








In numerous applications in microfluidics, cell growth, soft lithography, and molecular imprinting, the surface of
poly(dimethylsiloxane) (PDMS) is modified from a hydrophobic methyl-terminated surface to a hydrophilic hydroxylterminated
surface. In this study, we investigated molecular structural and orientational changes at the PDMS-air
interface in response to three commonly used surface modification processes: exposure to long-wavelength ultraviolet
light (UV), exposure to short-wavelength UV that generates ozone (UVO), and exposure to oxygen plasma (OP). The
surfaces of two PDMS compositions (10:1 and 4:1 of base polymer/curing agent) were probed during modification,
using monolayer-sensitive IR + visible sum frequency generation (SFG) vibrational spectroscopy, with two different
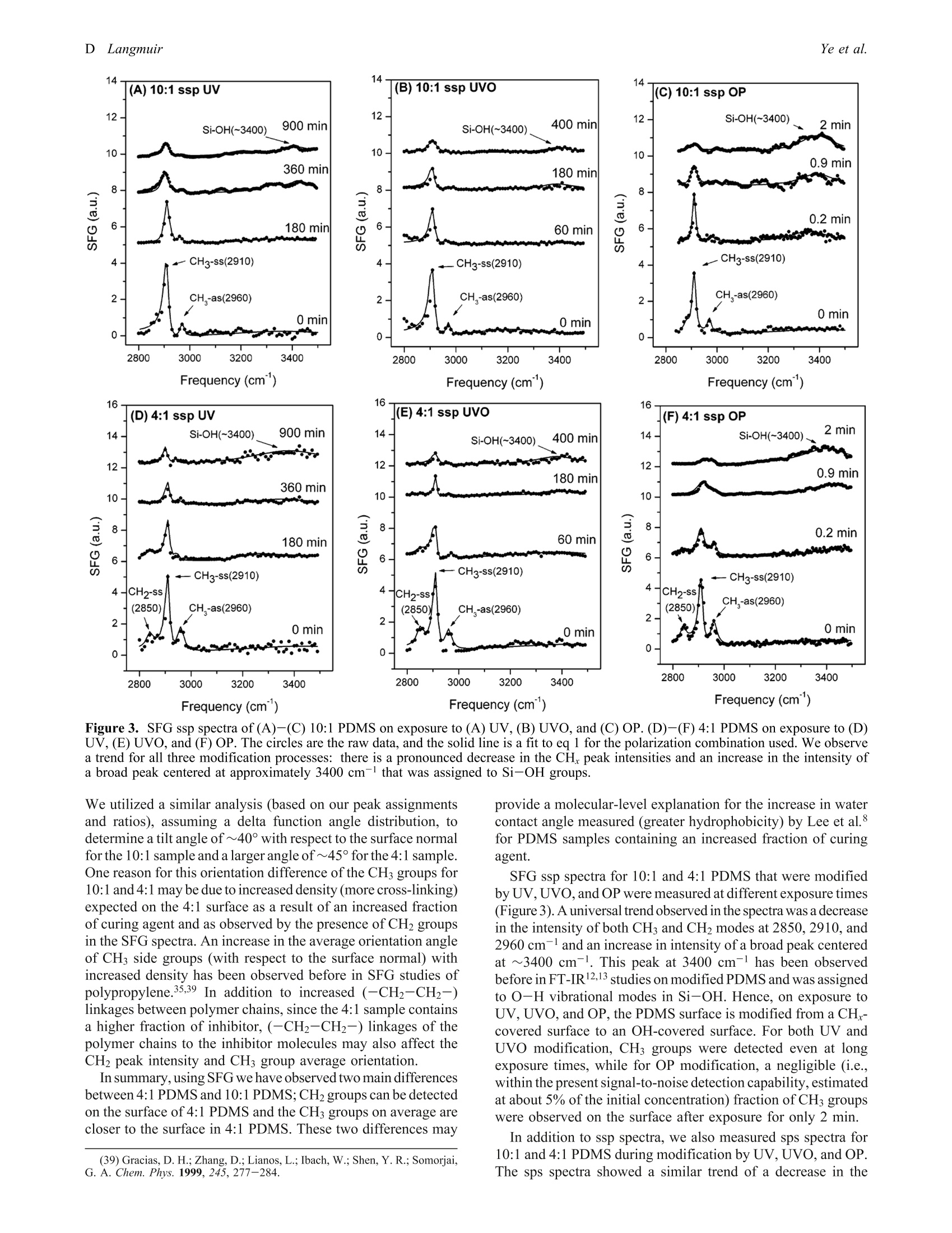
polarization combinations. During PDMS surface modification, the peak intensities of CH3 side groups and CH2
cross-link groups decreased, while peak intensities of Si-OH groups increased. There was no significant change in
the average orientation of the CH3 groups on the PDMS surface during modification. The concentration of CH3 groups
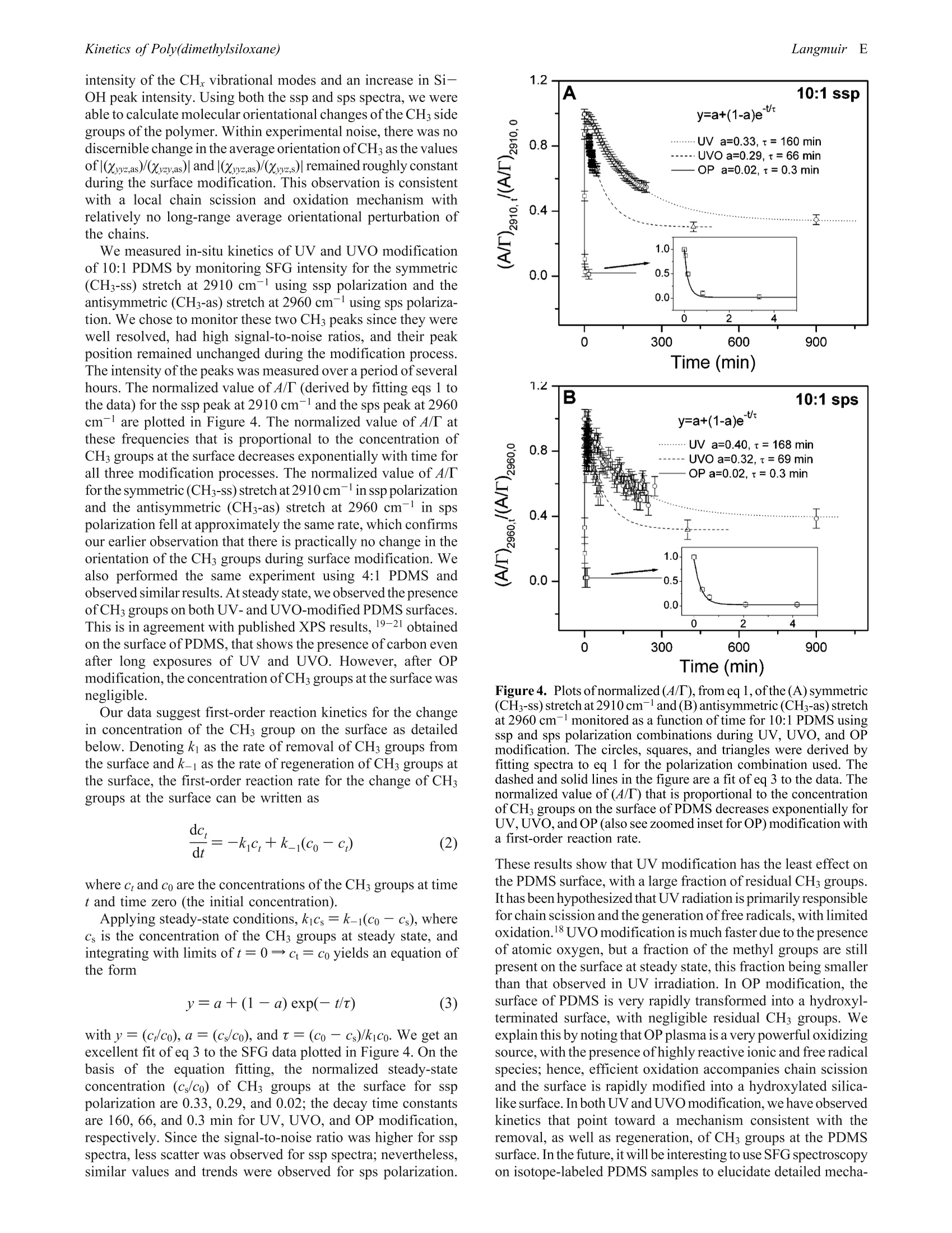
on the surface decreased exponentially with time, for all three UV, UVO, and OP modification processes, with first
order kinetics and time constants of approximately 160, 66, and 0.3 min, respectively. At steady state, residual CH3
groups were detected at the PDMS surface for UV and UVO treatments; however, there were negligible CH3 groups
detected after OP modification.
