








方案详情
文
采用新型的光学投影法热膨胀仪和压实粉体样品形式,使得对玻璃化转变过程中粘性流玻璃化转变点、构型平衡温度和激活能的非接触测量成为了可能。
本文介绍了采用这种非接触测试方法的测试结果,并与其它传统接触式热膨胀仪的测试结果进行了对比。通过对玻璃不同组分及各种结晶趋势的测试发现,这种非接触测量方法在烧结过程中的测试准确性方面有突出表现。对于具有低结晶趋势的玻璃,整个烧结阶段激活能随着温度上升而减小;而对于较高结晶稳定性玻璃,激活能在相变初期开始阶段会异常增加。做为结论,低速加热速率的稠化过程是部分禁止的。
方案详情

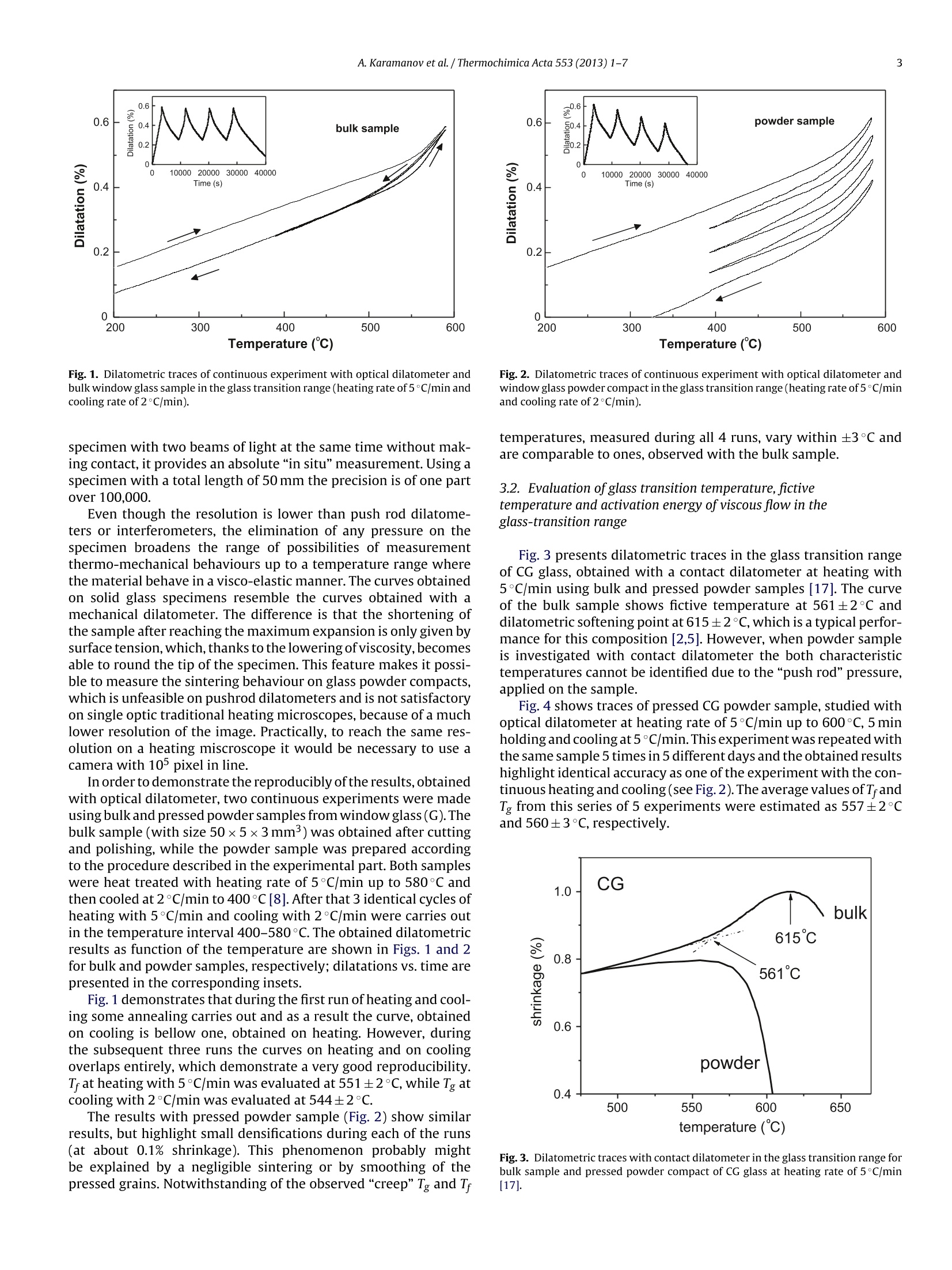
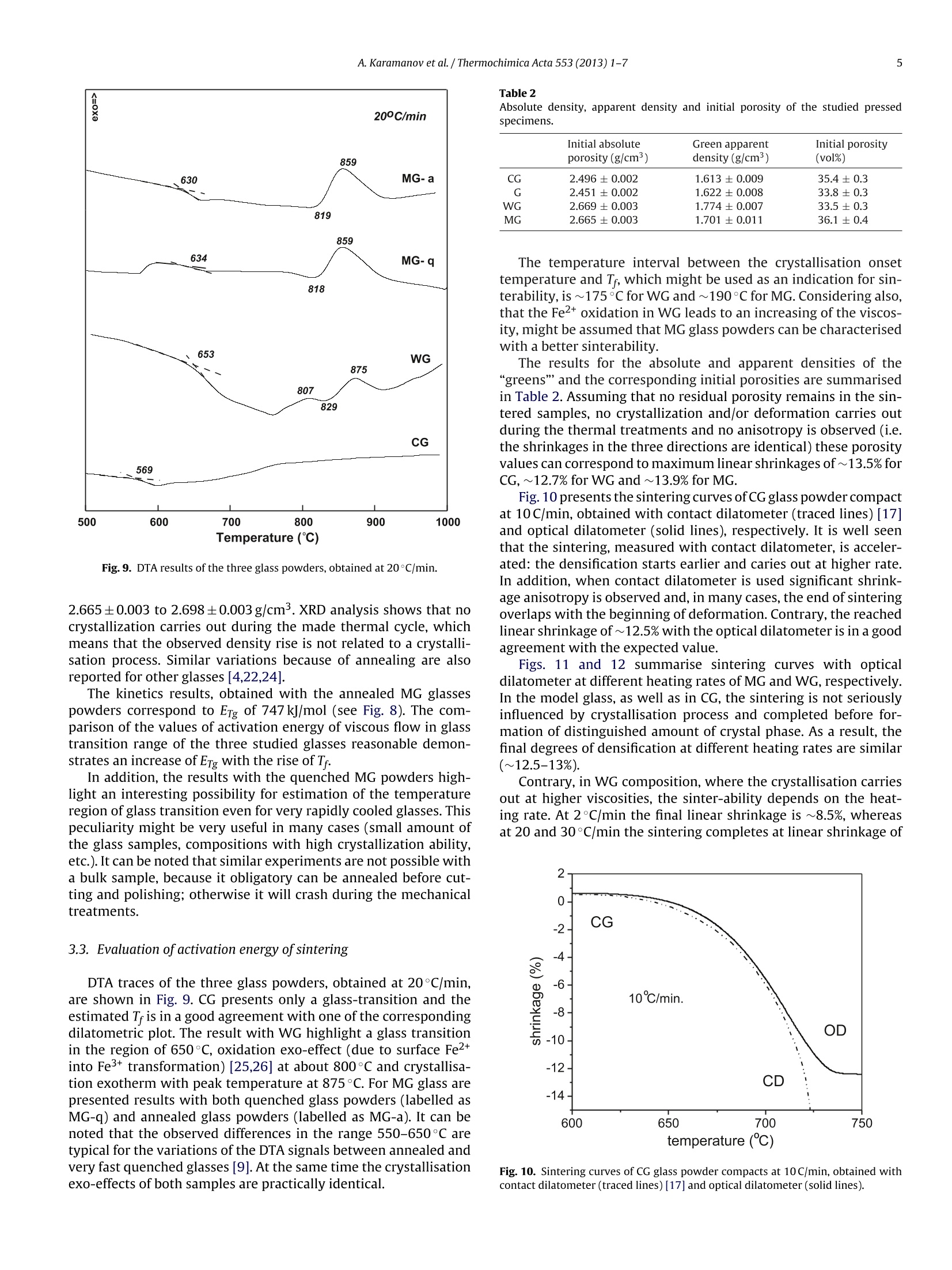
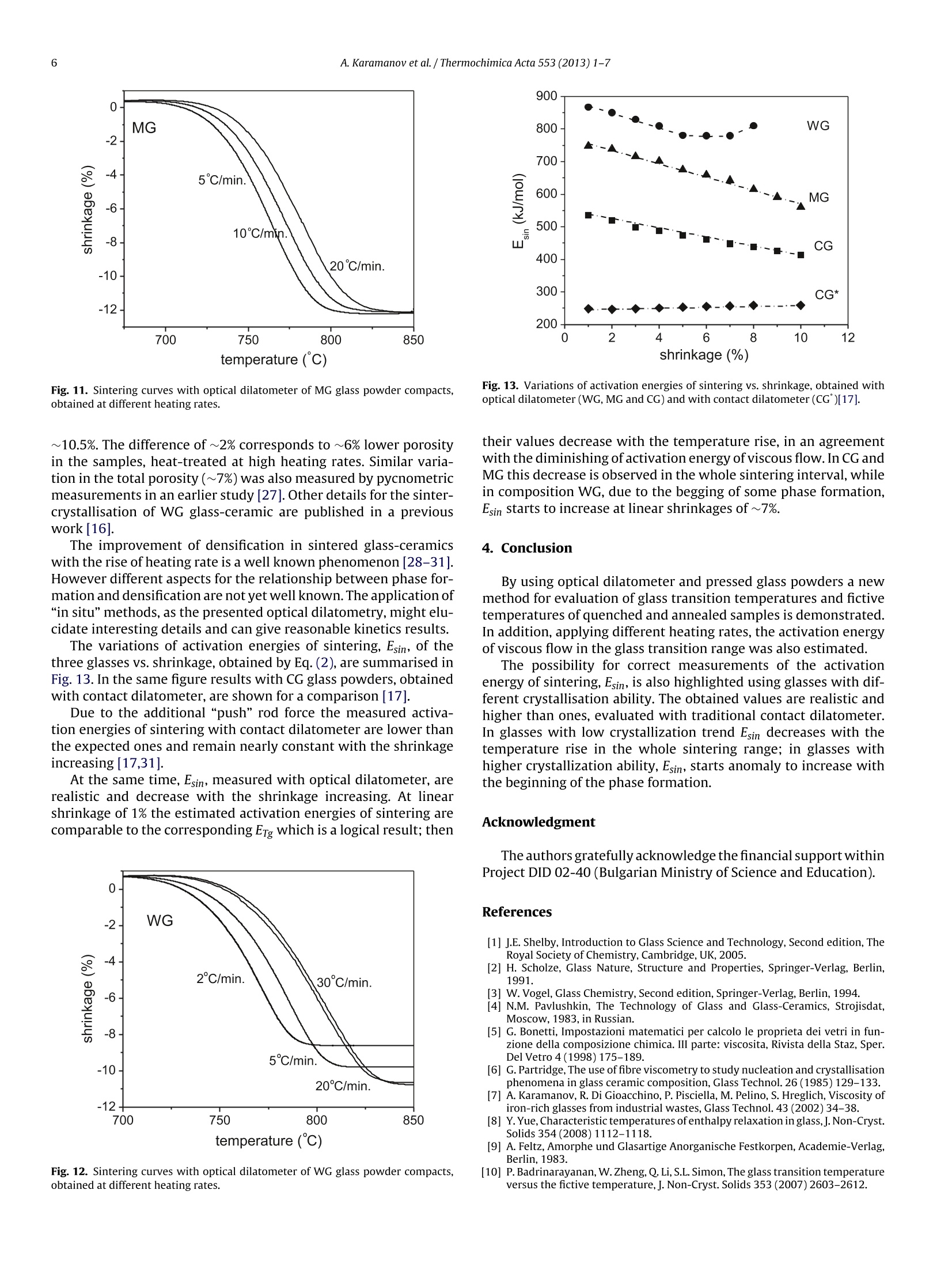
Thermochimica Acta 553 (2013)1-7Contents lists available at SciVerse ScienceDirect 2A. Karamanov et al. / Thermochimica Acta 553 (2013) 1-7 0040-6031/$ - see front matter O 2012 Elsevier B.V. All rights reserved.http://dx.doi.org/10.1016/j.tca.2012.10.006 Thermochimica Acta ELSEVIER journal homepage:www.elsevier.com/locate/tca Glass transition temperature and activation energy of sintering by opticaldilatometry Alexander Karamanova,*, Boris Dzhantov,Mariano Paganellib,Davide Sighinolfi aInstitute of Physical Chemistry, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., Block 11, 1113 Sofia, BulgariaDExpert System Solutions S.r.l. Viale Virgilio 58/L, 41123 Modena, Italy ARTICL EIN F O ABSTRACT Article history:Received 16 December 2011Received in revised form 2 0ctober 2012Accepted 4 October 2012Available online xxx Keywords:DilatometryGlass transitionSintering A new possibility for measurements of glass transition point, fictive temperature and activation energy ofviscous flow in the glass transition range is shown by using a contactless horizontal optical dilatometerand pressed powder samples. The results are compared with ones, obtained with bulk samples andtraditional differential contact dilatometer.The possibility for a correct measurement of the activationenergy of sintering, Esin, is also highlighted by means of glasses with different compositions and variouscrystallization trends. In glasses with low crystallization trend Esin decreases with the temperature risein the whole sintering range; in glasses with higher crystallization ability, Esin, starts anomaly to increasewith the beginning of the phase formation. As a result, at low heating rates the densification is partially inhibited. O 2012 Elsevier B.V. All rights reserved. 1.Introduction The viscosity variations of glass forming melts vs. temperaturehave fundamental significance for the glass science and industry.In order to characterise this viscosity-temperature curve a numberof specific viscosity values are selected as reference points [1-5].These particular viscosities are chosen due to their significance invarious aspects of the industrial and laboratory processing. Since the viscosity of glass forming melts vary by 12-13 ordersof magnitude between the strain point and the melting point itsmeasurements require the usage of different techniques, each ofwhich is restricted to a limited range of viscosity values [1-5]and sometime requires specific sample preparation. In addition,when nucleation and crystallisation or liquid-liquid immiscibilitytakes place during the measurement an increasing of the viscos-ity might be observed [1,6,7], which compromise the obtainedresults. Mostinteresting are the viscosity changes in the glass-transitionrange, where the glass transition temperature, Tg, is the most dis-cussed and studied characteristic temperature. However, despiteof its importance,the methods for determination of Tg are not yetunified in the glass society [1,8]. In a big part oftechnical literature Tg is associated with viscosityof 1013 dPa s or 1013.3 dPa s and its measurements usually is relatedto DSC or dilatometric measurements [1-3,8,9]. In the case of ( * Corresponding author. Tel.: +359 2 979 2552. ) ( E-mail address: kar a ma@ipc.bas . bg(A. Karamanov). ) evaluation by dilatometry well annealed monolithic bulk samplesare heated at 3-5 K/min and Tg is acquired as the onset from thekink in experimental curve; the error in this kind of measurementsis estimated as ±2 to 3 K. For example, Vogel suggest to measurethe glass transition