








摘要 评述了古陶器热释光测定年代的两个标准方法——“细粒混合矿物技术”和“粗粒石英技术”, 以及古瓷器热释光测定年代新技术——“前剂量饱和指数法”的研究和进展. 着重分析了年代测定中“古剂量”和“年剂量”这两个主要参数的测量原理、技术和方法, 对影响古剂量和年剂量测量准确性的一些复杂因素和解决的方法进行了分析和讨论. 近50年的研究结果表明, 热释光测定年代在古陶瓷真伪鉴定上已经取得了很大的成功, 但是年代测定的精确度和准确度还达不到理想的要求, 需要深入研究。
方案详情

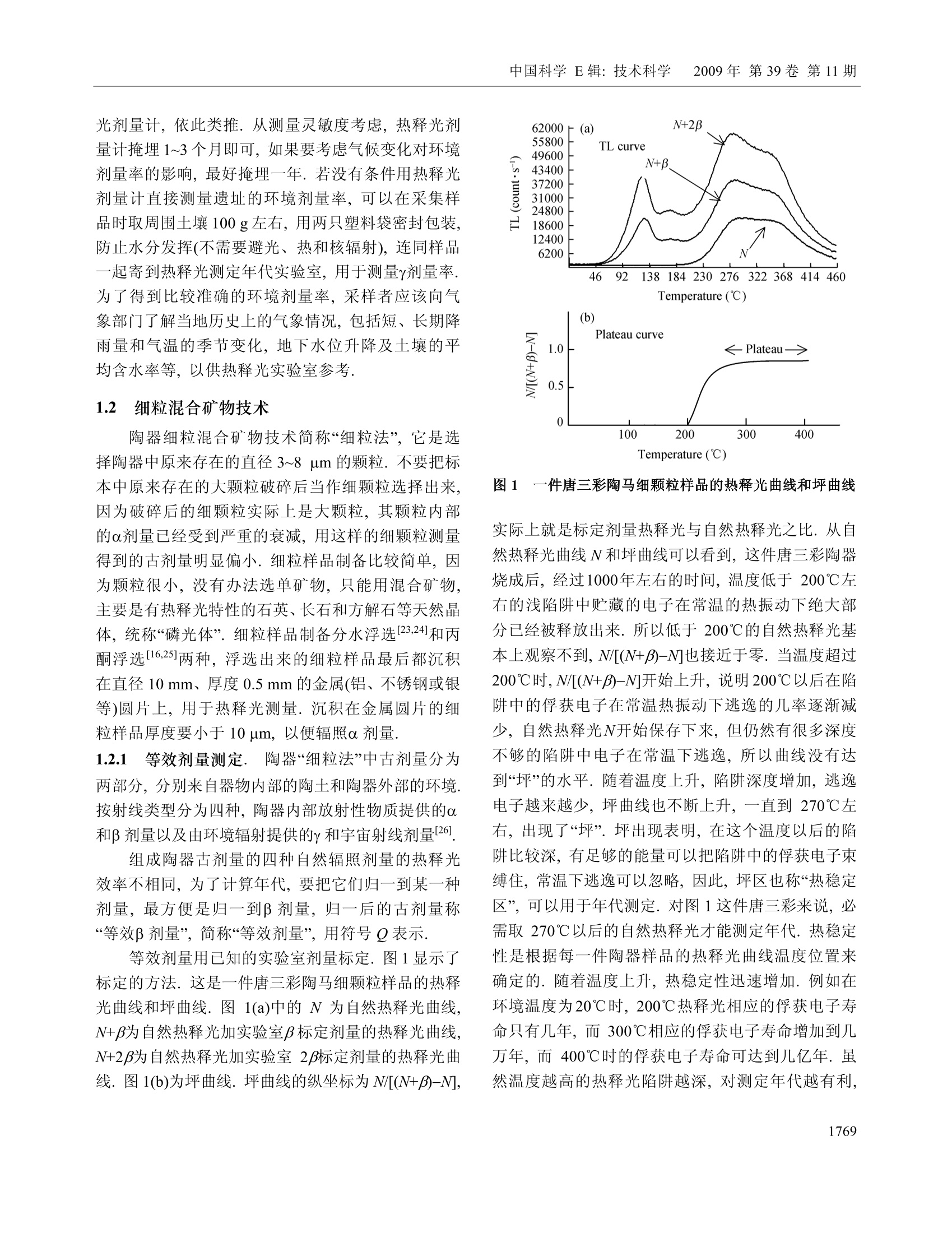
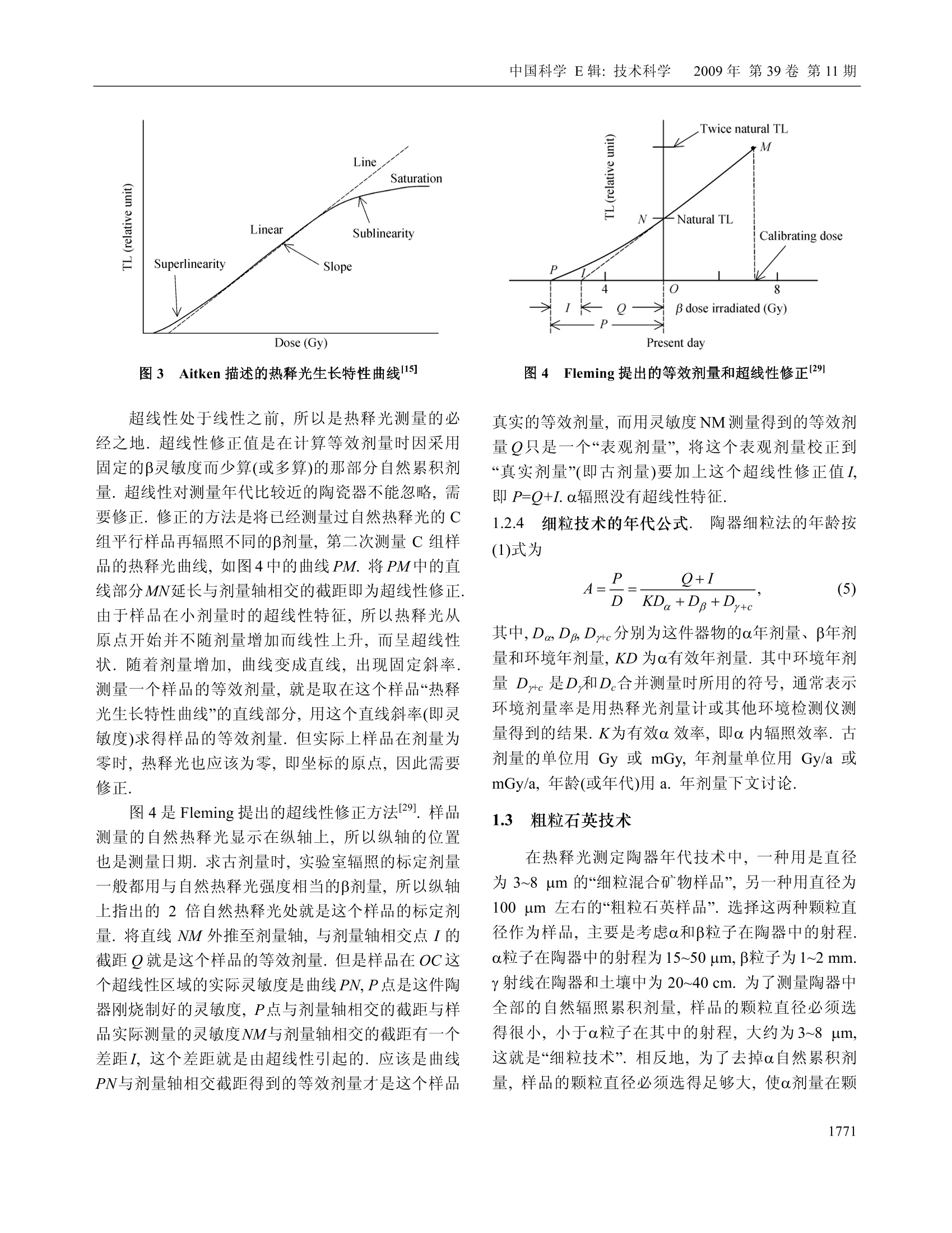
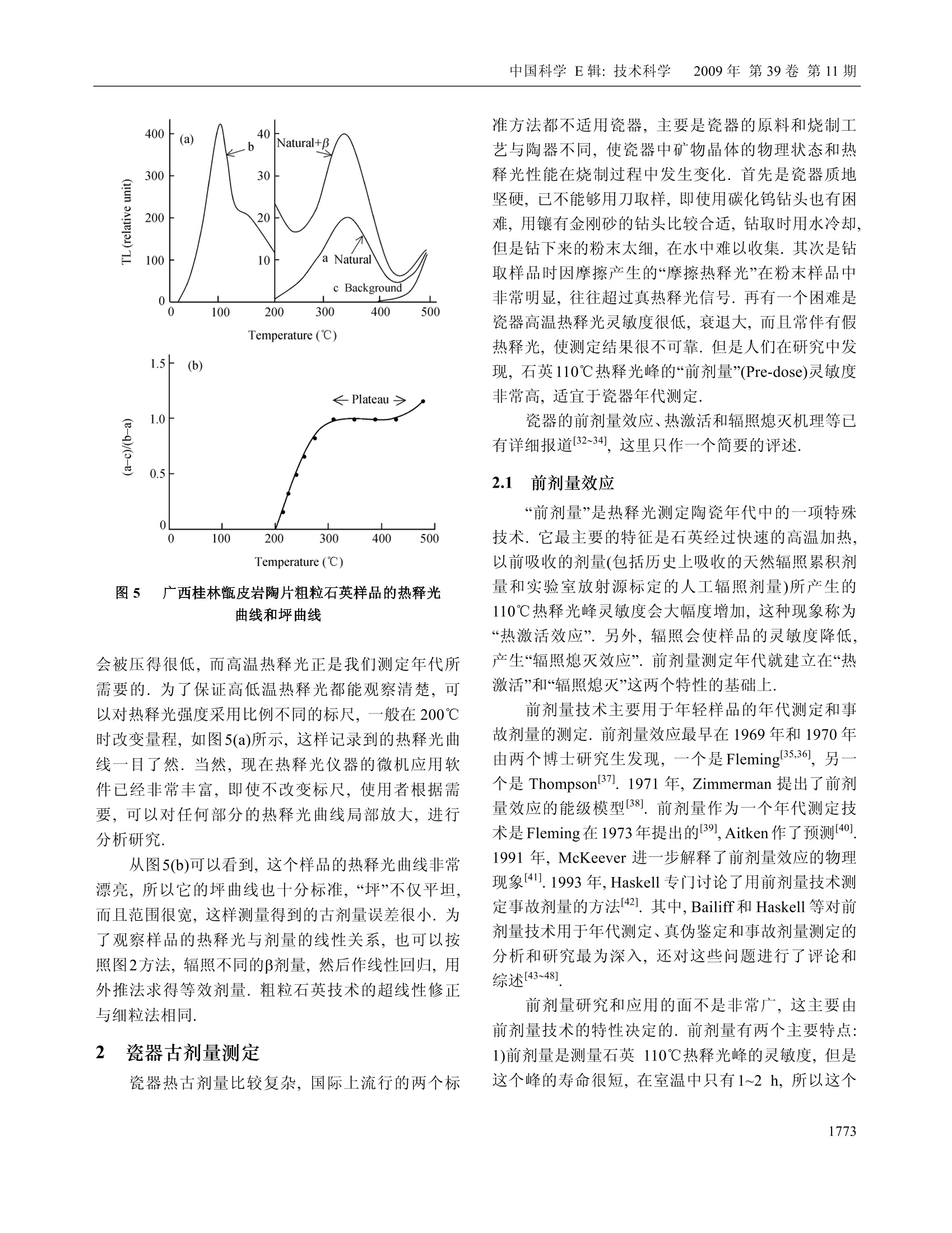
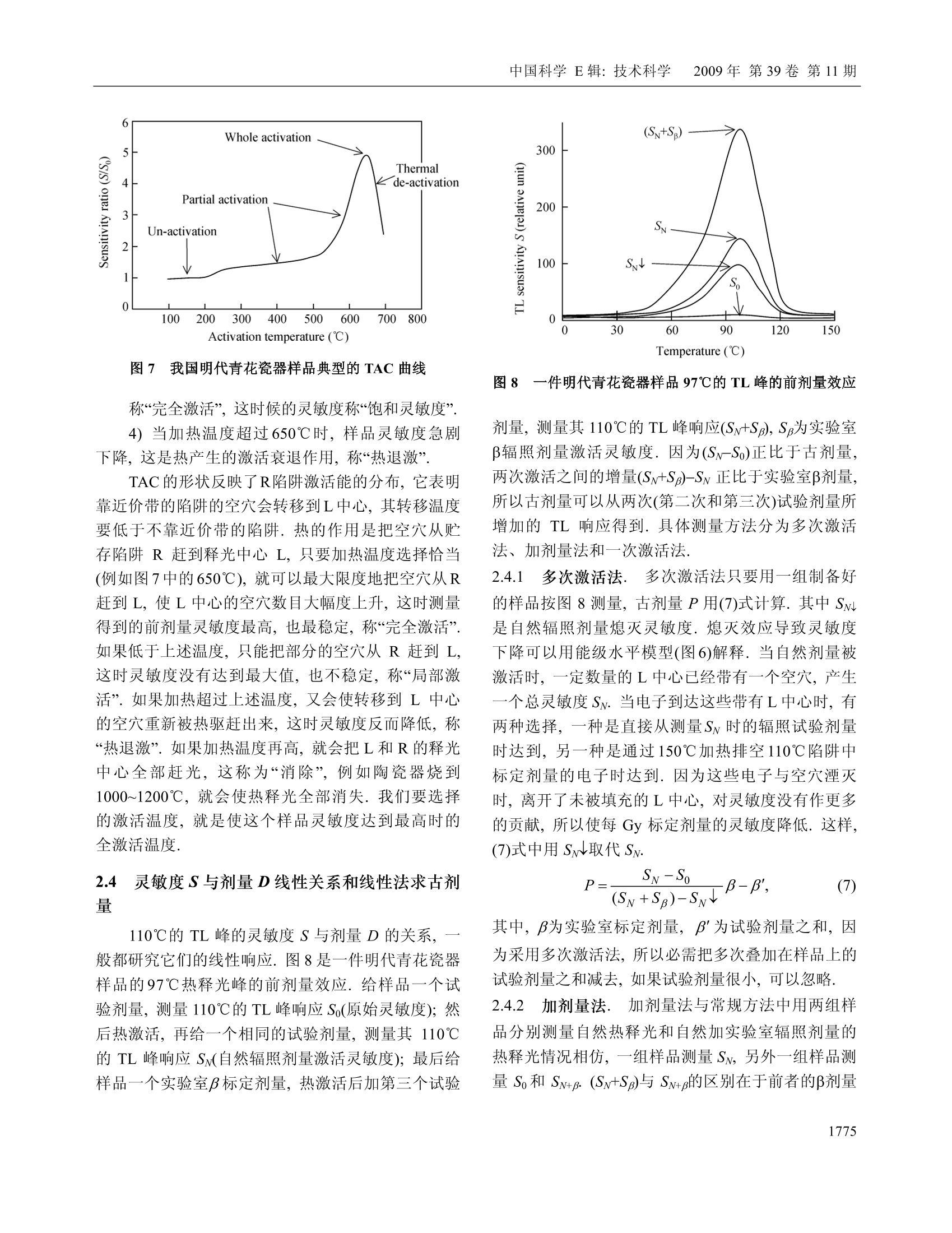
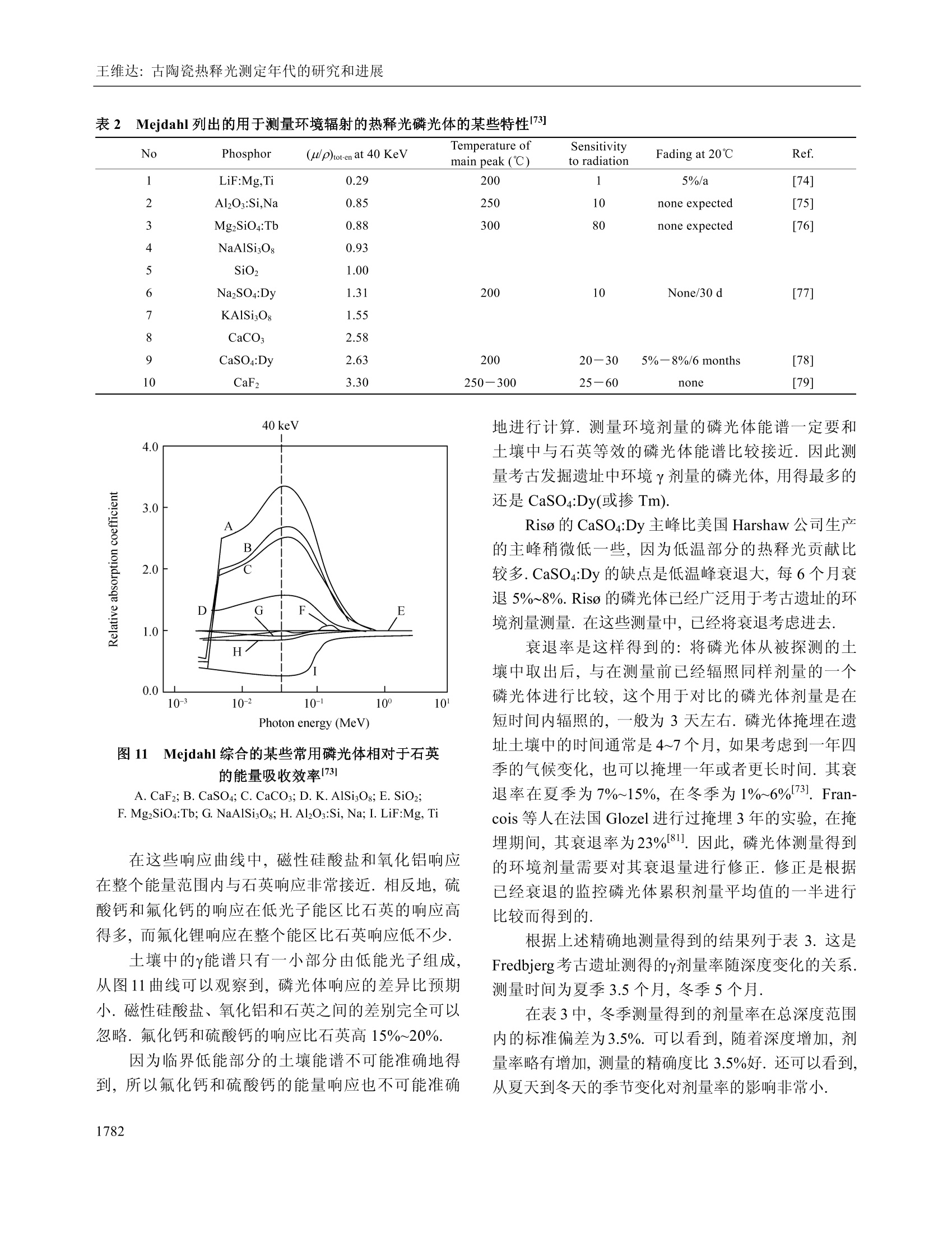
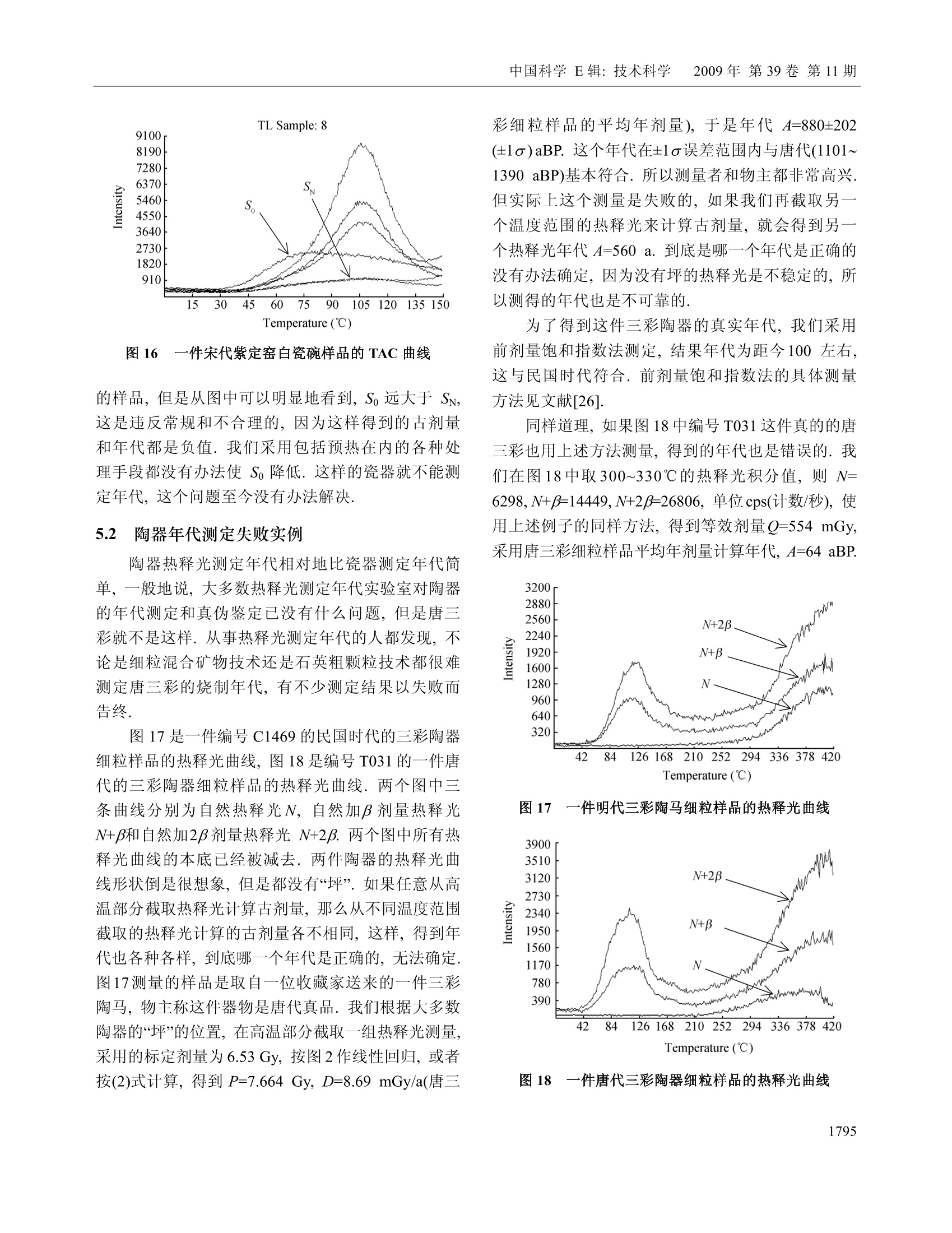
中国科学E辑:技术科学 2009年第39卷第11期:1767~1799www.scichina.com tech.scichina.com《中国科学》杂志社SCIENCE IN CHINAPRESS 王维达:古陶瓷热释光测定年代的研究和进展 古陶瓷热释光测定年代的研究和进展 王维达 上海博物馆文物保护与考古科学实验室,上海200050 *E-mail: labwangwd@163.com 收稿日期:2008-08-05;接受日期:2009-02-03 摘要 评述了古陶器热释光测定年代的两个标准方法—“细粒混合矿物技术”和“粗粒石英技术”以及古瓷器热释光测定年代新技术—“前剂量饱和指数法”的研究和进展.着重分析了年代测定中“古剂量”和“年剂量”这两个主要参数的测量原理、技术和方法,对影响古剂量和年剂量测量准确性的一些复杂因素和解决的方法进行了分析和讨论.近50年的研究结果表明,热释光测定年代在古陶瓷真伪鉴定上已经取得了很大的成功,但是年代测定的精确度和准确度还达不到理想的要求,需要深入研究. 关键词 热释光测定年代古陶器古瓷器 自1960年瑞士泊尔尼大学的 Grogler 等人和美国加利福尼亚大学的 Kennedy 等人第一次从古陶器粉末样品中检测到热释光(Thermoluminescence, TL)至今快50年了11.21.近50年来,热释光测定年代取得了很大的发展,在考古学和地质学上发挥了重要的作用[3~122].作为测定年代工具,热释光在考古学上的最大成功之处就是古陶瓷艺术品的年代测定和真伪鉴别,这是其他任何科学检测方法都不能比拟的. 热释光是一种物理现象,它是固体受到电离辐射激发而积蓄起来的辐射能在加热过程中以光子形式释放出来的一种磷光.利用这种现象,可以测定陶瓷器和砖瓦等物体的烧制年代,也能测定地质材料最后一次受热以来所经过的时间,是20世纪60年代发展起来的一项测定考古和地质年代的新技术.这项新技术的出现,首先对欧美博物馆的藏品产生了强烈的冲击,它以迅雷不及掩耳之势,一下子“揪出”了一批隐藏在国际著名博物馆中的古陶器赝品I3~5],揭开了争论几十年的古陶器真伪之谜,在文物收藏 界引起了巨大的轰动,被人们誉为“热释光革命”. 因为热释光是用辐射吸收剂量来量度的,所以热释光测定年代主要是测量一件器物的“古剂量”(即自陶瓷器烧成以来到测定年代为止所吸收的自然辐照累积剂量)和“年剂量”(即陶瓷器一年吸收的辐照剂量)这两个参数,只要将这件陶器的古剂量除以它自己的年剂量,就得到了这件器物最后一次受热以来所经过的时间或者烧制年代. 年代测定的原理虽然非常简单,但是要准确测得一件陶瓷器的古剂量和年剂量却不是一件容易的事情,因为影响这两个参数准确测定的因素很多.几十年来,科学家就是在为研究这些复杂因素而努力. 热释光测定年代主要是研究古剂量的测定,因为年剂量是用放射性和成分分析方法测量,当然也包括用人造热释光磷光体即热释光剂量计(TLD)测定 ( 引用格式:王维达.古陶瓷热释光测定年代的研究和进展.中国科学E辑:技术科学,2009,39(11):1767—1799 Wang W D. St u dy and pro g ress of the thermoluminescence dating of the anci e nt pottery and porcelain. Sci China Ser E -Tech Sci, 2009, 52(6):1613—1640, doi: 10.1007/s11431-009-0106-4 ) 器物内部和外部的辐照剂量率,但是这与用热释光测定器物古剂量的含义不同. 1 陶器古剂量测定 陶器热释光测定年代虽然从1960年就开始了,但在20世纪60年代,还是处于一个发展时期.20世纪60年代早期发表的热释光测定年代文章,主要强调这项技术的发展潜力,同时又报道了一些原来想象不到的复杂情况,世界上大多数热释光实验室到1966年才与测定地质年代与地层学发生关系.在以后的十几年中,突出表现在考古年代测定上的应用和测量方法的研究.其中,牛津大学考古研究室的Aitken 等人,京都大学的Ichikawa,威斯康星大学的Mazess, 费城大学的Ralph 和丹麦的 Riso 北欧热释光实验室的 Mejdahl 等人对古陶器自然辐照累积剂量测定、年剂量测定、引起年代误差的各种复杂因素的分析和修正进行了深入和广泛的研究,为古陶器热释光测定年代奠定了基础[7~15].当时,国际上有一半的论文是他们发表的.他们提出的“细粒混合矿物技术”(fine-grain technique)16]和“石英粗粒技术”(quartzinclusion technique)17,经过几十年的发展,已经被公认为古陶瓷热释光测定年代的两个标准方法.我国在20世纪60年代由中国科学院地质研究所率先研究了地质年龄测定.20世纪70年代由上海博物馆创建了国内第一个古陶瓷热释光测定年代实验室18~20]. 1.1 陶器标本采集 陶器标本是采集遗址中的陶器碎片,这在各个历史时期的考古遗址中是非常丰富的,样品采集应该在考古遗址发掘中同时进行,这样采集的样品能够比较真实的反映陶器及其和它长期共存的环境现状.样品采集正确与否,对热释光测定年代影响很大,所以一定要严格按照下列要求采集[15,21,22] 1.1.1 遗址. 被采集样品遗址的考古学性质一定要明确,遗址保存完好,没有经过扰乱而且在距地表30 cm以下. 1.1.2 标本数量和大小. 1)测定一个时代的样品必须采集同时代的陶器碎片3~6块.这3~6块碎片应该是同一时代的平行样品.采用平行样品可以减少测定一个孤立样品时可 能产生的差错. 2)同一时代的样品最好是不同类型结构,如粗陶、细陶、泥质陶、夹沙陶、黑陶和红陶等. 3)每一块残片的厚度应该超过 7 mm, 长、宽(或直径)超过 30 mm,尺寸大的更好,以保证标本内部β剂量的完整性. 1.1.3 采样要求. 1)样品应该在遗址发掘时采集,,一个遗址不同层位、探方或者坑位应分别采样,不能混和. 2)采集的碎片应该距离坑位棱角、不同土质交界处和底部岩石至少30 cm, 避免不同放射性物质提供不同y剂量,给确定环境年剂量造成困难. 3)碎片最好在均质土壤中采集,碎片周围要没有大石块、建筑物碎片或者贝壳和骨头的堆积物.因为大石块会对射线产生散射,石块越大,散射越严重,会扰乱环境剂量率的估计. 4)不要采集暴露在地表或者地表 30 cm以内的碎片,这些的样品会受到阳光照射,或者被人翻动,使样品古代累积起来的热释光受到影响. 1.1.4 样品贮藏和处理. 1)样品出土后,不要用水冲洗,应该立即用黑色塑料袋密封包装,再将其封装在第二只塑料袋内,以防止样品中水分挥发,因为样品的含水率对古剂量影响很大. 2)样品在包装、运输和贮藏过程中应该避免紫外线、红外线、X,y和β射线辐照,防止高温(超过100℃)加热. 3)过去出土的陶片,如果水分已经挥发掉或者没有避光包装且长期置于日光下的样品不能用于测定年代. 1.1.5 环境热释光剂量计的掩埋. 为了测定样品出土处的环境剂量率,必须把环境剂量计掩埋在样品出土处.掩埋的地方应该是原来样品出土处相同的周围土壤,不要把30 cm 以外的土壤覆盖在掩埋处,不然,测量得到的环境剂量率就不是被测样品原来接受的环境剂量率,因为y射线在土壤中的射程约30cm, 所以应该把热释光剂量计掩埋在土壤中有效探测距离为半径30 cm的球体范围内.凡是在这个范围内采集的样品,只要掩埋一个热释光剂量计,如在这个范围以外采集的样品,需要另外再掩埋一个热释 光剂量计,依此类推.从测量灵敏度考虑,热释光剂量计掩埋1~3个月即可,如果要考虑气候变化对环境剂量率的影响,最好掩埋一年.若没有条件用热释光剂量计直接测量遗址的环境剂量率,可以在采集样品时取周围土壤100g左右,用两只塑料袋密封包装,防止水分发挥(不需要避光、热和核辐射),连同样品一起寄到热释光测定年代实验室,用于测量y剂量率.为了得到比较准确的环境剂量率,采样者应该向气象部门了解当地历史上的气象情况,包括短、长期降雨量和气温的季节变化,地下水位升降及土壤的平均含水率等,以供热释光实验室参考. 1.2 细粒混合矿物技术 陶器细粒混合矿物技术简称“细粒法”它是选择陶器中原来存在的直径3~8 pm 的颗粒.不要把标本中原来存在的大颗粒破碎后当作细颗粒选择出来,因为破碎后的细颗粒实际上是大颗粒,其颗粒内部的α剂量已经受到严重的衰减,用这样的细颗粒测量得到的古剂量明显偏小.细粒样品制备比较简单,因为颗迷很小,没有办法选单矿物,只能用混合矿物,主要是有热释光特性的石英、长石和方解石等天然晶体,统称“磷光体”细粒样品制备分水浮选123,24]和丙酮浮选[16,25]两种,浮选出来的细粒样品最后都沉积在直径10mm、厚度0.5 mm 的金属(铝、不锈钢或银等)圆片上,用于热释光测量.沉积在金属圆片的细粒样品厚度要小于10um, 以便辐照α剂量. 1.2.1 等效剂量测定. 陶器“细粒法”中古剂量分为两部分,分别来自器物内部的陶土和陶器外部的环境.按射线类型分为四种,陶器内部放射性物质提供的α和β剂量以及由环境辐射提供的y和宇宙射线剂量[26]. 组成陶器古剂量的四种自然辐照剂量的热释光效率不相同,为了计算年代,要把它们归一到某一种剂量,最方便是归一到β剂量,归一后的古剂量称“等效β剂量”,简称“等效剂量”,用符号Q表示. 等效剂量用已知的实验室剂量标定.图1显示了标定的方法.这是一件唐三彩陶马细颗粒样品的热释光曲线和坪曲线.图1(a)中的 N 为自然热释光曲线,N+β为自然热释光加实验室β标定剂量的热释光曲线,N+2为自然热释光加实验室23标定剂量的热释光曲线.图1(b)为坪曲线.坪曲线的纵坐标为 N/[(N+B)-N], 图1 一件唐三彩陶马细颗粒样品的热释光曲线和坪曲线 实际上就是标定剂量热释光与自然热释光之比.从自然热释光曲线N和坪曲线可以看到,这件唐三彩陶器烧成后,经过1000年左右的时间,温度低于200℃左右的浅陷阱中贮藏的电子在常温的热振动下绝大部分已经被释放出来.所以低于200℃的自然热释光基本上观察不到, N/[(N+)-N]也接近于零.