








光催化H2O还原CO2还原为CH4是实现“碳中和”的有效途径之一。该反应中由于质子与CO2的耦合同质子与质子间的耦合存在着剧烈的竞争,最终导致产物中CH4的选择性比较低。
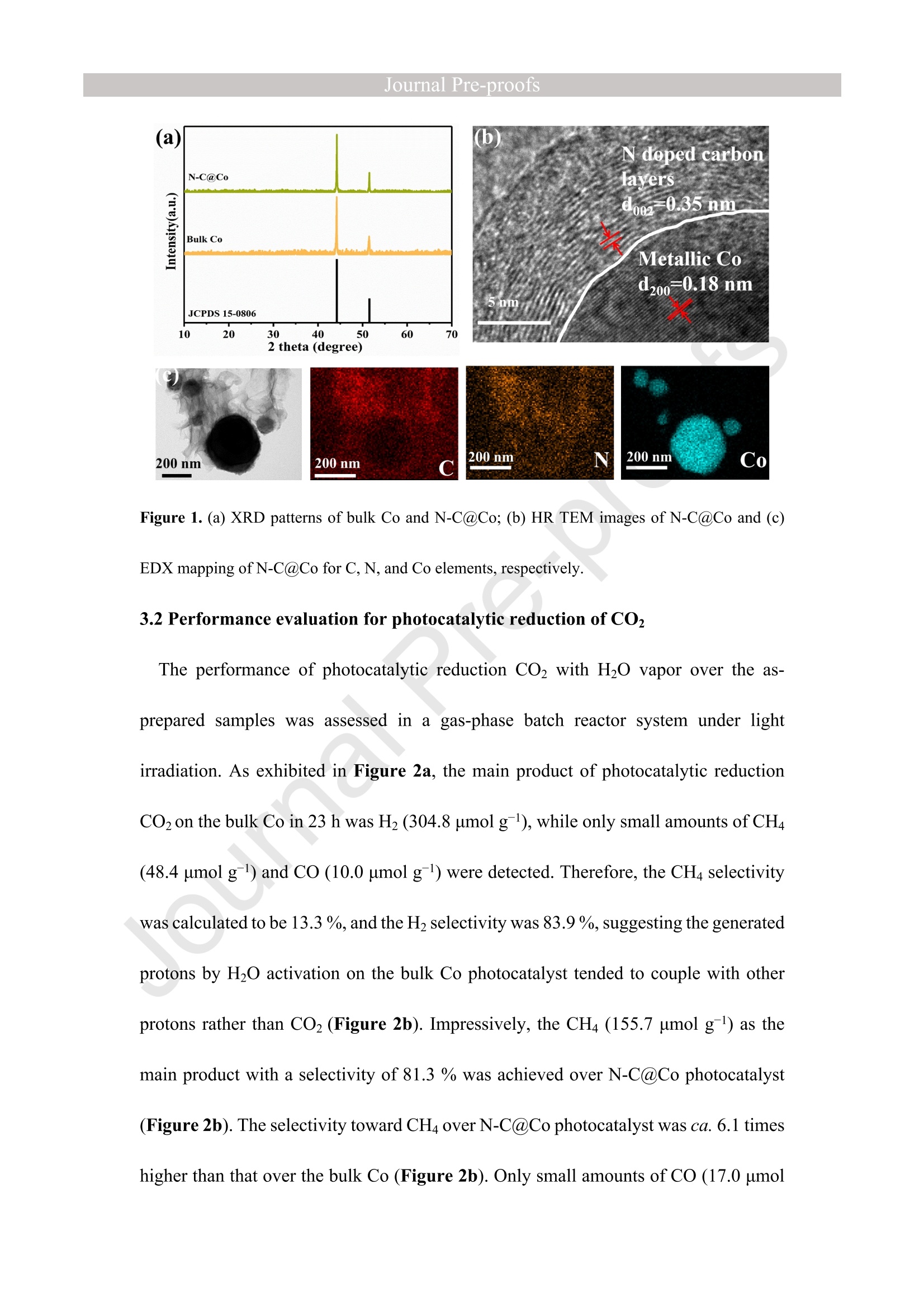
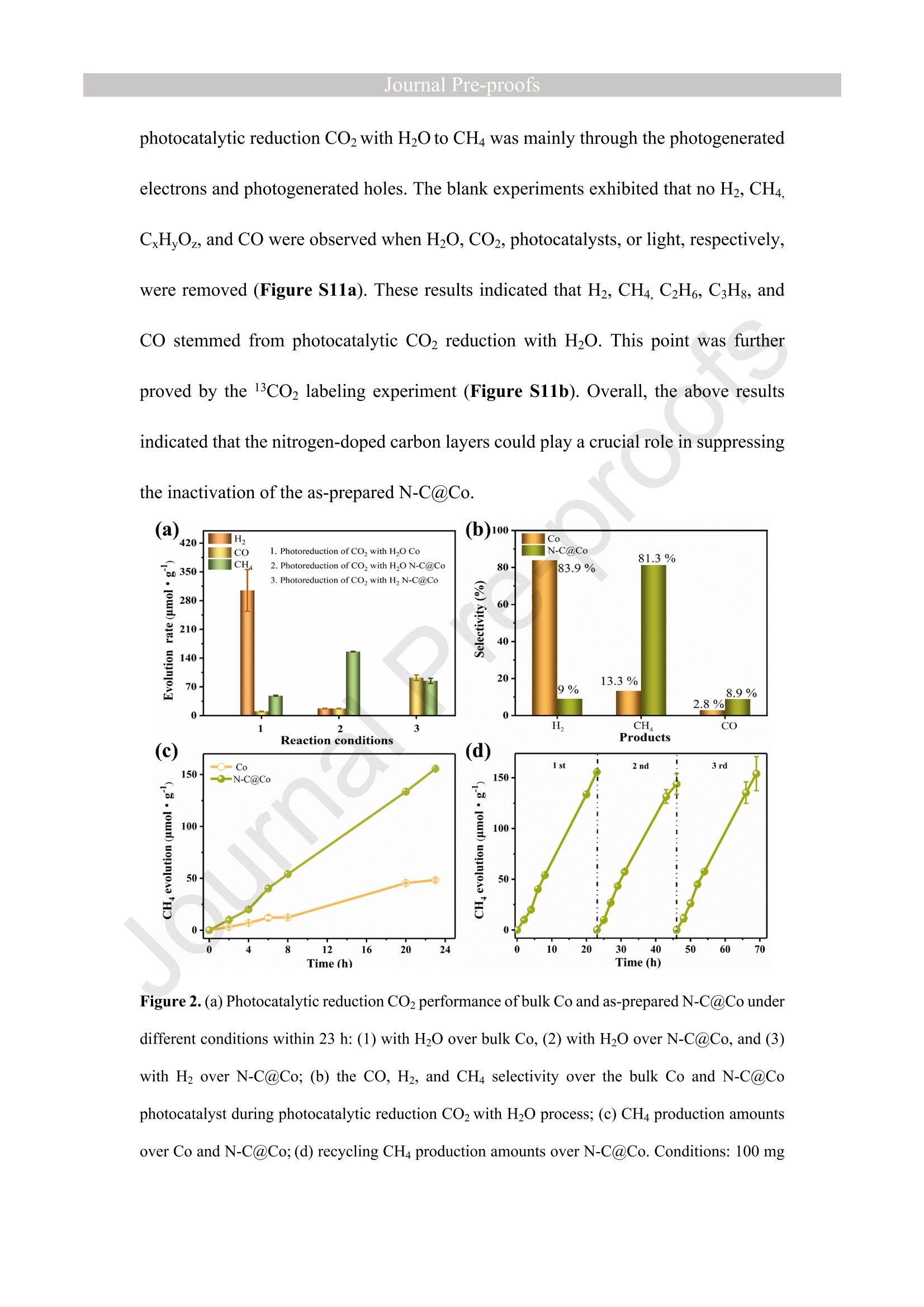
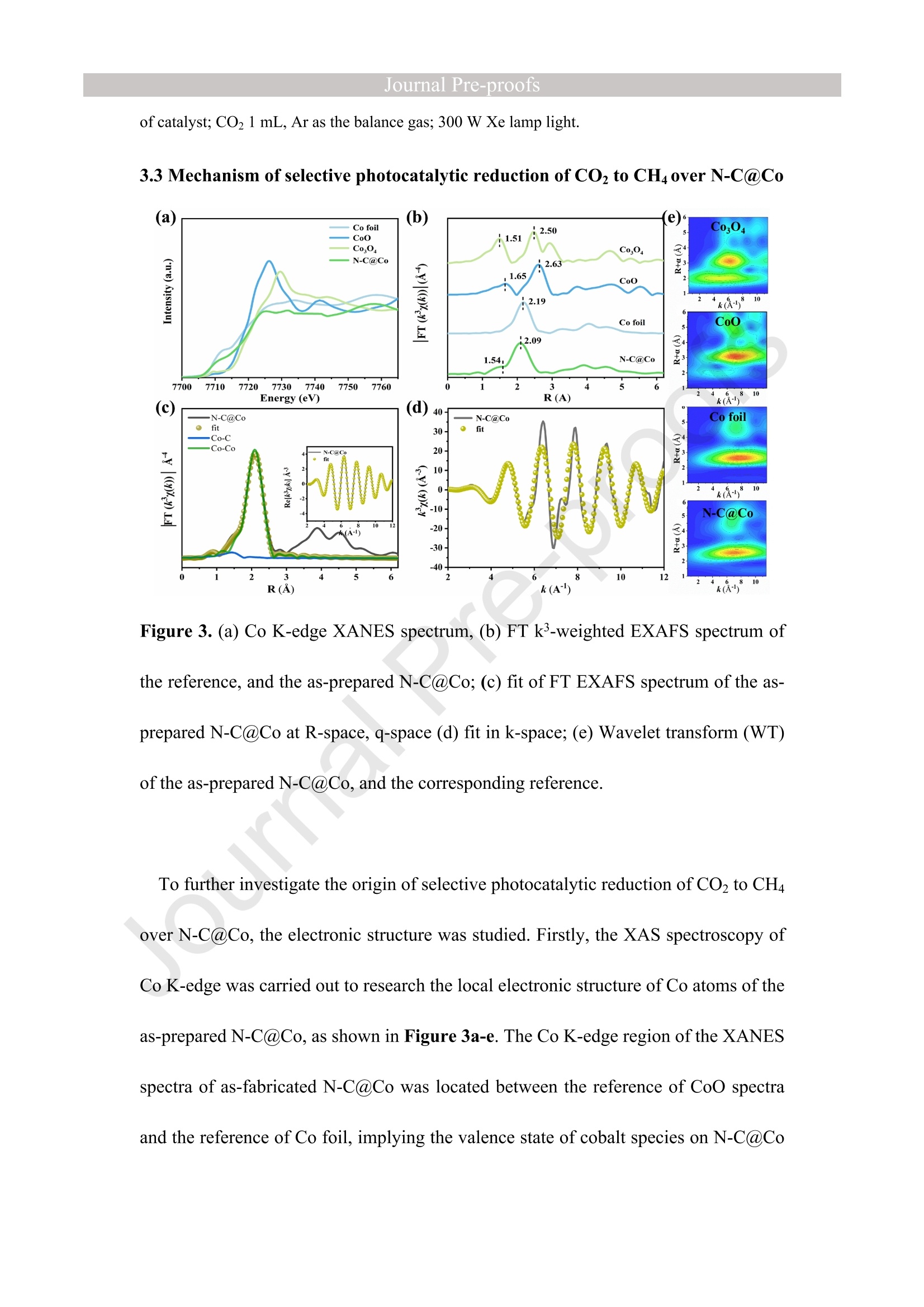
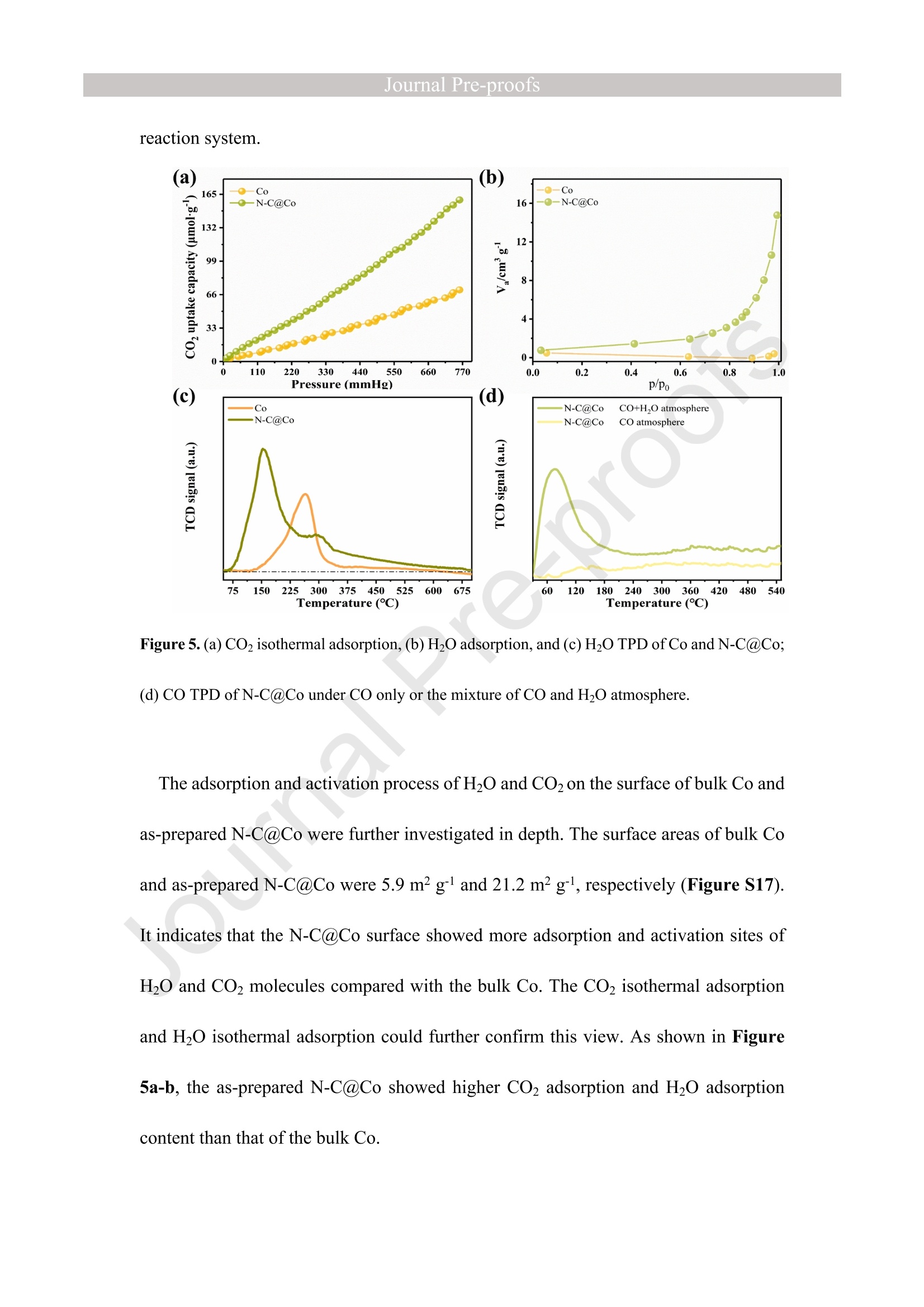
西南石油大学周莹教授课题组设计了氮掺杂碳包覆金属钴(N-C@Co)光催化剂,以提高光催化还原CO2制CH4的选择性。理论计算和实验表征分析表明在N-C@Co和H2O之间形成分子间氢键,有效抑制了生成的质子的迁移,促进了CO2分子的吸附和活化,并且有效抑制CO中间体的脱附(CO中间体通常被认为是生成CH4的关键物种)。这使得在光催化还原CO2的过程中质子可以选择性的与CO2耦合为CH4。最终,N-C@Co所对应的H2的选择性和活性相对于体相Co而言分别降低了17.6倍和9.3倍。而N-C@Co所对应的CH4的选择性和活性相对于体相Co而言分别提升了6.1倍和3.2倍。这项工作为设计出具有高CH4选择性的光催化材料提供了思路。其中X射线精细结构光谱(TableXAFS-500,安徽创谱仪器科技有限公司)对Co局域电子结构的判断是催化机理分析的重要依据。
方案详情

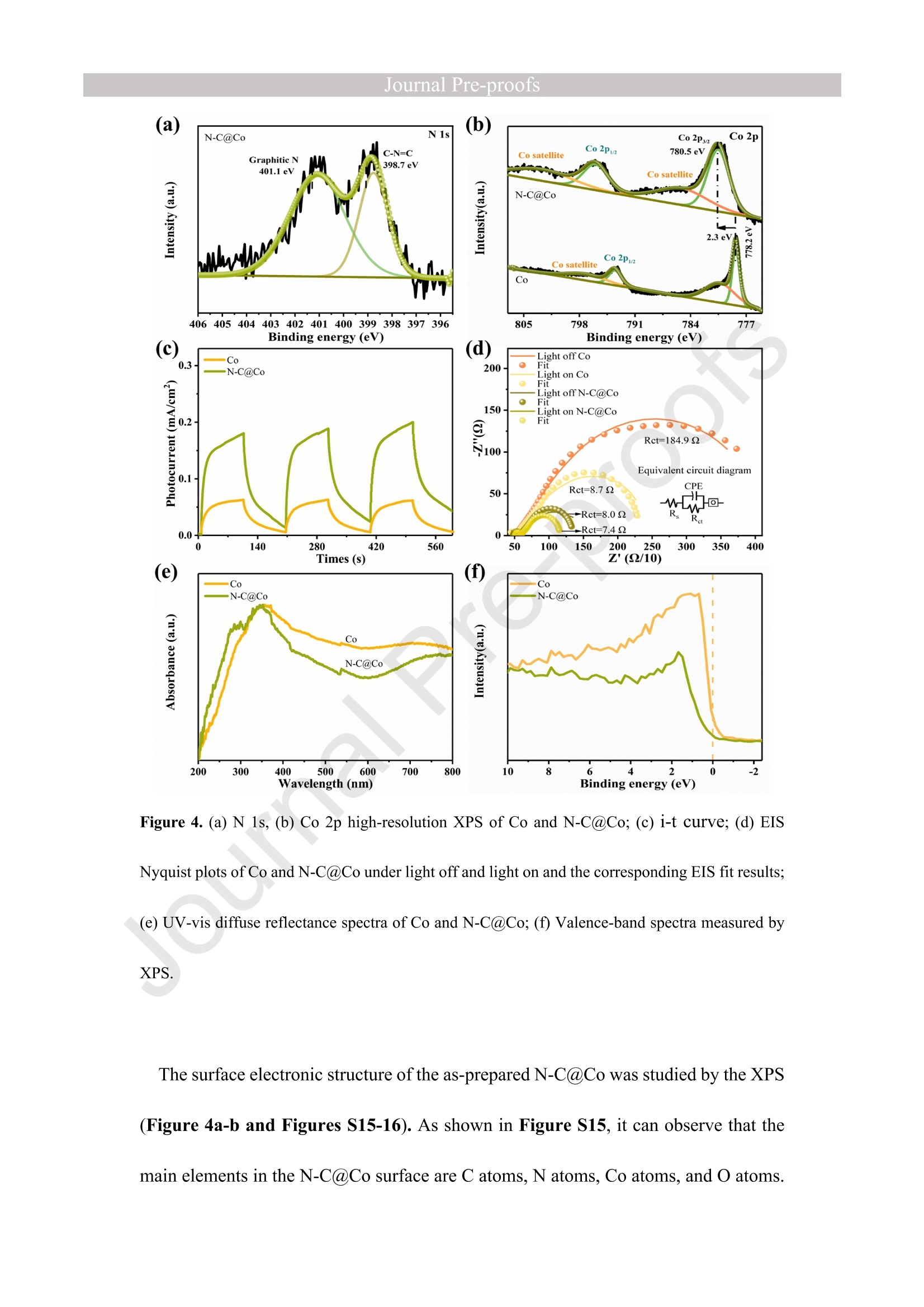
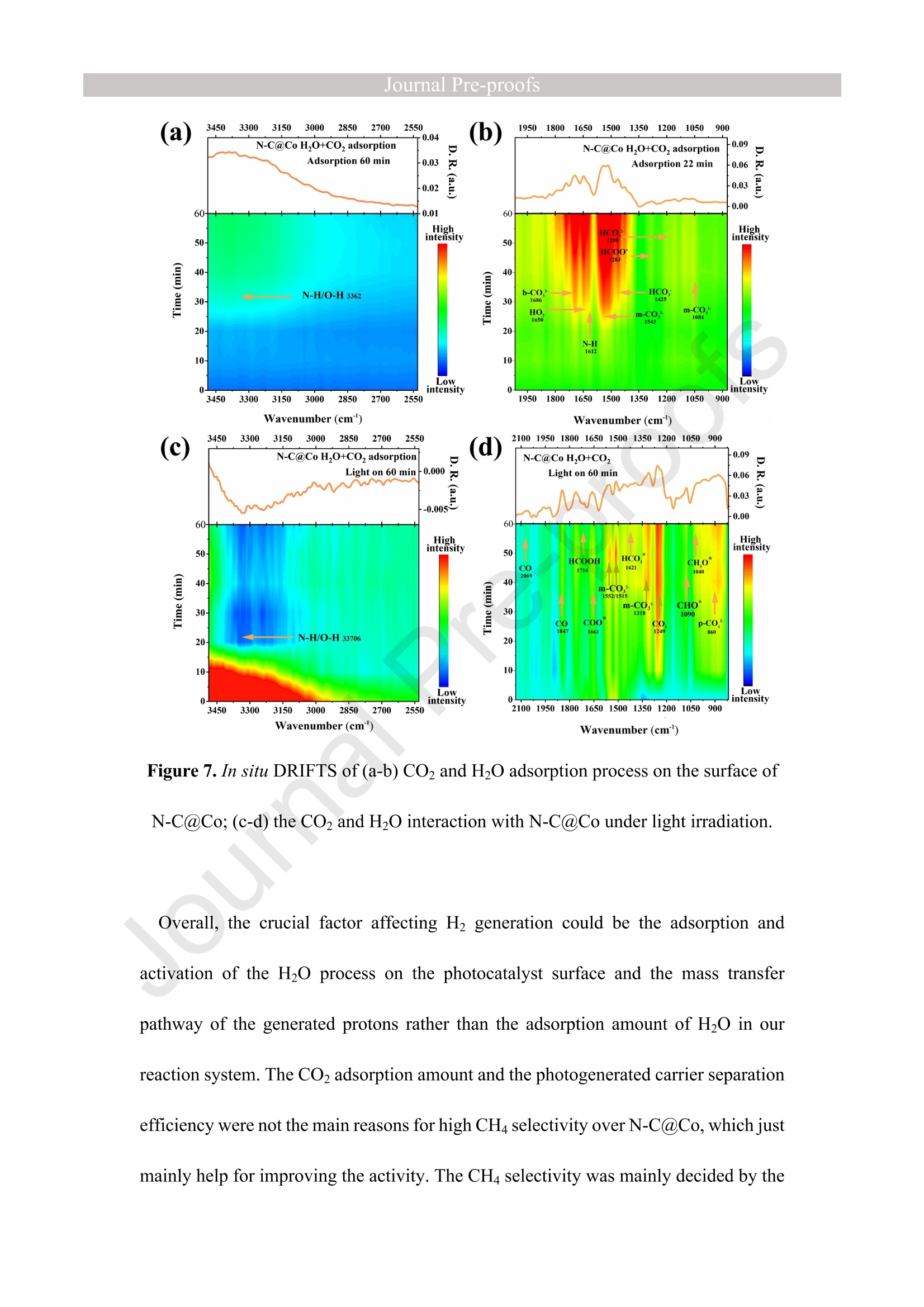
Journal ]nal Pre-proofs Journal Pre-proofs Intermolecular hydrogen bond modulating the selective coupling of protonsand CO, to CH4 over nitrogen-doped carbon layers modified cobalt Minzhi Ma, Jiahao Chen, Zeai Huang, Wenjun Fa, Fang Wang, Yuehan Cao,Yuantao Yang, Zhiqiang Rao, Rui Wang, Ruiyang Zhang, Yanzhao Zou,Ying Zhou PII: S1385-8947(22)02080-0 DOI: https://doi.org/10.1016/j.cej.2022.136585 Reference: CEJ 136585 To appear in: Chemical Engineering Journal Received Date: 24 January 2022 Revised Date: 14 April 2022 Accepted Date: 22 April 2022 Please cite this article as: M. Ma, J. Chen, Z. Huang, W. Fa, F. Wang, Y. Cao, Y. Yang, Z. Rao, R. Wang, R.Zhang, Y. Zou, Y.Zhou, Intermolecular hydrogen bond modulating the selective coupling of protons and CO, toCH4 over nitrogen-doped carbon layers modified cobalt, Chemical Engineering Journal (2022), doi: https://doi.org/10.1016/j.cej.2022.136585 This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a coverpage and metadata, and formatting for readability, but it is not yet the definitive version of record. This versionwill undergo additional copyediting, typesetting and review before it is published in its final form, but we areproviding this version to give early visibility of the article. Please note that, during the production process, errorsmay be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. C 2022 Published by Elsevier B.V. Intermolecular hydrogen bond modulating the selective coupling ofprotons and CO, to CH4 over nitrogen-doped carbon layers modifiedcobalt Minzhi Malal [b], Jiahao Chen b, Zeai Huanglb]*, Wenjun Fale, Fang WangIb, YuehanCaolbl, Yuantao Yanglb, Zhiqiang Raolbl, Rui Wanglb, Ruiyang Zhanglb, YanzhaoZoulb], and Ying Zhoulal, [b]. [d]* [a] State Key Laboratory of Oil and Gas Reservoir Geology and Exploitation,Southwest Petroleum University,Chengdu, 610500, China. [b] Institute of Carbon neutrality, School of New Energy and Materials, SouthwestPetroleum University, Chengdu, 610500, China [c] Key Laboratory of Micro-Nano Materials for Energy Storage and Conversion ofHenan Province & College of Advanced Materials and Energy, Xuchang University,Henan, 461000, China Corresponding authors: .ZeaiHuang (zeai.huang@swpu.edu.cn);;Ying Zhou(yzhou@swpu.edu.cn) Abstract: Photocatalytic reduction of CO2 with H2O to CH4 is a promising route tomigrate CO2 emission and complete the carbon neutrality goal. Nevertheless, one of thebiggest challenges for this elegant strategy is that the coupling of the protons and CO2to form CH4 is fiercely competed with proton-proton coupling to form H2, leading toextremely low CH4 selectivity. Herein, we designed and fabricated the nitrogen-dopedcarbon layers modified cobalt (N-C@Co) photocatalyst achieving the selectivecoupling of protons and CO2 to CH4 during photocatalytic reduction of CO2. Thesuccessful formation of intermolecular hydrogen bonds between the as-prepared N-C@Co and H2O molecule was found to suppress the mass transfer of the generatedprotons and promote the adsorption and activation of the CO2 molecule. More crucially,it was conducive to suppressing the desorption of the CO intermediate, which wastypically deemed as the decisive species for CH4 generation. As a result, the Hselectivity (9.0 %) and activity (17.3 umol g-l) of the as-prepared N-C@Co werereduced by a factor of 9.3 and 17.6, respectively, as compared to that of bulk Co. TheCH4 selectivity of N-C@Co was boosted 6.1 times from 13.3% of bulk Co to 81.3 %of N-C@Co with the generation rate of 155.7 umol gin 23 h. This work provides anew insight into the photocatalyst design for improving CH4 selectivity and suppressingthe competing H2 and CO generation. Keywords: Intermolecular hydrogen bond, Photocatalytic CO2 reduction, SuppressingH2 and CO generation, Selective CH4 generation. 1. Introduction The photocatalytic CO reduction with H20 to CH4 has recently attractedconsiderable attention because this strategy holds great promise for migrating COemissions and completing the carbon neutrality goal [1-6]. CH4 generation duringphotocatalytic CO2 reduction is thermodynamically favorable as compared to that ofH2 and CO generation (Eq.1-3) [7]. However, photocatalytic CO2 reduction with H2Otends to be fiercely competitive with H2 and/or CO generation reaction in most reportedsystems, decreasing the selectivity toward CH4 [7, 20]. This is primarily because thereduction of CO2 and H20 to CO and H2, respectively, are 2-electron processes (Eq.1-2), whereas CO2 reduction to CH4 is an 8-electron process (Eq.3) [8]. In addition, thegeneration potentials of CO and formate species (Eq.2 and Eq.4), which are regardedas the crucial intermediates for CH4 formation, are higher than the generation potentialof H2[9]. Therefore, achieving the selective photocatalytic reduction of CO2 to CH4 ishighly demanded but of great challenges. 2H+2e→H,E0= -0.41 V vs. NHE(1)CO,+2H++2e→CO+H2OE0= -0.52 V vs. NHEI(2)CO2+8H++8e→+CH4+2H2OE0=-0.24 V vs. NHE(3)CO2 +H++ 2e→HCOO-E0= -0.61 V vs. NHE(4) The general strategy to solve this issue has been mainly focused on modulating theadsorption strength or configuration of the CO2 molecules on the photocatalyst surface[7, 10-16]. Because CO2 adsorption and activation were typically considered as the decisive factor for inducing the persistent coupling of the protons and CO andsuppressing the desorption of intermediates during CH4 generation [7,15, 17]. However,the effect of interaction between H20 molecules and the photocatalyst surface on theproduct selectivity of CO2 reduction has received insufficient attention [18-20].Because the H20 molecule activation process on the photocatalyst surface wascommonly thought to be the critical limiting factor for the activity of photocatalyticCO2 reduction rather than the selectivity of the CO2 reduction product [15-17]. Butgiven that the generation of a CH molecule needs four protons to couple with a CO2molecule (Eq.3) [17]. Therefore, it could infer that the formation of sufficient protonsprobably helps to promote the CH4 formation during photocatalytic CO2 reduction.However, if the generated protons on the surface of photocatalysts hold high masstransfer mobility and high generation rate, it seems to increase the likelihood of proton-proton coupling compared to that of the protons and CO2 coupling (Eq.1-4), resultingin the production of H2 and/or CO dominantly [21, 22]. Hence, modulating theadsorption and activation process of H20 molecules on the surface of a photocatalyst isalso highly important to control the selectivity of CO2 reduction products. However,few researchers have focused on this crucial issue until now. Based on the above considerations, inducing protons generated by H20 activation tobe selectively coupled with CO2 is a key factor in achieving selective photocatalyticCO2 reduction to CH4. It was recently reported that the proton-proton coupling to formH2 was suppressed when the intermolecular hydrogen bonds were formed between H2O molecules and the surface of the catalyst [23-26].More crucially, it has been essentiallyconfirmed that the CO2 molecules could be captured by the intermolecular hydrogenbonds [25, 26]. Therefore, the rational design of the photocatalyst surface to formintermolecular hydrogen bonds during photocatalytic reduction of CO2 with the H20process could be an efficient route to realize the selective generation of CH4. Herein, as a proof ofconcept, we designed and prepared the N-C@Co photocatalystfor CO2 reduction to achieve the selective generation of CH4. Firstly, the surfaceplasmon resonance (SPR) of the cobalt metal supplied sufficient photogeneratedelectrons for meeting the needs of multielectron transfer in the process of CH4generation. Secondly, the hydrogen atoms of H20 molecules were easy to produceintermolecular hydrogen bonds with nitrogen atoms of high electronegativity on thesurface of the as-prepared N-C@Co. Then, it was found that the process of producingprotons by activating H2O molecules on the N-C@Co surface was directed byintermolecular hydrogennbonds. Finally, the massttiransfer of the protonswassuppressed and the selective coupling of protons and CO in time was achieved,accompanied by inhibiting desorption of the formed CO intermediate. As a result, theselective photoreduction of CO2 with H20 to CH4 (CH4 selectivity: 81.3 %) over theas-prepared N-C@Co was obtained and the H2 and CO generation were successfullysuppressed. 