point after cooling of the sample at 1K/minthought the glass transition region and subsequent heating at5 K/min [3]. Similar are also the standards, used in“ExperimentalGlass Station”-Murano (Venice)[5]. The variations of properties and structure in the glass transi-tion region depend not only on the thermal history, but also on thedirectionof heat-treatment (i.e. heating or cooling). Thus, in orderto distinguish the differences, observed at heating and at cooling,the idea for fictive temperature, Tr, has been introduced [8,10,11].According this statement Tg is measured on slow cooling, whilethe corresponding temperature, measured on heating, is definedas T. Practically, Tg is the temperature at which the extrapolatedlines of non-stable solid glass and meta-stable liquid cross, whileTr is the temperature at which the fixed glassy structure will be inan equilibrium. In principle, Tr can be estimated at a temperatureslightly lower than Tg, but in many cases the differences is compara-ble with the experimental error in the dilatometric measurements[8,10]. Although Tg can be measured correctly only on cooling, it oftencontinue to be measured on heating due to difficulties to control aconstant cooling rate (especially in the dilatometric experiments).In addition, the condition for slow cooling cannot be respectedwhen the studied composition is characterised by high trends ofliquid-liquid immiscibility or/and crystallisation. In these cases,instead slow cooling of the samples an annealing treatment at tem-perature lower than Tg, seems to be more appropriated. Table 1Chemical compositions (wt%) of the used glasses. CG G WG MG SiO2 72.0 71.4 49.8 57.7 Al203 0.2 0.6 11.1 B203 5 Fe203 0.2 0.1 4.5 CaO 10.1 9.8 19.5 18 CaF2 - - 2.8 MgO 2.8 3.3 4.0 14.4 Na2O 13.9 13.3 5.0 2.2 K20 0.7 1.3 1.8 P205 - 1.9 - 0thers - 1.4 2.7 The dilatometric measurements have traditionally been per-formed using monolithic samples and push rod dilatometer. Inorder to obtain highly reliable results in measurements above theglass-transition range (with a minimum effect associated with theviscous deformation of samples), it is necessary to use a dilatometerwith a weak measuring force and samples with a large cross-sectional area, so that the measuring pressure is kept at low values.A recent innovation in the field of dilatometry is the chance to carryout measurement of the dimensional variations without touch-ing the sample, but simply by observing its two tips with twohigh-definition cameras [12]. This new equipment gives results,somewhat similar to ones obtained by the widely used hot stagemicroscopy[13-15],but the accuracy of the measurements is con-siderably higher. This contactless dilatometric measurement allows to widen theapplication fields of the traditional contact technique and givesadvantages in terms of sample preparation(i.e. monolithic sampleswith different size can be investigated, as well as pressed powders).It is very important also that, from an instrumental point of view,the thermal shift between purely elastic behaving to visco-elasticdoes not represent an actual limitation for the acquisition of dilato-metric data. As a result, it becomes possible to realize also tests onsamples with a fully viscous behaviour. In this manner it is possi-ble to study correctly the kinetics of sintering processes driven byviscous flow, as well as to evaluate the actual influence of crystalli-sation on the densification process in the production of sinteredglass-ceramics. In the present work some detail for these new possibilities arehighlighted and discussed. 