当温度超过200℃时, N/[(N+J)-M]开始上升,说明200℃以后在陷阱中的俘获电子在常温热振动下逃逸的几率逐渐减少,自然热释光N开始保存下来,但仍然有很多深度不够的陷阱中电子在常温下逃逸,所以曲线没有达到“坪”的水平.随着温度上升,陷阱深度增加,逃逸电子越来越少,坪曲线也不断上升,一直到270℃左右,出现了“坪”坪出现表明,在这个温度以后的陷阱比较深,有足够的能量可以把陷阱中的俘获电子束缚住,常温下逃逸可以忽略,因此,坪区也称“热稳定区”可以用于年代测定.对图1这件唐三彩来说,必需取270℃以后的自然热释光才能测定年代.热稳定性是根据每一件陶器样品的热释光曲线温度位置来确定的.随着温度上升,热稳定性迅速增加.例如在环境温度为20℃时,200℃热释光相应的俘获电子寿命只有几年,而300℃相应的俘获电子寿命增加到几万年,而400℃时的俘获电子寿命可达到几亿年.虽然温度越高的热释光陷阱越深,对测定年代越有利, 但是温度再高,本底急剧上升,热释光很地快被掩盖,无法检测.图1(a)中的三条热释光曲线的本底已经被减掉.将图1中坪处的(N+B)/N比值乘实验室β辐照剂量,就是这件陶器的等效剂量. 石英等天然磷光体的热释光对热非常敏感,个样品第一次测量后再进行第二次测量,样品的热释光灵敏度会发生变化,所以在测量等效剂量时,不能把已经测量过自然热释光的样品再用于测量实验室辐照剂量的热释光,因此,一个标本需要制备很多的平行金属圆片样品.测量时,将平行样品分成A,B和C三组,分别测量自然热释光(C组)和自然热释光加实验室α(A组)和β (B组)辐照剂量热释光,然后将A组和B组的热释光分别减去C组热释光,得到这个样品实验室α和β辐照剂量的热释光. 为了观察样品的热释光与剂量的线性关系,般在求等效剂量时采用线性回归方法.把A组和B组样品分别辐照o,2a,3a,…和β,2B,3B,….不同的实验室α和β标定剂量,将热释光强度和剂量作线性回归,得到线性相关系数,同时从直线方程的常数项或者从图2的截距得到等效剂量◎和等效o剂量Q图2是用C组和B组样品的热释光求得的等效剂量O. 图2 加剂量法求等效剂量 Q 如果已经证实这个样品线性相关比较好,也可以用下面的关系式计算等效剂量Q, 其中,Gv是样品的自然热释光, GN+B是样品自然热释 光加实验室β剂量热释光,β是实验室90Sr/90Y源辐照的β剂量,f是沉积细粒样品金属圆片对β粒子的反散射系数,是实验室β源辐照样品时由沉积细粒样品金属对β粒子反散射而增加的剂量. 1.2.2 a效率K3.7测定. 因为古剂量已经等效成β剂量,所以年剂量也要等效成β剂量,不然无法计算年代.等效的方法是将其他剂量乘上相对于β剂量的热释光效率.y的热释光效率与β基本相同,可以直接相加,α和β的热释光效率相差悬殊,需要乘上α相对于β的热释光效率K,将α年剂量转换成“有效α年剂量”,但是K不能直接测定,要从K3.7换算.测量K3.7实际上就是测量等效α剂量Oc,方法与测量等效剂量相同.如果线性相关比较好,也可以用下面的关系式计算, 式中, Gv+a是自然热释光加实验室α剂量热释光,α是实验室241Am 源照照的α剂量,α粒子没有反散射现象.和O之比就是这个样品的α效率K3.7,即 式中,K3.7是能量为 3.7 MeV 的o粒子外辐照效率(见下文),实际上样品在陶器内部接受的都是α内辐照,所以计算年代时也要用α内辐照效率.外辐照效率乘内外辐照效率转换系数,可以得到α内辐照效率K,详见文献[27,28]. 1.2.3 超线性修正.各种磷光体的热释光与剂量关系不完全是线性的.典型的规律是小剂量时灵敏度上升,称“超线性”(supralinearity), 随着剂量增加,灵敏度固定,出现直线部分,当剂量再增加时,因为饱和开始,造成灵敏度下降,称“次线性”(sublinearity),当剂量增加到很大时,灵敏度达到饱和. Aitken 称这种关系为“热释光生长特性曲线”151,见图3.各种天然和人造磷光体的热释光生长特性是不同的,陶器和砖瓦等考古样品年代测定,绝大多数用在生长曲线的直线部分.年老的钟乳石等,多用在线性到次线性部分.在前剂量技术中,瓷器年代测定只能用在次线性到饱和部分. 图3 Aitken 描述的热释光生长特性曲线15] 超线性处于线性之前,所以是热释光测量的必经之地.超线性修正值是在计算等效剂量时因采用固定的β灵敏度而少算(或多算)的那部分自然累积剂量.超线性对测量年代比较近的陶瓷器不能忽略,需要修正.修正的方法是将已经测量过自然热释光的C组平行样品再辐照不同的β剂量,第二次测量C组样品的热释光曲线,如图4中的曲线PM. 将PM中的直线部分MN延长与剂量轴相交的截距即为超线性修正.由于样品在小剂量时的超线性特征,所以热释光从原点开始并不随剂量增加而线性上升,而呈超线性状.随着剂量增加,曲线变成直线,出现固定斜率.测量一个样品的等效剂量,就是取在这个样品“热释光生长特性曲线”的直线部分,用这个直线斜率(即灵敏度)求得样品的等效剂量.但实际上样品在剂量为零时,热释光也应该为零,即坐标的原点,因此需要修正. 图4是 Fleming 提出的超线性修正方法129.样品测量的自然热释光显示在纵轴上,所以纵轴的位置也是测量日期.求古剂量时,实验室辐照的标定剂量一般都用与自然热释光强度相当的β剂量,所以纵轴上指出的2倍自然热释光处就是这个样品的标定剂量.将直线NM外推至剂量轴,与剂量轴相交点Ⅰ的截距Q就是这个样品的等效剂量.但是样品在OC这个超线性区域的实际灵敏度是曲线 PN, P点是这件陶器刚烧制好的灵敏度,P点与剂量轴相交的截距与样品实际测量的灵敏度NM与剂量轴相交的截距有一个差距I,这个差距就是由超线性引起的.应该是曲线PN与剂量轴相交截距得到的等效剂量才是这个样品 图4 Fleming 提出的等效剂量和超线性修正29 真实的等效剂量,而用灵敏度NM 测量得到的等效剂量Q只是一个“表观剂量”将这个表观剂量校正到“真实剂量”(即古剂量)要加上这个超线性修正值I,即P=Q+I.α照照没有超线性特征. 1.2.4 细粒技术的年代公式. 陶器细粒法的年龄按(1)式为 其中,DaDpDrc分别为这件器物的0年剂量、β年剂量和环境年剂量, KD 为α有效年剂量.其中环境年剂量 Dy+c 是D,和D。合并测量时所用的符号,通常表示环境剂量率是用热释光剂量计或其他环境检测仪测量得到的结果.K为有效α效率,即α内辐照效率.古剂量的单位用 Gy 或 mGy,年剂量单位用 Gy/a 或mGy/a, 年龄(或年代)用 a. 年剂量下文讨论. 1.3 粗粒石英技术 在热释光测定陶器年代技术中,. 一种用是直径为3~8 um 的“细粒混合矿物样品”,另一种用直径为100 um 左右的“粗粒石英样品”.选择这两种颗粒直径作为样品,主要是考虑α和β粒子在陶器中的射程.α粒子在陶器中的射程为15~50 pm, β粒子为 1~2 mm.y射线在陶器和土壤中为20~40 cm. 为了测量陶器中全部的自然辐照累积剂量,样品的颗粒直径必须选得很小,小于α粒子在其中的射程,大约为3~8 um,这就是“细粒技术”.相反地,为了去掉α自然累积剂量,样品的颗粒直径必须选得足够大,使α剂量在颗 粒中严重衰减,仅作用于颗粒表面,如果颗粒表面再经过氢氟酸蚀刻,那么样品中的α剂量可以完全去除,这样测量得到的古剂量就没有α辐照的贡献.但是粗颗粒直径也不能太大,否则,β剂量也会受到严重衰减,所以一般选在100 um 左右,这就是“粗粒技术”,国际上称“石英夹杂物技术”因为它主要选择夹杂在陶胎中的石英颗粒作为样品. 石英热释光性能良好,是测定年代比较理想的天然矿物.而且粗粒技术的样品大部分是石英,其他热释光性能差的矿物比较少,所以热释光与年代的对应关系比较明确,受干扰的因素也相对地少.这个方法的缺点是要在陶器中选择直径100 um 粗粒石英有些困难.首先,不是所有陶器都有粗粒石英,泥质的陶器选不出合适的石英颗粒;其次,即使有粗粒石英,由于取样量很大,对陶器有严重破坏.当然,也有一些陶器含有比较丰富的石英颗粒.例如广西桂林曾皮岩遗址中的陶器,石英含量最高可以达到50%左右,其中直径80~120 um 的石英颗粒含量就有10~20%30.但是大多数的陶器中的粗粒石英还是少而大小不均匀.挑选石英的陶片以体积大为宜,石英少的陶片用量要多,反之则少.在一般情况中,粗粒石英含量有5%以上就可以采用,不过每一块陶片最终至少要能够制备出 50 mg 以上的石英样品才能用于古剂量测量. 把预检出来的陶片用硅胶干燥后,在研钵中轻轻地压碎,用分样筛选出直径80~120 um的颗粒几百毫克到几克.大约1g样品用10ml浓度为40%的氢氟酸在室温中蚀刻45 min, 然后用水清洗,把不能被氢氟酸溶解的浮在上层的深色物质去掉,留下白色样品.如果其中还有比较多的方解石等矿物,可以用稀盐酸再溶解一次.最后按蒸馏水→乙醇→丙酮的顺序把石英清洗出来,在室温中温置15 min 干燥.这时候如果发现样品中还残留少量带磁性物质,可以用强磁铁将其吸出.如果样品量比较多,也可以用磁选机进行分离.这样制成的样品,石英纯度比较高301. 从热释光测量中发现,如果粗粒样品中石英选得很纯,测量得到的热释光分散性比细粒法小,反之,分散性远比细粒法大.这可能与氢氟酸对不同发光物质的蚀刻结果不一致有关.因此,在制备样品的过程 中应尽可能提高石英的纯度,使样品符合下列要求. 1)除了石英以外,大多数不需要的矿物基本上都被溶解. 2)将陶器在烧制时扩散到石英表面层的金属氧化物颜色去掉,使石英透明度提高,热释光灵敏度增加,曲线形状比较一致. 3)石英颗粒表面的α剂量层已经被剥离,测量得到的自然热释光和与此相对应的等效剂量已经没有α的贡献. 因此,(5)式也改为 其中,0.90是β剂量衰减和损失的修正系数,它与石英样品的粒径和蚀刻条件有关.当石英粒径为100 um时,U系提供的α剂量衰减约89%,Th系提供的o剂量衰减约75%,仍然留有小部分o剂量,必须用氢氟酸蚀刻. Bell 的实验表明B31],石英颗粒经过上述条件处理后,表面大约有6 um 厚的一层被蚀刻,α剂量已所剩无几,可以忽略,但由此引起的β剂量损失却不能忽略.根据Bell计算, 6 um的蚀刻层相当于 Th 系的β剂量损失 5.7%, U 系的β剂量损失 4%, 4K的β剂量损失可以忽略.此外,直径100 um 石英颗粒粒径大小引起的β衰减系数,U系为 0.938, Th 系为0.910,40K为0.966,总共衰减约6%.将蚀刻引起的损失和粒径大小造成的衰减作用一并考虑时,石英的等效剂量约减少10%,因此,(6)式中D的修正系数取 0.90.如果石英样品的颗粒直径不是100 um 左右,不论是大于还是小于100 um, 这个修正系数都不是0.90,需要另外进行计算. 粗粒石英等效剂量测量是将制备得到的石英样品每次称取固定的重量(一般为 5~10 mg), 直接铺在热释光测量仪加热板上进行测量,也可以放在金属杯里,再将金属杯放在加热板上测量.为了准确测量每一粒石英发射的热释光,样品在加热板上只能铺一层,颗粒上下之间不要重叠. 图5(a)和(b)是广西桂林皮岩遗址中陶器粗粒石英样品的热释光曲线和坪曲线.石英有一个110℃低温峰,这个峰灵敏度很高,衰退很快.如果要观察这个样品热释光曲线全貌,高温部分热释光曲线就 图5 广西桂林n皮岩陶片粗粒石英样品的热释光曲线和坪曲线 会被压得很低,而高温热释光正是我们测定年代所需要的.