








方案详情
文
气相色谱分析农药残留的基质效应及其解决方法
气相色谱分析农残
气相色谱仪
方案详情

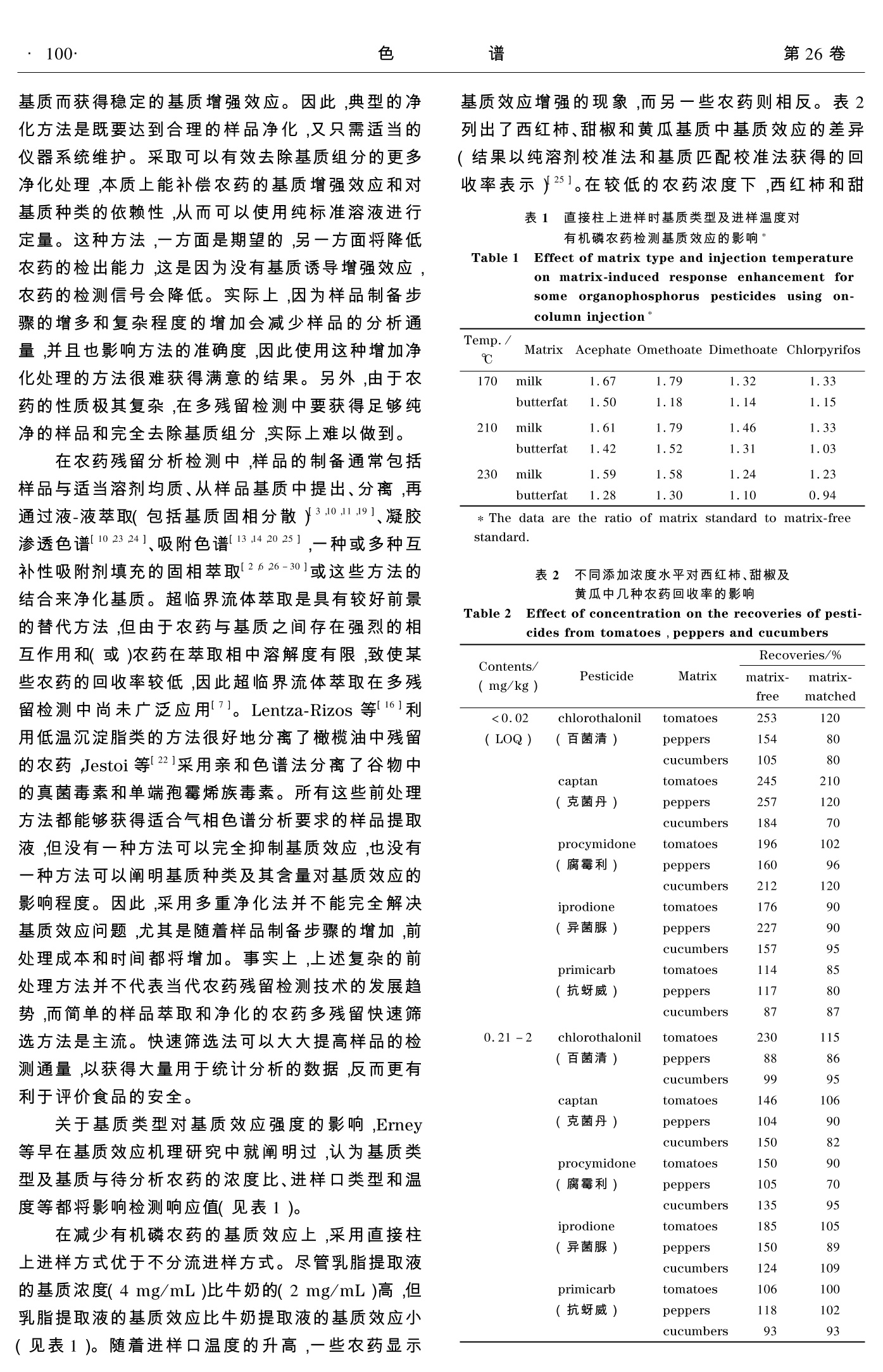
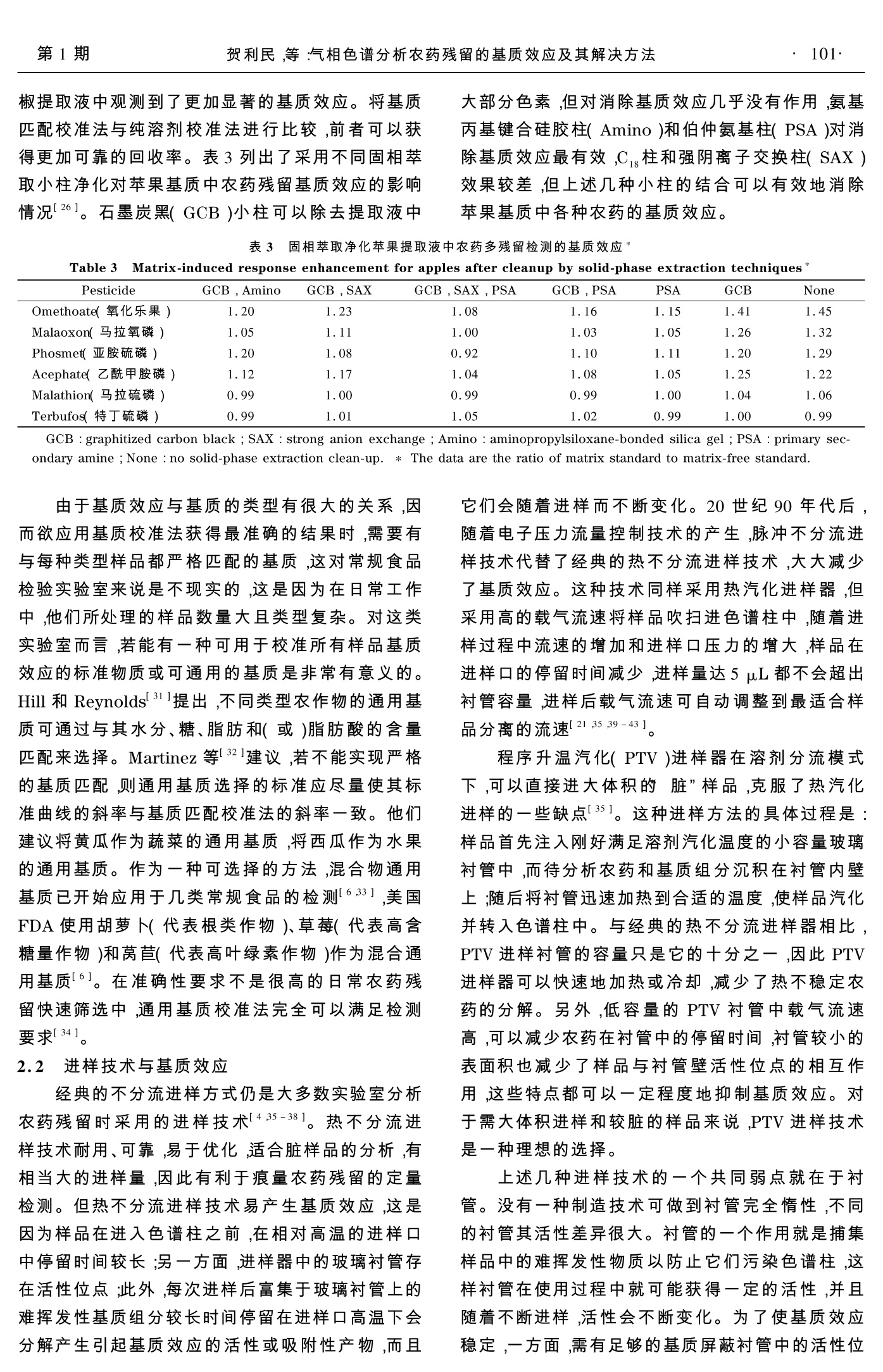
2008年1月January 2008色 谱Chinese Journal of ChromatographyVol. 26 No.198~104 第1期贺利民,等:气相色谱分析农药残留的基质效应及其解决方法·99· 气相色谱分析农药残留的基质效应及其解决方法 贺利民, 刘祥国, 曾振灵 (农业部畜禽产品质量监督检验测试中心(广州)华南农业大学兽医学院,广东广州510642) 摘要:对于相同浓度的农药,其在基质溶液中的色谱响应会比其在纯溶剂中的响应高。通过减少热不稳定农药的分解,以及屏蔽进样口的活性位点而减少极性农药在活性位点的吸附或分解,基质效应可增加从进样口传输到色谱柱中的农药残留量。各种进样方式和基质净化方法都可以减少但不能完全消除基质效应;基质匹配校准法和分析保护剂法是避免基质效应最有效的方法;在实际检测中,所采用的消除或补偿基质效应的方法应考虑减少仪器系统的维护。本文概述了农药残留分析检测中的基质效应及其解决方法。 关键词:气相色谱;农药残留;基质效应;解决方法 中图分类号:O658 文献标识码:A 文章编号:1000-8713(2008)01-0098-07 栏目类别:专论与综述 Solutions to matrix-induced response enhancement inpesticide residue analysis by gas chromatography HE Limin, LIU Xiangguo, ZENG Zhenling (Testing Center of Animal and Poultry Products Quality Control Inspection (Guangzhou) of Ministry ofAgriculture, College of Veterinary Medicine, South China Agricultural University, Guangzhou 510642, China) Abstract: The sample matrix can cause an enhancement in the observed chromatographic re-sponse for pesticide residues in a matrix extract compared with the same concentration in a ma-trix-free solution. The matrix increases the transfer of pesticides from the hot vaporizing injec-tors by reducing the thermal stress for labile compounds and by masking the active sites in theinjector responsible for the adsorption or decomposition of polar pesticides. The use of differ-ent injector types and matrix simplification procedures can reduce matrix-induced enhancementbut do not eliminate it. The most effective strategy is to use matrix-matched calibration stand-ards or analyte protectants which equalize the response enhancement for calibration standardsand sample extracts. From a practical point of view, it is important that the method used tocorrect for matrix-induced enhancement is compatible with low system maintenance. The dif-ferent approaches for correcting matrix-induced enhancement for calibration in pesticide resi-due analysis are discussed and compared in this review. Key words: gas chromatography (GC); pesticide residue; matrix-induced response enhance-ment; solutions 在20世纪90年代,人们就发现在用气相色谱分析有机磷农药时,无论是采用填充柱还是毛细管柱,检测信号的强度与试样基质的特性都非常相关。