方案详情

B LangmuirYe et al. LangmuirC Published on Web 01/24/2006PAGE EST: 5.3 Kinetics of Ultraviolet and Plasma Surface Modification ofPoly(dimethylsiloxane) Probed by Sum Frequency VibrationalSpectroscopy Hongke Ye, Zhiyong Gu, and David H. Gracias*,* Department of Chemical and Biomolecular Engineering and Department of Chemistry, The Johns HopkinsUniversity, 3400 North Charles Street, Baltimore, Maryland 21218 Received July 26, 2005. In Final Form: October 19, 2005 In numerous applications in microfluidics, cell growth, soft lithography, and molecular imprinting, the surface ofpoly(dimethylsiloxane) (PDMS) is modified from a hydrophobic methyl-terminated surface to a hydrophilic hydroxyl-terminated surface. In this study, we investigated molecular structural and orientational changes at the PDMS-airinterface in response to three commonly used surface modification processes: exposure to long-wavelength ultravioletlight (UV), exposure to short-wavelength UV that generates ozone (UVO), and exposure to oxygen plasma (OP). Thesurfaces of two PDMS compositions (10:1 and 4:1 of base polymer/curing agent) were probed during modification,using monolayer-sensitive IR+ visible sum frequency generation (SFG) vibrational spectroscopy, with two differentpolarization combinations. During PDMS surface modification, the peak intensities of CH side groups and CH2cross-link groups decreased, while peak intensities of Si-OH groups increased. There was no significant change inthe average orientation of the CH groups on the PDMS surface during modification. The concentration of CH groupson the surface decreased exponentially with time, for all three UV, UVO, and OP modification processes, with firstorder kinetics and time constants of approximately 160,66, and 0.3 min,respectively. At steady state, residual CHgroups were detected at the PDMS surface for UV and UVO treatments; however, there were negligible CH groupsdetected after OP modification. Introduction In recent years, the rapid miniaturization of structures anddevices in biology and electronics has resulted in the developmentof novel microfabrication strategies that feature rapid, low-costprototyping.Several ofthese strategies including soft-lithographyand decal transfer lithography (DTL)2 rely on the surfacemodification of a polymeric stamp using physical processes suchas exposure to ultraviolet light (UV) and oxygen plasmas (OP).The primary polymer used in these lithographic methods is thesilicon-based polymer poly(dimethylsiloxane) (PDMS) whichhas several attractive properties including optical transparency,chemical inertness, and elasticity.PDMS consists of repeating(-OSi(CH3)2-) units with a hydrophobic, low-surface-energy,methyl-terminated surface. The hydrophobic nature ofthe surfaceis advantageous since it is relatively inert with low surface energy.However, in many applications in microfluidics, bioengineering,and lithography, it is necessary to render the surface of PDMShydrophilic to improve wetting of aqueous solvents, decreasenonspecific binding of proteins and cells, and increase adhesion.It is also possible to irreversibly bond PDMS surfaces with eachother or with other surfaces, immediately after oxidation, bymere contact.4 This facile bonding of surface-modified PDMSis critical in pattern transfer in DTL and in the sealing ofmicrofluidic channels. The PDMS surface can be rendered hydrophilic using a varietyof physical techniques, including exposure to UV light (in DTL)or exposure to OP (in microfluidics). In this paper, we investigatedthe surface modification ofPDMS using the three most commonly ( * To whom correspondence sh o uld be ad d ressed. E-mail: d g racias@ ihu.edu. ) ( T Department of Chemistry. ) ( (1)Xia, Y . N.; Whitesides, G. M. Annu. R ev. M ater. S ci. 1998, 28, 153-184. ) ( (2) Childs, W. R.;Nuzzo,R. G. J. Am. Chem. Soc. 2002, 124, 1 3583-13596. ) ( (3) Sia, S . K. ; Whitesides, G. M . Electrophoresis 2003, 24,3563 -3 576. ) ( (4) McDonald, J. C.; Whitesides, G. M. Acc . Chem. Res . 2002,35,491-4 9 9. ) used physical methods (a) UV modification, exposure to long-wavelength UV light(A=315-400nm),(b)UVO modification,exposure to short-wavelength UV light which includes wave-lengths (1=185/254 nm), and (c) OP modification, exposureto an oxygen plasma. As opposed to long-wavelength exposure(UV), short-wavelength exposure (UVO) at 185 and 254 nmgenerates atomic oxygen-a very strong oxidizing agent.5.6 Atomicoxygen is generated in ambient air by the combination ofphotochemical processes at 185 and 254 nm. The 185 nm lineproduces ozone from molecular oxygen by a two-step photo-chemical process, while the 254 nm line converts the ozone toatomic oxygen. An oxygen plasma consists of a large numberof electrons and highly reactive ionic and free radical oxygenspecies that facilitate oxidation of surfaces. The PDMS samples used in our experiments (Sylgard 184, themost commonly used form of PDMS) were made up of a basepolymer and a curing agent. The components undergo ahydrosilylation