








电极/溶液界面单分子吸附层的统计力学处理
文中提出电极/溶液界面溶剂化层偶极取向分布模型,应用统计力学方法及热力学平衡条件导出普通化的单层吸附等温方程,其电解质溶液的溶剂组成可以使纯态的或混合物(多组分)的。
方案详情

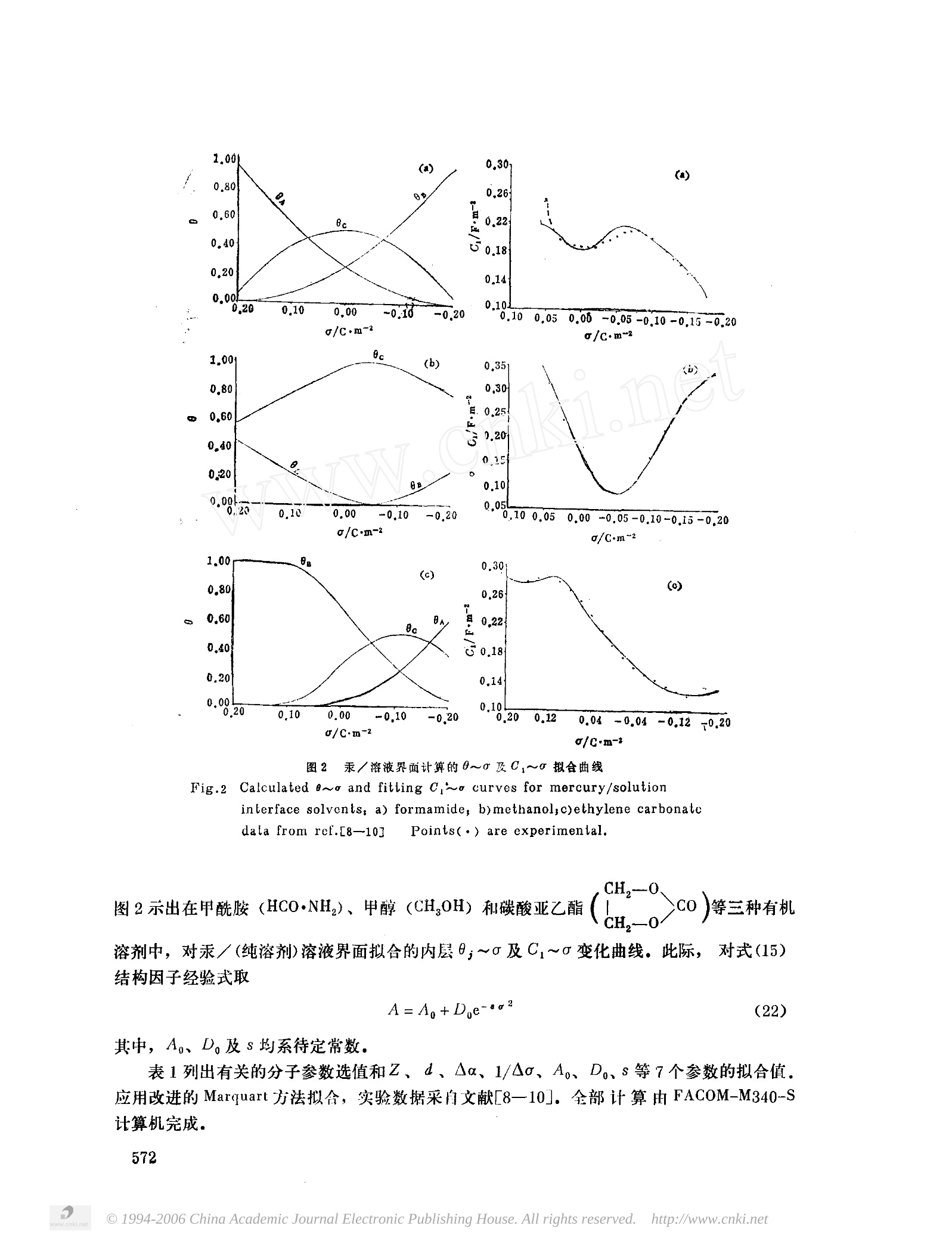
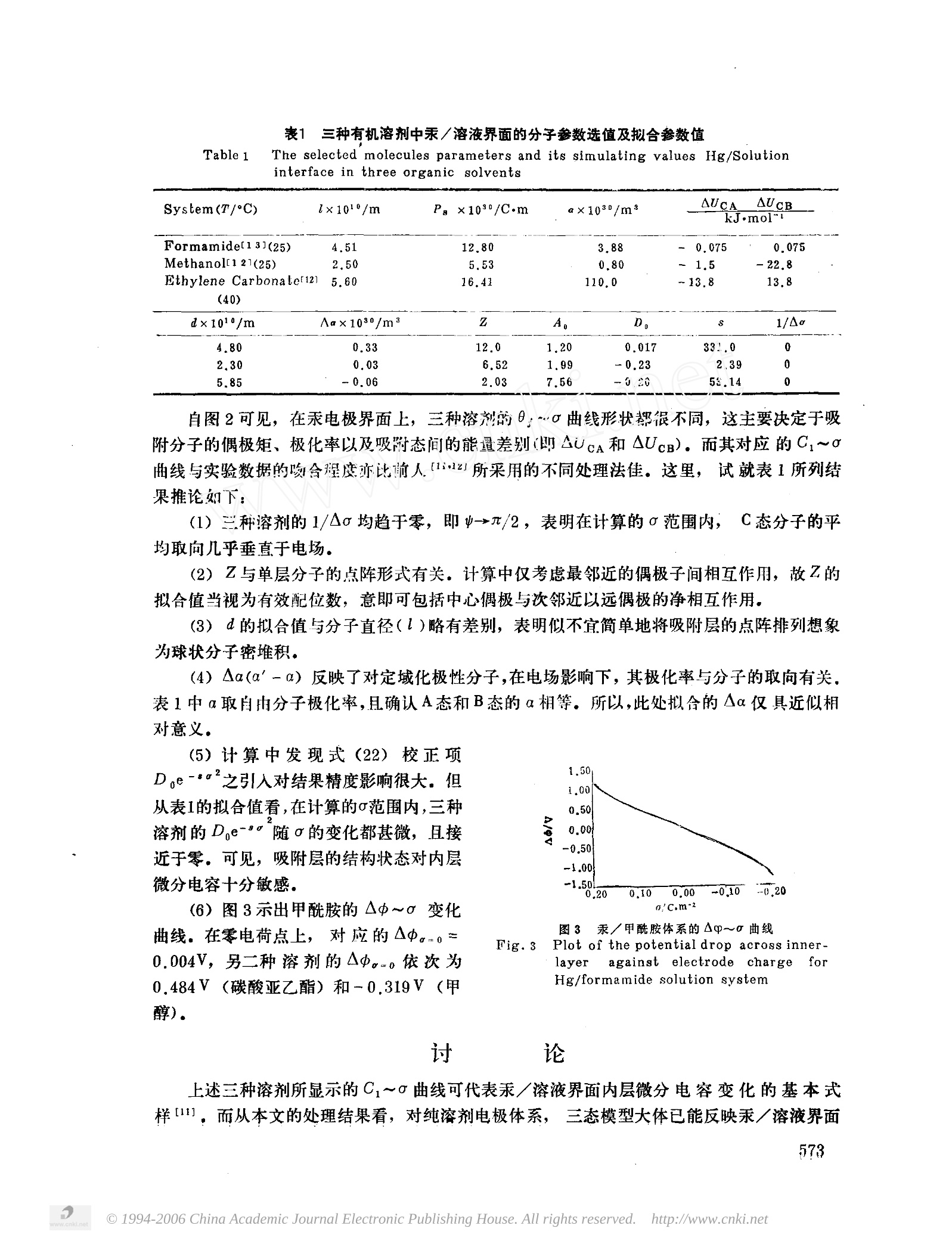
物 理 化学学报ACTA PHYSICO-CHIMICA SINICAVol.6,No.5Oct.,1990第6卷第5期1990年10月 二-- 电极/溶液界面单分子吸附层的统计力学处理 非水溶液中汞电极的内层微分电容 苏文煅* 周小林(厦门大学化学系) (厦门大学计算中心) 本文提出电板/溶液界面溶剂化层偶极取向分布模型,应用统计力学方法及热力学平衡件导出普遍化的单层吸附等温方程,其电解质溶液的溶剂组成可以是纯态的或混合物(多组份)的。文中分别以甲酰胺、碳酸亚乙酯和甲醇等三种纯溶剂的汞/溶液界面为例,采用曲线拟合计算内层微分电容随表面电荷变化关系。预计本模型处理对汞/水溶液或汞/(混合溶剂)溶液界面仍可适用 关于极性溶剂或溶质分子在电极/溶液界面的吸附已有许多理论模型,主要是从分子的性质剖析荷电表面的等温吸附以及微分电容变化特性。Watts-Tobin 训1]最先提出,对电极表面的溶剂化层,其溶剂分子可能存在以偶极正端朝向或背对电场的两种取向,,并由此解释汞/溶液界面的微分电容曲线。此后,又有三态、四态等多态模型的出现[2-6],虽然,各种理论模型在揭示界面电化学行为方面互有所长,但由于影响吸附层结构的作用因素相当复杂,目前关于它的微观情态以及处理方法都还没有一致的认识。 在界面双电层研究中,汞/溶液体系具有典型意义、并积累了丰富的知识,按 Parsons 分类内层微分电容(C)随表面电荷()的变化大体可归结为类水的(water-like)、单驼峰的(purely humped)和非水的(water-unlike)三种基本式样。现时普遍认为,对于象水一类的质子溶剂 (protic-solvent), 其在电极表面的单层铺展将以缔键缔合的方式连结并构成局部有序排列15],另一种处理法则设想吸附层上存在缔合的溶剂分子团(cluster)17但对非水溶剂,则通常多采用二态或多态模型处理之。我们认为,对金属表面的单分子层吸附,从热运动的原因考虑,即如汞上水分子层,也只在较低的温度下才可能形成以氢键方式连结的似冰结构有序排列;温度升高即趋无规分布状态。是以,有无希望建立一个能适用于各类溶剂体系的电极/溶液界面单(分子)层吸附模型,这正是有待尝试的问题。以下简要说明本文所设想的基本模型并分别以甲酰胺、甲醇和碳酸亚乙酯等三种有机溶剂的汞/溶液界面为例,给出对纯溶剂电极体系内层微分电容的处理结果。应用同一模型处理汞/水溶液或汞/(混合溶剂)溶液体系,将另文报导。 ( 1989年1月24日收到初稿,1990年4月20日收到修改商。 国家自然科学基金资助项目 ) 基 本模 型 (1) 吸附相正则配分函数 设电极表面不存在离子或其他溶质特性吸附,而电解质溶 液的溶剂由r种物质组成并同时在电极表面发生吸附。