








方案详情
文
LA-ICP-MS 相比于传统的环境样品分析方法(消解样品转化为溶液测试)更具吸引力。然而,利用LA-ICP-MS对样品定量需要利用标样,这些标样往往需要与样品基体匹配并且目标元素浓度要与未知样品相近。缺乏有效的标样限制了LA-ICP-MS更广泛应用。本工作提出了一种利用LA-ICP-MS准确分析环境粉末中微量元素含量的方法。为了进行LA-ICP-MS分析,样品粉末与内标(氧化银)还有粘合剂(四硼酸钠)混合,然后压饼成型。定量是通过使用具有不同基体组成和微量元素含量的参考物质确定的校准函数来完成的,这些参考物质与样品前处理一样。利用这种方法,由基体引起的剥蚀差异而导致的单个参考物质物理化学性质的改变可以消除。此外,与ICP相关的基体效应通过碰撞反应池技术可以降到*。该方法的适用性通过分析四种Cd, Cu, Ni, Zn含量不同的参考物质NIST SRM1648a(城市颗粒物)、NIST SRM 2709(圣华金土壤)、BCR 144(污泥)和BCR 723(道路灰尘)说明。信号的计算通过交替使用三个参考物质来计算校准函数进行,而其余的第四种参考物质作为未知样品,所有元素分析结果与标准值高度吻合
方案详情

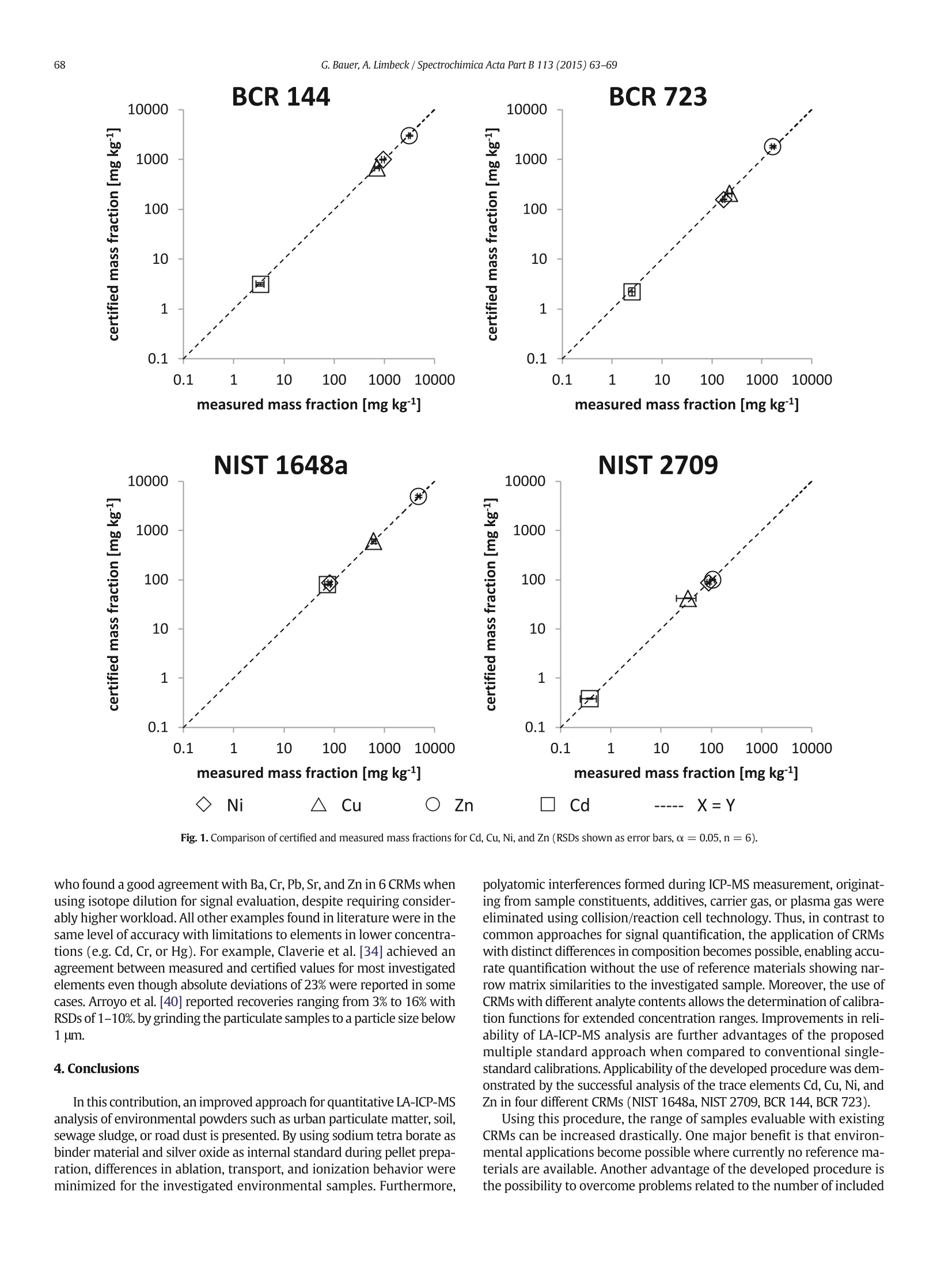
Spectrochimica Acta Part B 113(2015)63-69Contents lists available at ScienceDirectSpectrochimica Acta Part Bjournal homepage: www.elsevier.com/locate/sab 64G. Bauer, A. Limbeck / Spectrochimica Acta Part B 113(2015)63-69 Quantitative analysis of trace elements in environmental powders wwithlaser ablation inductively coupled mass spectrometry usingnon-sample-corresponding reference materials for signal evaluation Gerald Bauer, Andreas Limbeck* Institute of Chemical Technologies and Analytics, Division of Instrumental Analytical Chemistry, TU Wien, Getreidemarkt 9/164, 1060 Vienna, Austria Article history:Received 20 April 2015Accepted 7 September 2015Available online 16 September 2015 Keywords: LA-ICP-MSQuantitative analysisCertified reference materialsNon-matrix-matched standardsInternal standard correction Laser ablation mass spectrometry with inductively coupled plasma(LA-ICP-MS) has become a very powerful technique for direct analysisof trace metals in solid samples [1-3]. The main advantages of this tech-nique are the combination of easy access to solid samples without ex-tensive preparation steps and the high sensitivity of ICP-MS necessaryfor trace element analysis. By direct measurement of the solid sample,disadvantages related to conventional liquid ICP-MS analysis can beavoided. Namely, complex and often time-consuming sample diges-tions, as well as related problems like analyte losses or sample contam-ination during pre-treatment are circumvented when using solidsampling techniques. Another major benefit of LA-ICP-MS using “dryplasma”conditions is a considerable decrease of solvent-related inter-ferences [4-8].However, for analysis of powdered samples such asash, dust, or soil, the sample has to be transferred into a suitable formfor LA. This can be done by fusing [9], pelletizing [10-12], or embedding ( * Corresponding author. Tel . : +4 3 158801 15180; fax: +43 1 58801 1 5199.E-mail address: A .L im be ck@ t uw ien.a c .at ( A . Limbeck). ) ( h ttp: / / dx. d oi . org / 1 0 . 101 6/j . sab. 20 15. 0 9. 0 07 0584-8547/O2015 Els e vier B.V. All rights r eserved. ) [13-16] the powder in an appropriate matrix. All of these techniquescan be applied to meet the analytical needs. Although LA is without doubt an attractive technique for direct solidsampling with ICP, accurate sample quantification remains a challengingtask.Matrix composition greatly influences the interaction between laserbeam and sample affecting the quantity/size of generated particles/aerosol. Furthermore, quantification is not easily achieved due to differ-ences during sample ablation (nonstoichiometric generation of vaporspecies), variations in transfer efficiency (sedimentation and/or recombi-nation of aerosol), and differences in ionization efficiency in the plasma(aerosol composition/cooling effects, particle size/fragmentary atomiza-tion, plasma load) [4-7]. Although some effects can be minimized withthe use of femtosecond lasers instead of nanosecond lasers [2,17], certi-fied reference materials (CRMs) with a matrix matching the compositionof the samples are often required for reliable quantification. Although many CRMs are available, the differences in samplecomposition prove to be a limiting challenge [18].O'Connor et al.[8,10]emphasized that if the standard composition differs even slightly fromthe sample, no reliable information can be achieved. Even if there is aCRM with matching matrix composition available, also analyte concen-tration needs to be in the same order of magnitude. When the analyte concentration in the standard varies too much from the sample, quantifi-cation is possible although accuracy will be limited. In the analysis of en-vironmental samples (e.g. airborne particulate matter), this problematicis of special importance, since contents of the investigated target analytesmay vary over some orders of magnitude while available CRMs coveronly limited concentration ranges. A further crucial aspect of CRMs isthe limited number of elements with certified concentrations. Thus,besides sample type, also the elements of interest and their respectiveconcentration levels have to be considered. To overcome differences inmatrix and analyte composition of CRM and sample, several approacheshave been reported. Some effects can be compensated using an internalstandard if a feasible matrix element is available or can be added duringsample preparation [9,19,20]. Another possibility is to introduce nebu-lized liquid standards or aerosols mixed with the ablated aerosol intothe ICP [21].Other strategies focus on preparing matrix-matched stan-dards by pelletization [22-25] or fusion [26]. Also approaches based onmathematical ratio calculation can be used as is common for geologicalsamples [27]. However, not all of the before-mentioned aspects can betackled using most of these quantification methods. A detailed discussionof advantages and drawbacks of frequently applied quantification strat-egies in LA-ICP-MS analysis of