reaction upon curing that results in cross-linkingof the polymer chains. To investigate the effect of cross-linkingon the surface properties of PDMS, before and after surfacemodification, we investigated PDMS surfaces with differentratios(10:1 and 4:1 w/w) of base polymer to curing agent. The Sylgard184 base polymer is composed of dimethylsiloxane oligomerswith vinyl-terminated end groups, platinum catalyst, and silicafiller (dimethylvinylated and trimethylated silica), while the curingagent contains a cross-linking agent (dimethylmethylhydrogensiloxane) and an inhibitor (tetramethyltetravinyl cyclotretrasi-loxane). The cross-linking process occurs when vinyl and siliconhydride groups undergo a hydrosilylation reaction in the presence ( (5)Vig, J. R.; LeBus, J . W. U .S. P atent 4,028,135, 1 977. ) ( (6) Muisener , R. J . ; Koberstein, J. T. Polym. Mater . Sc i .Eng. 1997, 77,653. ) ( (7)Simpson, T. ; Jeynes, C.; Parbhoo, B.; Keddie, J. L.J. Polym. Sci. A 2004, 42.1421- 1 431. ) ( (8) Lee, J. N.; Jiang, X.; Ryan, D .; W hitesides, G. M . Langmuir 2004, 20,1 1 684- 1 1691. ) of the catalyst to form a Si-C bond, thereby facilitating (-CH2-CH2-) linkages between PDMS chains (Figure 1).9,10 Eventhough the 10:1 composition ofPDMS is widely used, a fractionof uncured oligomers as high as 5% of the total weight of thepolymer has been reported.Since 4:1 PDMS has a higher fractionof curing agent, the (-CH2-CH2-) cross-linking is expectedto be higher as compared to the 10:1 sample. Previous studies of the modification of the PDMS surfaceusing FT-IR spectroscopy,12,13 contact-angle goniometry,14,15chemical force microscopy,16 secondary ion mass spectrometry(SIMS),near-edge X-ray absorption fine structure (NEXAFS),18and X-ray photoelectron spectroscopy (XPS)19-21 have providedsome information regarding the mechanism of oxidation of thePDMS surface, but information regarding the kinetics of surfacemolecular changes is lacking. Additionally, it is difficult to detectsurface-specific molecular concentration and orientation changesat a polymer interface using FT-IR due to limited surfacesensitivity. While contact-angle goniometry does provide valuableinformation about the surface energy, it is not possible to get amolecular-level understanding. Although SIMS,NEXAFS,andXPS are extremely surface sensitive, they require vacuumoperation, and the studies done using these spectroscopiesinvolved a time delay between modification of the polymer andthe spectroscopic measurement. Hence, using these spec-troscopies, it has not been possible to study the PDMS surfacein-situ during modification, or in an ambient environment of airwhere PDMS is most commonly used. In this paper, we usedsurface-specific IR + visible sum frequency generation (SFG)spectroscopy2?to probe the structural changes on the PDMSsurface. SFG measurements ofUV and UVO modification wererecorded in situ as a function of time; measurements of OPmodification were done in air immediately after irradiation. Byusing different optical polarization combinations,23 we were alsoable to monitor molecular orientation at the surface of PDMSduring UV, UVO, and OP modification. SFG24 is a nonlinear surface-specific spectroscopic techniquethat allows nondestructive analysis of the surface of a samplein air. It has been convincingly demonstrated, both on the basisof theoretical and experimental studies, that SFG spectroscopyhas monolayer sensitivity for polymers25 that possess bulkinversion symmetry; the surface sensitivity is a result of the factthat this second-order nonlinear optical process vanishes under ( ( 9) Hammouch, S.O . ; Beinert, G. J .; Zilliox, J . G.; H erz, J. E. Polymer 1 9 95, 36.421 -4 26. ) ( (10) F u, P . F.; Glover, S.; King, R. K.; L e e, C. L . ; Pretzer, M. R.; Tomalia,M. K . P olym. P repr. 2003, 44, 1014-1015. ) ( (11) L ee, J . N .; P ark, C .; Whitesides, G. M . A nal. Chem.20 0 3, 75, 65 4 4-6554. ) ( (12) Berdichevsky, Y.; Khandurina, J.; Guttman, A.;Lo, Y. H. Sens. Actuators,B 2004,97,402-408. ) ( (13) Graubner, V.M. ; Jordan, R.; Nuyken,O.; Schnyder, B.; Lippert,T.; K ot z ,R.; Wokaun, A. Macromolecules 2004, 3 7,5936-5943. ) (14) Chaudhury, M. K.; Whitesides, G. M. Langmuir 1991, 7,1013-1025. (15) Chen, L.;Ren, J. C.; Bi, R.; Chen, D. Electrophoresis 2004, 25,914-921. (16) Hillborg, H.; Tomczak,N.;Olah, A.; Schonherr, H.; Vancso,G.J.Langmuir ( 2004,20,785-794 . (17) Childs, W. R .; Michael , M. J .; Lee, K. J.;Nuzzo, R. G. Langmuir 2005, 21, 1 0096-10105. (18) Efimenko, K. ; Wa l lace, W. E.; G enzer, J. J. Col l oid Interface Sci. 200 2 , ) ( 254,306-315. ) ( ( 19) Ouyang, M. ; Yu a n, C.; Muisener, R. J .; Boulares, A.; Koberstein, J. T.Chem. M ater. 2000 , 12, 1 591-1596. ) ( ( 20) Schnyder, B.;Lippert, T.; Kotz,R.;Wokaun, A.; Graubner, V. M. ; Nuyken,O. . Surf. Sc i . 