








方案详情
文
人造肉产品迅速兴起后,随之而来的食品口感、风味、营养、安全等问题也逐渐备受瞩目。为了应对人造肉企业相关用户的需求,岛津分析中心精心推出《人造肉检测整体解决方案》,汇编了口感检测、风味物质、营养成分、添加剂及有毒有害成分的检测报告,希望我们的工作能够对您有所帮助。
方案详情

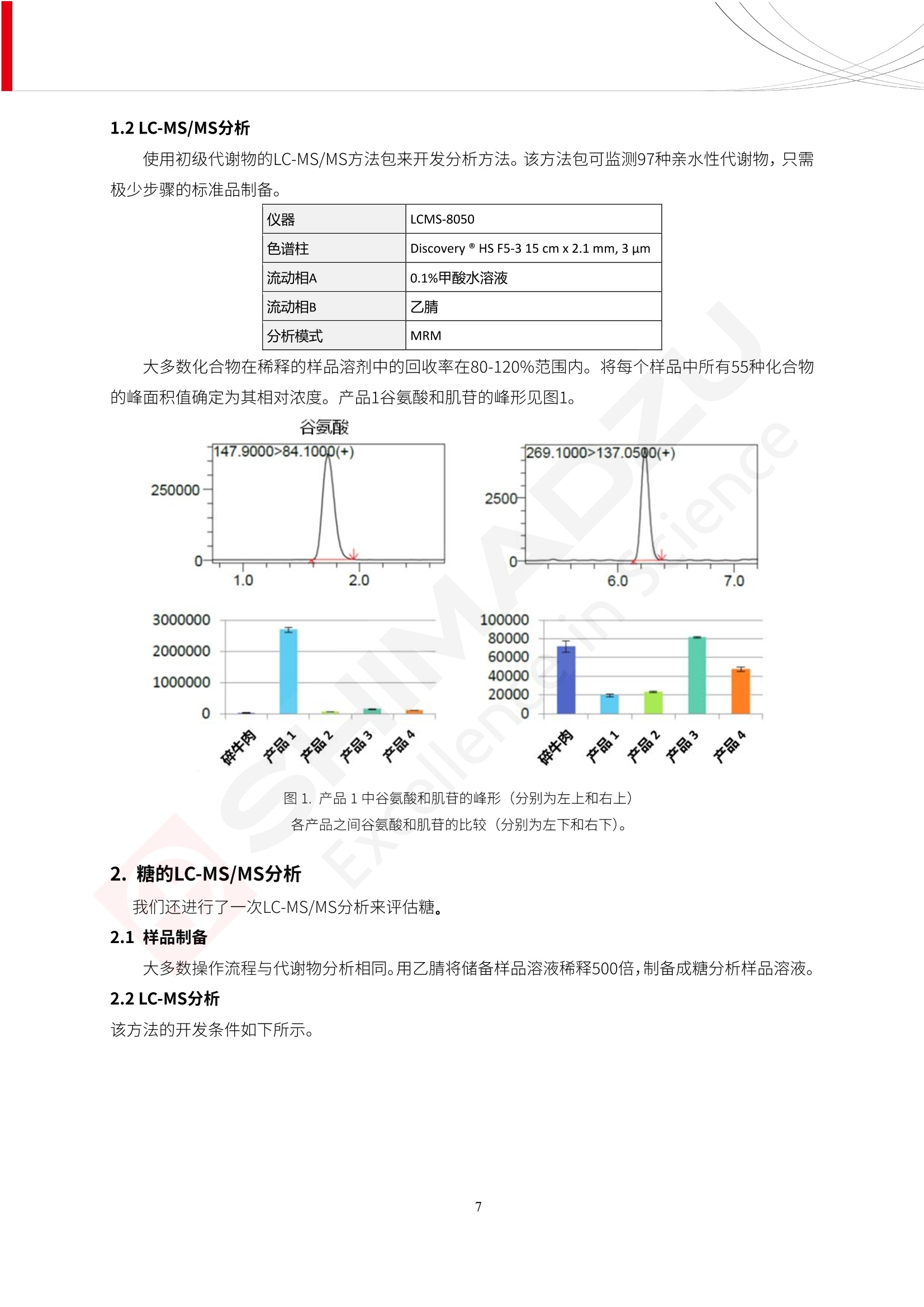
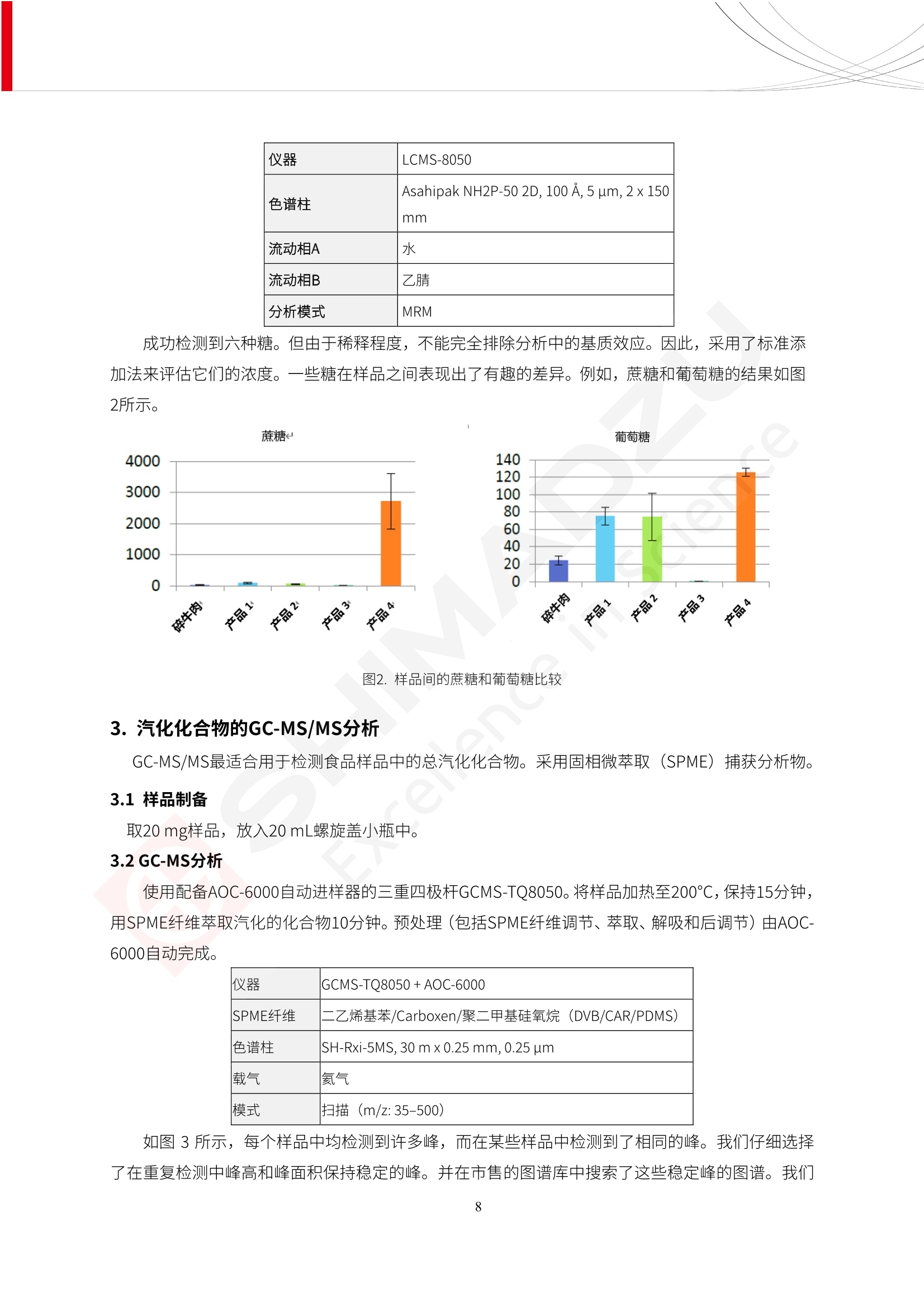
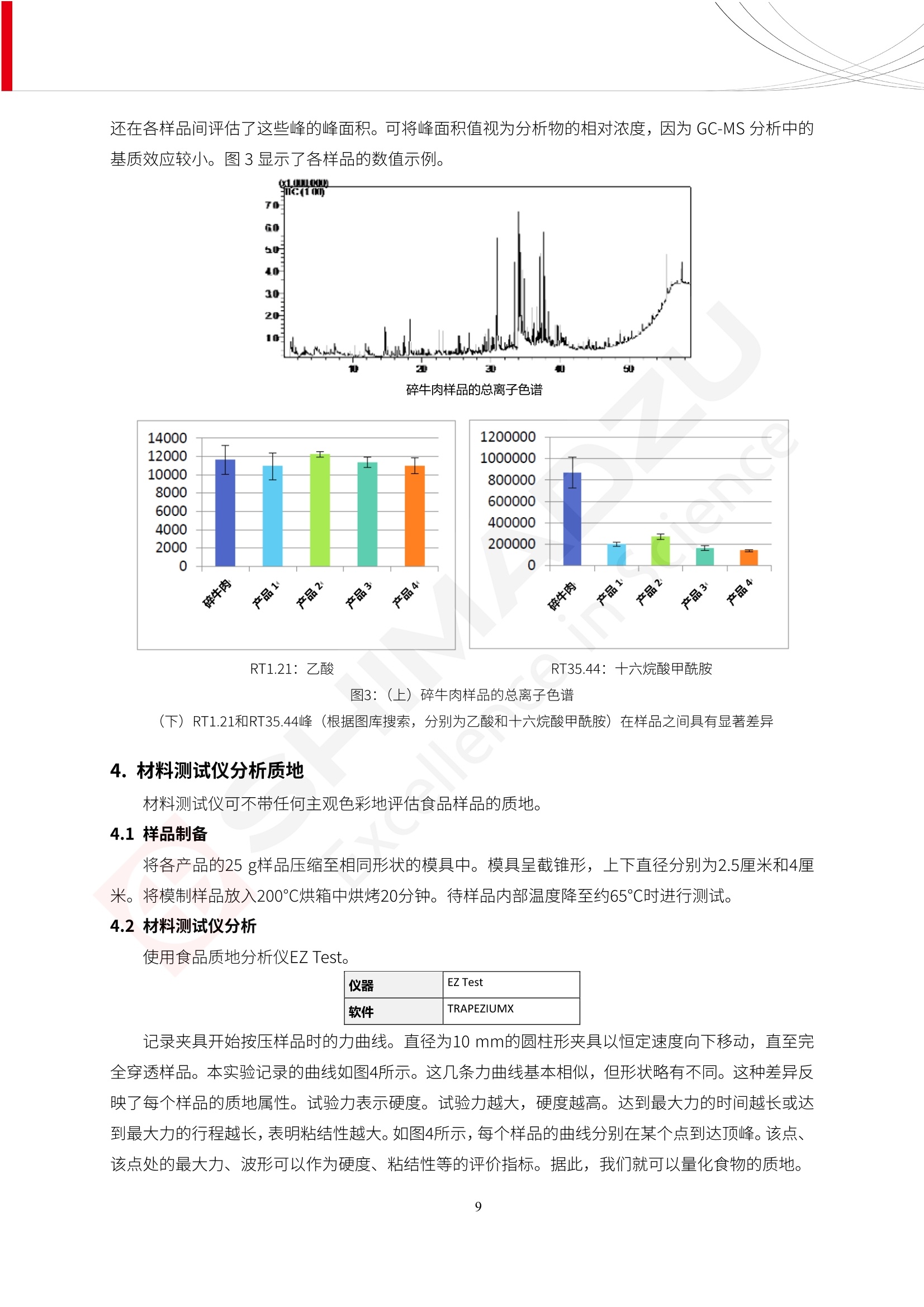
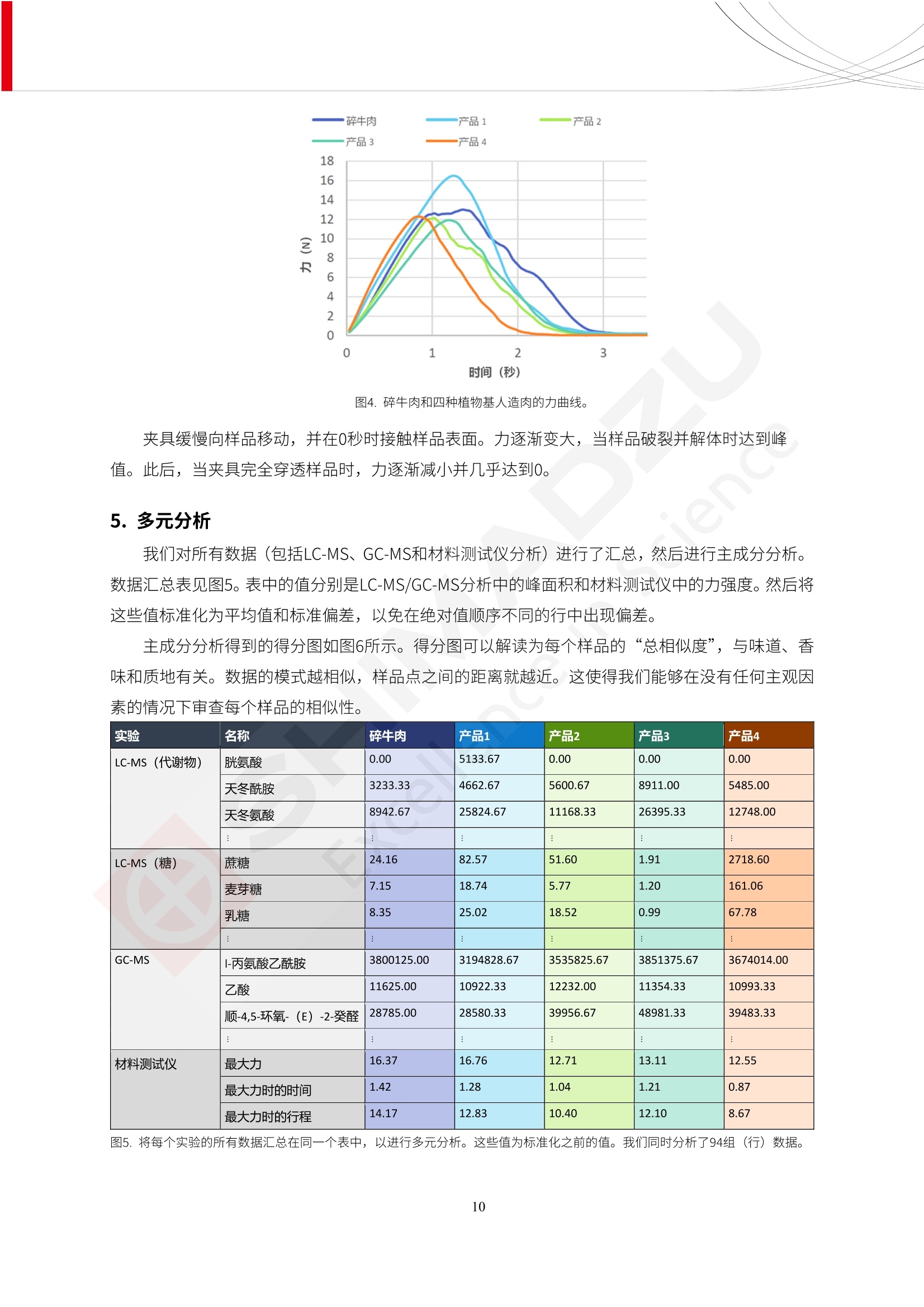
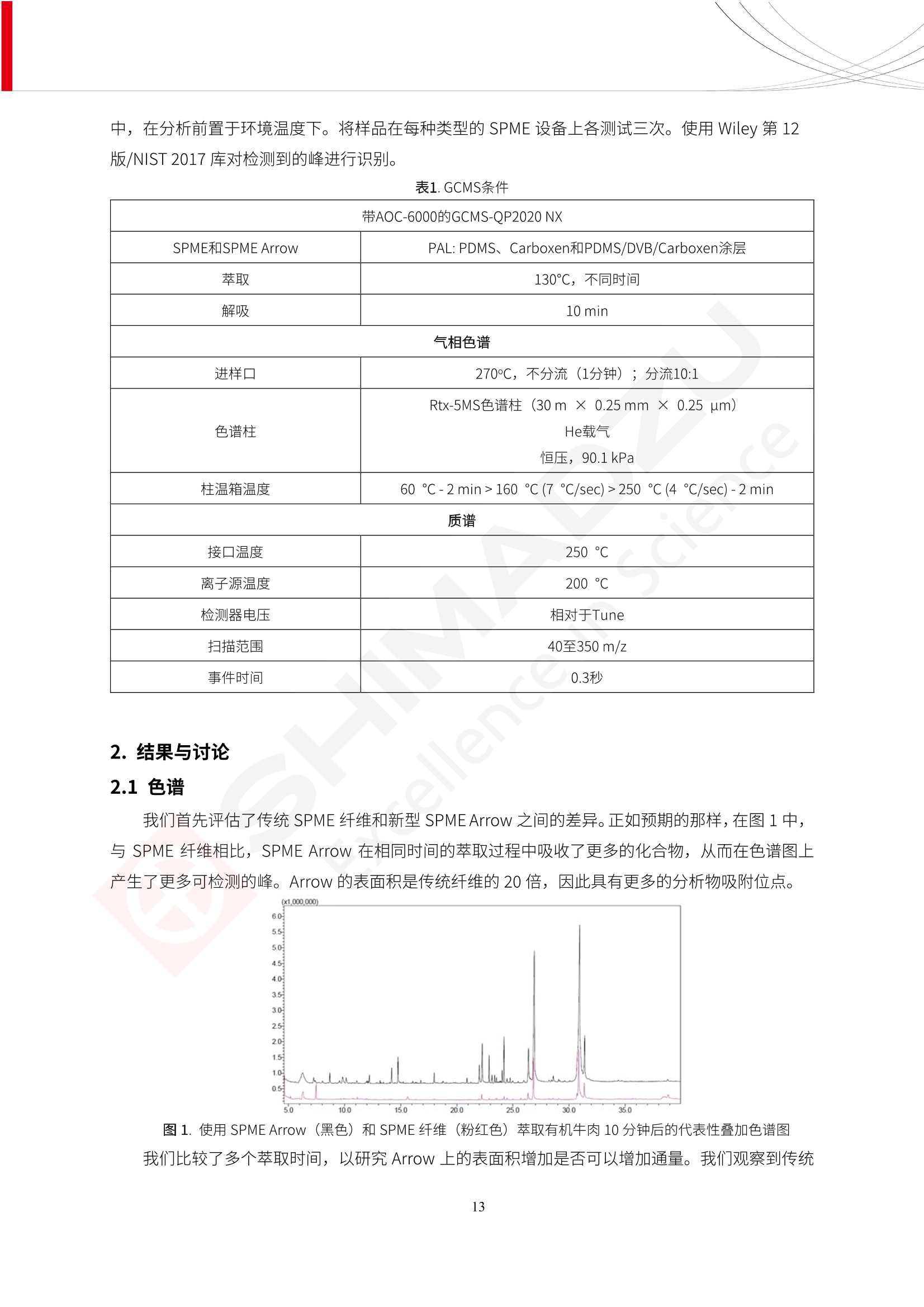
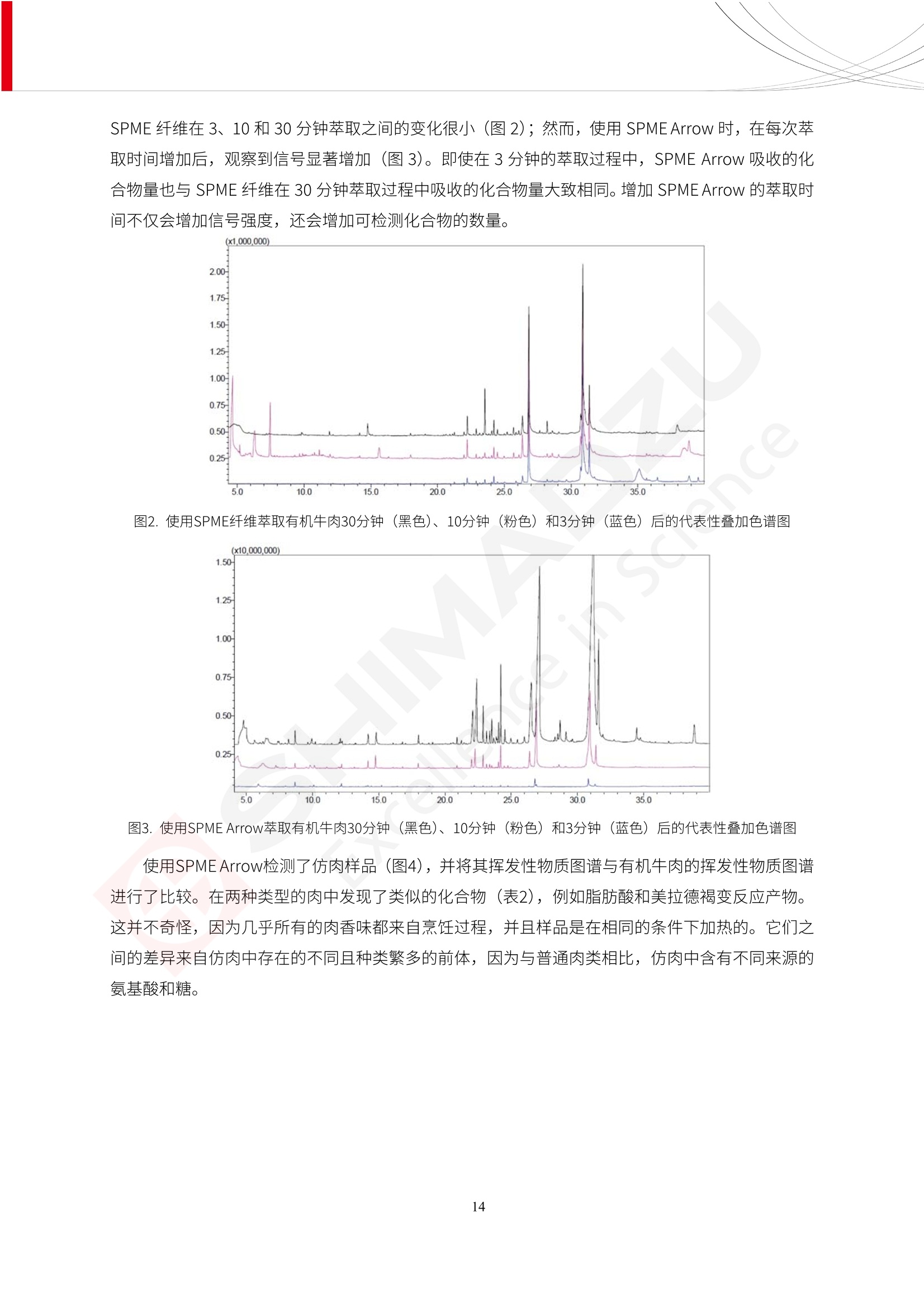
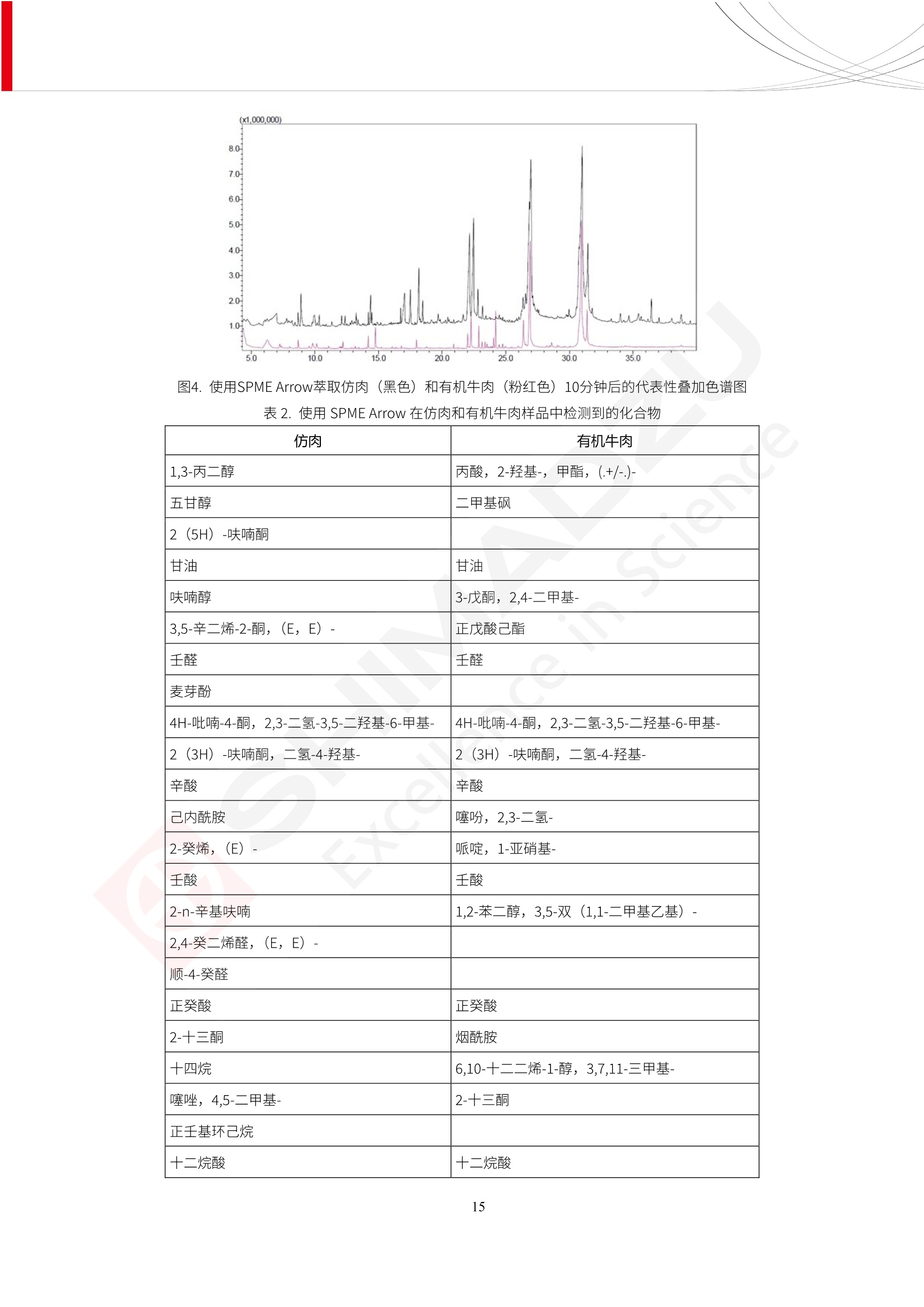
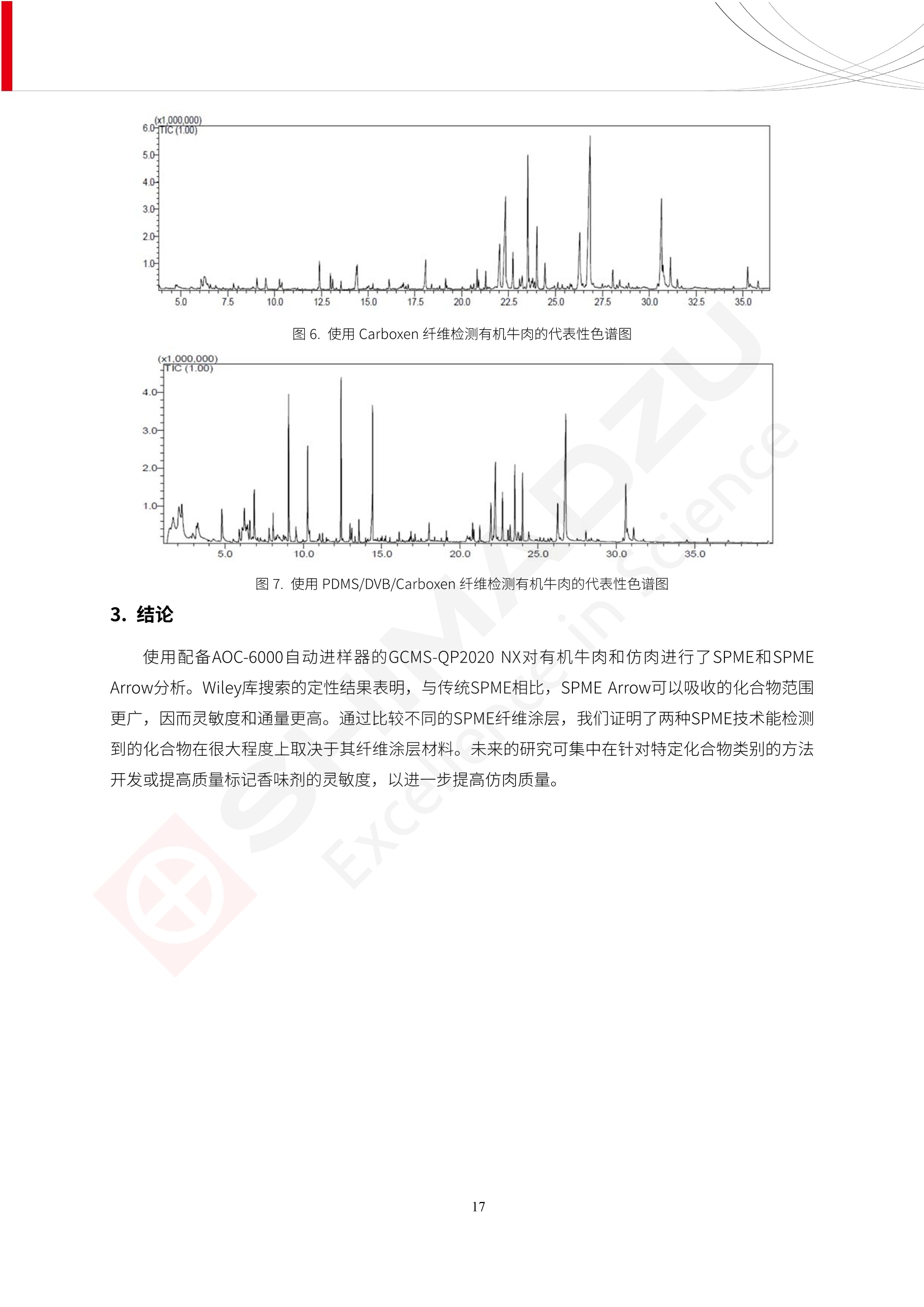
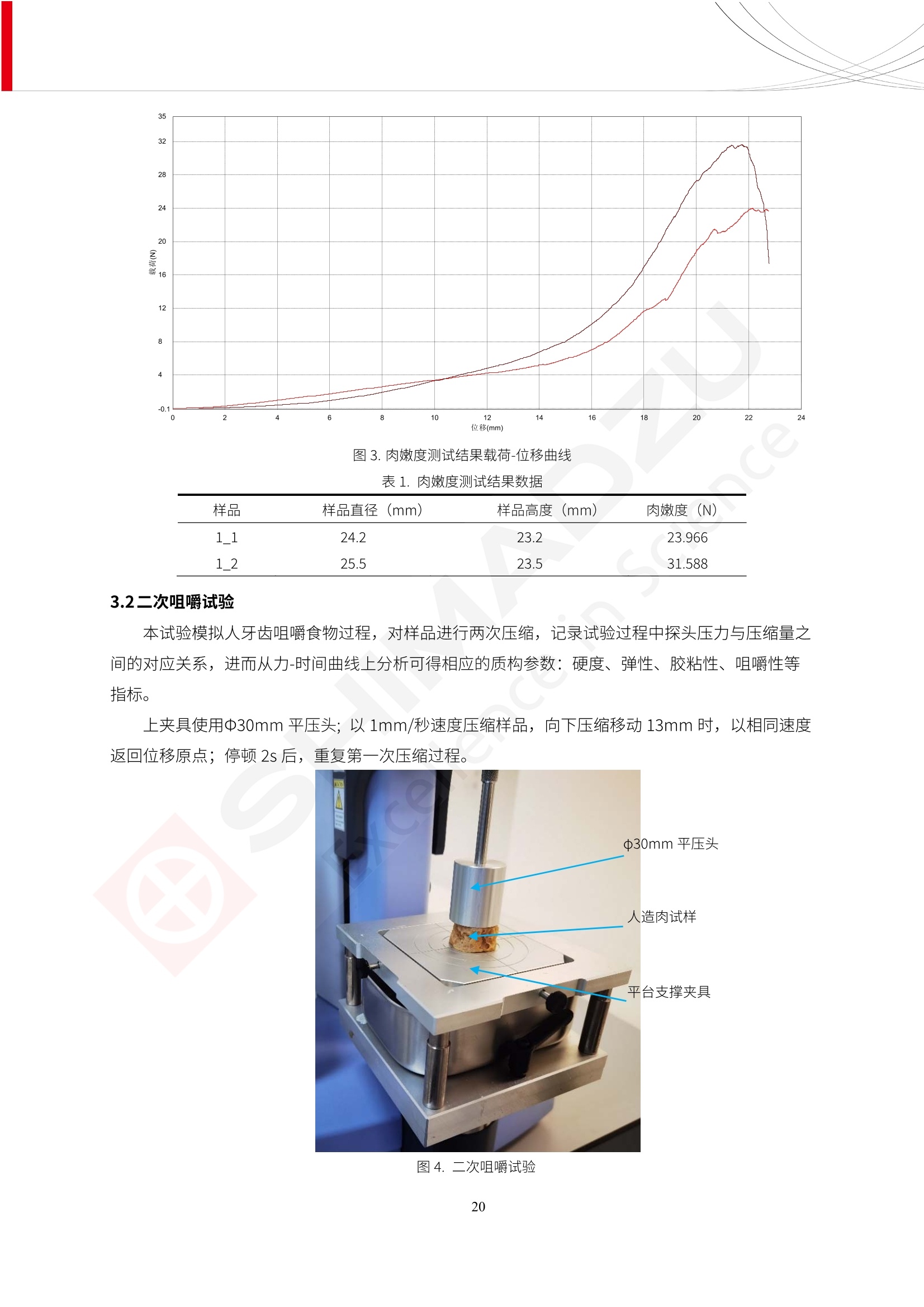
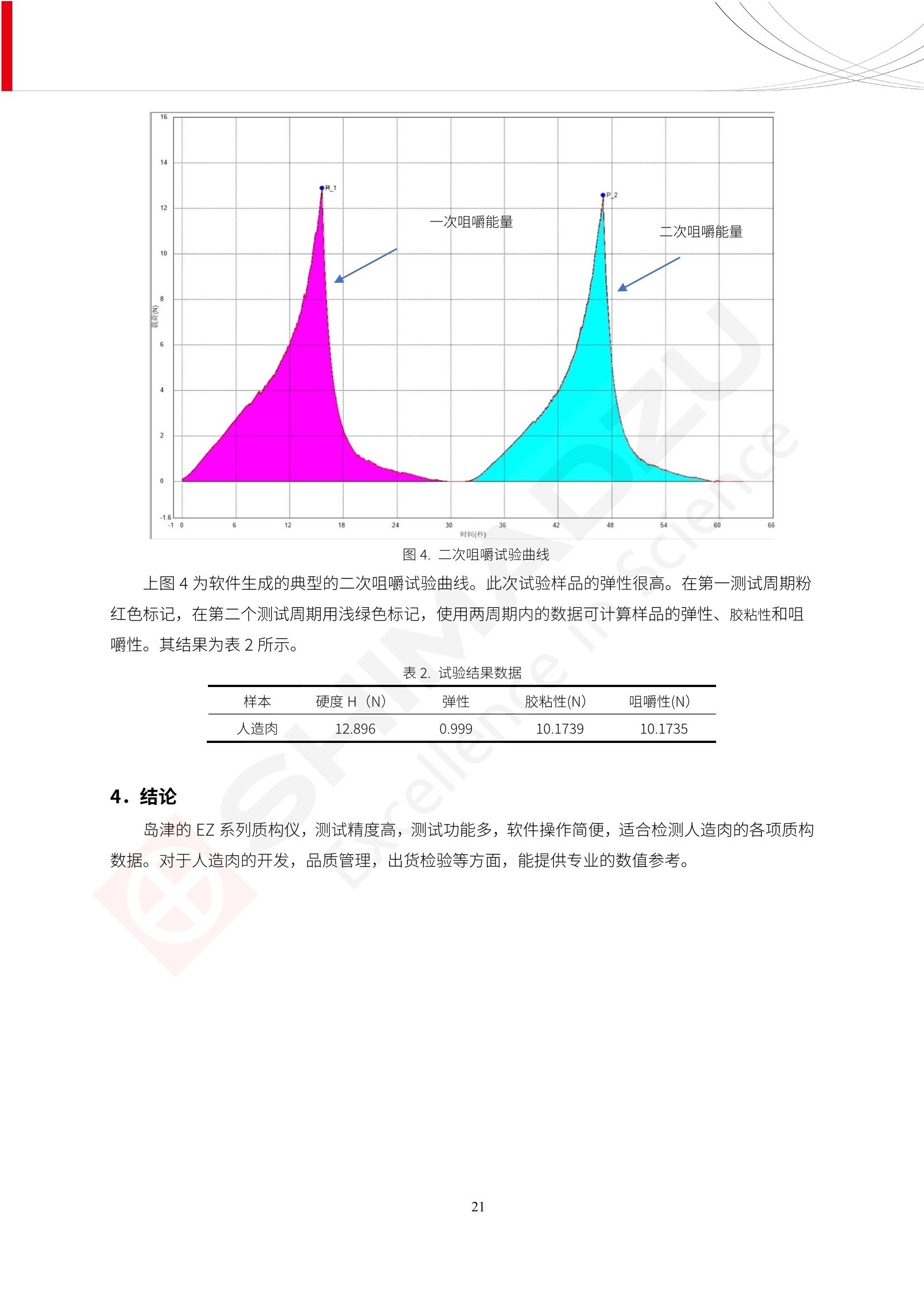
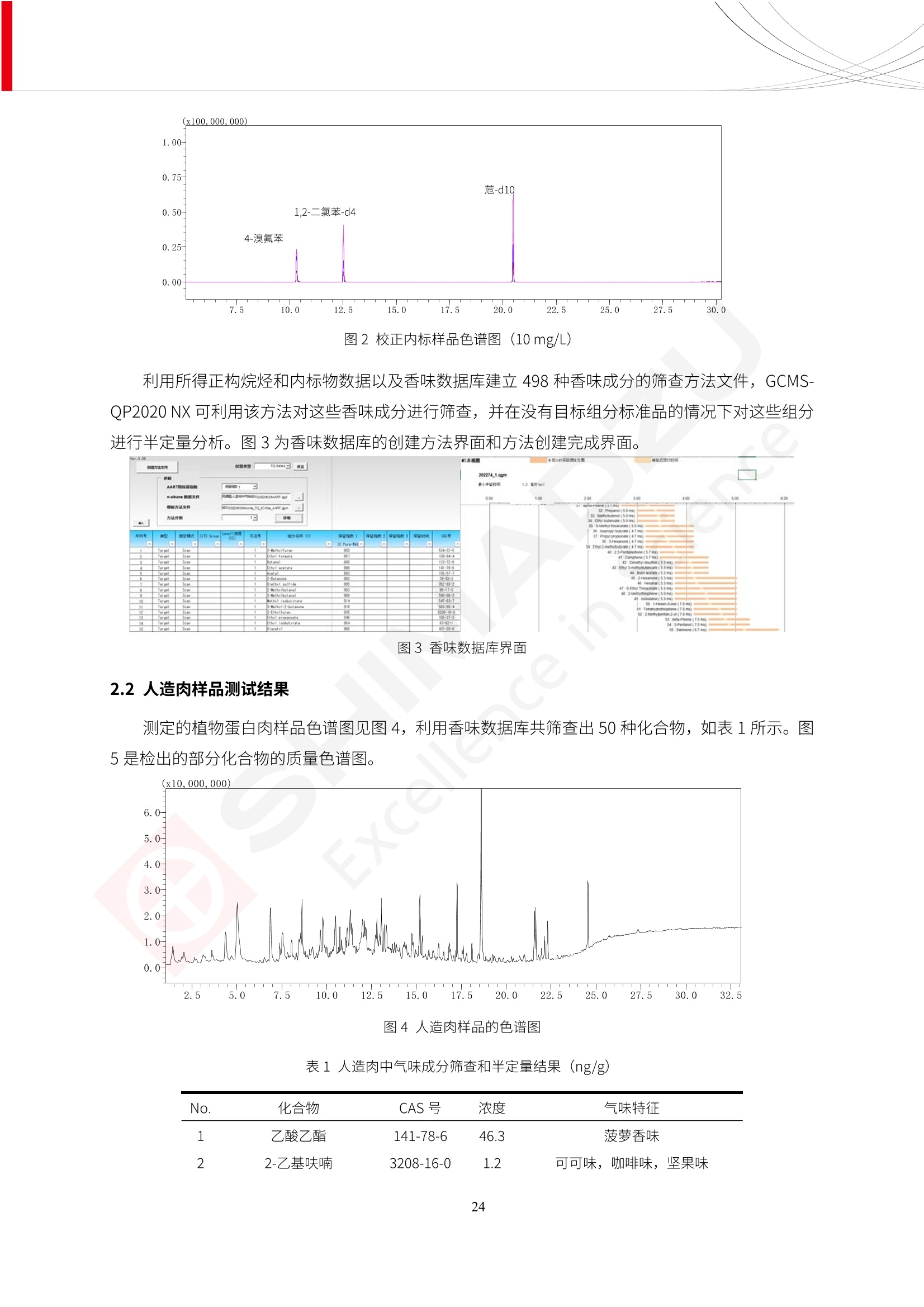
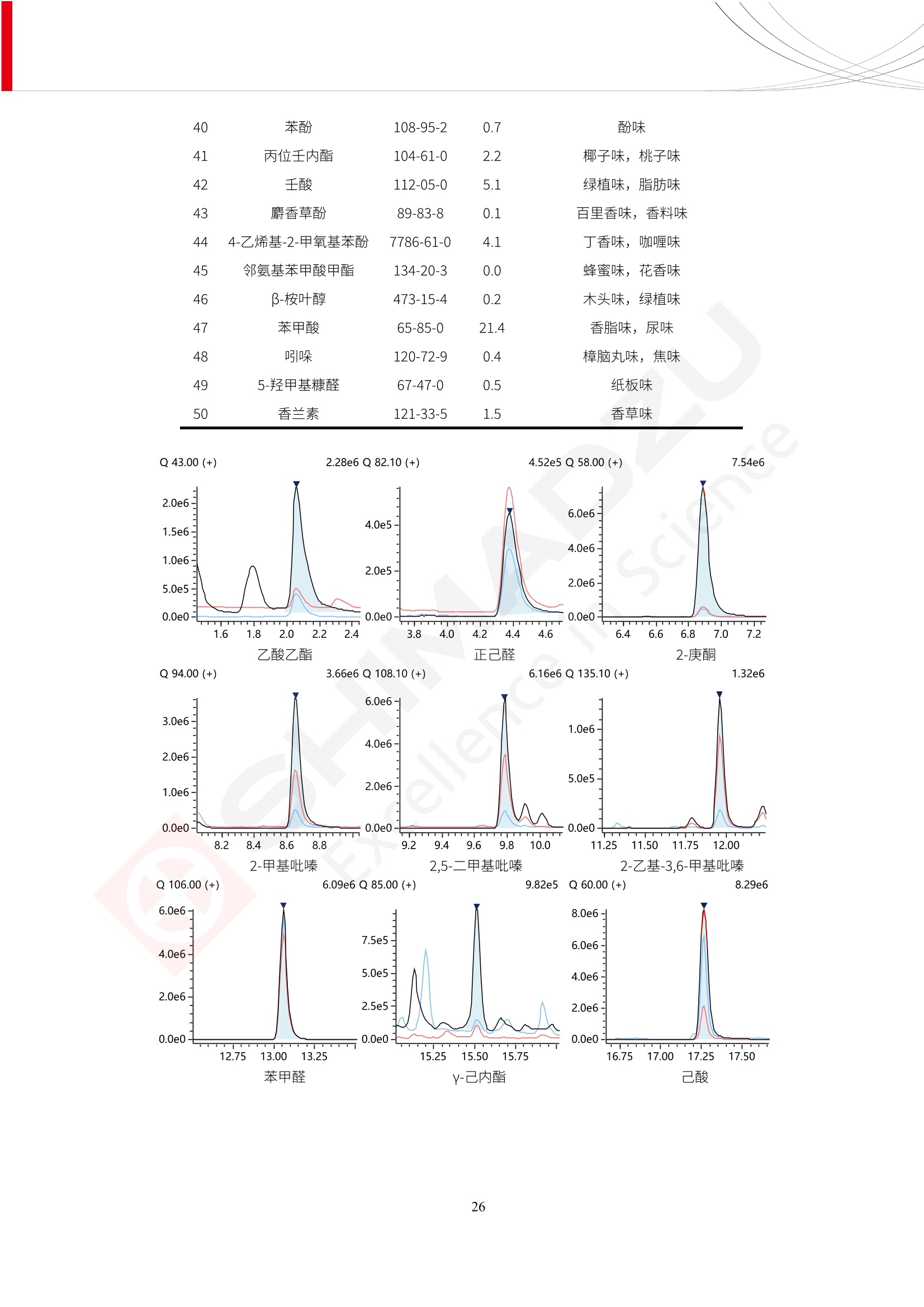
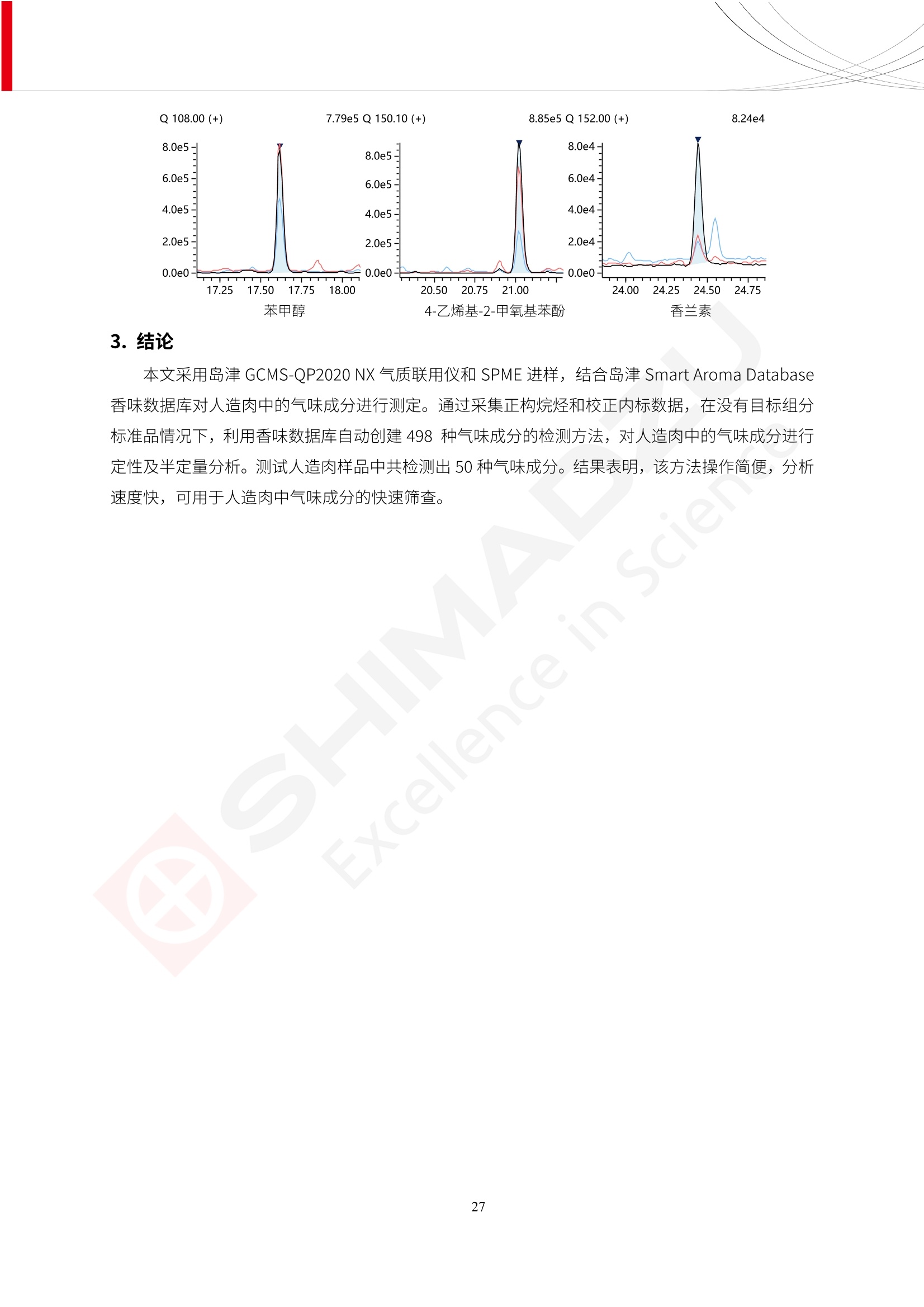
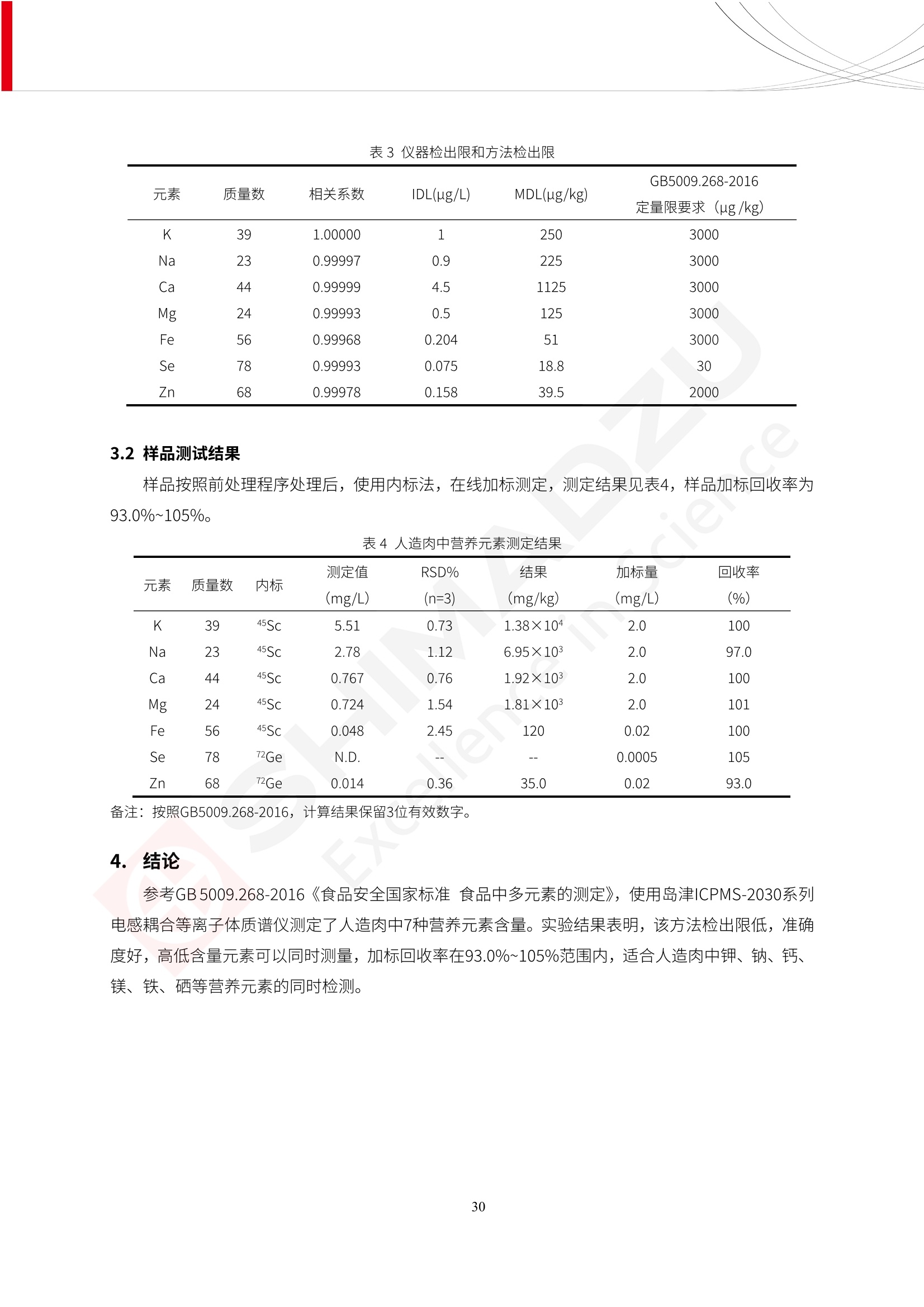
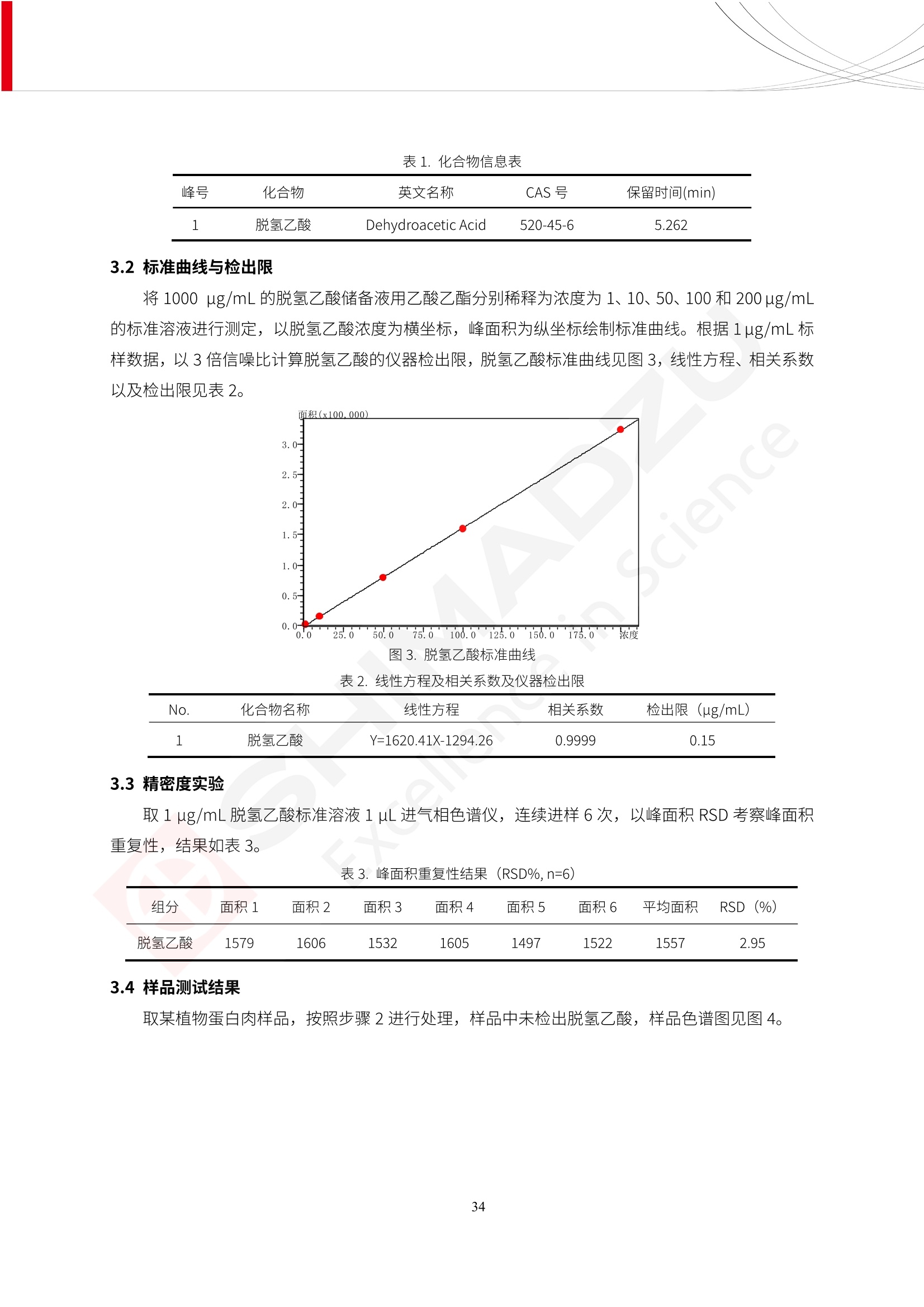
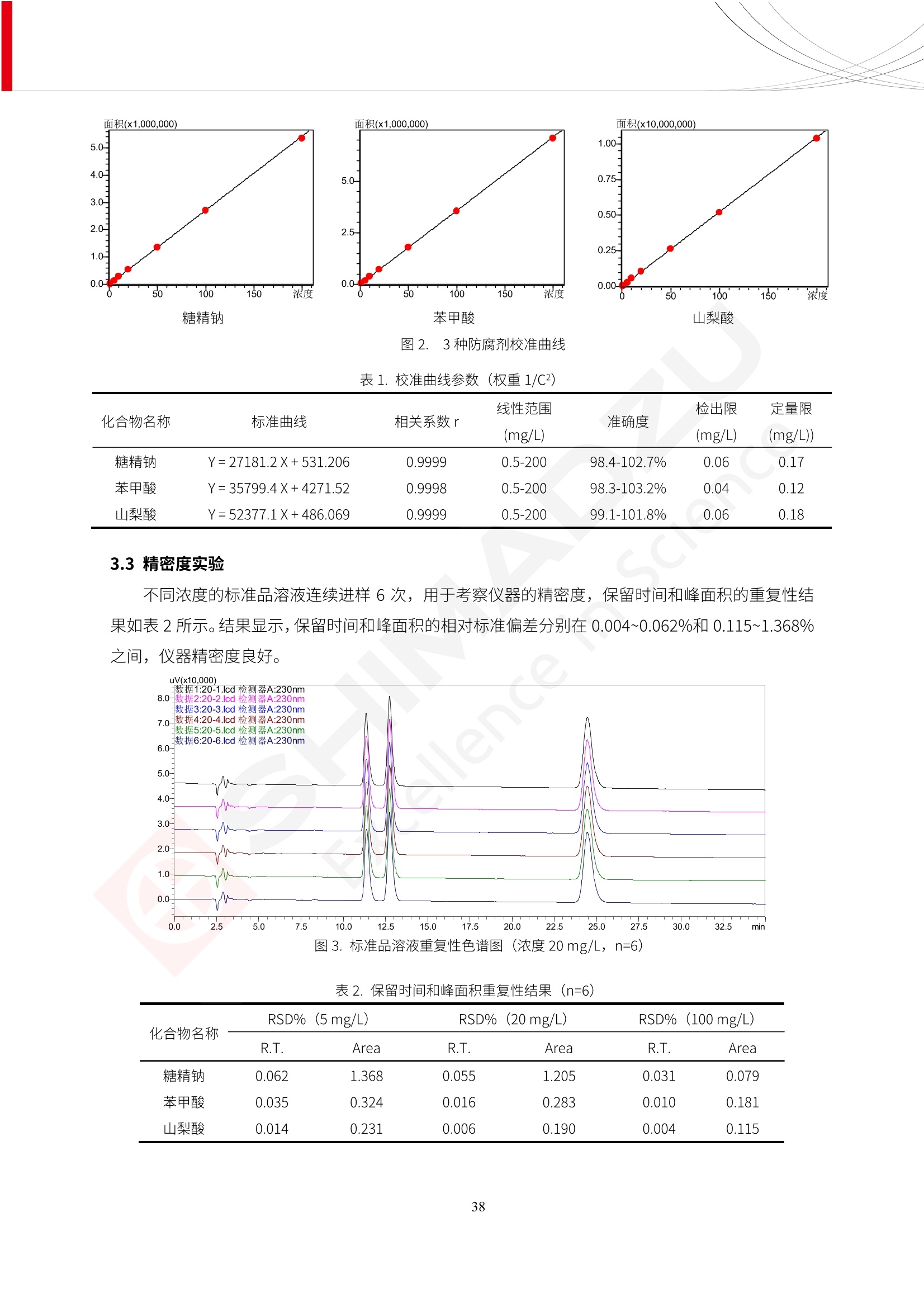
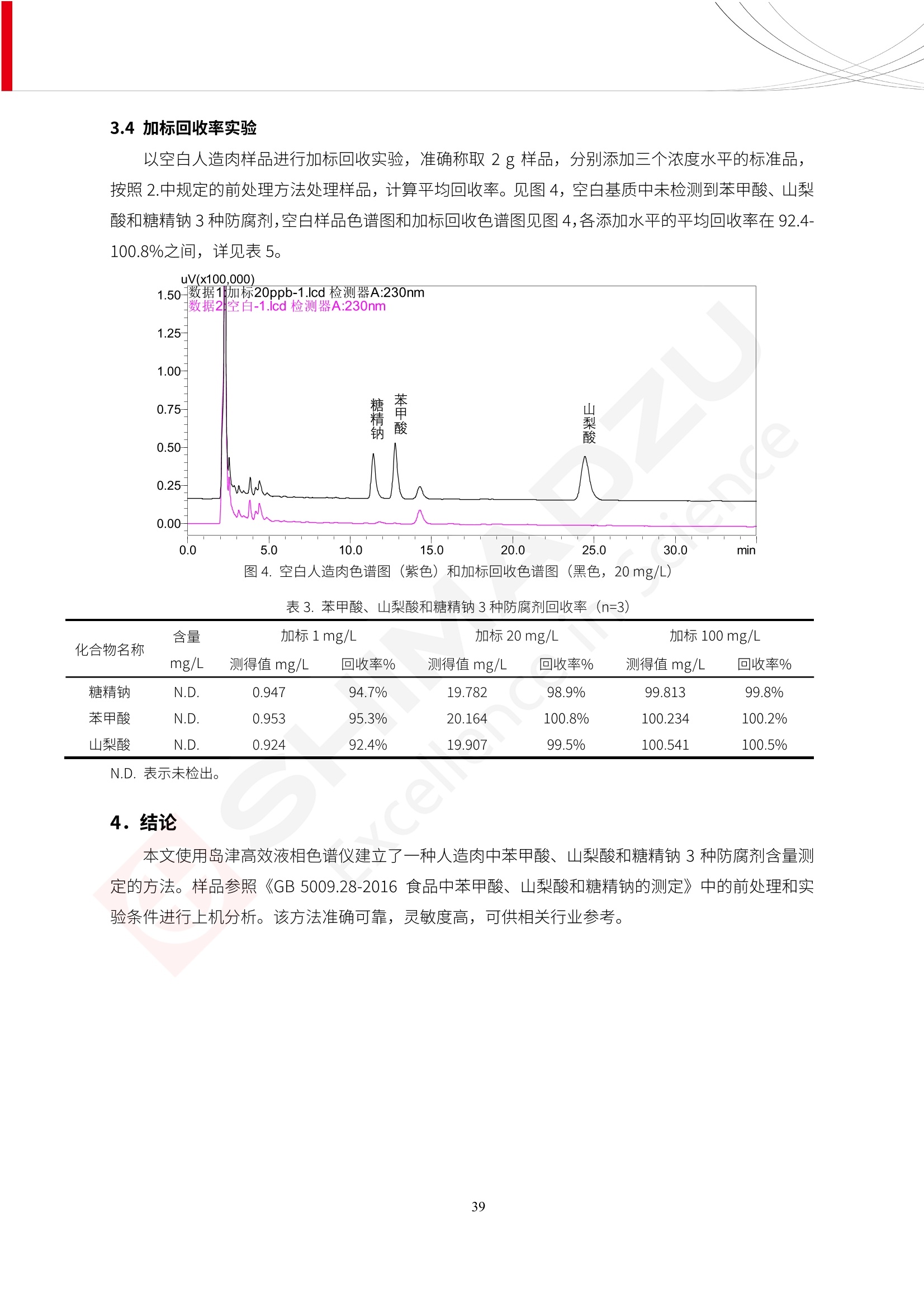
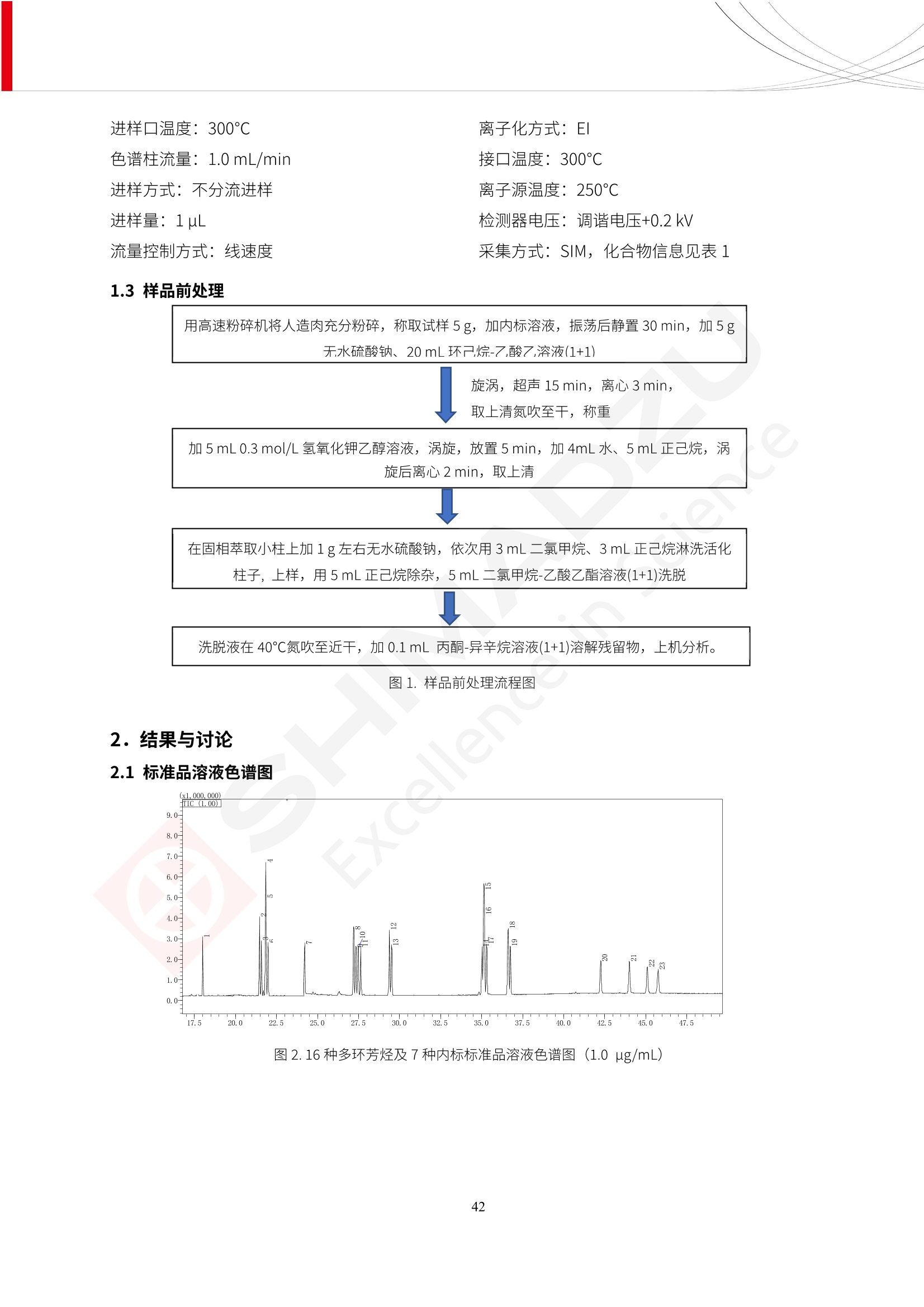
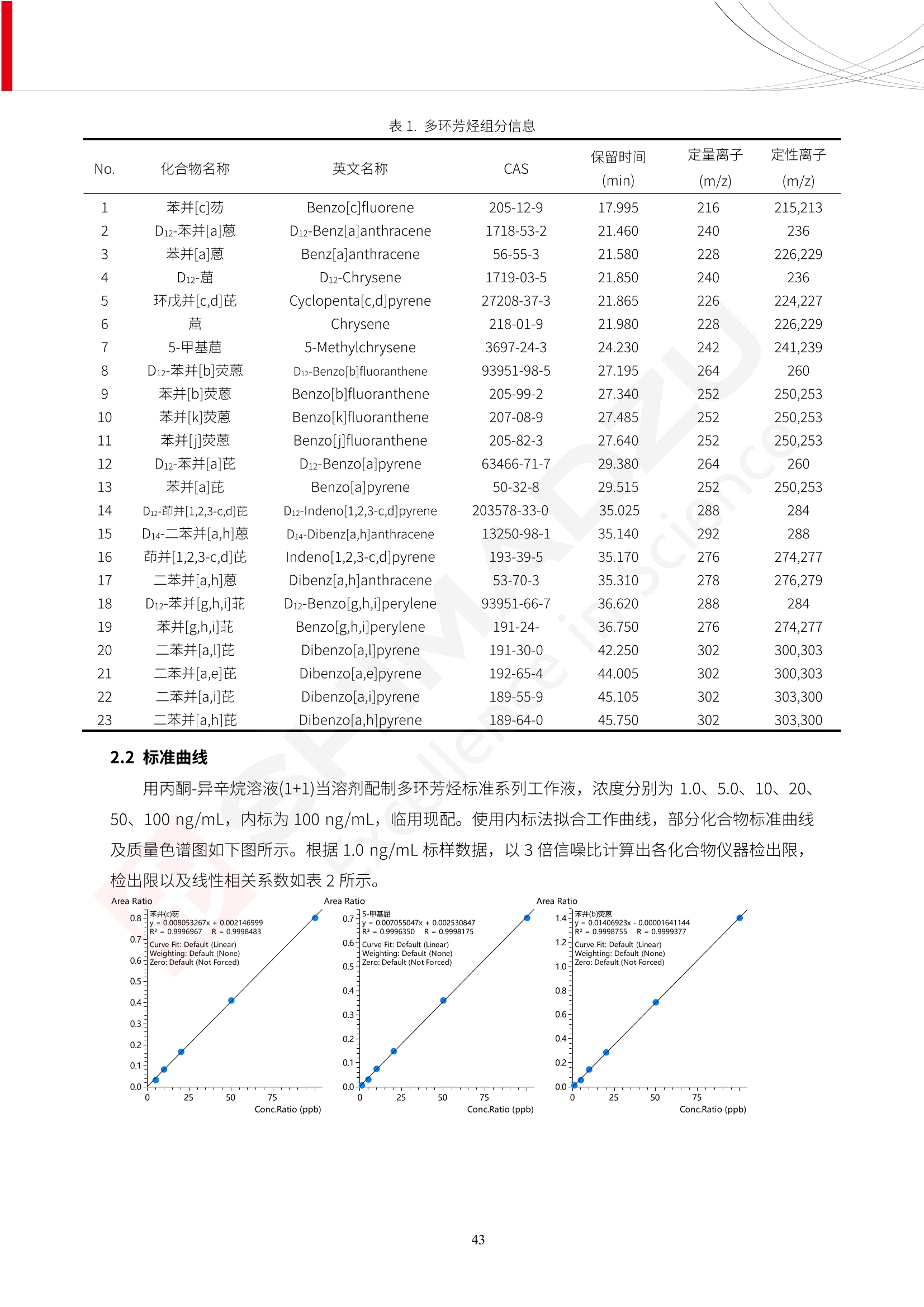
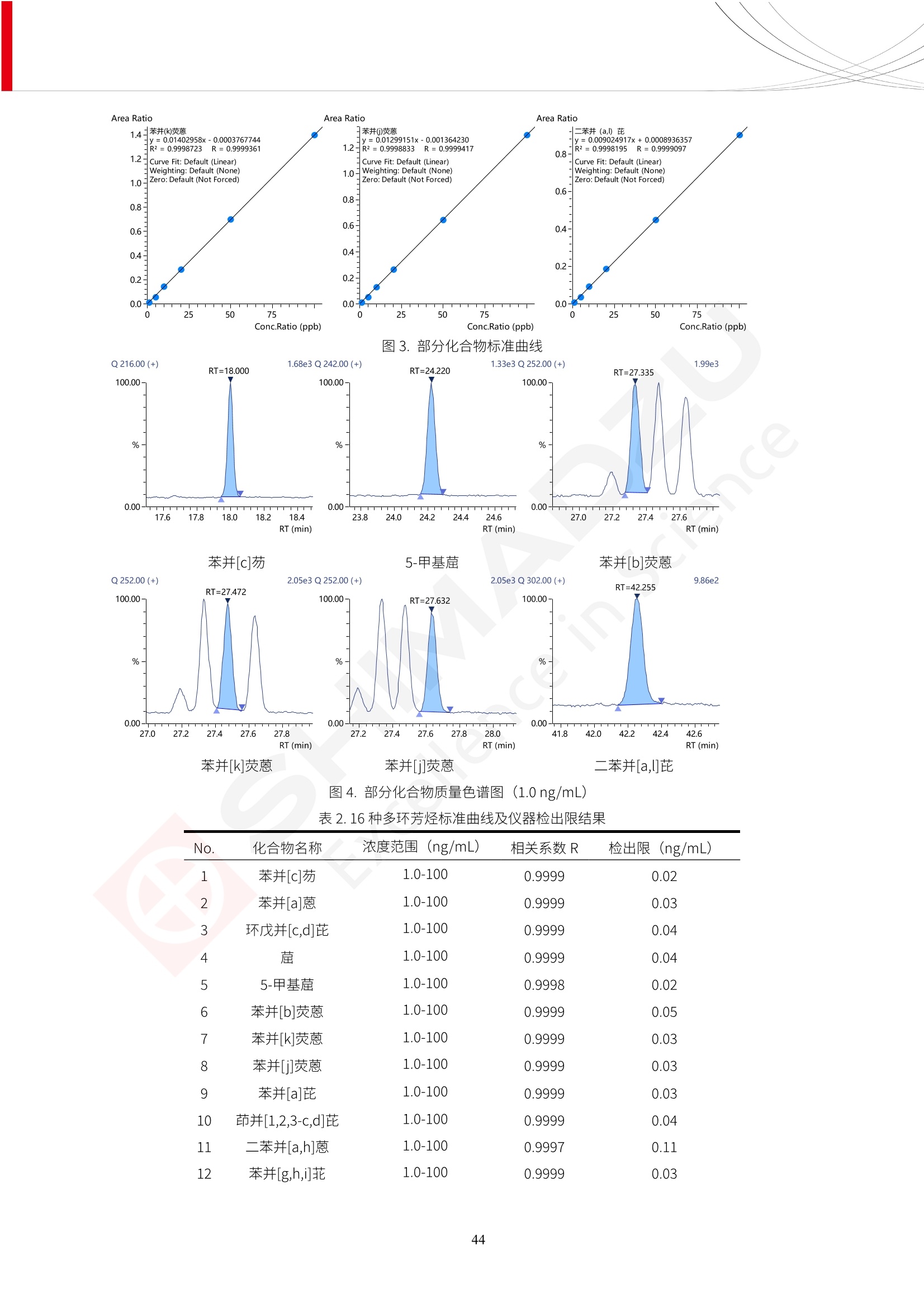
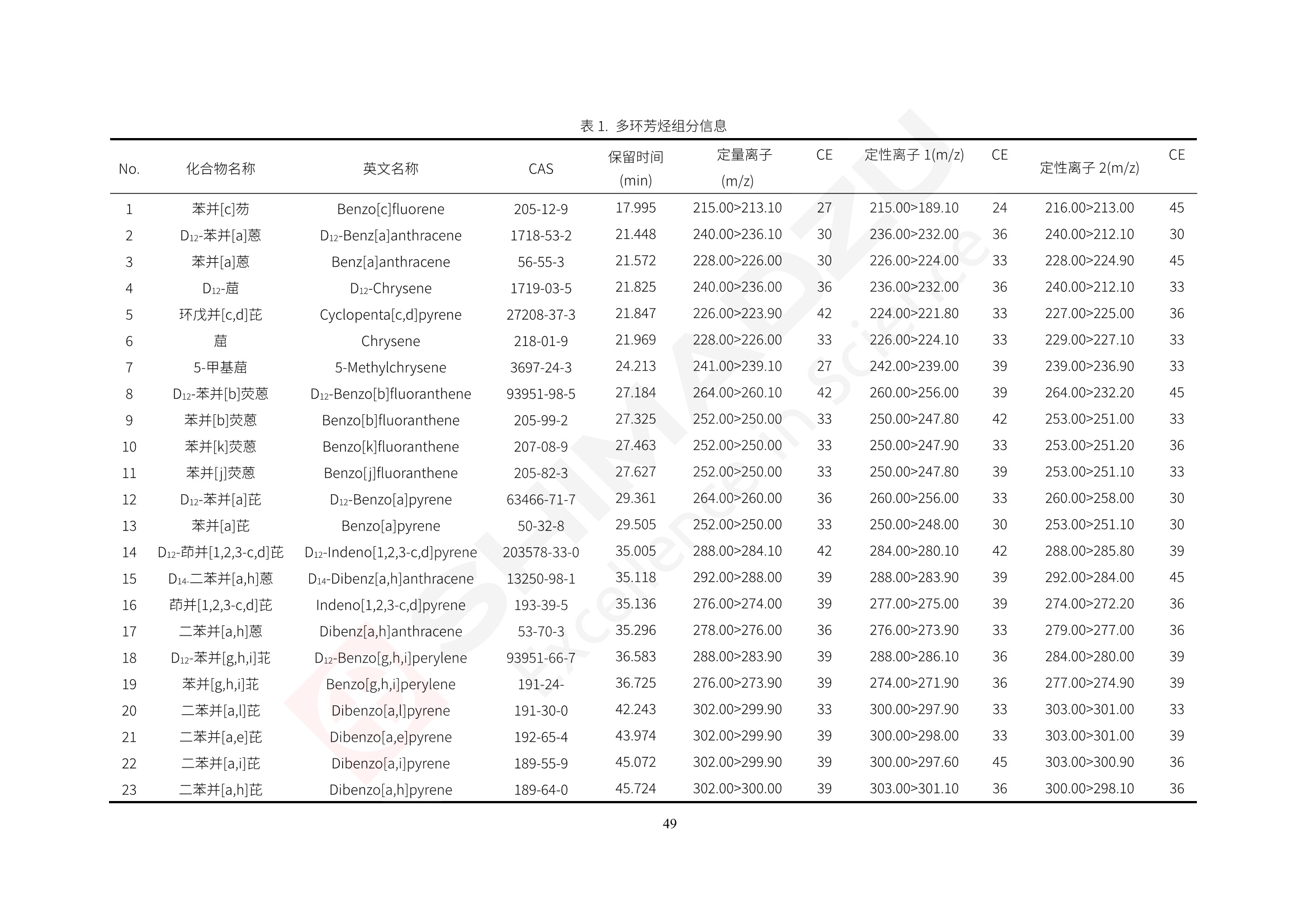
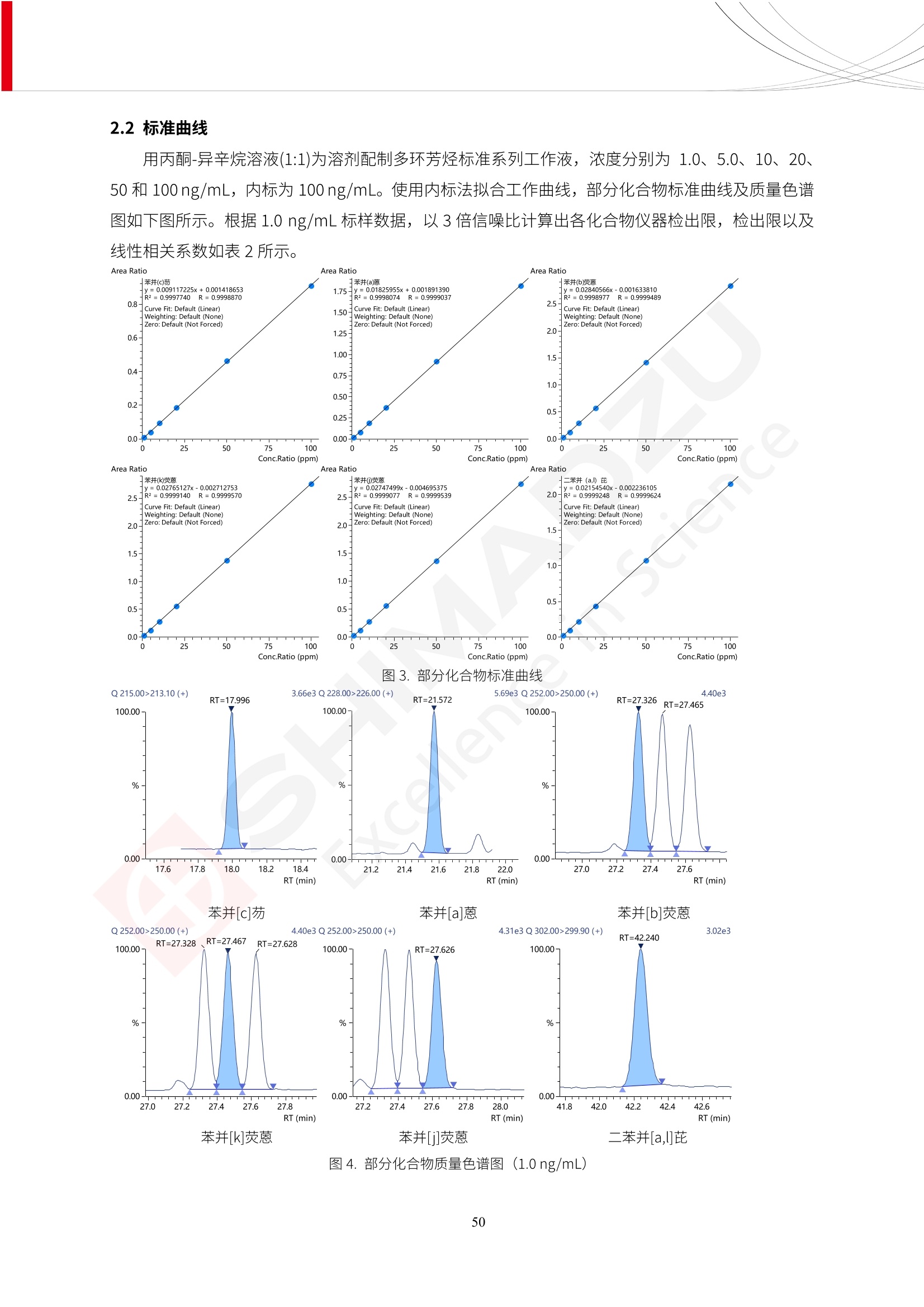
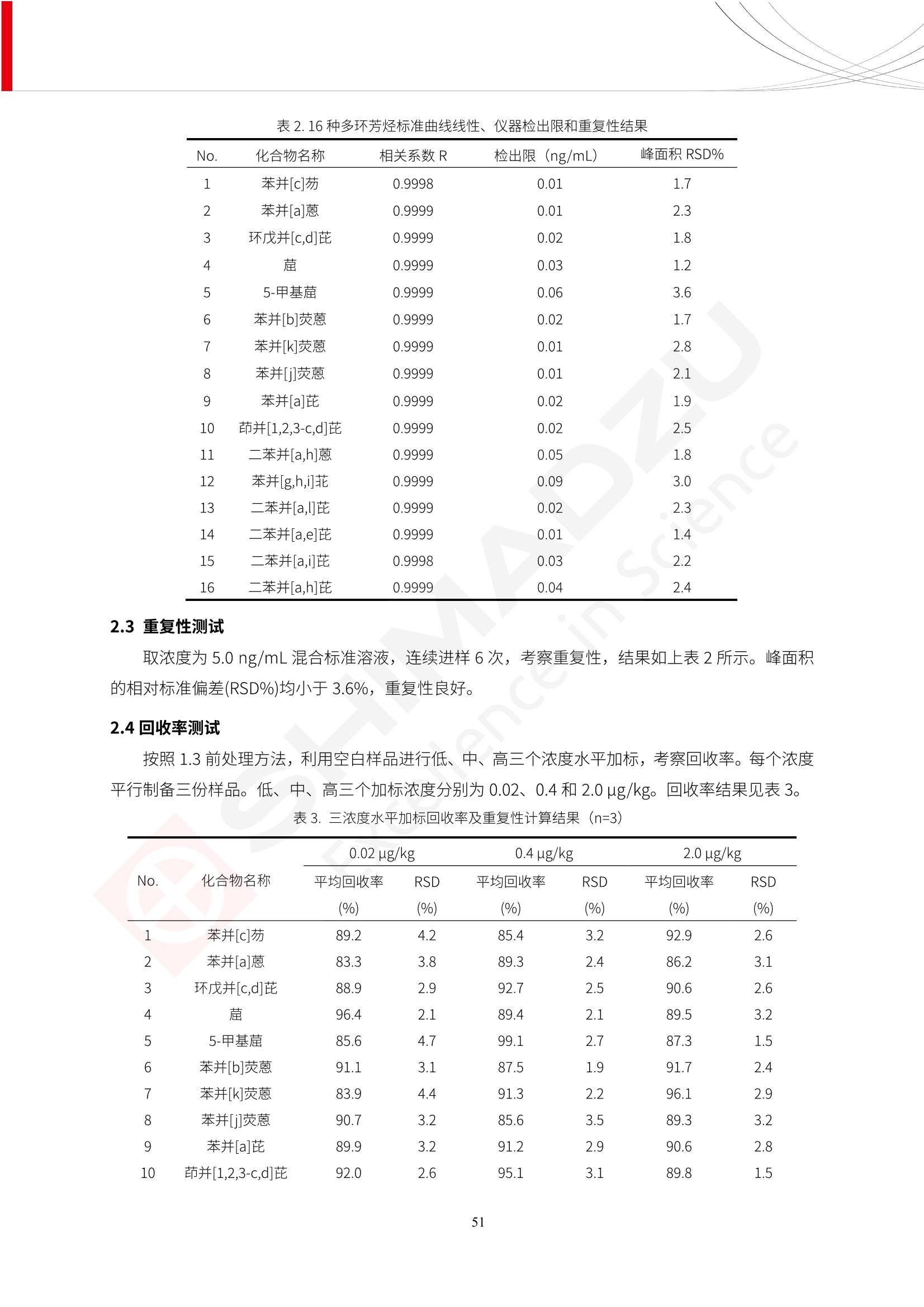
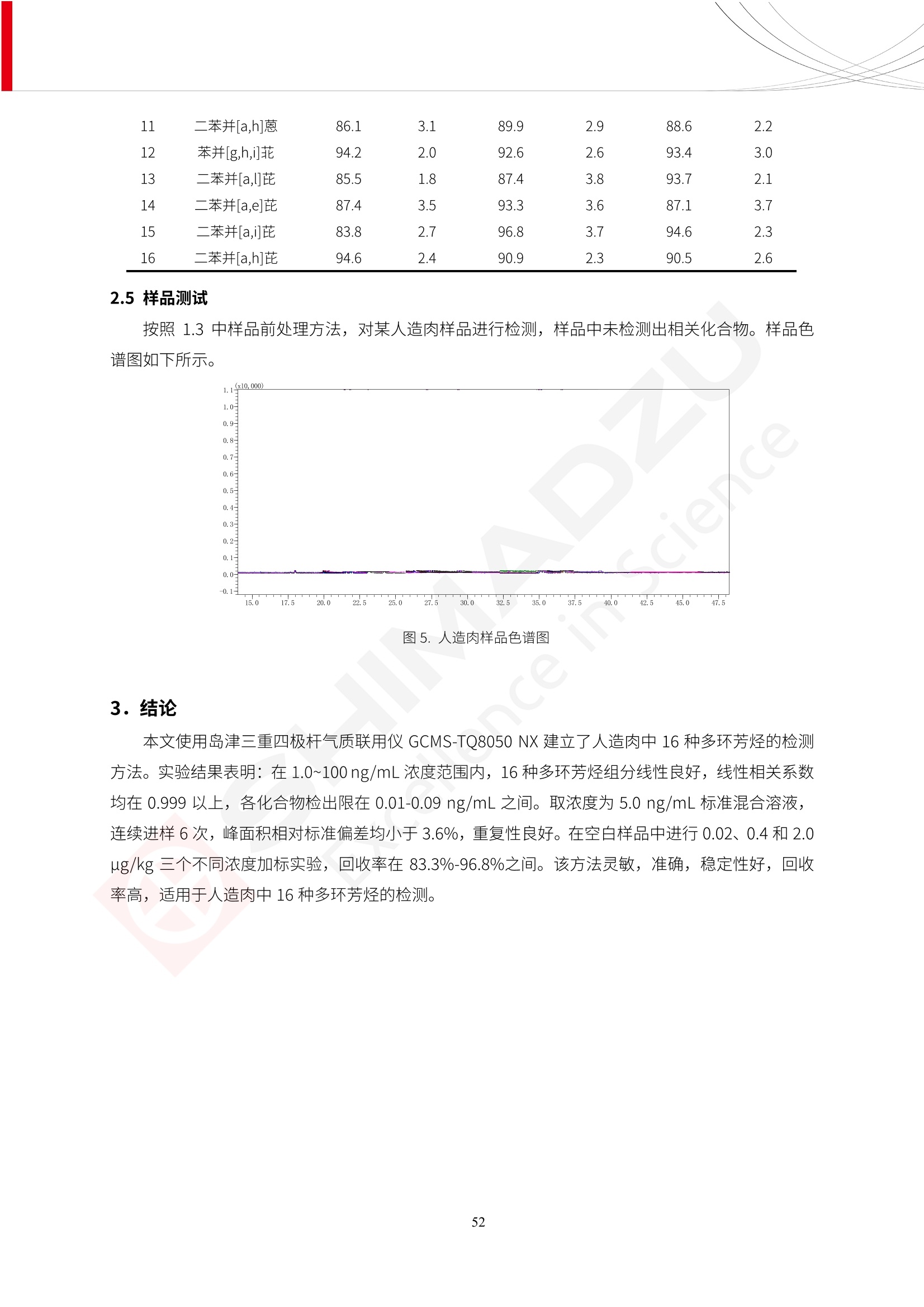
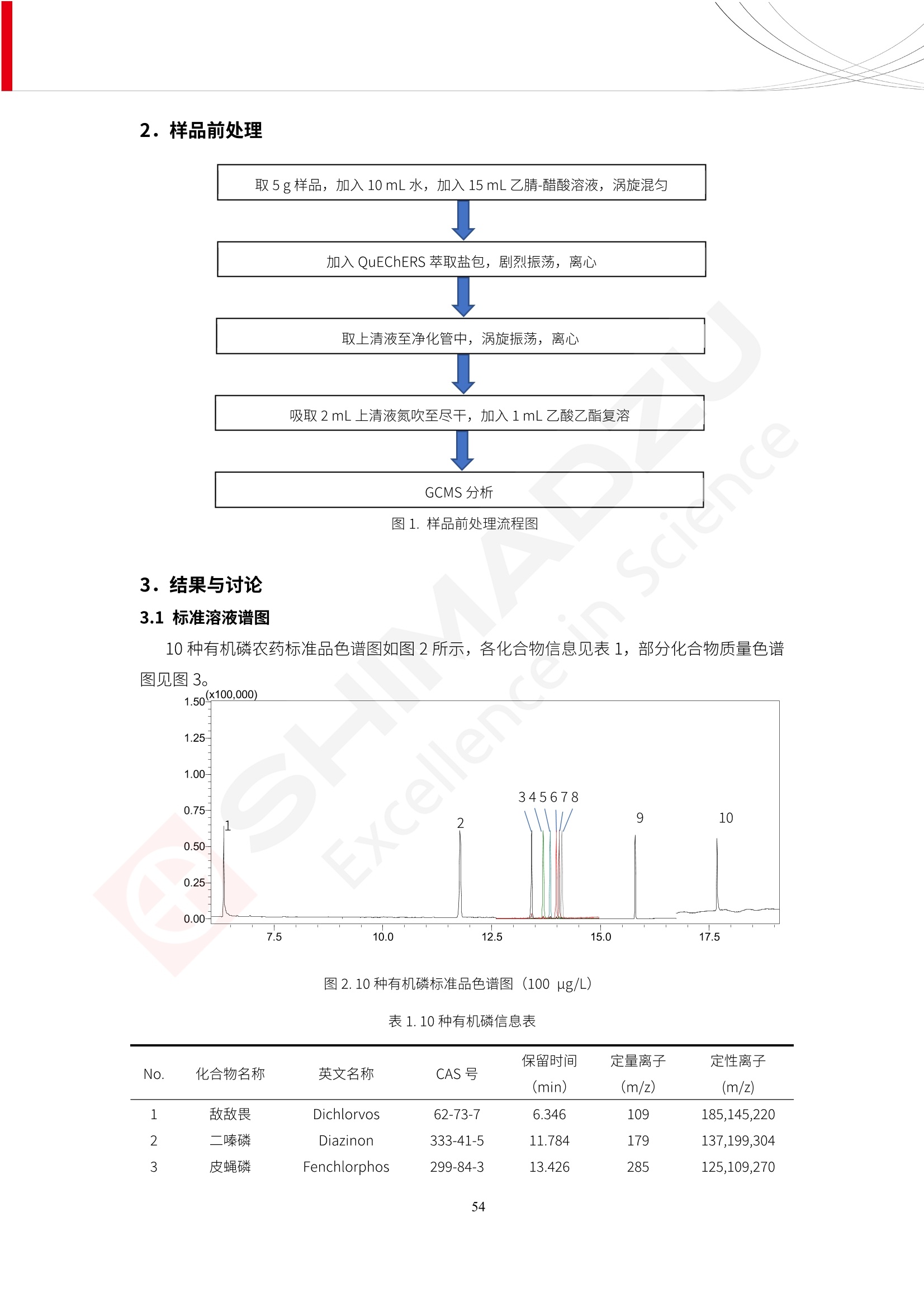
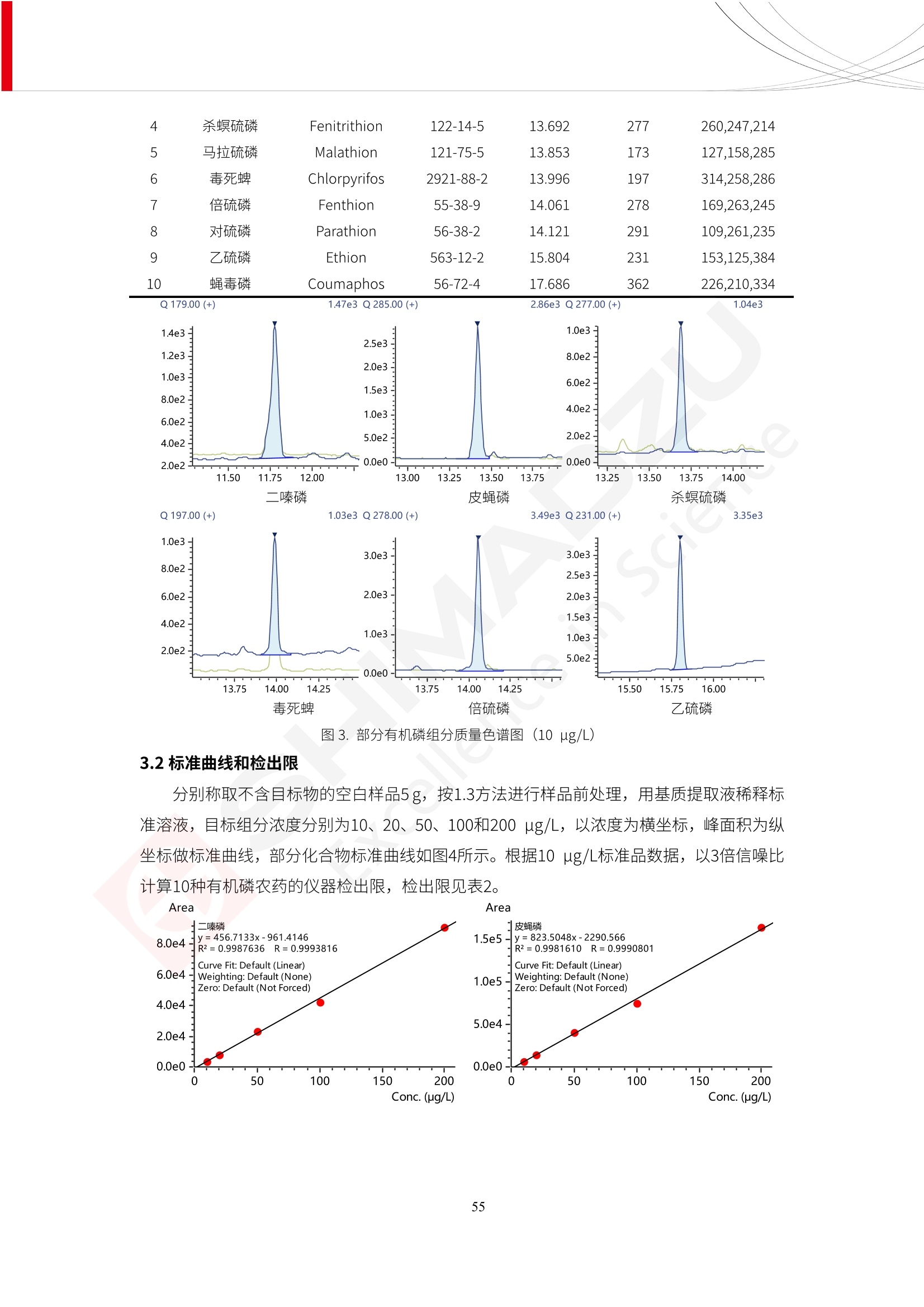
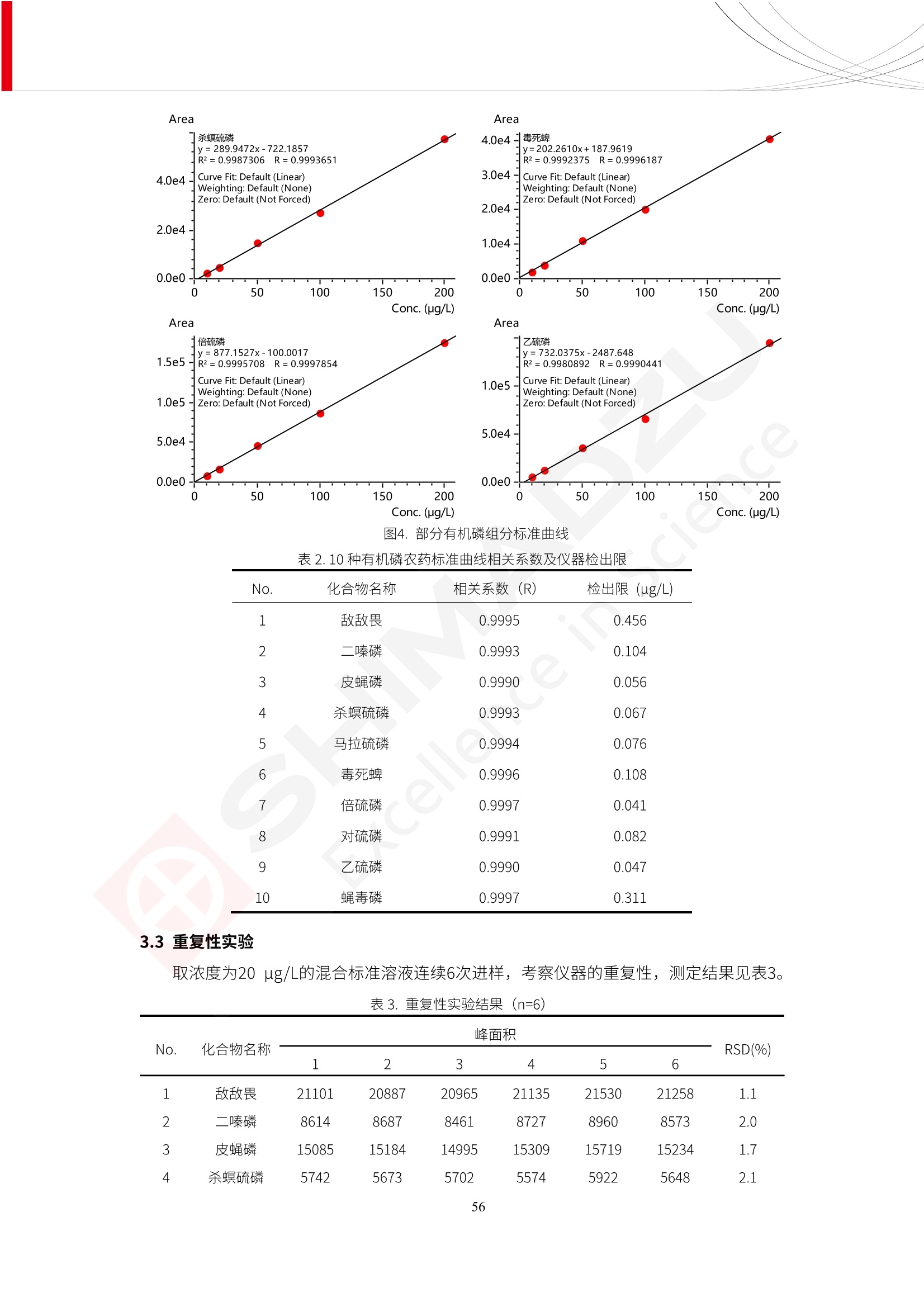
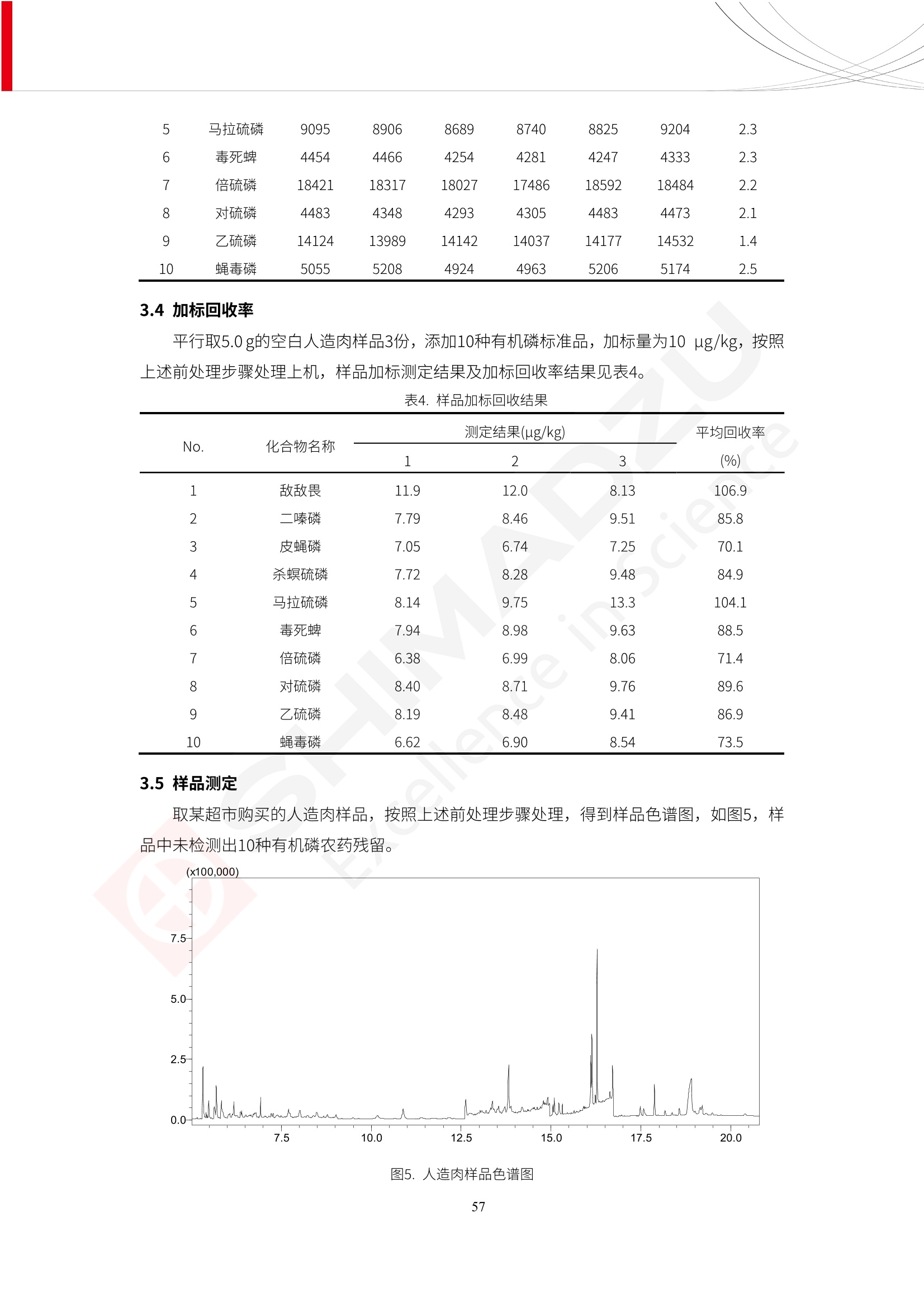
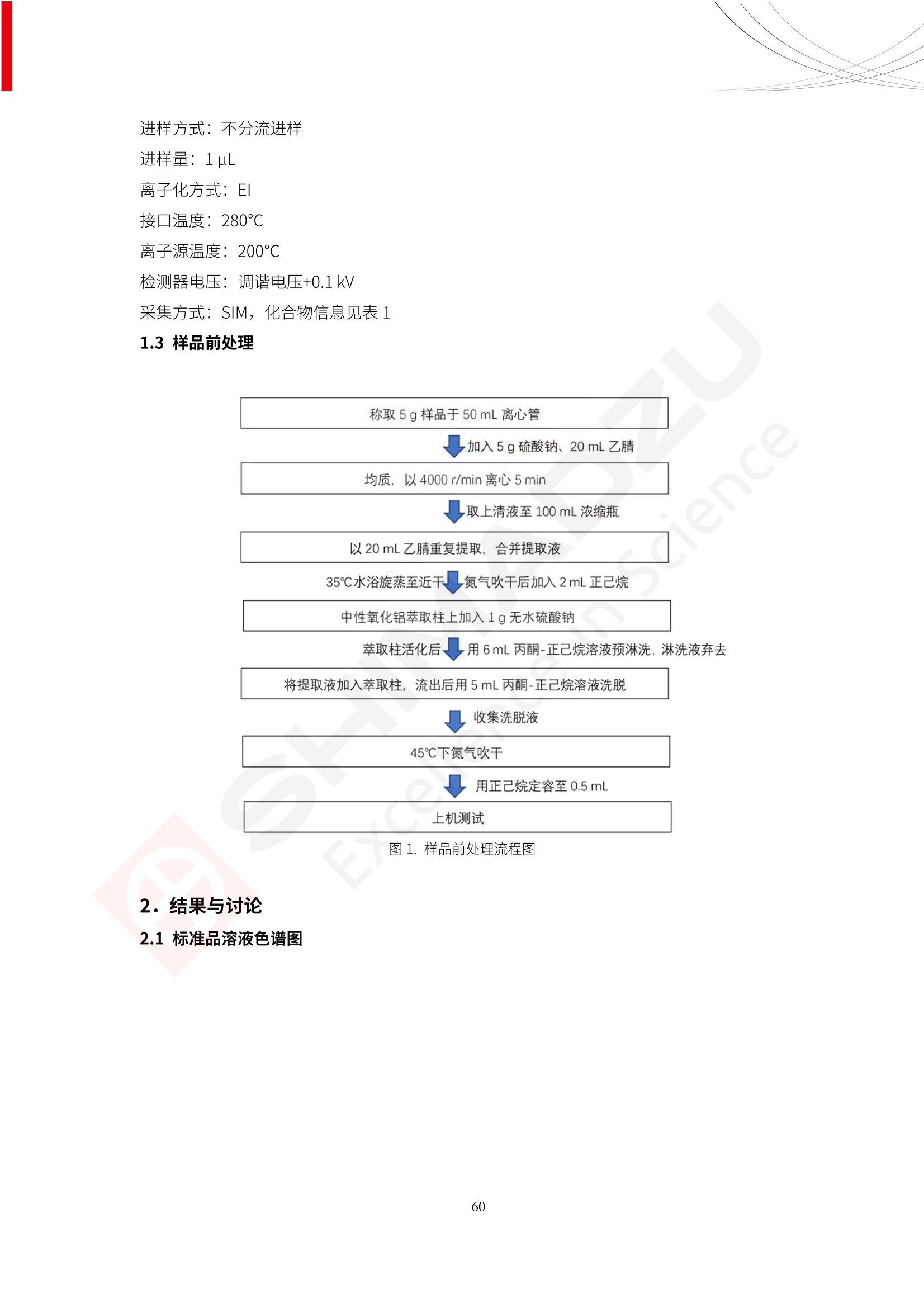
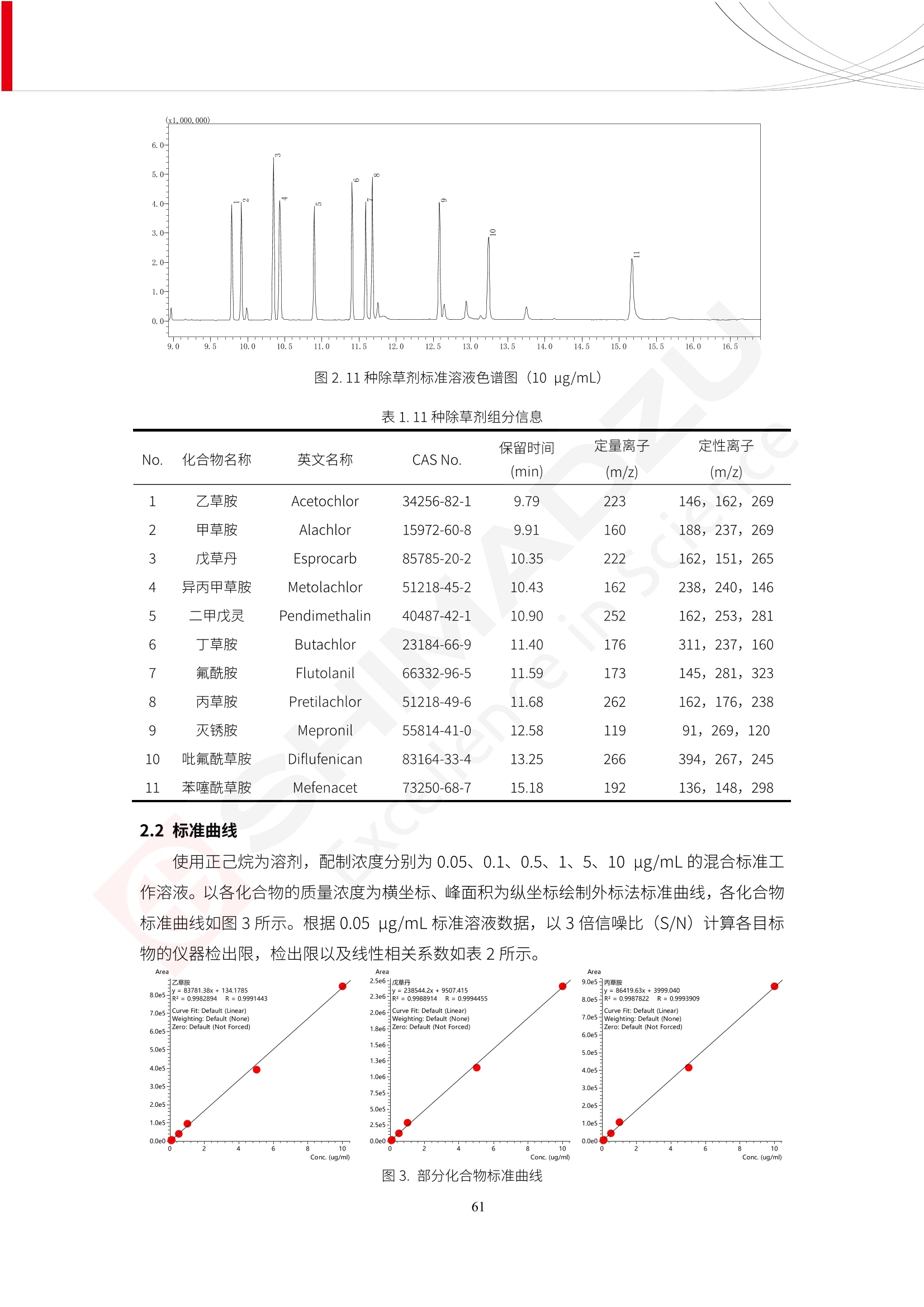
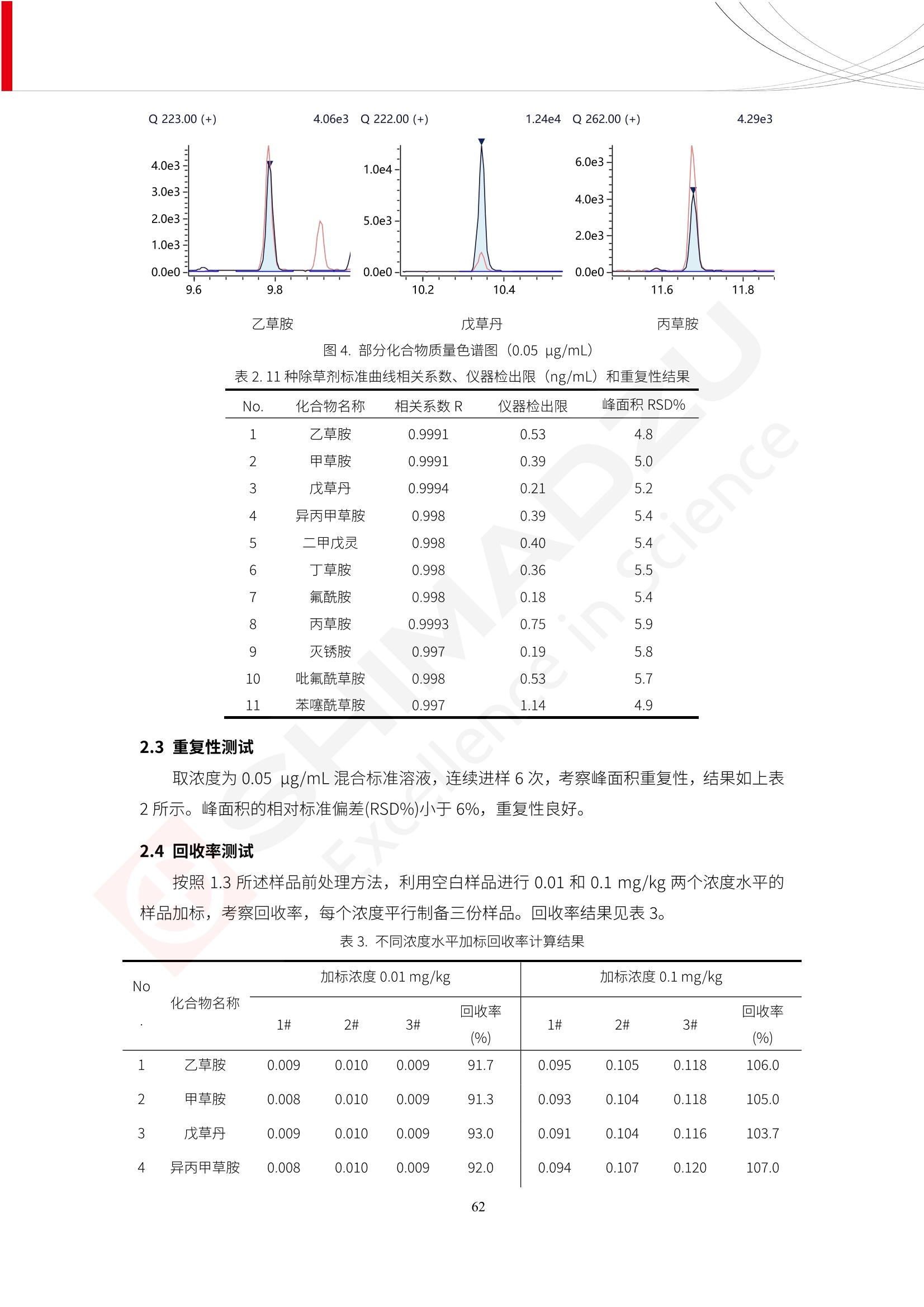
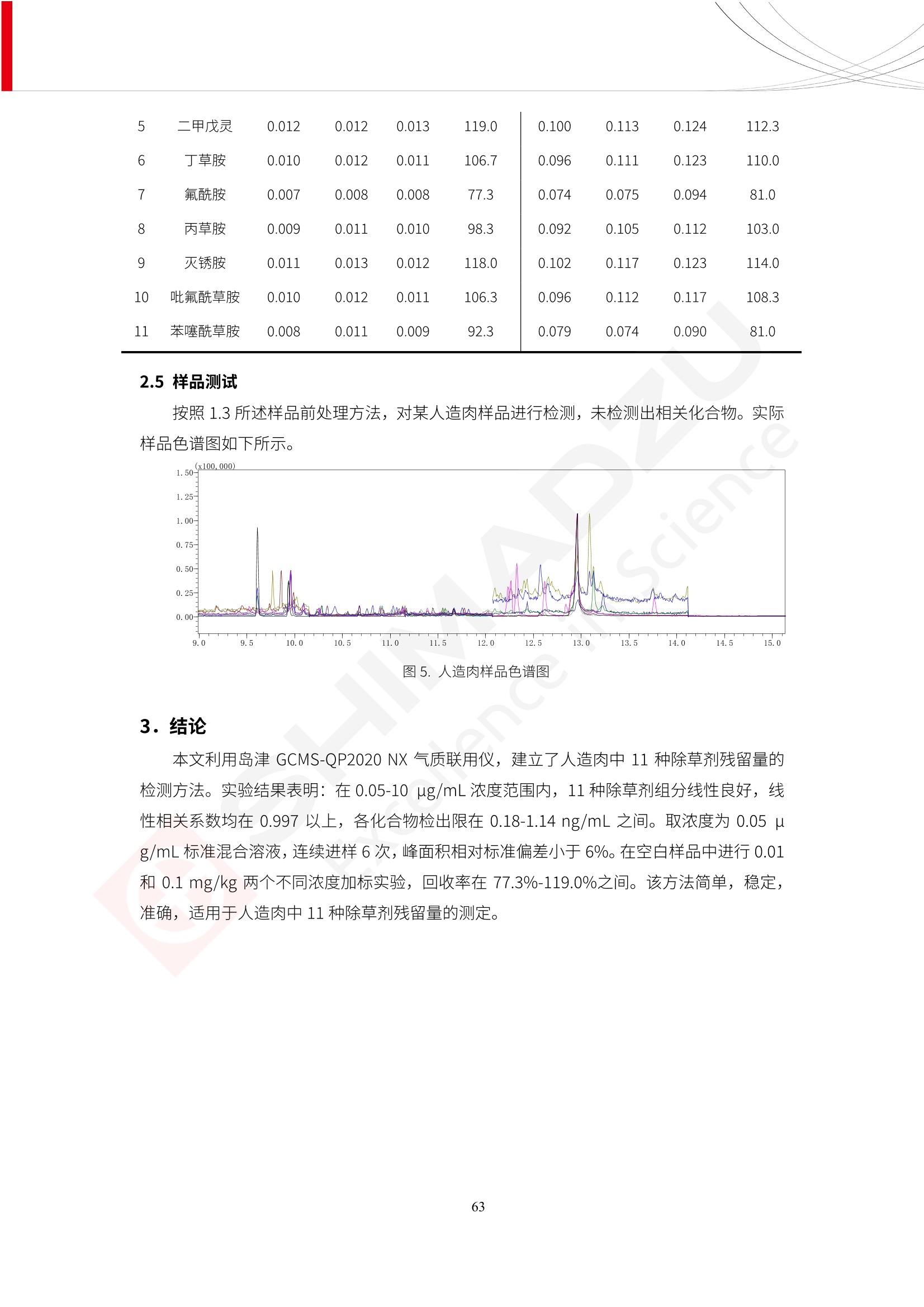
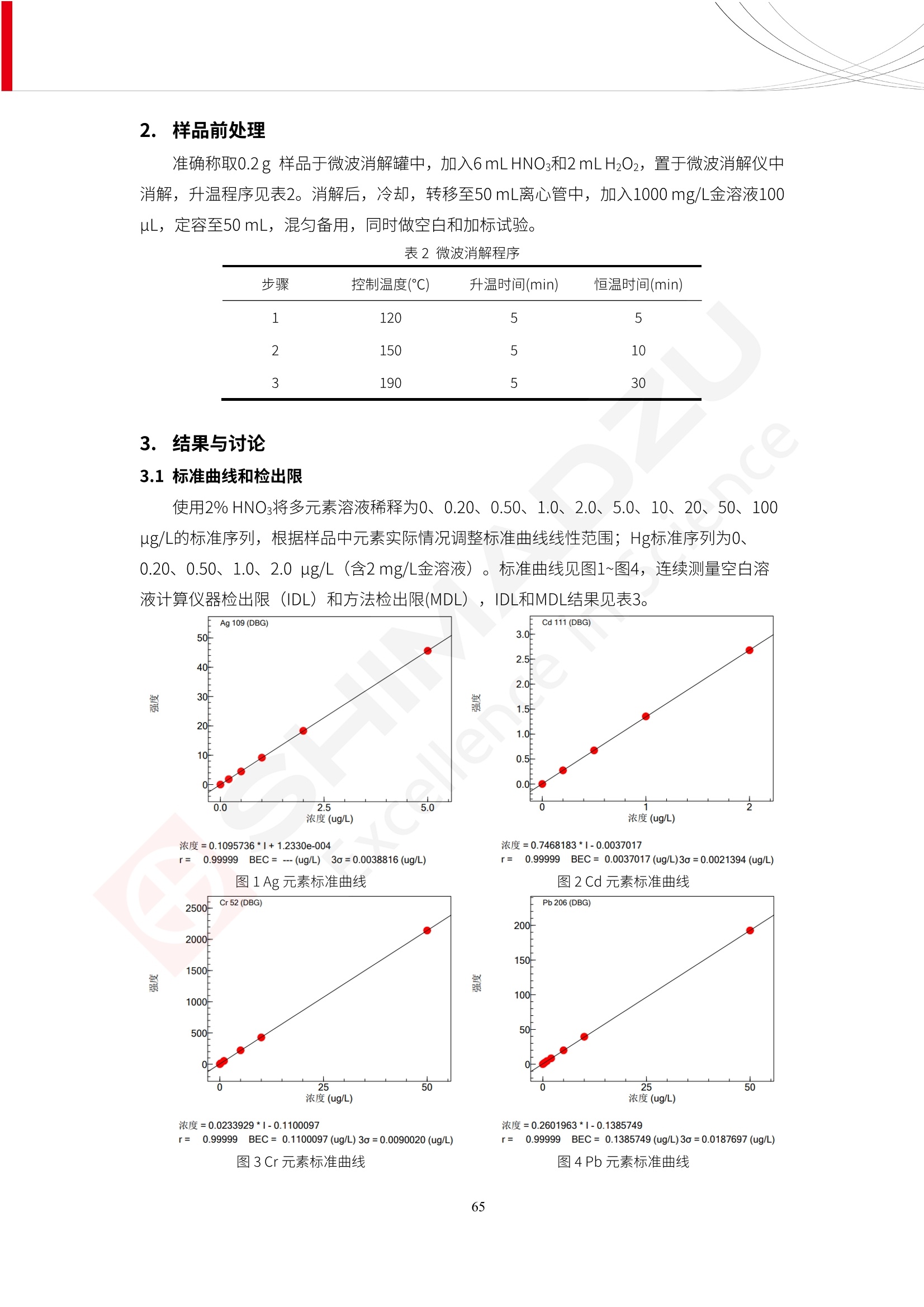
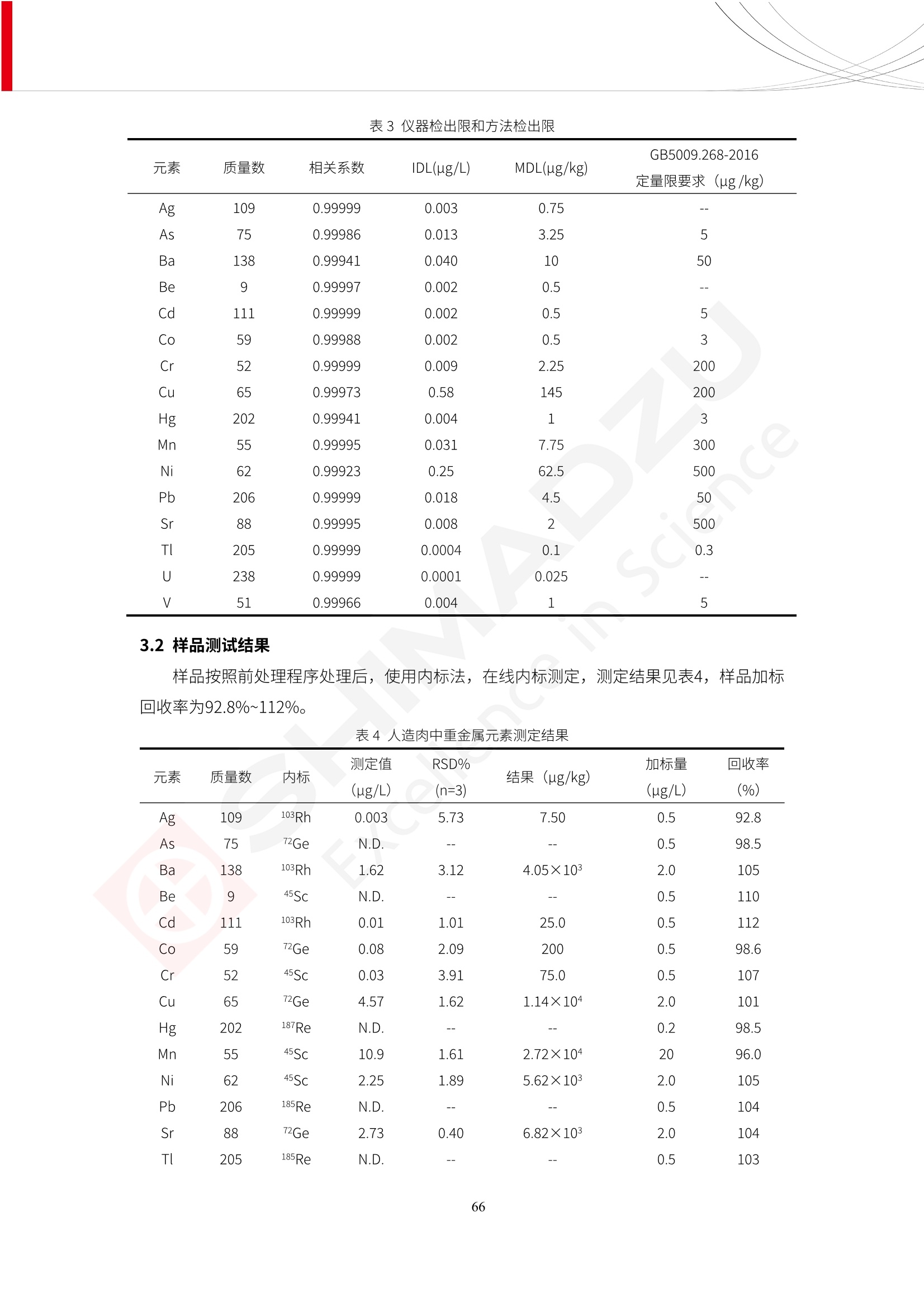
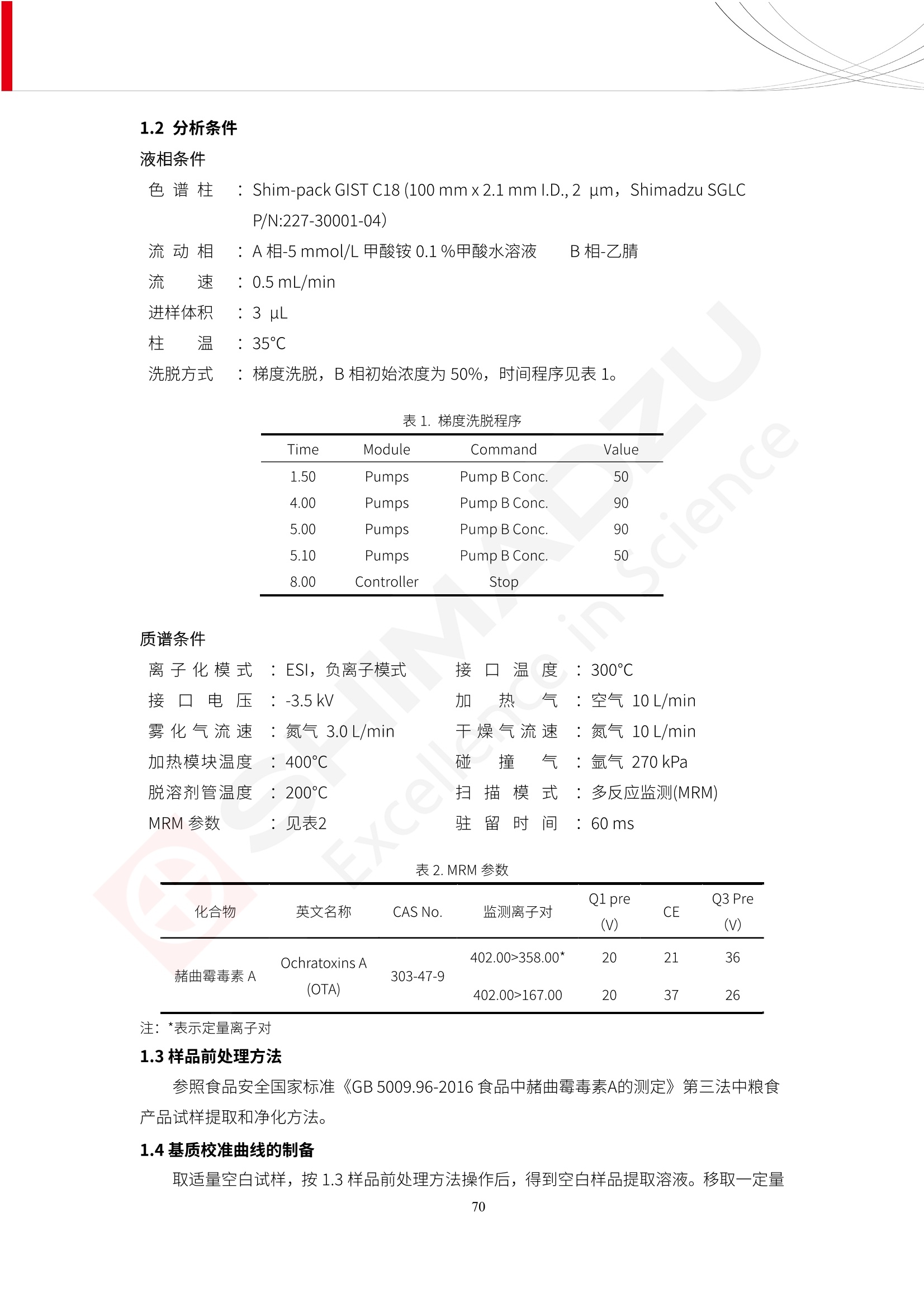
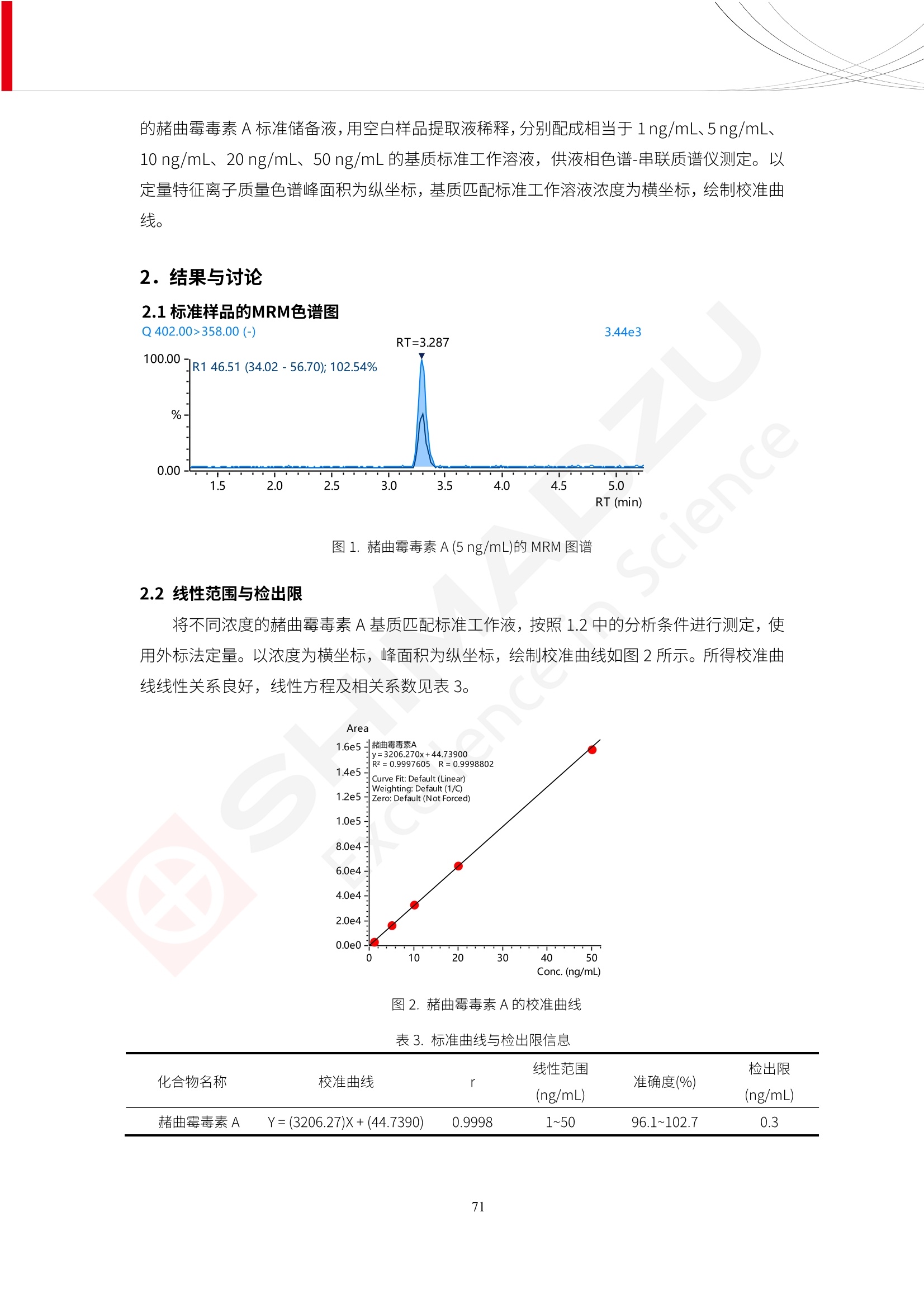
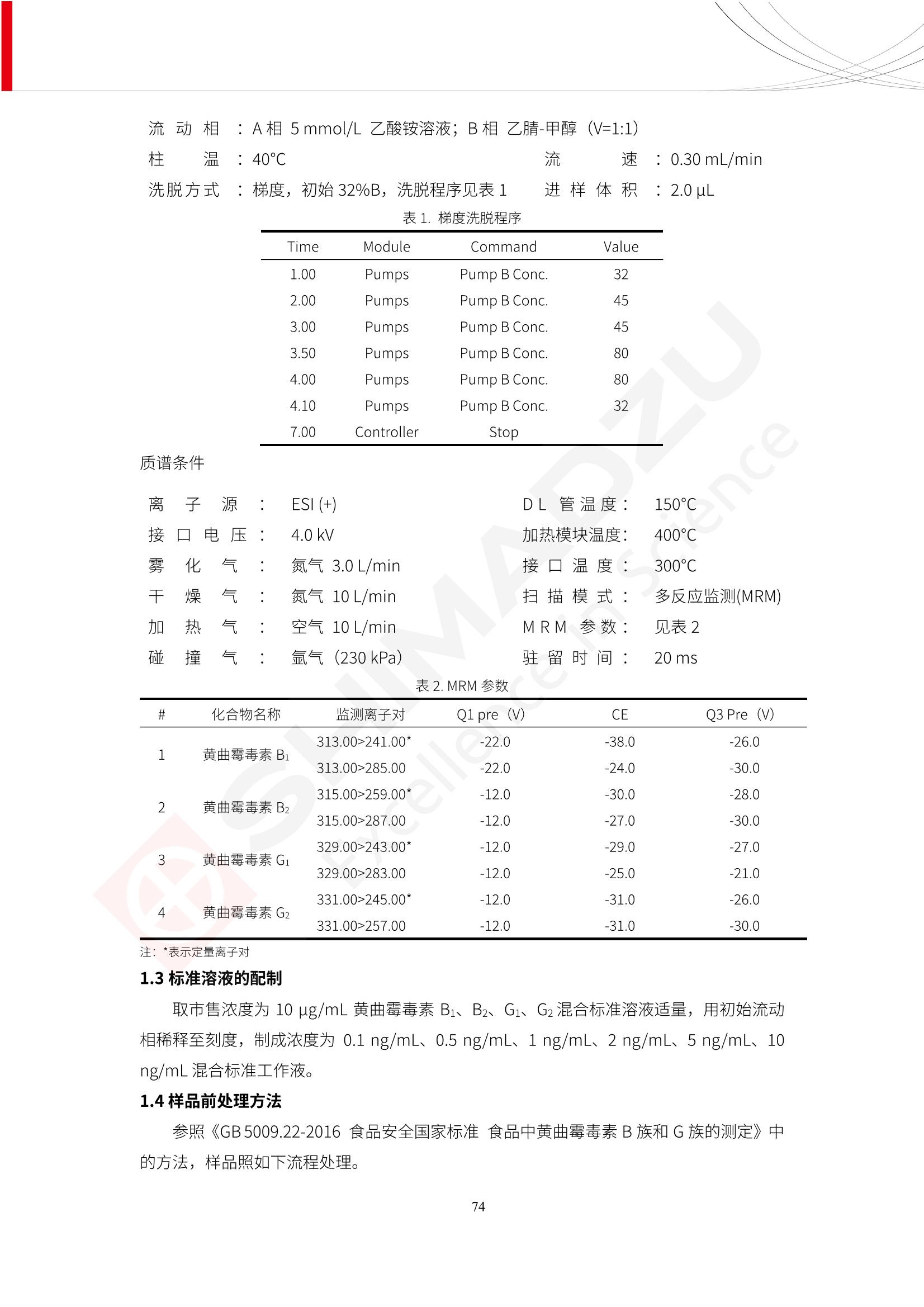
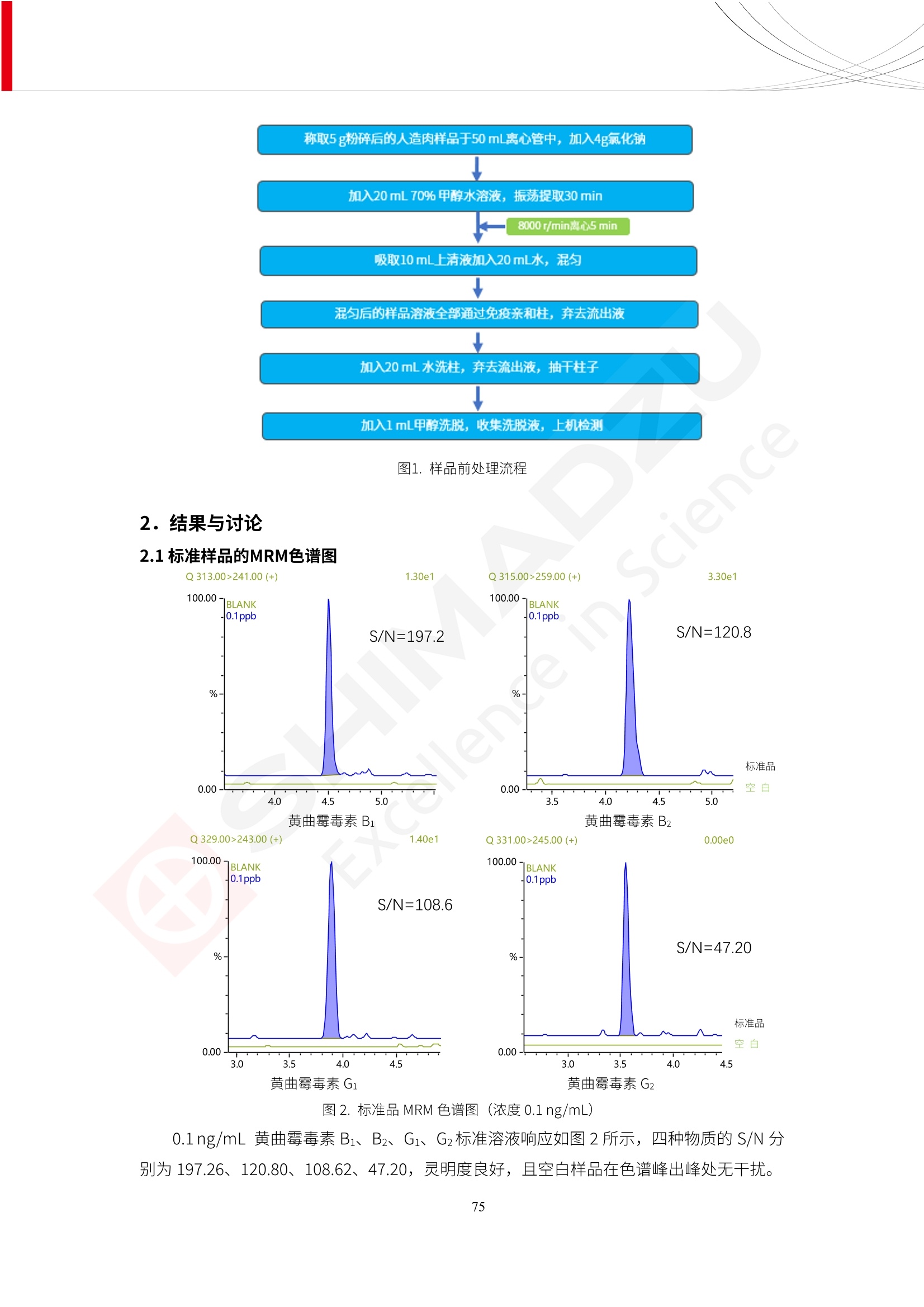
人造肉产品迅速兴起后,随之而来的食品口感、风味、营养、安全等问题也逐渐备受瞩目。为了应对人造肉企业相关用户的需求,岛津分析中心精心推出《人造肉检测整体解决方案》,汇编了口感检测、风味物质、营养成分、添加剂及有毒有害成分的检测报告,希望我们的工作能够对您有所帮助。SGCOE-22-27 Excellence in Science 人造肉检测整体解决方案 前言 随着现代社会的发展,肉制品作为摄入蛋白质的优良选择,其消耗量也是逐年增大。联合国粮农组织预计到2050年,人们对肉类制品需求量将会比现在的需求量增加70%。然而地球资源有限,如果仅依靠传统畜牧业供应,将无法满足市场需求。其次,畜牧业不可避免使用的抗生素、激素等兽药会导致在畜禽肉中的药物残留,引发食品安全事件。另一方面,禽畜肉中饱和脂肪酸含量较高,若长期大量食用容易引发“三高”。因此,人们将目光投向了非传统畜禽养殖业提供蛋白质来源的领域, “人造肉”应运而生。人造肉以其营养健康、节能减排、安全高效等优势受到广泛关注。 目前市场市的人造肉产品主要分为两种类型,第一种是以植物蛋白为原料制备的植物蛋白肉,此类产品可以最大限度地模拟真实肉品的外观和口感,在国内外文献中常见的名称有“植物素肉”、“plant-based meat”等,第二种是以动物细胞为原料,通过精准的细胞培养扩增制备得到细胞培养肉,此类产品可以绕开动物养殖过程而为人类提供真实的动物肌肉组织,主要的名称为“culturedmeat”。此外,还有除了前两种以外的肉类替代品,包括以真菌、乳蛋白、鸡蛋蛋白和其他新型蛋白质培养的人造肉产品。 人造肉产品迅速兴起后,随之而来的食品口感、风味、营养、安全等问题也逐渐备受瞩目。现在人造肉市场存在的问题集中在①口感、风味问题;②营养、安全性问题;③产品标准体系尚未建立。因此人造肉未来市场主要解决的问题,一是要出台并完善相关的食品安全法律法规,二是在产品组成成分上更接近真肉。 岛津公司作为全球著名的分析仪器厂商,自1985年创业以来,始终秉承创始人岛津源藏的创业宗旨“以科学技术向社会做贡献”和和“为了人类和地球的健康”理念,不断钻研领先时代、满足社会需求的科学技术。岛津中国一直关注国内食品安全以及新兴食品行业的发展以及相关标准法规的颁布与实施,积极应对并及时提供全国、快速有效的解决方案。为了应对人造肉企业相关用户的需求,岛津分析中心精心推出《人造肉检测整体解决方案》,汇编了口感检测、风味物质、营养成分、添加剂及有毒有害成分的检测报告,希望我们的工作能够对您有所帮助。 岛津企业管理(中国)有限公司分析中心 目5录 一、人造肉研究现状 1 二、人造肉检测应用方案...... 5 1、感官与营养.. .5 植物基人造肉质量评价. .6 传统 SPME 和 SPME Arrow 用于人造肉类香味定性分析的优化和评价 12 EZ-Test 人造肉质构评价..... 18 GCMS 结合岛津香味数据库分析人造肉中的气味成分.. 22 ICPMS-2030 系列测定人造肉中的营养元素. 28 2、食品添加剂...... 31 气相色谱法测定人造肉中脱氢乙酸的含量, .32 高效液相色谱法测定人造肉中苯甲酸、山梨酸和糖精钠的含量 36 3、污染物 40 GCMS 法测定人造肉中的多环芳烃含量 .41 GC-MS/MS 法测定人造肉中16种多环芳烃含量. .47 GCMS 法测定人造肉中10种有机磷农药残留量, 53 GCMS 法测定人造肉中11种除草剂残留量.. .59 ICPMS-2030 系列测定人造肉中的重金属元素, 64 4、真菌毒素 68 高效液相色谱-串联质谱法测定人造肉中赭曲霉毒素A...... 69 LC-MS/MS 测定人造肉中B族和G族黄曲霉毒素. .73 一、人造肉研究现状 长期以来,人们都认为肉类消费是健康饮食的重要组成部分,这也是社会发展的指标之一。随着人类经济社会快速发展,全球总人口估计在2050年将达到98亿,联合国粮农组织预计,那时人们对肉类制品需求量将会比现在的需求量增加70%。然而地球资源有限,如果仅依靠传统畜牧业供应全球庞大的市场,将无法满足需求,同时产生的环境问题和社会问题不容忽视。 传统畜牧业在养殖过程中需要占用大量的土地资源和水资源,禽畜产生的粪便会污染养殖场水土,且畜牧养殖业会排放出大量的温室气体,其排放量占到全球排放量的12%;在畜牧业中无法避免兽药和抗生素的使用,因此动物源性食品中残留的抗生素、激素和兽药会导致许多食品安全问题;禽畜肉中饱和脂肪酸含量较高,长期大量食用会引发高血糖、高血脂和高血压等病症。因此,人类将目光投向了非传统畜禽养殖业提供蛋白质来源的领域,“人造肉”应运而生。有统计显示植物基肉等人造肉替代食品的规模已超过50亿美元,未来市场潜力巨大。 1.人造肉种类与命名 目前市场上的人造肉产品主要分为两种类型,第一种是植物蛋白肉,是指利用植物性蛋白与其它植物性成分,通过合成、加工而形成的具有肉类营养价值、口感、风味的食品,也称为植物性肉。植此类产品可以最大限度地模拟真实肉品的外观和口感,在国内外文献中常见的名称有“植物素肉”、6“6.植物蛋白基肉制品”、 “plant-based meat”、“soy-based meat”等,在2020年中国公布的《植物基肉制品》团体标准中规定植物蛋白肉的命名应该采用“植物XX”、 “植物基XX”、“植物源XX”、 “植物蛋白XX”、 “植物制成的XX”等方式对植物蛋白肉产品进行命名。 第二种是以动物细胞为原料,通过精准的细胞培养扩增制备得到细胞培养肉,此类产品可以绕开动物养殖过程而为人类提供真实的动物肌肉组织,主要的名称为“cultured meat”、、“animalfree meat”、 “cultivated meat”、6“6in vitro meat”、“培育肉”、“试管肉”等。 2.人造肉的组成 2.1植物蛋白肉的组成 蛋白质是植物蛋白肉中最主要的成分,是构成植物蛋白肉的基础,它所具有的水化性、乳化性、凝胶性和质构性能等都对产品有着重要的影响。在植物蛋白肉产品中使用的植物性蛋白质主要为大豆蛋白、豌豆蛋白、小麦蛋白和花生蛋白等。为使各组分形成致密的稳定结构,并能赋予产品一定的黏弹性,需要添加黏合剂,常使用的黏合剂有海藻酸钠、卡拉胶、谷氨酰胺转氨酶、甲基纤维素和黄原胶等。植物蛋白肉不具有动物肉的风味,需要添加肉类风味物质模拟动物肉的风味,如乙基 麦芽酚,鸡肉香精、猪肉香精、呈味氨基酸等物质。除此之外,还可添加辣椒、大蒜等香辛料进行调味。油脂是植物蛋白肉中必不可少的组分,其使用量会影响产品的柔嫩度、风味和产品的加工性能,油脂的添加量应控制在 2%~5%。在设计人造肉产品颜色时应尽可能模拟动物肉的颜色及变化。目前立用于植物蛋白肉的着色剂有甜菜红、胡萝卜素、番茄红、焦糖色素和血红蛋白等,,它们能使植物蛋白肉呈现良好的“肉色”。 2.2细胞培养肉的组成 细胞培养肉的生产加工过程需要使用的材料主要有动物肌肉干细胞、细胞培养基、动物血清、分化诱导因子和抗生素等,使用的主要技术为无菌操作技术和细胞培养技术。 2.3菌体蛋白肉的组成 菌类蛋白是一种采用连续流动发酵技术发酵链孢霉菌 (Fusarium venenatum A3/5) 得到的菌丝体发酵产物,然后与鸡蛋清、风味剂、食用色素等辅料进行混合,再进行成型、加热、冷冻、质构化和包装等一系列加工步骤,最终就得到菌类蛋白肉的相关产品。其中鸡蛋清是作为黏合剂存在的,辅助产品形成类似肉的质构。并且在生产过程将蒸汽加热后的菌类蛋白肉迅速冷却到-18℃,便可通过控制冰晶的大小和生长速率使菌类蛋白肉内部形成类似肉的质构。 3.人造肉优势和劣势 与真肉相比,人造肉在营养价值、食品安全、能源消耗和环境友好方面都具有潜在的优势;人造肉的劣势则包括成本较高、质感欠佳(植物肉)、技术存在瓶颈(培养肉)等。 表1.人造肉的优势和劣势 优势 劣势 营养价值 同样可以提供养殖肉中所富含的蛋白质、 脂肪等营养元素,也可以控制其营养成 分,满足不同群体的需求。 植物性肉的成本比真肉稍高; 成本较高 培养肉目前无法量产,制作工艺尚为复 杂,未攻克难题还较多,因而成本较高。 食品安全 人造肉不具备养殖肉中出现的细菌、病 毒、抗生素、激素等问题。 质感次佳 植物肉在口感、色泽、风味上与传统肉类 出现较大的差异。 能源消耗 可以减少养殖畜牧业带来的原料、土地等 能源消耗。 技术瓶颈 培养肉量产上仍存在技术难题,规模生 产工艺、反应器都还不完善。 环境友好 可以缓解畜牧业生产过程中所产生的废 弃物和垃圾,减少温室气体的排放。 4.人造肉加工关键技术 人造肉在风味、口感和色泽等方面与传统肉类存在较大差异。 4.1风味 植物基肉的主要原料大豆蛋白具有浓烈的豆腥味,想要去除这种味道难度很大。有研究表明,将温度提高至80℃以上可以显著降低豆腥味,但非豆腥味成分损失较多;乳酸菌能够代谢已醛,却附带不良的酸腐味;添加缬氨酸和异亮氨酸能够降低不良酸腐味,但也会导致体系碱味增加、pH值 升高和严重褐变;添加肉味香精,可能附带一定安全隐患,而且包埋效果差、香气弱,粉味重,储存及加工过程难以保留其“肉味” 4.2口感 肉类的口感通常由其蛋白结构决定。植物肉需要使用挤压和纺丝等技术才能模仿动物蛋白的质地和口感,但受植物蛋白分子结构的限制,仍与传统肉有差距。培养肉主要特点是结构松散,无法产生肉的咀嚼感。近年来,可降解支架、食品3D 打印等技术的发展为培养肉模拟传统肉类结构提供了新的解决方案。培养肉可以选最优质的品种和最好的部位进行培养,并通过 3D 打印等技术将不同类型成分进行更好的配比,使其口感更好。 4.3色泽 色泽作为人造肉的重要感官指标之一,其色泽会直接影响消费者的食欲和购买欲。如何赋予植物基肉良好的色泽关系到消费者对素肉接受度。目前赋予其色泽的主要技术路径有两条,第一是直接向制品中添加食用级色素,包括天然色素(血红素、辣椒红)、人工合成色素(红曲红);第二是通过微生物发酵与植物蛋白合成类肉的颜色。 5.人造肉相关标准 早在2018年10月,美国农业部 USDA 和食品药物管理局 FDA 就举行了联合听证会,并发表声明共同对人造肉进行监管。FDA负责解决细胞在试验室的整个培育过程, USDA 把握次要的控制权,监督细胞的摘取与最后的出产和贴标签环节。欧盟则在新食品法规(REGULATION (EU) 2015/2283)中明确规定,由细胞培养物或源自动物的组织培养物产生的食物都将被视作一种新型食品,并启用全新的管理条例。 目前,我国人造肉相关已发布和正在制定的标准如下表: 表2.人造肉相关标准信息 标准名称 类别 状态 检测项目 备注 GB/T XXX-20XX 素肉制品术语与分 类 国家 征求意见 无规定 规定素肉制品的术 标准 语、定义及分类 T/CIFST 002-2021 团体 2021-08-23 无规定 原辅料要求、感官要求 规定植物基食品的分 类、基本要求和标签 植物基食品通则 标准 发布并实施 标识要求 (色泽、形状、 气味、杂 质)、理化指标(蛋白 该标准给出了各检项 T/CIFST 001-2020 团体 2020-12-25发布 质)、污染物、真菌毒 的方法标准和/或限 植物基肉制品 标准 2021-06-25实施 素、微生物(大肠杆菌、 沙门氏菌、金黄色葡萄球 量要求。 