








方案详情
文
The medicinal plant Atractylodes macrocephala (Baizhu in Chinese) has been widely used in traditional Chinese medicine for energy and stomach complaints, treatment of dyspepsia and anorexia, anti-inflammation, anticancer and for increasing assimilation.
A high-speed counter-current chromatography (HSCCC) method was developed for the preparative separation and purification of two main bioactive components,namely, atractylon and atractylenolide III from A. macrocephala by using light petroleum (60–908C)–ethyl acetate–ethanol–water (4 :1:4:1 v/v) as the two-phase solvent system in dual-mode elution. Compared with the separation using the normal-mode elution, the dual-mode HSCCC can be achieved with shorter elution time.Atractylenolide III (32.1 mg) at 99.0% purity and 319.6 mg atractylon at 97.8% purity could be obtained from 1000 mg crude sample in a single run. The recoveries of atractylenolide III and atractylon were 95.4 and 92.6%, respectively.
方案详情

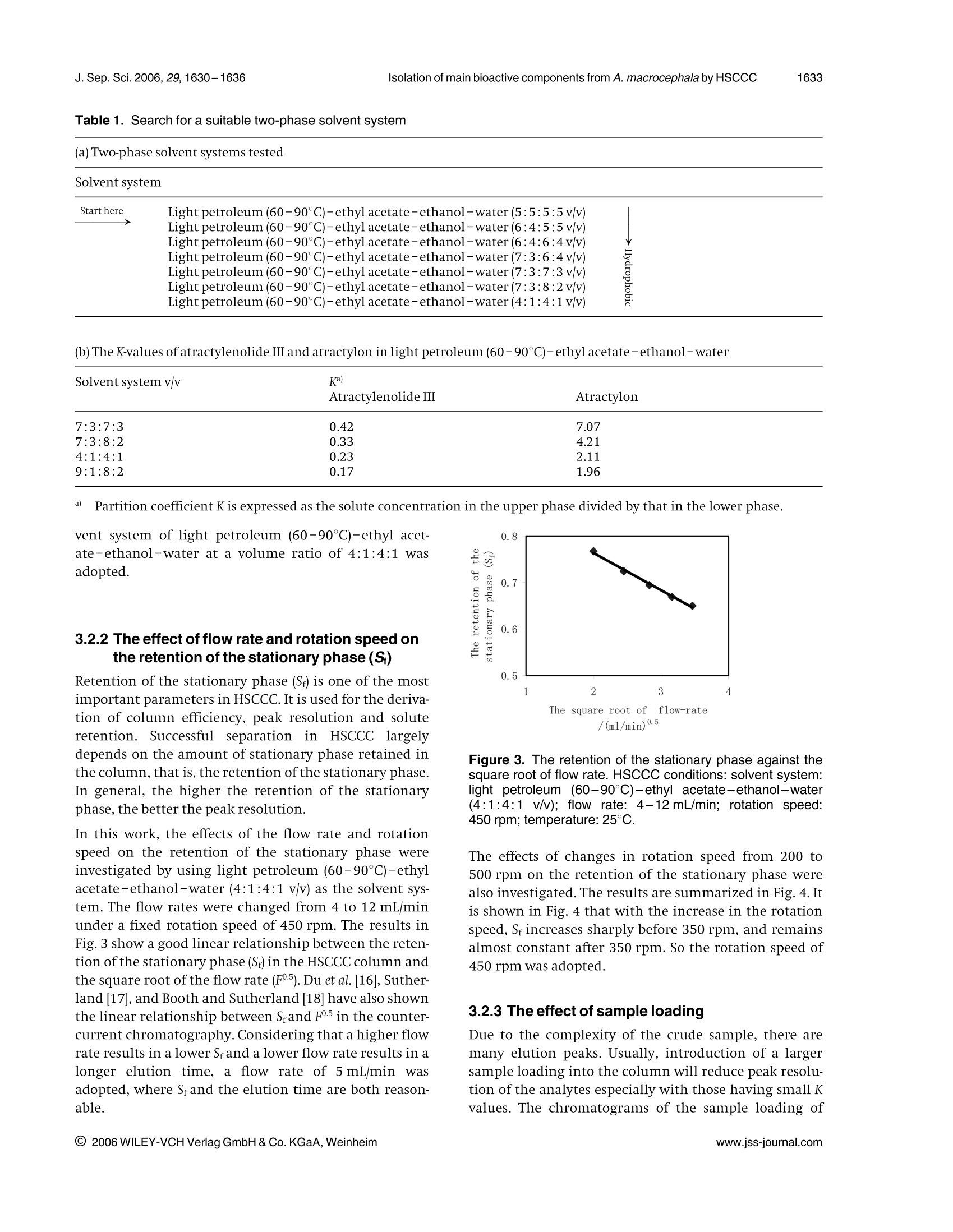
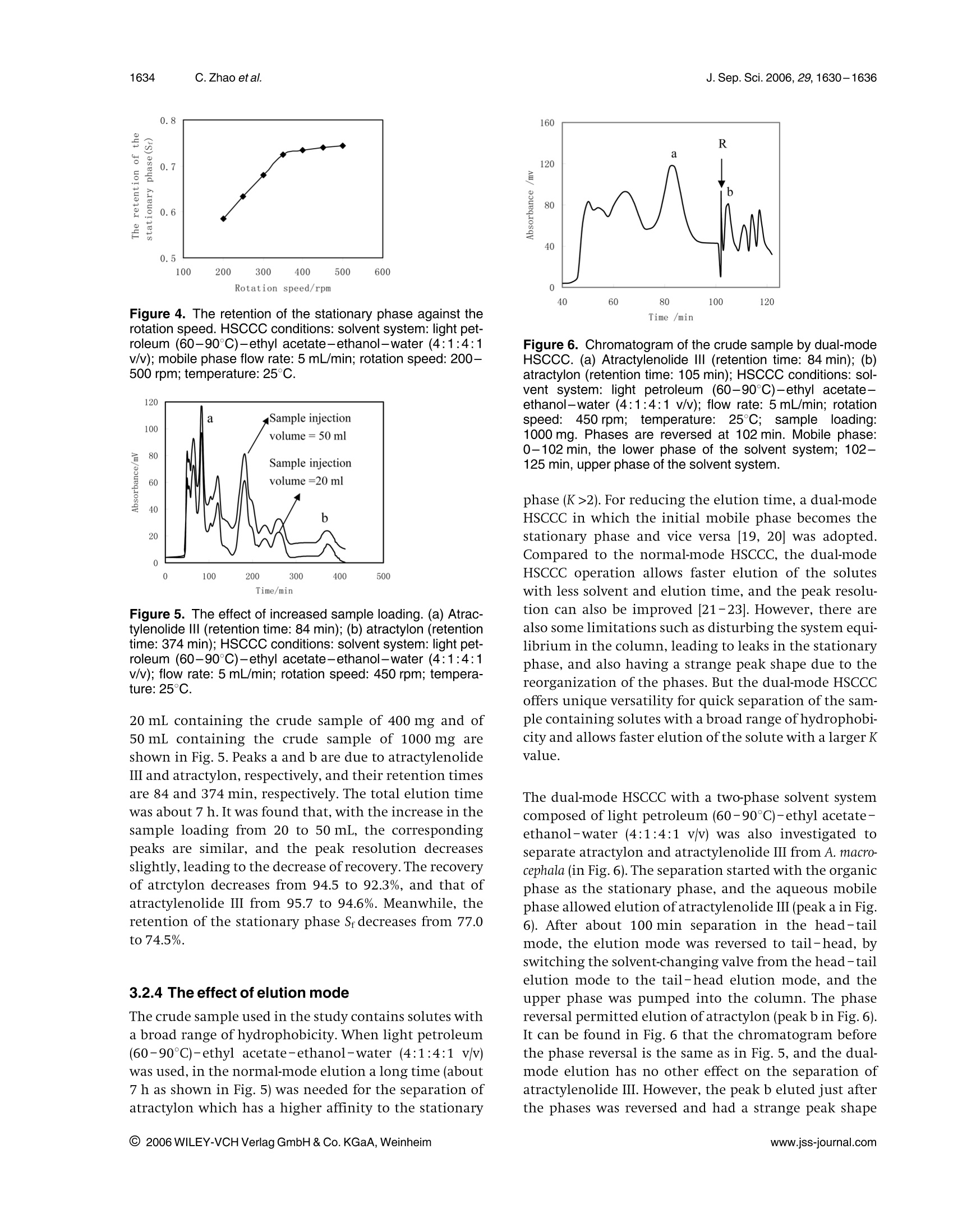
J. Sep. Sci. 2006, 29,1630-16361630 1631J. Sep. Sci. 2006, 29, 1630-1636Isolation of main bioactive components from A. macrocephala by HSCCC Original Paper Preparative isolation and purification of atractylonand atractylenolide Ill from the Chinese medicinalplant Atractylodes macrocephala by high-speedcounter-current chromatography The medicinal plant Atractylodes macrocephala (Baizhu in Chinese) has been widelyused in traditional Chinese medicine for energy and stomach complaints, treatmentof dyspepsia and anorexia, anti-inflammation, anticancer and for increasing assimi-lation. A high-speed counter-current chromatography (HSCCC) method was devel-oped for the preparative separation and purification of two main bioactive compo-nents, namely, atractylon and atractylenolide III from A. macrocephala by using lightpetroleum (60-90℃)-ethyl acetate-ethanol-water (4:1:4:1 v/v) as the two-phasesolvent system in dual-mode elution. Compared with the separation using the nor-mal-mode elution, the dual-mode HSCCC can be achieved with shorter elution time.Atractylenolide ⅢI (32.1 mg)at 99.0% purity and 319.6 mg atractylon at 97.8% puritycould be obtained from 1000 mg crude sample in a single run. The recoveries ofatractylenolide III and atractylon were 95.4 and 92.6%, respectively. Keywords: Atractylon |Atractylenolide III | Atractylodes macrocephala |High-speed counter-currentchromatography(HSCCC) Received: November 30, 2005; revised: March 24,2006;accepted: March 24, 2006 1 Introduction The rhizome of Atractylodes macrocephala (Baizhu in Chi-nese), as one of the most popular traditional Chinesemedicinal herbs, has been widely used for a long time. Itis reported as a nutrient for energy and stomach com-plaints and for treatment of dyspepsia and anorexia inthe Pharmacopoeia of People’s Republic of China [1].Atractylon, atractylenolide I, I, II and some othersequensenes are the major bioactive components of A.macrocephala [2, 3]. Many studies showed that these com-pounds had the effects of anti-inflammation, anticancer,and increasing of assimilation [4,5]. Usually, atractylon is the main bioactive component of A.macrocephala. The contents of atractylenolide I, II and IIIare small, and their contents vary with the productionarea and cropping season [6]. A. macrocephala used in thiswork is from Xinchang county in Zhejiang Province, ( Correspondence: