








方案详情
文
布鲁克在ASMS 2020发布了高通量dia-PASEF@方案,即将Evosep One LC与timsTOF Pro再次联合,最大程度发挥Evosep One LC快速分离和timsTOF Pro扫描速度和稳定性的优势。英国牛津大学Roman Fischer教授采用高通量dia-PASEF@方案,进行血液蛋白质组学大队列研究,43天完成4300针连续进样,总共采集2.5亿张二级谱图,整个采集过程只需一次离子传输管清洗。
方案详情

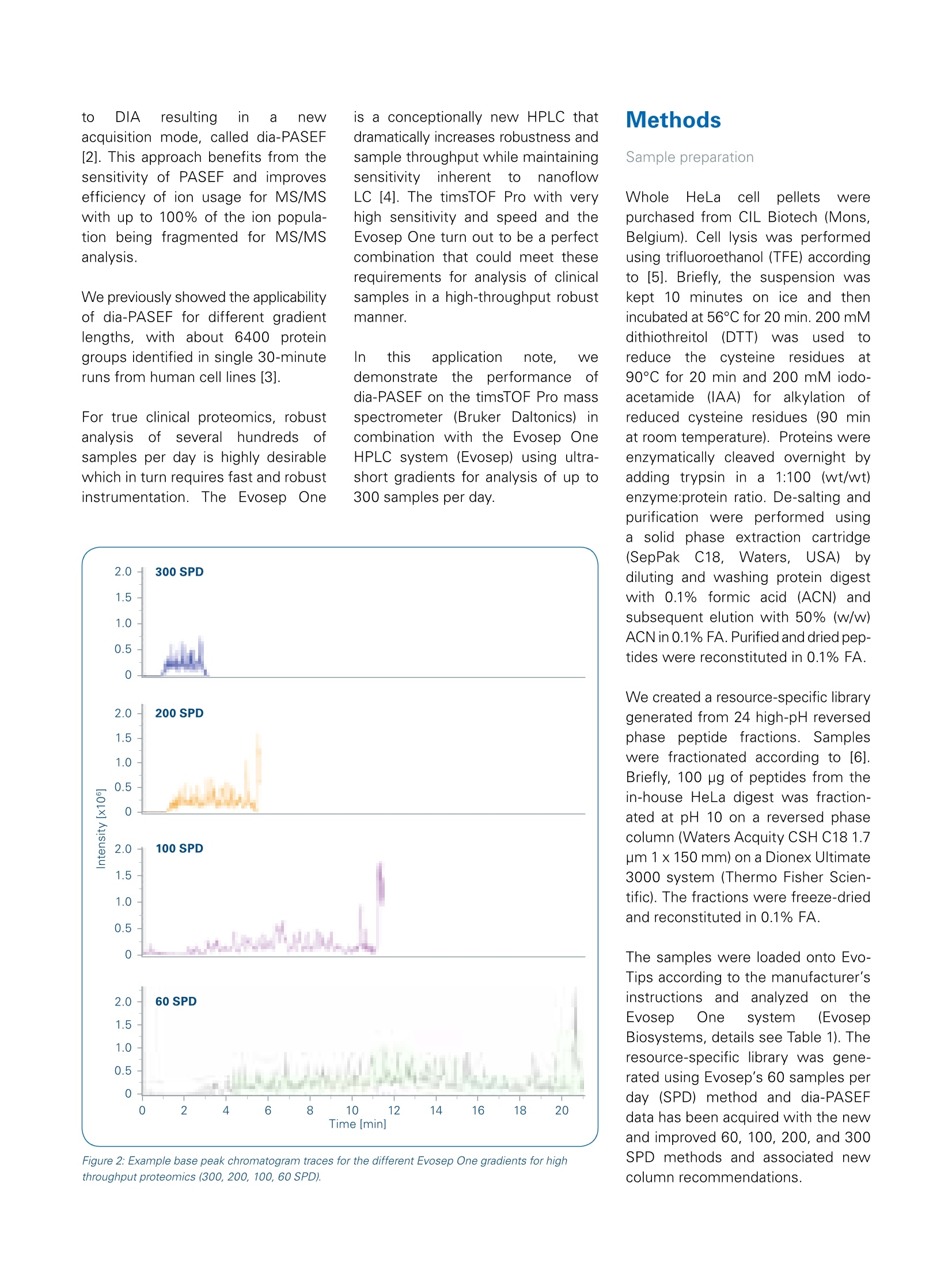
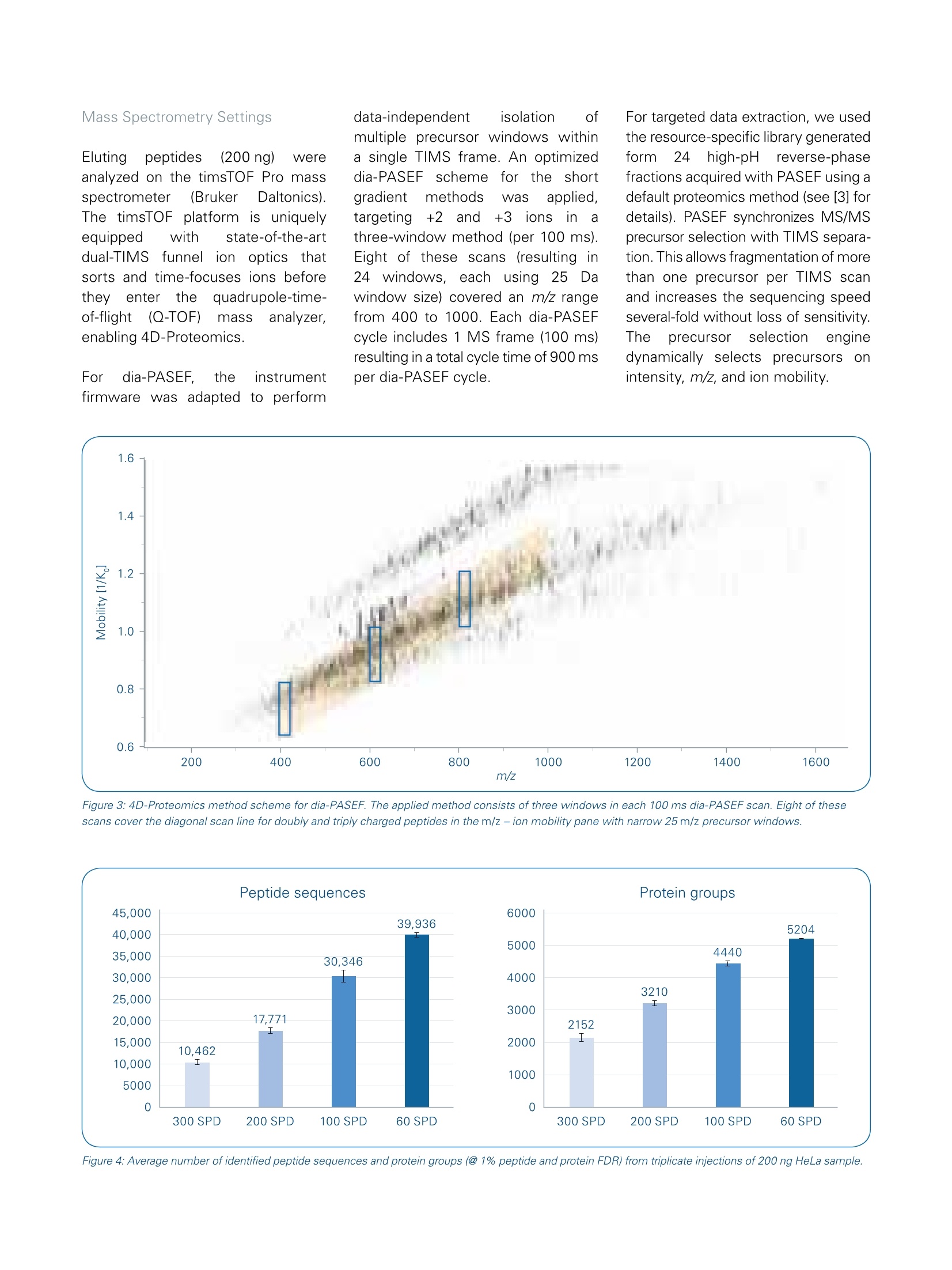
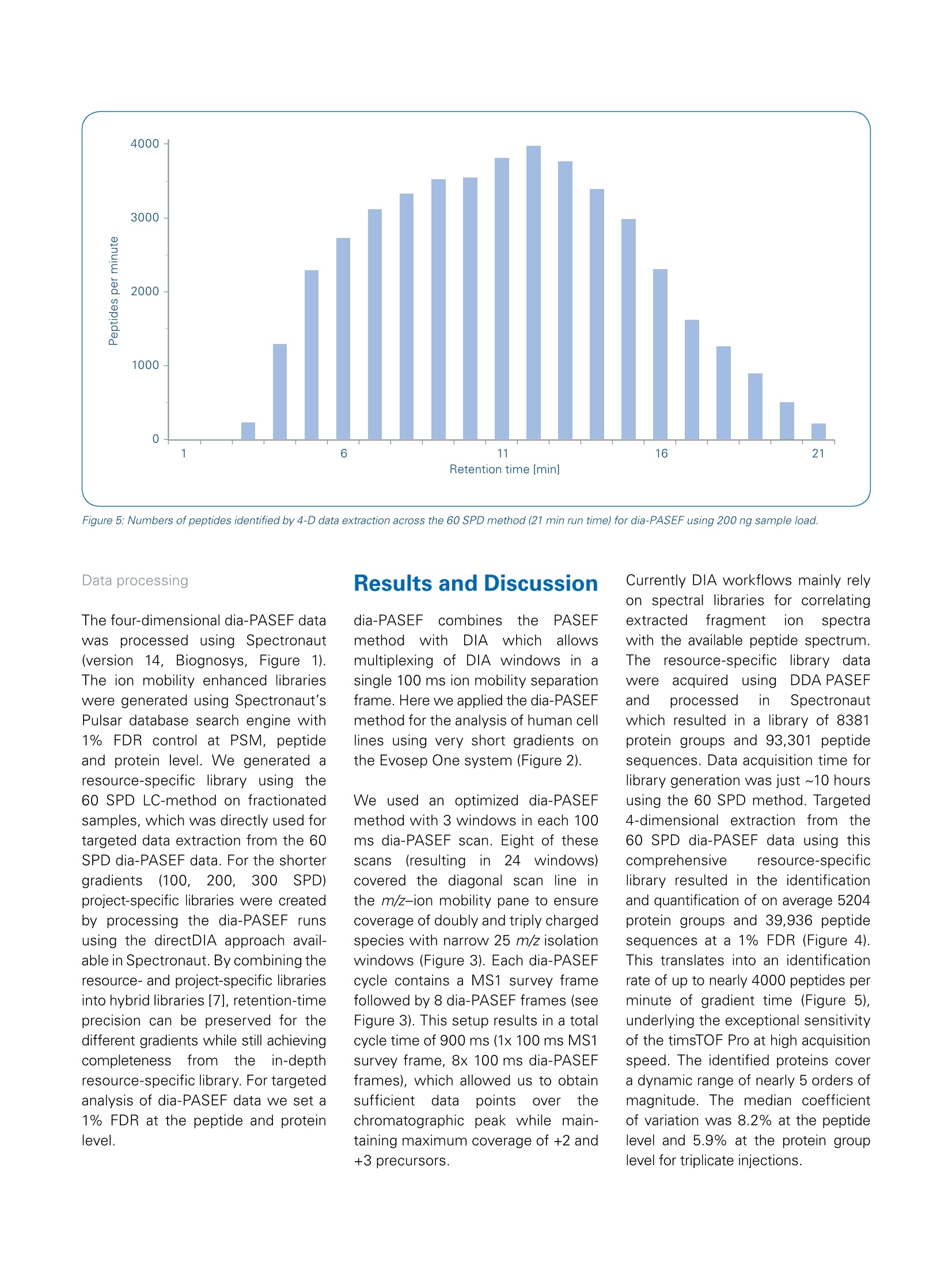
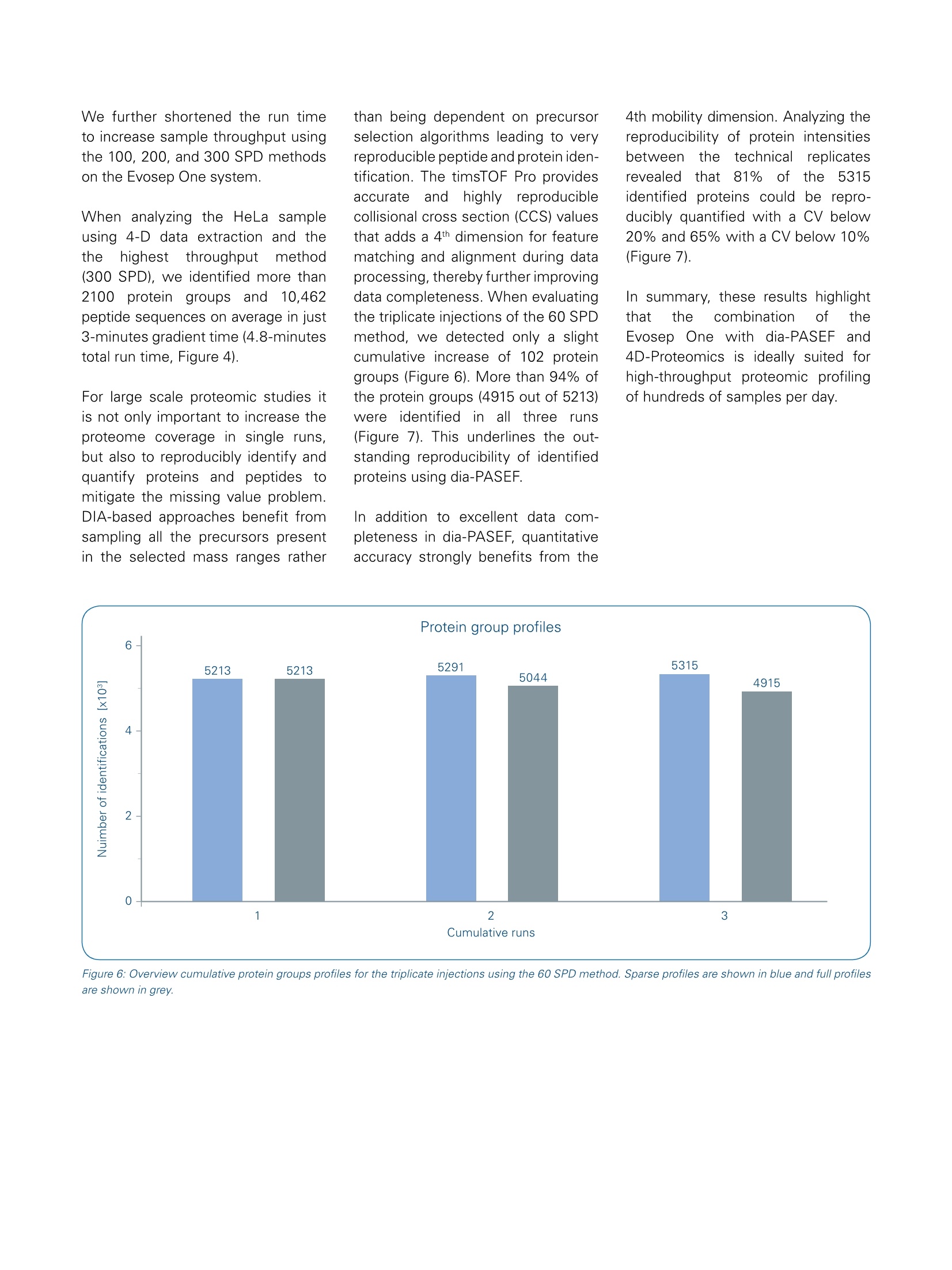
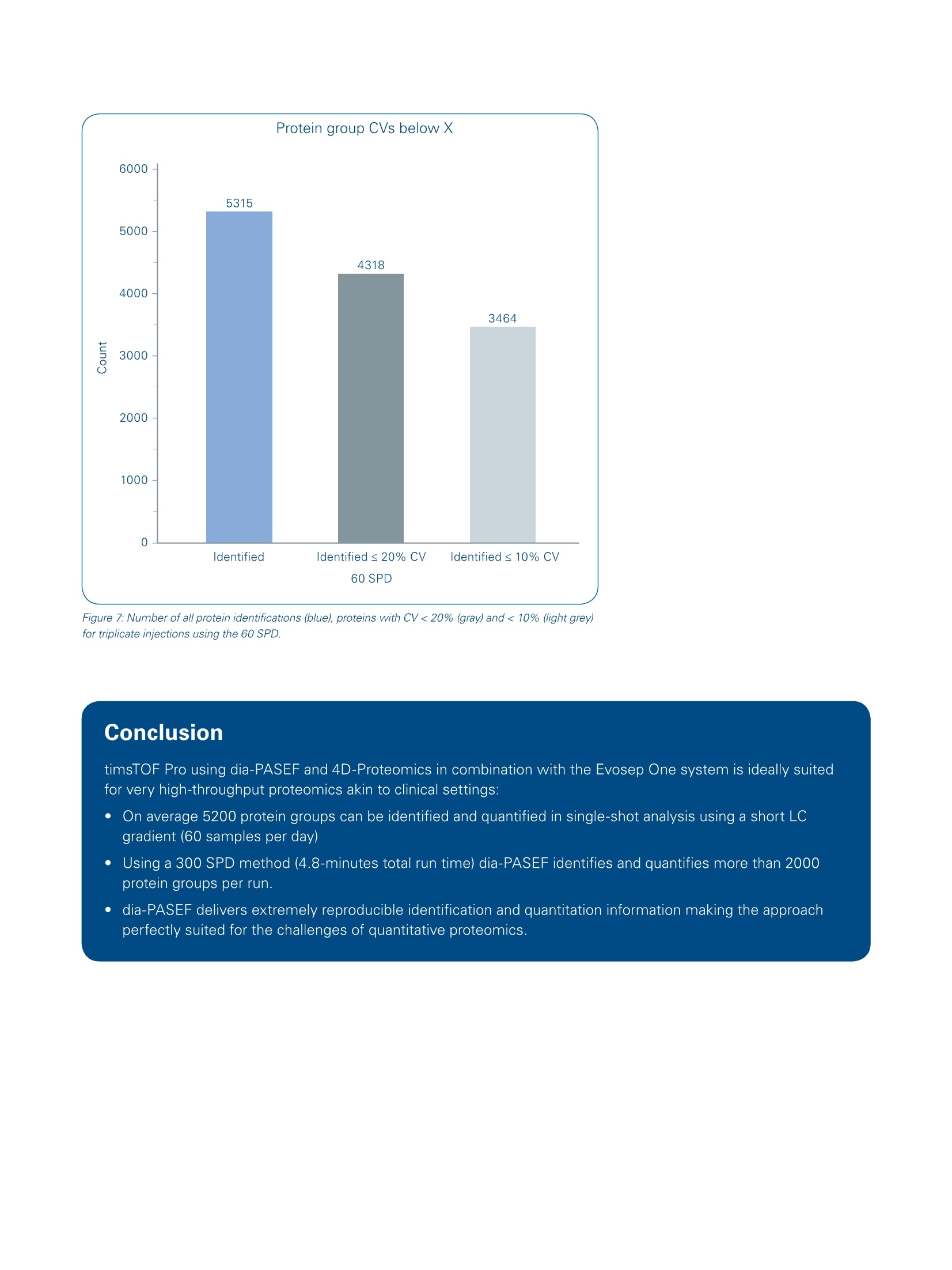
Mass Spectrometry Settings High throughput 4D-Proteomics -Application ofdia-PASEFQ;and the Evosep One for short gradients The timsTOF Pro offers a combination of two unique technologies, namely a 4thdimension provided by Trapped Ion Mobility Spectrometry (TIMS) to enhance ionseparation and sensitivity and Parallel Accumulation Serial Fragmentation (PASEF [1])to improve ion utilization efficiency and data acquisition speed. Abstract nthissapplicationnote Wedemonstrate theebenefitscofdia-PASEF technology onthetimsTOF Pro platform coupled to an Evosep One for high-through-put in-depth proteome analysisof up to 300 samples per day(SPD). We quantify about5200 protein groups in only 21-minutes run time. In 4.8-minutes ultra-high throughput runs(300.arSPD) we are still able to quantifymore than 2000 protein groupsand 8500 peptides. ( A uth ors : S tep ha n i e K as par-Sc h oe ne fe l d 1 ,T h o m a s Kosinskil, M ar ku s L u be ck 1, Ol iv e r Ra t h e r ' , G a ry Kru p p a1 , Ni co l a i Ba c h e ?, D o r t e B . B e k k e r-J e nse n ; B ruk er D al t on ik G mb H , Br e men, Germa n y ;2E v os ep B iosy s t e ms , O d e ns e , D a n m ar k . ) Figure 1: dia-PASEF data processing workflow using Spectronaut (v14). A resource-specific library was created from 24 fractions acquired using the 60 SPDEvosep method in combination with PASEF technology. Additionally, project-specific libraries were created for the 100, 