








方案详情
文
Abstract: High-speed counter-current chromatography methods,combined with solvent partition, were applied to the systematic separation and purification of chemical components from Chinese medicinal herb Polygonum multiflorum extract. The aim of this paper is summing up the rules of solvent system selection for diverse fractions of herbal extract, and establishing the systematic pattern to screen the bioactive constituents rapidly. Nine compounds including emodin, chrysophanol, rhein, 6-OH-emodin, emodin-8--d-glucoside, polygonimitin B, 2,3,5,4-tetrahydroxystilbene-2--d-glucoside, gallic acid and an unkown glycoside, which differed in quantity and polarity remarkably, were obtained. The purities of them were all
above 97% as determined by high-performance liquid chromatography (HPLC), and their structures were identified by H-NMR and electrospray ionization mass spectrometry (ESI–MS). The results demonstrated that HSCCC is a speedy and efficient technique for systematic isolation of bioactive components from traditional medicinal herbs.
方案详情

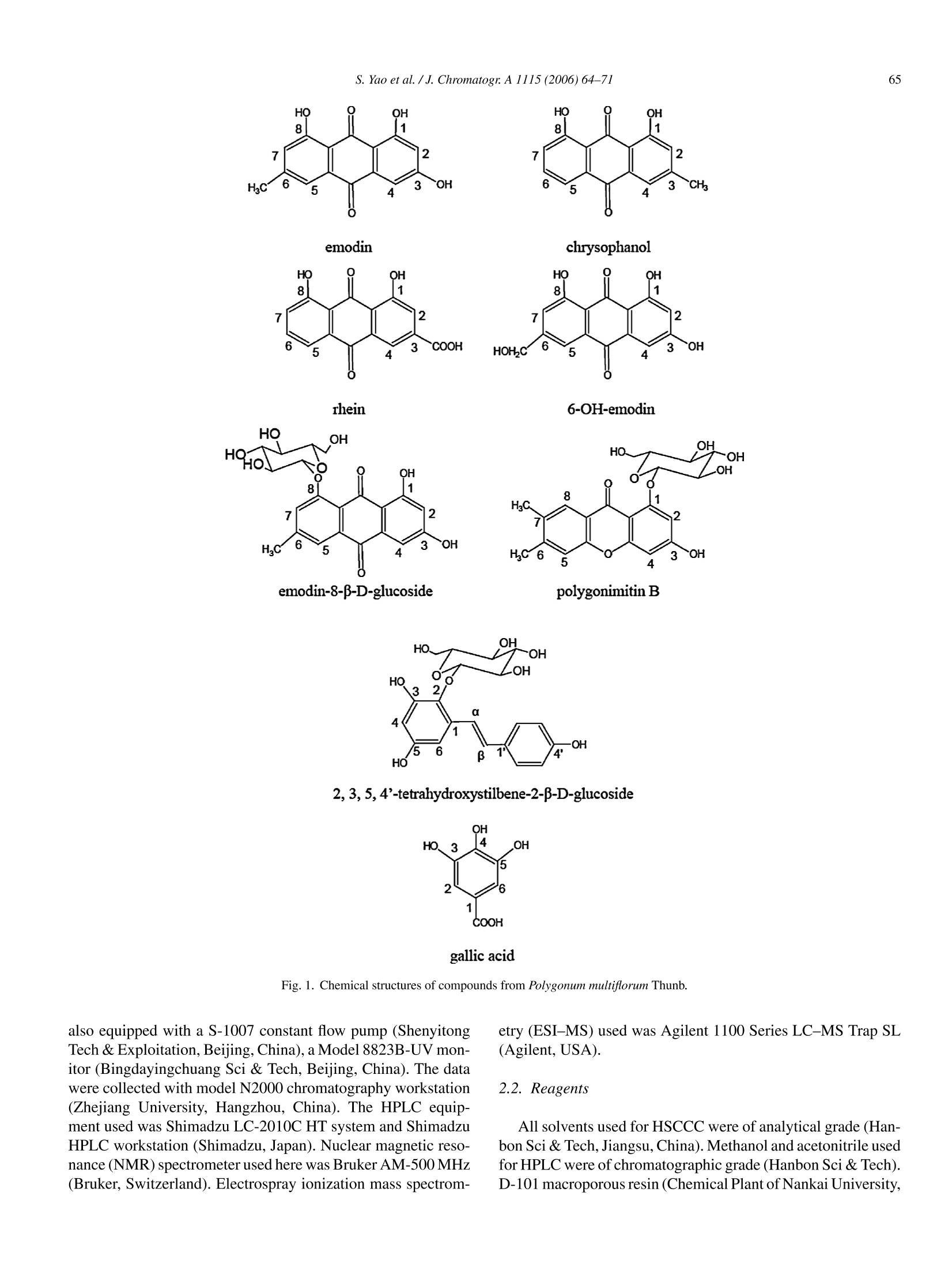
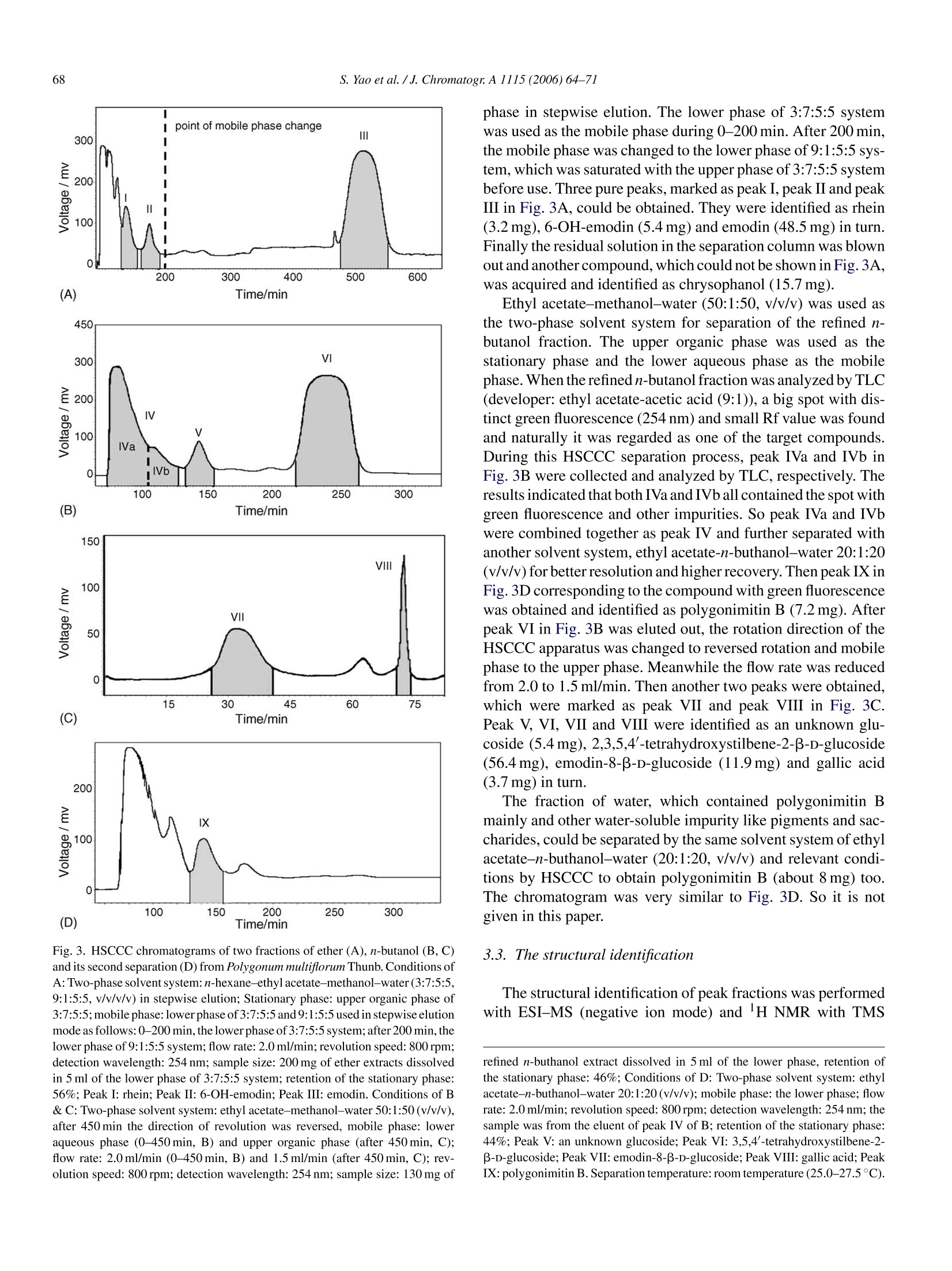
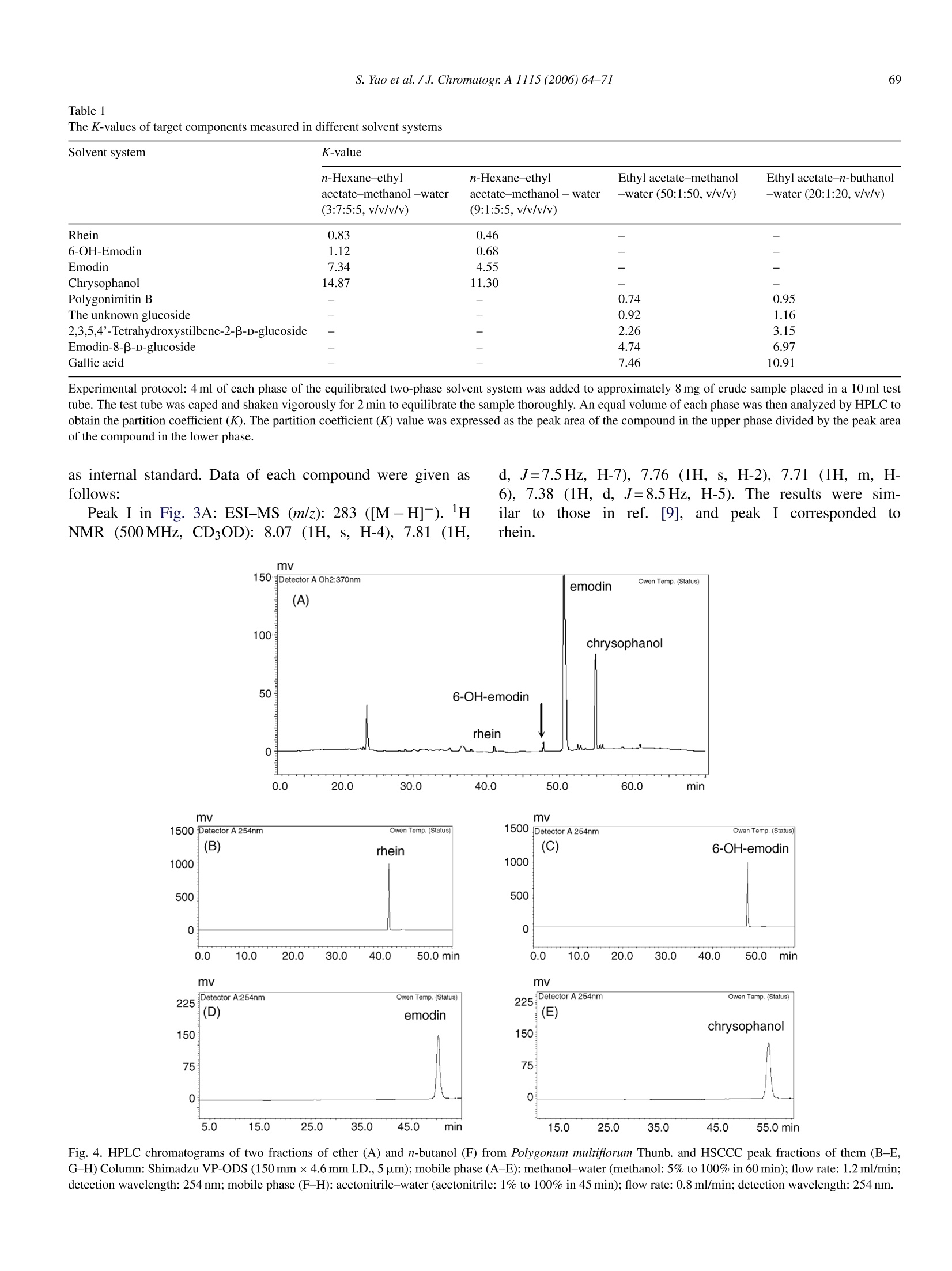
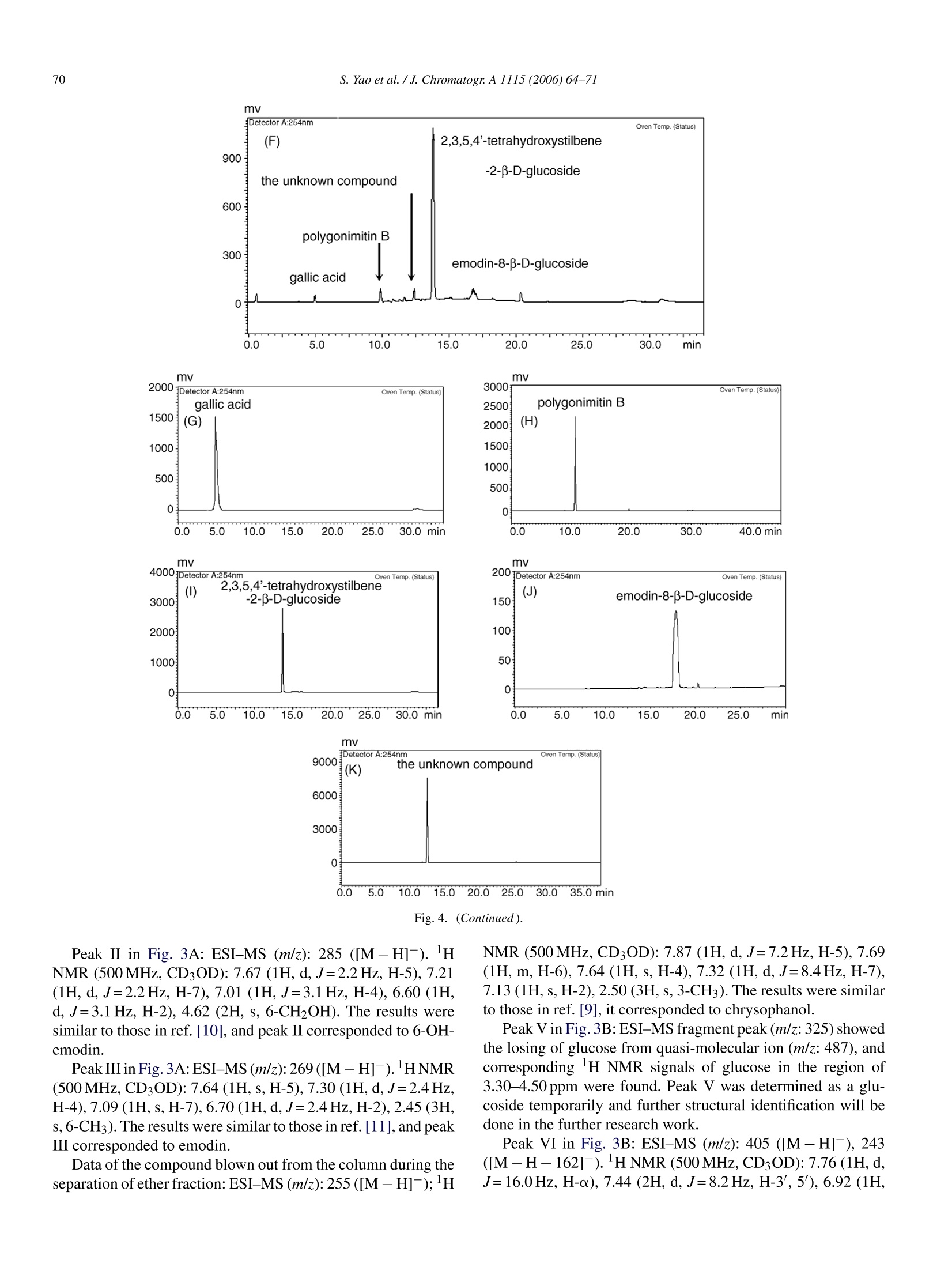
Available online at www.sciencedirect.comJOURNAL OFCHROMATOGRAPHY AJournal of Chromatography A, 1115 (2006)64-71 S. Yao et al./J. Chromatogr:A 1115(2006)64-7165 www.elsevier.com/locate/chroma Preparative isolation and purification of chemical constituents from the rootof Polygonum multiforum by high-speed counter-current chromatography Shun Yao, Yi Li, Lingyi Kong a.* Department of Natural Medicinal Chemistry, China Pharmaceutical University, Nanjing 210009, China Analytical & Testing Center, Nanjing Normal University, Nanjing 210097, China Received 30 December 2005; received in revised form 18 February 2006; accepted 23 February 2006Available online 27 March 2006 Abstract High-speed counter-current chromatography methods, combined with solvent partition, were applied to the systematic separation and purificationof chemical components from Chinese medicinal herb Polygonum multiflorum extract. The aim of this paper is summing up the rules of solventsystem selection for diverse fractions of herbal extract, and establishing the systematic pattern to screen the bioactive constituents rapidly. Ninecompounds including emodin, chrysophanol, rhein, 6-OH-emodin, emodin-8-B-D-glucoside,polygonimitin B, 2,3,5,4'-tetrahydroxystilbene-2-B-D-glucoside, gallic acid and an unkown glycoside, which differed in quantity and polarity remarkably, were obtained. The purities of them were allabove 97% as determined by high-performance liquid chromatography (HPLC), and their structures were identified by H NMR and electrosprayionization mass spectrometry (ESI-MS). The results demonstrated that HSCCC is a speedy and efficient technique for systematic isolation ofbioactive components from traditional medicinal herbs. O 2006 Elsevier B.V. All rights reserved. Keywords: High-speed counter-current chromatography; Polygonum multiflorum; Systematic isolation and purification 1. Introduction Dried root of Polygonum multiflorum Thunb. (Polygonaceae)well known as