2003,532 , 1067-1071. ) ( (21) Graubner, V. M .; Jordan , R. ; Nuyken,O.; Schnyder, B.;Li p pert, T . ;Kotz,R.; Wokaun, A. M acromolecules 2 004, 37,5936-5943. ) ( (22) Zhu, X. D.; Suhr, H.; Shen, Y. R. Phys. Rev. B. 1987, 35 , 304 7 -3050. (23) Guyotsionnest, P .; Hunt, J. H.; Shen, Y. R. P hys. R ev. L ett. 1987, 59,1597-1600. ) ( (24) Shen, Y. R. Nature 1989, 337,519-525. ) ( (25) G racias, D . H .; C h e n, Z.; Shen, Y. R . ;Somorjai, G. A. Acc. Che m . Res.1999,32,930- 9 40. ) centrosymmetry. SFG has been used to study the surface oxidationand UV-induced modification of polymers including polysty-rene,26,27 polyimide,28 and poly(ethylene terephthalate)29 withhigh surface sensitivity. The SFG spectroscopy involvedoverlapping a visible beam (@vis) and a tunable IR (WIR) beamat the surface of the PDMS. The surface vibrational spectrumwas obtained by tuning the IR beam over the relevant resonantvibrational modes and measuring the intensity of the reflectedsum frequency (SF) (WSF =WvIs +wIR) beam as a function ofIR frequency. This paper is the first SFG study of the modification ofPDMSusing either UV, UVO, or OP modification. The measurementswere conductedunder relevant ambient conditions and are surfacesensitive. We were able to monitor concentrations relative toorientations ofthe surface groups in-situ, which has allowed usto develop a quantitative kinetic model for the modificationprocesses. Experimental Section Materials Used. Two compositions of PDMS (Dow CorningSylgard 184,two-component kit) were prepared by vigorously mixing10:1 and 4:1 (w/w) ratios of the base polymer to curing agent. Afterthe mixture was degassed using vacuum, the polymer was spun onquartz substrates (1 in. diameter, 1/8 in. thick, Esco Products) at5000 rpm for 30 s and subsequently cured at 65℃ for 4 h. Thespin-coated films had a thickness of ~1.8 um as measured byprofilometry. We also cast thick films (1 cm) of PDMS by pouringthe mixture of base polymer and curing agent into a Petri dish andsubsequently curing it. Surface Modification. Three different surface-modificationstrategies were used. (a) UV modification by exposure to a longwavelength(1=315-400 nm,peaked at 365 nm), B-100A mercurylamp (UVP, Inc., www.uvp.com). The intensity at the sample wasmeasured using a UV power meter and was~4 mW/cm at =365nm. (b) UVO modification by exposure to a low-pressure, shortwavelength mercury PEN-RAY lamp (UVP, Inc., www.uvp.com)with line emissions at 185 and 254 nm. The intensity at the samplewas comparable to the UV exposure, ~4 mW/cm’at 1=254 nm.(c) OPmodification by exposure to an 18 W radio frequency HarrickScientific plasma source (PDC-32G, http://www.harricksci.com/plasma.cfm) in a background pressure of 200 mTorr. SFG. A tunable IR and visible laser beam were spatially andtemporally overlapped on the PDMS surface at incident angles of55° and 60°, respectively, relative to the surface normal. We useda solid-state mode-lockedNd:YAG laser (1064 nm, 31 ps pulse, 10Hz repetition rate, EKSPLA, Inc., www.ekspla.com) to generateboth the visible and the tunable IR beam. The fundamental laseroutput with a frequency at 1= 1064 nm was doubled using a typeI potassium dideuterium phosphate crystal to generate the visible a=532 nm beam. The tunable IR beam was generated using acombination of optical parametric generation (OPG)/optical para-metric amplification (OPA) based on a lithium triborate crystal anddifference frequency generation (DFG) based on a silver galliumsulfide (AgGaS2) crystal. The frequency of the IR beam was tunedbetween 2800 and 3500 cm-l; the output energy was ~200 uJ inthis frequency region. The SFG signal was measured using amonochromator in reflection geometry. Spectral Fitting. SFG spectra were obtained with two differentpolarization combinations ssp (s-polarized SF output, s-polarizedvisible input, and p-polarized infrared input) and sps. Using the twopolarizations it was possible to get information of relative orientationof chromophores at the surface. The SF beam consists of anonresonant term and a resonant term, so that the measured reflected ( (26) Zhang, D .; Dougal, S . M .; Y eganeh, M. S. Langmuir 20 0 0, 16, 45 2 8-4532. ) ( (27) L i, J.; Oh, K.; Y u , H . C hin. J. P olym . S c i. 2 0 05, 23 , 18 7 -196. ) (28) Oh-e, M.; Kim, D.; Shen, Y. R. J.Chem. Phys. 2001, 115, 5582-5588. ( (29) Miyamae, T. ; Y amada, Y.;Uyama,H.;Nozo y e, H. Appl. Surf . Sci. 20 0 1, 180,126-137. ) Figure 1. Schematic diagram of the PDMS curing process. Thebase polymer (containing dimethylsiloxane oligomers with vinyl-terminated end groups) reacts with the curing agent (containingdimethylmethylhydrogen siloxane) in the presence of a platinumcatalyst to form cross-links between oligomers. intensity from the sample, for the ssp and sps polarizationcombination, is given by, where x and x are the second-order nonresonant and resonantnonlinear optical susceptibilities. The frequency of the incident IRbeam, the strength, damping constant, and resonance frequency ofthe qth vibrational mode in a given polarization combination aredenoted by wR, Ag, Iy, and w, respectively. All SFG spectra werenormalized by the intensities of the IR and visible beams tocompensate for the effects