2. Experiments Four glasses having different crystallization abilities, whosechemical compositions are summarized in Table 1, were used.The first composition (labelled CG) and the second composition(labelledG)are similar compositions for container uncoloured glassand window glass, respectively, which was washed,crashed andmilled below 71 um. The third glass (labelled WG) comes fromas-it-is vitrification (i.e. without additives) of bottom ashes fromMunicipal Solid Waste Incinerator [16]. The fourth composition(labelled MG) is a model glass, which was melted at 1400°C for1 h and then instantly quenched in water. The WG and MG fritsalso were milled bellow 71 um. The crystallization trends ofobtained glass powders were inves-tigated by DTA (PerkinElmer Diamond apparatus)at heating rate of20°C/min and using samples of~20 mg. The glass powders were mixed with a 7%PVA solution, unaxiallypressed with pneumatic press"Nannetti”at 40 MPa into“greens”with size 50x5x3-4mm3 which after that were dried at room temperature. The initial total porosities, PT, of the samples (forseries of 5 specimens) were estimated by the relationship: where pa is apparent density and pas is absolute density. pa wascalculated by measuring samples volume with precise electronmicrometre,while pas - using the initial glass powders and argondisplacement Pycnometer (AccuPyc1330). After preliminary 30 min step at 280°C (in order to eliminate thePVA binder) the samples were heat-treated in optical dilatometer“MISURA HSML-ODLT1400"at different heating rates up to 1000°Cor to temperatures, where deformation of the sintered samplesstarts. The results, obtained with CG glass, were compared with earlierdata from analogous composition acquired with contact differen-tial dilatometer“Netzsch 402 ED"[17]. In this previous work bulkand pressed powder samples of container glass were heat-treatedat different heating rates in order to evaluate the glass transitionrange, the activation energy of viscous flow in the glass transitionrange, Erg, and the activation energy of sintering Esin at differentshrinkage values. The activation energies of sintering can be estimated by theCheng equation [17-19]: where Tx are temperatures corresponding to a fixed degree oftrans-formation(in the case of sintering-equal percentages of shrinkage)obtained at different heating rates v. The plot of ln(v/Tx2)versus 1/Txis a line, whose slope corresponds to the activation energy. Similar relationship (i.e. the Kissinger equation) is widelyapplied to study the phase transformation in glasses and othersolids by DTA or DSC [19-21]. In the Kissinger equation Tx corre-sponds to the temperature of maximum transformation rate, Tp,which at non-isothermal crystallisation always is related to thedegree of transformation α~0.61[22] (i.e. in this case the equa-tions of Cheng and Kissinger coincide). However when the degreesof transformation, reached at Tp, vary with the heating rate (whichis possible in some cases of sinter-crystallization)both equationsgive different results. Eq. (2) was also used for the evaluation of activation energy ofviscous flow in the glass transition range, Erg. It can be noted thatthe results, obtained with the Cheng equation, are very similar alsoto ones, calculated with the widely used relationship of Bartenev|22. 3. Results and discussion 3.1. Accuracy and reproducibility ofresults, obtained with opticaldilatometer The work principle of the double beam optical dilatometer isdirect vision of both the tip of specimen captured by two digitalcamera equipped with high magnification and long working dis-tance optical systems and controlled by a micrometric slide. Sinceit is not possible to push the optical magnification beyond the wave-length of light, the maximum resolution of the optical system canbeslightly higher than the wavelength of light. In the equipmentused for this research work the resolution is 0.5 micron per pixel. The horizontal optical dilatometer utilized in this work, uses~50 mm long sample, displaced horizontally into the furnace andlighted by two beams of blue light with a wavelength of 478nanometers. Two digital cameras capture the images of the last fewhundreds microns of each sample tip. Measuring both ends of the Fig. 1. Dilatometric traces of continuous experiment with optical dilatometer andbulk window glass sample