








采用立陶宛EKSPLA公司NT342B-20-SH-SFG型纳秒波长可调谐光学参量振荡器(OPO)对镁离子水合物(Mg(H2O)n]+, n = 20–70)的电子光谱与纳米量热法进行了测量研究,讨论了是否会自发形成水合电子的问题。
方案详情

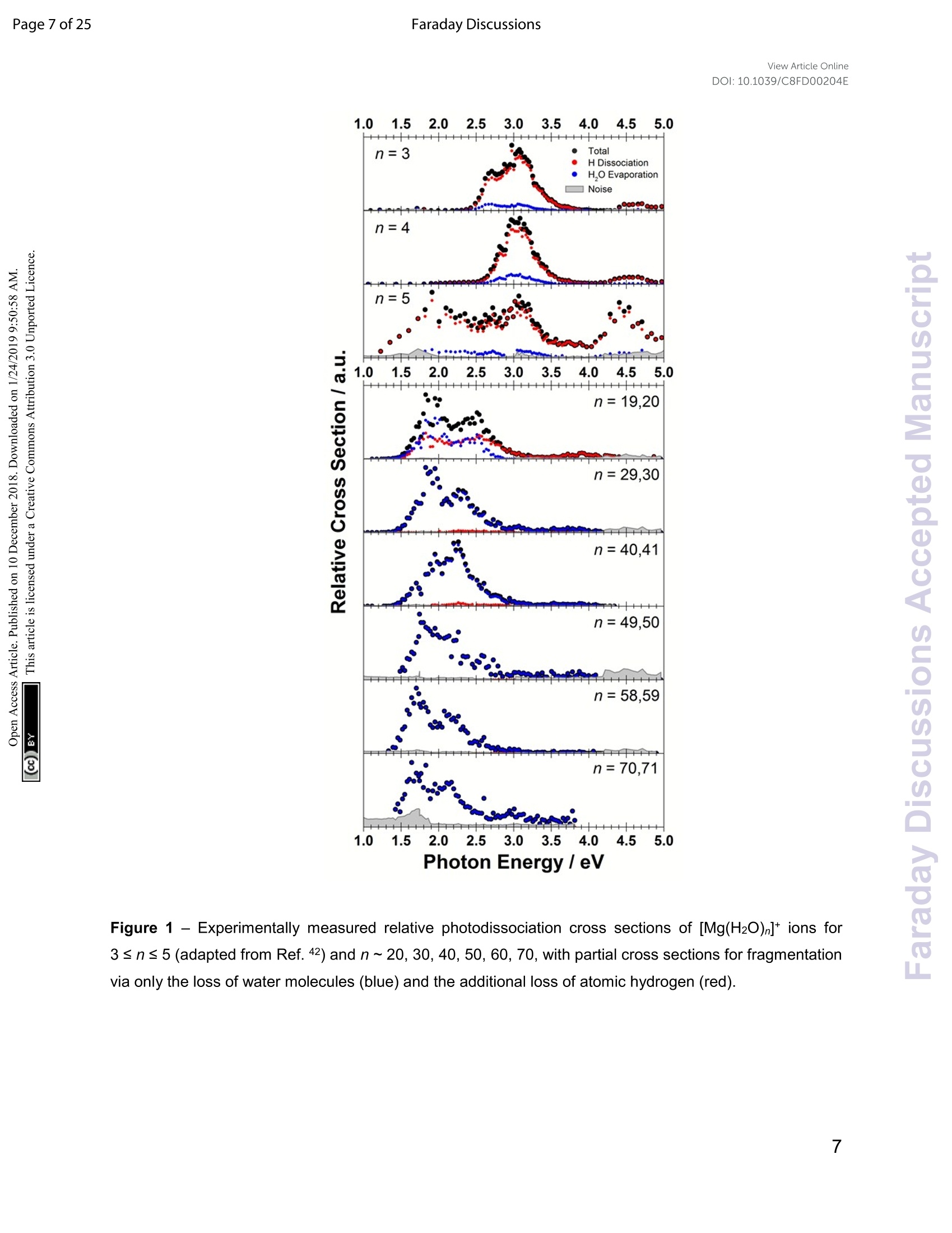
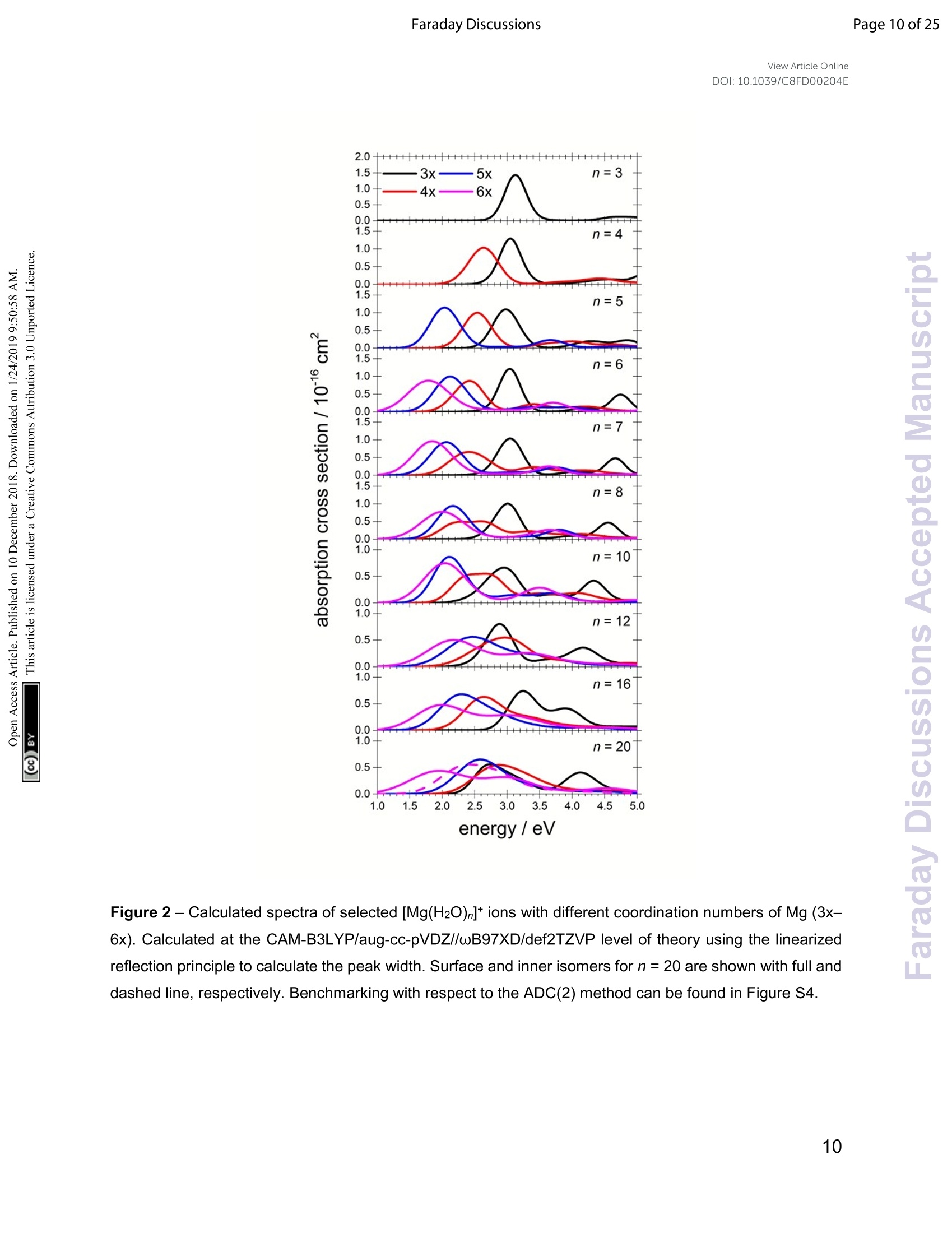
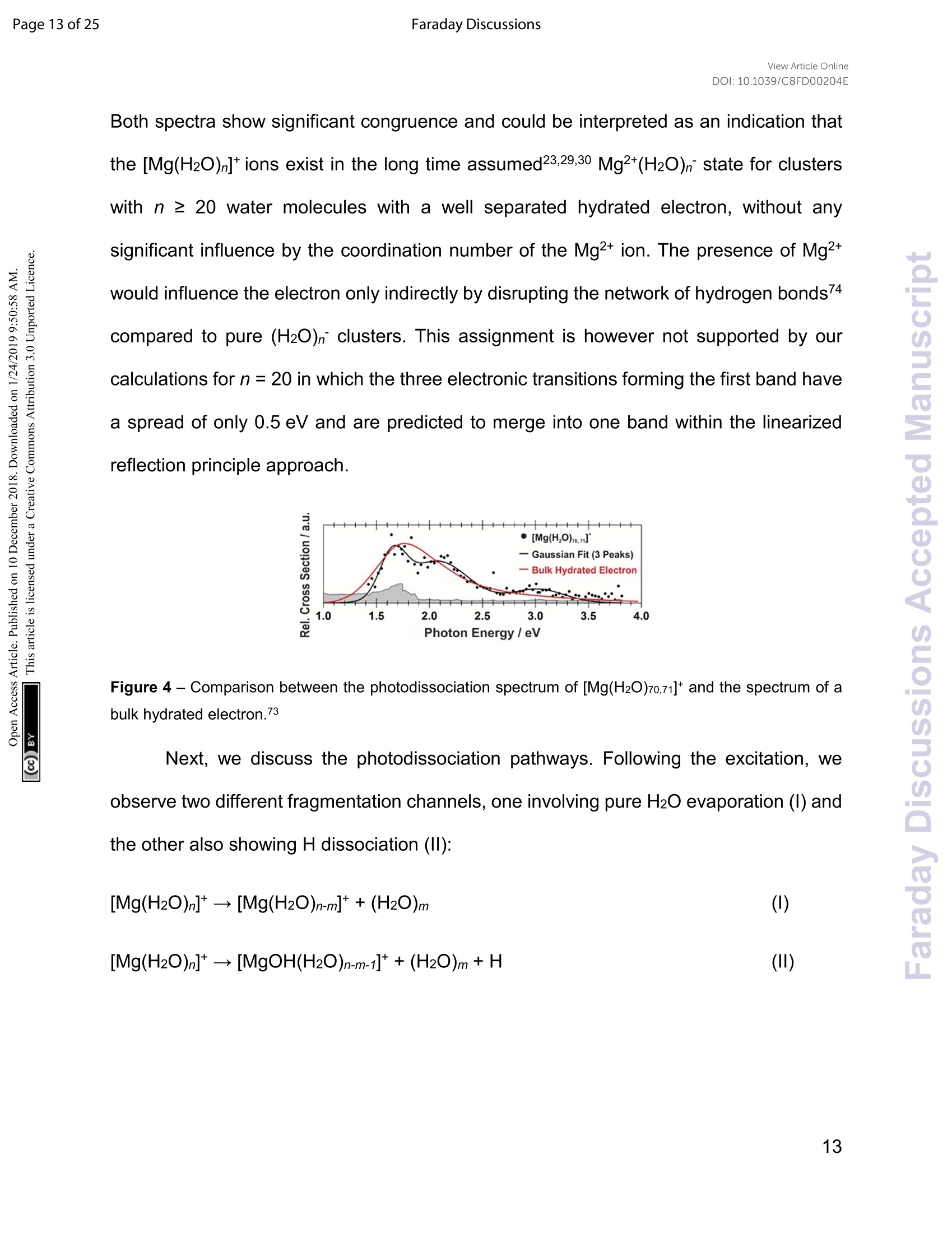
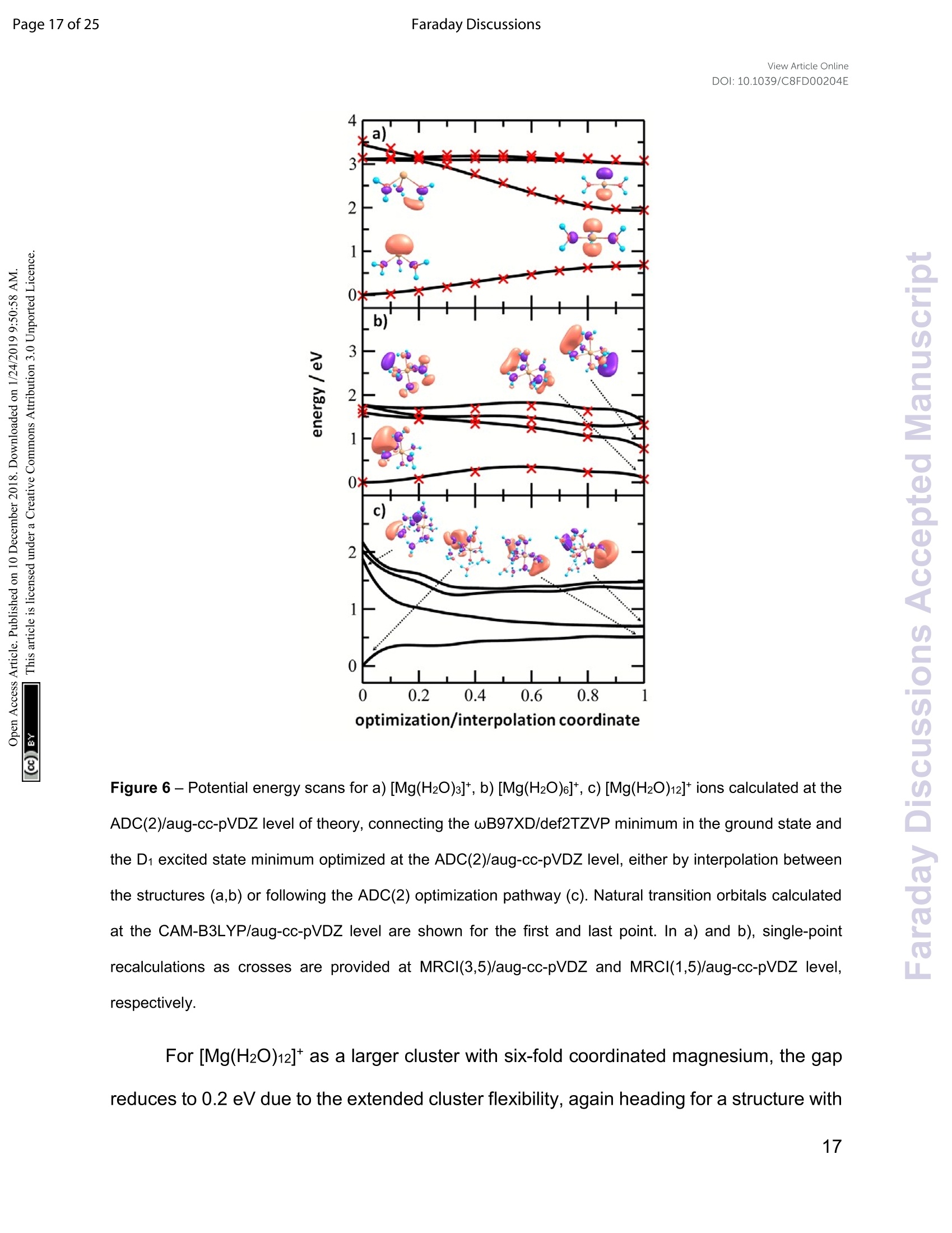
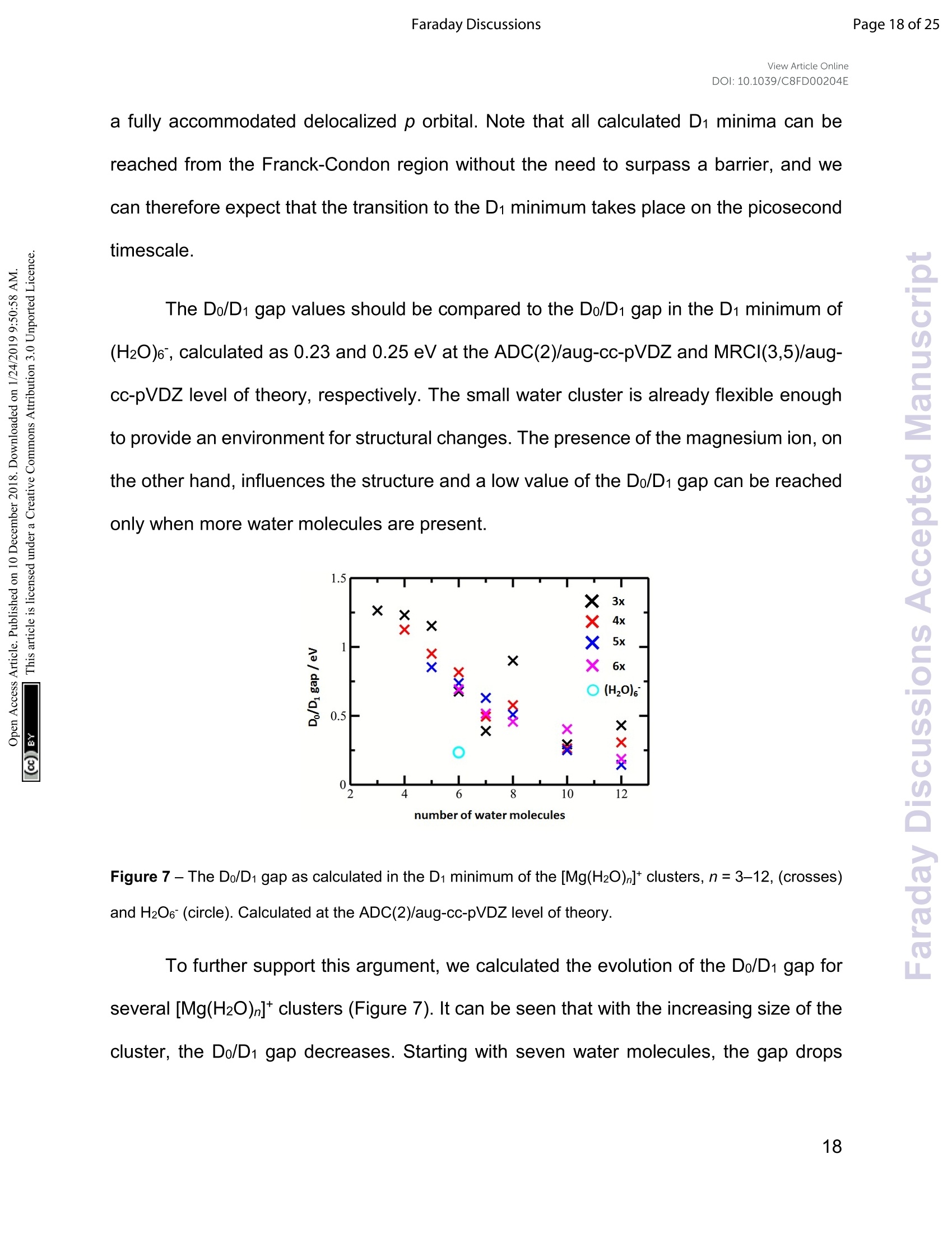
View Article OnlineView Journal Page 1 of 25Faraday DiscussionsView Article OnlineDOI: 10.1039/C8FD00204E FF.aradayDiscussions Accepted Manuscript This manuscript will be presented and discussed at a forthcoming Faraday Discussion meeting.All delegates can contribute to the discussion which will be included in the final volume. Register now to attend! Full details of all upcoming meetings:http://rsc.li/fd-upcoming-meetings This is an Accepted Manuscript, which has been through theRoyal Society of Chemistry peer review process and has beenaccepted for publication. Accepted Manuscripts are published online shortly afteracceptance, before technical editing, formatting and proof reading.Using this free service, authors can make their results availableto the community, in citable form, before we publish the editedarticle. We will replace this Accepted Manuscript with the editedand formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in theInformation for Authors. Please note that technical editing may introduce minor changesto the text and/or graphics, which may alter content. The journal'sstandard Terms 8 Conditions and the Ethical guidelines stillapply. In no event shall the Royal Society of Chemistry be heldresponsible for any errors or omissions in this Accepted Manuscriptor any consequences arising from the use of any information itcontains. This article can be cited before page numbers have been issued, to do this please use:T. Taxer, M. Oncak, E. Barwa, C.van der Linde and M. K. Beyer, Faraday Discuss., 2018, DOI: 10.1039/C8FD00204E. www.rsc.org/faraday_d Electronic Spectroscopy and Nanocalorimetry of Hydrated Magnesiumlons [Mg(H2O)n]*, n=20-70:Spontaneous Formation of a HydratedElectron? Thomas Taxer, Milan Oncak,* Erik Barwa, Christian van der Linde,Martin K. Beyer* Institut fur lonenphysik und Angewandte Physik, Universitat Innsbruck,TechnikerstraBe 25, 6020 Innsbruck, Austria * Corresponding authors: milan.oncak@uibk.ac.at; martin.beyer@uibk.ac.at Abstract Hydrated singly charged magnesium ions [Mg(H2O)n]t are thought to consist of an Mg2*ion and a hydrated electron for n> 15. This idea is based on mass spectra, which exhibita transition from [MgOH(H2O)n-1]* to[Mg(H2O)n]* around n = 15-22, black-body infraredradiative: dissociation. and quantumi Cchemical calculations.. FHere. we presentphotodissociation spectra of size-selected [Mg(H2O)n]* in the range of n = 20-70measured for photon energies of 1.0-5.0 eV. The spectra exhibit a broad absorption from1.4 to 3.2 eV, with two local maxima around 1.7-1.8 eV and 2.1-2.5 eV, depending oncluster size. The spectra shift slowly from n= 20 to n = 50, but no significant change isobserved for n=50-70. Quantum chemical modeling of the spectra yields severalcandidates for the observed absorptions, including five- and six-fold coordinated Mg2+ witha hydrated electron in its immediate vicinity, as well as a solvent-separated Mg2*/e pair. The photochemical behavior resembles the one of the hydrated electron, with barrierlessinterconversion into the ground state following the excitation. Keywords: hydrated electron, photochemistry, hydrogen evolution, photodissociation,nanocalorimetry I. Introduction Hydrated metal ions are a useful and well-defined model to examine fundamentalchemical reactions including hydrogen production or corrosion mechanisms and toinvestigate the link from small metal-water complexes to bulk aqueous solution.1-11 Amongvarious hydrated metal ions,[Mg(H2O)n]* has received continuous attention.12-40 Hydratedsingly charged magnesium ions can be found in two different species, depending onhydration number. For n≤5 and n≥ 17, hydrated magnesium ions [Mg(H2O)n]t aredominant and, in the range of 6≤n≤16, hydrated magnesium hydroxide ions[MgOH(H2O)n-i]* are almost exclusively found.18,19,28,29,41 In our previous work on this topic,42 we investigated the photochemistry of smallcluster sizes with 1≤n≤5, expanding on earlier experimental15,16,18,19,21,22,28,29 andtheoretical 14,23,24,43 results. In our measured photodissociation spectra, we could assignspecific isomers, rproviding insight into the electronic structure and dissociationmechanisms, which involve water evaporation and hydrogen dissociation. In the intermediate range of 6 ≤n≤16, lwata and co-workers proclaimed anegative energy for the hydrogen elimination process for n≥6, which explains theswitching to [MgOH(H2O)n-1]* in the ion formation in this size region.23 The re-switchingback to [Mg(H2O)n]+ for larger clusters was explained through the formation of amagnesium di-cation and a hydrated electron Mg?