200 and 300 SPD methods usingdia-PASEF data to allow for source-specific retention time alignment. Both libraries have been combined into "Hybrid Libraries" used for targeted dataextraction from the dia-PASEF data. Data-independent acquisition (DIA)facilitates reproducible and accurateproteinidentification and quantifi-cation across large sample cohorts.This is achieved by using isolationof wide quadrupole windows, ratherthan selecting individual precursors,to ensure that all pprecursor ionsare fragmentedineevery sample.Ion mobility provides an additionaldimension of separation for complexsamples, which can also be usedfor aligning precursor and fragmentions. By making use of reproduciblemobility values from the timsTOF Pro,Wee extenddtlthePASEFprinciple to DIA resulting in newacquisition mode, called dia-PASEF[2].This approach benefits from thesensitivity of PASEF and improvesefficiency of ion usage for MS/MSwith up to 100% of the ion popula-tion being fragmented for MS/MSanalysIs. We previously showed the applicabilityof dia-PASEF for different gradientlengths, with about 6400 proteingroups identified in single 30-minuteruns from human cell lines [3]. For true clinical proteomics, robustanalysiscofseveral hundredsofsamples per day is highly desirablewhich in turn requires fast and robustinstrumentation.The Evosep One is a conceptionally new HPLC thatdramatically increases robustness andsample throughput while maintainingsensitivityinherent to nanoflowLC [4]. The timsTOF Pro with veryhigh sensitivity and speed and theEvosep One turn out to be a perfectcombination that could meet theserequirements for analysis of clinicalsamples in a high-throughput robustmanner. In this application note, Wedemonstrate the performanceofdia-PASEF on the timsTOF Pro massspectrometer (Bruker Daltonics)incombination with the Evosep OneHPLC system (Evosep) using