Erney 等首先成功地解释了这种现象。他致力于含脂高的食品(如黄油、牛奶、植物油等)中农药残留检测的固相萃取净化技术研究[2,3]。从脂肪基质中有效地将农药残留分离提取出来,使进样口和色谱柱免受难挥发物和后流出物的影响是十分重要 的,而固相萃取技术可以显著地减少基质干扰成分,因而非常适合这种应用,但基质效应依然存在。在农药残留检测中,食品检验实验室通常采取开始进几针空白基质标准溶液,以获得重现性好的响应值,在随后的分析过程中,采取纯溶剂标准溶液与样品溶液交替进样,标准溶液的响应将缓慢而非常显著的增加。用纯溶剂标准溶液计算,有些农药的空白基质加标回收率可能比理论值高几倍,有些农药却 ( 收稿日期:2007-07-18 ) ( 第一 作 者:贺利民,副研究员. E-mail: liminokhe@ scau. edu.cn. ) ( 通讯联系人:曾振灵. Tel: (020)8 5 284896, E-mai l : zlze n g@ scau. edu. cn. ) ( 基金项目:农业行业标准制定项目(070106-69)及华南农业大学校长基金(2007B003)资助项目. ) 并非如此。基质的种类和含量都会影响回收率,即使在相同的仪器操作条件下,每种农药的回收率可能都不同,尤其在低残留水平时,随着进样的不断进行,一些农药的峰会变差而难以准确积分。 Erney 等的主要贡献就是提出了产生上述现象的基本原理,并且提出基质诱导响应增强的概念,尽管这一概念当时在科学界引起了广泛的争议11,4]。Erney和他的同事将基质效应归因于农药由热进样口向色谱柱的传输过程中有基质的参与。通过在汽化过程中减少热不稳定农药的分解,以及屏蔽进样口中的活性位点而减少极性农药在活性位点的吸附和解吸的速度,样品基质增加了待分析农药从进样口向色谱柱的传输量。他们提出的术语“基质诱导色谱响应增强现象”,现在称为基质诱导响应增强效应或简化为基质效应。他们通过实验后,提取了两类易产生基质增强效应的化合物,一类是在汽化温度下热不稳定的化合物,一类是样品进入色谱柱或检测器时易被吸附的化合物。这也暗示吸附活性位点不是缘于玻璃衬管表面的硅醇基或金属离子,就是因前一次进样的难挥发物或基质组分热分解产物沉积于进样口和色谱柱中所产生。进样口温度变化影响基质效应,一方面,温度增加可以减少对农药的吸附,有利于吸附在衬管中的农药释放;另一方面,温度增加会加速热不稳定农药的分解。通过与纯溶剂标准溶液比较,可以说明对基质诱导响应增强敏感的农药回收率增高是由于基质的保护作用。纯溶剂标准溶液的实际响应值低于其本应该进入色谱柱产生的值,是因为纯溶剂不能为待分析农药提供足够的保护,而在空白基质溶液中添加相同浓度的农药,可使农药更加完全地转移到色谱柱中,从而响应增加,在这个过程中基质起到了分析保护作用。 最实际的解决基质效应的方法是利用不含农药的空白基质匹配标准溶液校准,它可以同等程度地补偿标准溶液和样品溶液中农药的响应1,4,5]。美国环境保护署(EPA)和食品药品管理局(FDA)禁止采用这种方法,但美国官方实验室在农药残留检测中长期使用基质匹配校准法[6,71。因一些农药在接近残留限量被检出时,纯溶剂标准溶液校准法将导致测定浓度偏大,这在实际中是不希望的,为此,欧盟推荐利用基质匹配标准溶液校准法8,美国政府却希望通过其他技术手段解决这种问题。 农药残留基质效应中的一个普遍问题是不同实验室的研究结果差异较大,这主要是因为进样装置不同,尤其是进样口玻璃衬管活性差异以及进样器使用情况不同等。连续进样时,因前一次进样在衬管中沉积的活性组分对进样器的活性位点有潜在的 影响,因此不能认为每种农药都有一个稳定的基质效应。实验室内的研究表明,如果进样口没有受到严重污染,在同样的基质类型和组成情况下,同一仪器能够保持较长时间稳定、可靠的响应,但同样的样品在不同仪器上的基质效应强度不同,不同实验室使用不同的仪器,或在不同的实验条件下所获得的基质效应数值差异更大。本文主要综述植物样品农药残留分析检测中的基质效应及其解决方法。 1 基质效应较强的典型农药 不易受基质效应影响的农药常常是热稳定的,或在进样口中吸附作用小。而基质效应敏感的典型农药有:乙酰甲胺磷、α-六六六、谷硫磷、联苯菊酯、克菌丹、甲萘威、百菌清、毒死蜱、蝇毒磷、氯氰菊酯、p,p-滴滴涕、二嗪农、抑菌灵、狄氏剂、乐果、异狄氏剂、乙硫磷、氧嘧啶磷、杀螟硫磷、灭菌丹、异菌脲、马拉氧磷、马拉硫磷、卡比吗唑、甲胺磷、杀扑磷、久效磷、氧化乐果、甲拌磷、伏杀硫磷、亚胺硫磷、腐霉利、苯胺灵、三唑酮、三唑醇、磷酸三丁酯、敌百虫和单端孢霉烯族毒素类。这类农药大多具有极性和(或)能形成强氢键作用的酸性和(或)碱性化合物,一般带有磷酸基(-P=O)、羟基、氨基、咪唑基、苯并咪唑基、氨基甲酸酯基(-O-CO-NH-)和脲基(-NH-CO-NH-)官能团。具有-P=S基团的有机磷农药与具有-P=O 基团的有机磷农药相比,前者受基质效应的影响就小些。大多数研究表明,不只是植物试样存在基质效应,各种动物食品样品也存在基质效应。