








采用立陶宛EKSPLA公司研制的NL301HT型纳秒调Q Nd:YAG激光器输出的355nm激光进行选择性紫外光致解离,配合质谱仪,进行了气相交联测量,和波恩-奥本海默分子动力学计算。探测了非共价肽肽离子复合物中精氨酸-磷酸肽相互作用
方案详情

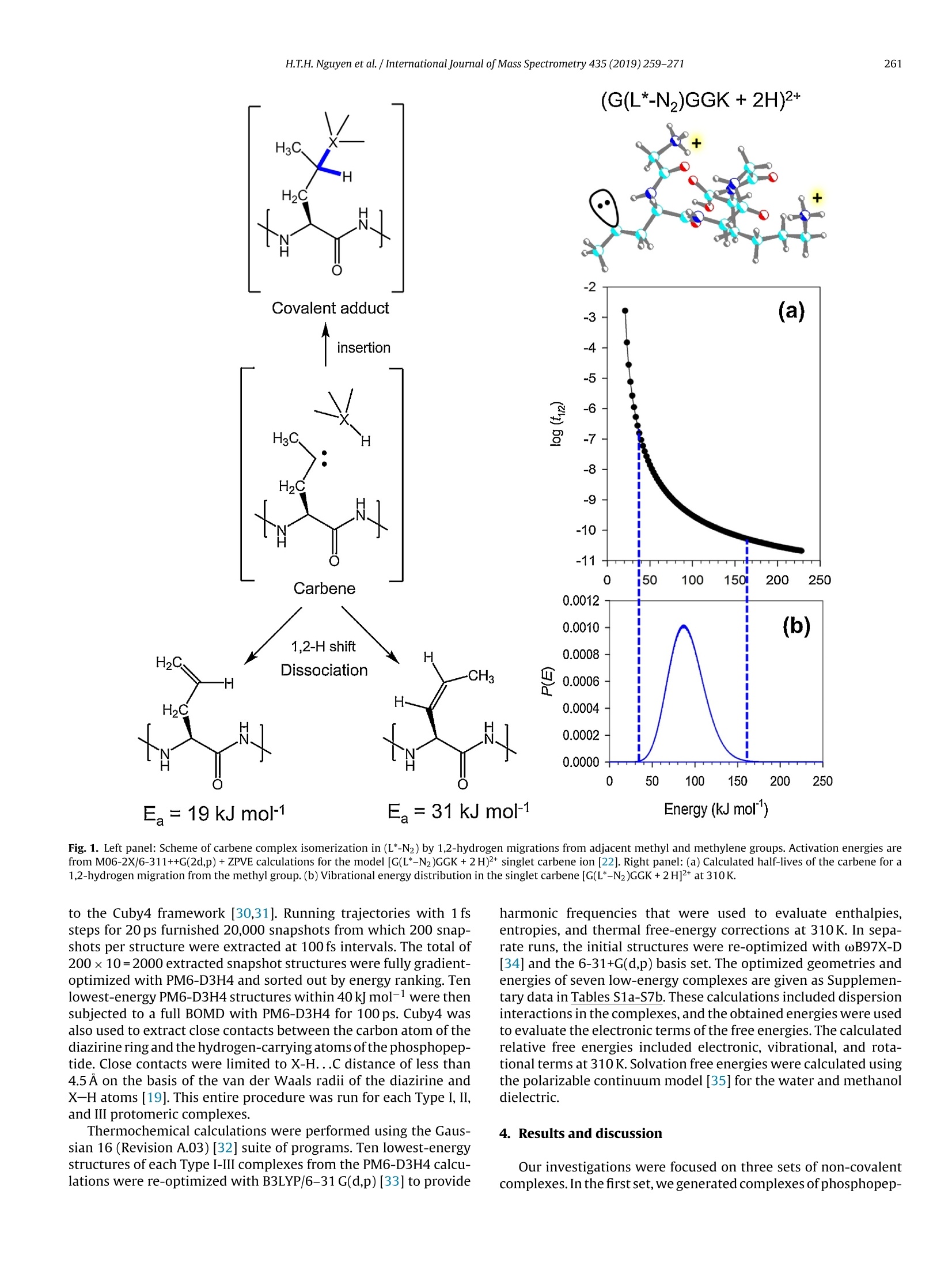
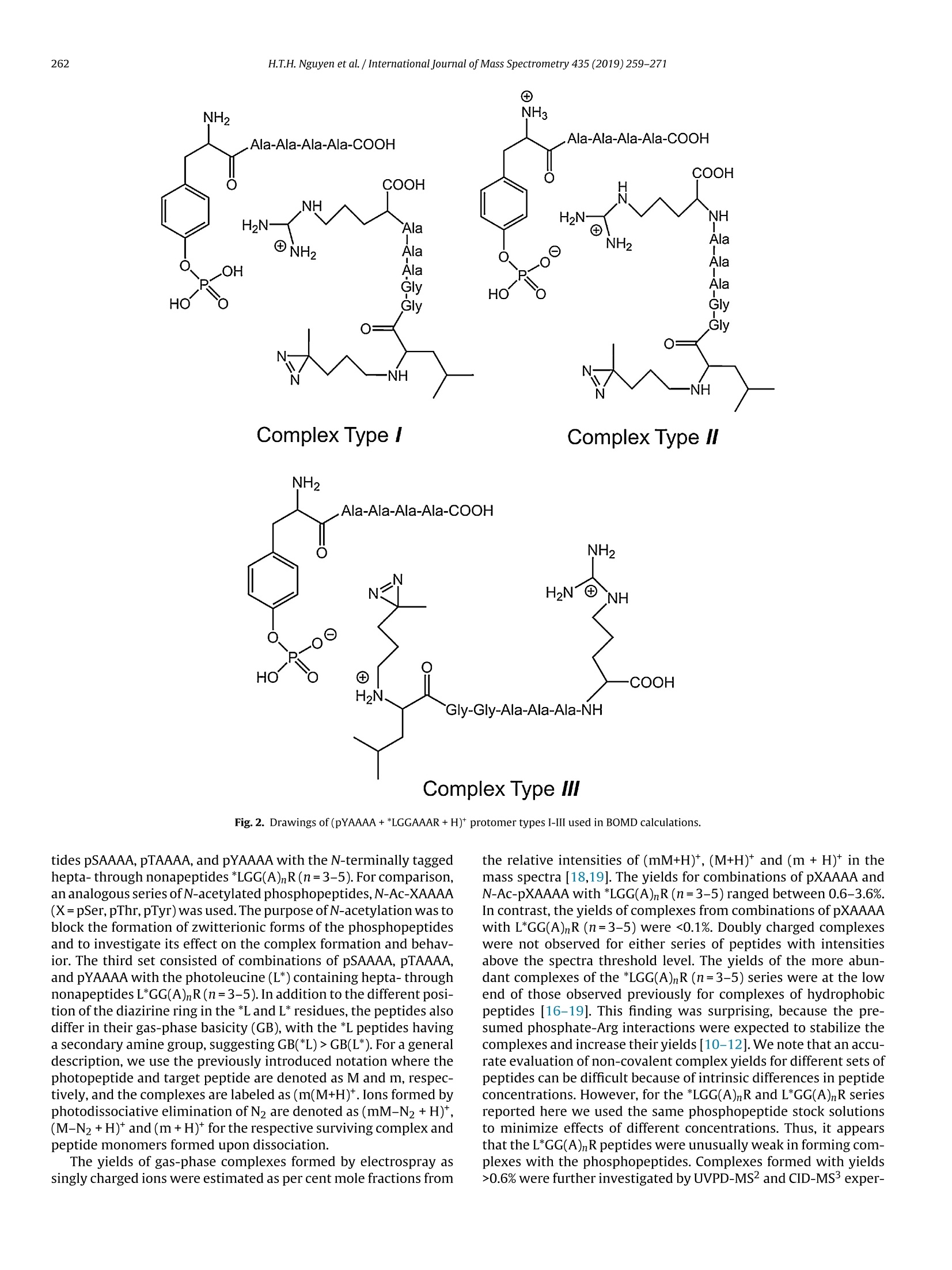
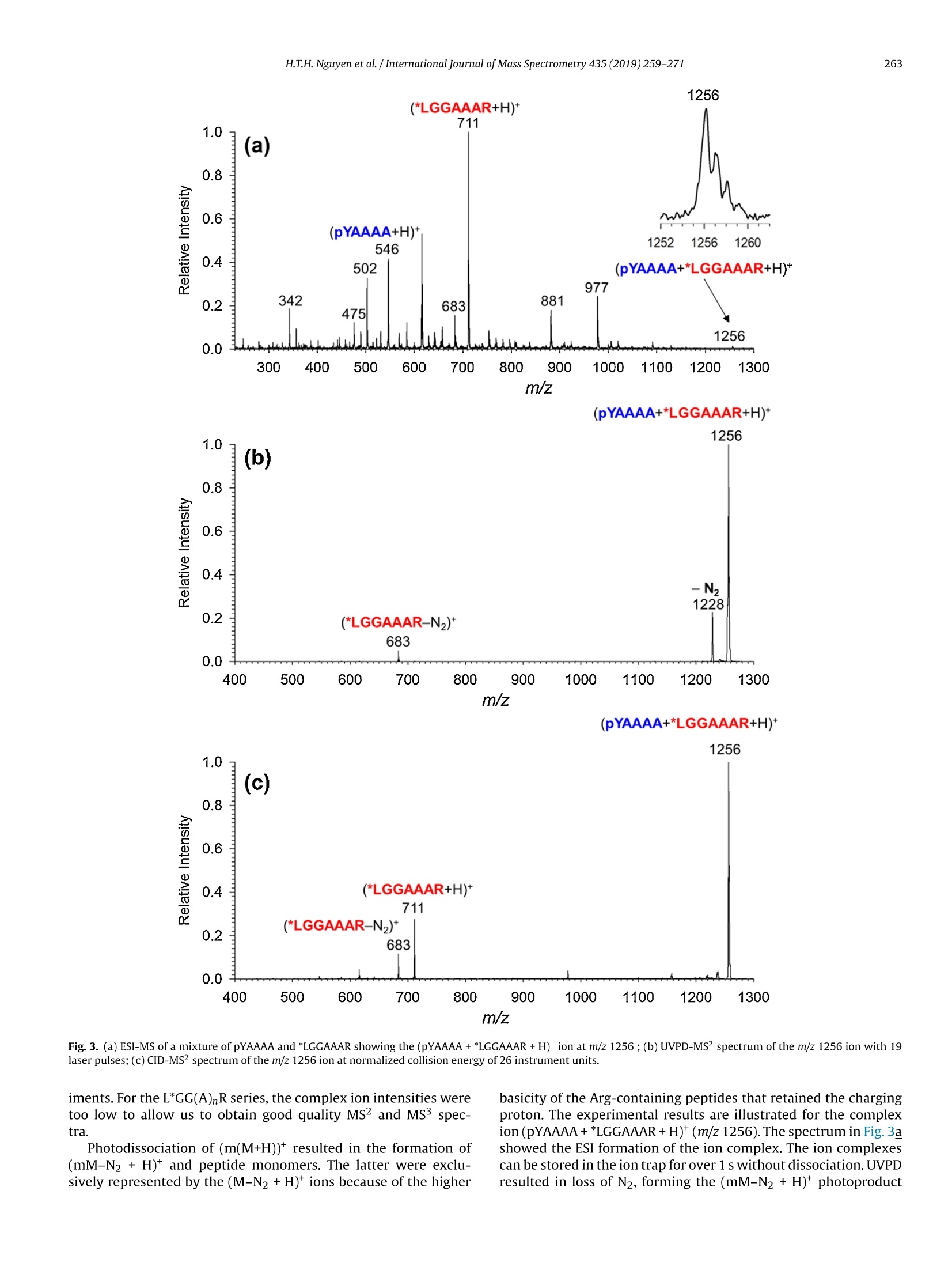
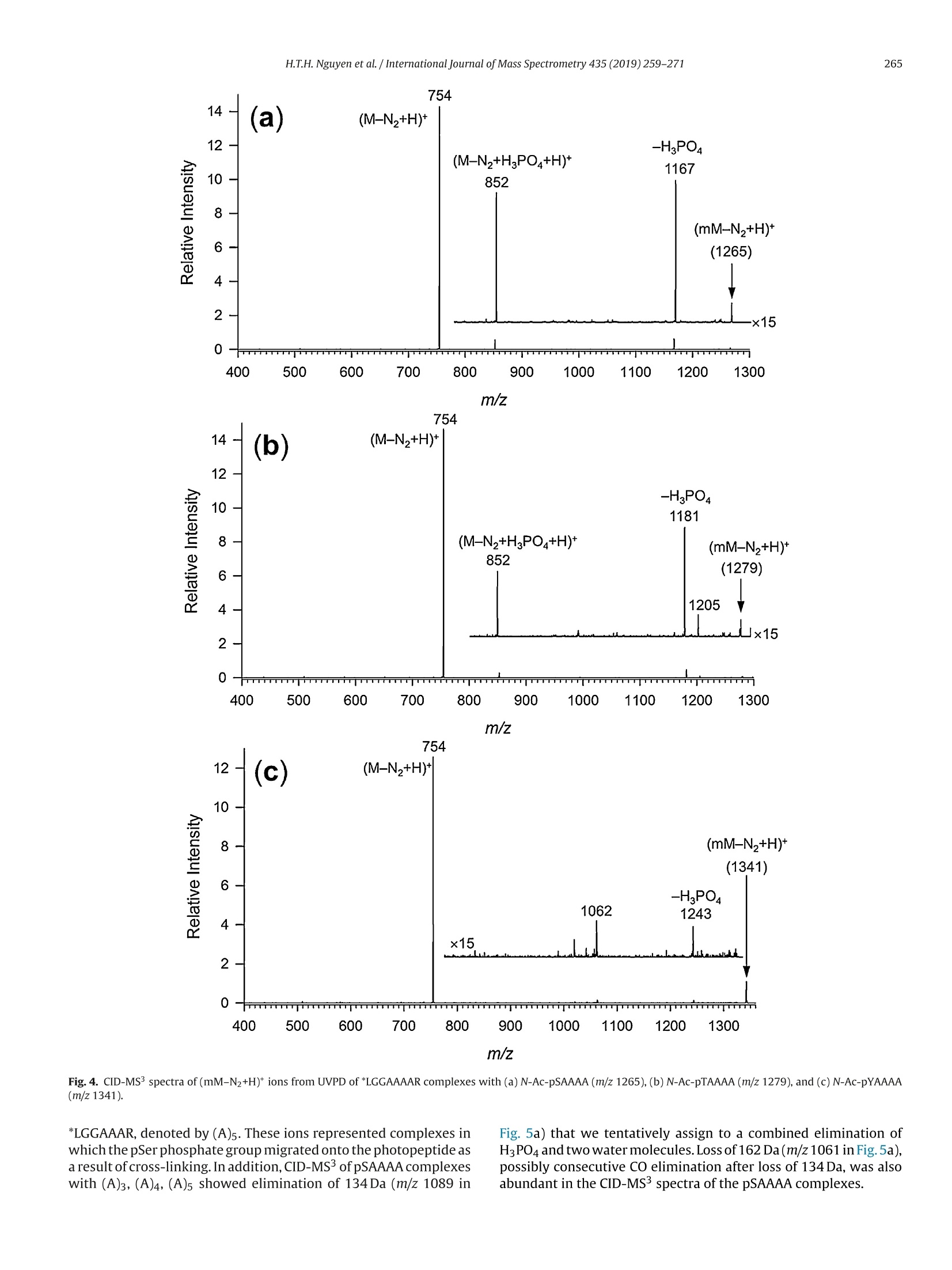
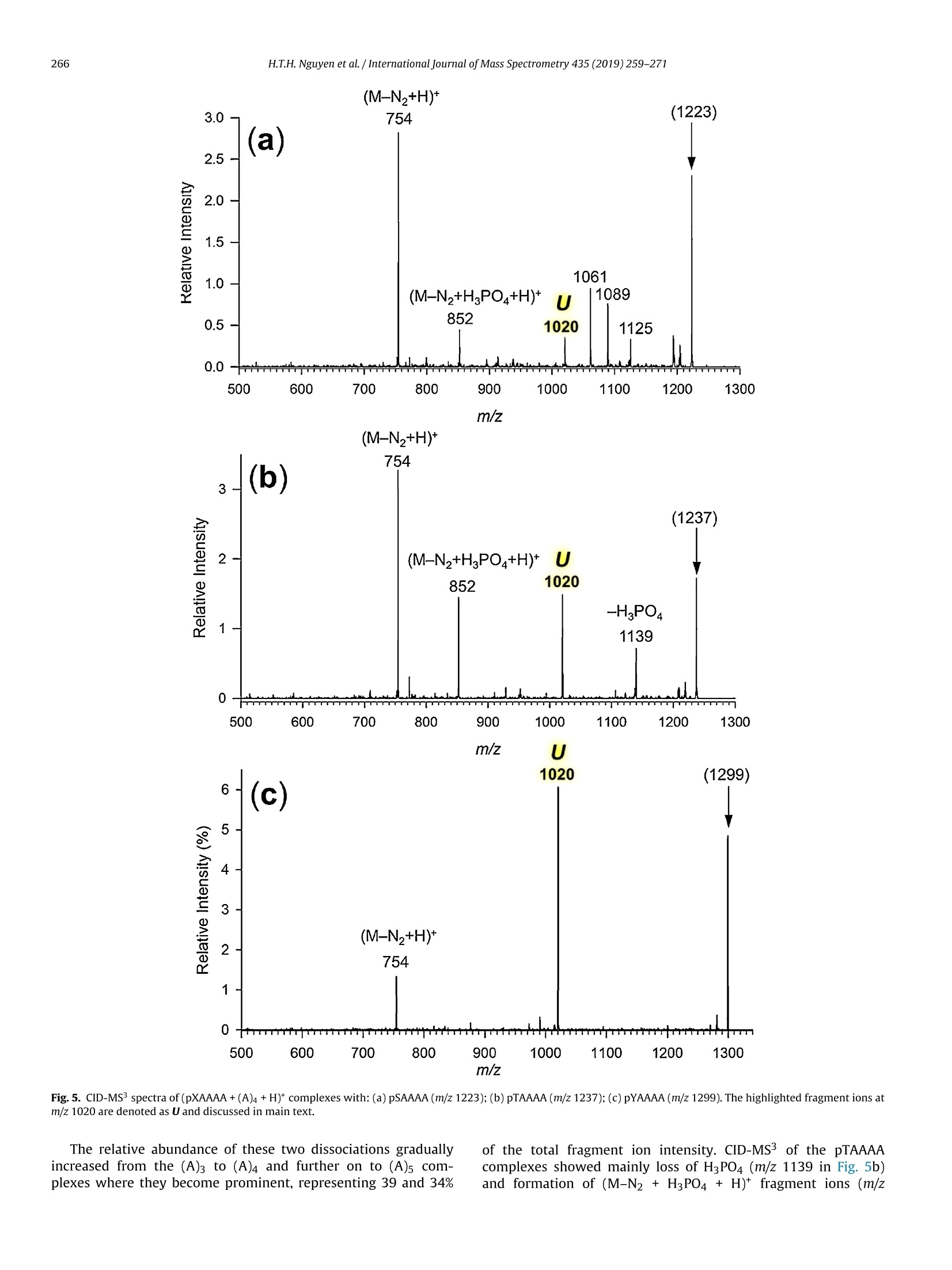
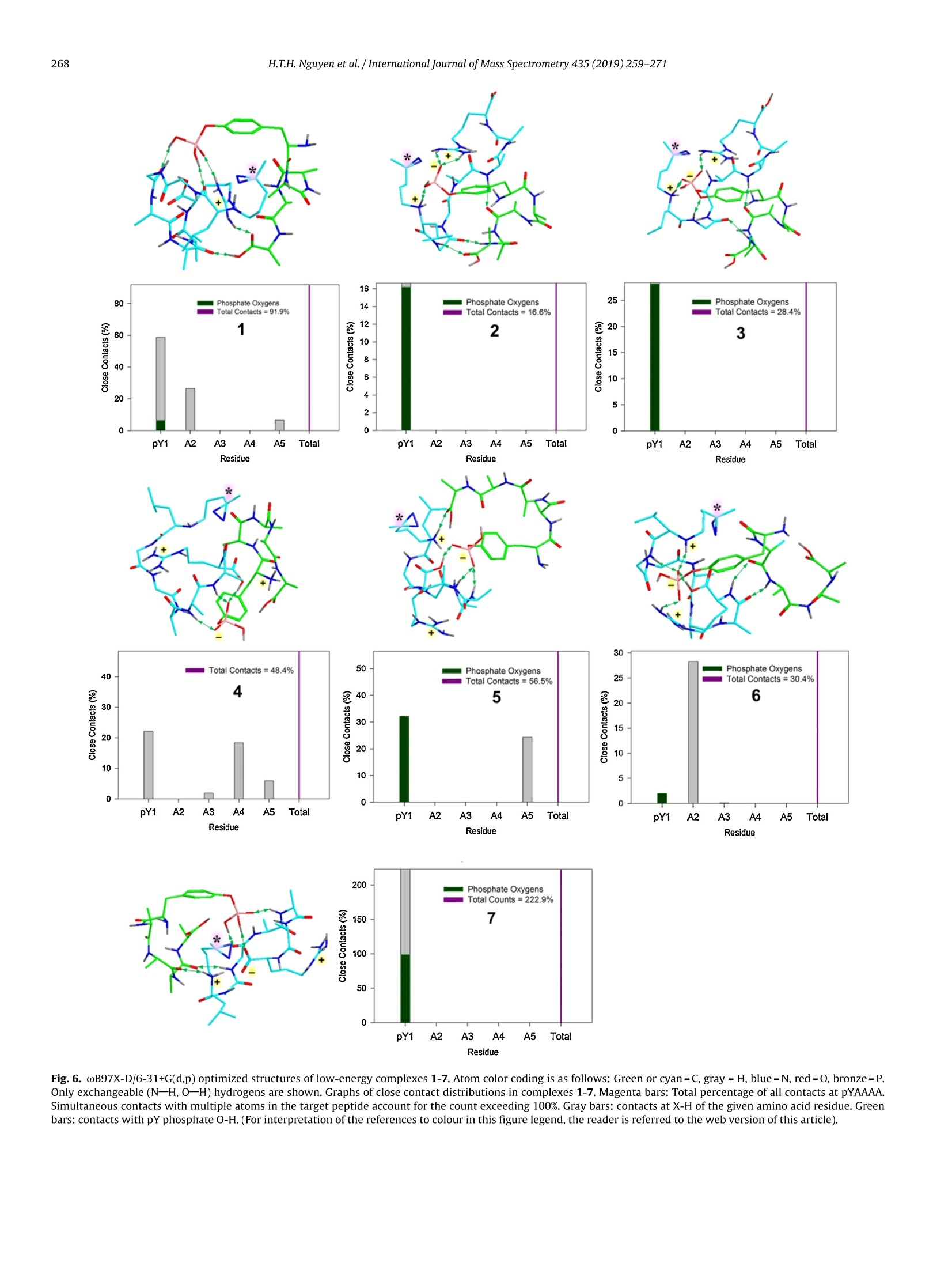
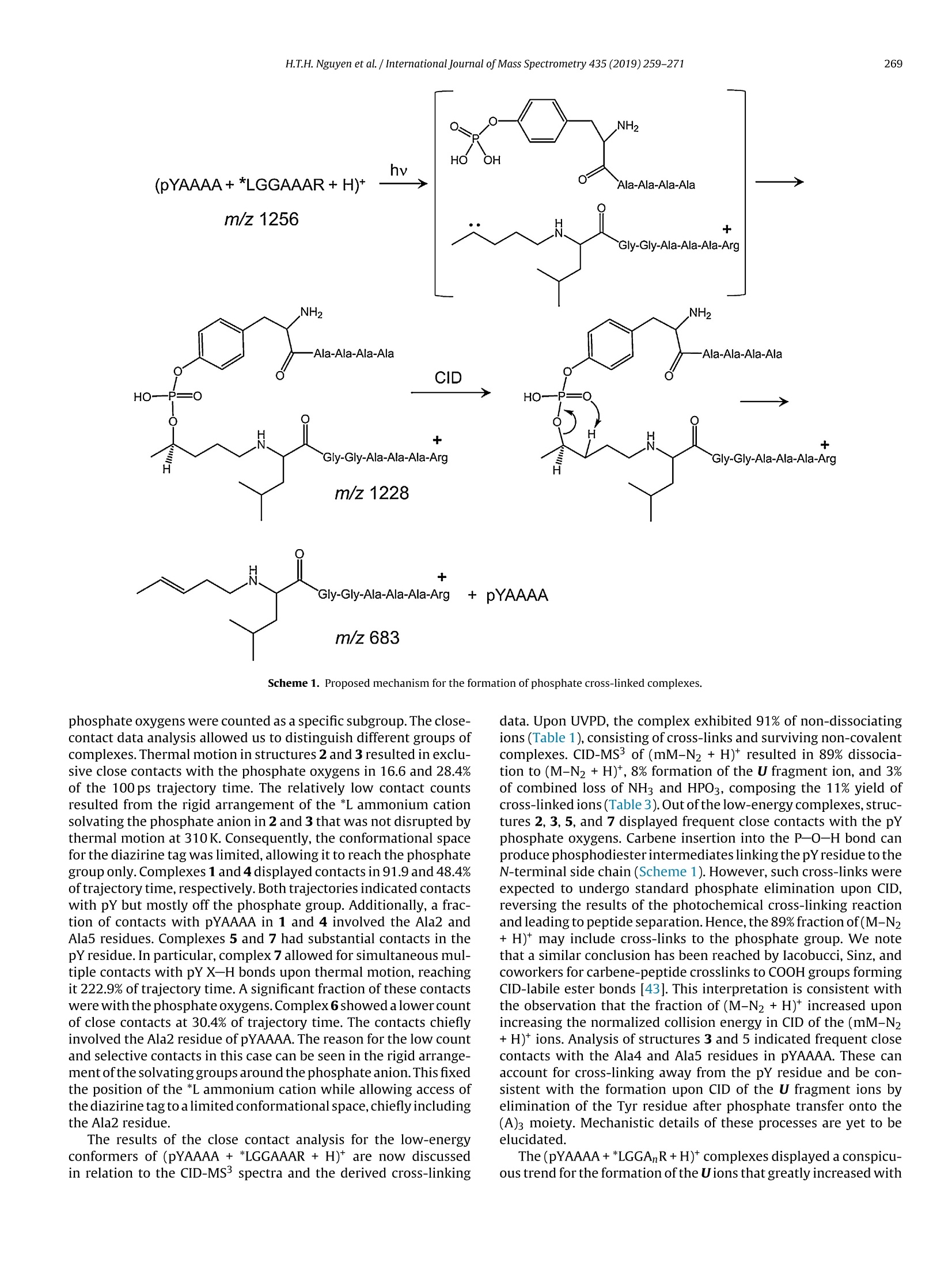
International Journal of Mass Spectrometry 435 (2019)259-271 H.T.H. Nguyen et al./International Journal of Mass Spectrometry 435 (2019) 259-271260 Contents lists available at ScienceDirect International Journal of Mass Spectrometry ELSEVIER journal homepage: www.elsevier.com/locate/ijms Full Length Article Probing arginine-phosphopeptide interactions in non-covalentpeptide-peptide ion complexes using gas-phase cross-linking andBorn-Oppenheimer molecular dynamics calculations Huong T.H. Nguyen, Shu Rong Huang, Yang Liu, Yue Liu, Joseph A. Korn,Frantisek Turecek* Department of Chemistry, Box 351700, University of Washington,Seattle, WA, 98195-1700, United States ARTICLEINFO Article history: Received 15 June 2018 Received in revised form 19 September 2018 Accepted 28 September 2018Available online 3 November 2018 Dedicated to Professor Helmut Schwarz onthe occasion of his 75th birthday. Keywords:Arginine-phosphopeptide complexesPhotodissociative crosslinkingSequence analysislon structuresBorn-Oppenheimer molecular dynamics ABSTRACT We report a study of non-covalent complexes of phosphopeptides pXAAAA and N-Ac-pXAAAA (X=Ser,Thr, Tyr) with arginine-containing peptides carrying diazirine 4,4-azipentyl tags at the N-terminus,*LGG(A)nR, or in the photoleucine residue, L*GG(A)nR(n=3-5). Complexes with *LGG(A)nR were suc-cessfully generated as singly charged ions in the gas phase in 0.6-3.5% yields. In contrast, complexeswith L*GG(A)nR were formed in negligible (<0.1%) yields. Selective photodissociation at 355 nm of thediazirine ring in [*LGG(A)nR+pXAAAA+ H]t ions resulted in loss of N2, producing large (75-95%) frac-tions of non-dissociating complexes that were further probed by collision-induced dissociation tandemmass spectrometry (CID-MS3).Covalent cross-links in the complexes produced by photoinduced carbeneinsertion were identified by specific CID-MS3dissociations involving H3PO4 transfer and backbone frag-mentations. N-acetylation in the phosphopeptides was found to have a substantial negative effect onthe formation of covalent cross-links. Amongst the phosphorylated amino acid residues, pTyrshowedthe highest tendency (up to 92%)to form covalent cross-links. The fractions of covalent cross-links sub-stantially increased with the length of the photopeptide chain. Born-Oppenheimer molecular dynamics(BOMD) calculations of canonical and zwitterionic protomers of the (pYAAAA+*LGGAAAR+H)*complexindicated multiple close contacts between the incipient carbene of the diazirine ring and the X-H bonds(X=C, N, O) in the phosphopeptide. BOMD in combination with structural analysis by density func-tional theory calculations were used to interpret the experimental data and explain the cross-linkingefficiencies. O 2018 Elsevier B.V. All rights reserved. 1. Introduction Reversible protein phosphorylation [1] is one of the most impor-tant post-translational modifications that plays a critical role inregulating many cellular processes including cell cycle, cell growth,and cell signal transduction pathways. Although the mechanismof reversible protein phosphorylation has been studied inten-sively [2], the importance for protein structure and functioning ofnon-covalent binding of phosphopeptides to lysine and arginineresidues in proteins [3] and simpler models continues to stimu-late investigations [4]. For example,the phosphotyrosine bindingregion SH2 of Src proteins, that has been characterized by high-resolution methods such as X-ray crystallography [5] and NMR ( * Corresponding author. ) ( E-mail address: turece k @chem.washin g ton.edu (F. Turecek). ) spectroscopy [6], has also been the subject of mass spectrome-try studies [7]. Loo and coworkers have shown that non-covalentphosphopeptide complexes with the Src homology 2 domain pro-tein could be produced by electrospray ionization and studied bymass spectrometry [8]. Likewise,binding of arginine-rich peptidesto RNA has been studied by electrospray ionization mass spectrom-etry and the complex ion formation in the gas phase was correlatedwith the known binding properties in solution [9]. Non-covalentcomplexes of arginine-rich peptides with phosphorylated peptideshave been produced by soft ionization methods and shown tobe stable in the gas phase [10,11]. In addition, collision-induceddissociation (CID) of arginine-phosphate non-covalent complexesproduced fragmentions by covalent-bond cleavages while the non-covalent interactions were in part preserved intact [12,13]. Theseresults pointed to the importance of strong non-covalent interac-tions of the arginine-phosphate type in the gas phase. However,interpretation of the mass spectrometric data did not provide spe- N=N Leu-Gly-Gly-(Ala)n-Arg H *L Recently, we have com- bined experimental studies of photodissociative cross-linkingin the gas phase with Born-Oppenheimer molecular dynamics(BOMD) calculations as a new approach to probing non-covalentbinding in peptide-peptide ion complexes produced by electro-spray ionization. The chemical nature of our photodissociativecross-linking method relies on diazirine tags that are