







在样品纯化过程中你是否遇到过五花八门的难点,这个问题对于做制备纯化的小孙来说有太多值得吐槽的地方,如样品极性太小正相模式不保留、样品极性太大硅胶固定相洗脱不下来、样品溶解度太差纯化过程析出或拖尾严重及洗脱不下来等情况。在分离纯化的过程中,难免会碰到溶解性比较差的物质,这类物质有时会对分离纯化造成一些负面的影响,如由于拖尾而影响分离度,甚至是直接析出导致管路堵塞,或者少量试剂不能完全溶解至澄清,而样品纯化之前一定要有解决溶解度的办法,否则带着问题上样纯化效果一定不会很理想。解决样品溶解度的问题没有固定答案,具体样品需要具体分析,本文主要和大家分享小孙在纯化过程中如何处理溶解度较差的样品。
方案详情

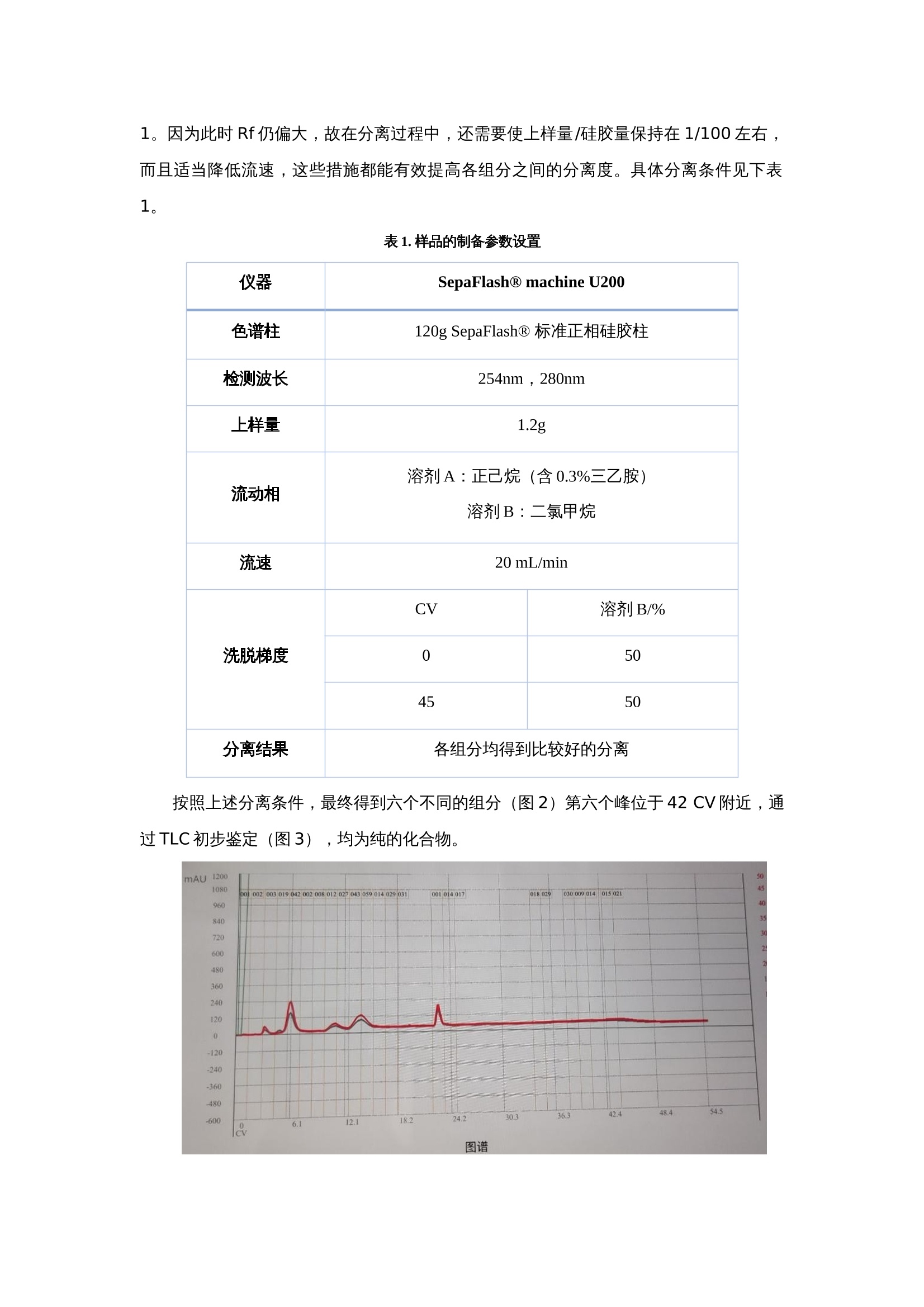
FLASH色谱纯化实验小讲堂 纯化时样品溶解度问题的解决方案 在样品纯化过程中你是否遇到过五花八门的难点,这个问题对于做制备纯化的小孙来说有太多值得吐槽的地方,如样品极性太小正相模式不保留、样品极性太大硅胶固定相洗脱不下来、样品溶解度太差纯化过程析出或拖尾严重及洗脱不下来等情况。在分离纯化的过程中,难免会碰到溶解性比较差的物质,这类物质有时会对分离纯化造成一些负面的影响,如由于拖尾而影响分离度,甚至是直接析出导致管路堵塞,或者少量试剂不能完全溶解至澄清,而样品纯化之前一定要有解决溶解度的办法,否则带着问题上样纯化效果一定不会很理想。解决样品溶解度的问题没有固定答案,具体样品需要具体分析,本文主要和大家分享小孙在纯化过程中如何处理溶解度较差的样品。主要常用以下几种方法: 加助溶试剂 解决溶解度问题最根本的是要找到样品最合适的溶解试剂,但是有时能溶解样品的试剂不适应于样品的分离模式。 如某样品极性很小,乙腈易溶解,但因极性太小在反相柱中不易洗脱,正相模式下正己烷-乙酸乙酯体系不能完全溶解样品导致拖尾致使样品没有分离度。最后解决办法添加第三路管路10%乙腈,虽然乙腈和正己烷不互溶,但是添加的10%乙腈解决了部分样品的溶解度问题,最终纯化结构是能提纯到一半纯品,一半交叉,回收交叉部分按此方法可继续纯化。 用极性相近的试剂置换 反相纯化时除了非水反相,其他体系如甲醇-水/乙腈-水等都会使用到水,但是很多样品用有机试剂溶解后遇水会析出,这种情况下即使可以正常实验也会使峰形拖尾或导致分离度降低,甚至有的样品会直接析出结块导致仪器超压不能运行等情况。 三泰科技的应用实验人员也遇到过上述情况,样品析出情况严重,如何解决这个问题,经过试验发现可以用一种有机试剂代替水相运行即甲酰胺,甲酰胺相对极性7.3,可以与水、甲醇等互溶,可以上样时临时代替水润柱,上样后就不会有析出的情况,甲酰胺在反相体系中没有洗脱能力,极性和水较相近可以代替水运行实验,考虑到成本问题成功运行后可以再用水置换过来。 