为了保证高低温热释光都能观察清楚,可以对热释光强度采用比例不同的标尺,一般在200℃时改变量程,如图5(a)所示,这样记录到的热释光曲线-一目了然.当然,现在热释光仪器的微机应用软件已经非常丰富,即使不改变标尺,使用者根据需要,可以对任何部分的热释光曲线局部放大,进行分析研究. 从图5(b)可以看到,这个样品的热释光曲线非常漂亮,所以它的坪曲线也十分标准,“坪”不仅平坦,而且范围很宽,这样测量得到的古剂量误差很小.为了观察样品的热释光与剂量的线性关系,也可以按照图2方法,辐照不同的β剂量,然后作线性回归,用外推法求得等效剂量.粗粒石英技术的超线性修正与细粒法相同. 2 瓷器古剂量测定 瓷器热古剂量比较复杂,国际上流行的两个标 准方法都不适用瓷器,主要是瓷器的原料和烧制工艺与陶器不同,使瓷器中矿物晶体的物理状态和热释光性能在烧制过程中发生变化.首先是瓷器质地坚硬,已不能够用刀取样,即使用碳化钨钻头也有困难,用镶有金刚砂的钻头比较合适,钻取时用水冷却,但是钻下来的粉末太细,在水中难以收集.其次是钻取样品时因摩擦产生的“摩擦热释光”在粉末样品中非常明显,往往超过真热释光信号.再有一个困难是瓷器高温热释光灵敏度很低,衰退大,而且常伴有假热释光,使测定结果很不可靠.但是人们在研究中发现,石英110℃热释光峰的“前剂量”(Pre-dose)灵敏度非常高,适宜于瓷器年代测定. 瓷器的前剂量效应、热激活和辐照熄灭机理等已有详细报道[32~34],这里只作一个简要的评述. 2.1 前剂量效应 “前剂量”是热释光测定陶瓷年代中的一项特殊技术.它最主要的特征是石英经过快速的高温加热,以前吸收的剂量(包括历史上吸收的天然辐照累积剂量和实验室放射源标定的人工辐照剂量)所产生的110℃热释光峰灵敏度会大幅度增加,这种现象称为“热激活效应”另外,辐照会使样品的灵敏度降低,产生“辐照熄灭效应”前剂量测定年代就建立在“热激活”和“辐照熄灭”这两个特性的基础上. 前剂量技术主要用于年轻样品的年代测定和事故剂量的测定.前剂量效应最早在1969年和1970年由两个博士研究生发现,-一个是 Fleming[35,36],另一个是 Thompson 37. 1971年, Zimmerman 提出了前剂量效应的能级模型[38].前剂量作为一个年代测定技术是Fleming 在1973年提出的39], Aitken 作了预测[40].1991年, McKeever 进一步解释了前剂量效应的物理现象411.1993年, Haskell 专门讨论了用前剂量技术测定事故剂量的方法[421.其中, Bailiff 和 Haskell 等对前剂量技术用于年代测定、真伪鉴定和事故剂量测定的分析和研究最为深入,还对这些问题进行了评论和综述[43~48] 前剂量研究和应用的面不是非常广,这主要由前剂量技术的特性决定的.前剂量有两个主要特点:1)前剂量是测量石英110℃热释光峰的灵敏度,但是这个峰的寿命很短,在室温中只有1~2h,所以这个 峰在石英的自然热释光中已经不存在,需要用加热的方法将其激活出来;2)前剂量110℃热释光峰的饱和剂量很小,可以测定的年龄比较轻,所以不能应用于新石器时代和地质年龄测定.但是石英110℃热释光峰的灵敏度非常高,只要用很小的试验剂量,就可以达到很高的测量灵敏度,适宜于测定距今几百年到一千多年烧过物质的年龄.而瓷器的石英含量很高,其制造历史特别是中国唐代以后的瓷器制造历史正好在这个范围内,因此,前剂量技术就成了热释光测定古瓷器年代的一个重要的方法.前剂量热释光与常规的热释光有些不同.常规热释光是研究热释光强度与剂量的关系,而前剂量是研究单位剂量的热释光(即热释光灵敏度)与剂量的关系.常规热释光中的古剂量是用样品陶瓷器烧成以来所吸收的自然辐照累积所释放的古热释光来测定的,而前剂量中的古剂量是用现在所加的试验剂量的热释光来测定的,所以两者在方法上有很大的不同.从目前研究情况来看,不是陶瓷器中所有矿物晶体的热释光都有前剂量效应,只有石英110℃的热释光峰有明显的前剂量效应,而且灵敏度很高,J,一旦经过热的激活,只要用0.01~0.1 Gy这样小的试验剂量,就能够把110℃的热光光峰激活出来,其灵敏度可以达到供精确测量的程度.此外,瓷器中的莫来石也有一定的前齐量效应. 2.2 石英110℃热释光峰热激活机理 石英110℃的 TL 峰之所以会有这种效应,可以用晶格中带空穴的释光中心复合率增加来解释.常规的热释光灵敏度变化是电子陷阱俘获几率的变化,而前剂量热释光灵敏度变化是L释光中心空穴被复合几率的变化.图6是 Zimmerman 和 Chen 提出的前剂量效应的能级模型[38,49].图6中T和Tz是电子陷阱,L和R是空穴陷阱,假定T非常浅,它俘获的电子寿命近1h左右,而T2则非常深,一般的加热温度不能使其排空,故称为“绝热”陷阱.在模型中引进Tz是为了维持电荷平衡.前剂量110℃的TL 峰是指由试验剂量提供的被陷阱T俘获的电子加热释放后被释光中心俘获并与其空穴复合时发出的光.因为T的电子寿命在常温下仅1h左右,所以这个晶体即使在 图6 Zimmerman 和 Chen 提出的石英110℃热释光峰的前剂量效应的能级模型138,49] 历史上积累了很多自然辐射剂量,在测量时仍然观察不到110℃的TL峰,但晶体一旦受到热的激活,情况就不同了.热使价带产生电子和空穴对,电子被激励到R陷阱,与受辐照时储藏在R中的空穴复合,使R中的空穴减少,而价带产生的空穴则被L 俘获,L俘获一个空穴后就成为被激活的释光中心,因为R减少的空穴等于L俘获的空穴,所以看起来好像是热激活把空穴从R赶到了L.L俘获的空穴越多,将来同一个试验剂量所产生的电子复合成光子的数目也越多.这些电子与因激活而增加了的释光中心L的空穴复合成光子,使发射光子的数目大幅度上升,这就是110℃热释光峰的激活. 2.3 热激活特性(TAC)曲线 瓷器的热释光特性是用热激活特性曲线来描述的.测量时取一个已经制备好的圆薄片样品(常用直径2mm,厚度0.2 mm), 从100℃开始,每隔50℃加热一次,直到700℃.测量每次加热后的激活灵敏度S, 作灵敏度S随加热温度T变化的曲线,就是这件瓷器的TAC曲线.图7是我国明代青花瓷器样品典型的TAC曲线,它由下列4个阶段组成. 1)当样品加热到100~200℃之间时,其灵敏度最低,也可以说基本上没有激活,故称“不激活”. 2)当样品加热温度高于200℃时,激活开始,随着加热温度上升,激活不断增加,灵敏度不断提高.在200~650℃时,称“局部激活”. 3)当样品加热到650℃时,灵敏度达到最大值, 图7 我国明代青花瓷器样品典型的TAC 曲线 称“完全激活”,这时候的灵敏度称“饱和灵敏度”. 4)当加热温度超过650℃时,样品灵敏度急剧下降,这是热产生的激活衰退作用,称“热退激” TAC 的形状反映了R陷阱激活能的分布,它表明靠近价带的陷阱的空穴会转移到L中心,其转移温度要低于不靠近价带的陷阱.热的作用是把空穴从贮存陷阱R赶到释光中心L,只要加热温度选择恰当(例如图7中的650℃),就可以最大限度地把空穴从R赶到L,使L中心的空穴数目大幅度上升,这时测量得到的前剂量灵敏度最高,也最稳定,称“完全激活”如果低于上述温度,只能把部分的空穴从R赶到L,这时灵敏度没有达到最大值,也不稳定,称“局部激活”.如果加热超过上述温度,又会使转移到L中心的空穴重新被热驱赶出来,这时灵敏度反而降低,称“热退激”如果加热温度再高,就会把L和R的释光中心全部赶光,这称为“消除”例如陶瓷器烧到1000~1200℃,就会使热释光全部消失.我们要选择的激活温度,就是使这个样品灵敏度达到最高时的全激活温度. 灵敏度S与剂量D线性关系和线性法求古剂 110℃的 TL 峰的灵敏度S与剂量D 的关系,般都研究它们的线性响应.图8是一件明代青花瓷器样品的97℃热释光峰的前剂量效应.给样品--个试验剂量,测量110℃的 TL 峰响应 So(原始灵敏度);然后热激活,再给一个相同的试验剂量,测量其110℃的 TL 峰响应Sy(自然辐照剂量激活灵敏度);最后给样品一个实验室β标定剂量,热激活后加第三个试验 图8 一件明代青花瓷器样品 97℃的TL峰的前剂量效应 剂量,测量其110℃的 TL 峰响应(Sy+Sg), Sg为实验室β辐照剂量激活灵敏度.因为(Sx-So)正比于古剂量,两次激活之间的增量(Sy+S)-Sv 正比于实验室β剂量,所以古剂量可以从两次(第二次和第三次)试验剂量所增加的 TL 响应得到.具体测量方法分为多次激活法、加剂量法和一次激活法. 2.4.1 多次激活法. 多次激活法只要用一组制备好的样品按图8测量,古剂量P用(7)式计算.其中Sw是自然辐照剂量熄灭灵敏度.熄灭效应导致灵敏度下降可以用能级水平模型(图6)解释.当自然剂量被激活时,一定数量的L中心已经带有一个空穴,产生-个总灵敏度 Sv. 当电子到达这些带有L中心时,有两种选择,一种是直接从测量Sy 时的辐照试验剂量时达到,另一种是通过150℃加热排空110℃陷阱中标定剂量的电子时达到.因为这些电子与空穴湮灭时,离开了未被填充的L中心,对灵敏度没有作更多的贡献,所以使每Gy 标定剂量的灵敏度降低.这样,(7)式中用Svv取代 Sy. 其中,β为实验室标定剂量,β'为试验剂量之和,因为采用多次激活法,所以必需把多次叠加在样品上的试验剂量之和减去,如果试验剂量很小,可以忽略. 2.4.2 加剂量法. 加剂量法与常规方法中用两组样品分别测量自然热释光和自然加实验室辐照剂量的热释光情况相仿, -一组样品测量Sv, 另外一组样品测量S和 Sv+p(Sw+Sp)与 SN+p的区别在于前者的β剂量 加在自然剂量激活之后,而后者的β剂量加在自然剂量激活之前.这样Sv+和Sy都只加热一次,但是试验剂量还是用了两次,不过其中一次是测量 So, 用在激活之前,可能影响比较小.加剂量法的P用下式计算 该方法避免了因为加热引起前剂量特性的变化和熄灭干扰.但是也存在一个缺点,即两组样品之间需要归一.归一可以用样品的重量,也可以用 So. 用重量归一精度比较差.用So归一比较好,但是对S很低的样品有些困难. 2.4.3 一次激活法. 次激活法是取前两种方法长处而避其短处.特点是采用三组样品,每组样品只加一次试验剂量,需要激活的两组样品也只要激活一次,保证三组样品测量条件基本一致.第一组样品测量 So, 第二组测量 Sv, 第三组测量 Sy+b.B·一次激活法的P也用(8)式计算. 因为石英110℃热释光峰的半衰期在室温中仅1~2h,所以测量要一气呵成,而且测量时不能按第一、第二、第三组样品顺序进行,而要先测量三个组的第一个平行样品,然后测量三个组的第二个平行样品,依此类推.如果每一个样品的试验剂量是分别辐照,辐照后马上测量S, 则110℃热释光峰的衰退影响可以忽略. 用线性法求古剂量的实验见文献[50].这个实验是用天然石英细粒样品用己Sr/0Y源辐照一个已知β剂量,模拟古剂量,用线性回归测量.结果发现,前剂量效应的线性范围比较小,仅相当于距今几十年到一百多年的范围I51. 