Heshouwu, is one of the most popular traditionalmedicinal herb in China, and is officially listed in the ChinesePharmacopoeia [1]. It is frequently used as a tonic and purga-tive in China and Japan. The main active constituents of theherb have been reported to be hydroxyanthraquinones,stilbenes,other phenolic compounds and their glycosides [2]. High-speed counter-current chromatography (HSCCC),which was first invented by Ito [3], is a form of liquid-liquidpartition chromatography. It is considered as a suitable alter-native for separation of phenolic compounds such as hydrox-yanthraquinones and stilbenes [4-6]. Only a few CCC studies[7] have targeted a range of compounds in a single procedure,and most reports focus on several main compounds in cer-tain fraction. In the present paper, diverse methods of HSCCCwere established for systematic separation of different frac- ( * Corresponding a u thor. T el. : +86 25 8 5391289. ) ( E-mail address: l y k ong @jl onli n e . com ( L . K ong). ) ( 0021-9673/$-see fro n t ma t ter @ 2006 Els e vier B.V. All ri g hts reserved. doi:10.1016/j. chro ma.2006.02.07 1 ) tions of Polygonum multiflorum Thunb.Nine compounds includ-ing emodin, chrysophanol, rhein, 6-OH-emodin,emodin-8-B-D-glucoside, polygonimitin B, 2,3,5,4'-tetrahydroxystilbene-2-B-D-glucoside, gallic acid and an unkown glycoside, were obtainedas shown in Fig. 1. 2. Experimental 2.1. Apparatus The HSCCC instrument employed in the present study wasTBE-300 high-speed counter-current chromatography (TautoBiotechnique, Shanghai, China) with three multilayer coil sep-aration column connected in series (I.D. of the tubing=1.5 mm,total volume=300 ml) and a 20 ml sample loop. The revolutionradius was 5 cm, and the values(B=r/R, where r is the rotationradius or the distance from the coil to the holder shaft, and Ris the revolution radius or the distances between the holder axisand central axis of the centrifuge) of the multilayer coil variedfrom 0.5 at internal terminal to 0.8 at the external terminal. Therevolution speed of the apparatus can be regulated with a speedcontroller in the range between 0 and 1000 rpm. The system was chrysophanol HOH,C 6-OH-emodin polygonimitin B 2, 3, 5, 4'-tetrahydroxystilbene-2-B-D-glucoside gallic acid Fig. 1. Chemical structures of compounds from Polygonum multiflorum Thunb. also equipped with a S-1007 constant flow pump (ShenyitongTech & Exploitation, Beijing, China), a Model 8823B-UV mon-itor (Bingdayingchuang Sci & Tech, Beijing, China). The datawere collected with model N2000 chromatography workstation(Zhejiang University, Hangzhou, China). The HPLC equip-ment used was Shimadzu LC-2010C HT system and ShimadzuHPLC workstation (Shimadzu, Japan). Nuclear magnetic reso-nance (NMR) spectrometer used here was Bruker AM-500MHz(Bruker, Switzerland). Electrospray ionization mass spectrom- etry (ESI-MS) used was Agilent 1100 Series LC-MS Trap SL(Agilent,USA). 2.2. Reagents All solvents used for HSCCC were of analytical grade (Han-bon Sci & Tech,Jiangsu,China). Methanol and acetonitrile usedfor HPLC were of chromatographic grade (Hanbon Sci & Tech)D-101 macroporous resin (Chemical Plant of Nankai University, Tianjin, China) was used for sample preparation and water usedwas distilled water. The dried roots of Polygonum multiflorum were purchasedfrom a local drug store and identified by Professor MingjianQing, Department of Medicinal Plant, China PharmaceuticalUniversity. 