ultra-short gradients for analysis of up to300 samples per day. Sample preparation WholeetHeLacellpellets werepurchased from CIL Biotech (Mons,Belgium). Cell lysis was performedusing trifluoroethanol (TFE) accordingto [5]. Briefly, the suspension waskept 10 minutes on ice and thenincubated at 56°C for 20 min. 200 mMdithiothreitol([(DTT)waslSused toreduce the cysteine residuesa90°C for 20 min and 200 mM iodo-acetamide(IAA) for alkylation ofreduced cysteine residues (90 minat room temperature). Proteins wereenzymatically cleaved overnight byadding trypsin in a1:100 (wt/wt)enzyme:protein ratio. De-salting andpurification were performed usinga solid phase extraction cartridge(SepPak C18,Waters, USA)bydiluting and washing protein digestwith 0.1% formic acid (ACN) andsubsequent elution with 50% (w/w)ACN in 0.1% FA. Purified and dried pep-tides were reconstituted in 0.1% FA. We created a resource-specific librarygenerated from 24 high-pH reversedphase peptide fractions. Sampleswere fractionated according to [6].Briefly, 100 pg of peptides from thein-house HeLa digest was fraction-ated at pH 10 on a reversed phasecolumn (Waters Acquity CSH C18 1.7um 1 x150mm) on a Dionex Ultimate3000 system (Thermo Fisher Scien-tific). The fractions were freeze-driedand reconstituted in 0.1% FA. The samples were loaded onto Evo-Tips according to the manufacturer’sinstructions; and analyzedonntheEvosep One system (EvosepBiosystems, details see Table 1). Theresource-specific library was gene-rated using Evosep's 60 samples perday (SPD) method and dia-PASEFdata has been acquired with the newand improved 60, 100, 200, and 300SPD methods and associated newcolumn recommendations. Elutingpeptides (200 ng) Wereanalyzed on the timsTOF Pro massspectrometer (Bruker Daltonics).The timsTOF platform is uniquelyequipped with state-of-the-artdual-TIMS 1funneleliionoptics