2.5 灵敏度S与剂量D指数关系和饱和指数法求古剂量 由于前剂量技术中的热释光灵敏度与剂量的线性和近似线性范围非常狭小,所以在测定瓷器年代时经常出现年代偏老的倾向,甚至把现代瓷器测定成古代瓷器,所以很早就有人研究灵敏度S与剂量D的指数关系,我们还专门研究了测定瓷器古剂量的“前剂量饱和指数法”关于这方面内容已有专门文章报道[33,51],本文只作简要的评述. 灵敏度变化的饱和指数形式最早由色列列 Tle-Aviv 大学的Chen提出[49].后来,英国牛津大学的 Aitken[15],Chen 等人[52~54],英国 Durham 大学的 Baliiff[55~56],香港城市大学的 Leung 等人7进一步研究了灵敏度变化的非线性响应和指数模式.为了瓷器年代测定,我们对瓷器前剂量非线性现象特别对剂量在几个 Gy 范围内的灵敏度响应做了详细的研究[32~34].结果发现瓷器的线性范围非常狭窄,即使剂量在几个 Gy 内,灵敏度与剂量也呈现出次线性关系, Leung 等人在石英上的实验也证明了这一点I58].只有在某些情况中,次线性才可以近似地当作直线.在这些情况中,把灵敏度和剂量的次线性关系当作线性关系来处理,误差不是很大,但是标定剂量受到限制,因为一个大的标定剂量将导致一个严重的非线性.相反地,一个小的标定剂量又不足以诱发一个能精确测量的热释光,特别对瓷器年代测定更是这样. 实验显示,瓷器的激活和熄灭灵敏度与剂量的指数关系非常明显.当一个瓷器样品按 Baliiff 的“辐照实验室β标定剂量→测量辐照熄灭灵敏度→热激活→测量激活灵敏度”顺序反复循环测量时,它们就出现了“热激活”和“辐照熄灭”特性,得到每一件瓷器的“热激活灵敏度饱和曲线”和“辐照熄灭灵敏度饱和曲线”[123].从这两条曲线可以清楚到看到,不论是激活还是熄灭,只有在辐照剂量小于几个 Gy 时,灵敏度和剂量的关系才可以近似地当作直线,超过这个剂量,就出现了次线性关系,并按指数规律上升,直至饱和.由于线性或近似线性范围非常小,所以当我们用线性回归代替指数回归时,测到的年代会严重偏老.这是因为线性法是用实验室辐照下的灵敏度变化率求实验室辐照前的剂量即古剂量,而灵敏度变化率是灵敏度对剂量的微商,这是一个单调下降函数,所以剂量增加以后的灵敏度变化率小于增加以前的灵敏度变化率,用低灵敏度求高灵敏度的古剂量,得到的结果肯定偏大,而且实验室辐照剂量越大,古剂量偏得也越大,就会把现代瓷器测定成古代瓷器.根据这些情况,我们研究了两条曲线的指数部分以及加热和辐照对激活和熄灭的关系,证明只要这两条曲线的灵敏度和剂量是指数函数,则前后两个灵敏度的变化与前一个的灵敏度一定是线性函数.利用这一个关系,可以将灵敏度与剂量的指数函数转换成线性函数.灵敏度变化的线性回归非常方便, 通过这一回归,得到指数函数的两个参数——饱和灵敏度 S..和常数B.于是,瓷器古古剂量P表示为 根据热激活和辐照熄灭特性,可以得到测定瓷器古剂量的两个方法,一个是“激活法”,另一个是“熄灭法”59].研究结果表明,这两个方法在测定距今100~1000年的年代时,得到的结果是相同的,但是在测定低端年龄(小于距今100年)时激活法比较准确,高端年龄(大于距今1000年)时熄灭法比较准确.另外,对少数不能用这两个方法测定年代的瓷器,提出两个特殊的处理方法,详见文献[59]. 3 陶瓷器年剂量测定 年剂量是热释光测定年代另一个重要参数.它的测量和估算主要使用固体物理和天然放射性技术,还需要计算样品吸收的辐射能量,这些能量是从天然放射性核素中发射出来的.在一个测定年代的实验室中,年剂量测量和估算可能要比古剂量测量和估算需要更多的核物理基础和放射性技术,同时还需要古陶瓷发掘者提供很多的遗址情况,例如遗址所在地历史上的气象变化,地下水位的波动以及土壤的水饱和程度等,因为埋葬环境特别是过去土壤和样品湿度变化对放射性和年剂量会产生明显的影响. 陶瓷器由自然界粘土烧制成,地球上的粘土有很多的天然放射性元素,原子序数Z大于84的重天然元素都具有天然放射性,轻天然元素也有一部分是天然放射性.除此以外,来自太空的宇宙射线也属于天然放射性辐射.样品的古剂量和年剂量都来自这些天然放射性物质产生的辐射. 重元素的天然放射性核素有三大系列——钍(Th)系、铀(U)系和锕(Ac)系.轻元素中的天然放射性核素一般都独立存在,不构成衰变系列.对测定年代来说,主要是两个核素——40K和Rb,特别是4K,它提供的剂量比分很大,而Rb几乎可以忽略.它们母体半衰期都很长,在10°~10年,远大于陶瓷器制造的年代跨度,可以作为每年提供一个固定剂量率的放射源. 陶瓷器年剂量与古剂量既对应又不同.对应的是古剂量本身就来自年剂量.不同的是,如果测量古剂量样品的物体和成分不同,或者测量方法不同,或 者样品的颗粒直径和薄片厚度不同,那么年剂量也是不同的.这一点非常重要,往往被从事热释光测定年代工作者忽视,致使测量得到的热释光年代出现错误和混乱. 如上所述,年剂量由器物内部的天然放射性物质提供的辐照剂量和外部提供的环境辐照剂量四部分组成.第一部分是器物内部的U系、Th 系提供的o年剂量,换算成与β等效的有效α年剂量后,大约占器物总年剂量的45%,比例最大;第二部分是器物内部的U系、Th 系、K和Rb提供的β年剂量,大约占器物总年剂量的30%;第三部分是环境提供的y年剂量,大约占器物总年剂量的22%;第四部分是来自太空的宇宙射线年剂量,大约占器物总年剂量的3%.以上的比率是陶瓷器或粘土中放射性含量在“典型”的情况下得到的.“典型”情况为K 含量 1%, Rb 含量50 pg/g, 天然Th含量10 ug/g和天然U含量3 ug/g15. 3.1 细粒技术和粗粒石英技术的年剂量组分 陶瓷器样品的年剂量可以直接测量,也可以间接测量.直接测量指测量得到的值就是剂量率,这有两种方法, -种是热释光剂量测量法,另月--种是闪烁计数法,后者只测量样品年剂量中的y分量.间接方法是指所测量得到的值不是剂量,而是样品中放射性比活度(即放射性强度,也称比放射性),然后再从测量得到的比活度换算成样品的年剂量.因为只要知道放射性母核每次衰变时所释放的能量,就能够算出样品中单位比活度提供的年剂量.将单位比活度年剂量乘以所测量到的比活度,即为样品的年剂量.设样品中某放射性母核的比活度为1 Bq/kg, 即每公斤样品中这个母核每秒衰变一次,而且每一次衰变释放的能量为1MeV,假定这个能量是α或者β粒立提供的.因为1 eV=1.602x10-19 J,1 Gy=1 J/kg,所以1 MeV~1.6×10-13 Gy, 这就是说,样品中这个母核每秒所提供的剂量为1.6x10-13 Gy=1.6x10-7 uGy.把1秒改为1年,样品中这个母核单位比活度的α或者β年剂量就为1.6×10-x60×60×24×365=5.04(uGy/a).只要测量出这个母核在样品中的比活度Q,就可以得到样品中由该母核提供的年剂量 Qx5.04 uGy/a. Ait-ken 给出的U系、Th系和K的年剂量资料可以参阅文献[60]. 表1列列 Aitken 的陶器和土壤中 Th 系、U系和K含量在典型情况下得到的α,β以及y年剂量的组分,用百分率表示. 3.2 β,y和α年剂量在陶瓷器中分布 1)β年剂量.β剂量主要来自器物内部.器物的胎是粘土,所以胎中放射性物质就是自然界粘土中的天然放射性物质 U 系、Th系、K和Rb.4K是提供β剂量的主要来源.它在天然K中的原子丰度为0.01%左右,所以在典型情况下,K提供的β年剂量的比率是16%,Th系和U系提供的β年剂量比率是14%,Rb在天然Rb 中的原子丰度为27.8%,它提供的β年剂量比率只有 0.5%,因为比率较低,所以一般都将其忽略.β射线是带电粒子,在陶瓷器中的射程为1~3mm, 所以器物的β剂量主要来自器物内部,外部环境(例如埋葬陶瓷器遗址中的土壤)提供的β剂量只作用于器物表面.由于器物表层与土壤接触时产生互相渗透和交换,造成表层剂量难以确定,因此在测定年代时,把器物外表层去掉厚度2~3 mm,土壤提供的β剂量就不予考虑. 2)y和宇宙射线年剂量.U系,Th系和K三者提供的y年剂量相差不大.y射线的电离作用比较弱,所以穿透本领很大,在土壤和陶器中的射程大约20~40cm, 而器物的器壁厚度远小于y射线的射程,所以陶 瓷器内部放射性物质产生的y能量主要向器物四周扩散,消耗在器物以外的环境中,器物本身吸收的y剂量很少,可以忽略.当然,如果物体的厚度达到或超过y射线在其中的射程时,例如古建筑的砖墙,就要考虑砖墙内部y射线提供的剂量.但是陶瓷器不可能有这样厚度的器壁,所以只考虑环境提供的y剂量和太空提供的宇宙射线剂量.在典型情况下,环境中K提供的y年剂量比率是5%,U系和 Th 系提供的y年剂量比率是17%,太空提供的宇宙射线年剂量比率是3%. 3)α年剂量. 0剂剂量只来自Th系和U系.在典型的情况下, Th 系提供的有效o年剂量占年剂量的比率为21%,U系提供的有效α年剂量占年剂量的比率为24%.0年剂量是三种年剂量中最复杂的一种.α是重粒子,电离密度大,在陶瓷器中的射程只有10~50um, 因此,α剂量全部由陶瓷器内部的放射性物质提供.因为电离密度大,所以在陶瓷器中通过时使位于α离子径迹的中心部位热释光陷阱已经得到饱和,致使α辐射电离产生的电子在单位体积中不能被陷阱俘获而造成浪费的电子数目比β和y辐射大得多,所以辐照同样剂量时,α产生的热释光远比和β或者y少,少的程度用α热释光效率表示.α效率的物理概念很强,比较复杂,随便使用,会使o年剂量和年代产生很大的误差161. Source of dose Effective a dose (%) B dose (%) y dose (%) Total dose (%) (a) Fine-grain dating K 16 5 Rb 0.5 Th: Full chain 21 6 (thorium and daughter later) (12) (22) U: Full chain 24 7 39 (radon and daughter later) (13) (7) (25) Cosmic ray 3 Total 45 30 100 (b) Quartz inclusion dating K 8 Th: Full chain 19 (thorium and daughter later) (11) (16) U: Full chain 12 28 (radon and daughter later) (11) (20) Cosmic ray 5 5 Total 56 44 100 α效率全称“α相对于β的热释光效率”,其符号为“K3.7”.K3.7是 Zimmerman 在1971年提出的116].其定义是辐照在样品上的能量为3.7 MeV 的α粒子产生的单位剂量热释光与辐照在同一样品上的β粒子产生的单位剂量热释光之比,即 式中,下标“3.7”表示这个效率是针对能量为 3.7 MeV的α粒子而言,如果能量不是 3.7 MeV,则这个效率又是另外一个数值,因为α粒子能量响应很大.α效率是用某单一能量的o束流辐照在样品上得到的,也就是通常所说的用α源作外照射标定的,所以这个效率又称“α外照射效率”.我们在测定年代时测量的就是这个效率.为什么热释光测定年代的α外照射效率一直固定在3.7 MeV的α粒子能量上?这没有什么原因,纯属偶然.可能是 Zimmerman 第一次用o源辐照样品时,o束流通过准直孔照在样品上的能量正好为3.7MeV,以后就一直用这个能量计算. 