2. Experiments 2.1 Materials All chemical reagents employed in experiments were analytical grade without anyfurther purification. The dicyandiamide, BN, CoO, NiO, Fe2O3, and Ni metal particleswere purchased from Aladdin Reagent Co., Ltd., the bulk Co was purchased fromMacklin Reagent Co., Ltd.. 2.2 Synthesis of N-C@Co and reference samples ec The g-CN4 nanosheets were prepared according to our previous work [27]. The as-prepared g-C3N4 nanosheets (2 g) (g-C3N4 NSs) was mixed with CoO (1.7g) indeionized water at room temperature under magnetic stirring. Then, the above mixturewas maintained at 230 C for 5 h. Finally, the N-C@Co was formed by in situcarbonizations at 950 ℃ for 1 h with an increasing rate of 10 C min under Arfollowing. In addition, the mixture of g-CN4 NSs and CoO was also annealed at 550℃,650℃, 750℃, and 850 C under an Ar atmosphere with the same heating method.The N-C@Ni was prepared by annealing the mixture of g-C3N4 NSs (2.0 g) and NiO(1.7 g) at 950 ℃ under identical conditions. 2.3 Characterizations X-ray powder diffraction (XRD) patterns were recorded by a X-ray diffractometer(DX-2700B) with Cu Ko radiation (40 kV, 40mA). The high-resolution TEM (HR-TEM) and the corresponding TEM mapping were obtained Tecnai G2 F30 electronmicroscope (200 kV). The scanning electron microscopy (SEM) was performed on aZEISS Gemini 300 microscope. XPS measurementss were conducted by ThermoESCALAB250Xi. The UV-vis diffuse reflectance spectra were recorded at room temperature on a Shimadzu UV-2600 spectrophotometer with an integrating sphere using BazSO4 as the reflectance standard. The CO adsorption isotherms and CO2temperature-programmed desorption (TPD) were conducted using ASAP 2020 V4.00and ASAP 2920.CO TPD of the N-C@Co were acquired using AutoChem1 II 2920.Before starting the experiment, the sample was pretreated at 300C under Heatmosphere. Then, the mixture gas of H2O and CO or pure CO were continuously fedinto the cell under 25 C for 1h. After that, He gas was fed into the cell for 1 h with 30mL min. At last, the sample was heated to 700℃ with 10℃ min. Before startingthe CO2 TPD experiment, the sample was pretreated at 300C under the He atmosphere.Then, the CO2 gas was continuously fed into the cell under 25 C for 1h. After that, theHe gas was fed into the cell for 1 h with 30 mL min. At last, the sample was heated to700 C with 10℃ min . The H2O TPD of the N-C@Co and bulk Co were acquiredusing AutoChem1 ⅡI 2920. The test method for H2O TPD was the same as that of CO2TPD. The BET of the as-prepared N-C@Co and bulk Co was obtained by ASAP2460.The elemental analysis (EA) was utilized to detect non-metallic elements (VarioUNICUBE). The thermogravimetric (TG, NETZSCH STA 449F3) was used to detectthe cobalt content of the as-prepared N-C@Co under a flow of Air. Contact angle datawas obtained by a video contact angle analyzer (Dataphysics OCA25, German). TheH2O adsorption of the as-prepared samples was conducted using Belsorp-MAX. Thequadrupole mass spectrometer (MS) was used in the isotopic labeling experiment(Pfeiffer Vacuum OmniStar GSD 320 01). The Raman spectra of the sample were analyzed on a micro-Raman 2000 system with a 785 nm high power laser. The X-ray absorption spectrum ((XAS)Wasic(onducted in Table XAFS-500(Specreation Instruments Co., Ltd.). This spectrum was composed by the X-rayabsorption near edge structure (XANES) and extended X-ray absorption fine structure(EXAFS). The Co K absorption edge of the as-prepared N-C@Co was obtained bytransmission mode. The N-C@Co was diluted with boron nitride before testing andthen it was pressed into pellets. XAS datawas normalized and subtracted thebackground by the ATHENA program (uncorrected for the phase shift) [28]. Thewavelet transformed (WT) of EXAFS spectra were obtained using the WTEXAFSprogram [29]. The ks-weighted x(k) data in the k-space ranging from 1.7-12.2 A-1(multiplied by a Hanning window function with dk=1.0 A-1) were Fourier transformedto obtain radial distribution functions (R space). Structural information parameters ofCo atoms in the as-prepared N-C@Co were obtained by the fit of the spectra of theFourier transformed kx(k) EXAFS (FT EXAFS), corresponding R-space between 1.2and 3 A using the ARTEMIS module of IFEFFIT. Effective backscattering amplitudesF(k) and phase shifts (k) of all fitting paths were calculated by the ab initio code FEFF6.0. During the fitting of the N-C@Co, the Debye-Waller factor (o2), and energy shift(8E) were allowed to vary. XAS spectra of the Co foil, CoO, and Co3O4 were selectedas the reference. 2.4 Photocatalytic CO2 reduction experiments The photocatalytic reduction of CO2 with H20 was conducted in a closed system without using neither sacrifice reagent or photosensitizer. The experimental conditionswere the same as in our previous work [30]. Typically, 100 mg of as-prepared sampleswere uniformly dispersed on a Petri dish, and then the Ar gas was purged to replace theair in the reactor. The photocatalyst was irradiated at a 300 W Xe lamp (PLS-SXE 300,Perfect Light Co., Ltd.) for 6 h under flowing argon conditions to remove the surfaceadsorbed CO2. Before photoirradiation, 1.0 mL of carbon dioxide and 0.4 mL ofwaterwere injected into the reactor. The CO, CH4, and H2 were detected by GC7900 gaschromatography (GC, Tian Mei Analytical Instrument Co., Ltd.), which was equippedwith MCF, TCD, and FID with a TXD-01 column. CxH,Oz products were detected bya 7890B GC (Agilent Technologies Inc.,HP/Plot-Q, USA). The performance of allsamples was tested at least three times. The 13CO2isotopic experiments were carriedout under the same reaction conditions, which were detected by OmniStar GSD-320,Pfeiffer Vacuum. CH4 selectivity was calculated as follows; CH4 selectivity=NcH,/N(cH+CO+H,)*100% l(5) H2 selectivity was calculated as follows; H2 selectivity=Nnz/N(CH4+CO+H2)*100% (6) 2.5 Photoelectrochemical measurements The i-t curve and EIS Nyquist plots were obtained on a CHI660E electrochemicalworkstation (Chenhua Instrument, Shanghai, China) using a three-electrode systemwith the working electrode, reference electrode, and counter electrode. 0.5 M NazSO4 was used as an electrolyte. Pt wire was selected as the counter electrode. The saturatedcalomel electrode (SCE) was selected as the reference electrode. The cleaned FTO glassdeposited with sample was utilized as the working electrode, which was prepared by adoctor blade method. In detail, the mixture of the photocatalyst and methylcellulosewas coated on a Fluorine-doped tin oxide (FTO) with a film thickness of ca.50 um and4 cm² area, which was further treated at 120 ℃ for 3 h under Ar flow. Photocurrent-time and electrochemical impedance measurements of sample films at open circuitpotential (OCP) were performed. 2.6In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS)investigation IZ The in situ DRIFTS measurementss were carried out using a Bruker Tensor IIinstrument equipped with a liquid nitrogen-cooled mercury-cadmium-telluride (MCT)detector in a three-window diffuse-reflectance cell (Harrick) with a nonmodified domecover [8,31,32]. Two IR measurement windows were made of ZnSe, while the siliconoxide window was used for light irradiation. Before the test, the N-C@Co and SiO(70-150 mesh) are fully mixed in a ratio of 1 to 5. 