菌)、食品添加剂、营养 强化剂。 T/QGCML 153- 理化指标(能量、蛋白 质、脂肪、碳水化合物、 本文件规定了植物肉 钠、氯化物、总砷、 的术语和定义、技术 团体 2021-08-11发布 铅)、微生物限量(大肠 2021 要求、检验方法、检 标准 2021-08-25实施 杆菌、沙门氏菌、金黄色 植物肉 葡萄球菌)、食品添加剂 验规则及标志、包 装、运输、贮存 (山梨酸及其钾盐、脱氢 Q/HSG 0001S- 乙酸及其钠盐)。 感官要求(色泽、形状、 气味、杂质)、理化指标 真菌毒素、污染物和 企业 2021-06-30 (水分、粗蛋白质、总 农药残留限量应分别 2021 标准 发布并实施 砷、铅、黄曲霉毒素 符合 GB 2761、2762 复合植物蛋白肉 b1)、真菌毒素、污染 和2763的规定要求 物、农药残留。 MISHExcellence in Sciel 二、人造肉检测应用方案 1、感官与营养 MISHExcellence in Science 植物基人造肉质量评价 随着人们对可持续发展和健康饮食生活方式兴趣的日益增加,植物基人造肉如今越来越受到关注。随着对植物基人造肉新产品的需求增加,产品的风味质量也相应受到关注。然而,有时很难精确评估包括味道和香味在内的风味,因为风味可能由许多化合物的高度复杂组合而决定。代谢物和芳香族化合物的广泛靶向分析是完成这项评估的一个好方法。LC-MS 和 GC-MS分别适用于亲水性代谢物和与味道相关的挥发性化合物的分析。 众所周知,决定食物品质的不仅仅是味道和香味。质地评估是评估食品质量的重要参数,尤其是在讨论仿肉产品时。装有适当夹具的材料测试仪可以研究与质地相关的特性,例如硬度。味道、香味和质地是相互独立的因素,因此应单独讨论。但是,我们有时想对食品的“整体质量”进行评分。多变量分析可以将每个样品在所有这些因素方面的“差异”可视化。因此,在我们的评估中,可以从食品固有的影响因素的角度,具体而全面地固化“质量”这个模糊的术语。 本文中,我们介绍了采用这种质量评估方法对四种不同植物基人造肉产品进行评估的结果。 C LC-MS/MS是研究食品样品中与味道相关的亲水化合物的最合适的仪器之一。 1.1样品制备 根据材料测试仪的分析要求(参见材料测试仪分析)烹制各产品,称取100mg样品,置于微量离心管中。取0.75mL甲醇,置于管中,用直径为5mm的氧化锆球在振动球磨机上研磨样品。离心后取上清液,用孔径为0.45mm的滤器过滤。将过滤后的溶液储存在-20℃下,作为原始储备样品溶液。用水将储备样品溶液稀释5000倍,制备代谢物分析样品溶液,以尽量减少基质效应。 1.2LC-MS/MS分析 使用初级代谢物的LC-MS/MS方法包来开发分析方法。该方法包可监测97种亲水性代谢物,只需极少步骤的标准品制备。 仪器 LCMS-8050 色谱柱 Discovery @ HS F5-3 15 cm x2.1 mm,3 um 流动相A 0.1%甲酸水溶液 流动相B 乙腈 分析模式 MRM 大多数化合物在稀释的样品溶剂中的回收率在80-120%范围内。将每个样品中所有55种化合物的峰面积值确定为其相对浓度。产品1谷氨酸和肌苷的峰形见图1。 图1.产品1中谷氨酸和肌苷的峰形(分别为左上和右上) 各产品之间谷氨酸和肌苷的比较(分别为左下和右下)。 2.糖的LC-MS/MS分析 我们还进行了一次LC-MS/MS分析来评估糖。 2.1样品制备 大多数操作流程与代谢物分析相同。用乙腈将储备样品溶液稀释500倍,制备成糖分析样品溶液。 2.2LC-MS分析 该方法的开发条件如下所示。 仪器 LCMS-8050 色谱柱 Asahipak NH2P-50 2D, 100 A, 5 um,2x150mm 流动相A 水 流动相B 乙腈 分析模式 MRM 成功检测到六种糖。但由于稀释程度,不能完全排除分析中的基质效应。因此,采用了标准添2所示。 图2.样品间的蔗糖和葡萄糖比较 3. 汽化化合物的GC-MS/MS分析 GC-MS/MS最适合用于检测食品样品中的总汽化化合物。采用固相微萃取 (SPME) 捕获分析物。 3.1样品制备 取20mg样品, 放入20 mL螺旋盖小瓶中。 3.2 GC-MS分析 使用配备AOC-6000自动进样器的三重四极杆GCMS-TQ8050。将样品加热至200℃,保持15分钟,用SPME纤维萃取汽化的化合物10分钟。预处理(包括SPME纤维调节、萃取、解吸和后调节)由AOC-6000自动完成。 仪器 GCMS-TQ8050+AOC-6000 SPME纤维 二乙烯基苯/Carboxen/聚二甲基硅氧烷 (DVB/CAR/PDMS) 色谱柱 SH-Rxi-5MS,30 mx 0.25 mm, 0.25 um 载汽 氦气 模式 扫描 (m/z:35-500) 如图3所示,每个样品中均检测到许多峰,而在某些样品中检测到了相同的峰。我们仔细选择了在重复检测中峰高和峰面积保持稳定的峰。并在市售的图谱库中搜索了这些稳定峰的图谱。我们 还在各样品间评估了这些峰的峰面积。可将峰面积值视为分析物的相对浓度,因为 GC-MS 分析中的基质效应较小。图3显示了各样品的数值示例。 RT1.21:乙酸 RT35.44:十六烷酸甲酰胺 图3:(上)碎牛肉样品的总离子色谱 (下)RT1.21和RT35.44峰(根据图库搜索,分别为乙酸和十六烷酸甲酰胺)在样品之间具有显著差异 4.材料测试仪分析质地 材料测试仪可不带任何主观色彩地评估食品样品的质地。 4.1样品制备 将各产品的25g样品压缩至相同形状的模具中。模具呈截锥形,上下直径分别为2.5厘米和4厘米。将模制样品放入200℃烘箱中烘烤20分钟。待样品内部温度降至约65℃时进行测试。 4.2材料测试仪分析 使用食品质地分析仪EZ Test。 记录夹具开始按压样品时的力曲线。直径为10mm的圆柱形夹具以恒定速度向下移动,直至完全穿透样品。本实验记录的曲线如图4所示。这几条力曲线基本相似,但形状略有不同。这种差异反映了每个样品的质地属性。试验力表示硬度。试验力越大,硬度越高。达到最大力的时间越长或达到最大力的行程越长,表明粘结性越大。如图4所示,每个样品的曲线分别在某个点到达顶峰。该点、该点处的最大力、波形可以作为硬度、粘结性等的评价指标。据此,我们就可以量化食物的质地。 图4.碎牛肉和四种植物基人造肉的力曲线。 夹具缓慢向样品移动,并在0秒时接触样品表面。力逐渐变大,当样品破裂并解体时达到峰值。此后,当夹具完全穿透样品时,力逐渐减小并几乎达到0。 5.多元分析 我们对所有数据(包括LC-MS、GC-MS和材料测试仪分析)进行了汇总,然后进行主成分分析。数据汇总表见图5。表中的值分别是LC-MS/GC-MS分析中的峰面积和材料测试仪中的力强度。然后将这些值标准化为平均值和标准偏差,以免在绝对值顺序不同的行中出现偏差。 主成分分析得到的得分图如图6所示。得分图可以解读为每个样品的“总相似度”,与味道、香味和质地有关。数据的模式越相似,样品点之间的距离就越近。这使得我们能够在没有任何主观因素的情况下审查每个样品的相似性。 实验 名称 碎牛肉 产品1 产品2 产品3 产品4 LC-MS(代谢物) 胱氨酸 0.00 5133.67 0.00 0.00 0.00 天冬酰胺 3233.33 4662.67 5600.67 8911.00 5485.00 天冬氨酸 8942.67 25824.67 11168.33 26395.33 12748.00 : : : : : LC-MS ((糖) 蔗糖 24.16 82.57 51.60 1.91 2718.60 麦芽糖 7.15 18.74 5.77 1.20 161.06 乳糖 8.35 25.02 18.52 0.99 67.78 : : : : : : GC-MS l-丙氨酸乙酰胺 3800125.00 3194828.67 3535825.67 3851375.67 3674014.00 乙酸 11625.00 10922.33 12232.00 11354.33 10993.33 顺-4,5-环氧-(E)-2-癸醛 28785.00 28580.33 39956.67 48981.33 39483.33 : : : : : : 材料测试仪 最大力 16.37 16.76 12.71 13.11 12.55 最大力时的时间 1.42 1.28 1.04 1.21 0.87 最大力时的行程 14.17 12.83 10.40 12.10 8.67 图5.将每个实验的所有数据汇总在同一个表中,以进行多元分析。这些值为标准化之前的值。我们同时分析了94组(行)数据。 图6主成分分析结果的得分图 6.结论 我们的 LC-MS/MS、GC-MS/MS 和材料测试仪可以分别评估植物基人造肉制品的整体质量,包括味道、香味和质地。然后结合这些数据,我们可以评估产品的整体质量。该策略不仅适用于植物基人造肉,也适用于所有食品的质量。 MISHExcellence in Scier 传统 SPME 和 SPME Arrow 用于人造肉类香味定性分析的优化和评价 摘要:随着公众对可持续蛋白质来源的兴趣越来越浓,植物基人造肉的热度越来越高,因为它与几种肉类产品的风味和质地非常相似。因为香气是风味的主要因素,所以在比较植物基人造肉与普通肉类产品时,挥发性气味的比较至关重要。为此,我们采用顶空固相微萃取纤维来吸收气味,然后通过气相色谱-质谱法对其进行分析。我们比较了几种光纤涂层和新的 Arrow 格式,发现它们在灵敏度和复合吸收方面存在差异。使用 Wiley 第 12 版/NIST 2017 库可以轻松识别检测到的化合物。 关键词: SPMESPME Arrow 人造肉 生肉本身几乎没有香味,因此几乎所有与“肉味”相关的香味都是在烹饪过程中通过氨基酸和还原糖之间的美拉德反应产生的。该反应决定了由哪些非挥发性前体释放挥发性芳香化合物。植物基人造肉是一种在外观和味道上都类似于动物肉的产品,正被越来越多的人所接受。它们提供了宝贵的营养,同时降低了某些健康风险,例如心脏病和II型糖尿病风险。此外,它们是最具环境可持续性的食物来源,因为它们的碳和水足迹小于动物性食物。植物蛋白(如大豆浓缩蛋白)加上色素、稳定剂和油可成功地模仿肉类的味道和质地。而且,就像在动物肉中一样,这种蛋白质的氨基酸也会发生美拉德反应。因此,肉类和肉类替代品都是复杂的基质,无需额外的样品制备即可通过气相色谱-质谱 (GC-MS) 进行分析。 固相微萃取 (SPME) 是一种无溶剂萃取技术,它利用吸附纤维从顶空或液体样品中吸附/吸收化合物。顶空固相微萃取提高了挥发性化合物的选择性和灵敏度,并减少了基质效应。与传统纤维相比,新的SPMEArrow包含更多的吸附相和更大的表面积,从而可以在更短的时间内萃取更多的分析物。结合重新设计的吸头和外护套,与传统的SPME分析相比,其通量和稳定性都有所提高。 1.实验部分 1.1仪器 带 AOC-6000 自动进样器的 GCMS-QP2020 NX 1.2样品与分析 表1总结了本项目中配备 AOC-6000 自动进样器的岛津 GCMS-QP2020 NX 的仪器条件。牛肉样品制备如下:称取约2.5克有机碎牛肉(85瘦肉:15脂肪),置于20mL标准钳口顶空玻璃瓶 中,在分析前置于环境温度下。将样品在每种类型的 SPME 设备上各测试三次。使用 Wiley 第 12版/NIST 2017 库对检测到的峰进行识别。 表1.GCMS条件 带AOC-6000的GCMS-QP2020 NX SPME和SPME Arrow PAL: PDMS、Carboxen和PDMS/DVB/Carboxen涂层 萃取 130℃,不同时间 解吸 10 min 气相色谱 进样口 270℃, 不分流(1分钟);分流10:1 色谱柱 Rtx-5MS色谱柱(30m × 0.25mm × 0.25um)He载气 恒压, 90.1kPa 柱温箱温度 60 ℃-2min>160℃(7℃/sec)>250 ℃(4℃/sec)-2 min 质谱 接口温度 250°℃ 离子源温度 200°℃ 检测器电压 相对于Tune 扫描范围 40至350m/z 事件时间 0.3秒 2.结果与讨论 2.1色谱 我们首先评估了传统 SPME 纤维和新型 SPME Arrow 之间的差异。正如预期的那样,在图1中,与 SPME 纤维相比, SPME Arrow 在相同时间的萃取过程中吸收了更多的化合物,从而在色谱图上产生了更多可检测的峰。Arrow 的表面积是传统纤维的20倍,因此具有更多的分析物吸附位点。 图 1. 使用 SPME Arrow (黑色)和SPME纤维(粉红色)萃取有机牛肉10分钟后的代表性叠加色谱图我们比较了多个萃取时间,以研究 Arrow 上的表面积增加是否可以增加通量。我们观察到传统 SPME 纤维在 3、10和30分钟萃取之间的变化很小(图2);然而,使用 SPME Arrow 时, 在每次萃取时间增加后,观察到信号显著增加(图3)。即使在3分钟的萃取过程中, SPME Arrow 吸收的化合物量也与 SPME纤维在30分钟萃取过程中吸收的化合物量大致相同。增加 SPME Arrow 的萃取时间不仅会增加信号强度,还会增加可检测化合物的数量。 图2.使用SPME纤维萃取有机牛肉30分钟(黑色)、10分钟(粉色)和3分钟(蓝色)后的代表性叠加色谱图 图3. 使用SPME Arrow萃取有机牛肉30分钟(黑色)、10分钟(粉色)和3分钟(蓝色)后的代表性叠加色谱图 图4. 使用SPME Arrow萃取仿肉(黑色)和有机牛肉(粉红色)10分钟后的代表性叠加色谱图 表 2. 使用 SPME Arrow 在仿肉和有机牛肉样品中检测到的化合物 仿肉 有机牛肉 1.3-丙二醇 丙酸,2-羟基-,甲酯,(.+/-.)- 五甘醇 二甲基砜 2(5H)-呋喃酮 甘油 甘油 呋喃醇 3-戊酮,2,4-二甲基- 3,5-辛二烯-2-酮,(E,E)- 正戊酸己酯 壬醛 壬醛 麦芽酚 4H-吡喃-4-酮,2,3-二氢-3,5-二羟基-6-甲基- 4H-吡喃-4-酮,2,3-二氢-3,5-二羟基-6-甲基- 2(3H)-呋喃酮,二氢-4-羟基- 2(3H)-呋喃酮,二氢-4-羟基- 辛酸 辛酸 己内酰胺 噻吩,2,3-二氢- 2-癸烯,(E)卜 哌啶,1-亚硝基- 壬酸 壬酸 2-n-辛基呋喃 1,2-苯二醇,3,5-双(1,1-二甲基乙基)- 2,4-癸二烯醛,(E,E)- 川顺-4-癸醛 正癸酸 正癸酸 2-十三酮 烟酰胺 十四烷 6,10-十二二烯-1-醇,3,7,11-三甲基- 噻唑,4,5-二甲基- 2-十三酮 正壬基环己烷 十二烷酸 十二烷酸 仿肉 有机牛肉 1-十五碳烯 氟膦酸,(1-甲基乙基)-,环己酯 8-十七碳烯 二十烷 2-十二酮 1-十六醇 甲酮,(1-羟基环己基)苯基- 十六碳烯酸,Z-11- 十四烷酸 十四烷酸 十四烷酸乙酯 1-十二烷醇,3,7,11-三甲基- 十八烷 十六烷,2,6,10,14-四甲基- 十四烷醛 十五醛- 十五烷酸 2-十七酮 delta-十二内酯 芥酸 芥酸 正十六烷酸 正十六烷酸 十七烷酸 2(3H)-呋喃酮,5-十二烷基二氢- 油酸 油酸 十八烷酸 十八烷酸 十六酰胺 8,11,14-二十碳三烯酸,(Z,Z,Z)- 角鲨烯 图5.