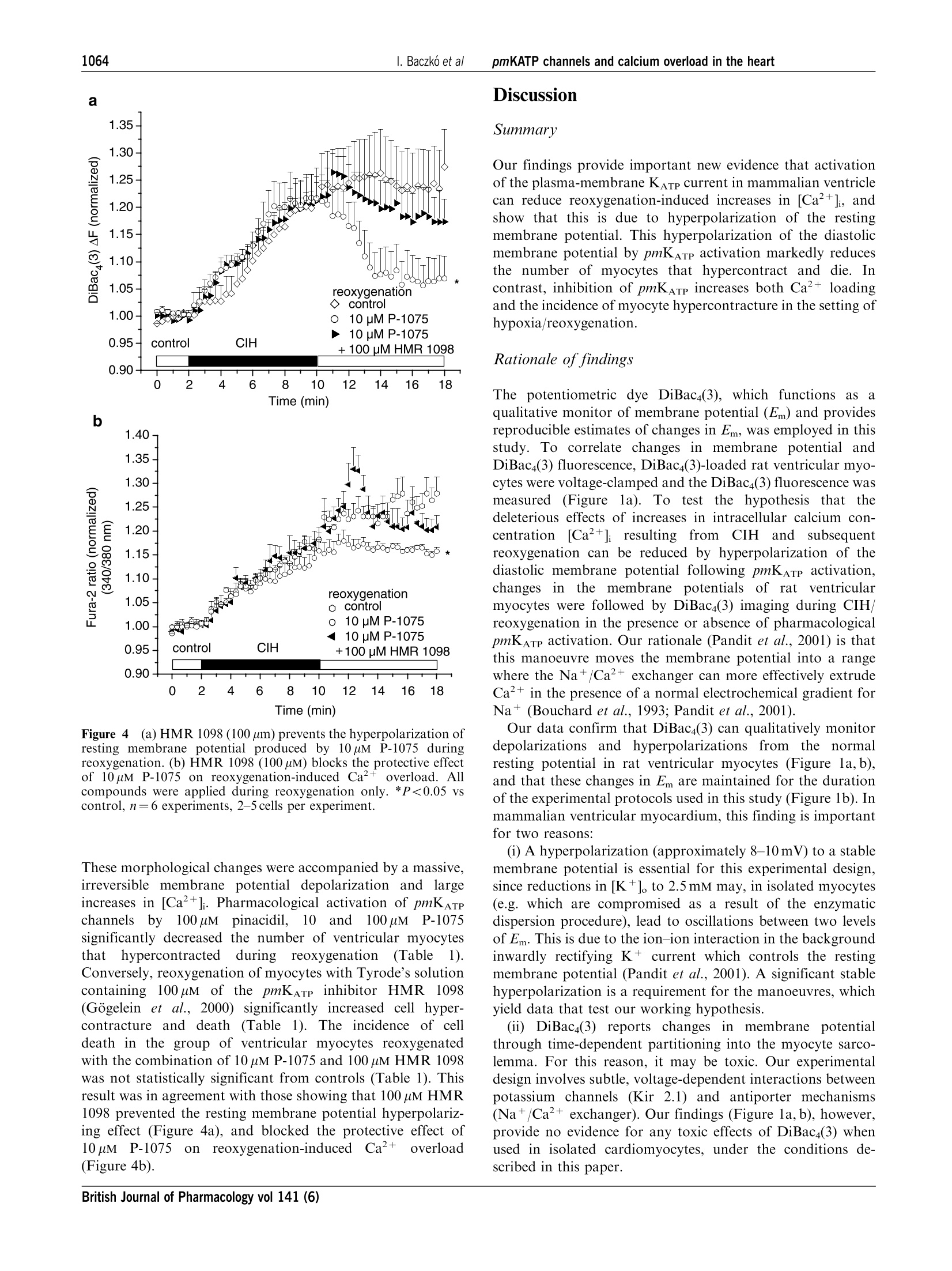
British Journal of Pharmacology (2004) 141, 1059-1067C 2004 Nature Publishing GrouppAll rights reserved 0007-1188/04 $25.00www.nature.com/bjp 1060I. Baczko et alpmKATP channels and calcium overload in the heart Pharmacological activation of plasma-membrane KATe channelsreduces reoxygenation-induced Ca2+overload in cardiac myocytesvia modulation of the diastolic membrane potential 1Istvan Baczk6, 1.2Wayne R. Giles & *Peter E. Light Department of Physiology and Biophysics, University of Calgary, Alberta, Canada; Department of Bioengineering, UCSD, LaJolla, CA 98195, U.S.A. and Department of Pharmacology,Faculty of Medicine and Dentistry, University of Alberta, Alberta,Canada T6G 2H7 1The opening of cardiac plasma-membrane ATP-sensitive K+ channels (pmKATp) can protect theheart against ischaemia/reperfusion injury. We recently demonstrated that the resting membranepotential (Em) of ventricular myocytes strongly modulates reoxygenation-induced Ca’+overload.This led to the hypothesis that activation of pmKATe can influence the extent of chemically inducedhypoxia (CIH)/reoxygenation Ca+overload via hyperpolarization of the diastolic membranepotential of ventricular myocytes. 2The membrane potential (Em) of isolated rat myocytes was determined using the perforated patch-clamp technique and DiBaca(3) imaging. Intracellular Ca²+([Ca²+]i) was monitored using FURA-2imaging. 3CIH/reoxygenation caused a significant depolarization of Em and a substantial increase in [Ca2+].The KATp opener pinacidil (100 uM) and the pmKATp opener P-1075 (100uM) hyperpolarized the Em ofnormoxic myocytes. Pinacidil (100uM) and P-1075 (10 and 100uM), applied during reoxygenation,hyperpolarized Em and prevented reoxygenation-induced increases in [Ca+]. 4Myocyte hypercontracture and death increased in parallel with an Em depolarization of10-15 mVand increases in [Ca+]. Under these conditions, the selective pmKATp channel inhibitor HMR 1098....further depolarized myocyte membrane potential and increased hypercontracture. 5In conclusion, activation of pmKATp channels can prevent CIH/reoxygenation-induced Caoverload via a mechanism that is dependent on hyperpolarization of diastolic membrane potential.Hyperpolarization toward normal resting membrane potential favours the Ca+extrusion mode ofNa+/Ca²+ exchange. British Journal of Pharmacology (2004) 141, 1059-1067. doi:10.1038/sj.bjp.0705702 Keywords: ATP-sensitive potassium channels; calcium overload; membrane potential; sodium/calcium exchange; heart Abbreviations: [Ca+], intracellular calcium concentration; CIH, chemically induced hypoxia; DiBaca(3), bis-(1,3-dibutylbarbi-turic acid)trimethine oxonol; Ek, equilibrium potential for potassium; Em, resting membrane potential; HEPES,4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid;HMR 1098, 1-[[5-[2-(5-chloro-o-anisamido)ethyl]-2-methoxy-phenyl]sulphonyl]-3-methylthiourea, sodium salt; KB-R7943, 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothioureamesylate; KO, knockout; mitoKATp channel, mitochondrial ATP-sensitive potassium channel; P-1075, N-cyano-N'-(1,1-dimethylpropyl)-N"-3-pyridylguanidine;pmKATp channel, plasma-membrane ATP-sensitive potassiumchannel In the time period following the discovery of ATP-sensitivepotassium (pmKATP) channels in cardiac tissue (Noma, 1983),many studies have demonstrated that pharmacological activa-tion of KATp channels under hypoxic or ischaemic conditionscan result in protection of the myocardium (Cole et al., 1991;McPherson et al., 1993; Hearse, 1995; Light et al., 2001). In themammaliannmyocardium,,two distinct classes of KATpchannels are expressed in (i) the plasma membrane (pm) andthe (ii) mitochondrium (mito). There is an ongoing debateregarding the mechanism(s) of action of potassium channelopeners that resultsi ni cnardioprotection. There is alscuncertainty and controversy surrounding the putative roles ( *Author f o r correspondence; E - mail: p e ter.light@u a lberta.caAdvance online publication : 1 March 2004 ) of pm and mitoK ATp channels in affording this protection (forrecent reviews, see Gross & Fryer, 1999; Gross & Peart, 2003).Activation of mitoKATp channels is thought to improvemitochondrial functioni viaregulation of mitochondrialvolume, calcium-handling and/or mitochondrial membranepotential (see the review by O'Rourke,2000). However,recentwork by Das et al. (2003) argues against a significantfunctionall rroollee of a sulphonylurea-inhibitable mitoKATpchannel. Several compelling lines of evidence indicate thatactivation of pmKATp channels is also cardioprotective. Forexample, pmKATp channel-deficient (KO) mouse hearts failedto exhibit preconditioning. In wild-type hearts, this ‘compro-mised’state could be mimicked by the selective pmKATpchannel inhibitor HMR1098 (Suzuki et al., 2002). Also, arecentwork by Suzuki et al. (2003) suggests that the cardioprotective effects of diazoxide, long considered to be aselective mitoKATe opener, is in fact mediated mainly by theactivation of pmKATp channels. Our recent findings demon-strate that activation of pmKATe channels can significantlyreduce hypoxia-induced calcium overload in rat ventriculaimyocytes (Light et al., 2001). Despite these findings, the.cellular ionic mechanisms by which pharmacological activa-tion of pmKATp in the heart elicits a cardioprotective effectremain to be established. Significant and maintained increases in intracellular calcium(Ca+overload) are generally thought to be a key determinantof the severity of myocardial damage during ischaemia/reperfusion (IR) (Marban et al., 1989; Park & Lucchesi,1999; Piper & Garcia-Dorado, 1999). Dysfunctional intracel-lular Ca+ homeostasis during reperfusion is thought tcrepresent a major determinant of the early phase of reperfu-sion injury, including stunning and cell hypercontracture/death (Marban et al., 1989; Tani, 1990; Bolli & Marbán, 1999)However, many of the underlying mechanisms responsible forthe observed calcium overload have not been demonstrated.Recent findings strongly suggest that alterations in the activityof the plasma-membrane Na+/Ca+ exchanger