2.3. Preparation ofcrude extract Five hundred grams of dry slice (thickness: 1.0cm, breadth:0.8 cm) of Polygonum multiflorum Thunb. was extracted with5 L 95% ethanol and then evaporated to form a syrup. The syrupwas dissolved in 300 ml of water by sonication and partitionedwith ether and n-butanol of equal volume three times succes-sively. Both ether and n-butanol solution was vacuum evaporatedat 65°C. About 8.0g residue of ether and 7.5 g residue of n-butanol were obtained, respectively. In order to enrich the targetcomponents, the residue of n-butanol was then loaded on D-101macroporous resin column (35 cm ×3.4 cm, the volume of thecolumn was 170 ml) and eluted with 1700 ml of distilled waterand 3400 ml of 30% ethanol, respectively. The effluent of 30%ethanol was collected and evaporated at 65°Cunder vacuum andabout 3.5 g residue was obtained. Finally the fraction of waterwas evaporated at 90°C under vacuum and about 10.0 g residuewas obtained. All the residues were stored in a refrigerator (5°C)for further use. 2.4. Preparation oftwo-phase solvent system and samplesolution In the present study, the two-phase solvent system composedof n-hexane-ethyl acetate-methanol-water (3:7:5:5,v/v/v/v),n-hexane-ethyl acetate-methanol-water (9:1:5:5, v/v/v/v), ethylacetate-methanol-water (50:1:50, v/v/v) and ethyl acetate-n-buthanol-water (20:1:20, v/v/v) were used for HSCCC sepa-ration. Each component of the solvent system was added to aseparatory funnel and thoroughly equilibrated at room temper-ature.The upper phase and the lower phase were separated anddegassed by sonication for 30 min shortly before use. For HSCCC separation of ether fraction, the sample solu-tion was prepared by dissolving 200 mg of ether extract in 5 mlof the lower phase of n-hexane-ethyl acetate-methanol-water(3:7:5:5, v/v/v/v). The sample solution for HSCCC separationof n-butanol fraction was prepared by dissolving 130 mg ofrefined n-buthanol extract in 5 ml of the lower phase of ethylacetate-methanol-water (50:1:50, v/v/v), and the sample solu-tion of water fraction was prepared with 100 mg extract in 5 mlof the lower phase of ethyl acetate-methanol-water (20:1:20,v/v/v). 2.5. HSCCCseparation In each separation process, the separation column of HSCCCwas first entirely filled with the upper organic phase (stationaryphase). Then the apparatus was rotated at 800 rpm, while thelower aqueous phase (mobile phase) was pumped into the col-umn at the flow rate of 2.0 ml/min. After the mobile phase front emerged (about half an hour later) and the system establishedhydrodynamic equilibrium, the sample solution was injectedinto the separation column through the injection valve. Theeffluent from the outlet of the column was monitored with theUV detector at 254 nm. The whole process of separation wasunder room temperature (25.0-27.5°C). Each peak fraction wasmanually collected according to the chromatogram and evapo-rated under reduced pressure. The residuals were dissolved inmethanol for subsequent HPLC analysis. 