使用PDMS纤维检测有机牛肉的代表性色谱图 图 6.使用 Carboxen 纤维检测有机牛肉的代表性色谱图 图7.使用PDMS/DVB/Carboxen纤维检测有机牛肉的代表性色谱图 3.结论 使用配备AOC-6000自动进样器的GCMS-QP2020 NX对有机牛肉和仿肉进行了SPME和SPMEArrow分析。 Wiley库搜索的定性结果表明,与传统SPME相比, SPME Arrow可以吸收的化合物范围更广,因而灵敏度和通量更高。通过比较不同的SPME纤维涂层,我们证明了两种SPME技术能检测到的化合物在很大程度上取决于其纤维涂层材料。未来的研究可集中在针对特定化合物类别的方法开发或提高质量标记香味剂的灵敏度,以进一步提高仿肉质量。 XE EZ-Test人造肉质构评价 摘要: texture profile analysis’(TPA),“全质构分析法”,作为一种食品质构分析的方法,正被广泛应用于食品质地、口感的数字化的评价。本文介绍了使用 EZ-SX 质构仪,对人造肉进行肉嫩度与全质构测试的例子,以评价其质地与口感。 关键词:质构仪 食品质构人造肉 随着全球人口增加,肉类短缺危机、健康与环保消费风潮、食品创新等因素推动了“人造肉”迅速兴起。 人造肉通常可以分为植物性肉、培养肉和其它肉类替代品。植物性肉通常以豌豆、大豆以及其他豆类为原料,由植物蛋白制成。培养肉也称为细胞肉,是指在特定的条件下,利用动物干细胞在培养基中培育出来。其他肉类替代品是指除了前两种以外的肉类替代品,包括以真菌、乳蛋白、鸡蛋蛋白、鱼蛋白、昆虫蛋白和其他新型蛋白质培养的人造肉产品。 与真肉相比,人造肉在营养价值、食品安全、能源消耗和环境友好方面都具有潜在的优势;人造肉的劣势则包括:成本较高、质感次佳(植物肉)、技术存在瓶颈(培养肉)等。 本试验我们选用植物性人造肉进行试验。使用质构仪进行肉嫩度与全质构测试,获取其质构数据。 1.。实验部分 1.1仪器 试验机: EZ-SX 试验类型:向下压缩/剪切试验 2.样品前处理 本试验使用的人造肉,配料:大豆分离蛋白、小麦粉、淀粉、植物油;直径约25mm, 高度21-23mm。 样品处理步骤:冷水浸泡10分钟-蒸锅蒸15分钟-冷却至室温(25度左右) 图1.试验用植物性人造肉外观图 3.结果与讨论 ecniec3.1肉嫩度测试 S 上夹具使用厚度 3mm, 60度刃口的专用剪切夹具,下夹具使用缝隙宽 4mm 的支撑夹具; 剪切速度:1mm/秒; 剪切距离:约20mm; 图2肉嫩度试验 图3.肉嫩度测试结果载荷-位移曲线 表1.肉嫩度测试结果数据 样品 样品直径(mm) 样品高度((mm) 肉嫩度(N) 11 24.2 23.2 23.966 1_2 25.5 23.5 31.588 3.2二次咀嚼试验 本试验模拟人牙齿咀嚼食物过程,对样品进行两次压缩,记录试验过程中探头压力与压缩量之间的对应关系,进而从力-时间曲线上分析可得相应的质构参数:硬度、弹性、胶粘性、咀嚼性等指标。 上夹具使用中30mm 平压头;以1mm/秒速度压缩样品,向下压缩移动13mm时,以相同速度返回位移原点;停顿2s后,重复第一次压缩过程。 图4.二次咀嚼试验 图4.二次咀嚼试验曲线 上图4为软件生成的典型的二次咀嚼试验曲线。此次试验样品的弹性很高。在第一测试周期粉红色标记,在第二个测试周期用浅绿色标记,使用两周期内的数据可计算样品的弹性、胶粘性和咀嚼性。其结果为表2所示。 表2.试验结果数据 样本 硬度 H (N) 弹性 胶粘性(N) 咀嚼性(N) 人造肉 12.896 0.999 10.1739 10.1735 4.结论 岛津的EZ 系列质构仪,测试精度高,测试功能多,软件操作简便,适合检测人造肉的各项质构数据。对于人造肉的开发,品质管理,出货检验等方面,能提供专业的数值参考。 GCMS结合岛津香味数据库分析人造肉中的气味成分 摘要:本文采用岛津气相色谱质谱联用仪GCMS-QP2020 NX,结合SPME进样和岛津Smart AromaDatabase香味数据库建立了人造肉中498种气味成分的分析方法,在SIM采集模式下,采用校准用标准样品生成的曲线对样品中的气味成分进行半定量分析。人造肉样品中共检测出50种气味成分。该方法操作简单,分析速度快,适用于人造肉中气味成分的筛查分析。 关键词:气相色谱-三重四极杆气质联用仪香味数据库人造肉 气味成分 人造肉通常可分为植物蛋白肉和动物蛋白肉两种。植物蛋白肉通常以豌豆、大豆以及其它富含植物蛋白的豆类为原料制成;动物蛋白肉则是在特定条件下,利用动物干细胞在培养基中培育得到。目前国内市场上能够购买到的人造肉食品基本都是植物蛋白肉。气味是食品的一个重要特征。研究人造肉中的气味成分,对优化人造肉加工工艺有着重要的意义。 GCMS 定性能力优异,常被用于食品中气味成分的分析。但大多数食品基质复杂,气味成分种类繁多,研究人员往往需要耗费大量精力处理数据。使用预先收录有化合物信息的数据库,可极大减少数据分析的工作量。 本文利用岛津 GCMS-QP2020 NX气质联用仪和 AOC-6000 多功能自动进样器,结合 Smart AromaDatabase 香味数据库分析人造肉中的气味成分,无需复杂设置就能快速创建498种气味成分的定性及半定量方法,可对人造肉中的气味成分进行快速的筛查分析。 1.实验部分 1.1仪器 GCMS-QP2020 NX气质联用仪 ellecAOC-6000 自动进样器x 1.2标准品 保留时间修改用标品:正枚烷烃(C7-C40) 混合标准品半定量校正用标品:对溴氟苯、1,2-二氯苯-d4、造-d10混合标准品 1.3分析条件 SPME 参数 SPME 纤维: CarboxenTM/PDMS/DVB 120um 老化温度:250℃ 老化时间(萃取前):10min 平衡温度:80℃ 平衡时间:10min 萃取时间:25min 进样口温度:250℃ 解吸时间: 3min 老化时间(萃取后):5min GC-MS 参数 色谱柱: InertCap-Pure Wax, 30 m×0.25 mm× 0.25um 柱温程序:50℃(5 min)_10℃/min_250℃ (15 min) 载气控制:恒压模式,83.5kPa 进样方式:分流进样 分流比:5:1 离子源温度:200℃ 接口温度:250℃ 检测器电压:调谐电压+0.2kV 采集方式:Scan/SIM ecn1.4样品前处理 iec 将顶空瓶置于150℃烘箱中烘烤 30 min, 精确称取待测样品1.0g装入20mL顶空瓶中,密封后按1.3条件上机分析。 2.结果与讨论 2.1气味系统方法建立流程 岛津香味数据库包含一系列的分析方法、质谱库等文件。使用 Aroma_TQ_IC-Wax_AART 方法测定正构烷烃标品,用于校正目标组分的保留时间。正构烷烃样品色谱图见图1.使用 Aroma_TQ_IC-Wax_Correct 方法测定4-溴氟苯、1,2-二氯苯-d4、芯-d10混合内标物标准品,用于建立半定量校正曲线。内标样品色谱图见图2。 图1正构烷烃色谱图 (x100,000,000) 图2校正内标样品色谱图(10mg/L) 利用所得正构烷烃和内标物数据以及香味数据库建立498种香味成分的筛查方法文件, GCMS-QP2020 NX 可利用该方法对这些香味成分进行筛查,并在没有目标组分标准品的情况下对这些组分进行半定量分析。图3为香味数据库的创建方法界面和方法创建完成界面。 2.2人造肉样品测试结果 测定的植物蛋白肉样品色谱图见图4,利用香味数据库共筛查出50种化合物,如表1所示。图5是检出的部分化合物的质量色谱图。 图4人造肉样品的色谱图 表1人造肉中气味成分筛查和半定量结果 (ng/g) NO. 化合物 CAS号 浓度 气味特征 1 乙酸乙酯 141-78-6 46.3 菠萝香味 2 2-乙基呋喃 3208-16-0 1.2 可可味,咖啡味,坚果味 C 40 苯酚 108-95-2 0.7 酚味 41 丙位壬内酯 104-61-0 2.2 椰子味,桃子味 42 壬酸 112-05-0 5.1 绿植味,脂肪味 43 麝香草酚 89-83-8 0.1 百里香味,香料味 44 4-乙烯基-2-甲氧基苯酚 7786-61-0 4.1 丁香味,咖喱味 45 邻氨基苯甲酸甲酯 134-20-3 0.0 蜂蜜味,花香味 46 3-桉叶醇 473-15-4 0.2 木头味,绿植味 47 苯甲酸 65-85-0 21.4 香脂味,尿味 48 吲哚 120-72-9 0.4 樟脑丸味,焦味 49 5-羟甲基糠醛 67-47-0 0.5 纸板味 50 香兰素 121-33-5 1.5 香草味 Q43.00 (+) 2.28e6Q82.10(+) 4.52e5Q58.00(+) 7.54e6 2.0e6- 6.0e6- 4.0e5 1.5e6- 4.0e6- 1.0e6- 2.0e5 5.0e5- 2.0e6- 0.0e0- 0.0e0- 0.0e0 TTT丁 厂T下 TTT TTT TTT 3.结论 本文采用岛津 GCMS-QP2020 NX气质联用仪和 SPME进样,结合岛津 Smart Aroma Database香味数据库对人造肉中的气味成分进行测定。通过采集正构烷烃和校正内标数据,在没有目标组分标准品情况下,利用香味数据库自动创建498种气味成分的检测方法,对人造肉中的气味成分进行定性及半定量分析。测试人造肉样品中共检测出50种气味成分。结果表明,该方法操作简便,分析速度快,可用于人造肉中气味成分的快速筛查。 MISHExcellence in Scie ICPMS-2030 系列测定人造肉中的营养元素 摘要:参考GB5009.268-2016《食品安全国家标准食品中多元素的测定》,加入硝酸和过氧化氢对人造肉进行微波消解,使用电感耦合等离子体质谱仪(ICP-MS)测定了人造肉中钾、钠、钙、镁等7种营养元素的含量。分析结果表明,方法检出限低,准确度好,加标回收率93.0%~105%,适合人造肉中高低含量营养元素的同时检测。 关键词:微波消解 ICP-MS人造肉营养元素 近年来,随着全球肉类短缺危机、健康与环保消费风潮、食品创新等因素推动,“人造肉”概念迅速兴起,据报道,全球肉类替代品市场正以每年68%的复合增长率增长,其中人造肉深受欢迎。人造肉通常可以分为植物性肉、培养肉和其它肉类替代品,同样可以提供养殖肉中所富含的蛋白质、脂肪等营养元素,也可以控制其营养成分,满足不同群体的需求。目前涉及人造肉的检测内容分四个部分:组学分析、营养成分分析、有毒成分分析、包装材料。其中营养成分分析可细分为营养物质检测、营养元素(如Se、Na、Mg、K、Ca等)检测等。 我国对食品安全有着严格的监控, GB 2762-2017《食品安全国家标准食品中污染物限量》明确规定了食品中重金属的限量要求,而相对应的, GB5009.268-2016《食品安全国家标准食品中多元素的测定》明确规定了食品中金属元素的检测方法,第一法即为电感耦合等离子体质谱法。 电感耦合等离子体质谱法 (ICP-MS) 具有灵敏度高、检出限低、线性范围广等特点,本文参考标准GB5009.268-2016,对人造肉样品进行微波消解后,使用岛津ICPMS-2030系列测定了人造肉中钾、钠、钙、镁、铁等营养元素的含量。 1.实验部分 表1ICP-MS分析条件 参 数 参数设定 参 数 参数设定 高频功率 1.20kW 等离子体气流速 9.0 L/min 辅助气流速 1.10 L/min 载气流速 0.70 L/min 炬管类型 Mini炬管 雾化器 同心雾化器 雾化室 旋流 雾化室温度 5℃ 采样深度 5.0mm 高频频率 27.12MHz 碰撞气体 He 碰撞气流速 6mL/min 池电压 -21V 能量过滤器电压 7.0V 2.样品前处理 准确称取0.2g样品于微波消解罐中,加入6 mL HNO3和2 mL H202,置于微波消解仪中消解,升温程序见表2。消解结束后,冷却,转移至50mL离心管中,定容至50mL, 混匀备用,同时做空白和加标试验。 表2微波消解程序 步骤 控制温度(℃) 升温时间(min) 恒温时间(min) 1 120 5 5 2 150 5 10 3 190 5 30 3.结果与讨论 3.1标准曲线和检出限 使用2% HNO3将铁、硒、锌标准储备溶液稀释为0、0.20、0.50、1.0、2.0、5.0、10、20、50、100 ug/L的标准序列,钾、钠、钙、镁标准储备溶液稀释为0、0.20、0.50、1.0、5.0、10 mg/L的标准序列,根据样品中元素实际情况调整标准曲线线性范围。标准曲线见图1~图4,连续测量空白溶液计算仪器检出限(IDL)和方法检出限(MDL),IDL和MDL结果见表3。 浓度=0.0053131*1+0.0126372r=1.00000BEC=-(ug/mL) 3a=0.0013114(ug/mL)图1K元素标准曲线 浓度=0.0021460*1+0.0155422 r日0.99997BEC= ---(ug/mLL))3g=9.9505e-004 (ug/mL) 图2Na元素标准曲线 浓度=8.4804e-004*1-0.0187901 r=0.99999BEC=0.0042737 (ug/mL)3a=0.0045796 (ug/mL) 0.99993BEC= 0.0187901 (ug/mL)3a=5.3093e-004 (ug/mL) 图3Ca元素标准曲线 图4Mg元素标准曲线 表3仪器检出限和方法检出限 元素 质量数 相关系数 IDL(ug/L) MDL(ug/kg) GB5009.268-2016 定量限要求(ug/kg) K 39 1.00000 1 250 3000 Na 23 0.99997 0.9 225 3000 Ca 44 0.99999 4.5 1125 3000 Mg 24 0.99993 0.5 125 3000 Fe 56 0.99968 0.204 51 3000 Se 78 0.99993 0.075 18.8 30 Zn 68 0.99978 0.158 39.5 2000 3.2样品测试结果 表4人造肉中营养元素测定结果 元素 质量数 内标 测定值 (mg/L) RSD% n=3) 结果 (mg/kg) 加标量 (mg/L) 回收率 (%) K 39 45Sc 5.51 0.73 1.38×104 2.0 100 Na 23 45Sc 2.78 1.12 6.95×103 2.0 97.0 Ca 44 45Sc 0.767 0.76 1.92×103 2.0 100 Mg 24 45Sc 0.724 1.54 1.81×103 2.0 101 Fe 56 45Sc 0.048 2.45 120 0.02 100 Se 78 72Ge N.D. 一一 0.0005 105 Zn 68 72Ge 0.014 0.36 35.0 0.02 93.0 备注:按照GB5009.268-2016,计算结果保留3位有效数字。 4.结论 参考GB5009.268-2016《食品安全国家标准食品中多元素的测定》,使用岛津ICPMS-2030系列电感耦合等离子体质谱仪测定了人造肉中7种营养元素含量。