Professor Chaohong He, Department of Che- mical Engineering, Zhejiang University , Hangzhou 310027, China. ) ( E-mail: chhezju@zju.edu.cn. ) ( Fax:+86-571-87951742. ) ( Abbreviation: HSCCC,high-speed counter-current chromatogra- phy ) Figure 1. Chemical structures of atractylon and atractyleno-lide ⅢIl. which is one of the main producing areas for A. macroce-phala in China. In this A. macrocephala, atractylenolide Iand II are too small and can be neglected. So the mainbioactive components are atractylon and atractylenolideI. The chemical structures of atractylon and atractyle-nolide III are shown in Fig. 1. Atractylon is very unstable, and also there are many iso-meric compounds existing together in A. macrocephala [7,8]. Thus, the separation of atractylon and atractylenolideIII from A. macrocephala is quite difficult. The conven-tional separation method of atractylon and atractyleno-lide III is the silica gel column chromatography undernitrogen protection, which is time-consuming and hasthe peril of loss of compounds [9]. Therefore it is of great interest to develop a new method to separate atractylonand atractylenolide III from A. macrocephala The high-speed counter-current chromatography(HSCCC) is a very effective tool for the preparative separa-tion and purification of natural products and Chinesetraditional herbs [10-12]. Compared with the traditionalliquid-solid column chromatography, HSCCC elimi-nates irreversible adsorption loss of samples and yieldshigher recovery and efficiency. So it is widely used in theseparation of active components from traditional Chi-nese medicinal herbs and natural products [13-15]. The present paper deals with the one-step isolation andpurification of atractylon and atractylenolide III from A.macrocephala by HSCCC. 2 Experimental 2.1 Apparatus The preparative HSCCC was carried out with a ModelTBE-1000A HSCCC (Tauto Biotech, Shanghai, China)equipped with a 1000 mL coil column made of PTFE tub-ing (1.8 mm) and a 100 mL sample loop. The p-value ofthe preparative column varied from 0.42 at the internallayer to 0.63 at the external layer (β=r/R, where r is thedistance from the coil to the holder shaft, and R is therevolution radius or the distance between the holderaxis and the central axis of the centrifuge). The rotationspeedof the apparatus can be regulated with a speed con-troller in the range between 0 and 600 rpm. The HSCCCsystem is equipped with a Model SD-9002 constant-flowpump, a HX-2050 water bath, a Model 8823B UV monitoroperating at 254 nm and a Model N2010 chromatogra-phy workstation. 2.2 Reagents and materials All organic solvents used for the preparation of the crudesample and for the HSCCC separation were of analyticalgrade (Hangzhou Datong Reagent factory, Hangzhou,China), and the water used was distilled water. The dried roots of A. macrocephala were from Xinchangcounty in Zhejiang Province,China. 2.3 Preparation of crude sample fromA. macrocephala The dried roots of A. macrocephala were powdered (about40 mesh) and extracted with ethyl acetate three times.The extraction time was 2,2 and 1 h, respectively. Theethyl acetate solutions were combined and evaporatedby rotary vaporization at 45℃ under reduced pressure.Subsequently, the extract was dissolved in ethanol andwas frozen at -4°C for 24 h. The deposit was separatedand removed. Then the residue was dissolved with asmall amount of ethyl acetate, and eight times the amount of light petroleum (60-90°C) was added to preci-pitate the residue. After removing the deposit, the crudesample was obtained. It was stored in a refrigerator forthe subsequent HSCCC separation. 