2.6. HPLC analysis and identification ofHSCCC peak. fractions The HPLC analysis of every fraction of Polygonum multi-florum Thunb. and HSCCC peak fraction were performed witha Shimadzu VP-ODS column (150 mm x4.6 mm I.D., 5 pm)at room temperature. For analysis of the fraction of ether, themobile phase was methanol and water in gradient mode as fol-lows: 5:95 to 100:0 in 60 min, The effluent was monitored at254 nm and the flow rate was set at 1.2 ml/min constantly. Whenthe fraction of n-buthanol purified by macroporous resin columnwas analyzed, the mobile phase was acetonitrile and water in gra-dient mode as follows: 1:99 to 100:0 in 45 min, The effluent wasmonitored at 254 nm and the flow rate was kept at 0.8 ml/minconstantlv 3. Results and discussion In the present study, D-101 macroporous resin was used topurify crude extract of n-butanol fraction. Water was used toremove the hydrosoluble chemicals, such as pigments and sac-charides which had no or little retention on D-101 macroporousresin, at first. Then 30% ethanol was used to elute the target com-pounds, which was prepared for further HSCCC isolation andpurification.At last, 95% ethanol was used to activate the macro-porous resin. Consequently, 3.5 g of refined n-butanol fractionwas obtained from 500 g Polygonum multiflorum. A roadmap ofthe whole work is given in Fig. 2 and the HSCCC chromatogramsare shown in Fig. 3. 3.1. HPLC analysis and identification of HSCCC peakfractions Each peak fraction of HSCCC was analyzed by HPLC.According to the adopted methods the purity of eight compoundswere all above 97%.The HPLC chromatograms were shown inFig. 4. Identification of the HSCCC peak fractions was based onretention time together with HNMR andESI-MS. The latter is atechnology of soft ionization used to give the exact mass/charge(m/z) of quasi-molecular ion and valuable fragment peaks oflosing residual group of glucose. 3.2. Selection oftwo-phase solvent system and otherconditions ofHSCCC Successful separation by HSCCC largely depends upon theselection of suitable two-phase solvent system. In this experi- Fig. 2. Roadmap of extraction and separation. Mobile phase 1: the lower phase of n-hexane-ethyl acetate-methanol-water (3:7:5:5, v/v/v/v); mobile phase 2: thelower phase of ethyl acetate-methanol-water (50:1:50, v/v/v); mobile phase 3: the lower phase of ethyl acetate-n-buthanol-water (20:1:20, v/v/v). ment, several kinds of solvent systems including n-hexane-ethylacetate-methanol-water (3:7:5:5,9:1:5:5,1:1:1:1,1:9:5:5,7:3:5:5.v/v/v/v), ethyl acetate-methanol-water (50:1:50,10:1:10,1:0:1, v/v/v), and ethyl acetate-n-buthanol-water(20:1:20, 4:1:5, v/v/v) were tested. The results indicated thatthe target compounds have appropriate K-values (0.5-2) basi-cally in four systems, which were listed in Table 1. In general,small K-values usually result in a poor peak resolution, whilelarge K-values tend to produce excessive sample band broad-ening. For the components of large K-values, other modes of elution, such as dual mode, reversed mode elution or expellingsolution from column could be employed to shorten separationtime [8]. For separation of the