每一溶剂分子占据一个吸附位,其偶极取向可归结为三种不同的态,即A(r)——偶极负端朝向金属;B(r)—偶极正端朝向金属和C(r)——偶极向量与电场斜交,其斜交角,乃一统计平均值。 图1金属/溶液界商吸附溶剂分子的模型 Fig.1 Modeling of adsorbedsolvent moleculesan metal/solution inierface 在仅考虑偶极-电极的静电吸引以 及最邻近的偶极子间相互作用下,采用 Bragg-Wiiiiams近似,给出单层吸附相的正则配分函数如下: 式中, N表示单位电极表面有效吸附位数, Nj(r)为第r种溶剂分子呈j1态取向的占据数, 且有Nj(r)=N; 4j(r)乃j(r)态吸附分子配分函数,可表示为 fJ 99(r)即该态分子内配分函数,Uj(r)相当于该吸附态分子与溶液中自由溶剂分子的基态能量之差。β=-(kT)-1;g(=0/e)指电场强度(e=8.85×10-12F·m-, 乃真空电容率)。Pj(r) 代表 j(r)态分子总偶极矩,如以Ps(r)表示分子的永久偶极,以α表示A(r)或 B (r)态的,以 a' 表示C(r)态的极化极化率,则依图1规定取向,分别有 式(1)中,指数因子内的Nj(r),i(r)项概括了吸附层上偶极间侧向相互作用能总和。其中, Nj(r),i(r)即该层单位表面最邻近的 j(r)-i(r')分子对的数目。已假定吸附分子的偶极取向是随机分布的,如以Z表示指定中心配位数,有 wj(r),i(r~)是最邻近的两偶极间的静电作用能,记d为其平均距离 (2)吸附相与溶液的组份化学势 对吸附相, 按统计力学,自由能F=-kT ln Q,故可导出j(r)态吸附分子化学势 lj(r)=(0F/aNj(r)) 0s(r)=Nj(r)/N,即该态分子覆盖度。 对溶液夜(s),据热力学, x, 和Y,分别表示混合溶剂中第r种物质的摩尔分数及其活度系数,并以纯r的*(s)作为儿,(s)的标准态。 (3) 平衡关系式 设吸附相(ads)中,对同种溶剂分子,其偶极取向各态可相互转化并达成如下平衡: 其平衡条件, 在溶液相中,r与r'分子也可因竞争吸附而存在以下平衡 这里, 以 Pj(r) 表示吸附层上r分子呈现j(r)态的相对几率,定义为 由此得 联立(8)、(11)两式及其约束条件20j(r)=1),便可求解单分子层多组吸吸附等温方程。 (4) Ao与C(o)计算式 按平行板模型,从电极面通过紧密层的电位降可确定为 (12) l和e.分别表示吸附层的平均厚度和介电常数,因e.之给值乃决定于吸附分子的极性, 可为解析为 此式右方第二项体现了吸附分子的偶极取向极化对电场产生的影响。而1/e'则概括了除此以外的其他极性因素,在近似处理下,令1/e'=1,则 单位表面有效吸附位数N显与单层排列点阵结构有关,可给出 A称点阵结构因子。由于吸附层的结构状态可能受到表面电荷影响,故设想N与o有关,但其表达式当视给定的电极体系而定。此外,图1中C(r)态斜交角亦假定随而变,且设 此处, Ao意含模型计算中可适用的勺范围。以式(12)对u求导,得 综上,合理选择分子参数便可依据求解的吸附等温方程以及(15)、(17)两式去计算C~o变化关系。· 纯溶剂电极体系 对纯溶剂电极体系,上述模型r=1且式(7)及(9)~(11)可略去不用。