等度洗脱 当样品在一方流动相中有溶解度时,可以考虑在最佳分离度的情况下等度洗脱,不需要从低比例慢慢加,减少析出的情况,案例如下: 待分离物质是一种卟啉类化合物的分离过程,该化合物溶解性稍差。通过TLC点板(图1)精确筛选流动相比例的过程中发现,当二氯甲烷的比例低于50%极易拖尾,且各组分之间没有明显的分离度,可能是由于此时二氯甲烷比例偏低,导致溶剂体系的溶解性降低,故拖尾严重;当二氯甲烷高于50%时,各组分之间有一定的分离度,且无明显拖尾现象,但Rf偏大。 图1.从左至右二氯甲烷比例依次增大,左三为50% 综合上述信息,我们决定采用等度淋洗,控制流动相恒定在正己烷:二氯甲烷=1:1。因为此时Rf仍偏大,故在分离过程中,还需要使上样量/硅胶量保持在1/100左右,而且适当降低流速,这些措施都能有效提高各组分之间的分离度。具体分离条件见下表1。 表1. 样品的制备参数设置 仪器 SepaFlash® machine U200 色谱柱 120g SepaFlash® 标准正相硅胶柱 检测波长 254nm,280nm 上样量 1.2g 流动相 溶剂A:正己烷(含0.3%三乙胺) 溶剂B:二氯甲烷 流速 20 mL/min 洗脱梯度 CV 溶剂B/% 0 50 45 50 分离结果 各组分均得到比较好的分离 按照上述分离条件,最终得到六个不同的组分(图2)第六个峰位于42 CV附近,通过TLC初步鉴定(图3),均为纯的化合物。 图2. 分离的色谱图 图3.分离前后对比点板(前四个产物) 上述3种方法是三泰科技技术人员遇到的样品纯化解决方案个例,相信大家在纯化时也会遇到这种情况,欢迎大家与我们分享解决方案。 如需进一步了解三泰产品订购信息,请访问ChemBeanGo在线商店:https://store.chembeango.com/。 在样品纯化过程中你是否遇到过五花八门的难点,这个问题对于做制备纯化的小孙来说有太多值得吐槽的地方,如样品极性太小正相模式不保留、样品极性太大硅胶固定相洗脱不下来、样品溶解度太差纯化过程析出或拖尾严重及洗脱不下来等情况。在分离纯化的过程中,难免会碰到溶解性比较差的物质,这类物质有时会对分离纯化造成一些负面的影响,如由于拖尾而影响分离度,甚至是直接析出导致管路堵塞,或者少量试剂不能完全溶解至澄清,而样品纯化之前一定要有解决溶解度的办法,否则带着问题上样纯化效果一定不会很理想。解决样品溶解度的问题没有固定答案,具体样品需要具体分析,本文主要和大家分享小孙在纯化过程中如何处理溶解度较差的样品。主要常用以下几种方法:1、加助溶试剂解决溶解度问题最根本的是要找到样品最合适的溶解试剂,但是有时能溶解样品的试剂不适应于样品的分离模式。如某样品极性很小,乙腈易溶解,但因极性太小在反相柱中不易洗脱,正相模式下正己烷-乙酸乙酯体系不能完全溶解样品导致拖尾致使样品没有分离度。