tthhatsorts and time-focuses ions beforetheyy enterttlhe quadrupole-time-of-flighitt(Q-TOF) mass analyzer,enabling 4D-Proteomics. For dia-PASEF, the instrumentfirmware was adapted to perform data-independent isolation ofmultiple precursor windows withina single TIMS frame. An optimizeddia-PASEF scheme for the shortgradient methods Was applied,targeting +2and ++3ions inathree-window method (per 100 ms).Eight of these scans (resulting in24windows, each using 255 Dawindow size) covered an m/z rangefrom 400 to 1000. Each dia-PASEFcycle includes 1 MS frame (100 ms)resulting in a total cycle time of 900 msper dia-PASEF cycle. For targeted data extraction, we usedthe resource-specific library generatedtorm24 high-pH reverse-phasefractions acquired with PASEF using adefault proteomics method (see [3] fordetails). PASEF synchronizes MS/MSprecursor selection with TIMS separa-tion. This allows fragmentation of morethan one precursor per TIMS scanand increases the sequencing speedseveral-fold without loss of sensitivity.The precursorselectionenginedynamically selects precursors onintensity, m/z, and ion mobility. Figure 3: 4D-Proteomics method scheme for dia-PASEF. The applied method consists of three windows in each 100 ms dia-PASEF scan. Eight of thesescans cover the diagonal scan line for doubly and triply charged peptides in the m/z -ion mobility pane with narrow 25 m/z precursor windows. Figure 4: Average number of identified peptide sequences and protein groups (@ 1% peptide and protein FDR) from triplicate injections of 200 ng HeLa sample. Figure 5: Numbers of peptides identified by 4-D data extraction across the 60 SPD method (21 min run time) for dia-PASEF using 200 ng sample load. The four-dimensional dia-PASEF datawass processedusing Spectronaut(versionn114, Biognosys, Figure 1).The ion mobility enhanced librarieswere generated using Spectronaut’sPulsar