incorpo-rated in synthetic peptides. The amino acid residues that can beselectively tagged in the side chain include leucine (L-2-amino-4,4-azi-pentanoic acid,called photoleucine, L*) [14], a methionineanalog (L-2-amino-5,5-azi-hexanoic acid, photomethionine, M*)[14], and lysine(L-2,6-diamino-4,4-azi-hexanoic acid)[15]. In addi-tion to these, the lysine e-amino group (K*),leucine N-terminus (*L)[16], and the cysteine thiol group (C*) in peptides [17] have beenselectively tagged with the 4,4-azi-1-pentyl group and used in gas-phase cross-linking [16-19]. The diazirine ring undergoes selectivephotodissociative loss of N2 by irradiation at 330-370nm [20],which is a region where natural peptide or protein chromophoresare transparent[21].Diazirinephotodissociation creates a transientsinglet carbene which either reacts by insertion into a proxi-mate X-H bond in the complex or competitively self-destructsby intramolecular isomerization by 1,2-hydrogen shift fromthe adjacent methyl or methylene groups, forming nonreactiveolefins. These competitive side reactions have very low activation ener-gies as established for peptide ions [22](19-31kJmol-1, Fig. 1,leftpanel), resulting in fast isomerization kinetics (Fig. 1, right panel a).For typical vibrational energies in gas-phase peptide ions, as illus-trated by the energy distribution curve for a model peptide(GL*GGK+2H)+(Fig. 1,right panel b)[22], the carbene lifetimes are shorterthan 10-’s,providing an internal clock for the timescale ofthe cova-lent bond formation. This time scale is commensurable with that forthe thermal conformational motion in the gas-phase complex thatcan be simulated by advanced molecular dynamics. In the absenceof covalent crosslinking, the vibrational excitation provided by thehighly exothermic carbene-to-olefin isomerization (Erxn> 200 kJmol-1,[22]) can drive complex dissociation [18,19]. In case a newcovalent bond is formed, its location can potentially be located bysequencing the products in the gas phase using collision-induceddissociation spectra (CID-MS3)[16-19]. We now apply gas-phase cross-linking to investigate non-covalent peptide-peptide complexes, consisting of phosphorylatedpentapeptides, pXAAAA where X=Ser, Thr, or Tyr and theirN-acetylated analogues, and diazirine tagged *LGG(A)nR andL*GG(A)nRn=3-5 photopeptides.Our goal was to investigate thebinding and structures of these complexes with regard to poten-tial interactions of the phosphate with a single arginine residue.To investigate the conformational space and achieve atomic-levelresolution in structural assignment, we used density functionaltheory calculations and Born-Oppenheimer molecular dynamics(BOMD) trajectory simulations with all-valence electron semiem- 2. Experimental Peptidesphoto-labeled at the N-terminus, *LGGAAAR,*LGGAAAAR,,*LGGAAAAAR, were synthesized on a CEM Lib-erty synthesizer. The tag was introduced by alkylating theselectively deprotected leucine N-terminus in a solid-phase boundpeptide with 1-iodo-4,4-diazirine pentane, as described in detailpreviously [16]. Phosphorylated pentapeptides pTAAAA,pSAAAA,pYAAAA, and their complementary N-acetylated peptides (>90%purity) were purchased from Genscript (Piscataway, NJ, USA)and used as received. The other peptide library, consisting ofL*GGAAAR, L*GGAAAAR, and L*GGAAAAAR, where L* was thephotoleucine (L-2-amino-4,4-azi-pentanoic acid) residue, wasalso synthesized using a standard Fmoc solid phase procedure[23], as described previously [19]. Solutions of the complexeswere prepared by mixing 50 uL of the phosphopeptide (5-10 pMin 50:50:1 methanol/water/acetic acid) and 100 pL of the pho-topeptide (5-10uM in 50:50:1 methanol/water/acetic acid).The mixed solution was electrosprayed from a pulled fused silicacapillary at a 1.5-2 uL/min flow rate into a linear ion trap tandemmass spectrometer, LTQ-XL ETD (ThermoElectron Fisher, San Jose,CA, USA). Photodissociation experiments at 355 nm were carriedout as described previously [24,25]. Briefly, the laser beam wasgenerated from an EKSPLA NL 301 HT (Altos Photonics, Bozeman,MT, USA) Nd-YAG laser operating at 20Hz frequency with a3-6ns pulse width. The laser was interfaced with the LTQ-XLETD mass spectrometer as described previously [26].The typicallight intensity used in the photodissociation experiments was18 mJ/pulse. Multiple (19-39) laser pulses were used to achievephotodissociation of mass-selectedion-molecule complexes.Collision-induced dissociation (CID) spectra were obtained at25-26 units of normalized collision energies (NCE) and withactivation times that were varied between the instrument defaultsetting of 30 ms for preliminary analysis and 1000 ms. The longactivation times were chosen to match the ion storage time usedin photodissociation. 