contributes toabnormal Ca+regulation during ischaemia/reperfusion (Eigel& Hadley,2001; Schafer et al., 2001), and it is known that theseverity of myocyte calcium overload is dependent ontransmembrane electrochemical gradients for Naand Ca+(Blaustein & Lederer, 1999). Our previous results have shownthat:(i) pmKATe channel activation by protein kinase Creduces reoxygenation-induced calcium overload (Light et al.,2001) and (ii))hyperpolarization of the diastolic/restingmembrane potential reduces the magnitude of calcium over-load mediated by reverse-mode Na+/Ca2+ exchange (Baczkóet al., 2003a). Accordingly, the main goal of the present study was todetermine the relationship between: (i) pharmacologicactivation of the repolarizing K+ current pmKATP, (ii) thediastolic membrane potential, (iii) calcium overload andmyocyte hypercontracture, in the setting of chemically inducedhypoxia (CIH)/reoxygenation. Our findings confirm thatactivation of pmKATe hyperpolarizes the diastolic membranepotential, and demonstrate that this is essential for reducinghypoxia/reoxygenation-induced calcium overload and irrever-sible myocyte injury. Part of this work has been recently presented in abstractform (Baczko et al., 2003b). Methods Resting membrane potential measurements usingperforated patch-clamp andDiBacy(3) fluorescence-imaging techniques Adult rats were euthanized with pentobarbital (150mgkg,i.p.) according to the University of Alberta Animal Policy andWelfare Committee and the Canadian Council on AnimalCare (CCAC) Guidelines. The hearts were removed undercomplete anaesthesia and right ventricular myocytes were thenobtained by enzymatic dissociation using the standard proto-cols described previously (Bouchard et al., 1993; Light et al.,1998). Changes in the resting membrane potential of freshlyisolated myocytes were estimated by monitoring changes in fluorescence intensity of the potentiometric bisoxonol dye bis-(1,3-dibutylbarbituric acid)trimethine oxonol (DiBaca(3)), ananionic probe that, upon changes in membrane potential,partitions into the myocyte sarcolemma and exhibits enhancedfluorescence on depolarization (Epps et al., 1994). Conversely,hyperpolarization leads to efflux of the probe and a decrease influorescence intensity. Right ventricular myocytes were in-cubated with 2uMDiBac4(3) for 20 min to achieve homo-genous probe distribution in the myocyte sarcolemma. Afterincubation, cells were placed on coverslips for observation at×200 with an inverted microscope (Olympus, CK40) and weresuperfused with the control solution containing 2uM Di-Bac4(3) and (in mM): NaCl 140, KCl 5, HEPES 10, CaCl, 2.0,MgCl, 1.4, glucose 10. A Photon Technology International(PTI, Lawrenceville, NJ, U.S.A.) imaging system with PTIImagemaster software (version 1.49))wasused for dataacquisition and analysis. DiBaca(3) was excited with 470 nmwavelength light and the emitted light intensity at 525 nm wasdigitized and stored. Normalized values were expressed as theratio of F(test)/F(control), where F(test)=525 nm wavelengthfluorescence intensity at a given time point in test conditionsand F(control) is the mean 525 nm fluorescence intensityduring the stable 2 min control period, before the applicationoftestsolutions. The restingmembranepotentialsISofDiBaca(3)-loaded right ventricular myocytes were measuredusing the perforated-patch technique similar to our previousstudy (Baczko et al., 2003a). The cardiomyocytes were thenvoltage-clamped at selected potentials (-90 to -50 mV, every10mV) and simultaneous DiBaca(3) fluorescence intensitymeasurements were carried out on the same cells at 35°C.The DiBaca(3) fluorescence intensity at the resting membranepotential was defined as 1.0 during normalization (Figure 1a). To demonstrate that the changes in Em observed usingDiBac4(3) imaging were maintained for the duration of ourexperimental protocols, ventricular myocytes were superfusedwith2.5,10mM[K+]。 for18 min and their DiBac4(3)fluorescence was monitored (Figure 1b). Membrane potentialmeasurementsts and DiBaca(3) imaging experimentswereperformed at 35±1°C. Measurements ofintracellular Ca²+([Ca+]) A sustained increase in [Ca+] has been reported to correlateclosely with cell mortality in the setting of ischaemia/reperfusion (for a review, see Bolli & Marban, 1999).Accordingly, continuous measurements of [Ca+]i have beenused as an indicator of calcium homeostasis in models of CIH/reoxygenation (Jovanovic et al., 1998). Right ventricularmyocytes from adult rats were loaded for 15min with theesterified form of the Ca+-sensitive fluorescent probe FURA-2-AM (2uM, dissolved in a 50% dimethyl sulphoxide and a50% vv- pluronic acid mixture (Molecular Probes, Eugene,OR, U.S.A.)). After loading, cells were washed and placed oncoverslips for observation at ×200 with an inverted micro-scope (Olympus, CK40), while being superfused with thecontrol solution containing (in mM): NaCl 140, KCl 5, HEPES10, CaCl, 2.0, MgCl2 1.4, or with selected test solutions. APhoton Technology International (PTI, Lawrenceville, NJ,U.S.A.) imaging system with PTI Imagemaster software(version 1.49) was used for data acquisition and analysis.FURA-2 was excited with alternating 3400and3380nmwavelengthss of light, and the emittedlliightintensity at 520 nm was digitized and stored. An estimate of [Ca²+]i wasobtained from the calculated 340/380nm ratio. Normalizedvalues were expressed as the ratio F(test)/F(control), whereF(test)= the image ratio of 340/380 nm at a given time point intest conditions and F(control) is the mean image ratio of 340/380 nm measured during the stable 2 min normoxic period,before the application of the solutions which resulted inhypoxic insult. In each successful protocol, myocytes weresubjected to 8 min of CIH (5mM 2-deoxy-glucose and 4mMNaCN) and then 8 min of reoxygenation in the normalsolution. All experimental solutions contained 100 uM probe-necid, an inhibitor of the uric acid transporter system, to slowFURA-2-AM leakage from the cells (Di Virgilio et al.,1990).Results from only those myocytes that did not develophypercontracture during CIH/reoxygenation are included inthe figures, which