实验结果表明,该方法检出限低,准确度好,高低含量元素可以同时测量,加标回收率在93.0%~105%范围内,适合人造肉中钾、钠、钙、镁、铁、、硒等营养元素的同时检测。 2、食品添加剂 SHIMADZU Excellence in Science 气相色谱法测定人造肉中脱氢乙酸的含量 摘要:本文使用 Nexis GC-2030建立了测定人造肉中脱氢乙酸含量的分析方法。样品经超声、离心分取和酸化后,加入乙酸乙酯萃取上机。结果表明,在1~200 ug/mL的浓度范围内,脱氢乙酸的线性相关系数R为 0.9999,线性良好。以3倍信噪比计算检出限,仪器检出限为 0.15 ug/mL, 满足标准要求。取浓度为 1ug/mL的标准溶液重复进样6次,峰面积的相对标准偏差为2.95%,精密度良好。对测试样品进行加标回收实验,加标平均回收率为104.0%。本方法操作简单、灵敏度高,可为人造肉中脱氢乙酸含量的测定提供参考。 关键词:气相色谱仪 人造肉 脱氢乙酸 人造肉是近年食品行业的一项研究热点。它对环境保护、解决粮食和蛋白质短缺等具有重要意义。人造肉按原料来源主要分为两种。一种是利用培养基培育动物干细胞得到的动物蛋白肉;另一种是利用植物蛋白和其它植物成分合成具有肉类特性的植物蛋白肉。 脱氢乙酸及其钠盐是食品行业中常用的广谱防腐剂,它对食品中的细菌、霉菌、酵母菌等都有较强的抑制作用。《GB2760-2014食品安全国家标准食品添加剂使用标准》规定各类食品中脱氢乙酸使用限量在 0.3 g/kg~1.0 g/kg 之间。一般按标准规定添加,脱氢乙酸不会对人体产生危害。但过量使用可能会导致一些皮肤过敏反应,甚至引起中枢神经中毒等问题。 本文参考《GB5009.121-2016食品安全国家标准食品中脱氢乙酸的测定》,使用岛津气相色谱仪 Nexis GC-2030 建立了人造肉中脱氢乙酸含量的测定方法。实验结果表明,该方法操作简单、灵敏度高、重复性好,可为人造肉中脱氢乙酸含量的测定提供参考。 1.实验部分 色谱柱: InertCap Pure-WAX, 30m×0.25 mm ×0.25 um 柱温程序: 150℃_10℃/min_210℃_20℃/min_240℃(2min) 进样口温度:240℃ 进样方式:分流进样,分流比5:1 进样量:1uL 载气:氮气 载气控制方式:恒线速度 (27.9cm/sec) 检测器: FID 检测器温度:300℃ 空气流量:200mL/min 氢气流量:32mL/min 尾吹气流量: 24mL/min 2.样品前处理 样品前处理步骤见图1所示 ceen 图1.样品前处理流程图 3.结果与讨论 3.1标准品谱图 脱氢乙酸标准品色谱图和化合物信息分别见图2和表1。 4.gcd 图2.脱氢乙酸标准溶液色谱图(浓度: 50ug/mL) 表1.化合物信息表 峰号 化合物 英文名称 CAS 号 保留时间(min) 脱氢乙酸 Dehydroacetic Acid 520-45-6 5.262 3.2标准曲线与检出限 将1000 ug/mL 的脱氢乙酸储备液用乙酸乙酯分别稀释为浓度为1、10、50、100和 200ug/mL的标准溶液进行测定,以脱氢乙酸浓度为横坐标,峰面积为纵坐标绘制标准曲线。根据1ug/mL标样数据,以3倍信噪比计算脱氢乙酸的仪器检出限,脱氢乙酸标准曲线见图3,线性方程、相关系数以及检出限见表2。 cience 图3.脱氢乙酸标准曲线 表2.线性方程及相关系数及仪器检出限 NO. 化合物名称 线性方程 相关系数 检出限(ug/mL) 1 脱氢乙酸 Y=1620.41X-1294.26 0.9999 0.15 3.3精密度实验 取 1 ug/mL 脱氢乙酸标准溶液1 uL 进气相色谱仪,连续进样6次,以峰面积 RSD 考察峰面积重复性,结果如表3。 表3.峰面积重复性结果 (RSD%,n=6) 组分 面积1 面积2 面积3 面积4 面积5 面积6 平均面积 RSD (%) 脱氢乙酸 1579 1606 1532 1605 1497 1522 1557 2.95 3.4样品测试结果 取某植物蛋白肉样品,按照步骤2进行处理,样品中未检出脱氢乙酸,样品色谱图见图4。 图4.植物蛋白肉样品色谱图 3.5回收率 将脱氢乙酸标准溶液添加到上述样品中,按步骤2进行处理,样品加标量为 100 mg/kg, 平行制样3次。回收率结果见表4。 表4.加标回收率结果 (mg/kg) 组分名称 测试值1 测试值2 测试值3 平均回收率(%) 脱氢乙酸 95.11 103.31 113.44 104.0% 4.结论 本文建立了使用 Nexis GC-2030 测定人造肉中脱氢乙酸含量的分析方法。结果表明,在1~200ug/mL的浓度范围内,脱氢乙酸的线性相关系数R为0.9999,线性关系良好,方法检出限为0.15ug/mL。取浓度为 1ug/mL的标准溶液重复进样6次,峰面积的相对标准偏差(RSD%) 为 2.95%,精密度良好。对空白样品进行加标回收实验,样品加标量为 100 mg/kg时,平均回收率为104.0%。本方法操作简单、灵敏度高、重复性好,可用于测定人造肉中脱氢乙酸的含量。 Excell 高效液相色谱法测定人造肉中苯甲酸、山梨酸和糖精钠的含量 摘要:本文建立了一种高效液相色谱法测定人造肉中苯甲酸、山梨酸和糖精钠含量的方法。样品参照《GB 5009.28-2016 食品中苯甲酸、山梨酸和糖精钠的测定》中的前处理和实验条件进行上机分析。3种防腐剂在0.5-200 mg/L 浓度范围内线性良好,相关系数>0.999,加标回收率在92.4-100.8%之间,连续6次进样保留时间 RSD%为 0.004~0.062%,峰面积 RSD%为 0.115~1.368%,系统精密度良好。方法准确可靠,灵敏度高,可用于实际人造肉样品的检测。 关键词:高效液相色谱 防腐剂 人造肉 人造肉分为两种:一种是以植物蛋白为原材料对肉类形色和味道进行模仿的植物肉制品;另一种是在培养基中利用动物干细胞进行一定条件培养,从而制造的人造肉。大豆蛋白是植物源人造肉常用的材料,豆制品可以给人体补充大量蛋白质满足人体营养需求。 为了延长植物源人造肉的保存时间,在生产加工中有时会加入一定量的苯甲酸、山梨酸或者糖精钠作为防腐剂来抑制微生物繁殖,若防腐剂添加过量被人体摄入后则会危害人体健康。因此,建立一种快速简单、准确、灵敏的检测防腐剂的方法意义重大。 目前测定防腐剂的方法主要有气相色谱法、液相色谱法等,我国《GB 5009.28-2016食品中苯甲酸、山梨酸和糖精钠的测定》中规定了2种检测这3种防腐剂的测定方法,分别是液相色谱和气相色谱检测方法。 本文使用岛津高效液相色谱仪,参照《GB 5009.28-2016食品中苯甲酸、山梨酸和糖精钠的测定》中规定的第一法,建立了一种可以准确测定人造肉中苯甲酸、山梨酸和糖精钠3种防腐剂的方法。 1.实验部分 1.1仪器 本实验采用岛津 Nexera LC-40 高效液相色谱仪,具体配置为: 液友泵:LC-40BX3 系统控制器 :CBM-40Lite 温箱 :CTO-40S 检 测 器 :SPD-40 自动进样器 :SIL-40C X3 色谱工作站 : LabSolutions Ver. 5.97 1.2分析条件 色谱柱: Shim-pack GIST C18 (250mmx4.6mml.D.,5 um),P/N:227-30017-08,岛津(上海)实验器材有限公司 流动相 : A-2 mmol/L甲酸+20 mmol/L乙酸铵溶液; B-甲醇 流 速 :1mL/min 进样体积 :10 uL 检测波长 :230nm 洗脱方式:等度洗脱, A/B=92/8(V/V) 1.3试剂配制 亚铁氰化钾溶液(92g/L):称取106g亚铁氰化钾,加入适量水溶解,用水定容至1000 mL。乙酸锌溶液(183g/L):称取 220 g 乙酸锌溶于少量水中,加入30mL冰乙酸,用水定容至1000mL。 甲酸-乙酸铵溶液(2 mmol/L 甲酸+20 mmol/L乙酸铵):称取1.54g乙酸铵,加入适量水溶解,再加入75.2 uL甲酸,用水定容至1000 mL, 经0.22 um 水相微孔滤膜过滤后备用。 2.样品前处理 准确称取2g样品于 50mL 具塞离心管中,加水约25mL,涡旋混匀,于50℃水浴超声 20min,冷却至室温后加入2mL亚铁氰化钾溶液和2mL乙酸锌溶液,混匀,于8000 r/min 离心 5 min, 将水相转移至 50mL 容量瓶中,于残渣中加水 20 mL, 涡旋混匀后超声 5 min, 于8000 r/min 离心5min, 将水相转移到同一50mL容量瓶中,并用水定容至刻度,混匀。取适量上清液过 0.22 um滤膜,待液相色谱测定。 3.结果与讨论 8 3.1标准品溶液色谱图 分别精密量取苯甲酸、山梨酸和糖精钠3种防腐剂标准品适量,用水稀释成浓度为0.5 mg/L、1mg/L、5mg/L、10 mg/L、20 mg/L、50 mg/L、100 mg/L、200mg/L八个浓度的混合标准系列工作溶液,按1.2中的分析条件进行测定,苯甲酸、山梨酸和糖精钠3种防腐剂标准品的溶液色谱图如图1所示。 图1.标准品溶液色谱图(10mg/L) 3.2线性范围和灵敏度 将不同浓度的标准品溶液,按1.2中的分析条件进行测定,以浓度为横坐标,峰面积为纵坐标,采用外标法建立标准曲线,结果如图2所示。苯甲酸、山梨酸和糖精钠3种防腐剂在0.5-200 mg/L浓度范围内,具有较好的线性关系,线性相关系数>0.999,具体结果见表1。 面积(x1,000,000) 5.0- 4.0 3.0- 2.0- 1.0 0.0-7 50 150 浓度 图2. 3种防腐剂校准曲线 表1.校准曲线参数(权重1/C2) 化合物名称 标准曲线 相关系数r 线性范围 (mg/L) 准确度 检出限 (mg/L) 定量限 (mg/L)) 糖精钠 Y=27181.2X+531.206 0.9999 0.5-200 98.4-102.7% 0.06 0.17 苯甲酸 Y=35799.4X+4271.52 0.9998 0.5-200 98.3-103.2% 0.04 0.12 山梨酸 Y=52377.1X+486.069 0.9999 0.5-200 99.1-101.8% 0.06 0.18 3.3精密度实验 不同浓度的标准品溶液连续进样6次,用于考察仪器的精密度,保留时间和峰面积的重复性结果如表2所示。结果显示,保留时间和峰面积的相对标准偏差分别在0.004~0.062%和 0.115~1.368%之间,仪器精密度良好。 图3.标准品溶液重复性色谱图(浓度20mg/L, n=6) 表2.保留时间和峰面积重复性结果(n=6) 化合物名称 RSD% (5mg/L) RSD% (20mg/L) RSD% (100 mg/L) R.T. Area R.T. Area R.T. Area 糖精钠 0.062 1.368 0.055 1.205 0.031 0.079 苯甲酸 0.035 0.324 0.016 0.283 0.010 0.181 山梨酸 0.014 0.231 0.006 0.190 0.004 0.115 3.4加标回收率实验 以空白人造肉样品进行加标回收实验,准确称取2g样品,分别添加三个浓度水平的标准品,按照2.中规定的前处理方法处理样品,计算平均回收率。见图4,空白基质中未检测到苯甲酸、山梨酸和糖精钠3种防腐剂,空白样品色谱图和加标回收色谱图见图4,各添加水平的平均回收率在92.4-100.8%之间,详见表5。 图4.空白人造肉色谱图(紫色)和加标回收色谱图(黑色, 20mg/L) 表3.苯甲酸、山梨酸和糖精钠3种防腐剂回收率(n=3) 化合物名称 含量 mg/L 加标1mg/L 加标20mg/L 加标100mg/L 测得值 mg/L 回收率% 测得值 mg/L 回收率% 测得值 mg/L 回收率% 糖精钠 N.D. 0.947 94.7% 19.782 98.9% 99.813 99.8% 苯甲酸 N.D. 0.953 95.3% 20.164 100.8% 100.234 100.2% 山梨酸 N.D. 0.924 92.4% 19.907 99.5% 100.541 100.5% N.D. 表示未检出。 SHIMADZU Excellence in Science GCMS法测定人造肉中的多环芳烃含量 摘要: 本文使用岛津气质联用仪 GCMS-QP2020 NX 建立了人造肉中16种多环芳烃的检测方法。实验结果表明:在 1.0-100 ng/mL 浓度范围内,16种多环芳烃组分线性良好,线性相关系数均在0.999以上,各化合物检出限在 0.02-0.15 ng/mL 之间。取浓度为5.0ng/mL标准混合溶液,连续进样6次,保留时间和峰面积的相对标准偏差(RSD%)分别在 0.01%-0.03%和1.5%-3.7%之间,精密度良好。在空白样品中进行0.02、0.4和2.0 ug/kg 三个不同浓度加标实验,回收率在83.5%-103.7%之间。该方法简单,稳定,准确,适用于人造肉中16种多环芳烃的测定。 关键词:气相色谱质谱联用仪多环芳烃圣人造肉 人造肉产品分为两大类,第一类是以植物蛋白为主要原料制备的植物基肉制品,第二类是通过动物细胞培养扩增制备的细胞培养肉。目前市场上主要以植物基肉制品为主,细胞培养肉还在研发期。植物基肉制品的植物原料主要包括豆类,谷物类,藻类,真菌类等。人造肉作为新兴的一种食品,其原料、生产流程、包装保藏等环节都面临污染的风险,其食品质量安全体系和标准一直是亟待明确和解决的。 多环芳烃 (PAHs) 是指含两个或两个以上苯环的芳烃,广泛分布于自然界中,其数量大、种类多,对人体危害极大,数种多环芳烃已被鉴定出具有较强的致癌、致畸和致突变作用,苯并[a]芘是其中毒性最大的一种强致癌物质。PAHs 主要对人体的呼吸系统、循环系统、神经系统、肝脏、肾脏等造成损害。GB2762-2017《食品安全国家标准食品中污染物限量》规定,谷物及其制品、肉及肉制品中苯并[a]芘的最高残留限量为 5 ug/kg。人造肉的生产环节复杂,容易受到多环芳烃的污染,因此建立和健全人造肉中多环芳烃的检测方法很有必要。 本文参考 GB5009.265-2021《食品安全国家标准食品中多环芳烃的测定》,采用岛津 GCMS-QP2020 NX气质联用仪,建立了人造肉中16种多环芳烃的检测方法。该方法简单,稳定,准确,分离度高,可供相关检测人员参考。 1.实验部分 1.1仪器 岛津气质联用仪 GCMS-QP2020 NX 1.2分析条件 色谱柱: DB-EUPAH, 20 m×0.18mm×0.14 um柱温程序:50℃(2 min)_15℃/min_220℃ _3.5℃/min_315℃(9 min) 进样口温度:300℃ 离子化方式:EI 色谱柱流量: 1.0mL/min 进样方式:不分流进样 进样量:1uL流量控制方式:线速度 接口温度:300℃离子源温度:250℃检测器电压:调谐电压+0.2kV采集方式: SIM,化合物信息见表1 1.3样品前处理 图1.样品前处理流程图 2.结果与讨论 2.1标准品溶液色谱图 图2.16种多环芳烃及7种内标标准品溶液色谱图(1.0ug/mL) 表1.多环芳烃组分信息 NO. 