有关基质效应研究的报道主要有:人尿中乙酰甲胺磷残留的检测91,坚果和水果汁中农药残留的测定[10-12],蜂蜜中农药残留的检测[131,葡萄汁和葡萄酒中杀螨剂、杀菌剂残留的检残[14,15],植物油中农药残留的检测[2,16],猪肉中农药残留的检测17,土壤和其他沉积物中农药及其他污染物的检测[18,191,玫瑰花及其产品中毒死蜱残留的检测[20],饮用水中卤代诱变剂的检测[21],谷物中真菌毒素检测[22]等。 2 基质效应的解决方法 2.1 基质净化法 对农药残留检测来说,基质净化是必须的。方面它可以使农药和基质组分完全分分;另一方面,可以减少难挥发性化合物或热不稳定化合物对仪器系统的污染。如果试样不经净化而直接进行分析检测,将增加仪器的维护频率,从而导致样品分析通量减少,分析效率降低。如上所述,虽然基质组分可弓起农药响应大大增加,但是实际中是希望用最少的 基质而获得稳定的基质增强效应。因此,典型的净化方法是既要达到合理的样品净化,又只需适当的仪器系统维护。采取可以有效去除基质组分的更多净化处理,本质上能补偿农药的基质增强效应和对基质种类的依赖性,从而可以使用纯标准溶液进行定量。这种方法,一方面是期望的,另一方面将降低农药的检出能力,这是因为没有基质诱导增强效应,农药的检测信号会降低。实际上,因为样品制备步骤的增多和复杂程度的增加会减少样品的分析通量,并且也影响方法的准确度,因此使用这种增加净化处理的方法很难获得满意的结果。另外,由于农药的性质极其复杂,在多残留检测中要获得足够纯净的样品和完全去除基质组分,实际上难以做到。 在农药残留分析检测中,样品的制备通常包括样品与适当溶剂均质、从样品基质中提出、分离,再通过液-液萃取(包括基质固相分散)[3,10,11,19]、凝胶渗透色谱[10,23,24]、吸附色谱[13,14,20,25],一种或多种互补性吸附剂填充的固相萃取[2,6,26-30]或这些方法的结合来净化基质。超临界流体萃取是具有较好前景的替代方法,但由于农药与基质之间存在强烈的相互作用和(或)农药在萃取相中溶解度有限,致使某些农药的回收率较低,因此超临界流体萃取在多残留检测中尚未广泛应用。Lentza-Rizos 等16]利用低温沉淀脂类的方法很好地分离了橄榄油中残留的农药,Jestoi等122]采用亲和色谱法分离了谷物中的真菌毒素和单端孢霉烯族毒素。所有这些前处理方法都能够获得适合气相色谱分析要求的样品提取液,但没有一种方法可以完全抑制基质效应,也没有一种方法可以阐明基质种类及其含量对基质效应的影响程度。因此,采用多重净化法并不能完全解决基质效应问题,尤其是随着样品制备步骤的增加,前处理成本和时间都将增加。事实上,上述复杂的前处理方法并不代表当代农药残留检测技术的发展趋势,而简单的样品萃取和净化的农药多残留快速筛选方法是主流。快速筛选法可以大大提高样品的检测通量,以获得大量用于统计分析的数据,反而更有利于评价食品的安全。 关于基质类型对基质效应强度的影响,Erney等早在基质效应机理研究中就阐明过,认为基质类型及基质与待分析农药的浓度比、进样口类型和温度等都将影响检测响应值(见表1)。 在减少有机磷农药的基质效应上,采用直接柱上进样方式优于不分流进样方式。尽管乳脂提取液的基质浓度(4mg/mL)比牛奶的(2mg/mL)高,但乳脂提取液的基质效应比牛奶提取液的基质效应小(见表1)。随着进样口温度的升高,一些农药显示 基质效应增强的现象,而另一些农药则相反。表2列出了西红柿、甜椒和黄瓜基质中基质效应的差异(结果以纯溶剂校准法和基质匹配校准法获得的回收率表示)[25]。在较低的农药浓度下,西红柿和甜 表1 直接柱上进样时基质类型及进样温度对有机磷农药检测基质效应的影响* Table 1 Effect of matrix type and injection temperatureon matrix-induced response enhancement forsome organophosphorusS]pesticides usingon·column injection* Temp./Matrix Acephate Omethoate Dimethoate Chlorpyrifos ℃ 170 milk 1.67 1.79 1.32 1.33 butterfat 1.50 1.18 1.14 1.15 210 milk 1.61 1.79 1.46 1.33 butterfat 1.42 1.52 1.31 1.03 230 milk 1.59 1.58 1.24 1.23 butterfat 1.28 1.30 1.10 0.94 *The data are the ratio of matrix standard to matrix-freestandard. 表22不同添加浓度水平对西红柿、甜椒及黄瓜中几种农药回收率的影响 Table 2 Effect of concentration on the recoveries of pesti-cides from tomatoes, peppers and cucumbers 253 120 椒提取液中观测到了更加显著的基质效应。