illustrate changes in [Ca+]i during CIH orreoxygenation. All Ca+-imaging experiments were carried outat 35+1°C. Drugs and chemicals 1-[[5-[2-(5-chloro-o-anisamido)ethyl]-2-methoxyphenyl]sulpho-nyl]-3-methylthiourea, sodium salt (HMR 1098) (Aventis,Frankfurt, Germany) was dissolved as a 5 mM stock solutionin distilled water. Pinacidil (Sigma Chemical Co., St Louis,MO, U.S.A.), P-1075 (Tocris, Ellisville, MO, U.S.A.) andProbenecid (Sigma Chemical Co., St Louis,MO, U.S.A.) weremade up as 10, 10 and 100 mM stock solutions in dimethylsulphoxide, respectively. Pluronic F-127 (Molecular Probes,Eugene, OR, U.S.A.) was prepared as a 20% wv- stocksolution in dimethyl sulphoxide. Each stock solution wasdiluted to the required concentration immediately before use. Statistical analysis Data are presented as means±s.e.m. Statistical significancewas evaluated using Student’s unpaired and paired t-test, asappropriate, with Origin 6.0 software. Myocyte hypercontrac-ture data were analysed using the x method with Yates’correction. A level of P<0.05 was considered to be statisticallydifferent. Results Effects of Em and selected [K+]。 on DiBac4(3)fluorescence Measurements using the perforated patch-clamp techniquefrom five adult rat ventricular myocytes loaded with 2uMDiBaca(3) yielded the following resting membrane potential:-74.25±0.86mV. So that changes in DiBac4(3) fluorescence inour imaging experiments could be correlated with changes inmembrane potential, these myocytes were voltage-clampedand changesin DiBaca(3)fluorescence were assessed(Figure la). To show that we were able to qualitatively monitor changesin resting membrane potential by measuring changes inDiBaca(3) fluorescence for the duration of our experiments,myocytes were superfused with Tyrode’s solution containing10 mM [K *]。 to elicit depolarization, and 2.5 mM [K +]。 toinduce hyperpolarization of the resting membrane potential. Membrane potential (mV) b 1.6- Time (min) Figure 1 Effects of diastolic membrane potential (Em) and selected[K+]。 on DiBaca(3) fluorescence in right ventricular myocytesisolated from adult rat hearts (a) Cardiomyocytes were voltage-clamped using the perforated patch-clamp technique. At selectedmembrane potentials, changes in DiBaca(3) fluorescence intensity(AF) were assessed and AF was used as an estimate of relative Emchanges in subsequent imaging experiments. AF was normalized ineach cell to F measured at the resting membrane potential, whichaveraged -74.25±0.86mV (n=5 cells). The line was fitted by linearregression. (b) Note that 2.5 mM [K +]。 hyperpolarized, and 10 mM[K+].depolarized the Em of quiescent, normoxic myocytes. Our data demonstrate that the changes in resting membranepotential of ventricular myocytes caused by the application ofselected[K*]oare wellmaintained and monitored byDiBac4(3) imaging (Figure 1b). Effect of pharmacological activation and inhibition ofIK-aTp on the Em ofadult rat ventricular myocytes Pharmacological activation of plasma-membrane KATp by100 uM pinacidil and P-1075 significantly hyperpolarized theresting membrane potential of normoxic rat ventricularmyocytes (Figure 2a). However, 10 uM P-1075 induced only a Time (min) b Figure 2Effect of pharmacological activation and inhibition ofpmKATp on resting membrane potential in right ventricular myocytesisolated from adult rat hearts. (a) Pinacidil (100 uM) and P-1075hyperpolarized, while 10 pM P-1075 and 100 uM HMR 1098 did notchange the Em of normoxic myocytes. (b) Pinacidil and the pmKATPselective P-1075 hyperpolarized the chemical hypoxia-induceddepolarized Em of myocytes towards Ek. Pinacidil, P-1075 andHMR 1098 were applied during reoxygenation only. *P<0.05 vsbaseline (a) or vs control (b), n=6 experiments, 2-5 cells perexperiment. a transient and very limited hyperpolarization in normoxicmyocytes. The time course of reaching the maximal hyperpo-larization(approximately1min)was similarin100 uMpinacidil- and 100 uM P-1075-treated groups (Figure 2a). Asexpected, HMR 1098 (100uM) did not change the restingmembrane potential of ventricular myocytes in normoxicconditions (Figure 2a). In the next set of experiments, myocytes were subjected to8 min of CIH and 8 min reoxygenation with test solutions. Allgroups of myocytes were superfused with a Tyrode’s solutionwith the same composition until the end of the CIH period.CIH was followed by reoxygenation by one of the testsolutions. Test solutions were control solution containing theKATp openers pinacidil and P-1075, the pmKATe inhibitorHMR 1098 or the combination of P-1075 and HMR 1098. Thecontrol group was reoxygenated with control solution only. Our rationale for administering these compounds only inreoxygenation was to pharmacologically activate or inhibitKATp only upon reoxygenation, since our previous worksuggested a distinct protective role for pmKATp duringreoxygenation (Light et al., 2001). CIH caused a similar, progressive depolarization ofmyocytes in each group (Figure 2b) No significant change in resting membrane potential wasobserved in myocytes superfused with the control solutionduring reoxygenation; the myocytes remained depolarized(increase in normalized F was 5.1±1.28% by the end ofreoxygenation vs the end of CIH, P>0.05; Figure 2b).However, application of the KATp openers pinacidil and thepmKATp selective P-1075 (10 and 100 pM) in the reoxygenationsolution significantly hyperpolarized the Em of myocytes closeto baseline values (normalized AF compared to baseline was0.98±0.02,1.07+0.05and0.96+0.03 VS 1.25±0.07 incontrols at the end of reoxygenation, respectively, P<0.05;Figure 2b). Effect of depolarization and hyperpolarization of theresting membrane potential on Ca+ loading during CIHand reoxygenation CIH induced a progressive increase in [Ca+] (increase inFura-2 ratio was 16.9±0.36%; Figure 3b, control group). Thiswas followed by a further rise in [Ca+]i during reoxygenation(increase in Fura-2 ratio was 9.8±0.18%;Figure 3b, controlgroup). As expected, superfusion of myocytes with Tyrode’s solutioncontaining15mM [K+]。 caused a depolarization, whilesubsequent application of 2.5 mM [K +]。