2.4 Selection of the two-phase solvent systems Lightpetroleum (60-90C)-ethyl acetate-ethanol-water was used as the two-phase solvent system. The com-position of the two-phase solvent system was selectedaccording to the partition coefficient (K). The K-valueswere determined by GC as follows: A suitable amount ofcrude sample was added to a test tube, to which 2 mL ofeach phase of the two-phase solvent system was added.After a thorough equilibration, the upper phase andlower phase were separated and analysed by GC. The par-tition coefficient (K) of the target component in the sam-ple was expressed as the peak area of the solute in theupper phase divided by that in the lower phase. 2.5 Preparation of two-phase solvent system andsample solutions Lightpetroleum(60-90°C)-ethyl1 acetate-ethanol-water (4:1:4:1 v/v) was prepared by adding all the sol-vents to a separation funnel according to the volumeratios and thoroughly equilibrated by shaking repeat-edly. Then the upper phase and the lower phase wereseparated 30 min prior to use. The sample solution was prepared by dissolving thecrude sample in the solvent mixture of the lower phaseand upper phase (1:1 v/v) of the solvent system, becausethe sample was not easily dissolved in either phase alone. 2.6 HSCCC separation procedure In each separation, the coiled column was first entirelyfilled with the upper phase (stationary phase), and thenthe apparatus was rotated at 450 rpm, while the lowerphase (mobile phase)was pumped into the column at aflow rate of 5.0 mL/min. After the mobile phase frontemerged and the hydrodynamic equilibrium was estab-lished in the column, the retention of the stationaryphase can be calculated; then the sample solution of50 mL, containing the crude extract of 1000 mg wasinjected through the injection valve. In the dual-modeHSCCC, after about 100 min separation in the head-tailmode, the elution mode was reversed to tail-head, byswitching the solvent-changing valve from the head-tailelution mode to the tail-head elution mode,and theupper phase was pumped into the column to obtaintail-head fractions. The effluent of the column was con-tinuously monitored with a UV detector at 254 nm. Frac-tions were collected every 2 min. Each peak fraction wasmanually collected according to the chromatogram andevaporated under reduced pressure. The residues were dissolved in ethanol for subsequent GC analysis. Afterthe separation, the solvents in the column were pushedout. 2.7 GC analysis and identification of fractionatedcompounds The crude sample and each HSCCC peak fraction wereanalysed by GC, and the partition coefficient was alsodetermined by GC. GC analyses were performed with aGC-1690 gas chromatography column (Hangzhou Ke-Xiao) equipped with a FID and SE-54 capillary column(30 mx0.25 mm; coating thickness 0.50 um). Oven tem-perature was programmed to 80°C for 2 min, and thenincreased to 260°℃ at a rate of 5°℃ min. Injector anddetector temperatures were 260 and 240°C, respectively.The carrier gas, nitrogen, was adjusted to1 mL/min. Thecrude sample and each peak fraction were diluted 20times and 0.5 uL of the diluted solution was injected intothe GC. The percentages of compounds were calculatedby the area normalization method. The