而由式(6)、(8)解出的单层吸附等温方程则简化为 4kfe.ds{P(8A-0B-0ccos?p) +P? c03(0-0g-0c) 以上, AUCA=Uc-UA, AUcB=Uc-UB, Aa=a’-a, g为q*(或4)/4之约简,或言C态对A(或B)态的权重因子。鉴于斜交角V与(2元一V)是等价的两种取向,故取g=2.相应的▲中与C亦依次还原为 (20) 上列诸式中,Z、Ac均无直接实验数据可凭。为此, 试由曲线拟合以期验证模型。 图2 汞/溶液界面计算的0~0及C~0拟合曲线 Fig.22Calculated e~o and fitting C~a curves for mercury/solutioninterface solvents: a) formamide, b)methanol;c)ethylene carbonatedata from ref.[8一10] Points(·) are experimental. CH,-0、 溶剂中,对汞/(纯溶剂)溶液界面拟合的内层0;~0及C~o变化曲线。此际,对式(15)结构因子经验式取 其中,Ao、D及s均系待定常数。 表1列出有关的分子参数选值和Z、dAa、1/Ao、A,、Do、s等7个参数的拟合值。应用改进的 Marquart 方法拟合,实验数据采自文献[8一10]。全部计算由FACOM-M340-S计算机完成。 表1 三种有机溶剂中汞/溶液界面的分子参数选值及拟合参数值 Table 1 The selected molecules parameters and its simulating values Hg/Solutioninterface in three organic solvents System(T/C) l×1010/m P: x108/C·m a×1030/m² AUCA AUcB kJ·mol^1 Formamide[13](25) 4.51 12.80 3.88 0.075 一 0.075 Methanolf1 21(25) 2.50 5.53 0.80 ~1.5 -22.8 Ethylene Carbonatef12] 5.60 16.41 110.0 ~13.8 13.8 (40) d×1010/m Aa×1030/m Z A, D, 1/A0 4.80 0.33 12.0 1.20 0.017 331.0 0 2,30 0.03 6.52 1.99 -0.23 2.39 0 5.85 -0.06 2.03 7.56 -020 53.14 自图2可见,在汞电极界面上,三种溶剂的0.~0曲线形状都很不同,这主要决定于吸附分子的偶极矩、极化率以及吸附态间的能量差别(即AUcA和AUcB)。而其对应的C1~0曲线与实验数据的吻合程度亦比前人前111z所所采用的不同处理法佳。这里,试就表1所列结果推论如下: (1)三种溶剂的1/Ao均趋于零,即y→n/2,表明在计算的o范围内, C 态分子的平均取向几乎垂直于电场。 (2)Z与单层分子的点阵形式有关。计算中仅考虑最邻近的偶极子间相互作用,故Z的拟合值当视为有效配位数,意即可包括中心偶极与次邻近以远偶极的净相互作用, (3) cd的拟合值与分子直径()略有差别,表明似不宜简单地将吸附层的点阵排列想象为球状分子密堆积。 (4)Aa(a'-a)反映了对定域化极性分子,在电场影响下,其极化率与分子的取向有关,表1中a取自由分子极化率,且确认A态和B态的α相等。所以,此处拟合的Aα仅具近似相对意义, (5)计算中发现式(22)校正项D小e-之引入对结果精度影响很大。但从表1的拟合值看,在计算的o范围内,三种溶剂的De-随o的变化都甚微,且接近于零。可见,吸附层的结构状态对内层微分电容十分敏感。 (6)图3示出甲酰胺的A山~a变化曲线。在零电荷点上,对应的▲中-0=0.004V, 另二种溶剂的Ao-o依次为0.484V(碳酸亚乙酯)和-0.319V(甲醇). 图3 汞/甲酰胺体系的Aq~o曲线 Fig.3Plot of the potential drop across inner- layer against electrode charge forHg/formamide solution system 上述三种溶剂所显示的Ci~曲曲线可代表汞/溶液界面内层微分电容变化的基本式样1],而从本文的处理结果看,对纯溶剂电极体系,,三态模型大体已能反映汞/溶液界面 内层的结构图象。表面电荷对吸附层结构状态可能产生的深刻影响不容忽视。.当0移向更正或更负的区域时,有可能由于溶液中的离子发生特性吸附或者由于强电场的极化致使C~曲线偏折上升、事实上, C,~r的形状走势明显与溶液的温度以及电解质的品种、浓度都有关【1014],所有这些,都是模型设想中有待改进的问题。此外,文献上提供的C1~o数据大多是基于 Gouy-Chapman-Grahame 模型并从实验的双层电容C(p)返算而得。因此,更严格的理论处理必同时考虑精确的分散层模式, ( 参 考 文 献 ) ( 11A Mott,N.T. and Waits-Tobin,R,J., Rectrochim, Aeta,1961,4,7. ) ( [2] Parsons,R., J. Electroanal.Chem,, 1981, 118,3. ) ( C3] Nikitas,P., J . C hem. S o c., F a raday Trans 1, 1 986,82.077. ) ( c4]Nikitas,P., Electrochim.Acta, 1987,32,205. ) ( [5] Guidelli,R., J.Electroanal.Chem.,1986,197.77. ) [6] iDutkiewicz,E.and Lamperski.S.,.Eleclroanal.Chem.. 1388,247,57. ( [7]Parsons,R., J.Eleetroanal, dhem, 1975,59:229. ) ( [8] Dutkiewiz,E, and P a rsons,R., J.Electroanal.Chem., 1 9 66, 1 1 ,196. ) ( [9] Fawcett,W.R, a n d Marckev,M.D., J.Chem.Soc., Faraday Trans,1973,69,634. ) ( [10]Gramhama,D.C., Z.Electrochem., 1955,59,740. ) ( [11] Parsons,R.,Electrochim. Acla, 1976,21,681. ) ( [12] Fawcett,W.R.,J.Phys.Chem., 