caused hyperpolariza-tion of the resting membrane potential, with respect to the Emestimated during superfusion with 5 mM [K+]。 (Baczko et al.,2003a; Figure 1b). In the next set of experiments, the influenceof resting membrane potential on [Ca +]i was evaluated. A4 min exposure to 15 mM [K+]。 significantly increased [Ca²+](increase in Fura-2 ratio was 7.0±0.58%vs baseline, P<0.01;Figure 3a). The CIH-induced Ca²+ loading was significantlylarger in the myocytes depolarized with15mM [K+].(36.1±0.94% vs 16.9±0.36% in controls superfused with5 mM [K *]。during CIH, P<0.001;Figure 3a, b). Importantly,reoxygenation-induced elevation of [Ca+] wasSreducedmarkedly in cardiac myocytes hyperpolarized with Tyrode’ssolution containing 2.5 mM [K+]. (change in Fura-2 ratio was-13.3±0.97 vs 13.5±1.1% in myocytes reoxygenated with15mM [K+]o, P<0.001; Figure 3a). Note that the Fura-2 ratioat the end of the reoxygenation period was reduced comparedto that at the end of the CIH period (1.31±0.0091.44±0.053, P<0.05; Figure 3a). These results are:1agreement with our recent study showing that the restingmembrane potential of cardiac myocytes is a critical determi-nant of CIH/reoxygenation-induced Ca+overload (Baczkoet al., 2003a). In the course of the imaging experiments, somemyocytes developed hypercontracture either during CIH orreoxygenation, which was preceded by a rapid increase in[Ca ]. These myocytes were no longer viable; thus, they wereexcluded from evaluation of [Ca+]i, but were included in theevaluation of cell hypercontracture (Table 1). a Time (min) b Time (min) Figure 3 Effect of depolarization and hyperpolarization of Emon CIH/reoxygenation (CIH/Reox) Ca2+overload. (a) De-polarization of myocytes (in 15mM [K+]o) caused a progressiveincrease in [Ca +]i during CIH/Reox. Hyperpolarization of themyocyte (following superfusion with 2.5 mM [K*]o) prevented thereoxygenation-induced increase in [Ca+]. (b) Pharmacologicalactivation of pmKATP by 100 uM pinacidil and P-1075 reduced,pmKATp inhibition by 100 uM HMR 1098 enhanced reoxygenation-induced increase in [Ca+]. All compounds were applied duringreoxygenation only.*P<0.05 vs control,n=6 experiments, 2-5 cellsper experiment. Table 11Percentage of cardiomyocyte hypercontrac-ture during reoxygenation Drug in reoxygenation Hypercontracted n is the number of cardiomyocytes subjected to reoxygena-tion. *P<0.05 vs control group. Pinacidil and P-1075 prevent and HMR 1098 increasesreoxygenation-induced Ca+ loading via a mechanisminvolving hyperpolarization of the diastolic restingmembrane potential In addition to causing resting membraneepotentialhyperpolarization upon reoxygenation (Figure 2b), pinacidiland P-1075 (10 and 1(100pM)ssignificantlyreducedreoxygenation-induced increase in [Ca+](increases in nor-malized Fura-2 ratio were 4.9±0.78,0.4±0.26 and 1.4±0.29vs 9.8±0.18% in controls, respectively, P<0.05; Figure 3band 4b). HMR 1098 (100uM)significantlyy increasedreoxygenation-induced Ca+ loading (increase in normalizedFura-2 ratio was 20.5±0.61VS9.8±0.18% in controls,P<0.05; Figure 3b). Both 2.5mM [K+]o and pmKATe activation resulted inhyperpolarization of the resting membrane potential (Figures1b and 2b) and consequent protection against increases in[Ca²+]i during reoxygenation. A difference between thefindings shown in Figures 3a and b can be noted, however.Reoxygenationi with2.5mM [K+]。 caused a significantdecrease in [Ca+]i compared to the end of the CIH period(normalized Fura-2 ratios were 1.31+0.01 at the end ofreoxygenation vs 1.44±0.05 at the end of CIH, P<0.05;Figure 3a). Reoxygenation with 100 uM P-1075 preventedfurther increase of [Ca ]i during reoxygenation (normalizedFura-2 ratios were 1.15±0.02 at the end of reoxygenation vs1.14±0.022attheenddof CIH, P>0.05;Figure3b).We calculate that in myocytes superfused with 15 mM [K+]oduring CIH and with 2.5 mM [K+] during reoxygenation(Figure 3a), the change in membrane potential is approxi-mately 30 mV by the end of reoxygenation (Ek at 15 mM [K +]。minus Ek at 2.5 [K]o). On the other hand, in myocytessuperfused with 5 mM [K +]。 both during CIH and reoxygena-tion (Figure 3b), 100 uM P-1075 hyperpolarizes the cell byapproximately 8-10 mV (Em during CIH (Figure 2b) minus Ekof -85 mV at 5mM [K+]). These results further suggest andare in agreement with our previous study (Baczko et al., 2003a)showing that changesSin restingmembranepotentialare critical in the development of reoxygenation-inducedCa2+ loading. HMR 1098 blocks the protective effect of P-1075 duringreoxygenation The presenceof 100uM HMR 1098 intthereoxy-genation solution prevented the hyperpolarizing effect of10 uM P-1075 (Figure 4a), and blocked the protective effectof 10 uM P-1075 on reoxygenation-induced Ca+ overload(Figure 4b). Activation and inhibition ofpmKarp and reoxygenation-induced myocyte hypercontracture The resting membrane depolarization and increased Ca+overload showed good correlation with the increased numberof hypercontracted cells in DiBac4(3) and Fura 2 imagingexperiments. We use the term hypercontracture to describemyocytes exhibiting completely irreversible pathophysiologicalbehaviour; this included dramatic cell shortening (>80% oforiginal cell length), and/or losing their rod-shaped cellularmorphology (rounding up), and blebbing of the sarcolemma. Figure 4(a) HMR 1098 (100 um) prevents the hyperpolarization ofresting membrane potential produced by 10 pM P-1075 duringreoxygenation. (b)HMR 1098 (100 uM) blocks the protective effectof 10 uM P-1075 on reoxygenation-induced Ca’+ overload. Allcompounds were applied during reoxygenation only. *P<0.05 vscontrol, n=6 experiments, 2-5 cells per experiment. These morphological changes were accompanied by a massive,irreversible membrane potential depolarization and largeincreases in [Ca+]. Pharmacological activation ofpmKATechannels by 100uMpinacidil, 10)and 100uMP-1075significantly decreased the number of ventricular myocytesthathypercontracteddduring rreeoxygenation (Table1).Conversely, reoxygenation of myocytes with Tyrode’s solutioncontaining 100uM of the pmKATe inhibitor HMR.1098(Gogelein et al., 2000) significantly increased cell hyper-contracture