fraction of ether, n-hexane-ethylacetate-methanol-water (3:7:5:5,v/v/v/v) was used as the two-phase solvent system of HSCCC. The anterior peaks wereseparated well, while the retention time of the remainder wasso long that the stepwise elution mode had to be employedThe lower phase of n-hexane-ethyl acetate-methanol-water(3:7:5:5,v/v/v/v) and (9:1:5:5,v/v/v/v) were used as the mobile (B) (C) Fig. 3. HSCCC chromatograms of two fractions of ether (A), n-butanol (B,C)and its second separation (D) from Polygonum multiflorum Thunb. Conditions ofA: Two-phase solvent system: n-hexane-ethyl acetate-methanol-water (3:7:5:5,9:1:5:5, v/v/v/v) in stepwise elution; Stationary phase: upper organic phase of3:7:5:5; mobile phase: lower phase of 3:7:5:5 and 9:1:5:5 used in stepwise elutionmode as follows: 0-200 min, the lower phase of 3:7:5:5 system; after 200 min, thelower phase of 9:1:5:5 system; flow rate:2.0 ml/min; revolution speed: 800 rpm;detection wavelength: 254 nm; sample size: 200 mg ofether extracts dissolvedin 5 ml of the lower phase of 3:7:5:5 system; retention of the stationary phase:56%; Peak I: rhein; Peak II: 6-OH-emodin; Peak II: emodin. Conditions of B& C: Two-phase solvent system: ethyl acetate-methanol-water 50:1:50 (v/v/v),after 450 min the direction of revolution was reversed, mobile phase: loweraqueous phase (0-450 min, B) and upper organic phase (after 450 min, C);flow rate: 2.0 ml/min (0-450min, B) and 1.5 ml/min (after 450 min, C); rev-olution speed: 800 rpm; detection wavelength: 254 nm; sample size: 130 mg of phase in stepwise elution. The lower phase of 3:7:5:5 systemwas used as the mobile phase during 0-200 min. After 200 min,the mobile phase was changed to the lower phase of 9:1:5:5 sys-tem, which was saturated with the upper phase of 3:7:5:5 systembefore use. Three pure peaks, marked as peak I, peak II and peakIII in Fig. 3A, could be obtained.They were identified as rhein(3.2mg),6-OH-emodin (5.4 mg) and emodin (48.5 mg) in turn.Finally the residual solution in the separation column was blownout and another compound, which could not be shown in Fig. 3A,was acquired and identified as chrysophanol (15.7mg). Ethyl acetate-methanol-water (50:1:50, v/v/v) was used asthe two-phase solvent system for separation of the refined n-butanol fraction. The upper organic phase was used as thestationary phase and the lower aqueous phase as the mobilephase. When the refined n-butanol fraction was analyzed by TLC(developer: ethyl acetate-acetic acid (9:1)), a big spot with dis-tinct green fluorescence (254 nm) and small Rf value was foundand naturally it was regarded as one of the target compounds.During this HSCCC separation process, peak IVa and IVb inFig. 3B were collected and analyzed by TLC, respectively. Theresults indicated that both IVa and IVb allcontained the spot withgreen fluorescence and other impurities. So peak IVa and IVbwere combined together as peak IV and further separated withanother solvent system,ethyl acetate-n-buthanol-water 20:1:20(v/v/v) for better resolution and higher recovery. Then peak IX inFig. 