我们在测定年代中,只能测得一个样品的α外辐照效率K3.7,但是计算年代时需要用α内辐照效率K,即有效α效率,因为测定年代用的样品吸收的α剂量都是在器物内部辐照条件下得到的.内辐照α效率K与外辐照α效率K3.7的关系为K/K3.7=0.85,0.85的理论计算和实际测量见文献[62]. 但即使如此,要测得样品的K3.7值也不容易.根据 Zimmerman 提出的α效率K3.7的定义,每一个热释光测定年代实验室,必须用准直孔从α源中获取能量为3.7 MeV的α束流,而且这束束流照在样品上的能量正好是3.7 MeV,这对一般的热释光测定年代实验室来说是一件非常困难的事情.为此, Aitken 提出了“a值系统”15].在a值系统中,α源辐照样品时可以不需要考虑其能量是否 3.7 MeV. a 值系统的本质是用α粒子经过石英时的单位时间(min)的总径迹长度密度(um/um)来标定α源的辐照场强度S,所以S的单位为(um/um’)/min 即um’/min. a值求法、a与K3.7的关系和K3.7的直接计算方法见文献[63].a值系统有特殊的意义,可对其作如下评价. 1)a值系统最大特点是用S来标定和传递α源辐照场强度,这项工作很有意义.在此以前,α源的标 定特别是传递剂量率一直非常困难,因为用 Gy 来标定和传递α源时,α的剂量率会强烈地依赖于α粒子的能量,而能量的变化将使α源的剂量率产生不确定性.现在用总径迹长度密度来标定和传递α源,能量因素可以不需要考虑,因为能量已被总径迹长度密度取代.这样,所有α源强度都可以用热释光进行标定和传递.另外,有了S值,可以计算任何物质任何能量的o真实吸收剂量率(在“K 值系统”中得不到α真实吸收剂量率,除非我们知道α射线辐照在样品上的能量,但这是很难做到的).对此,S值的独特之处,可以用一句话描述,我们能够不用 3.7 MeV的α粒子能量而得到能量为 3.7 MeV 的α效率. 2)因为a值是在石英中定义的,所以其物理意义也只有在石英中能够解释,在其他物质中就不能解释.对石英来说, a等于K3.7,所以a值可以直接应用.如果其他物质需要应用a值,必须通过 a=rK3.7的关系进行转换(r为其他物质与石英的质量阻止本领比率).而K3.7值适用于任何物质,包括石英.所以我们可以从α源的强度S直接求得包括石英在内的所有物质的α效率K3.7,不需要通过转换I63]. 3)从a计算K3.7有些问题.按照 a=rK3.7公式,石英与其他物质的α效率关系仅与两者质量阻止本领有关.事实上,效率还与α吸收剂量有关,而α吸收剂量又与α粒子经过被辐照的物质时的能量损失率有关.而能量损失率是物质的质量阻止本领与物质的密度的乘积.所以,α效率应该与被辐照的物质的密度有关.根据这一关系, a=rK3.7 应改为 a=rjrzK3.7,其中r为其他物质与石英的质量阻止本领比率; r2为其他物质与石英的密度比率. 3.3 年剂量测量方法 年剂量测量方法很多,但是归纳起来有三种,元素含量分析、厚源α粒子计数法和热释光剂量测定法..3.3.1 元素含量分析.元素含量分析.通过各种分析手段,获得器物和其埋藏地周围土壤中232Th, 238U(包括235U),4K和Rb的含量.如中子活化分析、裂变径迹分析以及各种仪器分析.仪器分析中常用的有y能谱分析、原子吸收光谱、X荧光分析仪、火焰光度计等.然后把含量换算成年剂量.这类方法用得很普遍,许多物理和化学实验室都有这些仪器.现代 分析仪器的精确度和准确度已经达到非常高的水平,所以采用含量分析,可以得到准确的结果.缺点是这些方法都假定放射系处于长期平衡状态.事实上不少陶瓷原料中有年轻的沉积物,其平衡状态并未建立,有些原来已经平衡的样品后来遭到破坏,都会给年剂量测定带来一定的误差.另外,有些含量分析设备昂贵,还有一些分析如中子活化必须送到有反应堆或者中子源的部门进行,这些都会给年代测定工作带来困难.因为成分分析在物理和化学实验室中已经普遍使用,不属于热释光测定年代中特有方法,所以不再赘述. 3.3.2 厚源α粒子计数法. 厚源α粒子计数法是测量年剂量最常用的方法,其优点是样品用量少(1g左右),仪器简单,成本低,使用方便,测量速度快,普通陶瓷类样品连续测量24小时即可.还有一个优点是可以从测量得到的α计数率直接得到年剂量.当一个α粒子从样品发射到ZnS 闪烁屏时, ZnS 产生一个闪烁,由光电倍增管光阴极接收放大后,在阳极输出一个脉冲.因为脉冲数与α粒子数成正比,所以用率表记录得到的脉冲数也是α粒子数.从α粒子计数率(包括“总α计数率”和“快对”或者“慢对”计数率)可以计算出这个样品的α,β和y剂量率. 图9是 Aitken 提出的 ZnS 屏上厚样品α计数的理想模型15].样品和 ZnS 的厚度都比样品发射的o粒子的射程R大,假定瞬时发射的α粒子都具有相同的初始能量,而且许多路线都是斜的.从图9可以看到,在靠近 ZnS 屏的发射核,大约有一半的α粒子产生闪烁(如图9中的A点). 离开ZnS 的核,产生的闪烁少得多(如图9中的B点).超过射程的核,没有一个α粒子能够达到 ZnS 屏(如图9中的C点).由此可见,在厚度为R的样品层内发射的α粒子,平均有1/4 图9 Aitken 描述的厚源α粒子计数的理想模型15] 达到ZnS 屏. Aitken从理论上推导了这个结果[15].如果在厚样品中取一个小体积元d4xdh(见图10),那么从这个体积元发射的α粒子是一个圆锥形立体角.圆锥体的母线R与重心线h的夹角为0,则从这个体积元发射的α粒子的立体角为2n(1-cos0). 实际上R和h是α粒子的射程,h也是体积元到闪烁屏的样品厚度.那么来自体积元的α粒子在4元立体角内达到屏的几率就为 Volume element of area dA. thickness dh 图10 Aitken 描述的在闪烁屏上一个厚样品的计数151 如果用C,表示单位时间和单位质量样品发射的α粒子数目,即放射性比度(Bq/kg),用p表示样品密度,则自面积为d4的圆柱体样品发射的o粒子达到屏的计数率ova可可以用下列积分计算. 因为屏的直径远大于α粒子的射程,故边界效应可以忽略.这样面积为A的整个样品达到屏的总α粒子计数率α应为 事实上,在每个平衡的放射系中,都含有 n个α粒子发射体.用C表示单位质量母核放射性比度,R表示各放射系成员发射的α粒子的平均射程,(12)式可以写成 根据U系和Th 系的o粒子数目以及它们的平均射程和陶瓷器样品的密度,钍系α粒子子数率om和铀系α粒子计数率 ou 以及它们之和的总α粒子计数率为 式中, CTh和Cu分别为钍系和铀系的放射性比度(Bq/kg), 0.85和0.82分别为α粒子计数仪对钍系和铀系的电子甄别阈因子. cpks 为每千秒计数.当样品和闪烁屏的直径为42mm时,从(14)式可以得到 Th 系和U系的α计数率omh和o, 即 oTh =0.123CTh (cpks), ou=0.132Cu(cpks). ((15)根据样品母核放射性比度和α粒子数目以及所带的能量总和,得到Th系和U系的o,β,y年剂量与母核放射性比度 CTh 和 Cu 的关系.将(15)式分别代入它们的关系式,则三个年剂量 Dc,D和D,分别为 上述公式推导见 Aitken 的 Thermoluminescence Dat-ingl151或者文献[64]. 从(16)~(18)式可以看到,只要测得一个样品的总α计数率a,就可以得到样品的a,β和y年剂量,这是非常方便的.但这是在样品中 Th 系和U 系的放射性相同的条件下得到的结果,如果Th/U比不同,就会产生比较大的误差.在极端情况下,即一个陶瓷样品中只有 Th 或者只有U, 那么(16)式得到的α年剂量 D的误差只有5%,换算成有效α年剂量时,误差上限在1%~2%,影响不大,这是因为只有钍的样品的α效率与只有铀的样品的α效率相比,仅高约8%.但是β和y的情况不同.在β剂量中,单位放射性比度的铀链剂量比钍链高,如果样品实际上没有铀,那么根据(17)式得到的β年剂量将比真实β年剂量高20%;反之,如果样品中没有钍,得到的旧年剂量比真值低20%.y年剂量对钍/铀比的依赖性与β相同,即在两种极端情况中,从(18)式得到的结果都与真实剂量率相差20%,但是高低正好相反.在y辐射中,没有钍高20%,没有铀低20%.因为β剂量是陶瓷器内部放射性物质提供的,而y剂量是器物的环境提供的,器物内部和外部不大可能有相同的钍铀比,所以这一高-低不能相互抵偿. 3.3.3 热释光剂量测量方法. 热释光剂量测量方法(TLD)是用人造磷光体测量陶瓷器内部和外部的环境剂量.最早使用的是测量环境y剂量率,其次是测量陶瓷器内部的β剂量率,最困难的测量器物内部的α剂量率. 1) TLD 测量环境剂量率.磷光体电离辐射效应早在20世纪上叶就开始研究,但是作为热释光磷光体测量辐照剂量大约从1950年开始65~671.1968年,Aitken 首先用热释光剂量计测量考古发掘遗址的环境剂量,他介绍了一个天然氟化钙技术[68,69].1968年Yamashite 等人发展了 CaSO4:Dy测量方法170.1970和1972年, Mejdahl 也发展了这项技术171,72].不久 CaF2和 CaSO :Dy 这两个磷光体得到广泛应用. Mejdahl 对测定环境剂量的磷光体提出如下要求. i)测量下限低,在0.01~0.05 mGy; ii) 1~2年内的衰退可以忽略; iii) 容易制备; iv) 可以重复使用; v)自我剂量可以忽略; vi) 退火过程简单; vii) 能量响应与测定年代的物质石英相同. 另外,如果磷光体对自然光、照明光和紫外线的灵敏度比较低的话,那么磷光体的操作更为方便,但是对热释光测定年代实验室来说,影响不大,因为陶瓷器年代测定都是在光线很暗的房间中进行.表2是Mejdahl 列出的可以用于测量环境辐射剂量的一些人造磷光体特性,同时列出了测定年代用的石英、长石、方解石的这些特性. 表2列出的这些特性,只能作为参考,因为这些特性会随磷光体制备和测量条件不同而异,例如光阴极、滤色片、峰读数与积分的关系等.相对于石英的能量响应值是根据(ulp)toten 进行计算. (ulp)toten是Storm等人提出的相对能量吸收效率符号[80].磷光体CaF2:Mn 和 CaF2:Dy 的灵敏度非常高,但是它们的衰退特性相当复杂,所有没有列在表2中. 图11是丹麦 Rise 国立实验室 Mejdahl 综合的某些常用磷光体的光子能量响应1731.这个图显示了磷光体能量响应对石英能量响应比率与光子能量的函数关系,光子能量范围在0.001~5.0MeV. 