化合物名称 英文名称 CAS 保留时间 定量离子 定性离子 (min) (m/z) (m/z) 1 苯并[c]芴 Benzo[c]fluorene 205-12-9 17.995 216 215,213 2 D12-苯并[a]蒽 D12-Benz[a]anthracene 1718-53-2 21.460 240 236 3 苯并[a]蒽 Benz[alanthracene 56-55-3 21.580 228 226,229 4 D12-䓛 D12-Chrysene 1719-03-5 21.850 240 236 5 环戊并[c,d]芘 Cyclopenta[c,d]pyrene 27208-37-3 21.865 226 224,227 6 䓛 Chrysene 218-01-9 21.980 228 226,229 7 5-甲基䓛 5-Methylchrysene 3697-24-3 24.230 242 241,239 8 D12-苯并[b]荧蒽 Di2-Benzo[b]fluoranthene 93951-98-5 27.195 264 260 9 苯并[b]荧蒽 Benzo[b]fluoranthene 205-99-2 27.340 252 250,253 10 苯并[k]荧蒽 Benzo[k]fluoranthene 207-08-9 27.485 252 250,253 11 苯并[j]荧蒽 Benzo[j]fluoranthene 205-82-3 27.640 252 250,253 12 D12-苯并[a]芘 D12-Benzo[a]pyrene 63466-71-7 29.380 264 260 13 苯并[a]芘 Benzo[a]pyrene 50-32-8 29.515 252 250,253 14 D12-茚并[1,2,3-c,d]芘 D12-Indeno[1,2,3-c,dlpyrene 203578-33-0 35.025 288 284 15 D14-二苯并[a,h]蒽 D14-Dibenz[a,h]anthracene 13250-98-1 35.140 292 288 16 茚并[1,2,3-c,d]芘 Indeno[1,2,3-c,d]pyrene 193-39-5 35.170 276 274,277 17 二苯并[a,h]蒽 Dibenz[a,h]anthracene 53-70-3 35.310 278 276,279 18 D12-苯并[g,h,i]花 D12-Benzo[g,h,ilperylene 93951-66-7 36.620 288 284 19 苯并[g,h,i]花 Benzo[g,h,ilperylene 191-24- 36.750 276 274,277 20 二苯并[a,]芘 Dibenzo[a,lpyrene 191-30-0 42.250 302 300,303 21 二苯并[a,e]芘 Dibenzo[a,e]pyrene 192-65-4 44.005 302 300,303 22 二苯并[a,i]芘 Dibenzo[a,ilpyrene 189-55-9 45.105 302 303,300 23 二苯并[a,h]芘 Dibenzo[a,h]pyrene 189-64-0 45.750 302 303,300 2.2标准曲线 用丙酮-异辛烷溶液(1+1)当溶剂配制多环芳烃标准系列工作液,浓度分别为1.0、5.0、10、20、50、100 ng/mL, 内标为 100 ng/mL,临用现配。使用内标法拟合工作曲线,部分化合物标准曲线及质量色谱图如下图所示。根据 1.0 ng/mL标样数据,以3倍信噪比计算出各化合物仪器检出限,检出限以及线性相关系数如表2所示。 图3.部分化合物标准曲线 苯并[c]芴 5-甲基䓛 苯并[b]荧蒽 图4.部分化合物质量色谱图 (1.0ng/mL) 表2.16种多环芳烃标准曲线及仪器检出限结果 NO. 化合物名称 浓度范围 (ng/mL) 相关系数R 检出限(ng/mL) 苯并[c]芴 1.0-100 0.9999 0.02 2 苯并[a]蒽 1.0-100 0.9999 0.03 3 环戊并[c,d]芘 1.0-100 0.9999 0.04 4 䓛 1.0-100 0.9999 0.04 5 5-甲基䓛 1.0-100 0.9998 0.02 6 苯并[b]荧蒽 1.0-100 0.9999 0.05 苯并[k]荧蒽 1.0-100 0.9999 0.03 8 苯并[j]荧蒽 1.0-100 0.9999 0.03 9 苯并[a]芘 1.0-100 0.9999 0.03 10 茚并[1,2,3-c,d]芘 1.0-100 0.9999 0.04 11 二苯并[a,h]蒽 1.0-100 0.9997 0.11 12 苯并[g,h,i]花 1.0-100 0.9999 0.03 13 二苯并[a,l]芘 1.0-100 0.9999 0.05 14 二苯并[a,e]芘 1.0-100 0.9996 0.04 15 二苯并[a,i]芘 1.0-100 0.9995 0.08 16 二苯并[a,h]芘 1.0-100 0.9999 0.15 2.3重复性测试 取浓度为 5.0 ng/mL混合标准溶液,连续进样6次,考察保留时间和峰面积的重复性,结果如下表3所示。保留时间和峰面积的相对标准偏差(RSD%)分别在 0.01%-0.03%和1.5%-3.7%之间,精密度良好。 NO. 化合物名称 RSD ((%) (5.0 ng/mL) R.T. Area 苯并[c]芴 0.03 2 苯并[a]蒽 0.02 3 环戊并[c,d]芘 0.03 4 䓛 0.02 1.5 5 5-甲基菌 0.01 2.6 6 苯并[b]荧蒽 0.02 ( 苯并[k]荧蒽 0.02 8 苯并[j]荧蒽 0.01 9 苯并[a]芘 0.02 2.4 10 茚并[1,2,3-c,d]芘 0.03 3.3 11 二苯并[a,h]蒽 0.02 2.8 12 苯并[g,h,i]花 0.02 1.7 13 二苯并[a,I]芘 0.03 3.6 14 二苯并[a,e]芘 0.03 2.9 15 二苯并[a,i]芘 0.02 3.4 16 二苯并[a,h]芘 0.02 3.7 2.4回收率测试 按照1.3前处理方法,利用空白样品进行低、中、高三个浓度水平加标,考察回收率。每个浓度平行制备三份样品。低、中、高三个加标浓度分别为0.02、0.4和2.0 ug/kg。回收率结果见表4。 表4.三浓度水平加标回收率及重复性计算结果(n=3) 0.02 ug/kg 0.4 ug/kg 2.0 ug/kg NO. 化合物名称 平均回收率 RSD 平均回收率 RSD 平均回收率 RSD (%) (%) (%) (%) (%) (%) 1 苯并[c]芴 91.3 3.1 92.1 3.6 93.8 2.3 2 苯并[a]蒽 87.4 3.3 94.3 2.1 91.0 3.5 3 环戊并[c,d]芘 95.0 2.3 89.6 3.0 85.3 3.6 4 䓛 103.7 2.6 90.4 2.3 96.5 2.2 5 5-甲基䓛 97.9 2.3 94.4 2.7 90.2 2.8 6 苯并[b]荧蒽 92.5 3.0 86.9 2.0 93.9 3.6 7 苯并[k]荧蒽 88.3 4.1 84.2 3.9 91.1 2.2 8 苯并[j]荧蒽 86.9 3.5 87.1 3.2 85.7 3.6 9 苯并[a]芘 92.7 2.6 93.2 4.2 92.4 3.3 10 茚并[1,2,3-c,d]芘 90.2 1.8 90.1 3.5 87.3 2.0 11 二苯并[a,h]蒽 83.5 3.5 85.0 2.6 85.6 2.5 12 苯并[g,h,i]花 90.4 3.7 89.8 2.9 84.7 3.6 13 二苯并[a,l]芘 87.7 2.9 93.5 2.7 92.0 3.2 14 二苯并[a,e]芘 93.0 3.3 95.4 3.4 96.3 4.2 15 二苯并[a,i]芘 84.7 3.5 92.8 4.1 94.6 2.8 16 二苯并[a,h]芘 93.5 2.8 88.6 3.5 92.1 3.4 2.5样品测试 按照1.3中样品前处理方法,对某人造肉样品进行检测,未检测出相关化合物。实际样品的色谱图如下所示。 图5.人造肉样品色谱图 3.结论 本文使用岛津气质联用仪 GCMS-QP2020 NX建立了人造肉中16种多环芳烃的检测方法。实验结果表明:在1.0-100 ng/mL 浓度范围内,16种多环芳烃组分线性良好,线性相关系数均在 0.999以上,各化合物检出限在 0.02-0.15 ng/mL 之间。取浓度为 5.0 ng/mL标准混合溶液,连续进样6次,保留时间和峰面积的相对标准偏差(RSD%)分别在 0.01%-0.03%和1.5%-3.7%之间,精密度良好。在空白样品中进行0.02、0.4和2.0 ug/kg 三个不同浓度加标实验,回收率在 83.5%-103.7%之间,该方法灵敏,准确,稳定性好,回收率高,适用于人造肉中16种多环芳烃的检测。 GC-MS/MS法测定人造肉中16种多环芳烃含量 摘要: 本文使用岛津三重四极杆气质联用仪 GCMS-TQ8050 NX 建立了人造肉中16种多环芳烃的检测方法。实验结果表明:在1.0~100ng/mL 浓度范围内,16种多环芳烃组分线性良好,线性相关系数均在0.999以上,各化合物检出限在 0.01-0.09 ng/mL 之间。取浓度为 5.0 ng/mL标准混合溶液,连续进样6次,峰面积的相对标准偏差均小于3.6%,重复性良好。在空白样品中进行0.02、0.4和 2.0 ug/kg 三个不同浓度加标实验,回收率在83.3%-96.8%之间。该方法简单,稳定,准确,适用于人造肉中16种多环芳烃的测定。 关键词:三重四极杆气质联用仪 多环芳烃 人造肉 人造肉产品分为两大类,第一类是以植物蛋白为主要原料制备的植物基肉制品,第二类是通过动物细胞培养扩增制备的细胞培养肉。目前市场上主要以植物基肉制品为主,细胞培养肉还在研发期。植物基肉制品的植物原料主要包括豆类,谷物类,藻类,真菌类等。人造肉作为新兴的一种食品,其原料、生产流程、包装保藏等环节都面临污染的风险,其食品质量安全体系和标准一直是亟待明确和解决的。 多环芳烃 (PAHs) 是指含两个或两个以上苯环的芳烃,广泛分布于自然界中,其数量大、种类多,对人体危害极大,数种多环芳烃已被鉴定出具有较强的致癌、致畸和致突变作用,苯并[a]芘是其中毒性最大的一种强致癌物质。PAHs 主要对人体的呼吸系统、循环系统、神经系统、肝脏、肾脏等造成损害。GB2762-2017《食品安全国家标准食品中污染物限量》规定,谷物及其制品、肉及肉制品中苯并[a]芘的最高残留限量为 5 ug/kg。人造肉的生产环节复杂,容易受到多环芳烃的污染,因此建立和健全人造肉中多环芳烃的检测方法很有必要。 1.实验部分 1.1仪器 岛津三重四极杆气质联用仪 GCMS-TQ8050 NX 1.2分析条件 色谱柱: DB-EUPAH, 20 m×0.18mm×0.14 um 柱温程序:50℃(2min)_15℃/min_ 220℃_3.5℃/min_315℃(9 min) 进样口温度:300℃ 色谱柱流量: 1.0mL/min 进样方式:不分流进样 接口温度:300℃ 进样量:1uL 离子源温度:250℃ 流量控制方式:线速度 检测器电压:调谐电压+0.6kV 离子化方式:El 采集方式: MRM, 化合物信息见表1 1.3样品前处理 图1.样品前处理流程图 2.结果与讨论 2.1标准品溶液色谱图 图2.16种多环芳烃及7种内标标准品溶液色谱图(1.0 ug/mL) 表1.多环芳烃组分信息 NO. 化合物名称 英文名称 CAS 保留时间 定量离子 CE 定性离子1(m/Z) CE 定性离子2(m/Z) CE (min) (m/z) 1 苯并[c]芴 Benzo[c]fluorene 205-12-9 17.995 215.00>213.10 27 215.00>189.10 24 216.00>213.00 45 2 D12-苯并[a]蒽 D12-Benz[a]anthracene 1718-53-2 21.448 240.00>236.10 30 236.00>232.00 36 240.00>212.10 30 苯并[a]蒽 Benz[alanthracene 56-55-3 21.572 228.00>226.00 30 226.00>224.00 33 228.00>224.90 45 4 D12-䓛 D12-Chrysene 1719-03-5 21.825 240.00>236.00 36 236.00>232.00 36 240.00>212.10 33 5 环戊并[c,d]芘 Cyclopenta[c,d]pyrene 27208-37-3 21.847 226.00>223.90 42 224.00>221.80 33 227.00>225.00 36 6 䓛 Chrysene 218-01-9 21.969 228.00>226.00 33 226.00>224.10 33 229.00>227.10 33 7 5-甲基䓛 5-Methylchrysene 3697-24-3 24.213 241.00>239.10 27 242.00>239.00 39 239.00>236.90 33 8 D12-苯并[b]荧蒽 D12-Benzo[b]fluoranthene 93951-98-5 27.184 264.00>260.10 42 260.00>256.00 39 264.00>232.20 45 9 苯并[b]荧蒽 Benzo[b]fluoranthene 205-99-2 27.325 252.00>250.00 33 250.00>247.80 42 253.00>251.00 33 10 苯并[k]荧蒽 Benzo[k]fluoranthene 207-08-9 27.463 252.00>250.00 33 250.00>247.90 33 253.00>251.20 36 11 苯并[j]荧蒽 Benzo[j]fluoranthene 205-82-3 27.627 252.00>250.00 33 250.00>247.80 39 253.00>251.10 33 12 D12-苯并[a]芘 D12-Benzo[a]pyrene 63466-71-7 29.361 264.00>260.00 36 260.00>256.00 33 260.00>258.00 30 13 苯并[a]芘 Benzo[ajpyrene 50-32-8 29.505 252.00>250.00 33 250.00>248.00 30 253.00>251.10 30 14 D12-茚并[1,2,3-c,d]e D12-Indeno[1,2,3-c,d]pyrene 203578-33-0 35.005 288.00>284.10 42 284.00>280.10 42 288.00>285.80 39 15 D14二苯并[a,h]蒽 D14-Dibenz[a,h]anthracene 