3D corresponding to the compound with green fluorescencewas obtained and identified as polygonimitin B (7.2mg). Afterpeak VI in Fig. 3B was eluted out, the rotation direction of theHSCCC apparatus was changed to reversed rotation and mobilephase to the upper phase. Meanwhile the flow rate was reducedfrom 2.0 to 1.5 ml/min. Then another two peaks were obtained,which were marked as peak VII and peak VIII in Fig. 3C.Peak V,VI, VII and VIII were identified as an unknown glu-coside (5.4 mg), 2,3,5,4'-tetrahydroxystilbene-2-B-D-glucoside(56.4mg), emodin-8-B-D-glucoside (11.9 mg) and gallic acid(3.7mg) in turn. The fraction of water, which contained polygonimitin Bmainly and other water-soluble impurity like pigments and sac-charides, could be separated by the same solvent system of ethylacetate-n-buthanol-water (20:1:20,v/v/v) and relevant condi-tions by HSCCC to obtain polygonimitin B (about 8 mg) too.The chromatogram was very similar to Fig. 3D. So it is notgiven in this paper. 3.3. The structural identification The structural identification of peak fractions was performedwith ESI-MS (negative ion mode) and HNMR with TMS ( refined n-buthanol extract dissolved in 5 ml of the lower phase, retention of t he stationary phase: 46%; C onditions of D: T wo -phase so l vent system: et h yl a cetate-n-buthanol- w ater 20:1:20 (v/v/v); mobile p hase: the lower phase; flowrate: 2.0 ml/min; revolution spee d :800 rpm; detection wavelength:254 nm; thesample was f r om the eluent o f p e ak I V of B; retention o f the stationary phase:44%; P e ak V: an unknown glucoside; Peak VI: 3 ,5, 4 ' -tetrahydroxystilbene-2-B- D -glucoside; Peak VII: emodin-8-B-D- g lucoside; Pe a k VIII: g a l lic acid; Peak I X: polygonimitin B. Separation temperature: room temperature(25.0-27.5°C) . ) Table 1The K-values of target components measured in different solvent systems Solvent system K-value n-Hexane-ethyl n-Hexane-ethyl Ethyl acetate-methanol Ethyl acetate-n-buthanol acetate-methanol-water acetate-methanol-water -water (50:1:50,v/v/v) -water (20:1:20,v/v/v) (3:7:5:5,v/v/v/v) (9:1:5:5, v/v/v/v) Rhein 0.83 0.46 6-OH-Emodin 1.12 0.68 Emodin 7.34 4.55 Chrysophanol 14.87 11.30 Polygonimitin B - 0.74 0.95 The unknown glucoside - - 0.92 1.16 2,3,5,4'-Tetrahydroxystilbene-2-B-D-glucoside - 2.26 3.15 Emodin-8-B-D-glucoside - 4.74 6.97 Gallic acid - 7.46 10.91 Experimental protocol: 4 ml of each phase of the equilibrated two-phase solvent system was added to approximately 8 mg of crude sample placed in a 10 ml testtube. The test tube was caped and shaken vigorously for 2 min to equilibrate the sample thoroughly. An equal volume of each phase was then analyzed by HPLC toobtain the partition coefficient (K). The partition coefficient (K) value was expressed as the peak area of the compound in the upper phase divided by the peak areaof the compound in the lower phase. as internal standard. Data of each compound were given asfollows: Peak I in Fig. 3A: ESI-MS (m/z): 283 ([M-H]-).HNMR (500MHz, CD3OD): 8.07 (1H, s, H-4), 7.81 (1H, d, J=7.5Hz, H-7), 7.76 (1H, s, H-2), 7.71 (1H, m, H-6), 7.38 (1H, d,J=8.5Hz, H-5). The results were sim-ilar to those in ref. [9], and peak I corresponded torhein. mV Fig.4. HPLC chromatograms of two fractions of ether (A) and n-butanol (F) from Polygonum multiflorum Thunb. and HSCCC peak fractions of them (B-E,G-H) Column: Shimadzu VP-ODS (150mm x4.6mm I.D.,5um); mobile phase (A-E): methanol-water (methanol: 5% to 100% in 60 min); flow rate: 1.2ml/min;detection wavelength:254 nm; mobile phase (F-H):acetonitrile-water (acetonitrile: 1% to 100% in 45 min); flow rate: 0.8 ml/min; detection wavelength: 254 nm. mv Detector A:254nm Oven Temp.