components separated from HSCCC were identifiedwith GC/EIMS, lH and 13C NMR. GC/EIMS analyses wereperformed with a Trace 2000 GC equipped with a HP-5capillary column (30mx0.25 mm; coating thickness0.25 um) and a Trace 2000 IT mass detector. The injectorand transfer line temperatures were 260 and 250°C,respectively.Oven temperature was programmed to 50°Cfor 2 min, and then increased to 240°C at a rate of 15°C/min. The carrier gas, nitrogen, was adjusted to 1 mL/min.The peak fractions of the target compounds were diluted10 times and 0.2 uL of the diluted solution was injected.The ionization energy was 70 eV with a scan time of1 sand a mass range of 30-500 amu. The percentages ofcompounds were calculated by the area normalizationmethod without considering response factors. H and13Cspectra were recorded in CDCl on a Bruker AMX 400spectrometer (Karlsruhe, Germany) using TMS as theinternalstandard. 3 Results and discussion 3.1 GC analysis of the crude sample The crude sample obtained from A. macrocephala was ana-lysed by GC, and the chromatogram is shown in Fig. 2.The contents of atractylon and atractylenolide III in thecrude sample are 34.7 and 3.36%, respectively. 3.2 Optimization of HSCCC conditions 3.2.1 Optimization of the two-phase solventsystem A successful separation by HSCCC depends upon theselection of a suitable two-phase solvent system. A sol-vent system composed of light petroleum (60-90℃)- Figure 2. GC chromatogram of the crude sample. Experi-mentalconditions: an SE-54 capillary column(30 mx0.25 mm; coating thickness 0.50 um) with a FID;injector temperature 260℃; detector temperature 240℃;oven temperature programmed from 80 (hold 2 min) to260℃ at 5°C/min, then held at 30 min; carrier gas nitrogen,at 1 mL/min; injection of 0.5 uL of the diluted crude sample. ethyl acetate-ethanol-water at different volume ratioswas investigated. This two-phase solvent system could beused to isolate and separate a broad range ofhydrophobi-city by modifying the volume ratio of the four compo-nents. Because the polarities of the target compounds inthe crude sample were unknown in the beginning, thesearch started with the two-phase solvent system com-posed of light petroleum (60-90℃)-ethyl acetate-etha-nol-water at a volume ratio of5:5:5:5which has a mod-erate degree of polarity. But the atractylon is mostly dis-tributed in the upper organic phase (K > 10) for the sol-vent system at a volume ratio of 5:5:5:5,which tends toresult in a too long elution time and a broad peak. So thevolume ratio was changed by reducing the volumes ofethyl acetate and water. The two-phase solvent systemstested in the present study are shown in Table 1(a). Thesearch is directed downwards along the arrow towardmore hydrophobic. Light petroleum (60-90℃)-ethylacetate-ethanol-water at the volume ratio of 6:4:5:5,6:4:6:4 and 7:3:6:4 were not suitable because themobile phases also had strong polarity, and atractyloncould not be eluted in an acceptable time. The partitioncoefficients of the target compounds in the other foursolvent systems were measured and are shown in Table1(b). Furthermore, the solvent systems listed in Table 1(b)were tested by HSCCC separation. The results indicatedthat when light petroleum (60-90°℃)-ethyl acetate-ethanol-water (7:3:7:3 or 7:3:8:2 v/v) was used as thesolvent system, the elution time was more than 18 or11 h, respectively. When light petroleum (60-90℃)-ethyl acetate-ethanol-water(9:1:8:2 