database search engine with1%FDR control at PSM, peptideand protein level. We generated aresource-specific library using the60 SPD LC-method on fractionatedsamples, which was directly used fortargeted data extraction from the 60SPD dia-PASEF data. For the shortergradients (100,200,:300SPD)project-specific libraries were createdby processing the dia-PASEF runsusing the directDIA approach avail-able in Spectronaut. By combining theresource- and project-specific librariesinto hybrid libraries [7], retention-timeprecision can be preserved for thedifferent gradients while still achievingcompleteness trom the in-depthresource-specific library. For targetedanalysis of dia-PASEF data we set a1% FDR at the peptide and proteinlevel. Results and Discussion dia-PASEF(combinestheeFPASEFmethodwith DIA whichallowsmultiplexing of DIA windows in asingle 100 ms ion mobility separationframe. Here we applied the dia-PASEFmethod for the analysis of human celllines using very short gradients onthe Evosep One system (Figure 2). We used an optimized dia-PASEFmethod with 3 windows in each 100ms dia-PASEF scan. Eight of thesescans (resultinginn224 windows)covered the diagonal scan line inthe m/z-ion mobility pane to ensurecoverage of doubly and triply chargedspecies with narrow 25 m/z isolationwindows (Figure 3). Each dia-PASEFcycle contains a MS1 survey framefollowed by 8 dia-PASEF frames (seeFigure 3). This setup results in a totalcycle time of 900 ms (1x 100 ms MS1survey frame, 8x 100 ms dia-PASEFframes), which allowed us to obtainsufficienttdataa Cpoints5over thechromatographic peak while main-taining maximum coverage of +2 and+3 precursors. Currently DIA workflows mainly relyon spectral libraries for correlatingextracted tragmentition spectrawith the available peptide spectrum.The resource-specific library cdatawereaacquired usinggDDA PASEFand processed