powdered samples has been presentedrecently [28]. Nevertheless, for the successful application of the men-tioned approaches, a CRM is required which matches sample composi-tion and analyte contents closely, a prerequisite which is not fulfilled forall sample types in the field of environmental sciences. In this work, we present a method for signal quantification inLA-ICP-MS analysis of trace elements in powdered environmental sam-ples using initially non-matrix matched CRMs. Preparation of pelletsusing a binder material and an internal standard helped to compensatedifferences in the physical properties of the used calibration standardsleading to reduced variations in ablation and transport efficiency. More-over, matrix-induced spectral interferences such as polyatomic ionswere overcome by using collision/reaction cell technology for q-ICP-MS analysis. With this combination, the establishment of a cross matrixcalibration with CRMs of different origin was possible. The developedprocedure allowed accurate quantification of Cd, Cu, Ni, and Zn in differ-ent environmental CRMs (fly ash, soil, dust, urban particulate matter). 2. Experimental 2.1. Chemicals and consumables All chemicals applied for pellet preparation (sodium tetra borate,caffeic acid, silver oxide) were purchased from VWR, Germany, orSigma-Aldrich, USA, with highest available grade. NIST SRM1648a(urban particulate matter) and NIST SRM2709 (San Joaquin Soil) aswell as EC JRC IRMM certified reference material BCR144 (sewagesludge) and BCR723 (road dust) were used as samples or standardsfor signal quantification. Powder mixtures were prepared and storedin 50 mL centrifuge vials from Sarstedt, Germany, and in 2 mL micro re-actions vials (Eppendorf vials) purchased from VWR, Germany. 2.2.Instrumentation A standard Nd:YAG nanosecond laser ablation system (NWR 213,ESI, USA), equipped with a fast-washout-cell, was used to ablate thesamples. For analysis, the sample aerosol was transferred via 60 cmPTFE tubing to an iCAP Q, a quadrupole ICP-MS with collision/reactioncell capabilities from ThermoFisher Scientific, Bremen, Germany.Helium was used as carrier gas and mixed with Argon after the ablationcell using a Y piece. Initial mixing of samples and additives was done using a shaker,Vortex Genie 2 from Scientific Industries, USA. Subsequent homogeni-zation was performed using a ball mill (MM 400, Retsch, Germany) toachieve sufficient homogeneity. A conventional laboratory press with 13 mm diameter press molds (Perkin-Elmer, Bodenseewerk, Germany)was used for preparation of sample or standard pellets. 2.3. Pellet preparation Prior to pellet preparation, the CRMs were dried to constant weightaccording to the procedures given in the reference data sheets andstored under dry conditions in an exsiccator. Since analyte losses byharsh pre-treatment conditions need to be considered, no milling orgrinding steps were performed prior to pellet preparation in order toavoid any changes in the CRM composition. Sodium tetra borate,d(0.9)<100 um, was chosen as binder material. The binder was previ-ously homogenized/grinded in the ball mill for 60 min using a pulsingrate of 15 pulses per second. Silver oxide, d(0.9)<80 um, was used asinternal standard. For facilitated handling, silver oxide has been dilutedwith the binder component in a 1-to-10 ratio. As optical absorbent atthe laser wavelength of 213 nm caffeic acid (CA), d(0.9)<60 um, wasintroduced without any pre-treatment. For pellet preparation, 50 mg of CRM which acted either as sample orstandard, 50 mg of the diluted internal standard (IS) mixture, and400 mg of the binder component were mixed for 30 min with a pulsingrate of 15 pulses s-1. When adding caffeic acid, 200 mg of binder wasreplaced by that component. Blank pellets were prepared without sam-ple addition to evaluate background levels and possible contaminations.Thus, all pellets consisted of 1% internal standard, 0 or 10% sample, and 0or 40% optical absorbant, with 49-99% binder. The homogenized powderwas filled into the press mold and pressed into a pellet (13 mm diameter,10 MPa for 30 s). The thickness of the obtained pellets was 2.0 mm. Thepellets were stored in Petri dishes under dry conditions until used foranalysis. Neither surface polishing nor pre-ablation processes were usedprior to analysis. 2.4. LA-ICP-MS analysis After inserting the samples, the ablation chamber was purged for30 min with He at a flow rate of 750 mL min-. Sample ablation wasperformed using different parameters. The spot diameter was adjustedto either 100 or 200 um. The laser output was varied between 30% and70% with a frequency of 10 Hz corresponding to a fluence ranging be-tween 0.65 J cm-2 to 15 Jcm-2. The scan speed was varied between50 and 300 um s-1. After sample ablation, the aerosol containing Heflow was diluted with an Ar gas flow of 0.4 L minn-1 and introducedinto the ICP-MS. A daily performance check of the ICP-MS was conducted by tuningfor maximum signal intensity(1In) while ablating NIST SRM 612.Experiments were done either under standard conditions or by usingcollision/reaction cell technology (CR-mode). For CR-mode,a collisiongas (He with 7%H2 purchased from Linde, Austria) was introduced at aflow rate of 4 mL min-1 into the flatpole of the instrument. The kineticenergy barrier was set to 3 V. Detailed instrumental parameters areshown in Table 1. Signals were recorded in transient signal mode(trquant) with Qtegra software (version 1.5.1189.31) provided by Ther-mo Scientific. Transient signals consist of the signals of