3. Calculations Born-Oppenheimer molecular dynamics (BOMD) trajectorieswere run with semiempirical all-valence-electron quantum chem-istry calculations using the Berendson thermostat algorithm [27].Temperature was set at 310 K to represent the experimental con-ditions in the ion trap. The ion system under study, (pYAAAA +*LGGAAAR+H), was represented by three different types of pro-tomers (Fig. 2). In Type I complexes, the peptides had canonical ion or neutralstructures with the charge located on the protonated guanidinegroup of the Arg residue whereas the phosphate group was neutral.Type lI complexes were constructed as zwitterions with two posi-tive charges located on the protonated guanidine group of the Argresidue and the N-terminus of the phosphorylated peptide whilethe phosphate group carried a negative charge. Type III complexwere also zwitterions with two positive charges located on the pro-tonated guanidine group of the Arg residue and the N-terminus ofthe photolabeled peptide while the phosphate group carried a neg-ative charge (Fig.2). For each complex type, ten initial structureswere constructed from PM6-optimized peptide subunits in whichthe diazirine tag and pTyr were in different spatial orientations, andthe complexes were optimized with semi-empirical PM6-D3H4calculations [28]. These complexes were subjected to a prelimi-nary BOMD using PM6-D3H4 under MOPAC [29] that was coupled Fig. 1. Left panel: Scheme of carbene complex isomerization in (L*-N2) by 1,2-hydrogen migrations from adjacent methyl and methylene groups. Activation energies arefrom M06-2X/6-311++G(2d,p)+ZPVE calculations for the model [G(L*-N2)GGK+2H)+ singlet carbene ion [22]. Right panel:(a) Calculated half-lives of the carbene for a1,2-hydrogen migration from the methyl group.(b) Vibrational energy distribution in the singlet carbene [G(L*-N2)GGK+2H]2+ at 310K. to the Cuby4 framework [30,31]. Running trajectories with 1 fssteps for 20 ps furnished 20,000 snapshots from which 200 snap-shots per structure were extracted at 100 fs intervals. The total of200×10=2000 extracted snapshot structures were fully gradient-optimized with PM6-D3H4 and sorted out by energy ranking. Tenlowest-energy PM6-D3H4 structures within 40 kj mol-1 were thensubjected to a full BOMD with PM6-D3H4 for 100 ps. Cuby4 wasalso used to extract close contacts between the carbon atom of thediazirine ring and the hydrogen-carrying atoms of the phosphopep-tide. Close contacts were limited to X-H...C distance of less than4.5 A on the basis of the van der Waals radii of the diazirine andX-H atoms [19]. This entire procedure was run for each Type I, II,and III protomeric complexes. Thermochemical calculations were performed using the Gaus-sian 16 (Revision A.03)[32] suite of programs. Ten lowest-energystructures of each Type I-III complexes from the PM6-D3H4 calcu-lations were re-optimized with B3LYP/6-31G(d,p)[33] to provide harmonic frequencies that were used to evaluate enthalpies,entropies, and thermal free-energy corrections at 310K. In sepa-rate runs, the initial structures were re-optimized with ωB97X-D[34] and the 6-31+G(d,p) basis set. The optimized geometries andenergies of seven low-energy complexes are given as Supplemen-tary data in Tables S1a-S7b. These calculations included dispersioninteractions in the complexes, and the obtained energies were usedto evaluate the electronic terms of the free energies.The calculatedrelative free energies included electronic, vibrational, and rota-tional terms at 310 K. Solvation free energies were calculated usingthe polarizable continuum model [35] for the water and methanoldielectric. 4. Results and discussion Our investigations were focused on three sets of non-covalentcomplexes. In the first set, we generated complexes of phosphopep- HO Fig. 2. Drawings of(pYAAAA+*LGGAAAR+H)*protomer types I-III used in BOMD calculations. tides pSAAAA,pTAAAA,and pYAAAA with the N-terminally taggedhepta-through nonapeptides *LGG(A)nR(n=3-5). For comparison,an analogous series ofN-acetylated phosphopeptides,N-Ac-XAAAA(X=pSer,pThr, pTyr) was used. The purpose of N-acetylation was toblock the formation of zwitterionic forms of the phosphopeptidesand to investigate its effect on the complex formation and behav-ior. The third set consisted of combinations of pSAAAA, pTAAAA,and pYAAAA with the photoleucine (L*) containing hepta- throughnonapeptides L*GG(A)nR(n=3-5). In addition to the different posi-tion of the diazirine ring in the *L and L* residues, the peptides alsodiffer in their gas-phase basicity (GB), with the *L peptides havinga secondary amine group,suggesting GB(*L)>GB(L*). For a generaldescription, we use the previously introduced notation where thephotopeptide and target peptide are denoted as M and m, respec-tively, and the complexes are labeled as (m(M+H)*. Ions formed byphotodissociative elimination of N2 are denotedas (mM-N2+H)+,(M-N2+H)*and(m+H)+ for the respective surviving complex andpeptide monomers formed upon dissociation. The yields of gas-phase complexes formed by electrospray assingly charged ions were estimated as per cent mole fractions from the relative intensities of (mM+H)+,(M+H)+ and (m +H)t in themass spectra [18,19]. The yields for combinations of pXAAAA andN-Ac-pXAAAA with *LGG(A)nR(n=3-5) ranged between 0.6-3.6%.In contrast, the yields of complexes from combinations of pXAAAAwith L*GG(A)nR (n=3-5) were <0.1%. Doubly charged complexeswere not observed for either series of peptides with intensitiesabove the spectra threshold level. The yields of the more abun-dant complexes of the *LGG(A)nR (n=3-5) series were at the lowend of those observed previously for complexes of hydrophobicpeptides [16-19]. This finding was surprising, because the pre-sumed phosphate-Arg interactions were expected to stabilize thecomplexes and increase their yields [10-12].We note that an accu-rate evaluation of non-covalent complex yields for different sets ofpeptides can be difficult because of intrinsic differences in peptideconcentrations. However, for the *LGG(A)nR and L*GG(A)nR seriesreported here we used the same phosphopeptide stock solutionsto minimize effects of different concentrations. Thus, it appearsthat the L*GG(A)nR peptides were unusually weak in forming