and death (Table 1). The incidence of celldeath in the group of ventricular myocytes reoxygenatedwith the combination of 10 uM P-1075 and 100 uM HMR 1098was not statistically significant from controls (Table 1). Thisresult was in agreement with those showing that 100 uM HMR1098 prevented the resting membrane potential hyperpolariz-ing effect (Figure 4a), and blocked the protective effect of10 uM P-1075onreoxygenation-induced Ca+overload(Figure 4b). Summary Our findings provide important new evidence that activationof the plasma-membrane KATp current in mammalian ventriclecan reduce reoxygenation-induced increases in [Ca+]i, andshow that this is due to hyperpolarization of the restingmembrane potential. This hyperpolarization of the diastolicmembrane potential by pmKATp activation markedly reducesthe number of myocytes that hypercontract and die. Incontrast, inhibition of pmKATe increases both Ca+ loadingand the incidence of myocytehypercontracture in the setting ofhypoxia/reoxygenation. Rationale of findings The potentiometric dye DiBaca(3), which functions as aqualitative monitor of membrane potential (Em) and providesreproducible estimates of changes in Em, was employed in thisstudy. To correlate changes in membrane potential andDiBaca(3) fluorescence, DiBaca(3)-loaded rat ventricular myo-cytes were voltage-clamped and the DiBac4(3) fluorescence wasmeasured (Figure la). To test the hypothesis that thedeleterious effects of increases in intracellular calcium con-centration [Ca+]i resulting from CIH and subsequentreoxygenation can be reduced by hyperpolarization of thediastolic membrane potential following pmKATp activation,changes irinthe membrane potentials of rat ventricularmyocytes were followed by DiBaca(3) imaging during CIH/reoxygenation in the presence or absence of pharmacologicalpmKATe activation. Our rationale (Pandit et al.,2001) is thatthis manoeuvre moves the membrane potential into a rangewhere the Na+/Ca2+ exchanger can more effectively extrudeCa2+ in the presence of a normal electrochemical gradient forNa+(Bouchard et al., 1993;Pandit et al., 2001). Our data confirm that DiBac4(3) can qualitatively monitordepolarizations and hyperpolarizations from the normalresting potential in rat ventricular myocytes (Figure la,b),and that these changes in Em are maintained for the durationof the experimental protocols used in this study (Figure 1b). Inmammalian ventricular myocardium, this finding is importantfor two reasons: (i) A hyperpolarization (approximately 8-10mV) to a stablemembrane potential is essential for this experimental design,since reductions in [K *]。 to 2.5 mM may, in isolated myocytes(e.g. which are compromised as a result of the enzymaticdispersion procedure), lead to oscillations between two levelsof Em. This is due to the ion-ion interaction in the backgroundinwardly rectifying K+ current which controls the restingmembrane potential (Pandit et al., 2001). A significant stablehyperpolarization is a requirement for the manoeuvres, whichyield data that test our working hypothesis. (ii) DiBaca(3) reports changes in membrane potentialthrough time-dependent partitioning into the myocyte sarco-lemma. For this reason, it may be toxic. Our experimentaldesign involves subtle, voltage-dependent interactions betweenpotassium channels (Kir 2.1) and antiporter mechanisms(Na+/Ca²+ exchanger). Our findings (Figure 1a, b), however,provide no evidence for any toxic effects of DiBac4(3) whenused in isolated cardiomyocytes, under the conditions de-scribed in this paper. Pharmacological activation and inhibition of plasma-membrane KaTe The main goal of this study was to determine whetherpharmacological activation of the population of ATP-sensitivepotassium channels which are expressed predominantly in thesarcolemma can ameliorate myocyte damage due to elevated[Ca+]i,following CIH/reoxygenation. For this reason, it wasessential to activate sarcolemmal KATp channels as opposed tothose expressed in the mitochondria. The available literaturesuggests that P-1075 functions in this capacity (Liu et al.,2001), and our findings are consistent with this supposition.Liu et al. have shown that P-1075, even in the highesconcentration used in this study (100 uM), does not activatenative mitoKATp channels. We also used pinacidil, a non-selective KATp channel opener with a chemical structure verysimilar to P-1075, for further confirmation of the effect ofKATp channel activation on reoxygenation-induced Ca+overload. The time course of the onset of the effects of KATP openerson resting membrane potential and reoxygenation-inducedCa2+ overload averaged approximately 1 min, which makes itunlikely that the hyperpolarization observed following P-1075application, even at this high concentration, was mediated bymitochondrial KATp channel activation. As shown by Sato et al.(2000), KATp channel openers elicit their effect on mitochon-drial function in 5-10 min. ATP-sensitive potassium channels consist of a hetero-octamer of four inwardly rectifying pore-forming potassiumchannel subunits (Kir6.2) together with four regulatorysulphonylurea receptor subunits (SUR) (Inagaki et al., 1995).Distinct isoforms of the SUR expressed in different tissues leadto well-defined pharmacological characteristics of the ex-pressed KATe channel. In the plasma membrane of cardiacmyocytes and skeletal muscle, SUR2A is the SUR isoform(Inagaki et al., 1996). Intheepresentstudy, HMR 10988., aacardioselectivesulphonylurea, was used for the inhibition of ventricularmyocyte KATe channels (Gogelein et al., 2000). The pharma-cological profile of mitochondrial KATp closely resembles theKir6.1/SUR1 (Liu et al., 2001). A recent study from ourlaboratory shows that HMR 1098 selectively inhibits thepmKATp isoform Kir6.2/SUR2A, as opposed to Kir6.1/SUR1(Manning Fox et al., 2002). This same study also shows thatHMR 1098 (10-1000uM) did not have an effect on Kir2.1current (Manning Fox et al., 2002). Also, evidence from recentwork using pinacidil and [’H]P-1075 demonstrates that thetarget site for potassium channel opener binding and selectivitys the C-terminal of the sulphonylurea receptor