100 mg of the mixture of SiO2 andthe as-prepared N-C@Co was pretreated at 300 °℃ for 30 min under 20 mL min-Argas (99.99%) flow. Subsequently,CO2(99.99%) and H20 vapor (introduced bybubbling the gas flow through a water saturator at 70 ℃) were continuously influx theHarrick cell reactor with a three-window diffuse-reflectance at 20 mL min- under darkconditions. The data was collected every four minutes. After the N-C@Co was pretreated at 300°C in the Ar atmosphere for 30 min, the background spectrum wasacquired at 30°℃ in the Ar atmosphere. 2.7 Theoretical calculation method The density functional theory (DFT) calculation was conducted using the Vienna Ab-initio Simulation Package (VASP) [33-36]. The generalized gradient approximation(GGA)functional and Perdew-Burke-Ernzerhof (PBE) functional were utilized for theexchange-correlation between electrons. Modifying Gaussian path and smearing widthwas 0.2 eV. Geometries were optimized until the energy and the force were convergedto 1.0×10-4 eV/atom and 0.02 eV/A, respectively. An energy cutoff was set as 380 eVfor the plane-wave expansion of the electronic wave function. A vacuum region of 15A was used to eliminate the interactions between the periodic slabs. In addition, k-pointmeshes are set to 2×2×1. The adsorption energy (Eads) of CO2 or H20 molecule on thesurface is calculated as follows: Eads-Etotal-Esurface-Eco, (EH,o) 1(7) Where Etotal is the energy of surface with adsorbed CO2 or HO molecule, Esurface andEco, or EH,o is the energies of isolated surface and CO2 or H20, respectively. 3 Results and discussion 3.1 Synthesis and characterization The N-C@Co nanostructure was prepared by a facile pyrolysis treatment of CoO andg-C3N4NSs. Typically, the g-C3N4 NSs will gradually decompose into nitrogen-dopedcarbon layers with the release of a large amount of reducing gas during the pyrolysis process at high temperatures [27]. Under this reductive atmosphere, the Coo wasreduced to Co metal nanoparticles. Simultaneously, the nitrogen-doped carbon layerswill grow in situ on the surface of Co metal nanoparticles. As shown in Figure 1a, theXRD patterns of bulk Co and N-C@Co showed two peaks at ca. 44.3°and 51.5°,assigned as Co metal (JCPDS: 15-0806)[37].No other peaks were detected, verifyingthat the cobalt species in the as-fabricated N-C@Co mainly existed as the metallic state(Figure 1a). ● The lattice fringes of N-C@Co in the HRTEM image were observed (Figure 1b), inwhich the 0.18 nm was assigned to the (200) plane of metallic Co, and the 0.35 nmcould be assigned to the (002) plane of layered materials [38]. As shown in Figure 1c,it could be observed that the as-fabricated N-C@Co was spherical. The correspondingSEM images could further prove this view (Figure S1). From the EDX mapping, it wasobserved that the carbon element was surrounded by the Co metal core, and the nitrogenelement was uniformly distributed (Figure 1c). The HR TEM and the correspondingEDX mapping images undoubtedly confirmed that the Co metal nanoparticles werewell encapsulated by nitrogen-doped carbon layers (Figure 1b-c). It could be furtherdemonstrated by the Raman spectra. As shown in Figure S2, the two characteristicpeaks at ca. 1310 cm (D band), and ca. 1580cm(G band) were observed, and theintensity ratio of the D and G bands (Ip/IG) was1.01. Figure 1. (a) XRD patterns of bulk Co and N-C@Co; (b) HR TEM images of N-C@Co and (c) EDX mapping of N-C@Co for C, N, and Co elements, respectively. 3.2 Performance evaluation for photocatalytic reduction of COz The performance of photocatalytic reduction CO2 with H20 vapor over the as-prepared samples was assessed in a gas-phase batch reactor system under lightirradiation. As exhibited in Figure 2a, the main product of photocatalytic reductionCO2 on the bulk Co in 23 h was Hz (304.8 umolg), while only small amounts of CH4(48.4 umolg) and CO (10.0 umol g-) were detected. Therefore, the CH4 selectivitywas calculated to be 13.3 %, and the H2 selectivity was 83.9 %, suggesting the generatedprotons by H2O activation on the bulk Co photocatalyst tended to couple with otherprotons rather than CO2 (Figure 2b). Impressively, the CH4 (155.7 umol gl) as themain product with a selectivity of 81.3 % was achieved over N-C@Co photocatalyst(Figure 2b). The selectivity toward CH4 over N-C@Co photocatalyst was ca. 6.1 timeshigher than that over the bulk Co (Figure 2b). Only small amounts of CO (17.0 umol g) and H2 (17.3 umol gl) were detected and the H2 selectivity was calculated to beonly 9.0% (Figure 2a and Figure 2b). In brief, the H2 activity and selectivity over N-C@Co photocatalyst were sharply decreased by 17.6 and 9.3 times, respectively, ascompared with that over the bulk Co.Other CO2 reduction products were also detected.As shown in Figure S3, C2H6(0.6umolg-) and C3Hg (0.9 umolg-l) were also detectedover N-C@Co photocatalyst. In addition, a small amount of CHOH and HCOOH werealso detected. The corresponding gas chromatographic signal is shown in Figure S4.Using bulk Co as the photocatalyst, only a small amount of CH;OH and HCOOH weredetected, and no C2H6 and CHg were detected (Figure S3). In addition, using H2 to replace H2O for photocatalytic reduction CO2 by using N-C@Co photocatalyst showed that the formation amount of CO and CH4 were 92.2 umolg-l and 84.5 umolg-l(Figure 2a), respectively. The selectivity of CH4 was only 47.8%,which is lower than using H20 as a protons source (81.3%). Additionally, the CH4selectivity and activity of photocatalytic reduction CO2 with H2O under similar catalyticreaction conditionswithout sacrificial reagent, photosensitizer, and noble-metalcocatalyst over recently reported were compared as exhibited in Table S1. For the as-prepared N-C@Co in our work, it exhibits the higher CH4 selectivity and activity.The 23-h long-term formation of H2, CO, and CH4 under light irradiation over N-C@Co and bulk Co were presented in Figure 2 and Figure S5. The CH4 generationoverN-C@Co photocatalyst grew approximately linearity with time increasing (Figure2c). The corresponding CH4 generation rate of the as-prepared N-C@Co was shown in Figure S6. The initial formation rate of CH4 was 5.1 umol g-h,reached the highest 8.5 umolg-h- at 6 h, and finally stabilized at ca. 7.0 umol g-h-. However, the bulkCo exhibited much lower stability for CH4 generation after 8 h reaction (Figure 2c),consistent with previous reports [39]. This is mainly because the bulk Co was readilyoxidized to low activity CoOx species [39, 40]. In addition, the CO generation over N-C@Co and bulk Co photocatalysts also increased approximately linearity with timeincreasing (Figure S5). However, the H2 generation over N-C@Co and bulk Cophotocatalysts firstly increased and then decreased with time increasing (Figure S5).As shown in Figure 2d, the CH4 production over N-C@Co in three consecutive cycleexperiments with a total of 69 hours still maintained excellent stability. In addition,there was no significant change in CO generation except H2 (Figure S7). Moreover,the CH4 initial reaction rates of the three cycles were almost the same (Figure S8).From the XPS and XRD, it could be clearly seen that the surface structure and bulkstructure of the as-prepared N-C@Co did not change significantly after 69 hours ofreaction (Figures S9-10). These results indicated that the as-prepared N-C@Coshowed a relatively stable activity for photocatalytic reduction CO2 to CH4, owing tothe as-prepared N-C@Co with a high chemical structure stability. In addition, thesurface temperature ofN-C@Co was about 120C during the process of photocatalyticCO2 reaction with H2O. However, the as-prepared N-C@Co did not show anyperformance of photocatalytic reduction CO2 with H20 at the same temperature or ashigh as 350 C. Therefore, it could infer that the as-prepared N-C@Co driven photocatalytic reduction CO2 with H2O to CH4 was mainly through the photogeneratedelectrons and photogenerated holes. The blank experiments exhibited that no H2,CH4,CxH,Oz, and CO were observed when H2O,CO2, photocatalysts, or light, respectively,were removed (Figure S11a). These results indicated that H2, CH4, C2H6, C3Hg, andCO stemmed from photocatalytic CO2 reduction with HO. This point was furtherproved by the 13Co labeling experiment (Figure S11b). Overall, the above resultsindicated that the nitrogen-doped carbon layers could play a crucial role in suppressingthe inactivation of the as-prepared N-C@Co. Figure 2. (a) Photocatalytic reduction CO2 performance of bulk Co and as-prepared N-C@Co underdifferent conditions within 23 h: (1) with H20 over bulk Co, (2) with H20 over N-C@Co, and (3)with H2 over N-C@Co; (b) the CO, H2, and CH4 selectivity over the bulk Co and N-C@Cophotocatalyst during photocatalytic reduction CO2 with H2O process; (c) CH4 production amountsover Co and N-C@Co;(d) recycling CH4 production amounts over N-C@Co. Conditions: 100 mg of catalyst; CO21 mL, Ar as the balance gas; 300 W Xe lamp light. 