com-plexes with the phosphopeptides. Complexes formed with yields>0.6% were further investigated by UVPD-MS2 and CID-MS3 exper- 1256 (*LGGAAAR+H)+ 711 1.0 (a) 0.0· 400 500 600 700 800 900 1000 1100 1200 1300 m/z Fig.3. (a) ESI-MS of a mixture of pYAAAA and *LGGAAAR showing the (pYAAAA+*LGGAAAR+H)*ion at m/z 1256 ; (b) UVPD-MSspectrum of the m/z 1256 ion with 19laser pulses;(c) CID-MS2 spectrum of the m/z 1256 ion at normalized collision energy of 26 instrument units. iments. For the L*GG(A)nR series, the complex ion intensities weretoo low to allow us to obtain good quality MS2 and MS3 spec-tra. Photodissociation of (m(M+H)) resulted in the formation of(mM-N2 + H)+ and peptide monomers. The latter were exclu-sively represented by the (M-N2 +H)ions because of the higher basicity of the Arg-containing peptides that retained the chargingproton.The experimental results are illustrated for the complexion (pYAAAA+*LGGAAAR+H)* (m/z1256). The spectrum in Fig. 3ashowed the ESI formation of the ion complex. The ion complexescan be stored in the ion trap for over 1 s without dissociation. UVPDresulted in loss of N2, forming the (mM-N2 +H) photoproduct Table 1 R(MS2) fractions of (mM-N2+H)* ions surviving photodissociation. *LGG(A)3R *LGG(A)4R *LGG(A)5R pSAAAA 75 81 88 pTAAAA 80 81 86 pYAAAA 91 92 95 N-Ac-pSAAAA 90 89 89 N-Ac-pTAAAA 88 89 88 N-Ac-pYAAAA 79 80 84 aPercent fractions, see text for definition. (m/z 1228, Fig. 3b) which underwent partial dissociation to the(*L-N2)GGAAAR+ H)* monomer (m/z 683, Fig. 3b). Remarkably,there was less than 1% elimination of H3PO4 from the (mM-N2 +H)+ions as illustrated in Fig. 3b. Similar results were obtained for allother pY, pS, and pT containing complexes. In contrast to pho-todissociation, collision-induced dissociation (CID) of (m(M+H))*resulted in complex dissociation to ((M+H))*(m/z 711, Fig.3c)thatwas accompanied by (M-N2+H)+ at m/z 683 while no (mM-N2+H) was detected. The yields of(mM-N2+H)* complexes survivingphotodissociation were expressed as R(MS2) values: R(MS2)=100x[mM-N2+H]*/{[mM-N2 +H]++[M-N2 +H]t} where squarebrackets indicate ion intensities in the UVPD-MS2 spectra (Table 1).The fractions of(mM-N2+H)t, expressed as R(MS2), combined con-tributions from non-covalent complexes surviving the exothermicloss of nitrogen [22] and their cross-linked isomers. The Table 1data indicated high levels of retention of non-covalent complexesthat exceeded 75%.Some minor trends of R(MS2) were observed independence on the photopeptide molecular size where increasingthe number of Ala residues resulted in a slight increase of the frac-tion of non-dissociating complexes. This trend and its magnitudeare consistent with the degree-of-freedom effect on the complexdissociation kinetics. For example, the internal degrees of freedomin (pYAAAA+*LGG(A)nR+H)* increased by 11% from 516 for n=3to 576 for n=5, a relatively minor change. The internal energiesin the (mM-N2+H)+ions immediately after photodissociation arecomposed of the precursor (m(M+H))*ion internal energy and theexothermicity of N2 elimination. The distribution of thermal vibra-tional energies at the ion trap temperature of 310 K was calculatedfor the (pYAAAA+*LGGAAAR+H)+complexes (Figure S1, Supple-mentary data), giving mean energy values of 258-261 kJ mol-1 fortwo conformers. An N2 elimination from the diazirine ring followedby carbene isomerization to an olefin is ca. 200 kJ mol-1 exothermic[22], increasing the mean vibrational energy of (mM-N2+H)ionsto ca.460 kJ mol-1 in the non-covalent complexes to drive theirdissociation. In spite of this substantial excitation, only a minorfraction of complexes dissociated upon UVPD (Table 1), raising thequestion of covalent versus non-covalent bonding in the photo-products. The fractions of non-covalent and covalently cross-linkedcomplexes were then analyzed by CID. The surviving(mM-N2+H)*photoproduct ion complexes wereselected by mass and further investigated by CID-MS3. The results Table 3 Summed relative intensities of covalently cross-linked fragment ions in the UVPD-CID-MS3spectra of(mM-N2+H)*ions from (pXAAAA+*LGG(A)nR+H)*complexes. Cross-Linked Fragment Ions (A)3 (A)4 (A)5" pSAAAA 48 80 89 DTAAAA 44 71 50 DYAAAA 11 87 92 Sums of relative intensities of fragment ions with m/z between that of the (M-N2+H)*and (mM-N2+H)* that originated from cross-linked complexes. (A)3 standsfor complexes with *LGG(A)3R; (A)4 stands for complexes with *LGG(A)4R; (A)sstands or complexes with *LGG(A)5R. for the (pXAAAA+*LGG(A)nR+H)+ ion complexes and their N-acetylated analogues are summarized in Tables 2 and 3.The(M-N2)photopeptides are abbreviated as (A)3,(A)4, and (A)5 according tothe number of Ala residues.The relative intensities of(M-N2+H)*monomer ions were taken as representing the fractions of non-covalent complexes. This followed from the general feature of CIDspectra (Fig. 3c) showing that non-covalent complexes exclusivelydissociated to monomers. Starting with the N-acetylated phosphopeptides, CID-MS3 analysis indicated that major frac-tions of the complexes (>90%) dissociated to monomeric peptides,as indicated by the data in the (M-N2 +H)+ columns in Table 2.As discussed below, these dissociations can chiefly originate fromnon-covalent complexes but also can include cross-links at thepX phosphate oxygens. The fractions of these complexes did notshow strong dependence on the length of the photopeptide chain.Regarding covalent cross-linked complexes, their minor fractionswere represented by fragment