*(H2O)n for n> 14.23 Berg et al. alsosuggested the formation of a solvent separated ion pair Mg2+and a hydrated electron forn> 17.29 Siu and Liu proposed an increase in the barrier when the solvated electronmoves beyond the third solvation shell as an explanation for the switch off for the hydrogenloss process.32 Reinhard and Niedner-Schatteburg proclaimed the formation of a hydratedelectron and a magnesium di-cation already for n≥8 and suggested the existence of acontact ion pair state for 6≤ n< 17 and a solvent separated ion pair for n≥17.30 Here, we focus on the spectroscopy of larger [Mg(H2O)n]* clusters. Hydratedmagnesium ions up to n= 80 in the ground electronic state were already examined byBerg et al.28,29 who measured black body infrared radiative dissociation (BIRD) rates upto n=41. They let larger clusters fragment down to smaller sizes and found waterevaporation as the only dissociation process for n≥ 22 and water evaporation andhydrogen dissociation in the range of 16 ≤ n≤ 21 whereas for n≤ 15, all clusters weretransformed into the [MgOH(H2O)n-1]* species. This was argued to happen as a result ofthe removal of the stabilizing solvent and the recombination of the Mg2+(H2O)n ion pair.We follow up on this work and study photodissociation spectra of the larger [Mg(H2O)n]tclusters, n=20-70. We describe various dissociation pathways and similarities to thebehavior of the hydrated electron, (H2O)n. Experimental results are supported by ab initiocalculations. Il. Methods A detailed description of the experimental setup can be found in earlier works.44-47A Fourier Transform lon Cyclotron Resonance Mass Spectrometer (FT-ICR-MS),equipped with a 4.7 T superconducting magnet was used to conduct the experiments.Mg*ions are generated in an ion source via laser vaporization of an isotopically enriched(99% 24Mg) rotating target disk, using the 2nd harmonic of a Nd:YAG laser. The ions thenare entrained by a gas pulse of helium seeded with water vapor at 20 bar and expandedinto high vacuum which leads to cluster formation of [Mg(H2O)n]. The ions are stored anddetected in a liquid nitrogen cooled ICR cell (T~90±5 K) to minimize the influence ofblackbody infrared radiative dissociation (BIRD).48-58 Ion selection is performed viaresonant ejection of ions with an unwanted mass to charge ratio. Because of the relativelyhigh BIRD fragmentation rates for these large cluster sizes, a combination of [Mg(H2O) ]+and [Mg(H2O)n-1]t (the primary BIRD fragment) was selected for the experiments. Alsopresent [MgOH(H2O)n-1, n-2]*species were not ejected to avoid unintentional excitation ofthe ions of interest to higher cyclotron radii and thus diminishing their overlap with thedissociation laser beam. The presence of these species does not influence our results asthey do not absorb photons in the wavelength range of interest (see Section 4 in the SIfor details).Photodissociation is induced via irradiation with the beam of a tunablewavelength, pulsed (20 Hz) UV/VIS/NIR laser system (Nd:YAG pumped OPO systemEKSPLA NT342 B-20-SH-SFG) with irradiation times up to 1 s. A more detaileddescription of the laser setup is available elsewhere.9 Due to the high absorption cross section of [Mg(H2O)n]t, sequential absorption ofseveral photons is frequently observed. Similar to earlier work,60,61 the average waterbinding energy was extracted from the photodissociation data using the equation=Eyy/m. Here, Ey is the photon energy for which the partial cross section of aspecific fragmentation channel [Mg(H2O)]*→[Mg(H2O)n-m]+ +(H2O)m reaches itsmaximum (see Section 6 in SI for details). This energy is then multiplied by the number ofphotons y involved in the dissociation and divided by the number of lost water moleculesm. The photon flux inside the cell is strongly dependent on the wavelength, which is atypical feature for OPO systems. In our experiments, it was on the order of 0.25-7 mJ/cm2.Directly after irradiation, mass spectra of fragment and parent ions were recorded. Fromtheparenttaandj fifragmentintensities,, along\with1the: laserpower,tthe relativephotodissociation cross sections were calculated using Lambert-Beer's law (see Section1 in the SI). The photochemistry of the [Mg(H2O)n]*,n=3-8,10,12,16,20,clusters was modeledusing methods of theoretical chemistry, with four types of clusters considered: with athree-, four-, five- and six-times coordinated Mg center (denoted also as 3x, 4x, 5x, and6x; see Figure S3 for all structures considered). The ground state structure of the clusterswas optimized at the wB97XD/def2TZVP level of theory.62 Excited states were calculatedusing time-dependent density functionaltheory (TDDFT) withthe : iCAM-B3LYPfunctional,63 completeactive space - self consistent field (CASSCF) with subsequentmulti-reference configuration interaction(MRCI)and ttlhe algebraic diagrammaticconstruction to the second order (ADC(2))64 methods. The ADC(2) method was used for benchmarking the CAM-B3LYP calculations. The results are shown in Figure S4. For theexcited states calculations, the aug-cc-pVDZ basis set was used. It was shown previouslythat this basis is able to capture correctly the first three excited states that are mostrelevant for the present study.42 Quantitative differences for higher-lying states might beexpected. Excited states up to 5 eV or to the limit of 35 excited states were considered. [Mg(H2O)n]+ clusters were created by sequential addition of water molecules andre-optimization. For the [Mg(H2O)12]* cluster, molecular dynamics (MD) to sample thepotential energy surface for structure search was run at the BLYP/6-31+g* level at 400 Kwith a time step of 30 a.u. (~0.73 fs). After a thermalization period of 3 ps, a productionrun of 111 pswasconducted, withh112 structures taken for reoptimization at thewB97XD/def2TZVP level. The Gaussian program65 was used for ground-state and TDDFT calculations,Molpro66 for CASSCF/MRCI, Turbomole67 for ADC(2). The spectrum width was modeledusing the linearized reflection principle.68-71 MD calculations were performed in the Abinprogram.72 5.0Photon Energy/eV Figure 1 -Experimentally measured relative photodissociation cross sections of [Mg(H2O)n]+ ions for3 ≤ n ≤ 5(adapted from Ref. 42) and n~ 20, 30, 40, 50, 60, 70, with partial cross sections for fragmentationvia only the loss of water molecules (blue) and the additional loss of atomic hydrogen (red). III. Results and Discussion Measured photodissociation spectra for [Mg(H2O)n]t, n~20, 30,40, 50,60, 70 at90±5 K are shown in Figure 1. Spectra for small clusters for n = 3-5 recorded at atemperature of 130±20 K, which were already discussed elsewhere,42 are shown forcomparison. The spectra for n=1, 2 show two separated peaks, corresponding to the 3s-3px,y and 3s-3pz electronic transitions of the isolated magnesium cation that are perturbedby the influence of the water molecules.42 For n≥3, these transitions are not separatedanymore. The spectra for n= 1-3 show a significant redshift, caused by the increasingcoordination of the magnesium cation with water molecules. The spectrum for n = 3 alsoshows the 3s-3d/4s electronic transitions at the higher energy end at about 4.5 eV. Forn = 4, the redshift stops, indicating that the observed isomer is still a triply coordinatedmagnesium cation with a fourth water molecule already in the second solvation shell. Forn=5, we assume three different isomers contributing to the broad shape of the spectrum,a three-, four- and five times coordinated magnesium cation. In the range of n ~ 6-18, magnesium hydroxide clusters [MgOH(H2O)n-1]t arepredominantly formed in the ion source. Therefore, no photodissociation spectra could bemeasured (see also Section 4 in the SI). The hydroxide formation reaction is suppressedagain for n > 18.28,29 While the photodissociation spectra change substantially with every coordinatedwater molecule for small clusters of n=1-5, the spectra of larger clusters are very similarin the whole n= 20-70 range explored in this study, with only limited changes in theposition and shape with the increasing size of the clusters. The spectrum for n~ 20 showsa broad absorption with a width of about 1.8 eV spanning from 1.4 eV to 3.2 eV, with two distinguishable bands peaking around 1.9 eV and 2.5 eV. There is also a long shallow tailon the higher energy side of the band, extending up to about 4.4 eV before vanishing inthe noise level. Compared to the spectrum forn~ 20, the higher-energy peak for n ~ 30 is shiftedto the red by about 0.2 eV, with the redshift continuously increasing with more watermolecules to about 0.4 eV for n~70. The lower-energy peak for n~30 is at about thesame position as for n~ 20, shifts slightly to the blue for n~ 40 and back to the red forn> 40. Forn~ 30, 40, 50, 70 there also seems to be a third, less intense band at~ 3.0 eV. To interpret the observations, we modeled the spectra with ab initio calculations,using various [Mg(H2O)n]* structures, n=3-20, with a three-, four-, five-and six foldcoordinated Mg ion (Figure 2). Due to the size of the system, we have to limit ourselvesto a relatively small selection of structures. However, we observe clear trends in theevolution of the photodissociation spectrum as we discuss below. Consistently withprevious studies,30Wee ppredictt tthat the electron delocalizeswiththe increasingcoordination as can be documented e.g. for n=20 where the gyration radius increasesfrom 1.9 A to 2.4 A for three- and six-fold coordinated Mg ion, respectively (see alsoFigure 3). For coordination numbers 3-5, the electron is localized next to the Mg ion. Fora coordination number of 6,we considered two [Mg(H2O)20]+isomers,with the[Mg(H2O)6]*unit on the surface and hydrated in the cluster. In these two structures, the electron residesnext to the Mg2+ion or is shifted to the second solvation layer in surface and innerstructures, respectively. Figure 2-Calculated spectra of selected [Mg(HzO)]* ions with different coordination numbers of Mg (3x- 6x). Calculated at the CAM-B3LYP/aug-cc-pVDZ//wB97XD/def2TZVP level of theory using the linearizedreflection principle to calculate the peak width. Surface and inner isomers for n = 20 are shown with full anddashed line, respectively. Benchmarking with respect to the ADC(2) method can be found in Figure S4. Figure 3 - Structures for n = 20 along with the spin density calculated at the CAM-B3LYP/aug-cc-pVDZ//wB97XD/def2TZVP level. See Figure S1 for other structures. Calculated photoabsorption spectra in Figure 2 show that the Mg coordinationnumber has the most important influence on the spectrum shape, with hydration in furthersolvation layers inducing only a minor shift of the absorption bands. For a three-foldcoordinated magnesium, the first absorption band composed of s-p transitions peaks atabout 3.2 eV forn=3. With the increasing number of water molecules, its maximum stayswithin 2.8-3.4 eV and shifts due to interactions in the second solvation layer. There is adistinct second absorption band that shifts to a lower energy with the increasing size andcan be assigned to transitions of 3s-3d and 3s-4s character. For other coordination numbers of the Mg ion, we see a similar trend for the firstabsorption band, with only limited change with increasing hydration, with the shift of themaximum within 0.5 eV. The same order of the excitation energies, i.e. lower excitationenergies for clusters with a higher Mg coordination number, is preserved for all clustersizes with the exception of three- and four-fold coordinated clusters for n = 20. The second absorption band localized in the 2.5-4.5 eV region shifts to lower