of intensity fluctuations and absorption.The SFG spectra presented in the paper include both the raw measureddata points and a solid line representing a fit of eq 1 to the data. Bymeasuring the SFG spectra with ssp and sps polarization combina-tions, and fitting the data to the eq 1, information about X andXRvzy were obtained that allowed us to get molecular orientationalinformation.30,31 Results and Discussion The SFG spectra obtained on spin-coated PDMS films usingssp and sps polarizations for 10:1 PDMS and 4:1 PDMS areshown in Figure 2. The circles represent the raw data, and thesolid line represents the fit to eq 1. The ssp spectra were dominatedby the symmetric stretch (CH3-ss) of the methyl side groups at2910 cm-1, while the sps spectra were dominated by theantisymmetric stretch (CH-as) at 2960 cm-. These results areconsistent and assigned on the basis of published SFG spectrafor PDMS3 and PDMS FTIR spectra.12,13,18 The strong polariza-tion dependence of the symmetric and antisymmetric peaksindicates that the methyl groups have their symmetric axes moreor less along the surface normal.33,34 Additional peaks observedin the SFG spectra of 4:1 PDMS at 2850 (in ssp) and 2915 cm-(in sps) were assigned35-37 to the symmetric (CH2-ss) and theantisymmetric (CH2-as) stretches expected in (-Si-CH2-CH2- ( (30) Hirose, C. ; Yamamoto,H.; A kamatsu, N . ; Domen, K. J. P h ys. Chem. 1993,97,10064- 1 0069. ) ( (31) Hirose, C. ; Akamatsu, N. ; Domen, K. J . Chem. Phys . 1992 , 96,997-1004. ) ( (32) Chen, C. Y.; Wang, J .; Chen, Z. Langmuir 2 004, 2 0,1 0 186-10 1 93.(33) Zhang, D.; Ward,R. S.; Shen, Y.R.; Somorjai, G. A. J. Phys. Chem. B1997,101,9060-9064. ) ( (34) Zhang, D.; Gracias, D. H.; Ward, R.; Gauckler, M.; T ian, Y.; Shen, Y . R.; Somorjai, G . A. J . Phys . Chem. B 1998, 102,6225-6230. ) ( (35) Zhang, D .; S hen, Y . R .; S omorjai, G. A. Ch e m. P h ys. Lett. 1997, 281, 394-400. ) ( (36)Lu,R . ; Gan, W.; Wu, B.;Zhang, Z.; Guo,Y.; Wang, H.-F.J. Phys. Chem. 2005,109, 1 4118-14129. ) ( ( 37) Zhuang, X . ; Miranda, P. B . ; Kim, D . ; Shen, Y . R. Phys. Rev. B 1999,59,12632 -1 2640. ) Figure 2. SFG spectra of 10:1 and 4:1 PDMS taken in air with thepolarization combinations (A) ssp (for s-polarized SF output,s-polarized visible input, and p-polarized infrared output) and (B)sps. The circles are the raw data, and the solid line is a fit to eq 1. Si-) linkages. The presence of CH2 peaks on 4:1 PDMS andtheir absence on 10:1 PDMS in both ssp and sps spectra leadsus to conclude that there is an increase in(-CH2-CH2-)linkageson the 4:1 PDMS surface as compared to the 10:1 PDMS surface.To confirm that the SFG signal being monitored on spin-coatedfilms was from the air-PDMS interface and not the PDMS-substrate interface, we also obtained spectra on very thick (1cm)PDMS films. For thick PDMS films, light from the PDMS-substrate interface was blocked from entering the PMT using anaperture. The SFG spectra of thick films were similar to thinspin-coated PDMS films, confirming that the SFG signal beingmonitored on spin-coated PDMS films was from the PDMS-airinterface. Although the spectra in both thin and thick films weresimilar to each other, the signal-to-noise ratio was higher forspin-coated PDMS films as their surfaces were smoother resultingin lower scattering. Hence, all the spectra reported in the paperwere obtained with thin (~1.8 um) spin-coated PDMS films. Itshould be noted that numerous SFG studies probing polymerinterfaces utilize spin coated films for similar reasons. In aprevious study by Wang et al., the authors obtained similar SFGspectra for thin and thick PMMA films, concluding that thesurface being probed was the air-polymer interface. They arguethat the IR beam intensity is depleted on passing through thepolymer bulk to the polymer-substrate interface (due toabsorption).38 The average value of the ratio 1(%yyz,as)/(%yzy,as)| for methylgroups on the surface of 10:1 and 4:1 PDMS was 0.76 and 0.89,while the ratio of|(yyz,as)/(Xyyz,s)| on the surface of 10:1 and 4:1PDMS was 0.42 and 0.58. These values of Xyyz,as, Xyzy,as, and Xyyz,swere obtained by measuring SFG spectra on 10:1 and 4:1 PDMSsurfaces using ssp and sps polarization combinations. We repeatedeach experiment on four different PDMS samples and computedthe ratios for each sample. Taking into account the effect of thebaseline noise level observed on the SFG spectra, the error ofthe mean value calculated for each ratio (from four samples) wasin the range of 0.02-0.04. A detailed analysis for determining the average orientationangle(0) of the methyl group vs the surface normal and the angledistribution parameter (a) based on the two ratios1(%yyz,as)/(%yzy,as)andl(yyz,as)/(%yyz,s)| has been outlined in prior publications30-32by treating the methyl side groups on PDMS with C3v symmetry. We utilized a similar analysis (based on our peak assignmentsand ratios), assuming a delta function angle distribution, todetermine a tilt angle of~40° with respect to the surface normalfor the 10:1 sample