ions formed either by loss of H3PO4from (mM-N2 + H)+, or resulted as H3PO4 adducts onto the(M-N2+H)* moiety, as shown for the (pXAAAA+(A)4 +H)* com-plexes (Fig.4a,b,c). Note that none of these reactions was observedin the absence of photodissociation and so they can be unambigu-ously assigned to covalently cross-linked products. Higher fractions of both the covalent cross-links and a varietyof dissociation reactions were observed for photodissociation ofcomplexes with pXAAAA having free N-termini. This was inferredfrom the CID-MS’spectra considering fragment ions with m/zbetween that of the (M-N2 +H)* and (mM-N2 +H)* that canbe unambiguously assigned to originate from covalent cross-links. Rather than listing the multitude of individual cross-linkedfragmentions for each combination of the phospho-and photopep-tide, we summed their relative intensities that are presented inTable 3. The CID-MS’ data differed for pSAAAA, pTAAAA, and pYAAAAin both the nature of CID fragmentations and dependence on thelength of the photopeptide. The fragmentations are illustrated byCID-MS’spectra of(pXAAAA+(A)4+H)+complexes(Fig.5a-c). CID-MS3 of the pSAAAA complexes resulted in loss of H3PO4 (m/z 1125in Fig. 5a) and formation of (M-N2 +H3PO4+ H)+ fragment ionsthat appeared at m/z 852 in Fig. 5a and were incrementally shiftedby 71 Da for the complexes with *LGGAAAR, denoted by (A)3, and Table 2 Relative fragment ion intensities in the UVPD-CID-MS’spectra of (mM-N2+H) ions from (N-Ac-pXAAAA+*LGG(A)nR+H)t complexes.Peptide Relative Intensity (%) (M-N2+H)+ (M-N2+H3PO4+H)+ (mM-N2-H3PO4+H)+ (A)3 (A)4 (A)5 (A)3 (A)4 (A)5 (A)3 (A)4 (A)5 N-Ac-pSAAAA 94 92 91 3.7 3.5 4.8 2.4 4.2 3.9 N-Ac-pTAAAA 96 94 96 2.2 1.8 1.9 1.8 3.5 1.8 N-Ac-pYAAAA 98 97 90 (A)3 stands for complexes with *LGG(A)3R. (A)4 stands for complexes with *LGG(A)4R.(A)s stands or complexes with *LGG(A)sR. 754 m/z Fig. 4. CID-MS’spectra of(mM-N2+H)*ions from UVPD of *LGGAAAAR complexes with (a) N-Ac-pSAAAA (m/z1265),(b)N-Ac-pTAAAA (m/z 1279), and (c)N-Ac-pYAAAA(m/z 1341). *LGGAAAR, denoted by (A)5. These ions represented complexes inwhich the pSer phosphate group migrated onto the photopeptide asa result of cross-linking. In addition, CID-MS3of pSAAAA complexeswith (A)3, (A)4, (A)s showed elimination of 134 Da (m/z 1089 in Fig. 5a) that we tentatively assign to a combined elimination ofHgPO4 and two water molecules. Loss of 162 Da(m/z1061 in Fig.5a),possibly consecutive CO elimination after loss of 134 Da, was alsoabundant in the CID-MS3spectra of the pSAAAA complexes. (M-N2+H)+ 3.0 754 (1223) (a)2.5 2.0 1.5 754 1 - TTM 500 600 700 800 900 1000 1100 1200 1300 m/z Fig.5. CID-M’ spectra of (pXAAAA+(A)4+H)* complexes with: (a) pSAAAA (m/z 1223); (b) pTAAAA (m/z 1237);(c)pYAAAA (m/z 1299). The highlighted fragment ions atm/z 1020 are denoted as U and discussed in main text. The relative abundance of these two dissociations graduallyincreased from the (A)3 to (A)4 and further on to (A)5 com-plexes where they become prominent, representing 39 and 34% of the total fragment ion intensity. CID-MS3 of the pTAAAAcomplexes showed mainly loss of H3PO4 (m/z 1139 in Fig. 5b)and formation of(M-N2 + H3PO4 + H)+ fragment ions (m/z 852 for the complex with (A)4). Interestingly, dissociations anal-ogous or homologous to the loss of 134 and 162Da neutralfragments in the pSer series were absent for the pThr com-plexes. A conspicuous feature of the CID-MS’spectra was the forma-tion of cross-linked fragment ions at m/z 949, 1020, and 1091 fromthe respective (A)3, (A)4, and (A)5 complexes (Figures S2-S4, Sup-plementary data). These fragment ions, which are highlighted andlabeled U in the CID-MS’spectra (Fig.5a,b,c), showed the same m/zwhen formed from the pSAAAA,pTAAAA, and pYAAAA complexes,indicating that the Ser, Thr and Tyr residues were eliminated bydissociation, so that covalent cross-linking must have taken placewithin the AAAA sequence of the phosphopeptides. At the sametime, the pertinent neutral losses of 203, 217, and 279 Da from thephosphopeptides were difficult to explain by including the intactpSer (168 Da), pThr (182Da) and pTyr (244Da) moieties, becausethere was no logical mass unit to account for the missing 35 Dadifference. Hence, we concluded that prior to or in the course ofdissociation, the (mM-N2 +H) ion complexes underwent HPO3migration onto the photopeptide. The recipient group c0ould not bed?etermined by CID-MS4 of the Uions (m/z 1020 from the complexeswith *LGG(A)4R) because of the substantially diminished ion countafter the MS3step. We tentatively suggest that HPO3 moved to oneof the potentially nucleophilic groups in thephotopeptides, whichare the Arg guanidine and the (*L-N2) secondary amine. The over-all dissociation leading to the formation of U fragment ions can betentatively explained as proceeding with backbone cleavage next tothe Ala-2 residue in the rearranged phosphopeptide. The formationof U fragment ions relatively increased from (A)3 to (A)4 complexesand then leveled off. In summarizing this section, we note that the CID-MS3 dissoci-ations of the photolyzed complexes indicated substantial fractionsof covalently cross-linked products with the phosphopeptides withfree N-termini. A common feature of the complexes was phosphatetransfer from the phosphopeptide to the photopeptide.Althoughdetailed mechanisms of the cross-linked ion dissociations remainto be elucidated, the above described reactive interactions betweenthe phosphopeptide and photopeptide moieties provide a firm evi-dence for covalent bond formation in the complexes upon diazirinephotodissociation. We note that CID-induced phosphate migration among serineresidues in phosphopeptide cations and anions has been reportedand discussed by Reid and coworkers [36], and its importance forphosphoproteome analysis has been debated [37-40]. Collision-induced transfer of HPO3 [41] and H2SO4[42] in non-covalentcomplexes of phosphorylated and sulfated peptides has beenreported. In contrast to the previous studies, we did not observephosphate transfer upon CID of non-covalent(pXAAAA+*LGG(A)nR+H)+complexes prior to photodissociative cross-linking. 