将基质匹配校准法与纯溶剂校准法进行比较,前者可以获得更加可靠的回收率。表3列出了采用不同固相萃取小柱净化对苹果基质中农药残留基质效应的影响情况[26]。石墨炭黑(GCB)小柱可以除去提取液中 大部分色素,但对消除基质效应几乎没有作用,氨基丙基键合硅硅柱(Amino)和伯仲氨基柱(PSA)对消除基质效应最有效,C柱和强阴离子交换柱(SAX)效果较差,但上述几种小柱的结合可以有效地消除苹果基质中各种农药的基质效应。 Table 3Matrix-induced response enhancement for apples after cleanup by solid-phase extraction techniques*3c Pesticide GCB, Amino GCB, SAX GCB, SAX, PSA GCB, PSA PSA GCB None Omethoate(氧化乐果) 1.20 1.23 1.08 1.16 1.15 1.41 1.45 Malaoxon(马拉氧磷) 1.05 1.11 1.00 1.03 1.05 1.26 1.32 Phosmet(亚胺硫磷) 1.20 1.08 0.92 1.10 1.11 1.20 1.29 Acephate(乙酰甲胺磷) 1.12 1.17 1.04 1.08 1.05 1.25 1.22 Malathion(马拉硫磷) 0.99 1.00 0.99 0,99 1.00 1.04 1.06 Terbufos(特丁硫磷) 0.99 1.01 1.05 1.02 0.99 1.00 0.99 GCB: graphitized carbon black; SAX: strong anion exchange; Amino: aminopropylsiloxane-bonded silica gel; PSA: primary sec-ondary amine; None: no solid-phase extraction clean-up.* The data are the ratio of matrix standard to matrix-free standard. 由于基质效应与基质的类型有很大的关系,因而欲应用基质校准法获得最准确的结果时,需要有与每种类型样品都严格匹配的基质,这对常规食品检验实验室来说是不现实的,这是因为在日常工作中,他们所处理的样品数量大且类型复杂。对这类实验室而言,若能有一种可用于校准所有样品基质效应的标准物质或可通用的基质是非常有意义的。Hill 和 Reynolds[31]提出,不同类型农作物的通用基质可通过与其水分、糖、脂肪和(或)脂肪酸的含量匹配已选择。Martinez 等321建议,若不能实现严格的基质匹配,则通用基质选择的标准应尽量使其标准曲线的斜率与基质匹配校准法的斜率一致。他们建议将黄瓜作为蔬菜的通用基质,将西瓜作为水果的通用基质。作为一种可选择的方法,混合物通用基质已开始应用于几类常规食品的检测[6,33],美国FDA使用胡萝卜(代表根类作物)、草莓(代表高含糖量作物)和莴苣(代表高叶绿素作物)作为混合通用基质61。在准确性要求不是很高的日常农药残留快速筛选中,通用基质校准法完全可以满足检测要求[34]。 2.2 进样技术与基质效应 经典的不分流进样方式仍是大多数实验室分析农药残留时采用的进样技术[4,35-38]。热不分流进样技术耐用、可靠,易于优化,适合脏样品的分析,有相当大的进样量,因此有利于痕量农药残留的定量检测。但热不分流进样技术易产生基质效应,这是因为样品在进入色谱柱之前,在相对高温的进样口中停留时间较长;另一方面,进样器中的玻璃衬管存在活性位点;此外,每次进样后富集于玻璃衬管上的难挥发性基质组分较长时间停留在进样口高温下会分解产生引起基质效应的活性或吸附性产物,而且 它们会随着进样而不断变化。20世纪90年代后,随着电子压力流量控制技术的产生,脉冲不分流进样技术代替了经典的热不分流进样技术,大大减少了基质效应。这种技术同样采用热汽化进样器,但采用高的载气流速将样品吹扫进色谱柱中,随着进样过程中流速的增加和进样口压力的增大,样品在进样口的停留时间减少,进样量达5 pL都不会超出衬管容量,进样后载气流速可自动调整到最适合样品分离的流速[21,35,39-43]。 程序升温汽化(PTV)进样器在溶剂分流模式下,可以直接进大体积的“脏”样品,克服了热汽化进样的一些缺点[35]。这种进样方法的具体过程是:样品首先注入刚好满足溶剂汽化温度的小容量玻璃衬管中,而待分析农药和基质组分沉积在衬管内壁上;随后将衬管迅速加热到合适的温度,使样品汽化并转入色谱柱中。与经典的热不分流进样器相比,PTV 进样衬管的容量只是它的十分之一,因此 PTV进样器可以快速地加热或冷却,减少了热不稳定农药的分解。另外,低容量的 PTV 衬管中载气流速高,可以减少农药在衬管中的停留时间,衬管较小的表面积也减少了样品与衬管壁活性位点的相互作用,这些特点都可以一定程度地抑制基质效应。对于需大体积进样和较脏的样品来说, PTV进样技术是一种理想的选择。 上述几种进样技术的一个共同弱点就在于衬管。