energies with increasingcluster size. At the same time, the spectral width calculated within the linearized reflectionprinciple is predicted to increase for larger clusters. With respect to the experimentalspectra, the bands for six-fold coordinated Mg at~1.9 eVand~3.0 eV are less separatedand the spectra are calculated to start already at 1.0 eV (compared to ~1.5 eV in theexperiment). This indicates that the width is overestimated by the linearized reflectionprinciple. For the surface isomer with n = 20, the absorption maximum is calculated at 2.0 eV,consistently with other clusters with a six-fold coordinated Mg ion. When the unpairedelectron is shifted to the second solvation layer with full hydration of the [Mg(H2O)6]unit,a shift of~0.4 eV to the blue is seen in the calculated spectrum. This shift is preserved inthe whole n = 17-20 series as we show in the SI (Figure S5). Comparing the calculated photoabsorption spectra to the measuredphotodissociation spectra, we can propose two interpretations of the results. We couldassign the two observed peaks in the experiment to five-fold and six-fold coordinatedmagnesium ions on the cluster surface. This would indicate that the coordinated Mg ionstays on the surface of the cluster in the whole investigated size range. Alternatively, wecould see the six-fold coordinated Mg ion in its two forms, i.e. with the unpaired electronin the vicinity of the Mg2+ion and in the second hydration layer. Still another possible interpretation of the experimental spectra would be to assignthe two bands to two and one s-p transitions of the hydrated electron, respectively,similarly to clusters with n=1, 2. A comparison of the spectrum of the largest measuredcluster with the spectrum of a hydrated electron in bulk water73 is provided in Figure 4. Both spectra show significant congruence and could be interpreted as an indication thatthe [Mg(H2O)]t ions exist in the long time assumed23,29,30 Mg2+(H2O)n state for clusterswith n ≥ 20 water molecules with a well separated hydrated electron, without anysignificant influence by the coordination number of the Mg2+ion. The presence of Mg2+would influence the electron only indirectly by disrupting the network of hydrogen bonds4compared to pure (H2O)n clusters. This assignment is however not supported by ourcalculations for n =20 in which the three electronic transitions forming the first band havea spread of only 0.5 eV and are predicted to merge into one band within the linearizedreflection principle approach. Figure 4-Comparison between the photodissociation spectrum of [Mg(H2O)70,71]* and the spectrum of abulk hydrated electron.3 Next, we discuss the photodissociation pathways. Following the excitation, weobserve two different fragmentation channels,one involving pure H2O evaporation (I) andthe other also showing H dissociation (II): [Mg(H2O)]+→[Mg(H2O)n-m]++(H2O)m (I) Figure 5 - Fragmentation pattern of a typical photodissociation experiment of [Mg(H2O)29,30]+. The twodifferent species of hydrated magnesium ions are represented by blue dots for [Mg(H2O) ]t ions and redsquares for [MgOH(H2O)n]tions. Color intensity represents measured ion intensity. Table 1- Average binding energy per water molecule (ineV). n\y 1 2 3 4 5 20 0.51 m 30 0.46 0.46 0.47 mmm m 41 0.49 0.48 0.47 0.47 0.48 50 0.45 0.45 0.47 0.47 0.47 59 0.47 0.46 0.46 0.46 0.47 71 0.43 0.45 0.46 0.46 0.47 Figure;5showsthe fragmentationpattern (of al typicallleexperimentfor[Mg(H2O)29,30]t, with fragmentations involving absorption of up to five photons (seeSection 3 in the SI for detailed spectra of all ions). The energetics of the channel (I) in theexperiment was analyzed through nanocalorimetry.44 We found that the average energy over all cluster sizes carried away by a water molecule is about 0.47(3) eV (Table 1), closeto the literature value for the binding energy of a water molecule to large ionic clusters (forn≥40) of about 0.447(4) eV.75 In other words, the absorbed photon energy is quantitativelyused for water dissociation, indicating that dissociation takes place in the electronicground state of the system. Following the excitation, the system thus reaches a conicalintersection and dissociates water molecules after funneling into the ground electronicstate. Note that this is a completely opposite behavior to the one observed for n=1-5.42We observe cases where more photons are absorbed than laser pulses applied duringthe irradiation time window, which means that several photons must be absorbed duringone laser pulse. As we will discuss below, it can be expected that internal conversiontakes place on the picosecond time scale. Thus, within the 5 ns of a typical laser pulse, itis possible for a cluster ion to absorb one or more photons in the electronic ground state.As dissociation of water molecules may take place faster than 5 ns, fragment ions createdwith more than a single photon could be direct fragments of the precursor ions as well assecondary products. Hydrogen atom dissociation, the second channel (Ⅱ), produces [MgOH(H2O)]*ions that do not absorb further photons in the considered wavelength range. Below a limitof n = 15 water molecules, only [MgOH(H2O)n]+ ions are observed as fragmentationproducts whereas above this value, [Mg(H2O)n]+ions are the dominant species (Figure 5).Formation of magnesium hydroxide species in this range agrees quantitatively with thebehavior of the [Mg(H2O)n]* clusters in the ground state investigated in previous massspectrometricexperiments.18,19,28,29Therefore.we(conclude