the gas blank, ablock-shaped plateau of stable elevated signal during sample ablationand again the gas blank signal after the laser is turned off. Each plateauwas divided into 6 regions of 10 s each. Also, a 10 s region was takenfrom the gas blank as background correction and automaticallysubtracted. For each pattern,average and relative standard deviations(RSDs) of these 6 (corrected) regions were calculated and used as nu-meric basis for all further investigations. At least 6 different areas were analyzed on each sample and/or sampledepth. For this purpose, each pellet was (fictively) divided into sixsections of equal surface area and a 600×600 um area was ablated inan“S-shaped”pattern in one sweep on each section. To determinenot only the lateral pellet composition but also surface vs. bulk com-position, the same spots were ablated 4 times in succession. These Table 1Instrument parameters for LA-ICP-MS analysis. Laser ablation system NWR213. ICP-MS iCAPQ ESI Thermo Scientific Type of laser Nd:YAG 213 nm RF power (W) 1550 Carrier gas flow rate (L min) 0.4 Ablation mode Raster (lines) Plasma gas flow rate(L min) 12 Auxiliary gas flow rate 0.8 Spot size (um) 100/200 Collision/reaction gas He with 7%H, CR gas flow rate (mL min-) 4 Fluence (J cm) 0.65-15 Kinetic energy barrier (V) 3 Acquisition time per isotope (ms) 10 Frequency (Hz) 110 Measured isotopes 58Ni,69Ni,63Cu,64Zn,65Cu,66Zn,107Ag,109Ag, cd,cd Scan speed (ums-) 300 He gas flow (L min) 0.75 Isotopes used for quantification 111Cd.60Ni, 63Cu, 66Zn, depth investigations were started by ablating a larger area at the surfacelevel while decreasing the margin by 100 um for each repeatedmeasurement, starting with a 900×900 um spot at the top level,resulting in a 600×600 um spot at the fourth depth level. Thus potential“wall effects” (repeated ablation of an already measured layer, togetherwith a lower layer) were reduced. For signal evaluation, the same600 ×600 um total spot size was used at all times. 3. Results and discussion 3.1. Selection of CRMs CRMs used for method development were selected because of theirdifferences in matrix composition, even though they are all categorizedas environmental samples. Since these materials originate from differ-ent sources, they show distinct deviations in their bulk as well as traceelement composition. Furthermore, they are available in powderedform, which is the only requirement to apply the procedure presentedin this contribution. According to the reference data sheets, the CRMparticle size distributions d(0.9) varied from 30 um for NIST1648a to90 um for BCR144 and BCR723. BCR144, trace elements in sewage sludge, is originating from watertreatment plants and therefore consists mostly of dried organic matterwith relatively high phosphorus and comparably low silicon content.NIST1648a, urban particulate matter, on the other hand, consists ofatmospheric particulates collected in an urban area with noticeable con-tents of elemental carbon.NIST2709,San Joaquin Soil, is agricultural soilwith a dominating silicon fraction and high aluminum content. Due topreparatory reasons, the total carbon content is only about 1.2% in thisCRM. Finally, BCR723, road dust, is a mixture of mainly silicon oxidesand different carbon species. When comparing these CRMs, the siliconcontent (calculated as silicon dioxide content for comparing reasons)varies from 13% for BCR144 to over 63% for NIST2709.Although detailedinformation about the contribution of organic material, elementalcarbon, and water-soluble ions is not available for all CRMs, severe dif-ferences were also expected for these principal constituents, substan-tially influencing the laser-sample interaction. In addition to thementioned deviations in bulk composition, the selected CRMs showalso distinct differences in the analyte concentrations. For example arethe trace metal concentrations in NIST2709 (San Joaquin soil) up totwo orders of magnitude below the levels of NIST SRM1648a (urbanparticulate matter). Table 2 [29] presents concentrations for targetanalytes as well as for selected matrix constituents. 3.2. Pellet investigation These reference materials are commonly applied for demonstrationof accuracy in liquid ICP-MS analysis using external calibration aftersample digestion. To make powders applicable to LA analysis, pelletiza-tion was chosen as sample preparation procedure since it is easy, timesaving, and the equipment is available in most analytical laboratories. To keep the procedure as simple, fast, and mild to the sample as possibleonly pre-drying of the materials, mixing and one pressing step wereapplied. Overall, the previously described pellet preparation routinetakes 40 min (mixing of sample with prepared binders/additives andpressing). The prepared CRM pellets showed no degradation over timeif stored under clean and dry conditions. One pellet can be used formore than 500 analyses, considering 600×600 um patterns, since thewhole surface and at least 50% of the bulk material can be utilized in ab-lation until the pellet integrity is lost. The most important property for successful analysis in LA-ICP-MS isthe homogeneous distribution of analytes throughout the pellet. Theaverage signals of each spot (within one sample layer, as well as withinthe 4 consecutive layers) were statistically evaluated for all investigatedelements (a =99%, data not shown).RSDs were around 10% forBCR144,NIST1648a,and BCR723, indicating general homogeneity ofthe pellets. For NIST2709, an enhanced RSD of around 20% was ob-served, which can be