没有一种制造技术可做到衬管完全惰性,不同的衬管其活性差异很大。衬管的一个作用就是捕集样品中的难挥发性物质以防止它们污染色谱柱,这样衬管在使用过程中就可能获得一定的活性,并且随着不断进样,活性会不断变化。为了使基质效应稳定,一方面,需有足够的基质屏蔽衬管中的活性位 点;另一方面,又不能有太多的难挥发性基质组分在衬管中沉积,以保证一定的样品分析通量和适当的仪器维护频率。 为了减少因基质组分在衬管中的沉积而需要的仪器维护,研究者们又开发了两种新的进样技术:直接样品导入(DSI)[44,45]技术和“脏”样品导入(DMI)I46,471技术。采用 DSI 技术进样,试样溶液预先注入已经安置在进样衬管的微管中,在较低的温度下汽化溶剂并排空,随后迅速加热衬管至足够样品汽化的温度。样品分析完成后,取出微管,换上新微管,准备下次进样。DMI 与 DSI 两种技术的不同之处在于每次进样后,DMI的衬管和微管都需要更换,而 DSI则只需要更换微管。两种进样技术的相同之处是都可以大体积进样。DSI 外优点主要是可以减少进样口的维护和样品净化,DMI的优点是可以除去从微管中溢出的难挥发性基质组分而免其进入仪器系统。从基质效应角度而言,DMI 与 DSI的主要问题是每次进样时,需要更换微管和(或)衬管,这会影响到检测结果的准确性。因为尚没有任何方法可以灭活与样品及样品蒸汽相互作用的玻璃表面的活性位点,所以 DSI 和 DMI进样技术同样也存在基质效应。 2.3 校正因子校准法 在一个较长时间或一段时期内能保持仪器稳定的系统中,可以采用纯溶剂校准曲线校正基质匹配校准曲线,并用纯溶剂校准曲线计算基质提取液中农药的准确浓度[48-50]。这首先需要获得纯溶剂校准曲线和基质匹配校准曲线的一些参数,且每条曲线都必须是线性的,并且校准曲线在大量样品和校准溶液进样的一定时间内保持稳定,后者可能是这种方法的关键所在,同时这也表明响应校正函数与仪器状态非常相关[50]。虽然方法的线性范围受与待分析农药浓度无关的基质效应的限制,但研究表明,在较宽的浓度范围内,基质效应依赖于待分析农药浓度与基质浓度的比。图1是牛奶提取液和纯溶剂中氧化乐果的典型校准曲线51。如果一种农药在某基质中的校准曲线建立起来,只要该校准曲线的斜率和截距没有统计学上的显著变化,就可以一直使用。这样,采用适当的校准曲线,通过简单地计算就可以获得试样中分析农药的含量。具体计算如下:假设某基质中某农药的响应值以Y表示,Y与其浓度Xsc(纯溶剂中的浓度)有关,依据方程(1)可以计算出Xvc(基质中的浓度)。 A是是距项(asc-aMc)/bMc,B 是斜率项bsc/bMc,a和b分别为最佳拟合校准曲线的截距和斜率。 图1 氧化乐果在牛奶基质匹匹液(MC)和乙腈溶剂(SC)中的校准曲线 Fig. 1 Calibration curves for omethoate in acetonitrile(SC) and matrix-matched milk extract (MC) 基于校正基质效应的方法还有标准加入法和稳定同位素稀释法。 标准加入法是分析化学中经常采用的一种方法,它适合于单个或少量样品的准确定量,虽然比较费时,但常在严格的统计学实验设计中采用[511。它的一般操作是:将每个试样提取液分成几份平行样,分别加入一定量的待分析农药的标准溶液,然后将所有平行样品稀释到相同的体积,用相同仪器条件进行分析,通过线性回归获得一定测量不确定度的检测农药浓度。采用标准加入法时,要求基质效应与待分析农药浓度无关或与待分析农药浓度成正比,且在添加浓度范围内校准曲线呈线性。但有时基质效应却显著依赖于待分析农药与基质浓度的比,且每个样品需测定几次,因而样品分析通量将受到影响,仪器的维护也要相应增加。鉴于此,在农药残留分析中,标准加入法常常成为校准基质效应的最后选择,一般只有当需要分析少量样品,而且分析结果要作为法律仲裁的依据时才使用此种方法。 当使用质谱法检测农药多残留时,稳定同位素标记的农药是非常合适的内标物24],可以用其有效校正基质效应。因为同位素标记的待分析农药标准品与待分析农药本身受到相同的基质效应影响,所以待分析农药与用同位素标记的农药标准品的响应值之比保持不变,但二者的浓度差应尽量小至使基质效应与待分析农药和基质浓度比不再相关。在进行大批量样品分析检测时,例如农药残留的快速筛选,通常是向所有试样中加入一个固定浓度的同位素标记的待分析农药标准品,因而欲获得准确的分析结果,便要求基质效应的影响保持不变或至少在同位素内标和待分析农药浓度的线性范围内。对于一些食品残留检测实验室而言,除了仪器设备的限制外,限制这种方法使用的主要是同位素标记的农药标准品不易获得且价格昂贵。 2.4 加入分析保护剂法 分析保护剂是仿效基质保护作用的单一化合物或简单的混合物[9,29,47,52-55]。当往纯溶剂标准溶液和样品溶液中加入相同量的保护剂时,它能同等程度地补偿标准溶液和样品溶液的基质效应。这种方法首先由 Erney 和 Poole 52]提出,但直到Lehotay和他的同事[29,47,53,54]对其进行更深入的研究之后,才在实际应用中得到重视。基质效应主要发生在从进样口到色谱柱以及色谱柱到检测器之间,可能包括待分析农药与硅醇基及其与玻璃衬管表面金属离子间的相互作用。