in the glass transition range (heating rate of5°C/min andcooling rate of 2°C/min). specimen with two beams of light at the same time without mak-ing contact, it provides an absolute“in situ”measurement. Using aspecimen with a total length of 50 mm the precision is of one partover 100,000. Even though the resolution is lower than push rod dilatome-ters or interferometers, the elimination of any pressure on thespecimen broadens the range of possibilities of measurementthermo-mechanical behaviours up to a temperature range wherethe material behave in a visco-elastic manner. The curves obtainedon solid glass specimens resemble the curves obtained with amechanical dilatometer. The difference is that the shortening ofthe sample after reaching the maximum expansion is only given bysurface tension,which, thanks to the lowering ofviscosity, becomesable to round the tip ofthe specimen. This feature makes it possi-ble to measure the sintering behaviour on glass powder compacts,which is unfeasible on pushrod dilatometers and is not satisfactoryon single optic traditional heating microscopes, because of a muchlower resolution of the image. Practically, to reach the same res-olution on a heating miscroscope it would be necessary to use acamera with 105 pixel in line. In order to demonstrate the reproducibly of the results, obtainedwith optical dilatometer, two continuous experiments were madeusing bulk and pressed powder samples from window glass (G). Thebulk sample (with size 50×5×3mm3) was obtained after cuttingand polishing,while the powder sample was prepared accordingto the procedure described in the experimental part. Both sampleswere heat treated with heating rate of 5°C/min up to 580°C andthen cooled at 2°C/min to 400°C[8]. After that 3 identical cycles ofheating with 5°C/min and cooling with 2°C/min were carries outin the temperature interval 400-580°C. The obtained dilatometricresults as function of the temperature are shown in Figs. 1 and 2for bulk and powder samples, respectively; dilatations vs. time arepresented in the corresponding insets. Fig. 1 demonstrates that during the first run of heating and cool-ing some annealing carries out and as a result the curve, obtainedon cooling is bellow one, obtained on heating. However, duringthe subsequent three runs the curves on heating and on coolingoverlaps entirely, which demonstrate a very good reproducibility.Tfat heating with 5°C/min was evaluated at 551±2°C, while Tg atcooling with 2°C/min was evaluated at 544±2°C. The results with pressed powder sample (Fig. 2) show similarresults, but highlight small densifications during each of the runs(at about 0.1% shrinkage). This phenomenon probably mightbe explained by a negligible sintering or by smoothing of thepressed grains. Notwithstanding of the observed “creep”Tg and Tf Fig. 2. Dilatometric traces of continuous experiment with optical dilatometer andwindow glass powder compact in the glass transition range (heating rate of5 °C/minand cooling rate of 2°C/min). temperatures, measured during all 4 runs, vary within ±3°C andare comparable to ones, observed with the bulk sample. 3.2. Evaluation of glass transition temperature, fictive. temperature and activation energy of viscous flow in theglass-transition range Fig.3 presents dilatometric traces in the glass transition rangeof CG glass, obtained with a contact dilatometer at heating with5°C/min using bulk and pressed powder samples [17]. The curveof the bulk sample shows fictive temperature at 561±2°C anddilatometric softening point at 615±2°C, which is a typical perfor-mance for this composition [2,5]. However, when powder sampleis investigated with contact dilatometer the both characteristictemperatures cannot be identified due to the “push rod”pressure,applied on the sample. Fig. 4 shows traces of pressed CG powder sample, studied withoptical dilatometer at heating rate of 5°C/min up to 600°C, 5 minholding and cooling at 5°C/min. This experiment