3.3 Mechanism of selective photocatalytic reduction of COz to CH4 0Ver N-C@Co Figure 3. (a) Co K-edge XANES spectrum, (b) FT k3-weighted EXAFS spectrum ofthe reference,and the as-prepared N-C@Co; (c) fit of FT EXAFS spectrum of the as-prepared N-C@Co at R-space,q-space (d) fit in k-space; (e) Wavelet transform (WT)of the as-prepared N-C@Co, and the corresponding reference. To further investigate the origin of selective photocatalytic reduction of CO2 to CH4over N-C@Co, the electronic structure was studied. Firstly, the XAS spectroscopy ofCo K-edge was carried out to research the local electronic structure of Co atoms of theas-prepared N-C@Co, as shown in Figure 3a-e. The Co K-edge region of the XANESspectra of as-fabricated N-C@Co was located between the reference of CoO spectraand the reference of Co foil, implying the valence state of cobalt species on N-C@Co was higher than 0 but lower than +2. This indicates that a strong interaction was formedbetween the nitrogen-doped carbon layers and Co metal. It might arise out of some Coatoms of N-C@Co coordination with N or C atoms. Because, based on our previousresearch, it found that the Co-O bond can be completely replaced via Co-N and Co-Cbond under reductive atmosphere conditions above 500 ℃[27]. This also can beconfirmed by further experiments. It was observed that the CoO was transformed intoCoNxCy by annealing of CoO and g-C3N4 NSs mixture at 550 ℃ under Ar atmosphereas shown in Figure S12. Moreover, nitrogen atoms were gradually removed from thestructure of CoNxCy and N-C@Co was ggradually formed when the annealingtemperature was higher than 650 °℃ (Figure S12). The FT k-weighted EXAFS spectrum of N-C@Co displays two peaks at around1.54 A and 2.09 A, respectively (Figure 3b). The peak of 2.09 A was assigned to theCo-Co bond, moving 0.1 A to the left compared with the Co-Co bonds of the Co foil.This further proved that a strong interaction between Co metal and nitrogen-dopedcarbon layers on the as-prepared N-C@Co was formed. Moreover, Co-(O)-Coscattering paths of the reference CoO (2.63 A) and Co3O4 (2.50 A) were not observedin the N-C@Co, suggesting that CoOx species were almost absent on N-C@Co. Theseresults were further confirmed by WT results. The intensity maximum ofN-C@Co wasat ca. 7.1 A-l that was deemed as cobalt neighbors. It was significantly different fromthe reference cobalt foil (7.8 A-1), CoO (7.8A-1), and Co3O4(6.2A-1), as shown in Fig.3e. To obtain the local coordination structure of cobalt atoms in the as-prepared N- C@Co, the corresponding FT EXAFS fitting was carried out to extract the electronicstructure information of Co atoms in the N-C@Co (Figure 3c). The C012Cicoordination moiety with 2.49 A of Co-Co and 1.95 A of Co-C average bond lengthsgave a reasonably good fit of the FT EXAFS spectrum of the as-prepared N-C@Co(Figure 3c-d, Table S2). This indicated that the peaks at around 1.54 A and 2.09 Awere mainly assigned to the Co-C and Co-Co bonds, respectively. According to theanalysis of non-metallic elements by EA, the contents of C, N, and O in N-C@Co are10.9 wt%, 0.9 wt%, and 0.2 wt%, respectively. This means that the probability ofbonding between carbon and cobalt is much higher. In addition, based on the analysisresults of EA and TG experiments under air atmosphere, indicated that the content ofmetal Co in the N-C@Co sample was ca. 88.4 wt% (Figure S13). All in all, the stronginteraction between nitrogen-doped carbon layers and Co in the as-prepared N-C@Cowas mainly caused by the formation of Co-C bonds in the interface. Moreover, basedon these results, we constructed the corresponding structural model of N-C@Co asshown in Figure S14. JoU Figure 4. (a) N 1s, (b) Co 2p high-resolution XPS of Co and N-C@Co;(c) i-t curve; (d) EIS Nyquist plots of Co and N-C@Co under light off and light on and the corresponding EIS fit results;(e) UV-vis diffuse reflectance spectra of Co and N-C@Co; (f) Valence-band spectra measured by XPS. The surface electronic structure of the as-prepared N-C@Co was studied by the XPS(Figure 4a-b and Figures S15-16). As shown in Figure S15, it can observe that themain elements in the N-C@Co surface are C atoms, N atoms, Co atoms, and O atoms. Binding energy was calibrated at 284.8 eV (Figure S16) [41]. The peak at 285.7 eV ofthe as-prepared N-C@Co was well fitted to the C-N bonds. As shown in Figure 4a,two peaks at 398.7 and 401.1 eV of the N-C@Co were well fitted to be C=N-C bondsand graphitic N, respectively [37]. As exhibited in Figure S16b, two peaks at 531.6and 529.9 eV were well fitted to be the adsorbed H2O species and the Co-O species.The surface Co species in the bulk Co were in metallic state with the binding energy ofCo 2p3/2 peak located at 778.2 eV (Figure 4b), consistent with the observation fromXRD (Figure 1a). However, the binding energy of Co 2p3/2 peak of the as-prepared N-C@Co was located at 780.5 eV, which was higher than that of the Co metal but lowerthan Co²+ [27]. This indicated that the valence state of surface cobalt species of the N-C@Co was located at 0 to +2. Based on the XAS fit results, this was mainly owing tothe coordination of cobalt atoms and carbon atoms (Co-C bonds) at the interfacebetween the Co metal and nitrogen-doped carbon layers. As exhibited in Figure 4c, compared with the bulk Co, N-C@Co showed the highesti-t intensity. The electrochemical impedance spectroscopy (EIS) was conducted toevaluate the photogenerated carriers transfer efficiencies, as shown in Figure 4d. Thecorresponding Nyquist plots exhibit that the radii of the as-prepared N-C@Codramatically reduce compared to the bulk Co, implying that the N-C@Co has thesmallest charge transfer resistance (Figure 4d). Moreover, the Nyquist plots were fittedto an equivalent circuit as shown in Figure 4d. It could be clearly observed that theresistance (Rct) of N-C@Co (184.9 Q) is less than that of bulk Co (18.7Q) under light off. In addition, the corresponding Ret values show a certain decrease under lightconditions, respectively. This may be due to the increase of carrier concentration duringillumination. As a result, it could conclude that the formation of Co-C bonds in theinterface between the nitrogen-doped carbon layer and Co metal could be used as thecharge transfer bridge to improve the charge density on the surfaceofN-C@Co. Notably, the photocurrent density of the bulk Co (ca. 0.5 mA cm) and N-C@Co(ca. 1.5 mA cm2) was two to three orders of magnitude higher than the conventionalsemiconductive materials (ca.≤10 uA cm²)[42-44]. This may be related to the surfaceplasmon resonance effect (SPR) of cobalt metal nanoparticles [39, 45]. As shown inFigure 4e, the bulk Co and as-synthesized N-C@Co exhibited two characteristic SPRabsorption peaks of cobalt with metallic states, which were around 350 nm and 750 nm[39, 46-48]. In addition, the position of the valence-band maximum ofthe bulk Co andN-C@Co was located near 0 eV (Figure 4f). These results indicated that both the bulkCo and N-C@Co could provide sufficient electrons with high mobility for the proton-coupled electron transfer reaction of the photoreduction CO2 to CH4 process. To studythe relationship between photogenerated carrier concentration and CH4 selectivity inour reaction system, a reference experiment was conducted. As shown in Figure 2a,the selectivity of CO reduction products is extremely different from the supply ofproton sources (H2 or H2O) by using the N-C@Co as the photocatalyst. These resultsindicate that the CH4 selectivity is independent of the concentration of photogeneratedcarriers and related to the reaction process of reactants on the N-C@Co surface in our reaction system. Figure 5.