分析保护剂有效地与待分析农药竞争衬管中的活性位点,以最大限度地提高纯溶剂中农药标准品的响应值,使之达到与基质中农药同等的响应。一般来说,具有这种保护作用的化合物通常有很强的形成氢键的能力,并与分析物具有相似的挥发性。 Lehotay 和他的同事证实,在色谱图一定保留时间窗口内,可以和待分析农药同时流出的化合物或其分解产物是比较合适的分析保护剂。这表明色谱柱(至少色谱柱的前端)、色谱柱与检测器连接处及检测器内表面都对基质效应的产生有一定的作用[53,54]。农药多残留分离分析常采用程序升温法,Lehotay 和他的同事的研究结果表明,当温度达到能使待分析农药在色谱柱中的分配系数很小之前,分析保护剂和待分析农药能停留在色谱柱的同一部位是很重要的。他们也证实,3-乙氧基-1,2-丙二醇是挥发性农药的合适分析保护剂,古洛糖酸y-内酯是半挥发性农药的合适保护剂,山梨醇可以作为低挥发性农药的合适保护剂,而使用上述三者的混合物可实现对性质差异较大的农药的满意定量。例如,25pL分析保护剂溶液(40 mg/mL 的3-乙氧基-1,2-丙二醇,4mg/mL 的古洛糖酸y-内酯和山梨醇,用水-乙腈(体积比为7:3)混合溶液配制)加入到1.0 mL乙腈提取的蔬菜或水果溶液中,便可起到很好的分析保护作用[47]。图2是混合水果乙腈提取液中应用分析保护剂测定农药残留的典型例子。其他有效的分析保护剂还有用于低挥发性农药残留测定的葡萄糖、果糖、木糖19,29]等糖类和玉米油或橄榄油551。一般而言,理想的农药残留检测用分析保护剂必须满足以下基本要求: 1)在溶液或进样过程中不与待分析农药发生化学反应; 2)在达到分析保护作用的浓度(有效补偿纯溶剂标准溶液的响应值)时,能溶于上机溶液中; 3)在色谱系统中不积累,以免增加系统的维护或缩短色谱柱的寿命; 4)不能干扰拟分析农药的检测。 图2 分析保护剂(a)添加前和(b)添加后对水果乙腈提取物中几种农药(100 ng/mL)响应的影响 Fig.2 Comparison of response enhancement for 100ng/mL of pesticides in a mixed fruit acetoni-trile extract (a) without and (b) with addi-tion of analyte protectants (3-ethoxy-1,2-propanediol, gulonolactone and sorbitol at10,1 andd1 mg/mL, respectively) in thesample 很多化学物质可以满足上述要求,但仅有少数化合物具有补偿复杂基质中农药多留检测的基质效应,使用这样的化合物作分析保护剂将有利于通用标准方法的开发。 3 小结 当使用纯溶剂标准曲线进行校准时,基质效应常常会引起农药残留分析结果的偏差和对样品制备过程回收率的错误估算。基质效应随不同的农药、不同的基质类型和基质浓度而变化,当然也与进样口的类型、设计及操作条件等仪器参数相关。有很多方法可以用来补偿基质效应,但作为校准溶液而言,最有效的方法是基质匹配标准溶液校准或加入分析保护剂校准。 从上面的综述可以看到,国外学者很早就注意到了农药残留测定中的基质效应问题,特别是20世纪90年代初以来,建立并发明了多种用于消除或补偿基质效应的方法和技术,而我国分析工作者对这一客观存在的现象尚未引起注意和重视,有关报道相对较少[56]。动物基质成分较植物更为复杂,在分析检测动物试样中的药物残留时,经常会遇到显著的基质增强效应[571,所以开展动物基质中药物残留 检测时的基质效应研究十分重要。开发可以消除或补偿动物试样基质效应的技术和方法,实现动物试样药物残留检测的准确定量,具有重要的理论和实践意义。 ( 参考文献: ) ( [1] Erney D R, Gillespie A M, Gilvydis D M, e t al. J C hroma- togr, 1993, 638:57 ) ( [2] Gillespie A M, Daly S L, Gilvydis D M, et al. J AOAC I nt, 1995,78:431 ) ( [3] Erney D R. J High Resolut Chromatogr,1995,1 8 : 59 ) ( Hajslova J, Z rostlikova J. J Chromatogr A, 2003, 1 0 0 0: 181 ) ( Erney D R, Pawlowski T M, P o ole C F . J High R e solut Chromatogr, 1997, 20:375 ) ( [6] Mercer G E . J AOAC I n t, 2005, 88:14 5 2 ) ( [7] Lehotay S J, Schaner A, Nemoto S , et al. .J J AOAC I nt, 2002,85:1148 ) ( [8] Hill A. Quality control procedures