was repeated withthe same sample 5 times in 5 different days and the obtained resultshighlight identical accuracy as one of the experiment with the con-tinuous heating and cooling (see Fig.2). The average values of T andTg from this series of 5 experiments were estimated as 557±2°Cand 560±3°C,respectively. Fig. 3. Dilatometric traces with contact dilatometer in the glass transition range forbulk sample and pressed powder compact of CG glass at heating rate of 5°C/min[17]. Fig. 4. Dilatometric traces with optical dilatometer and CG glass powder compactin the glass transition range at heating and cooling rates of 5°C/min. The made experiments with window and container glass pow-ders expand the application range ofoptical dilatometry in the glasstransition range. It is highlighted that the results, obtained withpressed powders and optical dilatometer,are comparable to onesof bulk samples, investigated with contact or optical dilatometers. The activation energy of viscous flow in the glass transitionrange, Erg, can be evaluated by Eq. (2) using data obtained at dif-ferent heating rates. Usually, these results are acquired with DTAor DSC, as well as with contact dilatometer and bulk glass samples.In this study for first time is demonstrated that similar data can becorrectly obtained also by using pressed glass powder and opticaldilatometer. The dilatometric traces at different heating rates of CG, WG andMG glass powders compacts are presented in Figs. 5,6 and 7,respec-tively. The CG and WG samples demonstrate an ordinary behaviour,typical for annealed glasses, and the Tf variations correspond toETg of585 kJ/mol for CG and 887 kJ/mol for WG (Fig. 8). These val-ues are in a good agreement with the data,obtained for similarcompositions with other techniques [2,5,23]. Due to the high quenching rate, the initial MG glass powdersshow results (presented with traced lines in Fig. 7), which aretypical for un-annealed glasses [1,9] and demonstrate some densi-fication after the region of strain point temperature. For this reasonadditional experiments were made, using preliminary annealed for Fig.5. Dilatometric traces in the glass transition range of CG glass powder compacts,obtained at different heating rates. Fig. 6. Dilatometric traces in the glass transition range of WG glass powder com-pacts, obtained at different heating rates. Fig. 7. Dilatometric traces in the glass transition range of MG glass powder com-pacts, obtained at different heating rates (solid lines - annealed glass powders;traced lines -quenched glass powders). 1 h at 600°C glass powders. The results are presented in Fig. 7 withsolid lines. The densities of glass powders, before and after annealing, weremeasured with“AccuPyc-1330"gas pycnometer and the resultelu-cidate that, due to the heat-treatment the density increases from Fig.8. Ln(v/Tx) versus 1/Tx plots and activation energies of viscous flow in the glasstransition range. Fig.9. DTA results of the three glass powders, obtained at 20°C/min. 2.665±0.003 to 2.698±0.003 g/cm3.XRD analysis shows that nocrystallization carries out during the made thermal cycle, whichmeans that the observed density rise is not related to a crystalli-sation process. Similar variations because of annealing are alsoreported for other glasses [4,22,24]. The kinetics results, obtained with the annealed MG glassespowders correspond to Erg of 747 kJ/mol (see Fig. 8). The com-parison of the values of activation energy of viscous flow in glasstransition range of the three studied glasses reasonable demon-strates an increase of Erg with the rise of Tr. In addition, the results with the quenched MG powders high-light an interesting possibility for estimation of the temperatureregion of glass transition even for very rapidly cooled glasses. Thispeculiarity might be very useful in many cases (small amount ofthe glass samples, compositions with high crystallization ability,etc.). It can be noted that similar experiments are not possible witha bulk sample, because it obligatory can be annealed before cut-ting and polishing; otherwise it will crash during the mechanicaltreatments. 