(Status) (F) 2,3,5,4'-tetrahydroxystilbene 900 -2-B-D-glucoside o.0 5.0 10.0 15.020.025.030.035.0 min Fig. 4. (Continued). Peak II in Fig. 3A: ESI-MS (m/z): 285 ([M-H]). HNMR (500 MHz, CD3OD): 7.67 (1H,d,J=2.2Hz, H-5), 7.21(1H, d, J=2.2Hz, H-7), 7.01 (1H,J=3.1Hz, H-4), 6.60 (1H,d, J=3.1Hz, H-2), 4.62(2H, s, 6-CH2OH). The results weresimilar to those in ref. [10], and peak II corresponded to 6-OH-emodin. Peak III in Fig. 3A:ESI-MS(m/z):269([M-H]-). HNMR(500 MHz, CD3OD): 7.64(1H,s,H-5), 7.30(1H, d,J=2.4Hz,H-4), 7.09 (1H, s, H-7), 6.70 (1H, d,J=2.4Hz, H-2), 2.45 (3H,s,6-CH3). The results were similar to those in ref. [11], and peakIII corresponded to emodin. Data of the compound blown out from the column during theseparation ofether fraction: ESI-MS (m/z): 255 ([M-H]-);H NMR (500MHz,CD3OD): 7.87 (1H, d,J=7.2Hz, H-5), 7.69(1H, m, H-6), 7.64 (1H, s, H-4), 7.32 (1H, d,J=8.4Hz, H-7),7.13(1H,s,H-2),2.50 (3H, s,3-CH3). The results were similarto those in ref. [9], it corresponded to chrysophanol. Peak V in Fig. 3B: ESI-MS fragment peak (m/z:325) showedthe losing of glucose from quasi-molecular ion (m/z: 487), andcorresponding H NMR signals of glucose in the region of3.30-4.50 ppm were found. Peak V was determined as a glu-coside temporarily and further structural identification will bedone in the further research work. Peak VI in Fig. 3B: ESI-MS (m/z): 405 ([M-H]-), 243([M-H-162]-). HNMR (500MHz, CD3OD): 7.76 (1H, d,J=16.0Hz, H-a), 7.44 (2H, d,J=8.2Hz, H-3',5'),6.92 (1H, d, J=16.0Hz, H-B), 6.80 (2H, d, J=8.2Hz, H-2', 6'), 6.62(1H, J=3.1Hz, H-6), 6.24 (1H, J=3.1Hz,H-4), 4.50 (1H, d,J=7.0 Hz, H-12-0-Glc), 3.40-3.80 (6H, m, protons of glucose).The results were similar to those in ref. [11],and peak VI cor-responded to 2,3,5,4'-tetrahydroxystilbene-2-B-D-glucoside. Peak VII in Fig. 3C: ESI-MS (m/z): 431 ([M-H]-), 269([M-H-162]-).HNMR(500MHz,CD30D): 7.45 (1H, br,s, H-4), 7.28 (1H,d,J=2.1Hz,H-5), 7.14 (1H,br, s, H-2), 6.99(1H, d, J=2.1Hz, H-7),5.04 (1H, d,J=6.2Hz, H-18-O-Glc),3.30-3.70 (6H, m, protons of glucose), 2.40 (3H, s, 6-CH3).Compared with the data given in ref. [11], and peak VII corre-sponded to emodin-8-B-D-glucoside. Peak VIII in Fig. 3C: ESI-MS (m/z): 169 ([M-H]-). DataofHNMR (500MHz, CD3OD) matched with the reported the data given in ref. [12], and peak VIII corresponded to gallic acid.Peak IX in Fig. 3D: ESI-MS (m/z): 417 ([M-H]-), 255([M-H-162]-).HNMR (500MHz, CD3OD): 7.26 (1H, s,H-8), 6.78 (1H, s, H-4), 6.74 (1H, d,J=2.2Hz,H-2), 6.17 (1H,s, H-5), 4.54 (1H,J=7.2Hz, H-11-0-Glc), 3.44~3.90 (6H, m,protons of glucose), 2.73 (3H, s, 7-CH3), 2.53 (3H, s,6-CH3).The results were similar to those in ref. [13], and peak IX cor-responded to polygonimitin B. 4. Conclusions Some of the chemical constituents of Polygonummultiflorum,one of the most important Chinese medicinal herbs, were iso-lated and purified systematically