能量吸收效率是采用 Storm 等人的资料I80] 表2Mejdahl 列出的用于环量环境辐射的热释光磷光体的某些特性73 No Phosphor (ul p)tot-en at 40 KeV Temperature of Sensitivity Fading at 20℃ Ref. main peak (℃) to radiation LiF:Mg,Ti 0.29 200 5%/a Al2Os:Si,Na 0.85 250 10 none expected Mg2SiO4:Tb 0.88 300 80 none expected 4 NaAlSisO; 0.93 5 SiO, 1.00 6 Na2SO4:Dy 1.31 200 10 None/30 d [771 / KAlSisO; 1.55 8 CaCO 2.58 9 CaSO4:Dy 2.63 200 20-30 5%一8%/6 months 10 CaF, 3.30 250-300 25-60 none 图11 Mejdahl 综合的某些常用磷光体相对于石英的能量吸收效率173] A. CaF2; B. CaSO4; C. CaCOs;D. K. AlSiO8; E. SiO2; F. MgzSiO4:Tb; G.NaAlSi;Os; H. Al2Os:Si,Na; I. LiF:Mg, Ti 在这些响应曲线中,磁性硅酸盐和氧化铝响应在整个能量范围内与石英响应非常接近.相反地,硫酸钙和氟化钙的响应在低光子能区比石英的响应高得多,而氟化锂响应在整个能区比石英响应低不少. 土壤中的y能谱只有一小部分由低能光子组成,从图11曲线可以观察到,磷光体响应的差异比预期小.磁性硅酸盐、氧化铝和石英之间的差别完全可以忽略.氟化钙和硫酸钙的响应比石英高15%~20%. 因为临界低能部分的土壤能谱不可能准确地得到,所以氟化钙和硫酸钙的能量响应也不可能准确 地进行计算.测量环境剂量的磷光体能谱一定要和土壤中与石英等效的磷光体能谱比较接近.因此测量考古发掘遗址中环境y剂量的磷光体,用得最多的还是 CaSO4:Dy(或掺 Tm). Riso 的 CaSO4:Dy 主峰比美国 Harshaw 公司生产的主峰稍微低一些,因为低温部分的热释光贡献比较多. CaSO4:Dy 的缺点是低温峰衰退大,每6个月衰退 5%~8%. Riso 的磷光体已经广泛用于考古遗址的环境剂量测量.在这些测量中,已经将衰退考虑进去. 衰退率是这样得到的:将磷光体从被探测的土壤中取出后,与在测量前已经辐照同样剂量的一个磷光体进行比较,这个用于对比的磷光体剂量是在短时间内辐照的,一般为3天左右.磷光体掩埋在遗址土壤中的时间通常是4~7个月,如果考虑到--年四季的气候变化,也可以掩埋一年或者更长时间.其衰退率在夏季为7%~15%,在冬季为1%~6%[73]. Fran-cois 等人在法国 Glozel 进行过掩埋3年的实验,在掩埋期间,其衰退率为23%811.因此,磷光体测量得到的环境剂量需要对其衰退量进行修正.修正是根据已经衰退的监控磷光体累积剂量平均值的一半进行比较而得到的. 根据上述精确地测量得到的结果列于表3.这是Fredbjerg考古遗址测得的y剂量率随深度变化的关系.测量时间为夏季3.5个月,冬季5个月. 在表3中,冬季测量得到的剂量率在总深度范围内的标准偏差为3.5%.可以看到,随着深度增加,剂量率略有增加,测量的精确度比3.5%好.还可以看到,从夏天到冬天的季节变化对剂量率的影响非常小. 表3Mejdahl用 Riso 的 CaSO:Dy 测量 Fredbjerg 考古遗址的y剂量率I73] Depth (cm) 30 40 50 60 70 80 90 100 Summer (mGya) 0.753 0.761 0.745 0.795 0.753 0.802 0.853 0.838 Winter (mGya) 0.752 0.772 0.769 0.754 0.783 0.795 0.799 0.836 Mejdahl 对辐射强度随季节变化已经做了很多调查研究1821,他以一个月为测量周期,,一共测量了三年时间,测量深度在30~70 cm. 在三个12月周期中,月剂量的标准偏差分别为3.9%,3.5%和3.0%.这些结果表明,当磷光体在遗址中测量几个月时,季节变化的影响可以忽略. 测量时一般将磷光体密封在聚乙烯胶囊里,胶囊放在一根不锈钢管中,管壁厚度为1.5 mm. 不锈钢管的作用是屏蔽光谱中低能部分,使 CaSO4:Dy 在低光子能区能量响应大的缺点得到补偿.经过这样补偿,不锈钢探测器几乎与石英等效. 总的来说,用热释光磷光体测量考古发掘遗址环境辐射测量已经取得成功,发现了一些适用于环境剂量测量的热释光磷光体,用得最广泛的是 CaF2和 CaSO4:Tm 或 Dy. 后来 Al203和 Mg2SiO4:Tb 制备成功,又增加了两个灵敏度和稳定性非常好的磷光体,这两个磷光体的最大优点是能量响应与石英非常接近.因此,它们在标定时不需要再等效成石英. CaSO4:Dy与石英的能量响应相差比较大,而Al203与石英的能量响应非常接近,所以CaSO4:Dy的能响修正因子可以与AlzO3进行比较而得到.Al2O3测量时是将其放在一个含铀的异性霞石正长岩的辐照单元中.结果发现, CaSO4:Dy 对 Al203 的能量响应比率为1.05.所以只要将 CaSO4:Dy 测量得到的环境剂量率修正5%,就可以与石英等效. 2) TLD 测量陶瓷器内部β剂量率.首先建议用TLD 测量标本内部剂量率是 Aitken 等人在1968年所做的工作183J.他将 CaSO4:Mn 剂量片夹在两块陶片之间进行测量.后来企图用细粒粘土和 CaSO:Mn 颗粒混和测量α和β吸收剂量,但是α剂量测不准确,因为o剂量在磷光体中被衰减. 把粘土和和光体混和的方法在1968,1970 和1972年已经被 Fleming 和 Aitken 用于粗粒石英测定年代中测量β剂量率[84,85].他们用的磷光体是 CaFz颗粒,其粒径与粗粒石英相仿,均为100 um左右.为了去掉o剂量,在CaFz的表面用水解的方法使灵敏度钝 化,然后将混和物用尼龙罐密封几个月,待氡气完全达到平衡后用分样筛选出 CaF2颗粒,再用实验室β源标定. Fleming 的方法虽然复杂,但是石英颗粒对β剂量的衰减已被自动校正. 1972年, Mejdahl 将被测量的粉末放在聚乙烯薄膜袋中,用CaSO4:Mn测量其表面β剂量率,剂量计是将磷光体固定在金属圆片上.这个方法可以准确地测量0.01 mGy 的吸收剂量,灵敏度非常高.缺点是剂量响应的标定有些问题,他是以60Co 的y剂量响应作对比,而这个对比中的一个参数,即屏蔽α剂量的薄膜厚度在测定已知年代时已经被固定.这个方法在粗粒长石测定年代中经过广泛试验,误差小于10%,偶尔有大的偏离.后来知道,上述β剂量测量方法对粗粒长石年代测定不适用,因为它不测定长石中钾提供的那部分剂量,但是它可以测量长石含钾量与标准样品中长石含钾量相同的岩石标本.遗憾的是长石含钾量变化很大,所以这个方法实际应用受到限制. 1974年, Bailiff 等人发展了一种可以给出β剂量绝对值的 TLD 系统,以供细粒和粗粒石英测定年代之用.这种系统是把粘土放在10 cm²的有机玻璃容器中,容器有一个厚度为125 um 的 melinex 薄膜窗.剂量计是一个薄的铜盘,直径30 mm, 大约500 mg的 CaF2用MS805 型硅胶将其封住,对大于 0.01 mGy的剂量误差小于3%,这个系统用标准样品进行标定.标准样品由KCl和 Th矿与U矿组成,同时用石英作为测量/辐射本底和磷光体的自身剂量的工具. Bail-iff 和 Aitken 在1980年一起发展了测量β剂量绝对值技术[86] 1976年, Mckerrell 等人又采用了一个类似的方法测定粘土的β剂量率.样品置于一个容量为5.3 cm的聚乙烯袋中,用量很少.剂量计中间有一个凹形的聚丙烯圆片,凹塘内放一块含有CaSO4:Mn 的聚四氟乙烯圆片(直径5 mm,厚度0.4mm). 圆片的β窗厚度为40 mg/cm.辐照时,两块圆片相互粘住,放在聚乙烯袋内的粘土中间,以4元几何条件接受辐照.封住聚乙 烯袋,防止氡气逃逸,然后将聚乙烯袋贮藏在-25℃中4个星期. 1978年, Mejdahl 又将 TLD 系统作了改进.改进后最大的特点是样品的β剂量是与被初级标准标定过的的次级标准粘土作比较而得到的[87].这个方法可以对标本内部剂量作绝对测定,误差大约5%. 1979年以来,我们先后用颗粒型和超薄型TLD(超薄型 TLD 见下文)对标本内部的β剂量率进行测定[88,89],并比较原块标本、粉末样品和粉末压制成型样品三种测量形式. 原块标本测量时把陶片或者砖瓦一剖为二,露出大约 4~16 cm²的内表面,将其磨平.把颗粒直径为100 um 左右或者超薄型的 CaSO4:Tm 用 8 mg/cm²厚度的聚乙烯薄膜封住后,夹在两块标本磨平的内表面中间,成4元几何状态接受样品辐照.把样品夹紧,再用聚乙烯袋密封.一般地说,整块标本如陶片、砖瓦等,质地紧密,氡逃逸几率很小,不密封对结果的影响也不是很大.但是为了统一测量条件,还是将其封住,并放置一个月以上,取出 CaSO4:Tm,测量热释光并用实验室β剂量标定它的吸收剂量. 测量结果表明,原块标本测量的重复性比较差,主要原因是标本中各种矿物颗粒分布不均匀,尤其在放射性含量相差悬殊的大夹杂物暴露在被测量表面时影响更为严重.此外,颗粒型 CaSO4:Tm 在测量时要铺得很薄,不能重叠,否则会使CaSO4:Tm对β剂量的吸收和衰减产生不均匀影响.颗粒型磷光体最大的优点是测量标本自然辐照剂量时,由石英颗粒大小引起的β剂量衰减已经被自动校正.但是石英颗粒在蚀刻过程中损失的β剂量还需要另外修正. 成型法是把测量过热释光的剩余样品研磨成粉末,再压制成型,与上述同样方法进行测量.此方法主要优点是避免样品的不均匀性,缺点是压制成型比较困难. 采用粉末样品测量时可以把测量过热释光的剩余样品研磨成粉末,将超薄型TLD直接夹在样品中间测量,这是一个比较简便和实用的方法,从制备样品、测量和标定等方面来看,都比其他两种方法方便.从上述实验发现一个重要问题.用超薄型TLD测量得到的Dg比颗粒型 TLD测量得到的D大37%.这说明颗粒型 TLD 测量得到的Dg小于真实剂量,因为入 射的β粒子进入有一定厚度的磷光体元件时,由于磷光体的阻止作用,使入射粒子的能量随元件的厚度增加而降低,使测量得到的剂量小于真实剂量.颗粒型CaSO4:Tm直径大约100 um, 这样的厚度其能量响应是明显的.而超薄型 CaSO4:Tm 的颗粒直径只有3~8 um,可以近似地看作无限薄元件,不少低能β粒子都可以穿透,因此发光效率不受影响,能够真实的反映样品的内部剂量.同样道理,如果样品内部β能量是变化的,对有一定厚度的元件也会产生类似的能量响应.考古样品中β剂量主要来自4K,其次来自Th系和U系.40K的β平均能量为 1.35 MeV, 而 Th 系、U系和其他天然放射性核素的β平均能量仅在 0.01~0.8 MeV之间.另外,元件受辐照时是夹在厚样品中间,由于介质的阻止作用,样品中发射的β粒子,从样品与元件的接触面到β粒子有效射程内,其能量由大到小,连续变化,因此, CaSO4:Tm 吸收的β剂量实际上是一个变化的能量. 由于上述两个原因,要用一定厚度元件直接和准确地测量标本内部剂量是不可能的.正因为如此,前文提到的从1968年以来不少实验室所测量的β剂量,只能是标本表面的吸收剂量,而无法直接测量内部剂量就是这个缘故. 3)超薄型 TLD 测量陶瓷器内部α剂量率.这是用TLD测量三种剂量率中最困难的一种. Aitken 早在1968年提出了这个问题,但是因为α剂量在磷光体上严重衰减和细粒磷光体难以从样品中取出等原因,使这个问题迟迟未能解决.