v/v) was used,atractylenolide III was eluted closer to the solvent frontwith lower resolution and could not get satisfactory sep-aration. When light petroleum (60-90℃)-ethyl acet-ate-ethanol-water (4:1:4:1v/v) was used as the solventsystem, good separation results could be obtained andthe separation time was acceptable. So the two-phase sol- Table 1. Search for a suitable two-phase solvent system (a) Two-phase solvent systems tested (a) Two-phase solvent systems tested Solvent system Start here Light petroleum (60-90°C)-ethyl acetate-ethanol-water(5:5:5:5v/v) Light petroleum ((60-90℃)-ethyl acetate-ethanol-water(6:4:5:5v/v) Light petroleum ((f60-90C)-ethyl acetate-ethanol-water((6:4:6:4v/v) Light petroleum (f(60-90C)-ethyl acetate-ethanol-water((7:3:6:4v/v) Light petroleum ((60-90℃)-ethyl acetate-ethanol-water(7:3:7:3v/v) Light petroleum (60-90C)-ethyl acetate-ethanol-water(7:3:8:2v/v) (60-90°C)-ethyl acetate-ethanol-water (4:1:4:1v/v) Light petroleum ( (b)The K-values of atractylenolide III and atractylon in light petroleum (60-90°C)-ethyl acetate-ethanol-water Solvent system v/v K) Atractylenolide III Atractylon 7:3:7:3 0.42 7.07 7:3:8:2 0.33 4.21 4:1:4:1 0.23 2.11 9:1:8:2 0.17 1.96 a) Partition coefficient K is expressed as the solute concentration in the upper phase divided by that in the lower phase. vent system of light petroleum (60-90℃)-ethyl acet-ate-ethanol-water at a volume ratio of 4:1:4:1 wasadopted. 3.2.2 The effect of flow rate and rotation speed onthe retention of the stationary phase (S;) Retention of the stationary phase (Sr) is one of the mostimportant parameters in HSCCC. It is used for the deriva-tion of column efficiency, peak resolution and soluteretention. Successful separation in HSCCC largelydepends on the amount of stationary phase retained inthe column, that is, the retention of the stationary phase.In general, the higher the retention of the stationaryphase, the better the peak resolution. In this work, the effects of the flow rate and rotationspeed on the retention of the stationary phase wereinvestigated by using light petroleum (60-90°C)-ethylacetate-ethanol-water (4:1:4:1 v/v) as the solvent sys-tem. The flow rates were changed from 4 to 12 mL/minunder a fixed rotation speed of 450 rpm. The results inFig.3 show a good linear relationship between the reten-tion of the stationary phase (Sr) in the HSCCCcolumn andthe square root of the flow rate (F5). Du et al. [16], Suther-land 17, and Booth and Sutherland [18] have also shownthe linear relationship between Srand F.5 in the counter-current chromatography.Considering that a higher flowrate results in a lower Sf and a lower flow rate results in alonger elution time, a flow rate of 5 mL/min wasadopted, where Sfand the elution time are both reason-able. /(ml/min)0.5 Figure 3. The retention of the stationary phase against thesquare root of flow rate. HSCCC conditions: solvent system:light petroleum (60-90℃)-ethyl acetate-ethanol-water(4:1:4:1 v/v); flow rate: 4-12 mL/min; rotation speed:450 rpm; temperature: 25C. The effects of changes in rotation speed from 200 to500 rpm on the retention of the stationary phase werealso investigated.The results are summarized in Fig. 4. Itis shown in Fig. 4 that with the increase in the rotationspeed, Srincreases sharply before 350 rpm, and remainsalmost constant after 350 rpm. So the rotation speed of450 rpm was adopted. 