complex(Schwanstecher et al., 1998). The results of these studiessuggest that it is very unlikely that the resting membranepotential changes seen in our experiments were due to theeffects of pinacidil, P-1075 or HMR 1098 on the backgroundinwardly rectifying K+ current (Iki). Activation ofplasma-membrane IK-ATe hyperpolarizes theresting membrane potential and provides cardioprotection It has been suggested that the activation of mitoKATp asopposed to pmKATP plays an important role in cardioprotec-tion (reviewed by Gross & Fryer, 1999). This suggestion isbased:con pharmacologicalstudies usinggdiazoxideand 5-hydroxydecanoatee(5-HD)) as aamitoKATp opener andblocker, respectively (Garlid et al., 1997; Liu et al., 1998).However, diazoxide has been shown to cause flavoproteinoxidation through a mechanism independent of its mitoKATpactivation (Grimmsmann & Rustenbeck, 1998; Hanley et al.,2002), and can open pmKATp at low concentration in thepresence of ADP (D’Hahan et al., 1999), and to protectcardiac myocytes during metabolic inhibition without causingmitochondrial depolarization or flavoprotein oxidation (Law-rence et al., 2001). Also, 5-HD can be a substrate and/orinhibitor of fatty acid p-oxidation (Hanley et al., 2003).Accordingly, these effects of diazoxide and 5-HD couldcontribute to their ability to mimic and/or block the effectsof ischaemic preconditioning. We are aware that a number of papers have been publishedrecently, suggesting a critical role for pmKATe in cardioprotec-tion in the setting of ischaemia/hypoxia and reperfusion/reoxygenation. Cotransfection of COS-7 cells with the Kir6.2/SUR2A genes encoding the cardiac pmKATp isoform resultedin decreased hypoxia/reoxygenation-induced Ca+ loading(Jovanovic et al., 1998). Also, Suzuki et al. (2002) have shownthat the infarct size-reducing effect of ischaemic precondition-ing is abolished in Kir6.2-deficient mice. Another recent studyfrom their laboratory suggests that diazoxide exerts itscardioprotective effects via the pmKATp channels due to itsability to significantly improve post-ischaemic functionalrecovery only in mice hearts expressing the pmKATp channelas opposed to Kir6.2 knockout hearts (Suzuki etal., 2003). Our results confirm that activation of pmKATp plays animportant role in protecting ventricular myocytes againsthypoxia/reoxygenation injury: in the present study, pharma-cological activation of pmKATe by pinacidil and P-1075, apmKATp selective pinacidil analogue, significantly decreasedreoxygenation-induced Ca+ overload (Figure 3b), an effectthat was blocked by the pmKATp inhibitor HMR 1098(Figure 4b). We also show that reoxygenation of myocytes inthe presence of Tyrode’s solution containing 2.5mM [K*].significantly reduced reoxygenation-induced Ca2+overload incardiac myocytes (Figure 3a). These results confirm our recentobservations, which demonstrate the beneficial effects ofhyperpolarization of thee resting membrane potential onreoxygenation-induced Ca²+ overload by altering Na+/Ca+exchanger activity (Baczko et al., 2003a). In agreement, themembrane potential hyperpolarizing effect of pmKATp openersobserved in this study in reoxygenation after hypoxia-inducedmembrane depolarization (Figure 2b) is accompanied by areduction in Ca+overload (Figure 3b). Based on our previousobservations and on the present study, we propose that themodulation of Na+/Ca+ exchanger-mediated Ca+entry byhyperpolarization of the resting membrane potential as aconsequence of pmKATp activation is an essential componentof the mechanism responsible for the cardioprotection offeredby ATP-sensitive potassium channel opening in ventricularmyocytes. Implications for cardioplegia The heart is subjected to elective global ischaemia duringcardiac surgery or cardiac transplantation. The application ofcardioplegic solutions arrests the heart and preserves itspostoperative function. However, some aspects of the post-operative cardiac dysfunction observed in the majority of patients can, at least in part, be attributed to the use ofhyperkalaemic cardioplegic solutions (Leung, 1993; Cohenet al., 1995). The results from our previous (Baczko et al.,2003a) and present study (Figure 1b, 3a) show that theincreased [K+]o, used in hyperkalaemic cardioplegic solutions(induction: 23 mM, maintenance: 13mM; Fukuhiro et al.,2000),depolarizes myocytes. This diastolic depolarization canlead to increased Ca2+ overload and cell hypercontracture/death upon reperfusion. A number of strategies have beenutilized to reduce Ca2+ overload during experimental cardi-oplegia. These include: (i) lowering [Ca²+]。 in a Mg?+-containing solution (Fukuhiro et al., 2000), (ii) addition ofthe sodium channel blocker tetrodotoxin (Snabaitis et al.,1997), (iii) application of the Na+/H+ exchanger inhibitorHOE-642 (Fukuhiro et al., 2000) and (iv) addition of KATpopeners. It is known that the KATp opener pinacidil canenhance cardioprotection compared to cardioplegic solutionalone (Hosoda et al., 1994). The observation that anotherKATe opener, lemakalim, protected rat hearts undergoinghypothermic cardioplegia when given alone, but failed toimprove post-ischaemic functional recovery when applied in ahyperkalaemic cardioplegic solution (Galinanes et al., 1992),can be rationalized by our results. Thus, it is very likely thatthe protective effect of lemakalim was reduced due todepolarization of the resting membrane potential in hyper-kalaemic milieu. Notably, recent studies on experimentalcardioplegia are in agreement with our results: solutionshyperpolarizing the diastolic membrane potential in rat heartsare effective during cardioplegia (Chambers & Hearse, 1999). Limitations The cellular events observed during metabolic inhibition in thisstudy closely replicate those observed in the setting ofischaemia/reperfusion injury; however, they might not be thesame. Therefore, our findings require further confirmation infuture whole-heart studies. In this study, DiBac4(3) proved to be a reliable qualitativereporter of changes in the resting membrane potential ofventricular myocytes. However, it seems that the dye is not anabsolutely accurate measure of