explained by the explicitly lower analyte concen-trations when compared to the other investigated CRMs. Since nosignificant difference in precision could be found when comparing theaverage signals obtained during depth analysis with the ones fromsurface analysis, all further precision and comparison measurementswere done directly on the surface (RSDs of 600 ×600 um spots, n=6). When examining the local analyte distribution of each 600×600 umspot, the transient signal characteristics showed some small fluctuationswithin the analyzed area. Since these fluctuations were identical for allmeasured analytes, sample in-homogeneities could be excluded as sourcefor the observed signal variations. Thus, local variations in ablation and/ortransport efficiency are more likely to account for differences in theamount of sample introduced into the detection system. To overcomethese problems and to further improve signal stability, silver oxide wasintroduced as internal standard during pellet preparation. The silveroxide content was set to 1% of total pellet mass. An excellent correlationbetween transient signal patterns of Ag (IS) and sample constituentswas observed, indicating a homogeneous distribution of all components. Table 2 CRM mass fractions for target analytes (values and uncertainties taken from the respectivereference data sheets). Reference material Cd Cu Ni Zn (mgkg) (mgkg) (mg kg) (mg kg) BCR 144 3.41 ±0.25 713 ±26 942 ±22 33143 ±103 NIST 1648a 73.7 ±2.3 610 ±70 81.1 ±6.8 4800 ±270 NIST2709 0.38 ±0.01 34.6 ±0.7 88 ±5 106 ±3 BCR 723 2.5 ±0.4 226 ±3** 171 :±3° 1660±100 Matrix constituents SiO2 (%) total carbon (%) IFe(%) Ca(%) BCR 144 13.6 4.43 4.06 NIST 1648a 27.4 12.7 3.92 5.84 NIST 2709 63.5 1.2 3.5 1.89 BCR 723 32.1 10* 3.29 6 * Indicative values. ** Uncertain value2 (confirmed within this work by microwave assisted digestion of thematerial using mineral acids and subsequent analysis with standard procedures). Thus Ag has been used as IS in all further investigations. After IS correc-tion, RSDs were decreased to below 8% for most elements in BCR144,NIST1648a, and BCR723. RSDs for NIST2709 remained unchanged, an out-come which could be explained with the low concentration levels prevail-ing in this CRM, thus measurement precision is not only influenced by theablation process but also by measurement uncertainty during ICP-MSanalysis. Summing up, by using a laser beam diameter of 100 um, a scanspeed of 300 um s-and by introducing an internal standard, sufficientreproducibilities for the LA-ICP-MS analysis of the prepared pelletscould be obtained. Further improvements were anticipated by prelimi-nary grinding of the samples, thereby existing differences in particle sizebetween binder, IS, and the investigated CRMs could be cleared, leadingto pellets with increased homogeneity. Similar findings for LA-ICP-MS investigations of sample pellets werereported in literature (Table 3) [30-38].O'Connor et al. [10] achievedRSDs of around 20% with their pelletization procedure using c and65 Cu as IS which is comparable to the results obtained for NIST2709.Shaheen et al. [13] investigated a large number of elements usingepoxy resins as matrix material, obtained RSDs varied from 2.4% to49% with most elements around 15% using silicon as IS. The higherRSDs corresponded to low analyte concentrations in the respective an-alyzed materials. Kleiber et al. [39] achieved RSDs of 10-20% whileperforming trace metal analysis of coal with various IS. In their researchthey compared continuous sampling (3 sites, 60 s line scan each) withsequential sampling (15 sites, 20 s line scan each) with RSDs of about10% for continuous and 10-20% for sequential sampling respectively. 3.3. Method development For signal quantification, the known CRM contents of the preparedpellets and the respective certified/indicative trace element concentra-tions (Table 2) were used. In each experiment, one CRM was investigat-ed while the other three were used to create a linear calibration curve. The concentration of the investigated CRM was calculated accordingto formula 1. Cunk Concentration of investigated CRM in mg kg(presumablyunknown). lunk Blank and IS corrected signal intensity corresponding to incounts s-1. dcal Axis intercept of calibration curve kcal Slope of calibration curve 3.4. Standard mode The prepared pellets were measured with ICP-MS using standardconditions as shown in Table 1. To determine the concentrations ofthe investigated sample, formula 1 was applied to the data after blankand internal standard correction. This process was repeated 4 times foreach data set changing the investigated CRM and calculating the calibra-tion curve with the remaining three standards. Linear calibration curveswere achieved for all combinations (R"between 0.90 and 0.95); the ele-ment concentrations found for the individual CRMs match the rightorder of magnitude. However, compared to the reference values, theresulting concentrations were well beyond the confidence region inmost cases. When calculating concentrations for BCR144 the deviationsbetween determined and certified content ranged from 40% to 133%and for NIST1648a from 47% to 174% of the certified contents. Lookingat NIST2709, deviations from 113% to 275% and at BCR723 from 36% to132% were found. Observed differences between measured and certified CRMs/SRMcontents indicate that matrix-induced variations in the ablation Table 3 Comparing recently published environmental work using pelletization and borate fusion with LA-ICP-MS regarding accuracy (relative standard deviation). Sample matrix Internal standard Pellet matrix Number of analytes Achieved RSD BCR144 Ag Sodium tetra borate 5 4-15% NIST1648a 2-8% NIST2709 5-20% BCR723 3-7% Sediment[91 None Li2B407/LiBO2 5 3% Sediment I[10] C, Cu No binder. 