for pesticide r esiduesanalysis-guidelines for residue monitoring in the EuropeanUnion, Document 7826/VI/97. B r ussels: European Com-mission, 1997 ) ( [9] LePage J T, Hebert V R, Tomaszewska E M , et al. J AOAC Int, 2005,88 : 1788 ) ( [10] Hu XZ, Yu JX, Yan Z G, et al. J AOAC Int, 2004, 87: 972 ) ( 111 Liapis K S, Aplada-Sarlis P , Kyriakidis N V. J Chromatogr A, 2003,996: 1 81 ) ( [12] Patel K, F ussell R J, Macarthur R, e t al. J C hromatogr A, 2004,1046:225 ) ( [13] Jimenez J J, Bernal J L, Nozal M J, et al . J Chromatogr A, 1998,823:381 ) ( 141 Bernal J L, del Nozal M J , Jimenez JJ, et al. J C h roma- togr A, 1997, 778: 111 ) ( [15] Holland P T, McNaughton D E, Malcolm C P. J Assoc Off Anal Chem I n t, 1994, 77: 7 9 ) ( [16] Lentza-Rizos C, Avramides E J, Cherasco F. J C hromatogr A, 2001, 912:135 ) ( [17] Garrido-Frenich A, Romero-Gonzalez R, Martinez Vidal J L, et al . J Chromatogr A, 2006,1 1 33:315 ) ( [18] Dabrowski L, Giergielewicz-Mozajska H, Gorski L, et a l . J Sep Sci, 2002, 25: 290 ) ( [19] Schmeck T ,Wenclawiak B. Chromatographia, 2005, 62: 159 ) ( [20] Kumar A, N adda G, Shanker A. J Chromatogr A, 2004, 1050:193 ) ( [21] Rantakokko P, Yritys M, Vartainen T. J Chromatogr A, 2004,1028: 1 79 ) ( [22] Jestoi M, Ritien I A, Rizzo A. J Agric Food Chem, 2004, 52:1464 ) ( [23] Hajslova J, Holadova K, Kocourek V, et al. J Chromatogr A, 1998,800: 283 ) ( [24] Ueno E, Oshima H, Saito I, et a l . J AOAC In t , 2004, 87 : 1003 ) ( [25] Menkissoglu-Spiroudi U, Fotopoulou A. I n t J Environ Anal Chem, 2004, 84:15 ) ( [26] Schenck F J, Lehotay S J. J Chromatogr A, 2000, 868: 51 ) ( [27] Podhorniak L V , Negron J F, G r iffith F D . J A OAC Int, 2001,84:873 ) ( [28] Schenck F J, Lehotay S J, Vega V. J Sep Sci, 2 0 02, 25: 883 ) ( [29] Anastassiades M, L ehotay S J, S t ainbaher D, et a l . . AOAC I n t, 2003, 86: 4 12 ) ( [30] Georgiou P P, Liapis K S, Miliadis G E, et al. Int J Envi-ron Anal Chem, 2 006, 86: 69 ) ( [31] Hill A R C, Reynolds S L. Analyst, 1999, 1 24: 9 53 ) ( [32] Martinez Vidal J L, Arrebola F J, G a rrido-Frenich A, et a l.Chromatographia, 2 004, 59:321 ) ( [33] Barwick V J, Ellison S L R, Lacey S J , et al. J Sci FoodAgric, 