1978,82,1385. ) ( [13] Borkowska,Z.et al, J. E l ectroanal. Chem., 1981,124,263. ) ( [14] Borkowska,Z., J,Electroanal. Chem,, 1988,244,1. ) STATISTICAL MECHANICAL TREATMENT OF ABSORBEDMONOLAYER AT ELECTRODE/SOLUTION INTERFACE I. The Inner-layer Differential Capacity forMercury Electrode in Non-aqueous Solution Su Wenduan* Zhou Xiaolin (Department of Chemistry, Xiamen University)(Computer Center, Xiamen University) ABSTRACT A model of dipole orientation distribution for solvate layeratt electrode/solutioninterface was suggested, and based on this model a generalizedmonolayer adsorptionisotherm was derived by using the method of statisticalmechanics andassuming theequilibrium condition of thermodynamics. The solvent of the electrolyte studiedmaybe single component or multic-components (mixture). The dependenceof inner-layerdifferential capacity upon snrface charge density at mercury electrode innfformamidemethanol and ethylene carbonate solutions was estimated by curve-fitting respectively.The present model is more suitable for aqueous and mixed-solventsystems will beexpeeted. China Academic Journal Electronic Publishing House. All rights reserved. http://www.cnki.net
确定





还剩5页未读,是否继续阅读?

产品配置单



武汉科思特仪器股份有限公司为您提供《电极/溶液界面单分子吸附层的统计力学处理》,该方案主要用于其他中--检测,参考标准--,《电极/溶液界面单分子吸附层的统计力学处理》用到的仪器有CS350M电化学工作站/电化学测试系统、CS2350H双单元电化学工作站(双恒电位仪)、CS150M电化学工作站/测试系统
推荐专场






相关方案
更多
该厂商其他方案
更多