that the hIydrogerdissociation reaction (II) also takes place in the ground electronic state. To calculate the partial cross sections of H2O evaporation and H dissociation inFigure 1, only [MgOH(H2O)n]t products created with the first absorbed photon areconsidered for the H dissociation channel and all fragments created by multiple photonsare counted for the H2O evaporation channel. For n=20, the partial cross sections of thetwo fragmentation pathways are about the same order of magnitude, but larger clustersfragment almost exclusively via the loss of water molecules. To analyze the photochemistry of thee[[Mg(H2O)n]+ ions,we investigatedphotochemical pathways using methods of theoretical chemistry. When a [Mg(H2O)n]+cluster is excited into one of the three close-lying electronic states (D1-D3), we can expectthat it quenches to the D1 state and evolves further on this potential energy surface. Forthe possible transfer back into the ground state as seen in the experiment, the size of theDo/D1 gap is crucial. We have already shown that for the [MgH2O] ion, the Do/D1 gap isalmost identical to the Do-D1 excitation energy due to negligible geometry relaxationfollowing the excitation.42 For n=2, 3, we already saw considerable relaxation with thetendency to linearize or planarize, decreasing the gap.42 Figure 6 shows the evolution of photochemical pathways for [Mg(H2O)n]t,n=3,6,12. For [Mg(H2O)3]*, optimization in the first excited :state D116leadsttoplanarization. This can be understood in terms of optimizing the molecular structure forthe 3p(Mg) orbital in which the electron is localized after excitation.The Do/D1 gap in theD1 minimum is about 1.3 eV. For [Mg(H2O)6]t, only OH bond orientation changes toprovide more suitable environment for an orbital of p character delocalized over the wholecluster. Calculations yield a Do/D1 gap of 0.6 eV. optimization/interpolation coordinate Figure 6- Potential energy scans for a) [Mg(H2O)3], b)[Mg(H2O)6]t,c)[Mg(H2O)12]t ions calculated at theADC(2)/aug-cc-pVDZ level of theory, connecting the wB97XD/def2TZVP minimum in the ground state andthe D1 excited state minimum optimized at the ADC(2)/aug-cc-pVDZ level, either by interpolation betweenthe structures (a,b) or following the ADC(2) optimization pathway (c). Natural transition orbitals calculatedat the CAM-B3LYP/aug-cc-pVDZ level are shown for the first and last point. In a) and b), single-pointrecalculations as crosses are provided at MRCI(3,5)/aug-cc-pVDZ and MRCI(1,5)/aug-cc-pVDZ level,respectively. For [Mg(H2O)12]* as a larger cluster with six-fold coordinated magnesium, the gapreduces to 0.2 eV due to the extended cluster flexibility,again heading for a structure with a fully accommodated delocalized p orbital. Note that all calculated D1 minima can bereached from the Franck-Condon region without the need to surpass a barrier, and wecan therefore expect that the transition to the D1 minimum takes place on the picosecondtimescale. The Do/D1 gap values should be compared to the Do/D1 gap in the D1 minimum of(H2O)6, calculated as 0.23 and 0.25 eV at the ADC(2)/aug-cc-pVDZand MRCI(3,5)/aug-cc-pVDZ level of theory, respectively. The small water cluster is already flexible enoughto provide an environment for structural changes. The presence of the magnesium ion, onthe other hand, influences the structure and a low value of the Do/D1 gap can be reachedonly when more water molecules are present. Figure 7 -The Do/D1 gap as calculated in the D1 minimum of the [Mg(H2O)n]tclusters, n=3-12, (crosses)and H2O6(circle). Calculated at the ADC(2)/aug-cc-pVDZ level of theory. To further support this argument, we calculated the evolution of the Do/D1 gap forseveral[Mg(H2O)n]* clusters (Figure 7). It can be seen that with the increasing size of thecluster, the Do/D1 gap decreases. Starting with seven water molecules, the gap drops below 0.5 eV and might reach values below 0.25 eV already for n = 12 (see Figure S6 foroptimized structures). We can thus propose that the photochemistry of larger [Mg(H2O)n]+clusters issimilar to the photochemistry of an electron on water clusters. After excitation, they reacha D1 minimum without any barrier and switch back to the Do ground electronic state thatlies energetically close to D1 in this region. Then, the clusters follow the known groundstate reactivity,28,29 i.e. evaporate water molecules or dissociate a hydrogen atom toproduce hydrated magnesium hydroxide for lower values of n. Conclusions Hydrated magnesium ions [Mg(H2O) ]+ exhibit a broad electronic absorptionspectrum from 1.4-3.2 eV, in the same range as the hydrated electron. The spectrum is,however, more structured, with two pronounced local maxima and possibly even a thirdone. Based on quantum chemical calculations, these maxima can be assigned to differentstructural motifs, in particular five- and six-fold coordinated magnesium, or six-foldcoordinated magnesium with two distinct hydration patterns. Due to the vast conformationspace of these species, however, only a limited number of structures could be investigatedcomputationally, which does not allow for a definitive assignment of the observed features.After excitation, the excitation energy is quantitatively used for dissociative reactions. Wepropose that the D1 minimum is reached on the picosecond timescale and the systemmight switch to the Do ground state due to a low value of the Do/D1 gap in the vicinity of the D1 minimum for larger clusters. This resembles the photochemistry of the hydratedelectron in (H2O)n clusters. Conflicts of interest There are no conflicts of interest to declare. Acknowledgement T.T. and M.K.B. acknowledge financial support from the Austrian Science Fund (FWF)through project No. P29174. M.O. acknowledges the support through the Lise MeitnerProgramme of the FWF project No. M2001-NBL. The computational results presentedhave been achieved using the HPC infrastructure LEO of the University of Innsbruck. Thetunable OPO system is part of the Innsbruck Laser Core Facility, financed by the AustrianFederal Ministry of Science, Research and Economy. 