and a larger angle of~45° for the 4:1 sample.One reason for this orientation difference of the CH3 groups for10:1 and 4:1 may be due to increased density (more cross-linking)expected on the 4:1 surface as a result of an increased fractionof curing agent and as observed by the presence of CH2 groupsin the SFG spectra. An increase in the average orientation angleof CH side groups (with respect to the surface normal) withincreased density has been observed before in SFG studies ofpolypropylene.35,39 In addition to increased (-CH2-CH2-)linkages between polymer chains, since the 4:1 sample containsa higher fraction of inhibitor, (-CH2-CH2-) linkages of thepolymer chains to the inhibitor molecules may also affect theCH2 peak intensity and CH group average orientation. In summary, using SFG we have observed two main differencesbetween 4:1 PDMS and 10:1PDMS;CH2 groups can be detectedon the surface of 4:1 PDMS and the CH3 groups on average arecloser to the surface in 4:1 PDMS. These two differences may ( (39) Gracias, D. H.; Zhang, D.;Lian o s, L.; Ibach, W.; Shen, Y. R.; Som o rjai, G. A. Chem. Phys. 1999, 245,277- 2 84. ) provide a molecular-level explanation for the increase in watercontact angle measured (greater hydrophobicity) by Lee et al.for PDMS samples containing an increased fraction of curingagent. SFG ssp spectra for 10:1 and 4:1 PDMS that were modifiedby UV,UVO, and OP were measured at different exposure times(Figure 3). A universal trend observed in the spectra was a decreasein the intensity of both CH and CH2 modes at 2850,2910, and2960 cm-l and an increase in intensity of a broad peak centeredat ~3400 cm-. This peak at 3400 cm- has been observedbefore in FT-IR12,13 studies on modified PDMS and was assignedto O-H vibrational modes in Si-OH. Hence, on exposure toUV, UVO, and OP, the PDMS surface is modified from a CHy-covered surface to an OH-covered surface. For both UV andUVO modification, CHs groups were detected even at longexposure times, while for OP modification, a negligible (i.e.,within the present signal-to-noise detection capability, estimatedat about 5% of the initial concentration) fraction of CHs groupswere observed on the surface after exposure for only 2 min. In addition to ssp spectra, we also measured sps spectra for10:1 and 4:1 PDMS during modification by UV, UVO, and OP.The sps spectra showed a similar trend of a decrease in the intensity of the CH, vibrational modes and an increase in Si-OH peak intensity. Using both the ssp and sps spectra, we wereable to calculate molecular orientational changes of the CH sidegroups of the polymer. Within experimental noise, there was nodiscernible change in the average orientation of CH3 as the valuesof|(%yyz,as)/(Xyzy,as)|and1%yyz,as)/(Xyyz,s)| remained roughly constantduring the surface modification. This observation is consistentwith a local chain scission and oxidation mechanism withrelatively no long-range average orientational perturbation ofthe chains. We measured in-situ kinetics of UV and UVO modificationof 10:1 PDMS by monitoring SFG intensity for the symmetric(CH3-ss) stretch at 2910 cm- using ssp polarization and theantisymmetric (CH3-as) stretch at 2960 cm-l using sps polariza-tion. We chose to monitor these two CH3 peaks since they werewell resolved, had high signal-to-noise ratios, and their peakposition remained unchanged during the modification process.The intensity of the peaks was measured over a period of severalhours. The normalized value of A/T (derived by fitting eqs 1 tothe data) for the ssp peak at 2910 cm-l and the sps peak at 2960cm-are plotted in Figure 4. The normalized value of A/T atthese frequencies that is proportional to the concentration ofCHs groups at the surface decreases exponentially with time forall three modification processes. The normalized value of A/Tfor the symmetric (CH3-ss) stretch at 2910 cm-l in ssp polarizationand the antisymmetric (CH-as) stretch at 2960 cm- in spspolarization fell at approximately the same rate, which confirmsour earlier observation that there is practically no change in theorientation of the CH3 groups during surface modification. Wealso performed the same experiment using 4:1 PDMS andobserved similar results. At steady state, we observed the presenceof CH3 groups on both UV-andUVO-modified PDMS surfaces.This is in agreement with published XPS results, 19-21 obtainedon the surface ofPDMS, that shows the presence of carbon evenafter long exposures of UV and UVO. However, after OPmodification, the concentration of CH3 groups at the surface wasnegligible. Our data suggest first-order reaction kinetics for the changein concentration of the CH3 group on the surface as detailedbelow. Denoting ki as the rate of removal of CHs groups fromthe surface and k-i as the rate of regeneration of CH3 groups atthe surface, the first-order reaction rate for the change of CH3groups at the surface can be written as where c and co are the concentrations of the CH3 groups at timet and time zero (the initial concentration). Applying