5. Complex ion structures and dynamics In order to shed light on the interactions in the complexes,we undertook a detailed computational study of the (pYAAAA +*LGGAAAR+H)+ion. BOMD of several Type I, II, and III protomericcomplexes (Fig. 2), followed by gradient geometry optimization,furnished several structures representing local energy minima atO K. The structures were sorted out by free energies to select low-est energy representatives of each protomeric type, as summarizedin Table 4. The gas-phase energies varied, depending on the DFTmethod used, and further adjustments were made by including gas-phase enthalpies, entropies, and solvation energies [35]. Regardingthe latter, solvation in the water and methanol dielectrics gavepractically identical results, and thus only the water data arereported.Because of all these effects, we used a rather broad energyrange of 40 kJ mol-1 to include protomeric complexes that can Table 4Relative energies of (pYAAAA +*LGGAAR+H)十 complexes in the gas phase andaqueous solution. Complex Type Relative Energy" B3LYPD ωB97X-DC AHg,0 AGg,310 AGaq8 AHg,o AGg,310 AGaq8 32 19 19 59 46 35 42 36 36 35 28 25 60 46 46 38 23 6 54 56 56 34 36 25 94 79 79 97 83 27 0 0 0 0 0 0 41 38 38 40 37 36 aIn kJ mol-1 including B3LYP/6-31 G(d,p) zero-point energies and referring to OKunless stated otherwise. Gas-phase structures fully optimized with the 6-31 G(d,p)basis set. ‘Gas-phase structures fully optimized with the 6-31+G(d,p) basis set. SeeFig.6 and Tables S1a-S7b forωB97X-D/6-31+G(d,p) optimized structures.See Fig.2for the Type definition.Relative free energies in the gas-phase at 310K.Includingsolvation free energies in a polarizable dielectric continuum of water.hRearrangedto Type III complexes upon gradient optimization. With these low-energy structures in hand, we investigated theirlong (100 ps) BOMD trajectories with the goal of determining closecontacts of the incipient carbene with X-H bonds in the targetpYAAAA peptide,resulting from intra-complex thermal motion attheion trap temperature(310K).Aclose contact ofthe carbene withan X-Hbond is prerequisite for the chemical reaction forming thenew covalent X-C bond and resulting in cross-linking.Alack of con-tact favors carbene isomerization to an unreactive olefin. Hence,thepercentage of time the diazirine spends in close contacts with X-Hbonds is indicative of the propensity for cross-linking. Close con-tacts obtained for the C-H, N-H, and O-H bonds in pYAAAA weregrouped by amino acid residues. With pY, close contacts with the Fig.6. wB97X-D/6-31+G(d,p) optimized structures of low-energy complexes 1-7. Atom color coding is as follows: Green or cyan=C, gray =H, blue=N, red=O, bronze=P.Only exchangeable(N-H,O-H) hydrogens are shown. Graphs of close contact distributions in complexes 1-7. Magenta bars: Total percentage of all contacts at pYAAAA.Simultaneous contacts with multiple atoms in the target peptide account for the count exceeding 100%. Gray bars: contacts at X-H of the given amino acid residue. Greenbars: contacts with pY phosphate O-H. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article). phosphate oxygens were counted as a specific subgroup. The close-contact data analysis allowed us to distinguish different groups ofcomplexes. Thermal motion in structures 2 and 3 resulted in exclu-sive close contacts with the phosphate oxygens in 16.6 and 28.4%of the 100 ps trajectory time. The relatively low contact countsresulted from the rigid arrangement of the *L ammonium cationsolvating the phosphate anion in 2 and 3 that was not disrupted bythermal motion at 310 K. Consequently, the conformational spacefor the diazirine tag was limited, allowing it to reach the phosphategroup only.Complexes 1 and 4 displayed contacts in 91.9 and 48.4%of trajectory time,respectively. Both trajectories indicated contactswith pY but mostly off the phosphate group. Additionally, a frac-tion of contacts with pYAAAA in 1 and 4 involved the Ala2 andAla5 residues. Complexes 5 and 7 had substantial contacts in thepY residue. In particular, complex 7 allowed for simultaneous mul-tiple contacts with pY X-H bonds upon thermal motion, reachingit 222.9% of trajectory time. A significant fraction of these contactswere with thephosphate oxygens.Complex6 showed alower countof close contacts at 30.4% of trajectory time. The contacts chieflyinvolved the Ala2 residue of pYAAAA. The reason for the low countand selective contacts in this case can be seen in the rigid arrange-mentofthe solvating groups around the phosphate anion. This fixedthe position of the *L ammonium cation while allowing access ofthe diazirine tag to a limited conformational space,chiefly includingthe Ala2 residue. The results of the close contact analysis for the low-energyconformers of (pYAAAA +*LGGAAAR+ H)+are now discussedin relation to the CID-MS’ spectra and the derived cross-linking