by HSCCC. The results of theresearch provided a successful pattern for isolation of chemicalcompounds from low polarity such as aglycones to high polaritysuch as glycosides by HSCCC. Because there are too many con-stituents present in natural herbs, and their polarity differed verymuch, it is comparatively difficult to isolate all the constituentsof crude extract with only one solvent system. So it is necessaryto obtain a few main fractions from low polarity to high polarityby using different solvent extraction. For low polarity fraction,such as aglycones, n-hexane could be used as the major solventof organic phase, and n-hexane-methanol (ethanol)-water or n-hexane-ethyl acetate-methanol (ethanol)-water could be used as the two-phase solvent system. For moderately polar fraction,just like a part of glycosides with single sugar, ethyl acetatecould be used as the major solvent of organic phase, and ethylacetate-methanol (ethanol)-water or ethyl acetate-n-buthanol-water could be used as the two-phase solvent system. If thepolarity of fraction was higher, just like general glycosides withpolysaccharide, n-buthanol could be used as the major solventof organic phase, and the volume ratio of every solvent shouldbe adjusted properly according to the actual separation demandto exert advantages of HSCCC. Acknowledgement This work was financially supported by the National Foundation for Outstanding Young Scientists (No. 30525032). References ( [ 1 ] China P harmacopoeia Co mmittee, Ph a rmacopoeia of th e Peo p le’sRepublic o f C hina, C hina C h emical In d ustry P r e ss, B e ijing, 2004, p .1 22 (the first div i sion of 2 005 e d ition). ) ( [2] H.Z. Zheng, Z.H. Dong, J. Se , Modern R esearch a nd A pplication of T raditional Chinese Medicine, Xue Yuan Pres s , Bei j ing, 1998 , p. 231 0 . ) ( [ 3] Y . I to, J. C hromatogr. 2 14 ( 1 981 ) 1 2 2. ) ( [4 ] T.Y. Zhang, X. Hua, R . X iao, S . K nog, J . L iq. Chromatogr. 11 (1988) 2 33. ) ( [ 5 ] R. M . Liu, A .F. Li, A .L. Sun, J. C hromatogr. A 10 5 2 (2004) 21 7 . ) ( [6] L. Chen, Y.S. H an, F.Q. Yang, T.Y . Zhang,J. Chromatogr. A 907 (2001)343. ) ( [7] R. Slimestad, A. M arston, K . Hostettmann, J . C hromatogr. A 71 9 (1996)438. ) ( [8] X.L. Cao, High-speed Counter-current Chromatography T echnology and i ts A pplication, China C hemical In d ustry P r e ss, Be i jing, 2 00 5, p. 44 . ) ( [9] R.M. L iu, A. F . Li, A.L. Sun, J. Chromatogr. A 1052 (2004) 2 17. ) ( [10] Materia Medica of Chinese Academy o f M e dical S c iences, M odernResearch o f C hinese T r aditional and Herbal D r ugs, 3 , P eking U n i ver-sity of Medical Science & P e king Union Med i cal College Pres s , Beijing,1997, p. 218. ) ( [11] J .B. L i , M. L in, C hin. Trad. H erb. D r ugs. 2 4 ( 1993) 11 5 . ) ( [ 12] R .H. L iu, L. Y. Kong, C hin. J. Chin. Mater. Med. 30 ( 2 005) 1 213. ) ( [ 1 3] L.X. Zhou, M . Lin, J .B. L i, S.Z. Li, Acta Pharma. S inica. 29 (1994)107. )
确定




还剩6页未读,是否继续阅读?

产品配置单



上海同田生物技术有限公司-高速逆流色谱仪HSCCC为您提供《何首乌根茎中化学成分分离纯化检测方案(高速逆流色谱)》,该方案主要用于中药材和饮片中含量测定检测,参考标准--,《何首乌根茎中化学成分分离纯化检测方案(高速逆流色谱)》用到的仪器有TBE300B+AKTA高速逆流色谱仪/离心分配色谱/萃取仪/制备色谱仪、TBE-200V 高速逆流色谱仪、TBE-1000A制备色谱仪/萃取仪/快速分离制备色谱仪
推荐专场





相关方案
更多
该厂商其他方案
更多