1 20% Polyvinyl alcohol, Vanillic acid, Nicotinic acid, Fly ash [12] None Pyrazinoic acid Cellulose. 10 1-117% Silver powder, (Pellets) Caffeine, 1-57% Maleic acid, (Fused beads) Cinnamic acid. Flue dust [24] Rh LizB407/LiBO2 Paraffin, 6 7% cellulose/N-methacrylate ZnO/Fe203 Soil [30] Sc, Lu No binder 12 15% Compost:[31] None No binder 9 10% Solid [[32] None No binder 3 6-21% Vanilla [33] None No binder 11 6-14% Environmental [34] None Li2B407/LiBO2 6 10-15% Soil and sediment [35] Ag, Ni No binder 8 5-14% Soil, sediment, ash [ 1[36] None Zn0/2-methoxy-4-(2-propenyl)phenol 3 <20% Road sediment [37] Ca No binder 3 10% Agricultural soil [38] Ge,Au, Sc, Pd Ag powder, 8 2-8% Al powder Coal [39] C, Rh, Mo No binder, KBr 10 10-20% Soil and sediment [40] Ge,Y No binder 16 2-10% behavior of the investigated samples are not or only partially correctedwith the use of an IS. Even though possible matrix effects have beenreduced due to the preliminary sample dilution by a factor of 10, theremaining matrix-induced variations which are still resulting in a limitedaccuracy. Consequently, the next development step focused on the abla-tion process. 3.5. Addition of optical absorbant The interaction of the laser beam with the sample depends on theoptical properties of the investigated sample (e.g.absorbance). Thus, sam-ples with different matrix composition might be ablated in a different wayeven when using identical conditions [4-7]. An attempt to overcome thisproblem is the introduction of a material with absorbance capabilities atthe used laser wavelength. Thus, differences in the absorbance of sampleand standard could be minimized.O'Connor et al. [10] reported positiveeffects when using vanillic acid, pyrazinoic acid, or nicotinic acid.Stankova et al. [12] achieved improvements with the addition of maleicacid. In this work, caffeic acid was chosen as modifier because of its goodabsorbing characteristics at a wavelength of 213 nm. To the knowledgeof the authors, caffeic acid has not been used as optical laser absorbantin LA-ICP-MS so far. The absorbant was added with 40% total pelletmass by replacing the respective part of the binder material. Otherwise,the same preparation procedure was applied and the pellets were againmeasured using the conditions described before. Data evaluation was performed as explained in the previous chapter.Overall, the addition of caffeic acid as optical absorbant did not signifi-cantly improve the results. Compared to experiments conducted with-out absorbant addition no improvements in accuracy were obtained,thus matrix-induced variations in ablation behavior and/or transportof different CRMs are not compensated by the addition of caffeic acid.No distinct improvements regarding the slopes or R2 of the calibrationcurves could be observed. Also differences between certified and mea-sured contents remain basically the same ranging from 39 to 123% forBCR144 and from 37 to 177% for NIST1648a. For NIST2709, deviationsfrom 115 to 267% were found and for BCR723 deviations ranged be-tween 39 and 112%. However, in the presence of caffeic acid, the analyteRSDs for NIST2709 were improved considerably below 6% for all inves-tigated elements. At the same time, the RSDs for NIST1648a increasedup to 60%. These changes in the reproducibility of measurement couldnot be attributed to insufficient sample in-homogeneity. Other possibleexplanations for these findings are impurities or interferences intro-duced by the addition of caffeic acid. Even though blanks were preparedand measured according to the described procedures, no significant im-provements in accuracy could be achieved. Thus, contaminations of theused chemicals could be excluded as source for poor results with insuf-ficient accuracy and reproducibility, the only remaining reason aretherefore spectral interferences in ICP-MS analysis. 3.6. Collision/reaction cell technology (CR-mode) The influence of spectral interferences on accuracy of analysis is wellknown from the field of liquid ICP-MS analysis. There, polyatomic ionswith m/z values non-resolvable (with the used mass analyzer) fromthe target analytes are created from the plasma gas Ar with solventand/or matrix constituents. In case of LA-ICP-MS measurements, thecontribution of the solvent is not present; nevertheless, the formationof interferences from sample components and carrier or plasma gas isstill possible. In the present study, binder (sodium tetra borate) andinternal standard (silver oxide), which were both introduced duringsample preparation, have to be considered as further sources for poly-atomic interferences. Simple blank measurements for correction of gen-erated interferences are considered to be not straightforward, since thepolyatomic ions formed during LA-ICP-MS analysis of the preparedsample pellets are assumed to be different for the investigated matrices. For example, vary the concentrations of potentially interference-forming elements such as C, N, O, Si, P, or S distinctly between the indi-vidual CRMs, thus affected m/z values and extent of the created interfer-ences are supposed to be different for each CRM. A successful way to overcome polyatomic interferences in quadrupoleICP-MS analysis is the use of collision or reaction cells [7]. In this study, acombined collision/reaction (CR) mode