in Spectronautwhich resulted in a library of 8381protein groups and 93,301 peptidesequences. Data acquisition time forlibrary generation was just ~10 hoursusing the 60 SPD method. Targeted4-dimensional extraction from the60 SPD dia-PASEF data using thiscomprehensive resource-specificlibrary resulted in the identificationand quantification of on average 5204protein groups and 39,936 peptidesequences at a 1% FDR (Figure 4).This translates into an identificationrate of up to nearly 4000 peptides perminute of gradient time (Figure 5),underlying the exceptional sensitivityof the timsTOF Pro at high acquisitionspeed. The identified proteins covera dynamic range of nearly 5 orders ofmagnitude. The median coefficientof variation was 8.2% at the peptidelevel and 5.9% at the protein grouplevel for triplicate injections. We further shortened the run timeto increase sample throughput usingthe 100, 200, and 300 SPD methodson the Evosep One system. When analyzing the HeLa sampleusing 4-D data extraction and thethe highest throughputnmethod(300 SPD), we identified more than2100 protein groups and10,462peptide sequences on average in just3-minutes gradient time (4.8-minutestotal run time, Figure 4). For large scale proteomic studies itis not only important to increase theproteome coverage in single runs,but also to reproducibly identify andquantify proteins and peptides tomitigate the missing value problem.DIA-based approaches benefit fromsampling all the precursors presentin the selected mass ranges rather than being dependent on precursorselection algorithms leading to veryreproducible peptide and protein iden-tification. The timsTOF Pro providesaccurateaandhighly reproduciblecollisional cross section (CCS) valuesthat adds a 4th dimension for featurematching and alignment during dataprocessing, thereby further improvingdata completeness. When evaluatingthe