13250-98-1 35.118 292.00>288.00 39 288.00>283.90 39 292.00>284.00 45 16 茚并[1,2,3-c,d]芘 Indeno[1,2,3-c,d]pyrene 193-39-5 35.136 276.00>274.00 39 277.00>275.00 39 274.00>272.20 36 17 二苯并[a,h]蒽 Dibenza,hlanthracene 53-70-3 35.296 278.00>276.00 36 276.00>273.90 33 279.00>277.00 36 18 D12-苯并[g,h,i]花 D12-Benzo[g,h,ilperylene 93951-66-7 36.583 288.00>283.90 39 288.00>286.10 36 284.00>280.00 39 19 苯并[g,h,i]花 Benzo[g,h,ilperylene 191-24- 36.725 276.00>273.90 39 274.00>271.90 36 277.00>274.90 39 20 二苯并[a,l]芘 Dibenzo[a,]pyrene 191-30-0 42.243 302.00>299.90 33 300.00>297.90 33 303.00>301.00 33 21 二苯并[a,e]芘 Dibenzo[a,e]pyrene 192-65-4 43.974 302.00>299.90 39 300.00>298.00 33 303.00>301.00 39 22 二苯并[a,i]芘 Dibenzo[a,ilpyrene 189-55-9 45.072 302.00>299.90 39 300.00>297.60 45 303.00>300.90 36 23 二苯并[a,h]芘 Dibenzo[a,h]pyrene 189-64-0 45.724 302.00>300.00 39 303.00>301.10 36 300.00>298.10 36 2.2标准曲线 用丙酮-异辛烷溶液(1:1)为溶剂配制多环芳烃标准系列工作液,浓度分别为1.0、5.0、10、20、50 和 100ng/mL, 内标为 100 ng/mL。使用内标法拟合工作曲线,部分化合物标准曲线及质量色谱图如下图所示。根据1.0 ng/mL标样数据,以3倍信噪比计算出各化合物仪器检出限,检出限以及线性相关系数如表2所示。 图3.部分化合物标准曲线 Q215.00>213.10(+) 3.66e3 Q 228.00>226.00(+) 5.69e3 Q252.00>250.00(+) 4.40e3 RT=17.996 RT=21.572 RT=27.326RT=27.465 100.00- 100.00 100.00- % % % 0.00- 0.00- 一 一 0.00- 1 1 11 1 17.6 17.8 18.0 18.2 18.4 21.2 21.4 21.6 21.8 22.0 27.0 27.2 27.4 27.6 RT (min) RT (min) RT (min) 苯并[c]芴 苯并[a]蒽 苯并[b]荧蒽 图4.部分化合物质量色谱图 (1.0ng/mL) 表2.16种多环芳烃标准曲线线性、仪器检出限和重复性结果 NO. 化合物名称 相关系数R 检出限(ng/mL) 峰面积RSD% 1 苯并[c]芴 0.9998 0.01 1.7 2 苯并[a]蒽 0.9999 0.01 2.3 3 环戊并[c,d]芘 0.9999 0.02 1.8 4 䓛 0.9999 0.03 1.2 5 5-甲基䓛 0.9999 0.06 3.6 6 苯并[b]荧蒽 0.9999 0.02 1.7 7 苯并[k]荧蒽 0.9999 0.01 2.8 8 苯并[j]荧蒽 0.9999 0.01 2.1 9 苯并[a]芘 0.9999 0.02 1.9 10 茚并[1,2,3-c,d]芘 0.9999 0.02 2.5 11 二苯并[a,h]蒽 0.9999 0.05 1.8 12 苯并[g,h,i]花 0.9999 0.09 3.0 13 二苯并[a,I]芘 0.9999 0.02 2.3 14 二苯并[a,e]芘 0.9999 0.01 1.4 15 二苯并[a,i]芘 0.9998 0.03 2.2 16 二苯并[a,h]芘 0.9999 0.04 2.4 C 2.3重复性测试 取浓度为 5.0 ng/mL混合标准溶液,连续进样6次,考察重复性,结果如上表2所示。峰面积的相对标准偏差(RSD%)均小于3.6%,重复性良好。 2.4回收率测试 按照1.3前处理方法,利用空白样品进行低、中、高三个浓度水平加标,考察回收率。每个浓度平行制备三份样品。低、中、高三个加标浓度分别为0.02、0.4和2.0 ug/kg。 回收率结果见表3。 表3.三浓度水平加标回收率及重复性计算结果(n=3) 0.02 ug/kg 0.4 ug/kg 2.0 pg/kg No. 化合物名称 平均回收率 RSD 平均回收率 RSD 平均回收率 RSD (%) (%) (%) (%) (%) (%) 1 苯并[c]芴 89.2 4.2 85.4 3.2 92.9 2.6 2 苯并[a]蒽 83.3 3.8 89.3 2.4 86.2 3.1 3 环戊并[c,d]芘 88.9 2.9 92.7 2.5 90.6 2.6 4 96.4 2.1 89.4 2.1 89.5 3.2 5 5-甲基䓛 85.6 4.7 99.1 2.7 87.3 1.5 6 苯并[b]荧蒽 91.1 3.1 87.5 1.9 91.7 2.4 7 苯并[k]荧蒽 83.9 4.4 91.3 2.2 96.1 2.9 8 苯并[j]荧蒽 90.7 3.2 85.6 3.5 89.3 3.2 9 苯并[a]芘 89.9 3.2 91.2 2.9 90.6 2.8 10 茚并[1,2,3-c,d]芘 92.0 2.6 95.1 3.1 89.8 1.5 11 二苯并[a,h]蒽 86.1 3.1 89.9 2.9 88.6 2.2 12 苯并[g,h,i]花 94.2 2.0 92.6 2.6 93.4 3.0 13 二苯并[a,I]芘 85.5 1.8 87.4 3.8 93.7 2.1 14 二苯并[a,e]芘 87.4 3.5 93.3 3.6 87.1 3.7 15 二苯并[a,i]芘 83.8 2.7 96.8 94.6 2.3 16 二苯并[a,h]芘 94.6 2.4 90.9 32 90.5 2.6 2.5样品测试 按照1.3中样品前处理方法,对某人造肉样品进行检测,样品中未检测出相关化合物。样品色谱图如下所示。 C 图5.人造肉样品色谱图 3.结论 GCMS法测定人造肉中10种有机磷农药残留量 摘要:本文采用QuEChERS 前处理方法,利用岛津气相色谱质谱联用仪 GCMS-QP2020 NX, 建立了人造肉中10种有机磷农药残留量的测定方法。在 10~200 ug/L 范围内建立标准曲线,各组分线性关系良好,相关系数均大于 0.999。取浓度为20 ug/L的混合标准溶液,连续6次进样,10种有机磷组分峰面积 RSD 均小于 2.3%,精密度良好。10种有机磷农药组分的仪器检出限为 0.041~0.456ug/L。对人造肉样品进行 10 ug/kg 的加标实验,平均加标回收率为70.1~106.9%。结果表明,该方法简单快捷,准确可靠,能够准确分析人造肉中有机磷农药的残留量。 关键词:气相色谱质谱联用法 人造肉有机磷农药 人造肉通常可分为植物性肉和培养肉两种。植物性肉通常以豌豆、大豆以及其他豆类为原料,由植物蛋白制成,培养肉也称为细胞肉,是指在特定的条件下,利用动物干细胞在培养基中培育出来。目前能够在市场上购买到的人造肉食品基本都是植物性人造肉。随着消费者对食品健康、环保意识的不断提高,植物性肉食品越来越受到消费者的喜欢。 有机磷农药是常用的含磷元素的有机化合物农药,主要用于蔬菜、农作物的虫害处理。有机磷农药进入人体后会引起不同程度的中毒现象,严重的可出现心动过速、血压升高、呼吸困难、意识不清等现象。因此,非常有必要对人造肉中的有机磷农药残留量进行检测,以保证产品的安全。 本文采用 QuEChERS 前处理方法提取净化人造肉产品中10种有机磷类农药,利用岛津气相色谱质谱联用仪 GCMS-QP2020 NX, 结合选择离子扫描方式 (SIM),建立了人造肉中10种有机磷农药残留量的测定方法,该方法操作简单方便,能够准确分析人造肉食品中10种有机磷农药的残留量。 1.实验部分 ecx1.1仪器 E 气相色谱质谱联用仪: GCMS-QP2020 NX 1.2分析条件 色谱柱: SH-Rxi-5Sil MS, 30 m ×0.25mm × 0.25um 柱温程序:50℃(2min)_30 ℃/min_180℃(5 min) _20 ℃/min_230℃_30℃/min_300℃(5 min) 进样口温度:280℃ 离子化方式:EI 载气控制模式:恒线速度 离子源温度:230℃ 进样方式:不分流进样 色谱质谱接口温度:300℃ 线速度: 39.7 cm/sec 检测器电压:调谐电压+0.3kV 进样量:1uL 采集模式: SIM,离子信息见表1 2.样品前处理 C 图1.样品前处理流程图 3.结果与讨论 3.1标准溶液谱图 10种有机磷农药标准品色谱图如图2所示,各化合物信息见表1,部分化合物质量色谱图见图3。 图2.10种有机磷标准品色谱图((100 ug/L) 表1.10种有机磷信息表 NO. 化合物名称 英文名称 CAS号 保留时间 (min) 定量离子 (m/z) 定性离子 (m/z) 1 敌敌畏 Dichlorvos 62-73-7 6.346 109 185,145,220 2 二嗪磷 Diazinon 333-41-5 11.784 179 137,199,304 3 皮蝇磷 Fenchlorphos 299-84-3 13.426 285 125,109,270 4 杀螟硫磷 Fenitrithion 122-14-5 13.692 277 260,247,214 5 马拉硫磷 Malathion 121-75-5 13.853 173 127,158,285 6 毒死蜱 Chlorpyrifos 2921-88-2 13.996 197 314,258,286 7 倍硫磷 Fenthion 55-38-9 14.061 278 169,263,245 8 对硫磷 Parathion 56-38-2 14.121 291 109,261,235 9 乙硫磷 Ethion 563-12-2 15.804 231 153,125,384 10 蝇毒磷 Coumaphos 56-72-4 17.686 362 226,210,334 图3.部分有机磷组分质量色谱图(10 ug/L) 3.2标准曲线和检出限 分别称取不含目标物的空白样品5g,按1.3方法进行样品前处理,用基质提取液稀释标准溶液,目标组分浓度分别为10、20、50、100和200 ug/L, 以浓度为横坐标,峰面积为纵坐标做标准曲线,部分化合物标准曲线如图4所示。根据10 ug/L标准品数据,以3倍信噪比计算10种有机磷农药的仪器检出限,检出限见表2。 图4.部分有机磷组分标准曲线 表2.10种有机磷农药标准曲线相关系数及仪器检出限 NO. 化合物名称 相关系数(R) 检出限 (ug/L) 1 敌敌畏 0.9995 0.456 2 二嗪磷 0.9993 0.104 3 皮蝇磷 0.9990 0.056 4 杀螟硫磷 0.9993 0.067 5 马拉硫磷 0.9994 0.076 6 毒死蜱 0.9996 0.108 7 倍硫磷 0.9997 0.041 8 对硫磷 0.9991 0.082 9 乙硫磷 0.9990 0.047 10 蝇毒磷 0.9997 0.311 3.3重复性实验 取浓度为20 ug/L的混合标准溶液连续6次进样,考察仪器的重复性,测定结果见表3。 表3.重复性实验结果(n=6) NO. 化合物名称 峰面积 RSD(%) 1 2 3 4 5 6 1 敌敌畏 21101 20887 20965 21135 21530 21258 1.1 2 二嗪磷 8614 8687 8461 8727 8960 8573 2.0 3 皮蝇磷 15085 15184 14995 15309 15719 15234 1.7 4 杀螟硫磷 5742 5673 5702 5574 5922 5648 2.1 5 马拉硫磷 9095 8906 8689 8740 8825 9204 2.3 6 毒死蜱 4454 4466 4254 4281 4247 4333 2.3 7 倍硫磷 18421 18317 18027 17486 18592 18484 2.2 8 对硫磷 4483 4348 4293 4305 4483 4473 2.1 9 乙硫磷 14124 13989 14142 14037 14177 14532 1.4 10 蝇毒磷 5055 5208 4924 4963 5206 5174 2.5 3.4加标回收率 表4.样品加标回收结果 NO. 测定结果(ug/kg) 平均回收率 (%) 化合物名称 1 2 3 1 敌敌畏 11.9 12.0 8.13 106.9 2 二嗪磷 7.79 8.46 9.51 85.8 3 皮蝇磷 7.05 6.74 7.25 70.1 4 杀螟硫磷 7.72 8.28 9.48 84.9 5 马拉硫磷 8.14 9.75 13.3 104.1 毒死蜱 7.94 8.98 9.63 88.5 7 倍硫磷 6.38 6.99 8.06 71.4 8 对硫磷 8.40 8.71 9.76 89.6 9 乙硫磷 8.19 8.48 9.41 86.9 10 蝇毒磷 6.62 6.90 8.54 73.5 3.5样品测定 取某超市购买的人造肉样品,按照上述前处理步骤处理,得到样品色谱图,如图5,样品中未检测出10种有机磷农药残留。 图5.人造肉样品色谱图 4.结论 本文采用 QuEChERS前处理方法,提取净化,利用岛津 GCMS-QP2020 NX气相色谱质谱联用仪,建立了人造肉中10种有机磷农残的测定方法。在10~200 ug/L 范围内, 10种有机磷组分线性良好,相关系数均大于0.999。取浓度为 20 ug/L 的标准溶液连续6次进样,各组分峰面积RSD 均小于2.3%,仪器重复性良好。对空白人造肉样品进行10 ug/kg的加标实验,平均加标回收率为70.1~106.9%。该方法操作简单方便,准确可靠,能够用于人造肉样品中有机磷农药残留量的测定。 UZDAIMHExcellence in Science GCMS法测定人造肉中11种除草剂残留量 摘要: 本文使用岛津 GCMS-QP2020 NX气质联用仪,建立了人造肉中11种除草剂残留量的检测方法。样品粉碎后经溶剂提取、浓缩、SPE 净化后上机测试。实验结果表明:在0.05-10 ug/mL 浓度范围内,11种除草剂组分线性良好,线性相关系数均在 0.997以上,检出限在 0.18-1.14ng/mL 之间。