was used by introducing a gasmixture (helium with 7%hydrogen) with simultaneous application ofthe kinetic energy discrimination (KED) approach. For optimization ofthe CR-mode conditions, the 40Ar2Na interference on63Cu has been se-lected, as this interference originates primarily from the added chemicalsand not from the investigated samples. Hence, the signals observed for63Cu and 65cu in blank pellets prepared with sodium tetra borate andsilver oxide were used for development of the CR method. While for65Cu, signals close to the background level were found, indicating the ab-sence of any Cu-contaminations in binder and internal standard, signifi-cant signals were found in experiments without collision/reaction gasand a KED value of 0 V at an m/z ratio of 63. These signals were assignedto the formation of 40Ar3Na ions. With increasing flow rate of the usedHe/hydrogen mixture and/or kinetic energy barrier value, a decrease ofthe 40Ar3Na signal was observed. Simultaneously, the signal of the silverisotopes dropped, but with a different extent, when compared to theexamined polyatomic interference. Best results in terms of minimumpolyatomic interference (signal at the m/z value of 63) with maximum in-tensity for the internal standard (17Ag signal) were reached when intro-ducing the CR gas at a flow rate of 4 mL min- and a KED value of3 V.Therefore, all sample measurements were performed using these condi-tions (detailed parameters in Table 1). For quantification, the same procedure for signal evaluation was usedas described previously.Again a linear calibration function was achievedfor all combinations with considerably increased coefficients of determi-nation (R²> 0.99 for all elements). Based on the signals derived for sampleand blank pellets, the respective limit of detection (LOD) was determinedfor each investigated element. LODs (3 o) were calculated from the stan-dard deviations of blank measurements (n =6), yielding results of1.7 ug kg for Ni, 9.5 ng kgfor Cu, 9.8 ug kg for Zn, and0.3 ng kgfor Cd. For all materials, a satisfying precision was accom-plished with average RSDs of 11.1% for NIST2709 and 7.6% for BCR144as well as 4% and below for NIST1648a and BCR723 for Cu, Ni, and Zn.For Cd in CR-mode, all RSDs were elevated to around 20% except forNIST1648a, which can be explained by the low concentrations of this ele-ment in the other CRMs and the reduced sensitivity of the ICP-MS analysisin the CR-mode. Comparison to recent LA-ICP-MS literature shows thatfindings for reproducibility of analysis expressed as RSD are similar oreven better to published data (Table 3); further improvements havebeen reported only when using isotope dilution for quantification. Using the developed procedure, all four investigated CRMs could beaccurately quantified; achieved results were in good agreement withthe certified values (Fig.1). Due to the wide range of the analyte concen-trations, the common logarithm of all values was calculated for compar-ison of the measured values with the certified concentrations. Slopes ofthe calculated linear regression functions (varying from 0.987 to 1.012)are not significantly different from the value of 1 for all four presentedcases(α=0.05), indicating no systematic error resulting from the cal-ibration with different CRMs. Intercepts of the respective functions donot differ significantly from 0(α=0.05). Obtained results were for all investigated elements and CRMs exceptNi in BCR 723 (road dust) within the uncertainty of the reference values,indicating that matrix effects in sample ablation and measurement werereduced to an extent not affecting the accuracy of LA-ICP-MS analysisanymore. Absolute deviations between measured and certified valuesvaried between less than 1% (Cu in NIST1648a) and approximately17% (Cd in BCR144). For most elements and samples, differences inthe order of 5% only were obtained; larger deviations were only ob-served for elements with concentrations close to the respective limitof detection. Similar findings were reported from Malherbe et al. [9], Fig. 1. Comparison of certified and measured mass fractions for Cd, Cu, Ni, and Zn (RSDs shown as error bars, α=0.05,n=6). who found a good agreement with Ba, Cr, Pb, Sr, and Zn in 6 CRMs whenusing isotope dilution for signal evaluation, despite requiring consider-ably higher workload. All other examples found in literature were in thesame level of accuracy with limitations to elements in lower concentra-tions (e.g. Cd, Cr, or Hg). For example, Claverie et al. [34] achieved anagreement between measured and certified values for most investigatedelements even though absolute deviations of 23% were reported in somecases. Arroyo et al. [40] reported recoveries ranging from 3% to 16% withRSDs of 1-10%. by grinding the particulate samples to a particle size below1 um. 4.Conclusions In this contribution, an improved approach for quantitative LA-ICP-MSanalysis of environmental powders such as urban particulate matter, soil,sewage sludge, or road dust is presented. By using sodium tetra borate asbinder material and silver oxide as internal standard during pellet prepa-ration, differences in ablation, transport, and ionization behavior wereminimized for the investigated environmental samples. Furthermore, polyatomic interferences formed during ICP-MS measurement, originat-ing from sample constituents, additives, carrier gas, or plasma gas wereeliminated using collision/reaction cell technology. Thus, in contrast tocommon approaches for signal quantification, the application of CRMswith