3.2.3 The effect of sample loading Due to the complexity of the crude sample, there aremany elution peaks. Usually, introduction of a largersample loading into the column will reduce peak resolu-tion of the analytes especially with those having small Kvalues. The chromatograms of the sample loading of Figure 4. The retention of the stationary phase against therotation speed. HSCCC conditions: solvent system: light pet-roleum (60-90°℃)-ethyl acetate-ethanol-water (4:1:4:1v/v); mobile phase flow rate:5 mL/min; rotation speed: 200-500 rpm; temperature: 25C. Figure 5. The effect of increased sample loading. (a) Atrac-tylenolide III (retention time: 84 min); (b) atractylon (retentiontime: 374 min); HSCCC conditions: solvent system: light pet-roleum (60-90°℃)-ethyl acetate-ethanol-water (4:1:4:1v/v); flow rate: 5 mL/min; rotation speed: 450 rpm; tempera-ture: 25°C. 20 mL containing the crude sample of 400 mg and of50 mL containing the crude sample of 1000 mg areshown in Fig.5. Peaks a and b are due to atractylenolideIII and atractylon,respectively, and their retention timesare 84 and 374 min, respectively. The total elution timewas about 7 h. It was found that, with the increase in thesample loading from 20 to 50 mL, the correspondingpeaks are similar, and the peak resolution decreasesslightly,leading to the decrease of recovery.The recoveryof atrctylon decreases from 94.5 to 92.3%, and that ofatractylenolide III from 95.7 to 94.6%.Meanwhile, theretention of the stationary phase S decreases from 77.0to 74.5%. 3.2.4 The effect of elution mode The crude sample used in the study contains solutes witha broad range of hydrophobicity. When light petroleum(60-90℃)-ethyl acetate-ethanol-water (4:1:4:1 v/v)was used, in the normal-mode elution a long time (about7 h as shown in Fig.5) was needed for the separation ofatractylon which has a higher affinity to the stationary Time /min Figure 6. Chromatogram of the crude sample by dual-modeHSCCC. (a) Atractylenolide Il (retention time: 84 min); (b)atractylon (retention time: 105 min); HSCCC conditions: sol-vent system: light petroleum (60-90℃)-ethyl acetate-ethanol-water (4:1:4:1v/v); flow rate: 5 mL/min; rotationspeed: 450 rpm; temperature: 25℃; sample: loading:1000 mg. Phases are reversed at 102 min. Mobile phase:0-102 min, the lower phase of the solvent system; 102-125 min, upper phase of the solvent system. phase (K>2). For reducing the elution time, a dual-modeHSCCC in which the initial mobile phase becomes thestationary phase and vice versa [19, 20] was adopted.Compared to the normal-mode HSCCC, the dual-modeHSCCC operation allows faster elution of the soluteswith less solvent and elution time, and the peak resolu-tion can also be improved [21-23]. However, there arealso some limitations such as disturbing the system equi-librium in the column, leading to leaks in the stationaryphase,and also having a strange peak shape due to thereorganization of the phases. But the dual-mode HSCCCoffers unique versatility for quick separation of the sam-ple containing solutes with a broad range of hydrophobi-city and allows faster elution of the solute with a larger Kvalue. The dual-mode HSCCC with a two-phase solvent systemcomposed of light petroleum (60-90°℃)-ethyl acetate-ethanol-water (4:1:4:1 v/v) was also investigated toseparate atractylon and atractylenolide III from A. macro-cephala (in Fig. 6). The separation started with the organicphase as the stationary phase, and the aqueous mobilephase