membrane potential at morenegative potentials, where the relationship between normalizedDiBac4(3) fluorescence intensity and actual membrane poten-tial becomes less steep (Figure 1a). Therefore, the changes in ( References ) ( BACZKO, I., GILES, W .R. & LIGHT, P . E. ( 2003a). Resting membrane potential regulate s Na +/ Ca+ e x change-mediated Ca’ a2+ overload during hypoxia/reoxygenation in rat ventricular m yocytes. J. Phy s iol.,550,889-898. ) ( BACZKO, I . , G ILE S , W.R. & LIGHT, P.E . (2003b). Activation o f sarcolemmal K A Tp ch a nnels reduces hy p oxia/reoxygenation-in- duced c alcium o v erload v i a hy p erpolarization o f the re s ting membrane potential (abstract). Biophys. J., 8 4, 422a. ) ( BLAUSTEIN, M.P. & L E DERER, W. J . (1999). Sodium/calcium e x- change: its p hysiological i m plications. Physiol. Re v ., 79 , 763-854. ) BOLLI, R. & MARBAN, E. (1999). Molecular and cellular mechanismsof myocardial stunning. Physiol. Rev., 79, 609-634. BOUCHARD, R.A., CLARK, R.B. & GILES, W.R. (1993). Role ofsodium-calcium exchange in activation of contraction inn ratventricle. J. Physiol., 472, 391-413. DiBaca(3) fluorescence can be used only for estimates inchanges in membrane potential as opposed to an exact voltagereadout. It must be noted that this does not affect the mainfinding of this study, showing that the cardioprotectivemechanism of pmKATe channel activation involves restingmembrane potential hyperpolarization. It is noted that there may be some additional effects ofP-1075 at lower concentrations on cellular processes other thanpmKATp channel activation (Jilkina et al., 2002). However, ourfinding that the selectiveepmKATe blocker HMR1098completely abolished the resting membrane potential hyper-polarizing as well as the protective effects of P-1075 stronglysupports our interpretations regarding a mechanism of actionof P-1075 being on the pmKATe in origin. Also, the findings ofthis paper confirm our previous observations regarding theimportant role of resting membrane potential hyperpolariza-tion in the prevention of reoxygenation-induced Ca+loading(Baczko et al.,2003a). Conclusions Results from this study show that the hyperpolarization of theresting membrane potential caused by pmKATp activation(Figure 2b) is sufficient to result in more effective Ca+extrusion in ventricular myocytes (Figure 3b). Recent findingsfrom our laboratory (Baczko et al., 2003a) suggest thathyperpolarization significantly reduces reverse-mode Na+/Ca2+ exchange activity, leading to a significant decreaseinreoxygenation-induced Ca+overload. Therefore, wesuggest that decreased Ca+ loading via the Na+/Ca2+exchanger may be the critical mechanism responsible for thebeneficial effects of pmKATp activation on Ca+ homeostasisduring reoxygenation. This work was supported by the Alberta Heritage Foundation forMedical Research (PEL, WRG), Heart and Stroke Foundation ofCanada (WRG) and the Canadian Institutes for Health Research(PEL, WRG). PEL is a Scholar of the Alberta Heritage Foundationfor Medical Research (AHFMR) and a Canadian Institutes of HealthResearch New Investigator. Salary support for Dr Baczko wasobtained from the Alberta Heart and Stroke Foundation EndowedResearch Chair held by WRG, and a CIHR Strategic TrainingInitiative. CHAMBERS, D.J. & HEARSE, D.J. (1999). Developments in cardio-protection: polarized’arrest as an alternative to depolarized'arrest. Ann. Thorac. Surg., 68, 1960-1966. COHEN,N.M., DAMIANO,R.J. & WECHSLER, A.S. (1995). Is there analternative to potassium arrest? Ann. Thorac. Surg., 60, 858-863. COLE, W.C., MCPHERSON, C.D. & SONTAG, D. (1991). ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ. Res., 69, 571-581. D'HAHAN, N., MOREAU, C., PROST, A.L., JACQUET, H., ALEKSEEV,A.E., TERZIC, A. & VIVAUDOU, M. (1999). Pharmacological plasticityof cardiac ATP-sensitive potassium channels toward diazoxiderevealed by ADP. Proc. Natl. Acad. Sci. U.S.A., 96, 12162-12167. ap (0DAS,M., PARKER, J.E. & HALESTRAP, A.P. (2003). Matrix volumemeasurements challenge the existence of diazoxide/glibencamide-sensitive KATp channels in rat mitochondria. J. Physiol., 547,893-902. ( DI VIRGILIO, F ., STEINBERG, T.H. & SILVERSTEIN, S . C. ( 1990). I nhibition of Fura-2 sequestration and secretion with organic aniontransport blockers.Cell Calcium, 11,57-62. ) ( EIGEL, B .N. & HADLEY, R.W. (2001). Antisense inhibition o f Na*/ Ca 2 + e xchange d u ring a n oxia/r e oxygenation i n v e ntricular m yo-cytes. Am. J.Physiol., 281 , H2184-H2190. ) ( EPPS, D.E., WOLFE, M.L. & GROPPI, V. (1994). Characterization of the steady-state a nd dynamic fluorescence pro p erties of t he potential- sensitive dye b is-(1,3-dibutylbarbituri c acid)trimethine oxonol(Dibac(4)(3)) in model systems a n d ce l ls. C h em. Ph y s. Li p ids, 69,137 - 150. ) ( FUKUHIRO, Y., WOWK, M. , OU, R . , R O SENFELDT, F. & PEPE, S. (2000). Cardioplegic strategies for calcium control:low Ca+, h ighMg, ci t rate, or Na* /H *e x c hange in h ibitor HOE-642. Circula- tion, 102, III319-III325. ) ( GALINANES,M. , SHATTOC K , M.J. & HEARSE, D.J. (1992). E f fects of potassium channel m o dulation during global ischaemia in isolated rat heart with and without cardioplegia.Cardiovasc. Res . , 26, 1063-1068 . ) GARLID, K.D.,PAUCEK, P., YAROV-YAROVOY,V.,MURRAY,H.N.,DARBENZIO,R.B.,D'ALONZO, A.J., LODGE,N.J.,SMITH, M.A. &GROVER,G.J. (1997). Cardioprotective effect of diazoxide and itsinteraction with mitochondrial ATP-sensitive K+ channels. Possi-ble mechanism of cardioprotection. Circ. Res., 81, 1072-1082. GOGELEIN, H., ENGLERT, H.C., KOTZAN, A., HACK, R., LEHR,K.H., SEIZ, W., BECKER, R.H.A., SULTAN, E., SCHOLKENS, B.A.& BUSCH, A.E. (2000). HMR 1098: an inhibitor of cardiac ATP-sensitive potassium channels. Cardiovasc. Drug Rev., 18,157-174. ( GRIMMSMANN,T. & RUSTENBECK, I. (1 9 9 8 ). Direct effects o f diazoxide o n mitochondria in pa n creatic p- c ells a n d on is o lated l iver mitochondria. Br. J. Pharmacol., 123,781- 7 88. ) GROSS,G.J. & FRYER,R.M.