1 V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Splitting Water with Cobalt, Angew. Chem. Int. Ed.,2011,50,7238. 2 C. Tard and C. J. Pickett, Structural and functional analogues of the active sites of the Fe-, NiFe-, andFeFe-hydrogenases, Chem. Rev.,2009,109,2245. 3 S. Canaguier, V. Artero and M. Fontecave, Modelling NiFe hydrogenases: nickel-based electrocatalystsfor hydrogen production, Dalton Trans., 2008, 25,315. 4 B. E. Barton, C. M. Whaley, T. B. Rauchfuss and D. L. Gray, Nickel-iron dithiolato hydrides relevant tothe NiFe-hydrogenase active site, J. Am. Chem. Soc., 2009, 131, 6942. 5 R. Brimblecombe, G. F. Swiegers, G. C. Dismukes and L. Spiccia, Sustained water oxidationphotocatalysis by a bioinspired manganese cluster, Angew. Chem. Int. Ed., 2008,47,7335. 6 M. K. Beyer, Hydrated Metal lons in the Gas Phase, Mass Spectrom. Rev., 2007, 26, 517. 7 M. A. Duncan, Spectroscopy of Metal lon Complexes: Gas-Phase Models for Solvation, Annu. Rev.Phys.Chem., 1997, 48,69. 8 K. Fuke, K. Hashimoto and S. Iwata, Structures, spectroscopies, and reactions of atomic ions with waterclusters, Adv. Chem. Phys., 1999,110,431. 9 V. E. Bondybey and M. K. Beyer, How Many Molecules Make a Solution?, Int. Rev. Phys. Chem., 2002,21,277. 10 G.Niedner-Schatteburg and V. E. Bondybey, FT-ICR studies of solvation effects in ionic water clusterreactions, Chem. Rev., 2000, 100,4059. 111\W. A. Donald, R. D. Leib, J. T. O'Brien, A. I. S. Holm and E. R. Williams, Nanocalorimetry in massspectrometry: A route to understanding ion and electron solvation, P. Natl. Acad. Sci. USA, 2008, 105,18102. 12 C. K. Siu and Z. F. Liu, Reaction mechanisms for size-dependent H loss in Mg*(H2O)n: solvationcontrolled electron transfer, Phys. Chem. Chem. Phys., 2005,7,1005. 13 T.-W. Lam, C. van der Linde, A. Akhgarnusch,Q. Hao, M. K. Beyer and C.-K. Siu, Reduction of Acetonitrileby Hydrated Magnesium Cations Mg*(H2O)n (n20-60) in the Gas Phase, ChemPlusChem, 2013, 78,1040. 14 C. W. Bauschlicher and H. Partridge, A Determination of Mg*-Ligand Binding Energies, J. Phys. Chem.,1991,95,3946. 15 K. F. Willey, C. S. Yeh, D. L. Robbins and M. A. Duncan, Photodissociation spectroscopy of Mg*CO2,Chem. Phys. Lett., 1992, 192,179. 16 K. F. Willey, C. S.Yeh, D. L. Robbins, J. S. Pilgrim and M. A. Duncan, Photodissociation spectroscopy ofMg*-H20 and Mg*-D2O,J. Chem. Phys., 1992, 97, 8886. ( 17 C . S. Yeh, K. F. Willey, D. L. Robbins and M. A . Duncan, Photodissociatio n of magnesium i on/moleculecomplexes in a reflectron time-of-flight mass spectrometer, Int. J. Mass Spectrom. Ion Process., 1994, 131,307. ) ( 18 F. Misaizu, M. Sanekata,K. Fuke and S. Iwata, Photodissociation Study on Mg*(H2O)n, n=1-5:Electr o nic Structure and P hotoinduced Intracluster Reaction,J. Chem. Phys.,1994,100,1161. ) 19 F. Misaizu, M. Sanekata, K. Tsukamoto, K. Fuke and S. Iwata, Photodissociation of size-selectedaquamagnesium Mg*(H2O)n ions for n=1 and 2, J. Phys. Chem., 1992,96,8259. 20 M.Sanekata,F. Misaizu, K. Fuke, S. Iwata and K. Hashimoto, Reactions of Singly Charged Alkaline-EarthMetal lons with Water Clusters: Characteristic Size Distribution of Product lons, J. Am. Chem. Soc.,1995,117,747. 21 Y. Inokuchi,K. Ohshimo, F. Misaizu and N. Nishi,Infrared photodissociation spectroscopy of[Mg(H2O)1-4]*and [Mg.(H2O)1-4Ar]t, J. Phys. Chem. A, 2004,108,5034. 22 Y. Inokuchi, K. Ohshimo, F. Misaizu and N. Nishi, Structures of [Mg(H20)1,2]* and [AI·(H2O)1,2]* lonsStudied by Infrared Photodissociation Spectroscopy: Evidence of [HO-AI-H]+ lon Core Structure in[AI(H2O)2]t, Chem. Phys. Lett., 2004,390,140. 23 H. Watanabe, S. Iwata, K. Hashimoto, F. Misaizu and K. Fuke, Molecular-Orbital Studies of theStructures and Reactions of Singly Charged Magnesium-Ion with Water Clusters, Mg*(Hz0)n, J. Am.Chem. Soc., 1995,117,755. 24 H. Watanabe and S. Iwata, Theoretical assignments of the photo-dissociation excitation spectra of Mg*ion complexes with water clusters. Multi-reference Cl studies, J. Chem. Phys., 1998,108,10078. ( 25 R.C. D u nbar, In f rared Radiative Cooling of Gas-Phase lons, Mass Spectrom. Rev., 1992, 11, 309. ) 26 R. C. Dunbar and S. Petrie, Magnesium monocationic complexes: a theoretical study of metal ionbinding energies and gas-phase association kinetics,J. Phys. Chem. A, 2005,109, 1411. 27 VDI-Warmeatlas, Springer, Berlin, Heidelberg, New York, 10th edn., 2006. 28 C. Berg, U. Achatz, M. Beyer, S. Joos, G. Albert, T. Schindler, G. Niedner-Schatteburg and V. E.Bondybey, Chemistry and charge transfer phenomena in water cluster cations, Int. J. Mass Spectrom.,1997,167,723. 29 C. Berg, M. Beyer, U. Achatz, S. Joos, G. Niedner-Schatteburg and V. E. Bondybey, Stability andReactivity of Hydrated Magnesium Cations, Chem. Phys., 1998, 239, 379. 