(a) CO2 isothermal adsorption, (b)H2O adsorption, and (c)H2O TPD of Co and N-C@Co;(d) CO TPD of N-C@Co under CO only or the mixture of CO and H2O atmosphere. The adsorption and activation process of H20 and CO2 on the surface of bulk Co andas-prepared N-C@Co were further investigated in depth. The surface areas of bulk Coand as-prepared N-C@Co were 5.9 m²g- and 21.2 m²gl, respectively (Figure S17).It indicates that the N-C@Co surface showed more adsorption and activation sites ofH20 and CO2 molecules compared with the bulk Co. The CO2 isothermal adsorptionand H2O isothermal adsorption could further confirm this view. As shown in Figure5a-b, the as-prepared N-C@Co showed higher CO2 adsorption and H20adsorptioncontent than that of the bulk Co. Generally, the adsorption amount of CO2 molecules on the photocatalyst surface ismainly related to the CO2 conversion efficiency and has little correlation to theselectivity of CO2 reduction products. Therefore, compared with the bulk Co, the highCH4 activity ofN-C@Co may relate to the high capacity of CO2 adsorption. Decreasing the adsorption amount of H20 on the photocatalyst surface commonlyinhibits the side reaction of H2 production [11]. As shown in Figure S18, N-C@Co wascompletely hydrophilic (contact angle: 0°), and its hydrophilicity was much higher thanthat of bulk Co (contact angle: 25.5). Moreover,the N-C@Co showed a much higherH2O adsorption amount than that of bulk Co (Figure 5b), and the corresponding H20adsorption content of the as-prepared N-C@Co (357.1 umol g-l) was 1.5 times that ofbulk Co (232.1 umol g-) via semi-quantitative analysis by H2O TPD (Figure 5c).However, it should be noted that the H2 selectivity and activity ofN-C@Co (9.0%;17.3umolg-) were significantly lower than that of the bulk Co (83.9 %; 304.8 umol g-)(Figure 2a). These results strongly indicated that the adsorption amount of H2O on thesurface of N-C@Co was negatively correlated with the quantity of H2 generation in ourreaction system. O Carbon Nitrogen CobaltO Hydrogen Oxygen Figure 6. Adsorption energy and free energy: adsorption energy of H2O on the surface of (a) bulkCo and (b)N-C@Co; free energy for dissociation of H2O on the surface of (c) bulk Co and (d) N-C@Co; (e) adsorption of protons on the surface of N-C@Co and the corresponding partial densityof states (PDOS); (f) the free energy for H reacting with H to H2; adsorption energy of CO2 on thesurface of (g) bulk Co and (h) N-C@Co; (i) adsorption energy of CO2 on the surface of N-C@Cowith adsorbed protons;(i) adsorption energy of CO on the surface of N-C@Co; (k) adsorptionenergy of CO on the surface of N-C@Co with adsorbed protons; the free energy for H reacting withCO2 to HCOO on the surface of (1) Co and (m)N-C@Co. The adsorption strength of H2O on the bulk Co and N-C@Co surface was conducted to study the activation process of H2O molecules and the reasons for suppressing competitive H2 evolution reaction. As shown in Figure 5c, it can be observed that thedesorption temperature of H2O on the as-prepared N-C@Co(155.7C) was much lowerthan that of the bulk Co (264.0C). In other words, the interaction strength betweenH20 molecules and cobalt was much higher than that between H20 molecules and N-C@Co. The DFT calculation could also prove this view. As shown in Figure 6a-b,cobalt adsorbs H20 mainly through the formation of Co-O bonds between cobalt atomsand oxygen atoms in H2O, and the corresponding adsorption energy of H20 wascalculated to be-0.53eV. However,the H20 was adsorbed on the surface of N-C@Coby forming the intermolecular hydrogen bonds between nitrogen atoms in N-C@Coand hydrogen atoms in H2O (Figure 5a). The adsorption energy of H2O on the N-C@Co was calculated to be -0.20 eV. The adsorption energy of H2O molecules on Cowas 2.7 times that of N-C@Co. Simultaneously, an intermolecular hydrogen bondbetween N-C@Co and H2O was observed by the in situ DRIFTS. As exhibited inFigure 7a and Figure S19, the adsorption peak from 3600 to 2800 cmwas ascribedas the vibration of the intermolecular hydrogen bond (N…H and/or O.H) over N-C@Co [49-51]. The absence of signals of intermolecular hydrogen bonds (the range of2800 cm- to 3600 cml) on the surface of Co was observed (Figure S21). In total, thesesuggested that the adsorption strength of H2O was diminished after the bulk Co wascovered by the nitrogen-doped carbon layers. It could infer that the ability ofN-C@Coto activate H20 molecules was lower than that of bulk Co. The free energy calculation could further prove this view (Figure 6c-d). It could be observed that the Gibbs freeenergy of H20 cracking (H2O→·OH+H) on Co(-0.68 eV) was lower than that of onthe N-C@Co(+1.36 eV). Based on the amount of CH,Oz and H2 formation as exhibited in Figure 2a andFigure S3, it was calculated that the capacity of generated protons from H2O activationover the as-fabricated N-C@Co was 83.2 %(668.2 pmol g) of that of the bulk Co(803.2 umol g). This demonstrated that although the adsorption strength andactivation ability of N-C@Co to H2O molecules were significantly lower than that ofbulk Co, the ability of N-C@Co to activate H20 molecules to generate protons was notsignificantly weaker than that of bulk Co (Figure 6a-b). Furthermore, it can find thatthe 1s orbital of the protons could hybridize with 2p orbitals of nitrogen atoms of theN-C@Co to form N-H bonds (Figure 6e). This indicates that the formation of H2 onthe surface of N-C@Co became more difficult owing to the mass migration of protonson the N-C@Co surface was suppressed. This point of view was further proved by theDFT calculation. As shown in Figure 6f, the Gibbs free energy of H reacting with H toH2 (H+e→1/2H2*) on N-C@Co (+0.35 eV) is higher than that of Co (-0.46 eV). Based on the above results, the reason for suppressing H2 generation and thecorresponding H2O molecules activation process over the as-prepared N-C@Co werediscussed in detail below. On the one hand, the mass migration of the protons wasgreatly weakened owing to the protons formed by H2O activation were captured by theN atoms on the surface of N-C@Co. On the other hand, the activation ability of intermolecular hydrogen bonds to H2O molecules was moderated, which effectivelyweaken the formation rate of protons but could not obviously reduce the amount of thegenerated protons. As a result, these will reduce the possibility of H2 generation by thecoupling of protons and protons and facilitate the coupling of protons with CO2. To study the effect of H20 molecular activation on CH4 selectivity in-depth, theCO2-TPD, in situ DRIFTS, and DFT calculationswere conducted. Generally,increasing the adsorption strength of carbon dioxide on photocatalysts was conduciveto the selective conversion of CO2 to CH4, because of suppressing the desorption of keyintermediate products during CH4 formation [7, 52]. Therefore, the CO2-TPD wasperformed to study the adsorption strength of carbon dioxide on bulk Co and N-C@Co.As shown in Figure S20, the peak at around 410 C of bulk Co was assigned to thechemisorption of CO2 [53],implying that bulk Co and CO2 showed strong interactions.However, this chemisorption peak of CO2 was not observed in the N-C@Co sample,indicating that the adsorption strength of N-C@Co for CO is lower than that of bulkCo. This was further supported by the DFT calculation. As shown in Figure 6g-h, CO2was adsorbed on the surface of Co mainly through chemical adsorption (-0.28 eV),while adsorbed on the surface of N-C@Co mainly through physical adsorption (-0.20eV). But, as exhibited in Figure 2a, the CH4 selectivity (81.3 %) of the as-prepared N-C@Co was much higher than that of the bulk Co (13.3%). This suggested that the N-C@Co has higher selectivity for CH4 generation, which is not due to its higheradsorption strength for CO2. However, it is still uncertain whether the adsorption strength between the N-C@Co and CO2 played a decisive role in the selectivephotocatalytic reduction of CO2 to CH4. To study this, we designed a referenceexperiment. As shown in Figure 2a, when H2 was used to replace H2O forphotocatalytic reduction of CO2, however, the CH4 selectivity and activity were onlyabout half. Therefore, this revealed that the adsorption strength ofCO2 on the N-C@Cosurface in our reaction system is not the main reason to determine the selectivity of CH4during the photocatalytic reduction CO2 process. The interaction between H2O andnitrogen-doped carbon