allowed elution ofatractylenolide III (peak a in Fig.6). After about 100 min separation in the head-tailmode, the elution mode was reversed to tail-head, byswitching the solvent-changing valve from the head-tailelution mode to the tail-head elution mode, and theupper phase was pumped into the column. The phasereversal permitted elution ofatractylon (peak b in Fig.6).It can be found in Fig. 6 that the chromatogram beforethe phase reversal is the same as in Fig. 5, and the dual-mode elution has no other effect on the separation ofatractylenolide III. However, the peak b eluted just afterthe phases was reversed and had a strange peak shape due to the reorganization of the phases. In the dual-mode HSCCC, 32.1 mg atractylenolide III at 99.0% purityand 319.6 mg atractylon at 97.8%6purity could beobtained from 1000 mg of crude sample. The recoveriesofatractylenolide III and atractylon were 95.4 and 92.6%,respectively. The recovery and purity in the dual-modeelution had no obvious change compared with thoseusing the normal-mode HSCCC. The separation in the normal-mode HSCCC would takeabout 7 h, while in the dual-mode HSCCC it only takesabout 2 h. Because the HSCCC apparatus used in theexperiments is a large scale preparative model with a1000 mL column volume, the separation usually needsmore than 6 h [24-26]. Even if the K value equals 1, theelution time would be 200 min at a flow rate of 5mL/min from the 1basic equations of chromatography(t=+K_), wh),e wrehe ret tu is the retention time of asolute, Vm is the mobile phase volume, Vs is the stationaryphase volume, and F is the flow rate of the mobile phase).So it is very difficult to reduce the elution time anyfurther considering the peak resolution. The use of thedual-mode HSCCC for the preparative purification ofatractylon and atractylenolide III is highlighted by com-parison with the results of the normal-mode HSCCC; andthe elution time can be greatly reduced. 3.3 Structural identification The chemical structures of the peaks a and b in Figs. 5, 6were identified according to their GC-MS,1H and 13CNMRdata. Peak a: EI-MS m/z: 248 (M*), 230,220,215,191,169,147 (100),121,109.1H NMR (400 MHz, CDCl3, TMS): 1.03 (3H,s, 14-H), 1.24 (1H, td, J=12.6 Hz, 1a-H), 1.54 (1H, d,J=14 Hz, 9-H), 1.60-1.70 (3H, m, 3-H), 1.87 (1H, d,J=13 Hz, 5-H), 1.97 (1H, t, J=10 Hz, 3a-H), 2.27 (1H, d,J=14Hz, 9-H), 2.37 (2H, d, J=13 Hz, 3b-H), 2.43 (1H,J=13 Hz,6-H), 2.62 (1H, dd,J=13, 3Hz, 6-H), 4.60 (1H, d,J=1 Hz, 15-Ha), 4.86 (1H,d,J=1Hz, 15-H).Peak b:EI-MS m/z:(216,M*),173,145,108(100),79,41.HNMR (400 MHz, CDCl3, TMS): 0.75 (3H, s,14-H), 1.3-1.7(5H,m), 1.94(3H,s, 13-H), 2.0-2.4(6H, m), 4.70 (1H, s,15-Ha), 4.85 (1H, s, 15H,), 7.04 (1H, s, 12-H). 13C NMR(400 MHz, CDCl3, TMS): 8.1 (C-13),17.5 (C-14), 20.9 (C-2),23.3(C-6), 36.6(C-10), 37.3 (C-3),39.2 (C-1), 41.9 (C-9), 45.7(C-5), 107.3 (C-15), 116.1 (C-7), 119.5 (C-11), 136.9 (C-12),149.3(C-4),149.8 (C-8). Compared with the data given in [27], peaks a and b wereidentified as atractylenolide III and atractylon. 