3.3. Evaluation ofactivation energy ofsintering DTA traces of the three glass powders, obtained at 20°C/min,are shown in Fig. 9. CG presents only a glass-transition and theestimated Tr is in a good agreement with one of the correspondingdilatometric plot. The result with WG highlight a glass transitionin the region of 650°C, oxidation exo-effect (due to surface Fe+into Fe+ transformation) [25,26] at about 800°C and crystallisa-tion exotherm with peak temperature at 875°C. For MG glass arepresented results with both quenched glass powders (labelled asMG-q) and annealed glass powders (labelled as MG-a). It can benoted that the observed differences in the range 550-650°C aretypical for the variations of the DTA signals between annealed andvery fast quenched glasses [9]. At the same time the crystallisationexo-effects of both samples are practically identical. Initial absolute Green apparent Initial porosity porosity (g/cm3) density(g/cm) (vol%) CG 2.496±0.002 1.613±0.009 35.4±0.3 G 2.451±0.002 1.622±0.008 33.8±0.3 WG 2.669±0.003 1.774±0.007 33.5±0.3 MG 2.665±0.003 1.701±0.011 36.1±0.4 The temperature interval between the crystallisation onsettemperature and T, which might be used as an indication for sin-terability, is ~175 °C for WG and ~190°C for MG. Considering also,that the Fe2+oxidation in WG leads to an increasing of the viscos-ity, might be assumed that MG glas powders can be characterisedwith a better sinterability. The results for the absolute and apparent densities of the“greens”and the corresponding initial porosities are summarisedin Table 2. Assuming that no residual porosity remains in the sin-tered samples, no crystallization and/or deformation carries outduring the thermal treatments and no anisotropy is observed (i.e.the shrinkages in the three directions are identical) these porosityvalues can correspond to maximum linear shrinkages of ~13.5% forCG,~12.7% for WG and~13.9%for MG. Fig. 10 presents the sintering curves of CG glass powder compactat 10C/min, obtained with contact dilatometer (traced lines) [17]and optical dilatometer (solid lines), respectively. It is well seenthat the sintering, measured with contact dilatometer, is acceler-ated: the densification starts earlier and caries out at higher rate.In addition, when contact dilatometer is used significant shrink-age anisotropy is observed and, in many cases, the end of sinteringoverlaps with the beginning of deformation. Contrary, the reachedlinear shrinkage of~12.5% with the optical dilatometer is in a goodagreement with the expected value. Figs. 11 and 12 summarise sintering curves with opticaldilatometer at different heating rates of MG and WG, respectively.In the model glass, as well as in CG, the sintering is not seriouslyinfluenced by crystallisation process and completed before for-mation of distinguished amount of crystal phase. As a result, thefinal degrees of densification at different heating rates are similar(~12.5-13%). Contrary, in WG composition, where the crystallisation carriesout at higher viscosities, the sinter-ability depends on the heat-ing rate. At 2°C/min the final linear shrinkage is ~8.5%, whereasat 20 and 30°C/min the sintering completes at linear shrinkage of Fig. 10. Sintering curves of CG glass powder compacts at 10 C/min, obtained withcontact dilatometer (traced lines) [17] and optical dilatometer (solid lines). Fig. 11. Sintering curves with optical dilatometer of MG glass powder compacts,obtained at different heating rates. ~10.5%. The difference of ~2% corresponds to~6% lower porosityin the samples, heat-treated at high heating rates. Similar varia-tion in the total porosity (~7%) was also measured by pycnometricmeasurements in an earlier study [27]. Other details for the sinter-crystallisation of WG glass-ceramic are published in a previouswork [16]. The improvement of densification in sintered glass-ceramicswith the rise of heating rate is a well known phenomenon [28-31].However different aspects for the relationship between phase for-mation and densification are not yet well known. Theapplication of“"in situ”methods, as the presented optical dilatometry, might elu-cidate interesting details and can give reasonable kinetics results. The variations of activation energies of sintering, Esin, of thethree glasses vs. shrinkage, obtained by Eq.