layers in the sample of N-C@Co played a decisive role inachieving selective CH4 generation. To have a further understanding of this phenomenon, the in situ DRIFTS, DFTcalculation, and CO-TPD were conducted. Firstly, the in situ DRIFTS adsorptionexperiment ofCO2 with H20 over the as-prepared N-C@Co was conducted. As shownin Figure 7b and Figure S19, the peaks at 1686 cm-l, 1543 cm-l, 1425 cm, and 1283cm-, 1200 cm-l, and 1084 cmwere assigned to the bidentate carbonates (b-C03),monodentate carbonates (m-CO3²), bicarbonate (HCO3) species, formate ion species(HCOO),bicarbonate] (HCO3) speciess,,Jmonodentate carbonates (m-CO32-),respectively [54-58]. The peak at 1650 cm-l could be assigned to the adsorbed HzO [54].As shown in Figure 7a and Figure S19, the peak at around 3362 cm was assigned tothe intermolecular hydrogen bonds between the N-C@Co photocatalyst and H20molecule. Notably, there were no obvious CO2-related adsorbedd species iandintermolecular hydrogen bonds on the N-C@Co surface in the absence of H2O as shown in Figure S21. These results indicated that the H2O molecules adsorbed on the surfaceof N-C@Co by intermolecular hydrogen bond played a very important role in theadsorption and activation of CO2. This point of view was further proved by the DFTcalculation. As shown in Figure 6i, the adsorption energy of CO on the N-C@Cosurface was improved almost 1.6 times after the introduction of species activated byH20 molecules on the surface of N-C@Co. As a result, it could be concluded that theintermolecular hydrogen bonds could promote the adsorption and activation of CO2molecules. As shown in Figure 5d, it is noteworthy that there is little adsorption of CO over N-C@Co, when H20 molecules were not adsorbed on the N-C@Co surface. In strongcontrast, it could observe that the CO adsorption amount (39.9 umolg) on the surfaceof the as-prepared N-C@Co was significantly increased, when the H2O was adsorbedon the surface of N-C@Co by intermolecular hydrogen bond (Figure 5d). DFTcalculation showed that when the species produced by the activation of H20 moleculeswere adsorbed on the surface of N-C@Co, the adsorption intensity of CO on the N-C@Co surface increased by 1.6 times (Figure 6j-k). In addition, when H2 was usedinstead of H2O in the photocatalytic reduction of CO2 process over N-C@Co (Figure2a), the increased activity (from 17.0 umol g-l to 92.2 umol g-) and selectivity (from8.9 % to 52.2%) toward CO were observed. Therefore, it could deduce that the formedintermolecular hydrogen bond between H20 molecules and N-C@Co photocatalystmight achieve to suppress the desorption of the CO intermediates. More importantly, it is generally assumed that CO was recognized as the crucial intermediate for the CH4generation, and inhibiting the desorption of CO helps to improve the selectivity of CH4during the photocatalytic reduction CO2 process [1, 59, 60]. The formation of the carboxyl intermediate (COOH*) is generally regarded as therate-determining step of CO2 activation, generated by CO2*+H++e→COOH*[61]. Asshown in Figure 6f, and l-m, the activation energy for H reacting with H to H2(0.35eV) was higher than the H reacting with CO2 to HCOO (0.27 eV) on the surface of N-C@Co. However, it showed the opposite phenomenon on the cobalt surface, indicatingthat protons tended to bind to CO2 on the surface ofN-C@Co, while easier to bind withother protons on the surface of Co. As a result, it was concluded that the formedintermolecular hydrogen bonds between the H20 and the N-C@Co could induce theselective coupling of the protons and CO and inhibit the desorption of the crucial COintermediate. The in situ DRIFTS experiments under light irradiation were used to detect the CH4generation process, as shown in Figure 7c-d and Figure S19c-d. The peak of theintermolecular hydrogen bond on the N-C@Co surface after illumination showed adecreasing trend (Figure 7c and Figure S19c). It was indicated that the activating ofH20 molecules on the N-C@Co surface was achieved by intermolecular hydrogenbonds. As shown in Figure 7d and Figure S19d, the peak at 2069 cm and 1847 cmwere due to the vibration of CO* species, and the peak at 1090 cm and 1040 cmcorresponded to the H-C=0 bending vibration and C-O stretching mode of *CHO species and CHgO* species, respectively [62]. The observed CO, *CHO and CHO*species are crucial intermediates for the conversion of CO2 to CH4 and thecorresponding intensity was increased with the time. It should be noted that CH;O*species were also detected with the continuous increase of irradiation time, however,CH;O* species were formed later than that of CO* and HCO* under light irradiation.[62]. Therefore, we infer that the photocatalytic reduction CO2 with H2O to CH4pathway on the N-C@Co surface may be CO2→COOH*→CO*-HCO*-HCHO*→CH;0*→CH4. Similar CO2 conversion pathway has also been demonstrated inpreviously reported work [7, 63]. Journal Pre-( Figure 7. In situ DRIFTS of (a-b) CO2 and H2O adsorption process on the surface ofN-C@Co; (c-d) the CO2 and H2O interaction with N-C@Co under light irradiation. Overall, the crucial factor affecting H2 generation could be the adsorption andactivation of the H20 process on the photocatalyst surface and the mass transferpathway of the generated protons rather than the adsorption amount of H2O in ourreaction system. The CO2 adsorption amount and the photogenerated carrier separationefficiency were not the main reasons for high CH4 selectivity over N-C@Co, which justmainly help for improving the activity. The CH4 selectivity was mainly decided by the formed intermolecular hydrogen bonds between the H2O molecules and N-C@Coduring photocatalytic CO2 reduction. Because it could modulate the generation rate ofactivation H2O to protons but did not significantly reduce the production number ofprotons. More crucially, the mass transferability of the generated protons was obviouslysuppressed by the strong interaction between the protons and the N-C@Co surface. Andthe H20 molecules adsorbed on the surface of N-C@Co to form intermolecularhydrogen bonds could inhibit the desorption of the CO intermediate. Then, thegenerated protons were inclined to couple with CO2 to form CH4 rather than combinewith other protons to generate H2 or inducing the generation of CO (Scheme 1). As aresult, it could conclude that the formed intermolecular hydrogen bond between H20and N-C@Co could achieve the control of the selective coupling of protons and CO toCH4. Based on this concept, the N-C@Ni was also prepared (Figure S22). As shownin Figure S23, it could observe that the CH4 selectivity of N-C@Ni duringphotocatalytic reduction CO2 with H2O was boosted 3.4 times from 22.4 % of bulk Nito 76.4 % of N-C@Ni. This implied that our proposed intermolecular hydrogen bondinduced selective coupling of protons and CO2 to CH4 was also feasible on the as-prepared N-C@Ni. Scheme 1. The illustration of CO2 photoreduction with H20 to CH4 over N-C@Co. 4 Conclusion In summary, the as-fabricated N-C@Co photocatalyst using pyrolysis g-C3N4nanosheets and CoO for photoreduction CO2 with H2O showed significantly enhancedCH4 selectivity together with the suppressed H2 selectivity compared with the bulk Co.The hydrogen atoms of H20 were found to be fixed on the surface of N-C@Co viaintermolecular hydrogen bonds for the adsorption and activation of H2O and COmolecules. Then, intermolecular hydrogen bonds were demonstrated to be beneficialfor suppressing the mass transfer of generated protons to form H2 and suppress thedesorption of crucial CO intermediate, and then it could induce the selective couplingof the generated protons and CO2 molecules to form CH4. This work provided a newidea to design the photocatalyst for improving the selectivity of CH4 and suppressingthe H2 and CO generation in the process of photocatalytic reduction of CO2 with H2O. Declaration of Competing Interest The authors declare that they have no known competing financial interests orpersonal relationships that could have appeared to influence the work reported in this paper. Acknowledgments We acknowledge the financial supported by the Sichuan Provincial InternationalCooperation Project (No. 2019YFH0164; No. 2021YFH0055; 2022YFH0084), theCheung KongScholars PIrogramof China, the SScciieennccee rproject of