triplicate injections of the 60 SPDmethod, we detected only a slightcumulative increase of 102 proteingroups (Figure 6). More than 94% ofthe protein groups (4915 out of 5213)were identified in all three runs(Figure 7). This underlines the out-standing reproducibility of identifiedproteins using dia-PASEF. In addition to excellent data com-pleteness in dia-PASEF, quantitativeaccuracy strongly benefits from the In summary, these results highlightthat the combination oftheEvosep One with dia-PASEF and4D-Proteomics is ideally suited forhigh-throughput proteomic profilingof hundreds of samples per day. 4th mobility dimension. Analyzing thereproducibility of protein intensitiesbetween thettechnicalreplicatesrevealedthat 881%ofthe5315identified proteins could be repro-ducibly quantified with a CV below20% and 65% with a CV below 10%(Figure 7). Figure 7: Number of all protein identifications (blue), proteins with CV<20% (gray) and <10% (light grey)for triplicate injections using the 60 SPD. Conclusion timsTOF Pro using dia-PASEF and 4D-Proteomics in combination with the Evosep One system is ideally suitedfor very high-throughput proteomics akin to clinical settings: On average 5200 protein groups can be identified and quantified in single-shot analysis using a short LCgradient (60 samples per day) Using a 300 SPD method (4.8-minutes total run time) dia-PASEF identifies and quantifies more than 2000protein groups per run. dia-PASEF delivers extremely reproducible identification and quantitation information making the approachperfectly suited for the challenges of quantitative proteomics. Learn More You are looking for further Information? Check out the link or scan the QR code for more details. www.bruker.com/timstofpro References [1] Meier F et al (2018). Mol Cell proteomics, doi: 10.1074/mcp.TIR118.000900 [2] Meier F. et