(2), are summarised inFig.13. In the same figure results with CG glass powders, obtainedwith contact dilatometer, are shown for a comparison [17]. Due to the additional“push"rod force the measured activa-tion energies of sintering with contact dilatometer are lower thanthe expected ones and remain nearly constant with the shrinkageincreasing [17,31]. At the same time, Esin, measured with optical dilatometer, arerealistic and decrease with the shrinkage increasing. At linearshrinkage of 1% the estimated activation energies of sintering arecomparable to the corresponding ETg which is a logical result; then Fig. 12. Sintering curves with optgcUical dilatometer of WG glass powder compacts,obtained at different heating rates. Fig. 13. Variations of activation energies of sintering vs. shrinkage, obtained withoptical dilatometer (WG, MG and CG) and with contact dilatometer (CG )[17]. their values decrease with the temperature rise, in an agreementwith the diminishing of activation energy of viscous flow. In CG andMG this decrease is observed in the whole sintering interval,whilein composition WG, due to the begging of some phase formation,Esin starts to increase at linear shrinkages of ~7%. 4. Conclusion By using optical dilatometer and pressed glass powders a newmethod for evaluation of glass transition temperatures and fictivetemperatures of quenched and annealed samples is demonstrated.In addition, applying different heating rates, the activation energyof viscous flow in the glass transition range was also estimated. The possibility for correct measurements of the activationenergy of sintering, Esin, is also highlighted using glasses with dif-ferent crystallisation ability. The obtained values are realistic andhigher than ones, evaluated with traditional contact dilatometer.In glasses with low crystallization trend Esin decreases with thetemperature rise in the whole sintering range; in glasses withhigher crystallization ability, Esin, starts anomaly to increase withthe beginning of the phase formation. Acknowledgment Theauthors gratefully acknowledge the financial support withinProject DID 02-40 (Bulgarian Ministry of Science and Education). ( References ) ( [1] J.E. Shelby, Introduction to Glass Science and Technology, Second ed i tion, Th e R oyal Society of Chemistry,Cambridge, UK, 2005. ) ( [ 2 ] H. S cholze, Glass Nature, S t ructure and Pr o perties, Springer-Verlag, Ber l in,1991. ) [3] W. Vogel, Glass Chemistry, Second edition, Springer-Verlag, Berlin, 1994. [4] N.M. Pavlushkin, The Technology of Glass and Glass-Ceramics, Strojisdat,Moscow, 1983, in Russian. [5] G. Bonetti, Impostazioni matematici per calcolo le proprieta dei vetri in fun-zione della composizione chimica. III parte: viscosita, Rivista della Staz, Sper.Del Vetro 4(1998)175-189. [6] G.Partridge, Theuse of fibre viscometry to study nucleation and crystallisationphenomena in glass ceramic composition, Glass Technol. 