distinct differences in composition becomes possible, enabling accu-rate quantification without the use of reference materials showing nar-row matrix similarities to the investigated sample. Moreover, the use ofCRMs with different analyte contents allows the determination of calibra-tion functions for extended concentration ranges. Improvements in reli-ability of LA-ICP-MS analysis are further advantages of the proposedmultiple standard approach when compared to conventional single-standard calibrations. Applicability of the developed procedure was dem-onstrated by the successful analysis of the trace elements Cd, Cu, Ni, andZn in four different CRMs (NIST 1648a, NIST 2709, BCR 144, BCR 723). Using this procedure, the range of samples evaluable with existingCRMs can be increased drastically. One major benefit is that environ-mental applications become possible where currently no reference ma-terials are available. Another advantage of the developed procedure isthe possibility to overcome problems related to the number of included elements or the sometimes inadequate concentration levels of availableCRMs. Furthermore, the proposed procedure is not limited to environ-mental samples; with some modifications, the concept should be alsoapplicable for LA-ICP-MS analysis of other sample materials such as ce-ment, ceramics, or plant material. The only requirements are samples inpowdered form and the existence of reference materials with suitableelement contents. ( References ) ( [1] B . Fer n a nd e z P is o ner o , D. G u nthe r, C r i t i c a l re visio n o f G D - MS, L A -IC P- MS an d S I M S a s i n organic m a s s spectr o me t ric t e c h niqu es fo r d i re c t s ol i d an a l y s i s, J. A n a l . At. S p ectr o m. 2 4 ( 200 9 ) 1 145 - 1 1 60 . ) ( [2] B . Ferna n dez, F. Cla v e rie , C. P ec h ey r an , O .F . X . D onard, F. Cl averi e , Di r e ct analy s i s o f ) solid samples by fs-LA-ICP-MS, TrAC Trends Anal. Chem. 26 (2007) 951-966. ( [3] J . Koch , D. G u n t h e r, R e view of t h e stat e -of - the-art of l a se r a b lati o n i nd u c tiv ely c o u p le d p lasma m a ss s p ectr o me t ry, Ap p l . Spec t ro sc. 65 (2 0 11) 1 5 5- 16 2. ) ( [4] N . J ak u b o w s ki , J o ha n n a Sabin e B ecker: in o rga nic ma s s sp e ct r ometry . P rin c i pl es and a ppl i c a tion s , Ana l . B ioan a l. Ch em . 3 9 2 (200 8)7 75- 7 76. A . Mo ntase r , I nd u cti v e ly Co upl e d P l asm a M a s s S pectrome t r y , Wi l ey , 1 9 9 8 . ) ( 6 R .D . S no ok, Ha n d bo ok o f i nd uc t iv e l y co u p led pl asma m a ss s pectr o m e try , Chr omat o graph i a 3 4 (19 9 2) (5 4 6- 5 4 6 ) . ) [7]S. Nelms, Inductively Coupled Plasma Mass Spectrometry Handbook, Blackwell,2005. [8] C. O' Connor, B.L. Sharp, P. Evans, On-line additions of aqueous standards for calibrationof laser ablation inductively coupled plasma mass spectrometry: theory and compari-son of wet and dry plasma conditions, J. Anal. At. Spectrom. 21 (2006) 556-565. [9] J.Malherbe, F. Claverie, A. Alvarez, B. Fernandez, R. Pereiro,J.L. Molloy, Elementalanalyses of soil and sediment fused with lithium borate using isotope dilutionlaser ablation-inductively coupled plasma-mass spectrometry, Anal. Chim. Acta793 (2013) 72-78. [10] C. O'Connor, M.R. Landon, B.L. Sharp, Absorption coefficient modified pressed pow-ders for calibration of laser ablation inductively coupled plasma mass spectrometry,J. Anal. At. Spectrom. 22 (2007)273-282. ( [11 ] M . Mo te lic a-H eino, O .F . X. D o n ar d , J.M . Mer met, L a s er a b la t ion of sy nt he tic geologi c al p ow de r s us i ng I C P- A E S d e t ection : e f f e c t s o f the ma t rix, c h e m i cal f o rm o f t h e analy te a nd lase r w ave l ength , J. A na l . A t . Spectro m . 1 4( 1 999) 675-68 2. ) ( [12] A . Sta n kova ,N . Gil o n, L . D ut r uch , V . Kani c ky, C om pa ri son of LA -I CP-MS and L A -I CP-O ES f or th e a na ly s i s of so m e ele me nt s i n fl y a sh es , J.A n a l. At . S pec tr o m. 2 6 ( 20 11 ) 4 43-4 49. ) ( [ 1 3] M .E. Sha h een, B.J . Frye r, A s i m p le s olution to e xp anding avai l able r e f e re n ce m a te- r ia ls f or las er ab l a t i o n i n duct iv e l y c o upl e d pl asm a m ass spe ct r o metry a n a lysi s: a p- p l i c ati ons to sedim e n t a ry m a teria l s, S p e ctr oc h im . A c ta P a rt B 66 ( 2 01 1 ) 627- 636 . ) [14] M.L. Viger,J.-F.Y.Gravel, D. Brouard, D. Beauchemin, D. Boudreau, Use of sol-gels assolid matrixes for trace analysis by uv laser ablation and laser-enhanced ionizationdetection, Anal. Chem. 77 (2005)706-710. [15] A.J. Fitzpatrick, T. Kurtis Kyser, D. Chipley, D. Beauchemin, Fabrication of solid cali-bration standards by a sol-gel process and use in laser ablation ICPMS, J. Anal. At.Spectrom. 23 (2008)244-248. [16]I. Hubova, M.Hola,J. Pinkas, V. Kanicky, Examination of sol-gel technique applicabil-ity for preparation of pellets for soil analysis by laser ablation inductively coupledplasma optical emission spectrometry,J. Anal. At. Spectrom. 22 (2007)1238-1243. [17] J. Koch, D. Giinther, Femtosecond laser ablation inductively coupled plasma massspectrometry: achievements and remaining problems, Anal. Bioanal. Chem. 387(2007) 149-153. [18] K. Stěpankova, K. Novotny, M. Vasinová Galiova, V. Kanicky, J. Kaiser, D.W. Hahn,Laser ablation methods for analysis of urinary calculi: comparison study based oncalibration pellets, Spectrochim. Acta Part B 81 (2013) 43-49. [19] C. Pickhardt, H.