最终解决办法添加第三路管路10%乙腈,虽然乙腈和正己烷不互溶,但是添加的10%乙腈解决了部分样品的溶解度问题,最终纯化结构是能提纯到一半纯品,一半交叉,回收交叉部分按此方法可继续纯化。2、用极性相近的试剂置换反相纯化时除了非水反相,其他体系如甲醇-水/乙腈-水等都会使用到水,但是很多样品用有机试剂溶解后遇水会析出,这种情况下即使可以正常实验也会使峰形拖尾或导致分离度降低,甚至有的样品会直接析出结块导致仪器超压不能运行等情况。三泰科技的应用实验人员也遇到过上述情况,样品析出情况严重,如何解决这个问题,经过试验发现可以用一种有机试剂代替水相运行即甲酰胺,甲酰胺相对极性7.3,可以与水、甲醇等互溶,可以上样时临时代替水润柱,上样后就不会有析出的情况,甲酰胺在反相体系中没有洗脱能力,极性和水较相近可以代替水运行实验,考虑到成本问题成功运行后可以再用水置换过来。3、等度洗脱当样品在一方流动相中有溶解度时,可以考虑在最合适的分离度的情况下等度洗脱,不需要从低比例慢慢加,减少析出的情况,案例如下:待分离物质是一种卟啉类化合物的分离过程,该化合物溶解性稍差。通过TLC点板(图1)精确筛选流动相比例的过程中发现,当二氯甲烷的比例低于50%极易拖尾,且各组分之间没有明显的分离度,可能是由于此时二氯甲烷比例偏低,导致溶剂体系的溶解性降低,故拖尾严重;当二氯甲烷高于50%时,各组分之间有一定的分离度,且无明显拖尾现象,但Rf偏大。 图1.从左至右二氯甲烷比例依次增大,左三为50%综合上述信息,我们决定采用等度淋洗,控制流动相恒定在正己烷:二氯甲烷=1:1。因为此时Rf仍偏大,故在分离过程中,还需要使上样量/硅胶量保持在1/100左右,而且适当降低流速,这些措施都能有效提高各组分之间的分离度。具体分离条件见下表1。表1. 样品的制备参数设置仪器SepaFlash® machine U200色谱柱120g SepaFlash® 标准正相硅胶柱检测波长254nm,280nm上样量1.2g流动相溶剂A:正己烷(含0.3%三乙胺)溶剂B:二氯甲烷流速20 mL/min洗脱梯度CV溶剂B/%0504550分离结果各组分均得到比较好的分离按照上述分离条件,最终得到六个不同的组分(图2)第六个峰位于42 CV附近,通过TLC初步鉴定(图3),均为纯的化合物。 图2. 分离的色谱图图3.分离前后对比点板(前四个产物)上述3种方法是三泰科技技术人员遇到的样品纯化解决方案个例,相信大家在纯化时也会遇到这种情况,欢迎大家与我们分享解决方案。
确定




还剩3页未读,是否继续阅读?

产品配置单


常州三泰科技有限公司为您提供《溶解度较差样品中纯化检测方案(制备液相色谱)》,该方案主要用于其他中组成分析检测,参考标准--,《溶解度较差样品中纯化检测方案(制备液相色谱)》用到的仪器有SepaBean machine T快速液相制备色谱、SepaBean machine快速液相制备色谱
推荐专场





相关方案
更多