26(1985) 129-133. ( [7] A. Karamanov, R. Di Gioacchino, P. Pi s ciella, M. P e lino, S . H reglich, Viscosity of i ron-rich g lasses from industrial w astes, Glass Technol.43 (2 0 02) 34-38. ) ( [8] Y. Yue, Characteristic temperatures ofenthalpy relaxation in glass, J. Non-Cryst. S olids 354 (2008)1112-1118. ) [9] A. Feltz, Amorphe und Glasartige Anorganische Festkorpen, Academie-Verlag,Berlin, 1983. [10] P.Badrinarayanan, W.Zheng, Q. Li, S.L. Simon, The glass transition temperatureversus the fictive temperature,J.Non-Cryst. Solids 353 (2007)2603-2612. ( [11] M .I. Ojovan, Viscosity and g l ass t r ansition in amorphous oxi d es, Adv . Cond. M atter Phys. 2008(2008)1-23. ) ( [12] M . P aganelli, Using t he o ptical dilatometer, Am. Ceram. S o c. B u ll 81 ( 2 002) 25-30. ) ( [13] A.R. Boccaccini, B. Hamann, In-situ h igh temperature optical microscopy. A r eview,J.Mater. Sci. 34(1999) 5419-5436. MI ) ( [14] M .J.Pascual,A.Duran,M.O.Prado,A new method for determining fixed viscosity p oints of glasses, Phys. Chem. Glasse s 46 (2005 ) 12-20. ) ( [15 ] ] F.G. Raether,Current state of in situ measuring methods for the control of firing p rocesses,J. Am. Ceram. Soc. 92 (2009)146-152. ) ( [ 1 6] L . Schabbach,F . Andreola,E. Karamanova, I. Lancellotti, A. Karamanov, L. Barbi- e ri, Integrated approach to establish the sinter-crystallisation ability of gla s ses f rom secondary r a w material, J. Non-Cryst. Solids 357 (2011) 1 0-17. ) ( [17] A . Karamanov, M. Aloisi, M. Pelino, Sintering behaviour of a glass obtained from M SWI ash,J. Eur. Ceram. Soc. 25 (2005)1531-1540. ) ( [18] H . Chen, A method for e v aluating viscosities of metallic glasses from the r ates o f thermal transformations, J. Non-Cryst. Solids 27 ( 1 978) 257-263. ) ( [19] M .J. Starink, The determination o f activation energy from linear heating rate e xperiments: a comparison of t h e a c curacy of i s oconversion m e thods, Ther- m ochim.Acta 404(2003)163-176. ) [20] H. Kissinger, Reaction kinetics in differential thermal analysis, Anal. Chem. 29(1957)1702-1706. [21] ]C.S. Ray, D.E. Day, Nucleation and crystallization in glasses as determined byDTA. Ceramic transactions.Nucleation and crystallization in liquids and glasses,Am. Ceram. Soc. 30 (1992)207-224. [22] J. Gutzow, Schmelzer, The Vitreus State, Thermodynamics,Structure, Rheologyand Crystallization, Springer-Verlag, Berlin, 1995. [23] L. Barbieri, A.C. Bonamartini, I. Lancellotti, Alkaline and alkaline-earth silicateglasses and glass-ceramics from municipal and industrial wastes,J. Eur. Ceram.Soc.20(2000)2477-2483. [24] A. Karamanov,R.Di Gioaccino,P. Pisciella,M. Pelino,Glass transformation rangeof iron-rich glass and glass-ceramics determined by different methods, GlassTechnol.42(2001)126-129. [25] A. Karamanov,M. Pelino, Crystallization phenomena in iron rich glasses,J.Non-Cryst. Solids 281(2001)139-151. [26] M.Pelino,A. Karamanov,Reply to"Comment on""The influence of the Fe+/Fe2+ratio on the crystallization of iron-rich glasses from industrial wastes", J. Am.Ceram. Soc. 84 (2001)2742-2743. ( [27] E . Karamanova, L. Schabbach, B. Ranguelov, G. Avdeev, I. Lancellotti, A. Ka r a- manov, Optimization of the h e at-treatment regime of a sintered gl a ss-ceramic from MSWA bottom ash, in: 10-th SGEM Con f erence, Alb e na, Bul g aria,vol. II, 2010, p p.893-900. ) [28] R. Muller, On the kinetics of sintering and crystallization of glass powders,Glastech. Ber. Glass. Sci. Technol. 67C (1994) 93-99. [29] A.R. Boccaccini, W. Stumpfe,D.M.R. Taplin, C.B. Ponton, Densification and crys-tallization of glass powder compacts during constant heating rate sintering,Mater.Sci. Eng.A219(1996)26-31. [30] E. Bernardo, L. Esposito, E. Rambaldi,A. Tucci, Y. Pontikes, G.N. Angelopou-los, Sintered esseneite-wollastonite-plagioclase glass-ceramics from vitrifiedwaste,J. Eur. Ceram. Soc.29(2009)2921-2927. [31] A. Karamanov, M. Pelino, Sinter-crystallization in the system diopside-albite,part II. Kinetics of crystallization and sintering, J. Eur. Ceram. Soc. 26 (2006)2519-2526. 采用新型的光学投影法热膨胀仪和压实粉体样品形式,使得对玻璃化转变过程中粘性流玻璃化转变点、构型平衡温度和激活能的非接触测量成为了可能。本文介绍了采用这种非接触测试方法的测试结果,并与其它传统接触式热膨胀仪的测试结果进行了对比。通过对玻璃不同组分及各种结晶趋势的测试发现,这种非接触测量方法在烧结过程中的测试准确性方面有突出表现。对于具有低结晶趋势的玻璃,整个烧结阶段激活能随着温度上升而减小;而对于较高结晶稳定性玻璃,激活能在相变初期开始阶段会异常增加。做为结论,低速加热速率的稠化过程是部分禁止的。
确定




还剩5页未读,是否继续阅读?

产品配置单

上海依阳实业有限公司为您提供《玻璃,晶体材料,固体,粉末中热膨胀,位移,变形检测方案(热膨胀仪)》,该方案主要用于玻璃中热膨胀,位移,变形检测,参考标准--,《玻璃,晶体材料,固体,粉末中热膨胀,位移,变形检测方案(热膨胀仪)》用到的仪器有光学投影法高温热膨胀仪
推荐专场






相关方案
更多
该厂商其他方案
更多