1999, 79 : 1190 ) ( [34] Kocourek V, Hajslova J, Holadova K. J Chromatogr A, 1998,800:297 ) ( [35] Zrostikova J, Hajslova J, G odula M. 1 . J C hromatogr A, 2001,937: 7 3 ) ( [36] Poole C F . The e ssence o f chromatography. Amsterdam: Elsevier, 2003 ) ( [37] Domotorova M, Kirchner M, Matisova E . J Sep Sci, 2006, 29: 10 51 ) ( [38] Mastovska K, L ehotay S J, Hajslova J. J C hromatogr A, 2001,926: 2 91 ) ( [39] Wylie P L, Uchiyama K. J AOAC Int, 1996, 79: 5 71 ) ( [40] Wylie P L , P hilips R J , K lein K J, et al. J High R e solut Chromatogr, 1991, 14: 649 ) ( [41] Godula R, Hajslova J, Alterova K. J High Resolut Chroma- togr, 1999, 22: 3 95 ) ( [42] Mastovska K, H ajslova J, L ehotay S J. J C hromatogr A, 2004,1054:335 ) ( [43] Kirchner M, Matisova E , Otrekal R , et al. J Chromatogr A,2005,1084:63 ) ( [44] Lehotay S J. J AOAC Int , 2000,83 : 680 ) ( [45] Jing H, Amirav A. Anal Chem, 1997,69: 1 426 ) ( [46] de Koning S, L ach G , Linkerhagner M, e t al. J C hroma-togr A, 2003, 1 008: 2 47 ) ( [47] Cajka T, M astovska E, Lehotay S J. J Sep Sci, 2005, 28: 1048 ) ( [48] Gonzalez F J E, Torres M E H, Cuadros-Rodriguez L C, et al. Analyst, 2002,127: 1 038 ) ( [49] Martinez-Galera M, Lopez-Lopez T, Gil-Garcia M D, et al. Anal Bioanal Chem, 2003,375:653 ) ( [50] Gonzalez F JE, Torres M E H, Lopez E A, et al. J Chrom- atogr A, 2002, 966: 1 55 ) ( [51 ] Massart D L, Vandeginste B G M , Buydens L M G, et al.Handbook of chemometrics and qualimetrics: Part A. A m-sterdam: E lsevier,1997 ) ( [52] Erney D R, Poole C F. J High Resolut Chromatogr, 19 9 3, 16:501 ) ( [53] Anastassiades M, Mastovska K , Lehotay S J. J Chromatogr A,2003, 1 015: 1 63 ) ( [54] Mastovska K , L ehotay S J, A nastassiades M. A nal C hem, 2005,77:8129 ) ( [55] Sanchez-Brunete C, Albero B, M a rtin G . Anal Sci, 2 0 05, 21:1291 ) ( [56] Huang B Y, Pan C P, W ang Y R, et al. Chemical Journal of Chinses Universities (黄宝勇,潘灿平,王一茹,等.高等 学校化学学报),2006,27(2):227 ) ( [57] He L M, S u Y J, Z e ng Z L, et al. A n i F e ed Sci Tech, 20 0 7, 132:316 )
确定





还剩5页未读,是否继续阅读?

产品配置单

苏州市莱顿科学仪器有限公司为您提供《蔬菜中农药残留检测方案(气相色谱仪)》,该方案主要用于蔬菜中农药残留检测,参考标准--,《蔬菜中农药残留检测方案(气相色谱仪)》用到的仪器有Agilent 7820A气相色谱仪
推荐专场






相关方案
更多