SSWPU(No.2021JBGS10), Central Government Funds of Guiding Local Scientific andTechnological Development for Sichuan Province (No. 2021ZYD0035), and theGraduate Scientific Research Innovation Foundation of SWPU (No.2021CXYB32).We acknowledge the BL11B beamline of the Shanghai Synchrotron Radiation Facility(SSRF) and Table XAFS-500 (Specreation Instruments Co., Ltd.) for the provision ofinstruments. This work was partly supported by the Ministry of Science and HigherEducation of the Russian Federation under agreement No. 075-15-2020-931 within theframework of the development program for a world-class Research Center“Efficientdevelopment of the global liquid hydrocarbon reserves" Appendix A. Supplementary data Supplementary data associated with this article can be found in the online version. References [1] N.N. Vu, S. Kaliaguine, T.O. Do, Critical Aspects and Recent Advances inStructural Engineering of Photocatalysts for Sunlight-Driven Photocatalytic Reductionof CO2 into Fuels, Advanced Functional Materials,29 (2019)1901825. ( [2] Y. Wang, Y. Qu, B . Q u , L. Bai, Y . Liu, Z . -D. Yang, W. Zh a ng, L. J i n g, H. Fu,Construction of Six-Oxygen-Coordinated Single Ni Site s on g-C3N4 with Boron-OxoSpecies f or P hotocatalyticc Water-Activation-Induce d CO Reduction, A dvancedmaterials, (2021 ) 2105482. ) ( [3] J . Xiong, M. Zhang, M. Lu, K. Zhao, C. Han, G. Cheng, Z. Wen, Achievingsimultaneous Cu particles anchoring in meso-porous TiO2 nanofabrication for ) ( enhancing photo-catalytic C O2 r e duction t h rough rapid charge se p aration, ChineseChemical Letters, (2021) 10.1016/j.cclet.2021.1007.1052. ) ( [4] Y. Wang, W . Gao, K. Li, Y . Zheng, Z. X ie, W. Na, J.G. Chen, H . W ang, StrongEvidence of the Role of H2O i n A f fecting M ethanol S electivity from CO2Hydrogenation over Cu-ZnO-ZrO2, Chem, 6 (2020) 419-430. ) ( [5]Z. Wang, J. H ong, S.-F. Ng, W. Liu, J. Huang, P. Chen, W.-J. Ong, Recent Progressof Perovskite Oxide in Emerging Photocatalysis L andscape: Water Splitting, CO2Reduction, and N2 Fixation, Acta Physico Chimica Sinica, 0 (2020) 2011033-2011030. ) ( [6] J.D. Xiao, H .L. Jiang, M etal-Organic F rameworks for P h otocatalysis a ndPhotothermal Catalysis, Acc C hem Res, 52 (2019) 356-366. ) ( [7]X. Li, Y. Sun, J. Xu, Y. Shao,J. Wu, X. Xu, Y. P a n, H . J u , J. Zhu, Y . X ie, Selectivevisible-light-driven photocatalytic CO2 reduction to CH4 m e diated by atomically thinCuIn5S8 layers, Nature Energy, 4 (2019) 690-699. ) ( [8]Y. Cao, L . Guo,M. Dan,D.E. Doronkin, C. Han, Z. Rao, Y. Liu, J. Meng,Z. Huang,K. Zheng, P. Chen, F. Dong, Y. Zhou, Modulating electron density of vacancy site bysingle Au atom for effective CO2 photoreduction, Nature Communications, 1 2 ( 2 021) 1675. ) ( [9] J.W. F u, J.G. Y u, C .J. Jiang, B . Cheng, g - C3N4-Based HeterostructuredPhotocatalysts, Advanced Energy Materials, 8 (2018) 1701503. ) ( [10] Y. Ma, Q. Tang, W .-Y. Sun, Z.-Y. Y ao, W. Zhu, T . L i , J. Wang, Assemblingultrafine TiO2 nanoparticles on UiO-66 octahedrons to promote selective photocatalyticconversion of CO2 to CH4 at a low concentration, Applied Catalysis B: Environmental,270 (2020)118856. ) [11]A. Li, Q. Cao, G. Zhou, B. Schmidt, W. Zhu, X. Yuan, H. Huo, J. Gong, M.Antonietti, Three-Phase Photocatalysis for the Enhanced Selectivity and Activity ofCO2 Reduction on a Hydrophobic Surface, Angewandte Chemie, 58 (2019) 14549-14555. ( [12]K. Wang, L . Zhang, Y. Su, D. Shao, S. Zeng, W. Wang, Photoreduction of carbondioxide of atmospheric c oncentration to methane with water over CoAl-layered doublehydroxide nanosheets, Journal of Materials Chemistry A, 6 (2018) 8366-8373. ) ( [13] W. Zhang, L. Wang, H. Liu, Y. Hao, H. Li, M .U. Khan, J . Zeng, Integration ofQuantum C o nfinement and Alloy Effect to Modulate Electronic Properties of RhWNanocrystals for Improved Catalytic Performance toward C O2 Hydrogenation, N a noLetters, 17 (2017)788-793. ) ( [14]R. Long, Y . L i, Y. Liu, S. Chen, X. Zheng,C. Gao, C. He, N. Chen,Z. Qi, L. Song,J. Jiang , J. Zhu, Y . Xiong , Isolati o n of Cu Atoms in Pd Lattice : Forming HighlySelective Sites for Photocatalytic Conversion of CO to CH4, J Am Chem Soc, 139(2017)4486-4492. ) ( [15] J. Wang, E. Kim, D.P. Kumar, A.P. Rangappa, Y. K im, Y. Z h ang, T.K. Kim,Highly Durable a nd F ully Dispersed C obalt Diatomic Site Catalysts for CO2Photoreduction to CH4, Angewandte Chemie, 61 ( 2022) e202113044. ) ( [16] R. Cheng, C.C. Chung, S. Wang,B. C ao, M. Zhang, C. Chen, Z. Wang, M. Chen, ) ( S. Shen, S.P. Feng, Three-dimensional self-attaching perovskite quantum dots/polymerplatform for efficient solar-driven CO2 reduction, Materials Today Physics, 1 7 (2021). ) ( [17] P. Liu, Z. Huang, X. Gao, X. Hong, J. Zhu, G. W ang, Y. W u ,J. Zeng, X. Zheng,Synergy Between Palladium Single Atoms and Nanoparticles Via Hydrogen Spilloverfor Enhancing CO2 Photoreduction to CH4, Adv.Mater., n/a (2022)2200057. ) ( [18] H. Yoshida, L. Zhang, M. Sato, T. Morikawa, T. K ajino, T. Sekito, S. Matsumoto,H. H irata, Calcium titanate photocatalyst prepared by a flux m e thod for reduction ofcarbon dioxide with water, Catalysis Today, 251 (2015) 132-139. ) ( [19] N.M. Dimitrijevic, B.K. Vijayan, O.G. Poluektov, T. Rajh, K.A. Gray, H. He, P.Zapol, Role of W ater and Carbonates i n Photocatalytic Transformation of CO2toCH4on Titania, Journal ofthe A merican C h emical Society, 133 (2011)3964-3971. ) ( [20] W. Ma, S. Xie, X .G. Zhang, F. Sun, J . Kang, Z. Jiang, Q. Z hang, D . Y. Wu, Y.Wang, P r omoting electrocatalytic CO2 reduction to formate via sulfur-boosting wateractivation on indium surfaces, Nat Commun, 1 0 (2019) 892. ) ( [21] Y. Yu, X. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Z h ou, F. Dong, SynergisticEffect of Cu Single Atoms and Au-Cu Alloy Nanoparticles on TiO for Efficient CO2Photoreduction, ACS nano, 15 (2021) 1 4453-14464. ) ( [22]T. Fan, H. L iu, S. Shao, Y.Gong,G. Li, Z. Tang, Cobalt Catalysts Enable SelectiveHydrogenation of CO2 toward Diverse Products: Recent Progress and Pe r spective, Th e Journal of Physical Chemistry Letters, (2021) 10486-10496. ) ( [23]N.Elgrishi, M.B. Chambers, M. Fontecave, Turning it off! Disfavouring hydrogenevolution to enhance selectivity for CO production during homogeneous CO2 reductionby cobalt-terpyridine complexes, Chem Sci, 6 (2015)2522-2531 . ) ( [24] L. Wang, W . Zhang,X. Zheng, Y. Chen, W. Wu, J . Qiu, X. Zhao, X. Zhao, Y. Dai,J. Zeng, I ncorporating nitrogen a t oms in t o cobalt na n osheets as a s t r a tegy to b oostcatalytic activity toward CO2 hydrogenation, Nature Energy, 2 (2017)869-876. ) ( [25] S. Wannakao, W . Jumpathong, K. Kongpatpanich, Ta i loring MetalloporphyrinFrameworks for an Efficient Carbon Dioxide Electroreduction: Selectively StabilizingKey Intermediates with H-Bonding Pockets, Inorg Chem, 56 (2017)7200-7209. ) ( [26] S. Roy, B. Sharma, J. Pecaut, P. Simon, M . Fontecave, P.D. Tran, E. Derat, V .Artero, Molecular Cobalt: Complexes with Pendan t IAmines for Selective Electrocatalytic Reduction of Carbon Dioxide to Formic Acid, Journal of the AmericanChemical Society, 139 (2017) 3685-3696. ) ( [27]M. Ma, Z. Huang, D.E. Doronkin, W. Fa, Z. Rao, Y. Zou, R. Wang, Y. Zhong, Y.Cao, R . Zhang, Y. Z h ou, Ul t rahigh surface density of Co-N2C single-atom-sites forboosting photocatalytic CO2 r eduction to m ethanol, Applied Catalysis B: Environmental, 300 (2022) 1 20695. ) [28] B. Ravel, M. Newville, ATHENA, ARTEMIS,HEPHAESTUS: data analysis forX-ray absorption spectroscopy using IFEFFIT, J Synchrotron Radiat, 12 (2005) 537-541. ( [29] Z.M. Xia, H. Zhang, K . C . Shen, Y.Q. Qu, Z. Jiang, Wa v elet analysis of extendedX-ray absorption fine structure data: Theory, application, Physica B, 542 (2018) 12-19. ) ( [30] B. Tan, Y. Ye, Z. Huang, L. Ye, M. Ma,Y. Z hou, P r omotion of photocatalyticsteam reforming of methane over Ag/Ag-SrTiO3, Chinese Chemical Letters, 31(2020) 1530-1534. ) ( [31]Y. Cao, R. Zhang, T . Zhou, S. Jin, J. H uang,L. Ye, Z. Huang, F. Wang, Y. Z hou,B-O Bonds in Ultrathin B o ron Ni t ride Nanosheets to Promote Photocatalytic CarbonDioxide Conversion, ACS applied materials & interfaces, 12 (2020) 9935-9943. ) ( [32] Z. Rao, Y. Cao, Z. Huang, Z. Yin, W. Wan,M. Ma, Y. Wu, J. Wang, G. Yang, Y.Cui, Z. Gong, Y. Zhou, Insights into the Nonthermal Effects of Light in Dry Reformingof Methane to Enhance the H2/CO R atio Near Unity over Ni/Ga2O3, ACS Catalysis, 11 (2021)4730-4738. ) ( [33]G. Kresse, D . Joubert,From ultrasoft pseudopotentials to the projector augmented-wave method, Physical Review B, 59 (1999) 1758-1775. ) ( [34] G. K resse, J. Furthmiiller, Efficient iterative schemes for ab in i tio t o tal-energycalculations using a plane-wave basis s e t, Physical Review B, 54 (1996) 11169-11186. ) ( [35] D. Lerch, O. Wieckhorst, G.L.W. Hart, R.W. Forcade, S. M -l l er, UNCLE: a code for constructing cluster expansions for arbitrary lattices with minimal user-input,Modelling and Simulation i n Materials Science and Engineering, 17 (2009) 055003. ) ( [36]Y. Le Page,P. Saxe, Symmetry-general least-squares extraction of elastic data forstrained materials from ab initio c alculations of stress, Physical Review B, 65 (2 0 02) 1104104. ) ( [37] S. Ning, H. Xu, Y . Q i, L. S ong, Q. Zhang, S. Ouyang, J. Y e , M i crostructureInduced Thermodynamic and K i netic Modulation to Enhance C O 2 Ph o tothermalReduction: A Case of Atomic-Scale Dispersed Co-N Species Anchored Co@C Hybrid,ACS Catalysis, 10 (2020) 4726-4736. ) [38] D. Zhao, Y. Zhu, Q. Wu, W. Zhou, J. Dan, H. Zhu, W. Lei, L.