al. (2019). bioRxiv, doi:https://doi.org/10.1101/656207 [3] Application Note LCMS 167: dia-PASEF@applied on different gradient lengths, https://www.bruker.com/fileadmin/user upload/8-PDFDocs/Separations MassSpectrometry/Literature/ApplicationNotes/1874392 LCMS-167 dia-PASEFapplied on different gradient lengths ebook.pdf [4] Bache N. et al. (2018). Mol Cell proteomcis, doi: 10.1074/mcp.TIR118.000853 [5] Wang et al. (2015). J Proteome Res, 4(6):2397-2403 [6] Kelstrup et al. (2018). J. Proteome Res., 17, 1,727-738 [7]Muntel et al. (2019). Mol. Omics, 15,348 For Research Use Only. Not for Use in Clinical Diagnostic Procedures. Bruker Daltonik GmbH Bruker Scientific LLC Bremen · GermanyBillerica,MA·USAPhone +49 (0)421-2205-0Phone +1(978) 663-3660 ms.sales.bdal@bruker.com-www.bruker.com 布鲁克与苏黎世联邦理工学院、德国马普研究所和多伦多大学合作,将PASEF与DIA(Data-Independent Acquisition,数据非依赖采集)的优势相结合,产生了一种新的采集模式称为dia-PASEF@。在dia-PASEF@扫描模式中,母离子在进入四级杆之前已经通过淌度进行了累积和分离,根据碰撞截面积(CCS)大小依次洗脱(与m/z有一定相关性),这样四级杆就可以根据洗脱离子的m/z进行离子选择,并且每批PASEF都会有多个窗口进行扫描,从而提高离子的利用率,避免了传统DIA方法中离子利用率低的问题。此外,使用离子淌度和质量数双重隔离窗口来触发MS/MS,提高了母离子选择和匹配的准确性,并降低了二级混合谱图的复杂性。并且在一级热图中,可以选择多电荷区域作为dia-PASEF@的母离子窗口,有效屏蔽单电荷杂质的干扰。dia-PASEF@具有更深的蛋白质组覆更高的分析通量和更优异的灵敏度,适合于高通量、微量样本的蛋白质组定量分析。采用95min梯度,单针进样200ng的HeLa,利用dia-PASEF@可以鉴定超过7,600种蛋白质、66,000种肽段。采用不同长度的梯度,30min可以鉴定6395个Hela细胞蛋白、4032个Yeast蛋白,达到快速、深度覆盖。即使在微量样本时,如单针进样10ng的Hela,仍能鉴定3000种蛋白质。布鲁克在ASMS 2020发布了高通量dia-PASEF@方案(图9A),即将Evosep One LC与timsTOF Pro再次联合,最大程度发挥Evosep One LC快速分离和timsTOF Pro扫描速度和稳定性的优势。该方案目前有4种方法(图9B),分别采用4.8min、7.2min、14.4min和24min色谱方法,对应的每天可以分析300、200、100和60蛋白质组学样本,把蛋白组学分析通量提升到一个全新的高度。分析结果(图9C,9D)显示出此方案在保证分析通量的同时,蛋白覆盖深度也有很好的保证,4.8min的分析单针可以鉴定2158蛋白,24min可以得到与传统蛋白组学长梯度分析相当的结果。图9. 高通量dia-PASEF@方案图10. Roman Fischer教授采用dia-PASEF@技术进行大队列研究
确定



还剩6页未读,是否继续阅读?

布鲁克·道尔顿(Bruker Daltonics)为您提供《HeLa细胞中肽段检测方案(液质联用仪)》,该方案主要用于其他中肽段检测,参考标准--,《HeLa细胞中肽段检测方案(液质联用仪)》用到的仪器有
相关方案
更多
该厂商其他方案
更多