data. Upon UVPD, the complex exhibited 91% of non-dissociatingions (Table 1), consisting of cross-links and surviving non-covalentcomplexes. CID-MS3 of(mM-N2+H)* resulted in 89% dissocia-tion to (M-N2 + H)t, 8% formation of the U fragment ion, and 3%of combined loss of NH3 and HPO3, composing the 11% yield ofcross-linkedions (Table 3). Out ofthe low-energy complexes,struc-tures 2, 3, 5, and 7 displayed frequent close contacts with the pYphosphate oxygens. Carbene insertion into the P-O-H bond canproduce phosphodiesterintermediates linking the pY residue to theN-terminal side chain (Scheme 1). However, such cross-links wereexpected to undergo standard phosphate elimination upon CID,reversing the results of the photochemical cross-linking reactionand leading to peptide separation.Hence, the 89% fraction of (M-N2+ H)* may include cross-links to the phosphate group. We notethat a similar conclusion has been reached by lacobucci, Sinz, andcoworkers for carbene-peptide crosslinks to COOH groups formingCID-labile ester bonds [43]. This interpretation is consistent withthe observation that the fraction of (M-N2 +H)+ increased uponincreasing the normalized collision energy in CID of the (mM-N2+H)ions. Analysis of structures 3 and 5 indicated frequent closecontacts with the Ala4 and Ala5 residues in pYAAAA. These canaccount for cross-linking away from the pY residue and be con-sistent with the formation upon CID of the U fragment ions byelimination of the Tyr residue after phosphate transfer onto the(A)3 moiety. Mechanistic details of these processes are yet to beelucidated. The(pYAAAA+*LGGAnR+H)*complexes displayed a conspicu-ous trend for the formation of the U ions that greatly increased with the length of the photopeptide (Figure S2, Supplementary data),as also illustrated by the 11% to 92% increase of the total frac-tion of cross-linked fragment ions (Table 3). The U ions involvecross-linking at the A3-A5 residues in pYAAAA. Although we didnot carry out detailed calculations of these larger systems, theabove-described analysis of the (pYAAAA+*LGGAAAR+H)+ struc-tures can be used for interpretation. The formation of U fragmentions competes with cross-linking to the pY phosphate group thatis facilitated by hydrogen bonding to the Arg and N-terminalcharged groups in the complexes. As the photopeptide sequencelength is increased, simultaneous coordination to the pY phosphateof the Arg and N-terminal charged groups becomes entropicallydisfavored, because there is a large number of alternative hydro-gen bonding sites for pY, forming conformers of comparable freeenergy. This should increase close contacts of the diazirine groupwith the C-terminal residues in pYAAAA and favor cross-linking inpositions remote from the pY phosphate. Another conspicuous result revealed by the present study wasthe large negative effect on cross-linking of N-terminal acetylationin all pXAAAA peptides (Table 2). This may result from a combina-tion of a steric effect of the N-acetyl group, hindering hydrogenbonding at the phosphopeptide N-terminus, and its diminishedbasicity that would disfavor zwitterionic protomers of the phos-phopeptide. 6. Conclusions Photodissociative loss of N2 from non-covalent complexes ofpXAAAA phoshopeptides with peptides tagged at the N-terminusand containing a single arginine residue produced large (75-95%)fractions of stable surviving adducts. The distribution of non-covalent and covalently cross-linked photoproducts was found todepend on several factors, such as the photopeptide chain length,nature of phosphorylated amino acid residue, and phosphopeptideN-terminal modification. CID of the photoproducts revealed H3PO4transfer onto the photopeptide followed by unusual backbonedissociations in the phosphopeptide residue. Structure analysisby density functional theory and Born-Oppenheimer moleculardynamics trajectory calculations indicated dominant polar interac-tions in the complexes involving the charged arginine,N-terminal,and phosphate groups. Acknowledgments Financial support from the Chemistry Division of the NationalScience Foundation (Grants CHE-1661815 for experiments andCHE-1624430 for calculations) is gratefully acknowledged. F.T.thanks the Klaus and Mary Ann Saegebarth Endowment for generalsupport. Appendix A. Supplementary data Supplementary material related to this article can be found,in the online version, at doi:https://doi.org/10.1016/j.ijms.2018.09.037. ( References ) ( [1] E.H. Fisch e r, E.G. Kre b s,J. B iol. Chem.2 1 6(195 5 ) 1 2 1 - 1 32 . ) ( [2] P . Cohen,Nat. Cell Biol. 4(2002)E127-E130. ) ( 3 3] M 1 .A.McKercher, X . Guan, Z. Tan, D.S. Wuttke, Proteins 86 (2018)164- 1 76. 4 K .A. Schug, W. Li n dner, Chem. R ev. 105 (2005)67-1 1 3. 5 ) ( G . W aksman , D. K o m in o s, S.C . Ro b ertson , N. Pant, D. Baltimore, R.B. B i rge, D . ) ( Cowburn, H . Ha n afusa, B.J. Mayer, M. Ov e rduin, et al . , Nature 3 5 8(1992) 646-653. ) ( [ 6] S .M. P ascal, T . Yamazaki, A.U. Singer,L. E . Kay, J.D . F o rman- K a y , Biochemistry 3 4(1995)11353- 1 1362. ) ( ] J . A. Loo, Mass Spectrom. R ev. 1 6 ( 1 997)1-23. ) ( J . A. Loo, P . Hu, P . McCo n nell , W.T . Muell e r, T . K. Sawyer, V. T hanabal,J. A m . Soc. M ass S pectrom. 8 ( 1997)234-243. ) ( [9] K .A. S annes - L o w ery, P. H u , D.P . Mack, H . - Y . M e i , J.A. Loo, Anal. C hem. 69 ) (1997)5130-5135. ( [10] A.S.Woods, S. Ferre, J . Proteom e Res. 4(2005 ) 1 397 - 1 402. ) ( [11] S .N. J ackson , S.C . Moyer, A.S. Woods, J . Am. Soc. Mass Spectrom. 