30 B. M. Reinhard and G. Niedner-Schatteburg, Co-Existence of Hydrated Electron and Metal Di-Cation in[Mg(H2O)n]t, Phys. Chem. Chem. Phys., 2002, 4, 1471. ( 31 1 B. M . Reinhard and G. Ni e dner-Schatteburg, loni z ation energies and s patial volumes of the singlyoccupied molecular orbital i n hydrated magnesium clus t ers [Mg,nH2O]t, J. C h em. Phys., 2003, 118, 3571. ) ( 32 C. K . Siu and Z. F. Liu, Ab Init i o Studies o n the Mechanism of th e Size-Dependent Hydrogen-LossReaction i n Mg*(H20)n, Chem. E ur.J., 2002,8,3177. ) ( 33 C . v an der Linde, A . Akhgarnusch, C.-K. S iu and M . K. B eyer, Hydrated Magnesium Cations Mg* ( H20)n, n20-60, Exhibit Chemistry of the Hydrated Elec t ron in Reactions with O2 and CO2, J. Phys . Chem . A, 2011,115,10174. ) ( 34 N . F. Dalleska, B . L. Tjelta and P . B . Armentrout, Sequential Bond E n ergies of Water to Na* (3s0), Mg* (3s1), and Al* (3s2), J. Phys. Chem . , 1994,98,4191. ) 35 C. L. Whalley, J. C. G. Martin,T. G. Wright and J. M. C. Plane, A kinetic study of Mg*and Mg-containingions reacting with O3, 02, N2, CO2, N20 and H2O: implications for magnesium ion chemistry in the upperatmosphere, Phys. Chem. Chem. Phys., 2011,13, 6352. ( 36 T. A sada a n d S. I w ata, H y brid procedure of ab initio molecular orbital calculation a n d Monte Car l o simulation for studying intracluster reactions. A p plications to Mg * (H2O)n(n=1-4), Chem. Phys. Lett., 1996,260, 1. ) 37 C. K. Siu and Z. F. Liu, Reaction mechanisms for size-dependent H loss in Mg*(H20)n. solvationcontrolled electron transfer, Phys. Chem. Chem. Phys., 2005,7,1005. 38 A. Atrens, G.-L. Song, F. Cao, Z. Shi and P. K. Bowen, Advances in Mg corrosion and researchsuggestions, Journal of Magnesium and Alloys, 2013,1,177. 39 C. J. Johnson, L. C. Dzugan, A. B. Wolk, C. M. Leavitt, J. A. Fournier, A. B. McCoy and M. A. Johnson,Microhydration of Contact Ion Pairs in M²+OH(H2O)n=1-5(M=Mg, Ca) Clusters: Spectral Manifestationsof a Mobile Proton Defect in the First Hydration Shell, J. Phys. Chem. A, 2014,118,7590. 40 E. Miliordos and S. S. Xantheas, Unimolecular and hydrolysis channels for the detachment of waterfrom microsolvated alkaline earth dication (Mg*, Ca, Sr2+, Ba?) clusters, Theor. Chem. Acc., 2014,133. 41 A. C. Harms, S. N. Khanna, A. B. Chen and A. W. Castleman,Dehydrogenation Reactions in Mg*(H2O)nClusters, J. Chem. Phys., 1994,100, 3540. 42 M. Oncak, T. Taxer, E. Barwa, C. van der Linde and M. K. Beyer, Photochemistry and Spectroscopy ofSmall Hydrated Magnesium Clusters Mg*(H2O)n,n=1-5,J. Chem. Phys.,2018, 149, 44309. 43 R. J. Plowright, T. J. McDonnell, T. G. Wright and J. M. C. Plane, Theoretical Study of Mg*-X and [X-Mg-Y]+Complexes Important in the Chemistry of lonospheric Magnesium (X,Y=H20, CO2, N2, O2, and O),J. Phys. Chem. A, 2009, 113, 9354. 444R. F. Hockendorf, O. P. Balaj, C. van der Linde and M. K. Beyer, Thermochemistry from lon-MoleculeReactions of Hydrated lons in the Gas Phase: A New Variant of Nanocalorimetry Reveals ProductEnergy Partitioning, Phys. Chem. Chem. Phys., 2010,12,3772. 45 P. Caravatti and M. Allemann, The infinity cell. A new trapped-ion cell with radiofrequency coveredtrapping electrodes for fourier transform ion cyclotron resonance mass spectrometry, Org. MassSpectrom., 1991, 26, 514. 46 P. Kofel,M. Allemann, H. Kellerhals and K. P. Wanczek, Time-Of-Flight ICR Spectrometry, Int. J. MassSpectrom. lon Process., 1986,72, 53. 47 C. Berg, T. Schindler, G. Niedner-Schatteburg and V. E. Bondybey, Reactions of Simple Hydrocarbonswith Nbn: Chemisorption and Physisorption on lonized Niobium Clusters, J. Chem. Phys., 1995, 102,4870. 48 O.P. Balaj, C. B. Berg, S. J. Reitmeier,V. E. Bondybey and M. K. Beyer, A Novel Design of a Temperature-Controlled FT-ICR Cell for Low-Temperature Black-Body Infrared Radiative Dissociation (BIRD) Studiesof Hydrated Ions, Int. J. Mass Spectrom., 2009, 279,5. 49 R. C. Dunbar, BIRD (Blackbody Infrared Radiative Dissociation): Evolution, Principles, and Applications,Mass Spectrom. Rev.,2004,23, 127. 50 B.S. Fox, M. K. Beyer and V. E. Bondybey, Black body fragmentation of cationic ammonia clusters, J.Phys. Chem. A, 2001,105,6386. 51 R. C. Dunbar and T. B. McMahon, Activation of unimolecular reactions by ambient blackbody radiation,Science,1998,279,194. 52 O. Hampe, T. Karpuschkin, M. Vonderach, P. Weis, Y. M. Yu, L. B. Gan, W. Klopper and M. M. Kappes,Heating a bowl of single-molecule-soup. structure and desorption energetics of water-encapsulatedopen-cage [60] fullerenoid anions in the gas-phase, Phys. Chem. Chem. Phys., 2011, 13, 9818. 53 S. W. Lee, P. Freivogel,T. Schindler and J. L. Beauchamp, Freeze-dried biomolecules: FT-ICR studies ofthe specific solvation of functional groups and clathrate formation observed by the slow evaporation of water from hydrated peptides and model compounds in the gas phase, J. Am. Chem.Soc., 1998,120,11758. 54T. Schindler, C. Berg, G. Niedner-Schatteburg and V. E. Bondybey, Protonated Water Clusters and theirBlack Body Radiation Induced Fragmentation, Chem. Phys. Lett., 1996,250,301. 55 P.D. Schnier, W. D. Price, R. A. Jockusch and E. R. Williams, Blackbody Infrared Radiative Dissociationof Bradykinin and its Analogues: Energetics, Dynamics, and Evidence for Salt-Bridge Structures in theGas Phase,J. Am. Chem.Soc.,1996,118,7178. 56 M. Sena and J. M. Riveros, The Unimolecular Dissociation of the Molecular Ion of AcetophenoneInduced by Thermal-Radiation, Rapid Commun. Mass Spectrom., 1994,8, 1031. 57 D.Tholmann,D. S. Tonner and T. B. McMahon, Spontaneous Unimolecular Dissociation of Small Clusterlons, (H3O*)Ln and Cl(H20)n (n=2-4), under Fourier Transform Ion Cyclotron Resonance Conditions, J.Phys. Chem., 1994, 98,2002. 