-J. Ma, L. Li, Low-loading Ir decorated cobalt encapsulated N-doped carbon nanotubes/porous carbonsheet implements efficient hydrogen/oxygen trifunctional electrocatalysis, ChemicalEngineering Journal, 430 (2022). [39] K. Feng, S. Wang, D. Zhang, L. Wang, Y. Yu, K. Feng, Z. Li, z. Zhu, C. Li, M.Cai, Z. Wu, N. Kong, B. Yan,J. Zhong, X. Zhang, G.A. Ozin, L. He, Cobalt PlasmonicSuperstructures Enable Almost 100% Broadband Photon Efficient CO2 Photocatalysis,Advanced materials, 32 (2020)e2000014. [40] P. Huang, J. Huang, S.A. Pantovich, A.D. Carl, T.G. Fenton, C.A. Caputo, R.L.Grimm, A.I. Frenkel, G. Li, Selective CO2 Reduction Catalyzed by Single Cobalt Siteson Carbon Nitride under Visible-Light Irradiation, J Am Chem Soc, 140 (2018) 16042-16047. ( [41] R. Wang, H. Xu, K. Zhang, S . Wei, W. D eyong, High-quality Al@Fe-MOFprepared using Fe-MOF a s a m i cro-reactor to i mprove adsorption p erformance for selenite, Journal of hazardous materials, 364 (2019)272-280. ) ( [42] R.Y. Zhang, P .H. L i, F. Wang, L.Q. Ye, A . Gaur,Z.A. Huang, Z.Y. Zhao, Y. Bai, Y . Zhou, Atomically dispersed Mo atoms on amorphous g-C3N4 promotes visible-lightabsorption and charge carriers transfer, Appl Catal B-Environ, 250 (2019)273-279. ) ( [43] L. Jiang , K. Wang, X. Wu, G. Zhang, Highly Enhanced F ull Solar Spectrum -Driven P hotocatalytic CO2 Reduction Performance in Cu2-xS/g - C 3 N4 Composite:Efficient Charge Transfer and Mechanism Insight, Solar RRL, 5 (2020) 2000326. ) ( [44] J. Barrio , D . Mateo, J. Albero, H. Garcia , M. Shalom, A Heterogeneous CarbonNitride- N ickel P hotocatalyst for Efficient Low - T emperature C O2 M e thanation,Advanced Energy Materials, 9 (2019) 1902738. ) ( [45] S. Kim, J.-M. Kim, J.-E . Park, J.- M . Nam , Nonnoble-Metal-Based PlasmonicNanomaterials: Recent A dvances a n d F u ture Perspectives, A dvanced materials, 30(2018)1704528. ) ( [46] Z. Kaminskiene, I. P rosycevas, J. S tonkute, A . G uobiene, Evaluation of OpticalProperties of Ag, Cu, and Co Nanoparticles Synthesized in Organic Medium, Acta PhysPol A, 123 (2013)111-114. ) ( [47] Y .C. Jiang,J.F. Wang, J. Gao, Giant photoconductivity induced by plasmonic Conanoparticles in Co-doped amorphous car b on/silicon het e rostructures, Ca r bon, 72(2014) 106-113. ) ( [48] O. Yeshchenko,I. Dmitruk, A. A lexeenko, A. D mytruk, V. Tinkov, O pticalproperties of sol-gel fabricated Co/SiO2 nanocomposites , Physica E: Low-dimensionalSystems and Nanostructures, 41 (2008)60-65. ) ( [49] J. Fu, B . Zhu, C. Jiang, B. Cheng , W. You, J. Yu, Hierarchical Porou s O-Dopedg-C3N4 with Enhanced Photocatalytic CO2 Reduction Activity, Small, 13 ( 2 017). ) ( [50] D. Pakhare, J. Spivey , A review of dry (CO2) reforming of methane over noblemetal catalysts,Chemical S ociety reviews, 43 (2014) 7813-7837. ) ( [51 ] S.N. Talapaneni, G. Singh, I.Y. Kim, K. AlBahily, A.H. Al-Muhtaseb, A .S.Karakoti, E. Tavakkoli, A. V inu, Nanostructured Carbon Nitrides for CO2 Capture andConversion, A dvanced materials, (2019) e1904635. ) ( [52] T.H. Tan, B. Xie, Y.H. Ng, S .F.B. A bdullah, H .Y.M. Tang, N. Bedford, R.A.Taylor, K.-F. Aguey-Zinsou, R. Am a l, J. S cott, Unlocking the potential of t he formatepathway in the photo-assisted Sabatier reaction, Nature Catalysis, 3 (2020)1034-1043.[53] C.H. Li, X. Tong, P. Yu, W. Du, J. Wu, H. Rao, Z.M.M. Wang, Carbon dioxidephoto/electroreduction with cobalt, Journal of Materials Chemistry A, 7 (2019)16622- 116642. ) ( [54] L. Liu, H. Zhao, J.M. A ndino, Y. L i , Photocatalytic CO2 Reduction with H2O onTiO, N anocrystals: C omparison of Anatase, Rutile, and B r ookite P o lymorphs andExploration of Surface Chemistry, ACS Catalysis,2(2012)1817-1828. ) ( [55] C. Han, R. Zhang, Y. Ye, L. Wang, Z. Ma, F. Su, H. Xie, Y. Zhou, P.K. Wo n g, L.Ye, Chainmail co-catalyst of NiO shell-encapsulated Ni for improving photocatalyticCO2 reduction over g-C3N4, Journal of Materials Chemistry A, 7 ( 2019) 9726-9735. ) ( [56] M.M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika,F. Girgsdies,D. Teschner,F. Seitz, A. Tarasov, S.V. Levchenko, R. Schlogl, E. F r ei, Ni Single Atom Catalysts forCO2 Activation, J Am Chem Soc, 141 (2019) 2451-2461. ) ( [57] S. Neatu, J.A. Macia-Agullo, P. Concepcion, H . Garcia, Gold-copper nanoalloyssupported on TiOz as photocatalysts for CO2reduction by water, J Am Chem Soc, 136 ) ( (2014) 1 5969-15976. ) ( [58] N. Ojha, A. Bajpai, S. Kumar, Visible light-driven enhanced CO2 reduction bywater over Cu modified S-doped g-C3N4, Catalysis S c ience & Te c hnology, 9 (2019) 4598-4613. ) ( [59] Y. Fang, X. Wang, Photocatalytic CO2 conversion by polymeric carbon nitrides,Chemical communications, 54 (2018) 5674-5687. ) ( [60] S.N. Habisreutinger, L. Schmidt-Mende,J.K. Stolarczyk, Photocatalytic reductionof CO2 on TiO2 and other semiconductors, Angewandte Chemie, 52 (2013)7372-7408. ) [61] P. Chen, B. Lei, X. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang, F.Dong, Rare-Earth Single-Atom La-N Charge-Transfer Bridge on Carbon Nitride forHighly Efficient and Selective Photocatalytic CO2 Reduction, ACS nano, 14 (2020)15841-15852. [62]X. Jiang, J. Huang, Z. Bi, W. Ni, G. Gurzadyan, Y. Zhu, Z. Zhang, PlasmonicActive "Hot Spots"-Confined Photocatalytic CO2 Reduction with High Selectivity forCH4 Production, Advanced materials, n/a (2022)e2109330. ( [63] P. Liu, Z. Huang, X. Gao, X. Hong, J. Zhu, G. W ang, Y. W u , J. Zeng, X. Zheng,Synergy Between P a lladium Single Atoms and Nanoparticles Via Hydrogen Spilloverfor Enhancing CO2 Photoreduction to CH4, Advanced materials, n/a2200057. ) Highlights: Intermolecular hydrogen bond was formed between N-C@Co and H2O molecule Intermolecular hydrogen bond induced selective coupling of protons and CO ●N-C@Co with intermolecular hydrogen bond suppressed the desorption of CO The selective CH4 generation from CO2 was achieved over N-C@Co photocatalyst Conflict of interest The authors declare that they have no conflict of interest. Pre-proofsnal Jour 光催化H2O还原CO2还原为CH4是实现“碳中和”的有效途径之一。该反应中由于质子与CO2的耦合同质子与质子间的耦合存在着剧烈的竞争,最终导致产物中CH4的选择性比较低。西南石油大学周莹教授课题组设计了氮掺杂碳包覆金属钴(N-C@Co)光催化剂,以提高光催化还原CO2制CH4的选择性。理论计算和实验表征分析表明在N-C@Co和H2O之间形成分子间氢键,有效抑制了生成的质子的迁移,促进了CO2分子的吸附和活化,并且有效抑制CO中间体的脱附(CO中间体通常被认为是生成CH4的关键物种)。这使得在光催化还原CO2的过程中质子可以选择性的与CO2耦合为CH4。最终,N-C@Co所对应的H2的选择性和活性相对于体相Co而言分别降低了17.6倍和9.3倍。而N-C@Co所对应的CH4的选择性和活性相对于体相Co而言分别提升了6.1倍和3.2倍。这项工作为设计出具有高CH4选择性的光催化材料提供了思路。其中X射线精细结构光谱(TableXAFS-500,安徽创谱仪器科技有限公司)对Co局域电子结构的判断是催化机理分析的重要依据。要点摘要:1. H2O分子和N-C@Co表面之间形成的分子间氢键可以调控H2O分子活化后生成质子的速度以及质子的质量迁移。2. 分子间氢键可以促进CO2分子的吸附和活化,并且还能够实现抑制CO中间体的脱附。最终实现质子选择性的与CO2分子耦合为CH4。3. 通过XAS(TableXAFS-500,安徽创谱仪器科技有限公司)研究N-C@Co上CO2选择性光催化还原为CH4的机理。根据N-C@Co的XAS谱图获取Co原子的局域电子结构信息分析表明,氮掺杂碳层和金属钴之间通过Co-C键紧密的结合到一起。并且Co-C键作为电子传输通道促进了光生电子和光生空穴的分离效率,进而提高了样品N-C@Co表面的光生电子浓度。样品N-C@Co以及对应的参比样品的XAS图谱(a)Co K-edge XANES 光谱;(b)N-C@Co的FT k3加权 EXAFS 光谱;(c)N-C@Co 的R-空间,q-空间拟合FT EXAFS光谱结果;(d)k-空间拟合结果;(e)N-C@Co以及所对应的参比样品的小波变换数据(WT)。
确定





































还剩41页未读,是否继续阅读?

产品配置单

安徽创谱仪器科技有限公司为您提供《含Co催化剂中结构分析检测方案(X射线吸收精细结构(XAFS))》,该方案主要用于其他中其他检测,参考标准--,《含Co催化剂中结构分析检测方案(X射线吸收精细结构(XAFS))》用到的仪器有桌面式X射线吸收精细结构谱仪(XAFS)
推荐专场





相关方案
更多