取浓度为 0.05 ug/mL标准溶液,连续进样6次,峰面积相对标准偏差小于6%,重复性良好。在空白样品中进行0.01 和 0.1mg/kg 两个不同浓度加标实验,回收率在77.3%-119.0%之间。该方法灵敏度高,重复性好,适用于人造肉中11种除草剂残留量的测定。 关键词:气相色谱质谱联用仪 除草剂 人造肉 人造肉分为两种,一种以动物细胞为基础,在培养皿中复制培育而成;另一种是从植物中提取蛋白、经加工模拟真肉味道和质地的植物蛋白肉,通常由豆类、真菌类、谷物类等原料制成。目前国内的人造肉产品主要为后一种类型。作为一种新型食品,人造肉的食品质量和安全标准一直受到广泛关注。2020年12月,中国食品科学技术学会发布首个关于植物基肉制品的团体标准 《T/CIFST 001-2020植物基肉制品》,对植物基肉制品的原料要求、污染物限量、食品添加剂等做出了明确规定。 乙草胺、甲草胺、异丙甲草胺等为一类常见的酰胺类除草剂,具有除草活性高、应用范围广、价格低廉等优点,广泛用于大豆、玉米、花生、棉花等旱田农作物上。人造肉的生产环节复杂,其原料的培育过程中也可能存在多种除草剂污染物残留的危险,因此建立和健全人造肉中除草剂残留量的检测方法很有必要。 本文参考 GB 23200.24-2016《食品安全国家标准粮谷和大豆中11种除草剂残留量的测定气相色谱-质谱法》,使用岛津 GCMS-QP2020 NX气质联用仪,建立了人造肉中11种除草剂的检测方法。该方法灵敏度高,稳定性好,可供相关检测人员参考。 1.实验部分 1.1仪器 岛津气质联用仪 GCMS-QP2020 NX 1.2分析条件 色谱柱: SH-Rxi-5 Sil MS, 30m×0.25mm×0.25um 柱温程序:100℃(1 min)_15℃/min_ 260℃ (15 min) 进样口温度:260℃ 色谱柱流量: 1.0mL/min 进样方式:不分流进样 进样量:1uL 离子化方式:EI 接口温度:280℃ 离子源温度:200℃ 检测器电压:调谐电压+0.1kV 采集方式: SIM,化合物信息见表1 1.3样品前处理 ecen 图1.样品前处理流程图 2.结果与讨论 2.1标准品溶液色谱图 图2.11种除草剂标准溶液色谱图(10ug/mL) 表1.11种除草剂组分信息 保留时间 定量离子 定性离子 NO. 化合物名称 英文名称 CAS NO. (min) (m/z) (m/z) 1 乙草胺 Acetochlor 34256-82-1 9.79 223 146,162,269 2 甲草胺 Alachlor 15972-60-8 9.91 160 188,237,269 3 戊草丹 Esprocarb 85785-20-2 10.35 222 162,151,265 4 异丙甲草胺 Metolachlor 51218-45-2 10.43 162 238,240,146 5 二甲戊灵 Pendimethalin 40487-42-1 10.90 252 162,253,281 6 丁草胺 Butachlor 23184-66-9 11.40 176 311,237,160 7 氟酰胺 Flutolanil 66332-96-5 11.59 173 145,281,323 8 丙草胺 Pretilachlor 51218-49-6 11.68 262 162,176,238 9 灭锈胺 Mepronil 55814-41-0 12.58 119 91,269,120 10 吡氟酰草胺 Diflufenican 83164-33-4 13.25 266 394,267,245 11 苯噻酰草胺 Mefenacet 73250-68-7 15.18 192 136,148,298 2.2标准曲线 使用正己烷为溶剂,配制浓度分别为0.05、0.1、0.5、1、5、10 ug/mL的混合标准工作溶液。以各化合物的质量浓度为横坐标、峰面积为纵坐标绘制外标法标准曲线,各化合物标准曲线如图3所示。根据0.05 ug/mL标准溶液数据,以3倍信噪比(S/N)计算各目标物的仪器检出限,检出限以及线性相关系数如表2所示。 图3.部分化合物标准曲线 图4.部分化合物质量色谱图 (0.05 ug/mL) 表2.11种除草剂标准曲线相关系数、仪器检出限 (ng/mL)和重复性结果 NO. 化合物名称 相关系数R 仪器检出限 峰面积RSD% 1 乙草胺 0.9991 0.53 4.8 2 甲草胺 0.9991 0.39 5.0 3 戊草丹 0.9994 0.21 5.2 4 异丙甲草胺 0.998 0.39 5.4 5 二甲戊灵 0.998 0.40 5.4 6 丁草胺 0.998 0.36 5.5 7 氟酰胺 0.998 0.18 5.4 8 丙草胺 0.9993 0.75 5.9 9 灭锈胺 0.997 0.19 5.8 10 吡氟酰草胺 0.998 0.53 5.7 11 苯噻酰草胺 0.997 1.14 4.9 enc 2.3重复性测试 取浓度为 0.05 ug/mL混合标准溶液,连续进样6次,考察峰面积重复性,结果如上表2所示。峰面积的相对标准偏差(RSD%)小于6%,重复性良好。 2.4回收率测试 按照1.3所述样品前处理方法,利用空白样品进行 0.01和0.1 mg/kg 两个浓度水平的样品加标,考察回收率,每个浓度平行制备三份样品。回收率结果见表3。 表3.不同浓度水平加标回收率计算结果 加标浓度 0.01 mg/kg NO 加标浓度 0.1 mg/kg 化合物名称 回收率1# 2# 3#(%) 回收率1# 2# 3# (%) 1 乙草胺 0.009 0.010 0.009 91.7 0.095 0.105 0.118 106.0 2 甲草胺 0.008 0.010 0.009 91.3 3 戊草丹 0.009 0.010 0.009 93.0 0.091 5 二甲戊灵 0.012 0.012 0.013 119.0 0.100 0.113 0.124 112.3 6 丁草胺 0.010 0.012 0.011 106.7 0.096 0.111 0.123 110.0 ( 氟酰胺 0.007 0.008 0.008 77.3 0.074 0.075 0.094 81.0 8 丙草胺 0.009 0.011 0.010 98.3 0.092 0.105 0.112 103.0 9 灭锈胺 0.011 0.013 0.012 118.0 0.102 0.117 0.123 114.0 10 吡氟酰草胺 0.010 0.012 0.011 106.3 0.096 0.112 0.117 108.3 11 苯噻酰草胺 0.008 0.011 0.009 92.3 0.079 0.074 0.090 81.0 2.5样品测试 按照1.3所述样品前处理方法,对某人造肉样品进行检测,未检测出相关化合物。实际样品色谱图如下所示。 图5.人造肉样品色谱图 3.结论 ICPMS-2030系列测定人造肉中的重金属元素 摘要:参考 GB 5009.268-2016 《食品安全国家标准食品中多元素的测定》,加入硝酸和过氧化氢对人造肉进行微波消解,使用电感耦合等离子体质谱仪(ICP-MS)测定了人造肉中砷、汞、镉、铅等16种重金属元素的含量。分析结果表明,方法检出限低,准确度好,加标回收率92.8%~112%,适合人造肉中高低含量重金属元素的同时检测。 关键词:微波消解 ICP-MS人造肉重金属元素 近年来,随着全球肉类短缺危机、健康与环保消费风潮、食品创新等因素推动, “人造肉”概念迅速兴起,据报道,全球肉类替代品市场正以每年68%的复合增长率增长,其中人造肉深受欢迎。人造肉通常可以分为植物性肉、培养肉和其它肉类替代品。人造肉同样可以提供养殖肉中所富含的蛋白质、脂肪等营养元素,也可以控制其成分,满足不同群体的需求。目前涉及人造肉的检测内容分四个部分:组学分析、营养成分分析、有毒成分分析、包装材料。其中有毒成分分析可细分为食品添加剂检测、重金属元素(如As、Hg、Cd、Pb等)检测等。 我国对食品安全有着严格的监控, GB 2762-2017《食品安全国家标准食品中污染物限量》明确规定了食品中重金属的限量要求,而相对应的, GB5009.268-2016《食品安全国家标准食品中多元素的测定》明确规定了食品中重金属检测方法。第一法即为电感耦合等离子体质谱法。 电感耦合等离子体质谱法(ICP-MS)具有灵敏度高、检出限低、线性范围广等特点,本文参考标准GB5009.268-2016,对人造肉样品进行微波消解前处理,使用岛津ICPMS-2030系列测定了人造肉中砷、汞、镉、铅等重金属元素的含量。 1.实验部分 十 1.1仪器 岛津ICPMS-2030系列电感耦合等离子体质谱仪。 1.2仪器分析条件 ICP-MS仪器分析条件见表1。 表 1ICP-MS分析条件 参 数 参数设定 参 数 参数设定 高频功率 1.20kW 等离子体气流速 9.0 L/min 辅助气流速 1.10 L/min 载气流速 0.70 L/min 炬管类型 Mini炸管 雾化器 同心雾化器 雾化室 旋流 雾化室温度 5℃ 采样深度 5.0mm 高频频率 27.12MHz 碰撞气体 He 碰撞气流速 6 mL/min 池电压 -21V 能量过滤器电压 7.0V 2.样品前处理 准确称取0.2g样品于微波消解罐中,加入6mLHNO3和2mLH202, 置于微波消解仪中消解,升温程序见表2。消解后,冷却,转移至50mL离心管中,加入1000 mg/L金溶液100uL, 定容至50mL,混匀备用,同时做空白和加标试验。 表2微波消解程序 步骤 控制温度(℃) 升温时间(min) 恒温时间(min) 1 120 5 5 2 150 5 10 3 190 5 30 3.结果与讨论 3.1标准曲线和检出限 使用2% HNO3将多元素溶液稀释为0、0.20、0.50、1.0、2.0、5.0、110、20、50、100ug/L的标准序列,根据样品中元素实际情况调整标准曲线线性范围;Hg标准序列为0、0.20、0.50、1.0、2.0 ug/L (含2mg/L金溶液)。标准曲线见图1~图4,连续测量空白溶液计算仪器检出限(IDL) 和方法检出限(MDL),IDL和MDL结果见表3。 浓度=0.1095736*|+1.2330e-0040.99999 BEC= --(ug/L) 3a=0.0038816 (ug/L) 浓度=0.7468183*1-0.0037017 厂三 0.99999BEC= 0.0037017 (ug/L)3o=0.0021394 (ug/L) 图1Ag元素标准曲线 图2Cd元素标准曲线 浓度=0.0233929*1-0.11000970.99999BEC= 0.1100097 (ug/L) 3a=0.0090020 (ug/L) 浓度=0.2601963*1-0.1385749厂吕0.99999BEC= 0.1385749 (ug/L)3a=0.0187697 (ug/L) 图3Cr元素标准曲线 图4Pb元素标准曲线 表3仪器检出限和方法检出限 元素 质量数 相关系数 IDL(ug/L) MDL(ug/kg) GB5009.268-2016 定量限要求 (ug/kg) Ag 109 0.99999 0.003 0.75 一一 As 75 0.99986 0.013 3.25 5 Ba 138 0.99941 0.040 10 50 Be 9 0.99997 0.002 0.5 一一 Cd 111 0.99999 0.002 0.5 5 Co 59 0.99988 0.002 0.5 3 Cr 52 0.99999 0.009 2.25 200 Cu 65 0.99973 0.58 145 200 Hg 202 0.99941 0.004 1 3 Mn 55 0.99995 0.031 7.75 300 Ni 62 0.99923 0.25 62.5 500 Pb 206 0.99999 0.018 4.5 50 Sr 88 0.99995 0.008 2 500 TI 205 0.99999 0.0004 0.1 0.3 U 238 0.99999 0.0001 0.025 一- V 51 0.99966 0.004 1 5 3.2样品测试结果 样品按照前处理程序处理后,使用内标法,在线内标测定,测定结果见表4,样品加标回收率为92.8%~112%。 表4人造肉中重金属元素测定结果 测定值 RSD% 加标量 回收率 元素 质量数 内标 (ug/L) (n=3) 结果 (ug/kg) (ug/L) (%) Ag 109 103Rh 0.003 5.73 7.50 0.5 92.8 As 75 72Ge N.D. 一一 一一 0.5 98.5 Ba 138 103Rh 1.62 3.12 4.05×103 2.0 105 Be 9 45Sc N.D. 一一 一一 0.5 110 Cd 111 103Rh 0.01 1.01 25.0 0.5 112 Co 59 72Ge 0.08 2.09 200 0.5 98.6 Cr 52 45Sc 0.03 3.91 75.0 0.5 107 Cu 65 72Ge 4.57 1.62 1.14×104 2.0 101 202 187Re N.D. 0.2 98.5 55 45SC 10.9 1.61 2.72×104 20 96.0 Ni 62 45SC 2.25 1.89 5.62×103 2.0 105 Pb 206 185Re N.D. 0.5 104 Sr 88 72Ge 2.73 0.40 6.82×103 2.0 104 丁I 205 185Re N.D. 0.5 103 U 238 185Re N.D. 112 V 51 72Ge N.D. 108 备注:按照GB5009.268-2016,计算结果保留3位有效数字。 4.结论 参考GB5009.268-2016《食品安全国家标准食品中多元素的测定》,使用岛津ICPMS-2030系列电感耦合等离子体质谱仪测定了人造肉中16种重金属元素含量。实验结果表明,该方法检出限低,准确度好,高低含量元素可以同时测量,加标回收率在92.8%~112%范围内,适合人造肉中砷、汞、镉、铅等重金属元素的同时检测。 in SciencecelenExcel 4、真菌毒素 SHIMADZU Excellence in Science 高效液相色谱-串联质谱法测定人造肉中赭曲霉毒素A 摘要:本文建立了使用岛津三重四极杆液质联用系统测定人造肉中赭曲霉毒素A含量的方法。赭曲霉毒素A在优化后的色谱及质谱条件下,采用负离子模式进行电离,通过多反应监测(MRM)模式进行测定。结果表明:使用外标法定量,赭曲霉毒素A在1ng/mL~50ng/mL浓度范围内峰面积与其质量浓度线性关系良好,所得校准曲线线性相关系数均在0.999 以上,各校准点准确度在 96.1%~102.7%之间,且精密度和回收率实验结果良好。 关键词:三重四极杆质谱 人造肉 赭曲霉毒素A 人造肉是近几年在欧美国家提出的一种肉类替代品方案,主要包括人工培养肉和植物性肉。