1 9 ( 2008) ) 1535-1541. ( [ 1 2] A .S. Woods, S.C. Moyer, S.N. Jackson J. Proteome Res. 7 (2008) 3 423 - 3427. ) ( [ 1 3] J . Lask in , Z . Y a ng, A.S. Woods, Phys. C hem. Chem. P h y s . 1 3 ( 2011) 6936-694 6 . ) ( [14] M. Suchanek, A. Radz i kowska, C. T hiele, Nat . Met h ods 2 (2005)261-26 8 . ) ( [ 1 5] T . Ya ng ,X.-M. L i , X . Bao, Y.M.E. Fung, D. L i , Nat . C he m . B i o l .12 (2 0 16) 7 0. ) ( |16| R . P ep i n, C . J . Shaffe r , F . Turece k, J. Mass S pec t rom . 52 (2017)557-5 6 0 . ) ( [ 1 7] Y . L iu , Z . Ramey, F . Tu r e c ek, Chem. E ur . J . 24 ( 201 8 ) 9 259 -9 263. ) ( [18| H .T.H. N guyen, P.C. Andriko p oulos, L. R u lisek, C.J. Shaffer, F . T urecek , J . Am. Soc.Mass Spec t rom.30 (2018)1706- 1 720. ) ( [ 19] C . J. S haffer, P.C. A nd r iko p oulos, J . Rezac , L. R ul i sek, F. Tu r ecek, j. Am . Soc. Mass Spectrom.27 (201 6 ) 633- 6 45. ) ( [20] M .T.H . Liu, Chemistry of Dia z irin e s, Vols. I a nd II , CRC P r e s s , B o c a Ra t on , FL,1987. ) ( [21] C.J. Shaffer, J . Martens, A. Mar e k, J . Oomens, F. Tu r ecek, J . Am. S oc.M a s s Spectrom. 27 (20 1 6 ) 1 1 76- 1 1 85. ) ( [22 A. Marek, F.Tu r ecek, J . Am. Soc. Mass Spectrom. 25 (2014)778-789. ) [231J.M. Janz, Y. Ren,R. Looby, M.A. Kazmi, P. Sachdev, A. Grunbeck, L. Haggis, D. ( C h i nnapen, A . Y . Lin , C . Seibert, T . M cMurry, K. E . C a r ls o n, T.W . Mui r , S. H unt , T . P .Sakmar, J . Am. Chem. Soc . 133 (2011) 1 5878-1 5 881. ) [24]C.J. Shaffer, A. Marek, R. Pepin, K. Slovakova, F. Turecek, J. Mass Spectrom. 50(2015)470-475. ( [25 H .T.H. Nguyen,C . J . S h affe r , F.Turecek, j. Phys. Che m . B 1 1 9 (2015)3948 - 3 961 . ) ( [26 C .J. S ha ffer, A . Marek,H.T.H. Ngu y e n , F. T urecek, J . Am. Soc. Mass Sp e ctrom . 2 6 (2015)1367-1381. ) ( [27] H .J . Berendsen,J . V. P ostma, W.F. van G uns t eren, A.R.H.J. D iNola,J . R. H aa k ,J . Chem. Phys.81 ( 1984)3684 - 3 690. ) ( [ 2 8] J . Rezac , J . Fanfrl i k, D . S a l a h ub , P. Hob z a,J. Chem . Theory Com p ut. 5 (2009)1 7 49- 1 760. ) ( [ 2 9] J.J.P. Stewar t , MOPAC 16; Stewart Computational Chemistry: ColoradoSprings, CO, USA. ) ( [30] J.Rezac, Cuby - Ruby Framework for Computational Chemistry, Version 4 h t t p://cu b y4 . m o lecul ar .cz. ) ( [31] J . Rezac , j. Comput. C hem. 37 (2016)1230- 1 237. ) ( [32] M .J . F risch, G . W. Trucks, H.B. Schleg e l, G . E. S c useria, M.A . Robb, J . R. C heeseman, G. Scalmani,V. Ba r o ne, G.A. Pe t ersson, H. Na k atsuji, M. C a ricato, A .V. M a re ni ch , J. B loino, B .G. Janesko,R. Gomperts , B . M e nnuc c i , H.P . H ratchian, J.V. Ortiz, A.F. Izm ay l ov ,J .L. So n nenberg, D. Williams- Y oung, F . D ing, F . Lipparini, F. Egidi, J. Goings , B . Peng , A. Petron e , T . H e nderson, D . R anasinghe, V . G. Z ak r zewski,J. Gao, N . R ega,G. Z h eng, W. Li a n g , M. H ada,M. E hara, K. T oyota, R . Fukuda,J. Hasegawa,M. I shida , T. N a kajima , Y . Honda,O. K itao, N . N a k ai,T. Vrev e n, K. T hrossell, J . A . M o ntgom e r y J r , J.E. Pe r a lta,F.Ogliaro, M. J . Bearpark, J . J . H ey d,E.N. Brothers, K.N. Kudin,V.N. Staroverov,T. A . K eit h ,R. K obayashi, J. N ormand, K . Raghavachari, A .P. Re n dell, J.C. Bu r ant, S.S. I yengar,J . T omasi, M . Cossi, J.M. Millam,M. Klene,C . Adamo, R. Cammi,J.W. Ocht e rski, R . L. Martin, K . M o rokuma , O . F ar k a s , J.B. Fo r esman, J .D. Fox, G a u ssian 1 6 , Revisio n A 01, Gaussian, In c ., Wallingford CT, 2 0 16. ) ( [ 3 3] A .D. B ec k e, P hy s. Rev . A 3 8 (1988)3098-3100. ) ( [34] J . D . C h ai , M. Head - Gordon, Ph y s. C h em. Chem.Phys . 1 0 (2008) 6 6 15-6620. ) We report a study of non-covalent complexes of phosphopeptides pXAAAA and N-Ac-pXAAAA (X = Ser,Thr, Tyr) with arginine-containing peptides carrying diazirine 4,4-azipentyl tags at the N-terminus,*LGG(A)nR, or in the photoleucine residue, L*GG(A)nR (n = 3–5). Complexes with *LGG(A)nR were successfully generated as singly charged ions in the gas phase in 0.6–3.5% yields. In contrast, complexes with L*GG(A)nR were formed in negligible (<0.1%) yields. Selective photodissociation at 355 nm of the diazirine ring in [*LGG(A)nR + pXAAAA + H]+ ions resulted in loss of N2, producing large (75–95%) fractionsof non-dissociating complexes that were further probed by collision-induced dissociation tandem mass spectrometry (CID-MS3). Covalent cross-links in the complexes produced by photoinduced carbene insertion were identified by specific CID-MS3 dissociations involving H3PO4 transfer and backbone fragmentations. N-acetylation in the phosphopeptides was found to have a substantial negative effect on the formation of covalent cross-links. Amongst the phosphorylated amino acid residues, pTyr showed the highest tendency (up to 92%) to form covalent cross-links. The fractions of covalent cross-links substantially increased with the length of the photopeptide chain. Born-Oppenheimer molecular dynamics (BOMD) calculations of canonical and zwitterionic protomers of the (pYAAAA + *LGGAAAR + H)+ complex indicated multiple close contacts between the incipient carbene of the diazirine ring and the X H bonds (X C, N, O) in the phosphopeptide. BOMD in combination with structural analysis by density functional theory calculations were used to interpret the experimental data and explain the cross-linking efficiencies.
确定






还剩11页未读,是否继续阅读?

产品配置单

北京欧兰科技发展有限公司为您提供《非共价肽肽离子复合物中气相交联检测方案(激光产品)》,该方案主要用于其他中理化分析检测,参考标准--,《非共价肽肽离子复合物中气相交联检测方案(激光产品)》用到的仪器有NL300系列高能量电光调Q激光器
推荐专场






相关方案
更多
该厂商其他方案
更多