58 R.L.Wong, K. Paech and E. R. Williams, Blackbody Infrared Radiative Dissociation at Low Temperature:Hydration of X+(H2O)n, for X=Mg, Ca, Int. J. Mass Spectrom., 2004, 232,59. 59 A. Herburger, C. van der Linde and M. K. Beyer,Photodissociation Spectroscopy of Protonated LeucineEnkephalin, Phys. Chem. Chem. Phys., 2017, 19, 10786. 60 C. Hock, M. Schmidt, R. Kuhnen, C. Bartels, L. Ma, H. Haberland and B. von Issendorff, CalorimetricObservation of the Melting of Free Water Nanoparticles at Cryogenic Temperatures,Phys. Rev. Lett.,2009, 103, 73401. 61 W.A.Donald, R. D. Leib, M. Demireva, B. Negru, D. M. Neumark and E. R. Williams, "Weighing" PhotonEnergies with Mass Spectrometry. Effects of Water on lon Fluorescence,J.Am. Chem. Soc., 2010, 132,6904. 62 J.-D. Chai and M. Head-Gordon, Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections, Phys. Chem. Chem. Phys.,2008, 10, 6615. 63T.Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004,393,51. 64 A. Dreuw and M. Wormit, The algebraic diagrammatic construction scheme for the polarizationpropagator for the calculation of excited states, WIREs Comput. Mol.Sci., 2015, 5,82. 65 M.J. Frisch, G. W. Trucks, H. B. Schlegel,G. E. Scuseria, M. A. Robb,J. R. Cheeseman, G. Scalmani, V.Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji,M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J.Bloino, G. Zheng, J. L. Sonnenberg,M. Hada, M. Ehara, K. Toyota,R. Fukuda, J. Hasegawa, M. Ishida, T.Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery,J. E. Peralta, F. Ogliaro, M.Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K.Raghavachari,A. Rendell,J. C. Burant,S.S. lyengar,J. Tomasi, M. Cossi,N. Rega,J.M. Millam, M. Klene,J. E. Knox, J. B. Cross,V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J.Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth,P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J.Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2013. 66 H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schutz, P. Celani, T. Korona, A. Mitrushenkov,G.Rauhut, T. B. Adler, R. D. Amos,A. Bernhardsson,A. Berning, D. L. Cooper, M.J.O.Deegan,A.J. Dobbyn,F. Eckert, E. Goll, C. Hampel,G. Hetzer, T. Hrenar, G. Knizia, C. Koppl, Y. Liu, A. W. Lloyd, R. A. Mata, A.J. May, S. J.McNicholas, W. Meyer, M. E.Mura, A. NicklaB, P. Palmieri, K. Pfluger, R. Pitzer, M. Reiher, U.Schumann, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, version2009.1, a package of ab initio programs, MOLPRO, version 2009.1, a package of ab initio programs,see http://www.molpro.net, Stuttgart, 2009. 67 TURBOMOLE GmbH, TURBOMOLE, University of Karlsruhe and Forschungszentrum Karlsruhe GmbH,2010. ( 68 W. Domcke, R. S c hinke. Photodissociation Dynamics, C ambridge M o nographs on Atomic, Molecular and C hemical P h ysics. Cambridge University P r ess, ISBN 0 - 521-38368-4, Ber. Bunsen. Ph y s. Chem.,1995, 99, 690. ) 69 S. Y. Lee, R. C. Brown and E. J. Heller, Multidimensional reflection approximation. Application to thephotodissociation of polyatomics, J. Phys. Chem.,1983,87, 2045. ( 70 S .-Y.Lee, Energy shift correction for the reflection approximation,J. Chem. Phys., 1985,82,4588. ) ( 71 M . K . Prakash, J. D . Weibel and R . A. Marcus, Isotopomer fractionation in th e UV photolysis of N20. C omparison of theory and experiment, J.Geophys. Res.,2005, 110, 380. ) ( 72 H ollas, D.; Svoboda, O.: Oncak, M.: Slavicek. P.. ABIN, Source code available at https://github.com/PHOTOX/ABIN. ) ( 73 F .-Y. Jou and G. R . Freeman, Temperature and isotope effects on the shape of t h e optical absorption spectrum of s o lvated electrons in water, J. P hys. C hem., 1 9 79,83 , 2383. ) 74 M. F.Bush,J.T. O'Brien,J. S. Prell, C.-C. Wu,R. J. Saykally and E. R. Williams, Hydration of Alkaline EarthMetal Dications: Effects of Metal lon Size Determined Using Infrared Action Spectroscopy,J. Am. Chem.Soc., 2009,131,13270. 75 W.A. Donald, R. D. Leib, M. Demireva, B. Negru, D. M. Neumark and E.R. Williams, Average SequentialWater Molecule Binding Enthalpies of M(H2O)19-124* (M = Co, Fe, Mn, and Cu) Measured withUltraviolet Photodissociation at 193 and 248 nm,J. Phys. Chem. A, 2011, 115, 2. Hydrated singly charged magnesium ions [Mg(H2O)n]+ are thought to consist of an Mg2+ion and a hydrated electron for n > 15. This idea is based on mass spectra, which exhibita transition from [MgOH(H2O)n-1]+ to [Mg(H2O)n]+ around n = 15–22, black-body infraredradiative dissociation, and quantum chemical calculations. Here, we presentphotodissociation spectra of size–selected [Mg(H2O)n]+ in the range of n = 20–70measured for photon energies of 1.0–5.0 eV. The spectra exhibit a broad absorption from1.4 to 3.2 eV, with two local maxima around 1.7–1.8 eV and 2.1–2.5 eV, depending oncluster size. The spectra shift slowly from n = 20 to n = 50, but no significant change isobserved for n = 50–70. Quantum chemical modeling of the spectra yields severalcandidates for the observed absorptions, including five- and six-fold coordinated Mg2+ witha hydrated electron in its immediate vicinity, as well as a solvent-separated Mg2+/e- pair. The photochemical behavior resembles the one of the hydrated electron, with barrierlessinterconversion into the ground state following the excitation.
确定





















还剩24页未读,是否继续阅读?

产品配置单

北京欧兰科技发展有限公司为您提供《镁离子水合物(Mg(H2O)n]+, n = 20–70)中电子光谱与纳米量热法测量检测方案(激光产品)》,该方案主要用于无机盐中可靠性能检测,参考标准--,《镁离子水合物(Mg(H2O)n]+, n = 20–70)中电子光谱与纳米量热法测量检测方案(激光产品)》用到的仪器有Ekspla NT340 高能量可调谐激光器(OPO)
推荐专场






相关方案
更多
该厂商其他方案
更多