-J.Dietze, J.S. Becker, Laser ablation inductively coupled plasma massspectrometry for direct isotope ratio measurements on solid samples, Int. J. MassSpectrom.242(2005)273-280. [20] Y.-L. Lee, C.-C. Chang, S.J. Jiang, Laser ablation inductively coupled plasma massspectrometry for the determination of trace elements in soil, Spectrochim. ActaPart B 58(2003) 523-530. [21]JJ.J. Leach, L.A. Allen, D.B. Aeschliman, R.S. Houk, Calibration of laser ablation induc-tively coupled plasma mass spectrometry using standard additions with dried solu-1AA0AAtion aerosols, Anal. Chem. 71 (1999) 440-445. [22]M1.A.O. da Silva, M.A.Z. Arruda, Laser ablation (imaging) for mapping and determin-ing Se and S in sunflower leaves, Metallomics 5 (2013) 62-67. ( [23] H . Z hou, Z . Wang,Y . Z h u, Q. L i , H . j . Z ou, H.- Y . Q u , Y . - R . C hen , Y . - P . Du, Qu a nt i tat i ve d e te r mina t io n o f t r ac e m e ta ls i n high - purity si l i c on c a r b ide p o w de r b y laser abla- t i on i ndu c tiv e ly c ou ple d p l a sma mass s p e ctro m e tr y wi t h o ut b i n ders , Spectrochim . A cta P art B 90 (2013 ) 55- 6 0. ) [24] A.G. Coedo, I. Padilla, M.T. Dorado, Determination of minor elements in steelmakingflue dusts using laser ablation inductively coupled plasma mass spectrometry,Talanta 67 (2005)136-143. [25]PI.Su,P. Ek, A. Ivaska, Determination of metal ions in single wood fiber by LA-ICP-MS2012,p.833. [26]A/.I. Helal, N.F.Zahran, R.A. Mohamed, H.T. Mohsen, J.S. Becker, A.P. Kobzev, A.H.Hashad, Trace elements analyses of zircon sample by integration of the LA-ICP-MS, EDS and RBS methods, Int. J. Mass Spectrom. 221 (2002)139-146. [27] W.E. Halter, T. Pettke, C.A. Heinrich, B. Rothen-Rutishauser, Major to trace elementanalysis of melt inclusions by laser-ablation ICP-MS: methods of quantification,Chem. Geol. 183 (2002) 63-86. [28]A/. Limbeck,P. Galler, M. Bonta, G. Bauer, W. Nischkauer, F. Vanhaecke, Recent ad-vances in quantitative LA-ICP-MS analysis: challenges and solutions in the life sci-ences and environmental chemistry, Anal. Bioanal. Chem. 407 (2015)6593-6617. ( [ 29] M .Z i sc hk a, T he c er ti f i c at io n o f th e co nten t s (m a s s fr ac ti o n s) o f p alla d ium, pla t inu m a nd r hodium in roa d dust : BC R -7 2 3, EUR-OP, L u x embourg, 20 0 2. ) [30]| S.C. Jantzi, J.R. Almirall, Elemental analysis of soils using laser ablation inductivelycoupled plasma mass spectrometry (LA-ICP-MS) and laser-induced breakdownspectroscopy (LIBS) with multivariate discrimination: tape mounting as an alterna-tive to pellets for small forensic transfer specimens, Appl. Spectrosc. 68 (2014)963-974. [31]MI.S. Jimenez,M.T. Gomez, J.R. Castillo, Multi-element analysis of compost by laserablation-inductively coupled plasma mass spectrometry, Talanta 72 (2007)1141-1148. [32] B. Fernandez, P. Rodriguez-Gonzalez, J.I. Garcia Alonso, J. Malherbe, S. Garcia-Fonseca,R. Pereiro, A. Sanz-Medel, On-line double isotope dilution laser ablation in-ductively coupled plasma mass spectrometry for the quantitative analysis of solidmaterials, Anal.Chim. Acta 851 (2014) 64-71. ( [33] E . M. H o n dro g ianni s, E . E h r li nge r , A . Pop la s k i , M . L is l e, Us e of l as e r a bl a tion - i nd uct ively c ou pl e d p l a s ma- t ime of fl ig ht- m a s s s pec tr o m e try to i de nt i fy th e ele -m ental c omp o si ti on o f v anil la a nd det e rmine the g e o g r a ph i c o r i g i n by di s c r i m i nan t f un ct ion a n alys i s, J . Agri c . F o o d C h e m. 6 1 (2013 ) 11 332 - 1133 7 . ) [34|F. Claverie, J.Malherbe, N. Bier, J.L. Molloy, S.E. Long, Standard addition method forlaser ablation ICPMS using a spinning platform, Anal. Chem.85 (2013)3584-3591. [35]S.A. Baker,M. Bi,R.Q. Aucelio, B.W. Smith,J.D.Winefordner, Analysis of soil and sed-iment samples by laser ablation inductively coupled plasma mass spectrometry,J.Anal. At. Spectrom. 14 (1999) 19-26. [36]M1. Pakiela,M. Wojciechowski, B. Wagner, E. Bulska, A novel procedure of powderedsamples immobilization and multi-point calibration of LA ICP MS, J. Anal. At.Spectrom. 26 (2011) 1539-1543. [37] M.Motelica-Heino, S. Rauch, G.M. Morrison, O.F.X. Donard, Determination of palladium,platinum and rhodium concentrations in urban road sediments by laser ablation-ICP-MS, Anal. Chim. Acta 436 (2001)233-244. [38]|1 P. Musil, V.T. Otruba, V. Kanicky, J.-M.Mermet, Determination of elements in agri-cultural soils using IR laser ablation inductively coupled plasma atomic emissionspectrometry, Spectrochim. Acta Part B 55 (1747-1758). [39]L. Kleiber, H. Fink, R. Niessner, U. Panne, Strategies for the analysis of coal by laserablation inductively coupled plasma mass spectroscopy, Anal. Bioanal. Chem. 374(2002)109-114. [40]L1. Arroyo, T. Trejos, P.R. Gardinali,J.R. Almirall, Optimization and validation of a laserablation inductively coupled plasma mass spectrometry method for the routineanalysis of soils and sediments, Spectrochim. Acta Part B 64 (2009) 16-25. LA-ICP-MS 相比于传统的环境样品分析方法(消解样品转化为溶液测试)更具吸引力。然而,利用LA-ICP-MS对样品定量需要利用标样,这些标样往往需要与样品基体匹配并且目标元素浓度要与未知样品相近。缺乏有效的标样限制了LA-ICP-MS更广泛应用。本工作提出了一种利用LA-ICP-MS准确分析环境粉末中微量元素含量的方法。为了进行LA-ICP-MS分析,样品粉末与内标(氧化银)还有粘合剂(四硼酸钠)混合,然后压饼成型。定量是通过使用具有不同基体组成和微量元素含量的参考物质确定的校准函数来完成的,这些参考物质与样品前处理一样。利用这种方法,由基体引起的剥蚀差异而导致的单个参考物质物理化学性质的改变可以消除。此外,与ICP相关的基体效应通过碰撞反应池技术可以降到最小。该方法的适用性通过分析四种Cd, Cu, Ni, Zn含量不同的参考物质NIST SRM1648a(城市颗粒物)、NIST SRM 2709(圣华金土壤)、BCR 144(污泥)和BCR 723(道路灰尘)说明。信号的计算通过交替使用三个参考物质来计算校准函数进行,而其余的第四种参考物质作为未知样品,所有元素分析结果与标准值高度吻合
确定






还剩5页未读,是否继续阅读?

产品配置单

上海凯来仪器有限公司为您提供《环境粉末中微量元素检测方案(激光剥蚀进样)》,该方案主要用于土壤中(类)金属及其化合物检测,参考标准--,《环境粉末中微量元素检测方案(激光剥蚀进样)》用到的仪器有ESL213 灵活的激光剥蚀系统
推荐专场





相关方案
更多
该厂商其他方案
更多