直到1983年由上海市放射医学研究所周智新研究成功的超薄型热释光剂量计才解决了样品内部α剂量率测量问题190~95] 超薄型热释光剂量计材料是 CaSO4:Tm 细颗粒,厚度只有1 mg/cm²,是一种定型的 TLD. 超薄型 TLD制备方法与细粒样品制备方法相似.将细粒CaSO4:Tm 沉积在厚度为10 um、面积可以根据需要的一张大的铝膜上,用硅油作粘结剂,在高温中将CaSO4:Tm和铝膜粘结在一起.粘结剂用量很少,保证铝膜上 CaSO4:Tm 和硅油的厚度只有几个微米.根据需要可以将制备好的大张超薄型 TLD 冲成不同直径的圆片,或者剪成不同形状的剂量片195].超薄型热释光剂量计可以在一次测量中同时得到样品内部的α和β剂量率,十分方便,测量方法如下(见图12). 图12 超薄型 TLD 测量样品α和β剂量率示意图 i) 取直径8 mm 超薄型 TLD12枚,在400℃中退火 60 min, 分成 a, b, c三组,每一组4枚 TLD.b组TLD正反面用厚度为 8 mg/cm²聚乙烯薄膜屏蔽.a组TLD仅在其铝膜的一面用同样厚度的聚乙烯薄膜屏蔽. ii) 将制备细粒和粗粒石英样品后剩余的标本粒屑研磨成粉末,在400℃中退火2h,将其装入直径55mm、高 20mm的称量瓶内,粉末高度大约20 mm. iii) 用聚乙烯薄膜屏蔽后的 a, b 两组TLD一起嵌入被测量的粉末样品之中.b组 TLD 测量样品4元方向的β和环境y加宇宙射线的剂量率(α粒子已经被薄膜阻挡). a组 TLD 仅测量样品2元方向α剂量率. iv)c组 TLD 以与全与b组TLD相同的条件嵌入另 -个称量瓶内的 CaSO4或者 SiOz 粉末的空白样品之中,用于测量环境辐射本底. v) 两个称量瓶置于同处,经过时间 t, 取出三组TLD, 分别用实验室β源标定其吸收剂量率 Da, D,和DC..于是,样品的o年剂量D,和3年剂量Dg就可以用下面两个公式计算. 其中 D=Dr+c. 对一个固定场所, Dphc可以作为一个常数,不需要每次测量. 4 一些复杂的因素和存在的问题 古陶瓷热释光测定年代研究从20世纪60年代开始,经过近50年的发展,已经在年代测定和真伪鉴 别上取得了很大的成功,成为古陶瓷科学鉴定一个重要的手段,但是年代测定的精确度和准确度尚不理想,这是因为古剂量和年剂量测量中存在着许多复杂因素和尚未解决的问题,主要有自然热释光的热稳定性、热释光的异常衰退、等效剂量的超线性修正、光衰退和光激发热释光、非辐射激发热释光、含水率对剂量率的衰减作用、放射系中的氡逃逸、Th/U比的变化和地下水的化学作用,还有α热释光相对效率等. 4.1 热稳定性 热稳定性是古陶瓷热释光测定年代中一个最基本和理论性比较强的问题,需要用动力学方法测量和估算年代测定中所用的天然矿物在不同温度和不同陷阱中的俘获电子的寿命. 古陶瓷中的古剂量是自陶瓷器烧成以来所积累的自然辐照剂量之和,这个剂量与陷阱中俘获电子的热稳定性密切相关.热稳定性好的陷阱是深陷阱.所谓“深”就是陷阱的能级比较低,离导带远,束缚电子的能力比较强,电子逃逸几乎可以忽略.反之,就是“浅”陷阱.这些陷阱束缚电子的能力比较弱,电子很容易从陷阱中逃逸,一些浅陷阱热释光在常温环境中已经部分或全部释放,有的甚至还没有到考古发掘时已经释放完了,这对测定年代来说是十分不利的.陷阱“深”和“浅”反映在热释光释放时所需要的温度.自然热释光曲线温度比较高的部分,就是热稳定性比较好的陷阱中俘获电子释放的结果,这部分热释光能比较真实的反映陶瓷器烧成以来所经过 的时间,所以测定年代一定要选择这部分的自然热释光.热释光的稳定与否,比较可靠的方法是测量这些热释光峰的寿命,最简单的方法是做热释光的坪曲线试验.“坪”表示这部分的热释光是由深陷阱中电子释放的,在常温下几乎没有逃逸,或者逃逸可以忽略,对测定年代来说是稳定的.没有坪的陶瓷器不能作年代测定,如果勉强测定,得到的年代是不可靠的. Aitken 根据动力学理论,在 ThermoluminescenceDatingy 一书中估算了与测定年代有关的主要矿物的陷阱参数和电子寿命115]. 每一个样品在测量前要先进行坪曲线试验,预先测量一个矿物样品热释光峰的寿命.另外,还需要做异常衰退试验.这是将一个样品分成为几份平行样品,将辐照过剂量的一份样品避光贮藏一段时间,与“新鲜”辐照的样品比较热释光水平.如果这个峰没有异常衰退影响,就可以用一级动力学计算它它寿命t, 其中, s(s )是频率因子, E(eV)是陷阱深度,T是绝对温度.k是波尔兹曼常数(在17℃时, kT=0.025 eV). 能够在实验室中测定E和s的方法很多, Aitken介绍了在测定年代中可以采用的三种方法.1)测定热释光曲线开始上升时其强度与温度的关系(简称“起始点上升法”).2)等温衰减.3)热释光峰随加热速率位移.另外还有一些方法是根据参数定义的峰形状和曲线位移技术等.详见文献[96~100]. 在所有方法中,准确测量样品温度是非常重要的.由于热迟后影响,样品的真实温度往往与热释光测量仪加热板底部热电偶显示的温度不一样,大多数比热电偶显示的温度低.这时候可以先用人造磷光体做试验,仔细观察温度迟后现象,并测量其迟后量.测量时选择磷光体曲线中某一个峰,观察这个峰的热电偶温度,然后将磷光体换成矿物样品,比较两个峰的热电偶温度,就可以得到测量矿物样品时的温度迟后量. Aitken用天然萤石(MBLE型)测量,当加热速率为1℃/s时,天然萤石的热释光曲线主峰在260℃;加热速率为5℃/s时,主峰在283℃;加热速率为10℃时,主峰在294℃15]. 4.1.1 起始点上升法. 在一个热释光峰起始地方,热释光正比于 exp(-E/kT),这时候可以不考虑是否满足一级动力学方程.这个温度响应一直持续 到被俘获的剩余电子大量减少为止.然后,作热释光强度随T变化的曲线,从直线的斜率得到E值.为了避免由被俘获的剩余电子减少而引起的误差,需要限制温度的范围,同时,使用的热释光强度不要超过峰强度的十分之一,对低端的重叠峰要用预热处理将其去掉. 这个方法的缺点是如果相关的释光中心受到热熄灭影响,得到的E 值会偏低很多,虽然它能够给出一个寿命的数值,但可能是错误的. Wintle 在测量石英325℃时得到一个E值为 0.64 eVl101,这个E值比其他方法得到的都低.后来用等温衰减和热释光峰随加热速率位移方法测定,得到与s相对应的E是1.69 ev[101] 4.1.2等温衰减法. 如果一个被辐照的样品维持在上升温度T,那么磷光体将按下式随时间衰减, 其中I是磷光体在t=0时的强度,t是俘获电子在T时的寿命.重复测量几个温度,作 log又对T的坐标图.根据(21)式,得到一条斜率为 E/k的直线,这条直线在T轴上的截距为 logs .如果一些陷阱出现不同的r值,那么这个图就是一条曲线.如果寿命相差很大,可能需要采用逐次代入法进行分析.究其原因,一个可能性是这个峰不满足一级动力学方程;另外一个可能性是这个方法对这样一个简单的形式不适用.在这种情况下,可以作一个Ⅰ和t乘积对 logt 图,这个乘积在t=r时达到其最大值[102,103],它与动力学级别无关.困难的是,只有在靠近这个磷光体峰的温度,强度还可以,其他地方的强度都比较低. 4.1.3 热释光峰随加热速率位移法. 从不同加热速率的热释光峰曲线可以看到, ·个热释光峰最大值的温度Tm是与加热速率β有关的.陷阱参数公式通过移项,可以变成 先测量不同加热速率的Tm, 再用(23)式左边的对数项与1/Tm作线性回归,从这条直线的斜率得到E值,从截距得到s值.这个方法与起始点上升法和磷光法比较,其优点是能够处理强度比较弱的热释光峰,还有一点就是它对其他峰的依赖性比较小.然而严格 地说,它只有在峰的位置才满足一级动力学方程,但是Chen等人得到的E值有非常好的近似性[103].另外,Wintle 在石英研究中发现,热熄灭对这个方法没有明显的影响[101]. 一级动力学最简单的试验是检验峰值最大时的温度 Tm 对剂量的依赖性,在正常的情况下应该是没有依赖性的.在更高级的动力学中, Tm向着温度下降方向移动,就像剂量增加一样.在等温衰减法的试验中,时间响应是一个指数函数,就像(22)式那样.这些试验能够直接用于一个孤立的狭窄的峰,例如石英110℃峰.当热释光曲线由重叠峰组成时,情况比较复杂,需要采用曲线拟合技术. Aitken列出了一些已经报道的与测定年代有关的陷阱深度E、频率因子s和电子寿命r的数值[15],这些数据有很大的研究和实用价值.但是,其中有些问题还要引起注意,当从短期的实验室测量得到那些非常长寿命的参数时会出现不符合实际的情况,因为某些不确切的因素会影响推断结果.例如,热熄灭会影响E值测量结果,异常衰退会影响有效的寿命,使E值和s值测量值小于预期值,所以,在实际应用时,对每一个样品进行坪曲线试验还是非常重要的. Aitken 将各个温度的热释光峰相应寿命列于表4.这个寿命不分何种矿物,所以比较粗糙,但是一目了然. 表4 Aitken 列出的不同热释光峰温的相应寿命115l Peak temperature (℃) 100 200 300 400 500 Lifetime (Pottery buried at 10℃) 2 h10a 600×10'a 3×1010a2×10a Lifetime (Pottery buried at 20℃) 0.5h 2a 70×10a3×10°a1×10a 动力学方法和坪曲线试验相互配合,起到了相辅相成作用,在解决热稳定性问题上,取得了比较大的成功.但是即使这样,热稳定性问题仍没有完全解决.主要表现下列三个方面. 1)动力学公式计算的陷阱深度、频率因子和电子俘获寿命都是在最简单和最理想的条件下得到的,同时以许多假设为前提,但是实际存在的情况都比理想条件复杂,还有不少假设并不成立,因此,用动力学方法测量和计算得到的热释光峰的寿命,有时候会与实际情况有比较大的出入,这在测定年代中要引起注意. 2)在热释光坪曲线试验中发现,很多陶瓷器好像有“坪”,但是这个“坪”不平坦,不明显,这说明这部分的热释光还是不稳定,用这样坪区的热释光测量得到的年代误差比较大,这些问题至今还没有办法解决. 3)在陶器热释光测定年代中,大多数用“细粒混合矿物技术”混合矿物中各种矿物的成分和比分都不知道,所以没有办法用动力学方法测量和计算细粒样品热释光峰的寿命,当其中有不少寿命短、热稳定性差的矿物的热释光混和在一起时,测量得到的古剂量会明显偏小,这也是陶器热释光测定年代准确性提不高的重要原因之一. 4.2 异常衰退 异常衰退是古剂量测量中经常出现的问题.陷阱中俘获电子除了热衰退外,即使在坪区还可能存在异常衰退,这是非热引起的衰退,是年代测定中比较难解决的问题. Aitken 介绍过一个异常衰退典型例子.这是从一块现代砖头中得到的细粒样品.一组样品在辐照后几分钟内立即测量热释光,另一组样品辐照后在-18℃中避光贮藏15h 后再测量热释光,结果发现,后面一组样品有严重的衰退,在热释光曲线的300℃处衰退40%,在400℃处也要衰退20%15. 异常衰退对测定年代的影响由Wintle等人发现[104,105],这个现象与月岩样品非常类似. Hoyt 等人在研究月岩样品时发现地球上的长石和拉长岩存在强烈的异常衰退.后来Hoyt等人又测量单颗粒月岩样品的热释光,结果15个样品中只有一种样品能测出自然热释光,其余14个样品均测量不出.