(1999). Sarcolemmal versus mitochondrialATP-sensitive K+ channels and myocardial preconditioning. Circ.Res., 84,973-979. GROSS, G.J. & PEART, J.N. (2003). KATp channels and myocardialpreconditioning: an update. Am. J. Physiol., 285, H921-H930. HANLEY, P.J., GOPALAN, K.V., LAREAU, R.A., SRIVASTAVA, D.K.,VON MELTZER, M. & DAUT, J. (2003). Oxidation of 5-hydro-xydecanoate, a putative blocker of mitochondrial ATP-sensitivepotassium channels. J. Physiol., 547,387-393. ( HANLEY, P . J., MICKEL, M., LOFFLER, M., BRANDT, U. & DAUT,J. (2002). KATp c hannel-independent t argets o f d iazoxide and 5 - hydroxydecanoate in the heart. J. Physiol., 542, 735-741. ) ( HEARSE,D.J . (1995). Activation of ATP-s e nsitive po t assium channels:a novel p harmacological approach t o m yocardial p rotection? Cardiovasc. Res., 30, 1 -17. ) ( HOSODA, H., SUN A MORI, M. & SUZUK I , A . (1994). Ef f ect of pinacidil on r at h earts undergoing hypothermic c a rdioplegia. Ann. T h orac. Surg., 58, 1631 - 1636. ) ( INAGAKI, N., GONOI,T., CLEMENT,J.P., NAMBA, N ., INAZAWA,J., GONZALEZ, G., AGUILAR-BRYAN, L., SE I NO, S. & BRYAN, J.(1995). Reconstitution of IKATe: an inward rectifier subunit plus the sulfonylurea receptor. Science,270, 1 166-1170. ) ( INAGAKI. N ., GONOI, T., CLEMENT, J .P., WANG, C . Z.AGUILA R -BRYAN,L., BR Y AN, J. & SEINO, S. (1 9 96). A f a milyof sulfonylurea r eceptors d etermines t h e p h armacological proper- ties of ATP-sensitive K * channels. Neuron, 16, 1011 - 1017. ) ( JILKINA, O., KUZIO, B., GROVER, G.J. & K U PRIYANOV,V.V. (2002). Effects of KATp c hannel openers, P-1075, p i nacidil, and diazoxide, on energetics a nd contractile function in i solated r at hearts. J. Mol. Cell. Cardiol., 34, 427-440. ) ( JOVANOVIC, A., JOVANOVIC, S., LORENZ, E.& TERZIC, A. (1998).Recombinant c ardiac ATP-sensitive K c hannel s ubunits c onfer r esistance to chemical hypoxia-reoxygenation injury. Ci r culation,98. 1 548-1555. ) ( LAWRENCE, C.L . , BILLUP S , B., RODRIGO, G.C. & STANDEN, N.B. (2001). The K ATp c hannel o pener d i azoxide p rotects cardiac myocytes during metabolic inhibition w ithout c ausing m itochon-drial depolarization o r f lavoprotein o x idation. B r .J. Ph a rmacol., 134,535-542. ) LEUNG, J.M. (1993). Clinical evidence of myocardial stunning inpatients undergoing CABG surgery. J. Cardiac. Surg., 8 (Suppl),220-223. LIGHT, P.E., KANJI, H.D., MANNING FOX,J.E. & FRENCH, R.J.(2001). Distinct myoprotective roles of cardiac sarcolemmal andmitochondrial KATP channels during metabolic inhibition andr73ecovery. FASEB J., 15, 2586-2594. ( L I GHT, P.E., SHIMONI, Y. , HARBISON, S. , GILES, W .R. & FRENCH, R .J. (1998). Hypothyroidism d e creases t h e A T P sensitivity of KAT p channels from r a t h e art. J. M embr. Bi o l., 1 6 2, 2 1 7-2 23 . ) ( LIU, Y.G., REN, G.F., O’ROURKE, B., MARBAN, E. & SEHARASEYON, J. (2001). Pharmacological comparison of native mitochondrial KATechannels w ith m olecularly d efined s urface K ATe c h annels. Mol. Pharmacol., 59,225-230. ) ( U , Y.G., SATO, T. , O'ROURKE, B . & M ARBAN, E. (1998). Mitochondrial A TP-dependent potassium ch a nnels: novel effectorsof cardioprotection? Circulation, 97, 2 463-2469. ) MANNING FOX, J.E., KANJI, H.D., FRENCH, R.J. & LIGHT, P.E.(2002). Cardioselectivity of the sulphonylurea HMR 1098: studieson native and recombinant cardiac and pancreatic KATp channels.Br.J. Pharmacol., 135,480-488. MARBAN, E., KORETSUNE, Y., CORRETTI, M., CHACKO, V.P. &KUSUOKA, H. (1989). Calcium and its role in myocardial cell injuryduring ischemia and reperfusion. Circulation, 80, IV17-IV22. MCPHERSON, C.D., PIERCE, G.N. & COLE, W.C. (1993). Ischemiccardioprotection by ATP-sensitive K+ channels involves high-energy phosphate preservation. Am. J.Physiol.,265,H1809-H1818. NOMA, A. (1983). ATP-regulated K* channels in cardiac muscle.Nature, 305, 147-148. ( O'ROURKE, B. (2000). Pa t hophysiological an d pro t ective rol e s of mitochondrial ion channels. J. P hysiol., 15, 23-36. ) ( PANDIT, S.V., C LA RK, R.B., GILES, W .R. & DEMIR, S.S. (2001). A m athematical m o del of action potential h e terogeneity i n adult rat l eft ventricula r myocytes. Biophys. J., 8 1 ,3029-3051. ) PARK, J.L. & LUCCHESI, B.R. (1999). Mechanisms of myocardialreperfusion injury. Ann. Thorac. Surg., 68,1905-1912. PIPER, H.M. & GARCIA-DORADO, D. (1999). Prime causes of rapidcardiomyocyte death during reperfusion. Ann. Thorac. Surg., 68,1913-1919. ( SATO, T ., SASAKI, N., SEHARASEYON, J ., O'ROURKE, B . . & MARBAN, E. ( 20 0 0). Sel e ctive ph a rmacological age n ts implicate mitochondrial but n ot sarcolemmal K A Tp ch a nnels in is c hemic cardioprotection. C irculation, 101,2418-2423. ) ( SCHAFER, C., LADILOV, Y., INSERTE,J., SCHAFER,M., HAFFNER,S., GARCIA-DORADO, D. & P IPER, H.M. (20 0 1). Ro l e of th e reverse mode o f the Na+/ C a+ exchanger in reoxygenation-inducedcardiomyocyte i n jury. Cardiovasc. Res., 51, 241 - 250. ) ( SCHWANSTECHER, M., SIEVERDING, C., DORSCHNER,H., GROSS,I. , AG U ILAR-BRYAN, L.,SCHWANSTECHER, C . & B RYAN, J.(1998). Potassium c hannel o peners require A TP to b i nd t o and a c tthrough sulfonylurea receptors . EMBO J., 17, 5529-5535. ) ( SNABAITIS, A.K., SHATTOCK, M .J. & CHAMBERS, D. J . (1997). Comparison of polarized and depolarized arre s t in t h e i sol a ted rat h eart for long-term p reservation. C irculation, 96, 3148-3156. ) ( SUZUKI, M . , SAITO, T., S ATO, T., TAMAGAWA, M., MIK I , T., SEINO, S. & N AKAYA, H. (2003). Ca r dioprotective effect o f diazoxide i s mediated by activation o f sarcolemmal b u t n o t mitochondrial ATP- s ensitive potassium channels i n mice. C i rculation, 107, 682-6 8 5. ) ( SUZUKI, M ..SASAKI. N., M IKI. T ., SAKAMOTO. N.. OHMOTO-SEKINE, Y., TAMAGAWA, M., SEINO, S ., MARBAN, E . & N AKAYA, H. (2002). Ro l e of sarcolemmal KA T p channels in cardioprotection against ischemia/reperfusion injury in mice. J . C lin. I nvest., 109, 509-516. ) ( T ANI, M. (1990). Mechanisms of Ca+ove r load in rep e rfused ischemic myocardium. A n nu. Rev. P h ysiol., 52,543-5 5 9. ) (ReceivedOctober 18, 2003 ( Revised December 2, 2003 ) British Journal of Pharmacology vol
确定





还剩7页未读,是否继续阅读?

海德创业(北京)生物科技有限公司为您提供《生物膜电位荧光染料 DiBAC4(3)在心肌细胞中应用》,该方案主要用于其他中--检测,参考标准--,《生物膜电位荧光染料 DiBAC4(3)在心肌细胞中应用》用到的仪器有
相关方案
更多
该厂商其他方案
更多