4 Concluding remarks An efficient HSCCC method for the separation and purifi-cation of atractylon and atractylenolide III from A. macro- cephala was developed by using light petroleum (60-90C)-ethyl acetate-ethanol-water (4:1:4:1v/v) as thetwo-phase solvent system in dual-mode elution. Atracty-lenolide III (32.1 mg) at 99.0% purity and 319.6 mg atrac-tylon at 97.8% purity could be obtained from 1000 mgcrude sample in a single run. The recoveries of atractyle-nolide III and atractylon were 95.4 and 92.6%, respec-tively. Compared with the normal-mode elution, thedual-mode elution can greatly reduce the elution time,which is an effective method for quick separation of thesample containing solutes with a broad range of hydro-phobicity. Financial support from the National Natural Science Foundationof the People's Republic of China (project no: 20576113) is grate-fully acknowledged. 5 References ( [1] China P harmacopoeia Co m mittee, Pharmacopoeia of t h ePeople's Republic of China, the f irst d ivision of 2000 edi- tion, China Chemical I n dustry P r ess, Beijing 1999, p .84. ) ( 2 | E E ndo, K., Toguchi,T., Toguchi, F., Hi k ino, H., et al., Chem. Pharm. Bull. 1979,12,755-760. ) ( 3 1 T T ang, D . F., H a o, Y. H., Liu, Z. Y ., Miao, S. L., et al., Chin. Pharm. Bull. 1984,19,43. ) ( 4 VWang, C. C., Chen, L. G., Yang, L. L., Planta. Medica. 2002, 68,204-208. ) ( [5] Wen, H. M., Zhang,A. H., Ge, J. H.,Wu, H., Chin. J. Pharm. Anal.2001,21,170- 1 72. ) [6] Zhao, Z. F., Shen, J. W., Gong, F., Liang, Y. Z., Qiu, X. M.,Chin. Tradit. Herb. Drugs. 2004,35,5-8. ( 7]E Hikino, H ., H ikino, Y . , Yosioka, I., Chem. P h arm. Bu l l. 1964,12,755-758. ) ( [8 W en, H. M . , Li, W., Chin. Materia. Medica. 1 997,22,662- 663. ) ( [9] Chen, J. M., Yu, M. Q., S h en, Y. Z., Liu, Z. Y., Huang, Z.J., Acta Bot. Si n . 1991,33,164-167. ) ( [10] L iu,R. M., Lei, F., Sun, A. L. , Kong, L. Y., J. C h romatogr. A 2004,1057,89-94. ) ( [11] W u,S. J., Sun, A. L., Liu, R.M., J. C hromatogr. A 2005, 1066 , 243-247. ) ( [12] Liu,R. M., Sun, Q. H., Sun , A. L., Cui, J. C . , J. Chromatogr. A 2005,1072,195-199. ) [13] Li, H. B., Chen,F., J. Chromatogr.A 2005,1074,107-110. [14] Liu,R. M., Chu, X., Sun, A. L., Kong, L. Y., J. Chromatogr. A2005,1074,139-144. ( [15] Ma, C. J., Li, G. S., Zhang,D. L., Liu, K . , Fan, X., J. Chroma- togr.A 2005,1078,188-192. ) ( [16] Du, Q., Wu, C., Quian, P., Ito, Y . , J. Chromatogr. A 1999, 835,231-235. ) ( [17]Sutherland, I.A., J. Chromatogr.A 2000,886,283-287 . ) ( [18] B o B oth, A . J., S utherland, I. A . , Lye, G. J. , Biotechnol. Bioeng. 2003,81,640-649. ) ( [19] Slacanin, I., Marston, A., Hostettmann, K. , J. Chromatogr. 1989,482,234-237. ) ( [20] Marson, A., Borel, C. , Hostettmann, K . J. Chromatogr. 1988,450,91-95. ) ( [21] Agnely, M., Thiebaut, D., J. Chromatogr. A 199 7 ,790,17- 30. ) ( [22 ] Gluck, S. J., M a rtin, E. J, J. L i q. C h romatogr. 19 9 0, 13, 3559-3564. ) ( [23] Menges, R. A., Bertrand, G. L., Armstrong,D. W., J. Liq.Chromatogr.1990,13,3061-3065. ) ( [24] F an,J. P . , H e, C . H . , J. L iq. Chromatogr. Re l . Technol. 2006,29, 815-826. ) ( [25] Aman,R., C arle,R., Conrad,J., Beifuss,U., Schieber,A.,J. Chromatogr.A 2005,1074,99-105. ) ( [26 P eng, J.Y.,Fan, G. R., Hong, Z. Y., C hai, Y. F., W u, Y. T., J.Chromatogr.A2005,1074,111-115. ) ( [27] Huang, B. S . , Sun,J. S . ,Chen, Z. L., Acta Bot. Sin. 1992, 31, 614-616. ) @WILEYInterScience°isco HG GRtATC WILEY-VCH Verlag GmbH & Co. KGaA, Weinheimwww.jss-journal.com @ WILEY-VCH Verlag GmbH & Co. KGaA, Weinheimwww.jss-journal.com
确定





还剩5页未读,是否继续阅读?

产品配置单




上海同田生物技术有限公司-高速逆流色谱仪HSCCC为您提供《白术中分离纯化苍术酮和苍术烯丙酯Ⅲ检测方案(高速逆流色谱)》,该方案主要用于中药材和饮片中含量测定检测,参考标准--,《白术中分离纯化苍术酮和苍术烯丙酯Ⅲ检测方案(高速逆流色谱)》用到的仪器有高速逆流色谱仪/高效逆流色谱仪、TBE-200V 高速逆流色谱仪、TBE300B+AKTA高速逆流色谱仪/离心分配色谱/萃取仪/制备色谱仪、TBE-1000A制备色谱仪/萃取仪/快速分离制备色谱仪
推荐专场






相关方案
更多
该厂商其他方案
更多