steady-state conditions, kqcs=k-i(co-Cs), wherecs is the concentration of the CH groups at steady state, andintegrating with limits of t=0一ct=co yields an equation ofthe form withy= (c,/co), a=(c/co), and t= (co -cs)/kjco. We get anexcellent fit of eq3 to the SFG data plotted in Figure 4. On thebasis of the equation fitting, the normalized steady-stateconcentration (cs/co) of CH groups at the surface for ssppolarization are 0.33, 0.29, and 0.02; the decay time constantsare 160, 66, and 0.3 min for UV, UVO, and OP modification,respectively. Since the signal-to-noise ratio was higher for sspspectra, less scatter was observed for ssp spectra; nevertheless,similar values and trends were observed for sps polarization. Time (min) Figure 4. Plots of normalized(A/T), from eq 1, ofthe (A) symmetric(CH-ss)stretch at 2910 cm-and(B) antisymmetric (CH3-as) stretchat 2960 cm-1 monitored as a function of time for 10:1 PDMS usingssp and sps polarization combinations during UV, UVO, and OPmodification. The circles, squares, and triangles were derived byfitting spectra to eq 1 for the polarization combination used. Thedashed and solid lines in the figure are a fit of eq 3 to the data. Thenormalized value of (A/T) that is proportional to the concentrationof CHs groups on the surface of PDMS decreases exponentially forUV,UVO, and OP (also see zoomed inset for OP) modification witha first-order reaction rate. These results show that UV modification has the least effect onthe PDMS surface, with a large fraction of residual CHs groups.It has been hypothesized that UV radiation is primarily responsiblefor chain scission and the generation of free radicals, with limitedoxidation.18 UVO modification is much faster due to the presenceof atomic oxygen, but a fraction of the methyl groups are stillpresent on the surface at steady state, this fraction being smallerthan that observed in UV irradiation. In OP modification, thesurface of PDMS is very rapidly transformed into a hydroxyl-terminated surface, with negligible residual CH3 groups. Weexplain this by noting that OP plasma is a very powerful oxidizingsource, with the presence of highly reactive ionic and free radicalspecies; hence, efficient oxidation accompanies chain scissionand the surface is rapidly modified into a hydroxylated silica-like surface. In both UV and UVO modification, we haveobservedkinetics that point toward a mechanism consistent with theremoval, as well as regeneration, of CH3 groups at the PDMSsurface. In the future, it will be interesting to use SFG spectroscopyon isotope-labeled PDMS samples to elucidate detailed mecha- nistic pathways since it is possible that multiple multistep chemicalreactions may be occurring at the same time. At the present time,we can only speculate on possible mechanisms for regenerationof CH groups on the basis of our results and publishedliterature.15-20,40 It has been suggested that the attack of radicalscontaining CHs groups, the reorientation of PDMS molecules atthe surface during modification, and migration of PDMSmolecules (or oligomers) from the bulk during modification couldall contribute to CH group regeneration. In our SFG experiments,we observe no significant change in reorientation of PDMSmolecules at the surface during modification. Conclusion In conclusion, we have monitored the surface modification ofPDMS by UV,UVO, and OP using SFG. We observed that CHand CH2 groups on the surface decreased during all threemodification processes and Si-OH groups were detected. Therewas no discernible change in the average orientation of the side ( (40) West, J. K. J . Biomed. Mater. Res . 1997,35, 505 - 511 . ) groups ofthe polymer during the modification process. The changeof CH groups at the surface follows first-order kinetics and isfastest for OP >UVO > UV. At steady state,OP modificationtransforms the PDMS surface from a CH3-terminated surface toone that has negligible CH3 groups, whereas UV and UVOmodification leave a fraction of CHs groups on the surface. Onthe basis of our results, we believe that it is possible to tailor thefraction of hydroxyl groups at the surface precisely using UV,UVO, or OP modification. With increasing oxidation, it has beenobserved14,15,18 that the PDMS surface has a much lower watercontact angle and is significantly more mechanically rigid (SiO2-like) as compared to the hydrophobic, native PDMS surface.Tailoring the surface properties is critical in many applicationsof PDMS in microfluidics, cell engineering, and lithography. Acknowledgment. We acknowledge funds received from aNational Science Foundation (NSF- MRI- CHE-0421010)equipment grant that was used to acquire and set up the SFGlaser spectroscopy system. LA052030R
确定




还剩4页未读,是否继续阅读?

产品配置单



北京欧兰科技发展有限公司为您提供《二甲基硅油Poly(dimethylsiloxane)中紫外和表面等离子体改性动力学检测方案(其它光谱仪)》,该方案主要用于其他中紫外和表面等离子体改性动力学检测,参考标准--,《二甲基硅油Poly(dimethylsiloxane)中紫外和表面等离子体改性动力学检测方案(其它光谱仪)》用到的仪器有Ekspla SFG 表面和频光谱分析系统、Ekspla PL2230型高能量皮秒激光器、Ekspla CARS 相干反斯托克斯拉曼显微光谱仪
推荐专场












相关方案
更多
该厂商其他方案
更多