目前市场上主要以植物性肉为主,其原料不局限于大豆蛋白,还会用豌豆、燕麦等粮食作物原料,制作上经过拉丝等技术将植物蛋白分子结构重构成肉类的纤维状结构,再通过交联酶、风味蛋白酶改进,使其口感质地及风味接近真肉。赭曲霉毒素 (Ochratoxins) 是一种有毒真菌代谢的产品,其中毒性最大、与人类健康关系最密切、对粮食作物污染最普遍的是赭曲霉毒素A (Ochratoxin A, OTA)。研究表明该种毒素可以损害动物的肾脏和肝脏,有致畸和致癌作用。植物性人造肉的质量安全涉及到原料、生产加工、储存流通及烹饪加工等各方面,如果受到赭曲霉毒素A的污染,必然会给消费者的健康带来风险。因此,建立快速、准确、高灵敏度检测人造肉中赭曲霉毒素A的分析方法意义重大。 目前,赭曲霉毒素A的检测方法主要采用的有薄层色谱法、酶联免疫吸附法以及液相色谱法等。高效液相色谱-串联质谱(LC-MS/MS)法由于具有检测灵敏度高、结果选择性好的特点,已成为粮食类复杂基质中痕量物质检测的主流方法。本文使用岛津超高效液相色谱仪和三重四极杆质谱联用系统,参照《GB5009.96-2016食品中赭曲霉毒素A的测定》中的方法,建立了一种快速准确测定人造肉中赭曲霉毒素A的方法。 1.实验部分 1.1仪器 岛津LCMS-8045 三重四极杆液质谱联用系统。具体配置为: 系统控制器: CBM-20A 脱气机: DGU-20A5R 输液泵 : LC-30AD×2 自动进样器: SIL-30AC柱温箱: CTO-20AC 检测器: LCMS-8045 色谱工作站: LabSolutions Ver. 5.99 1.2分析条件 液相条件 色谱柱 : Shim-pack GIST C18 (100 mmx2.1 mml.D.,2 um, Shimadzu SGLCP/N:227-30001-04) 流动相 : A相-5mmol/L甲酸铵0.1%甲酸水溶液 B相-乙腈 流 速 :0.5mL/min 进样体积 :3pL 柱 温 :35°℃ 洗脱方式 :梯度洗脱,B相初始浓度为50%,时间程序见表1。 表1.梯度洗脱程序 Time Module Command Value 1.50 Pumps Pump B Conc. 50 4.00 Pumps Pump B Conc. 90 5.00 Pumps Pump B Conc. 90 5.10 Pumps Pump B Conc. 50 8.00 Controller Stop e2nce 质谱条件 离子化模式:: ESI,负离子模式接口温度度:300°℃接口电压:-3.5kV加 热 气:空气10L/min雾化气流速:氮气 3.0L/min干燥气流速:氮气 10L/min加热模块温度:400°℃碰 撞 气:氩气 270kPa脱溶剂管温度:200°℃扫描模式:多反应监测(MRM)MRM 参数 :见表2驻留时间:60 ms十 表 2.MRM 参数 化合物 英文名称 CAS NO. 监测离子对 Q1 pre CE Q3 Pre (V) (V) 赭曲霉毒素A Ochratoxins A 303-47-9 402.00>358.00* 20 21 36 (OTA) 402.00>167.00 20 37 26 注:*表示定量离子对 1.3样品前处理方法 参照食品安全国家标准《GB5009.96-2016食品中赭曲霉毒素A的测定》第三法中粮食产品试样提取和净化方法。 1.4基质校准曲线的制备 取适量空白试样,按1.3样品前处理方法操作后,得到空白样品提取溶液。移取一定量 的赭曲霉毒素A标准储备液,用空白样品提取液稀释,分别配成相当于1ng/mL、5ng/mL、10 ng/mL、20 ng/mL、50 ng/mL 的基质标准工作溶液,供液相色谱-串联质谱仪测定。以定量特征离子质量色谱峰面积为纵坐标,基质匹配标准工作溶液浓度为横坐标,绘制校准曲线。 2.结果与讨论 2.1标准样品的MRM色谱图 图1.赭曲霉毒素A(5ng/mL)的 MRM图谱 ecneic2.2线性范围与检出限 S 将不同浓度的赭曲霉毒素A基质匹配标准工作液,按照1.2中的分析条件进行测定,使用外标法定量。以浓度为横坐标,峰面积为纵坐标,绘制校准曲线如图2所示。所得校准曲线线性关系良好,线性方程及相关系数见表3。 图2.赭曲霉毒素A的校准曲线 表3.标准曲线与检出限信息 化合物名称 校准曲线 r 线性范围 (ng/mL) 准确度(%) 检出限 (ng/mL) 赭曲霉毒素A Y=(3206.27)X+(44.7390) 0.9998 1~50 96.1~102.7 0.3 2.3精密度实验 对不同浓度的赭曲霉毒素A标准工作液连续测定6次,考察仪器的精密度,保留时间和峰面积的重复性结果如表4所示。结果显示:不同浓度样品溶液中赭曲霉毒素A的保留时间和峰面积相对标准偏差分别在 0.09%~0.31%和1.91%~5.14%之间,显示仪器精密度良好。 表4.保留时间(R.T.)和峰面积(Aera)重复性结果(n=6) 名称 Conc.(ng/mL) RSD% (R.T.) RSD%(Area) 赭曲霉毒素A 5 0.19 3.29 10 0.12 1.25 20 0.11 0.88 2.4加标回收率实验 称取25g空白试样,加入少量赭曲霉毒素A的标准储备溶液,使得赭曲霉毒素A的加标浓度分别为 10 ug/kg、20 ug/kg 和 40 ug/kg。加标样品经过1.3样品前处理操作后,测得赭曲霉毒素A的加标回收率在87.1%~91.6%之间,加标回收率结果见表5。 表5.赭曲霉毒素A的加标回收率结果(n=3) 化合物 加标水平(ug/kg) 平均回收率(%) 赭曲霉毒素A 10 91.6 20 87.1 40 89.5 3.结论 本文建立了一种使用岛津超高效液相色谱仪 LC-30A 和三重四极杆质谱仪 LCMS-8045联用测定人造肉中赭曲霉毒素A含量的方法。使用外标法定量,赭曲霉毒素A在1ng/mL~50ng/mL 浓度范围内峰面积与其质量浓度线性关系良好,所得校准曲线线性相关系数均在0.999以上,各校准点准确度在 96.1%~102.7%之间,且精密度和回收率实验结果良好。方法学结果表明,本方法操作简便,且准确度高,可用于人造肉中赭曲霉毒素A含量的快速测定。 LC-MS/MS测定人造肉中B族和G族黄曲霉毒素 摘要:本文建立了一种使用岛津超高效液相色谱-三重四极杆质谱联用仪测定人造肉中B族和G族黄曲霉毒素的方法,该方法可在 7 min 内完成对目标物的检测。黄曲霉毒素B1、B2、G1、G2在 0.1ng/mL~10.0 ng/mL 浓度范围内线性良好,校准曲线线性相关系数r在0.99以上,且精密度和回收率实验结果均符合标准要求。该方法灵敏度高,分析时间短,结果准确,可用于人造肉中黄曲霉毒素B1、B2、G1、G2的快速检测。 关键词:三重四极杆质谱 人造肉黄曲霉毒素 人造肉是近些年来食品行业的一项研究热点。人造肉按原料来源主要分为两种:一种是以植物蛋白为原材料对肉类形色和味道进行模仿的植物肉制品;另一种是在培养基中利用动物干细胞进行一定条件培养,从而制造的人造肉。大豆蛋白是植物源人造肉最常用的材料,豆制品可以给人体补充大量蛋白质,满足人体营养需求。 黄曲霉毒素是黄曲霉菌和寄生曲霉菌等产毒菌株产生的次生代谢产物,是一种强毒性物质。其衍生物有约20种,分别命名为 B1、B2、G1、Gz等。其中Bi的毒性最大,致癌性最强,黄曲霉毒素是一种剧毒的致肝癌物质。 植物性人造肉如果受到黄曲霉毒素的污染,必然会给消费者的健康带来风险。因此,建立快速、准确、高灵敏度检测人造肉中黄曲霉毒素的分析方法意义重大。 目前,黄曲霉毒素的检测方法主要采用的有薄层色谱法、酶联免疫吸附法、液相色谱法等以及高效液相色谱-串联质谱(LC-MS/MS)法。本文使用岛津超高效液相色谱仪和三重四极杆质谱联用系统,参照《GB5009.22-2016 食品安全国家标准食品中黄曲霉毒素B族和G族的测定》中的方法,建立了一种快速准确测定人造肉中黄曲霉毒素B1、B2、G1、G2的方法。 1.实验部分 1.1仪器 本实验使用超高效液相色谱仪 LC-40B X3 与三重四极杆质谱仪 LCMS-8045 联用系统。具体配置为: 系统控制器 :CBM-40 自动进样器 :SIL-40CX3 输 液 泵 :LC-40BX3×2 质 谱 仪 :LCMS-8045 柱 温 箱 :CTO-40S 色谱工作站 : LabSolutions Ver. 5.99 SP2 1.2分析条件 液相条件 色谱柱 : Shim-pack GISS C18 (100mm×2.1 mml.D.,1.9 um)岛津(上海)实验器材有限公司,P/N:227-30048-02 流动相 :A相 5mmol/L 乙酸铵溶液;B相乙腈-甲醇(V=1:1) 洗脱方式 :梯度,初始32%B, 洗脱程序见表1 柱 温 :40°℃ 流 速 :0.30mL/min进样体积 :2.0uL 表1.梯度洗脱程序 Time Module Command Value 1.00 Pumps Pump B Conc. 32 2.00 Pumps Pump B Conc. 45 3.00 Pumps Pump B Conc. 45 3.50 Pumps Pump B Conc. 80 4.00 Pumps Pump B Conc. 80 4.10 Pumps Pump B Conc. 32 7.00 Controller Stop 质谱条件 C 离子源:ESI(+)DL管温度:150°℃接口电压:4.0kV加热模块温度:400°C雾化气:氮气 3.0L/min接口温度:300°℃干燥气:氮气 10L/min扫描模式:多反应监测(MRM)加热气:空气 10L/minMRM 参数:见表2碰撞气:氩气(230kPa)注留时间:20ms 表2.MRM 参数 # 化合物名称 监测离子对 Q1 pre (V) CE Q3 Pre (V) 1 黄曲霉毒素Bi 313.00>241.00* -22.0 -38.0 -26.0 313.00>285.00 -22.0 -24.0 -30.0 2 黄曲霉毒素B2 315.00>259.00* -12.0 -30.0 -28.0 315.00>287.00 -12.0 -27.0 -30.0 3 黄曲霉毒素 G1 329.00>243.00* -12.0 -29.0 -27.0 329.00>283.00 -12.0 -25.0 -21.0 4 黄曲霉毒素 G2 331.00>245.00* -12.0 -31.0 -26.0 331.00>257.00 -12.0 -31.0 -30.0 注:*表示定量离子对 1.3标准溶液的配制 取市售浓度为 10 ug/mL 黄曲霉毒素 B1、B2、G1、G2混合标准溶液适量,用初始流动相稀释至刻度,制成浓度为 0.1 ng/mL、0.5 ng/mL、1 ng/mL、2 ng/mL、5 ng/mL、10ng/mL 混合标准工作液。 1.4样品前处理方法 参照《GB5009.22-2016 食品安全国家标准食品中黄曲霉毒素B族和G族的测定》中的方法,样品照如下流程处理。 图1.样品前处理流程 2.结果与讨论 ecne2.1标准样品的MRM色谱图 i cS 图2.标准品 MRM 色谱图 (浓度0.1ng/mL) 0.1 ng/mL 黄曲霉毒素 B1、B2、G1、G2标准溶液响应如图2所示,四种物质的 S/N 分别为197.26、120.80、108.62、47.20,灵明度良好,且空白样品在色谱峰出峰处无干扰。 2.2线性范围与检出限 精密量取 10 ug/mL混合标准溶液适量,用初始流动相溶液溶解,配制成浓度为0.1ng/mL、0.5 ng/mL、1 ng/mL、2 ng/mL、5 ng/mL、10 ng/mL 系列标准溶液。以待测物峰面积为纵坐标,浓度为横坐标,外标法绘制校准曲线,所得校准曲线线性关系良好,如图3所示。根据 MDL=3.3 S/N 计算检出限,线性方程及相关系数见表3。 黄曲霉毒素 G2 图3.校准曲线 表3.校准曲线与检出限信息 # 化合物 R 线性范围(ng/mL) 检出限(ug/kg) 准确度(%) 1 黄曲霉毒素Bi 0.9986 0.1~10.0 0.000669 80.1-112.5 2 黄曲霉毒素B2 0.9991 0.1~10.0 0.00109 89.2-109.8 3 黄曲霉毒素 G1 0.9988 0.1~10.0 0.00121 81.9-110.9 4 黄曲霉毒素 G2 0.9991 0.1~10.0 0.00279 84.4-108.1 2.3精密度 对浓度为 0.5 ng/mL的黄曲霉毒素B1、B2、G1、G2标准工作液连续测定6次,考察仪器的精密度,保留时间和峰面积的重复性结果如表4所示。结果显示:黄曲霉毒素B1、B2、G1、G2保留时间RSD<0.2%, 峰面积的 RSD<5.0%,仪器精密度良好。 2.4回收率 称取空白人造肉样品,加入黄曲霉毒素 B1、B2、G1、Gz标准溶液,使加标浓度为 0.20 ug/kg。按照1.4样品前处理方法提取净化后,测定黄曲霉毒素B1、B2、G1、G2的加标回收率。空白人造肉样品和加标人造肉样品 MRM 色谱图如图4所示,加标回收率结果见表4。 图4.空白人造肉样品和加标浓度为 0.20 ug/kg 人造肉样品 MRM 色谱图 表4.重复性和回收率结果* # 中文名称 保留时间 RSD(%) 峰面积 RSD (%) 平均回收率 回收率 RSD(%) % 1 黄曲霉毒素B1 0.11 2.68 90.6 2.84 2 黄曲霉毒素B2 0.08 3.82 85.4 4.02 3 黄曲霉毒素G1 0.06 4.53 85.0 4.82 4 黄曲霉毒素 G2 0.12 4.85 84.2 4.96 *重复性 n=6, 回收率n=3 3.结论 本文建立了一种使用岛津超高效液相色谱-三重四极杆质谱联用仪测定人造肉中黄曲霉毒素B族和G族的方法,该方法可在7 min内完成对目标物的检测。黄曲霉毒素B1、B2、G1、G2在0.1ng/mL~10.0ng/mL浓度范围内线性良好,校准曲线线性相关系数r在0.99以上,黄曲霉毒素B1、B2、G1、G2保留时间RSD<0.2%,峰面积的RSD<5.0%,精密度和回收率实验结果均符合标准要求。该方法灵敏度高,分析时间短,结果准确,可用于人造肉中黄曲霉毒素B1、B2、G1、G2的快速检测。 cCMin SIence SHExcell JQA-0376本公司三条工厂获得 ISO 认证 田岛津企业管理(中国)有限公司/岛津(香港)有限公司 http://www.shimadzu.com.cn 岛津企业管理(中国)有限公司
确定







































还剩80页未读,是否继续阅读?

产品配置单

岛津企业管理(中国)有限公司为您提供《人造肉检测整体解决方案》,该方案主要用于豆制品中真菌毒素检测,参考标准--,《人造肉检测整体解决方案》用到的仪器有岛津三重四极杆型气相色谱质谱联用仪GCMS-TQ8050 NX
推荐专场






该厂商其他方案
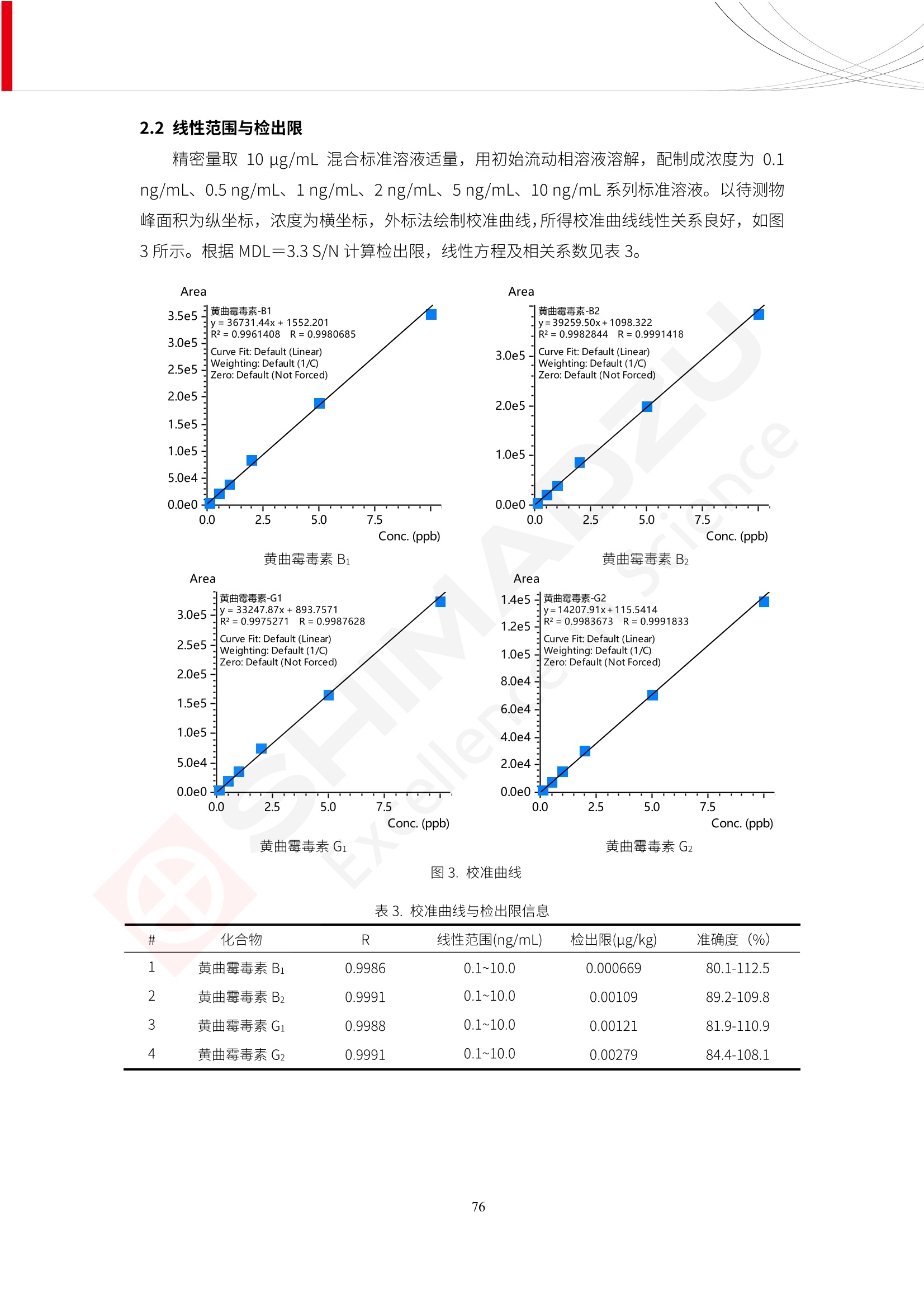
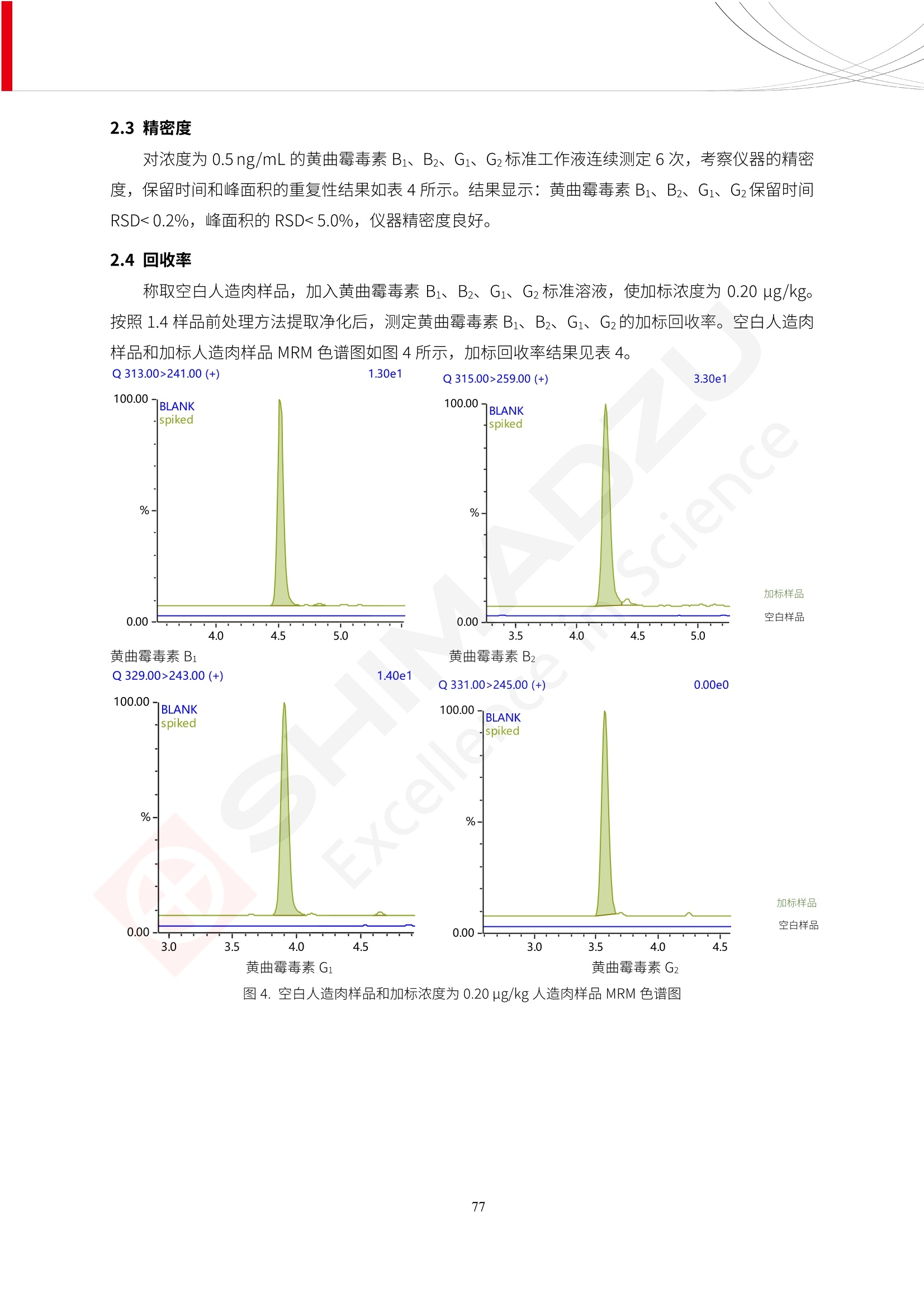
更多