同时, Sutton 等人也发现从陶器中选择出来的锆石有一半存在异常衰退.于是, Wintle 等人专门对长石、石、萤石、磷灰石和浮石的异常衰退进行了深入研究I106~108],结果发现这些矿物均有严重的异常衰退.异常衰退与贮藏的温度关系不大.在热衰退中,贮藏温度上升10℃,衰退相应增加一个数量级.但是在异常衰退中,必须使贮藏温度升高100℃才发生相同的变化.这种无热衰退特征在低温时尤为明显.把拉长岩贮藏在-200℃(液氮)时,虽然衰退减少了,但是仍有部分存在.即使将温度降低至-250℃(液氢)也无法使衰退进一步减少. 石英热释光性能良好,没有异常衰退,所以用粗颗粒石英测定年代时可以不考虑这个问题,但是在细粒法中,因为样品是混合矿物,许多有异常衰退的矿物都混杂其中,所以需要做异常衰退试验.这个试验一般采用贮藏方法.将一组辐照过的样品在一定的温度中贮藏,每隔一段时间取一个样品,与立即辐照的样品比较其热释光强度就可以发现异常衰退及其衰退程度. 减少异常衰退的影响没有很好的办法,一般只能用退火处理,把不稳定和有异常衰退的热释光去掉.我们测量了安徽油库西面剧烈变形后大玄武岩中的长石样品的热释光曲线和坪曲线I109],结果发现辐照过实验室β剂量的热释光中含有大量不稳定成分,很容易衰退,坪曲线上的“坪”不明显.但是辐照过实验室β剂量的样品在180℃中经过 3 min 退火处理,“坪”出现了,而且比较平坦.退火温度的选择非常重要.各种样品的热释光性能差异很大,它们的热稳定性和异常衰退的程度也各不相同,所以每一个样品的退火温度要进行反复的试验.温度过高,会把稳定的热释光也去掉,温度过低,没有把不稳定的热释光退尽,所以退火温度选择的原则是既要去掉实验室标定剂量(包括β和α)中的不稳定热释光和会产生异常衰退的热释光,又要不影响正常的热释光,这是比较困难的.事实上,要做到十全十美是不可能的,只能两者兼顾一下.退火的时候,应该将辐照实验室β剂量、α剂量和测量自然热释光的三组样品同时进行,保证一致的退火条件. 异常衰退一般都认为是隧道效应产生的. Viso-cekas等人对此作过详细的阐述[110,111].隧道效应不能用经典物理解释,只能用量子力学加以阐明.按量子论观点,波函数使得俘获的电子有一个超越能量垒的几率,当然,这个几率小而有限.但是当这个波函数与邻近带正电的中心重叠时,电子就从陷阱中逃逸出来,被中心吸引住,这就是隧道效应.图13描述了俘获电子从陷阱中逃逸的三条途径11.其中a是非热的隧道效应, b是热与隧道相互作用的结果,c与d是纯粹的热驱徒.虽然隧道效应与温度关系不大,但是陷阱越深,隧道效应的几率也越小,所以利用深陷阱即高温部分热释光测定年代,对减少异常衰退影响还是有好处的. 图 13 Visocekas 等提出的俘获电子从陷阱中逃逸的三条途径1111 4.3 超线性修正 热释光的超线性现象在陶器矿物中大量存在,特别是各种类型的长石,例如正长石、微斜长石和透长石等.石英也存在超线性现象. Fleming对超线性现象作了详细的研究[112].图14是一个刚退火的物质,辐照实验室剂量后,热释光对剂量呈现出超线性现象. 从原点0开始,如果热释光灵敏度保持不变,生长曲线应该是直线,即图中的虚线OA,但实际上不是这样.随着剂量增加,灵敏度上升,热释光呈超线性生长,出现曲线 OC. C 是灵敏度转折点,从C点开始,灵敏度保持不变,出现直线CF.古陶瓷的自然辐照累积剂量至少在几十 Gy 以上,所以标定等效量时,热释光和剂量的关系都用在直线CF部分,也就是直线EF,用EF的斜率(即热释光灵敏度)计算等效剂量. 图14 Fleming 观察到的一个刚退火物质辐照剂量后热释光灵敏度变化的超线性现象1121 但是样品的实际灵敏度并不是直线,它们是先曲线(超线性)后直线.曲线OC各点的切线斜率和直线EF的斜率不同,所以灵敏度也不同.用直线EF斜率得到的等效剂量与用曲线OC各点的切线斜率得到的等效剂量各不相同,它们之间的差异由超线性引起,故称其为“超线性修正” 热释光灵敏度超线性生长一般用辐照以后复合中心剩余的空穴进行解释.早在1978年, Chen 和Bowman就提出了一个竞争模型13],他们认为超线性现象是样品在加热和激励时,空穴和释光中心之间相互竞争的结果.后来, Lee和Chen又进一步作了解释.假定激励前有一定数量的空穴从释光中心离开,那么,竞争陷阱中就留下了相同数量的剩余电子.根据这一个假定,可以得到不同参数的热释光响应曲线.如果热释光强度用曲线下的面积表示,那么陷阱中剩余电子数量大的,在小剂量时,热释光曲线下面积(即热释光强度)与剂量的关系是线性的;陷阱中剩余电子数量小的,在小剂量时,热释光曲线下面积与剂量的关系是一个二次函数.在竞争陷阱早期饱和情况中,剂量响应更趋向二次函数.这个模型解释了为什么小剂量时热释光灵敏度上升的道理. Fain 等人在1994年也提出一个深陷阱竞争模型,比较详细地解释了超线性与饱和等一系列非线性现象114] 超线性修正前提是样品第一次测量自然热释光时的加热不会引起样品灵敏度变化,事实上不少样品受热后灵敏度发生变化,因此会给修正值Ⅰ引入一定的误差. 4.4 非辐射引起热释光 非辐射引起热释光是在测量正常热释光时伴随着的一种光,它严重地干扰了热释光信号,有时候甚至超过真热释光信号.非辐射引起的热释光种类很多,有压力热释光,这是样品在制备过程中因挤压而诱发的光;摩擦热释光,是样品在研磨时因细粒之间相互摩擦或者细粒与容器壁摩擦而诱发的光;还有样品在热释光测量前受过光的照射,会诱发光热释光.光激发产生的释光曲线形状与正常的热释光曲线形状很难分辨,而其数量相当几百年甚至几千年累积的辐照剂量.光激发影响最明显的是短波长的 光,所以样品制备和热释光测量必须在红灯或者黄灯下进行.陶瓷器是不透光的,一般只要去掉外表面2mm后,里面的样品基本上不受光激发影响,但是白色薄胎瓷器会受到影响. 在年代测定中,一般将辐射诱发的热释光称为“真热释光”而将非辐射诱发的热释光称为“假热释光”假热释光是一个通称,因为假热释光的产生的原因很多,所以在不同的场合,会有更加确切的定义和专用名词,例如热辐射信号,称为“红一热发光”或“本底”不用“光热释光”这个名词.又如化学热释光伴随着热激发化学变化和相位变换,我们给它的名词为“化学释光”.有时候在室温中,由于某些化学作用也会产生这样的光,它有别于“化学热释光”.类似的情况还有“生物释光”等. 从大量的检测结果来看,假热释光出现最多的是摩擦热释光和化学释光两种.摩擦热释光主要发生在热释光测量仪加热板上已经发光的颗粒或者气流经过它们的时候.其机理已经引起人们的关注和研究,但是没有明确的结论.对测定年代来说,最重要的是采用抑制手段,使所有的假热释光降低到最小程度.在热释光测量仪的发光炉中充满惰性气体,情性气体中的氧气和水蒸气含量要小于几个ug/g.这种方法主要是减少样品颗粒的表面效应.细颗粒样品表面积大,所以产生假热释光的几率也大,反之,粗颗粒样品产生的假热释光的几率比较小.但是惰性气体并不能抑制所有的假热释光,所以假热释光的问题还是比较难解决的. 4.5 光晒退影响 实际上,一个古陶瓷的自然辐照累积剂量,在漫长的岁月中,是--边累积, -边衰退,我们希望积累远大于衰退.释放温度越低的热释光,积累/衰退比率越小,反之,积累/衰退比率越大.衰退作用除了影响最大的热衰退和异常衰退外,光漂白(或称光晒退)作用也是不可忽视的. 光有双重作为,既能引起假热释光,又能晒退真热释光,所以在热释光测定年代中,样品不要暴露在各种光(包括自然光和人工光)源下.非辐射热释光中有一种假热释光是由光激发引起的热释光,这就是上一节讨论的光激发影响.当陶瓷粉末样品暴露在 光源下时,其贮藏的热释光又可能会减少,这就是光晒退作用.在测定年代中,特别是测定年龄比较轻的古陶瓷样品时,影响尤为明显,因为它们累积的自然辐照剂量比较小,, -一旦受到光晒退作用,测量得到的古剂量就会明显偏小. 英国伯明翰大学物理系的 Jayaprakash 等对氟磷灰石(FAP)辐照引起色中心的光晒退进行了详细的研究151.他们采用的氟磷灰石是用固态扩散合成的.FAP用X射线辐照后,测量热释光,三个发光峰分别为150,250和345℃左右.大约450℃,所有X射线引起的色中心完全晒退了.实验发现,即使样品反复进行“辐照一加热一晒退”热释光特性也没有发生严重的变化.~150℃的热释光在室温中慢慢地衰减下去.在暗室中常温贮藏24h后,强度衰减小于1/20. FAP在辐照X射线后,用汞汽灯进行光晒退退验.他们采用波长为320~400 nm或者400~480 nm 的滤色片让光通过,对 FAP 晒退 30 min, 晒退后,~150℃热释光强度是没有被晒退样品的1/10左右.~250℃热释光强度是没有被晒退样品的1/6左右.高温热释光峰(~345℃)是三个峰中晒退影响最少的,大约减少到原来强度的一半.由X射线引起的热释光,其晒退程度与暴露光的波长有关.暴露于光波长为320~400 nm或者400~480 nm 时引起的热释光晒退比暴露于光波长为560~600 nm 或者250 nm 时少. Aitken介绍了一个光晒退比较典型的例子15].图15是一个风形成的沉积物样品,其中a是自然热释光曲线;b是阳光照射1h以后的热释光曲线; c是阳光照射24h后的热释光曲线.从图中可以看到光晒退的作用是很强烈的. 光晒退在古陶瓷测定年代上不利之处就是光照引起自然热释光的减少.从上述实验可以看到,陶瓷器中的磷灰石对光晒退的反应最强烈,如果陶器中含有比较多的磷灰石,那么测量得到的年代就会轻得多,特别在细粒样品测定年代时影响更大.现在唯一的方法就是在样品制备和热释光测量过程中避免自然光和人工光源的照射,所有操作都应该在红灯或者黄灯下进行.但即使这样,也不能完全避免光晒退的影响,尤其是白色的薄胎瓷器,既要受到光激发影响,又要受到光晒退影响,而且影响非常严重. 图 15 Aitken 得到的一个风形成的沉积物由光照引起的自然热释光晒退1151 4.6 水分对剂量率的影响 水分对剂量率的影响非常大,在热释光测定年代中一定要对其修正.因为陶器品种繁多,质地紧密程度和空隙率相差悬殊,所以在遗址中,地下水渗透到陶器中的程度也各不相同.由于水对放射性辐照的阻止本领比陶器和土壤高,所以当地下水渗透到陶器和土壤中时,由于水、陶器和土壤对辐射能吸收率不同,致使用于测量热释光的颗粒样品的吸收剂量也因含水率不同而异.在无限基体中,由于空隙中水分吸收了放射性,使单位质量的放射性比干燥的空隙降低不少. 水对剂量率的影响有两个方面..一个方面是当地下水渗透到陶器和土壤中时,稀释了放射性浓度,使陶器吸收剂量率降低;另一方面是水的阻止本领比陶器和土壤高,水吸收辐射能比陶器和土壤多,所以水分“抢”走了一部分辐射能.这两个因素导致陶器内部磷光体吸收剂量减少,使实际吸收的辐射剂量小于年剂量测量时得到的数值,因为实验室测量年剂量时都是使用干燥样品.把干燥样品的α,β和y年剂量转换成潮湿样品的年剂量可以用 Zimmerman 提出的含水率修正公式116], 在(24)~(26)式中, DaDg和D,为别为潮湿样品的α, β和y年剂量, Da(d), DB(a)和 Dy(a)分别为干燥样品的α,β和y年剂量,W为陶器含水率, W'为陶器出土处土壤含水率.1.50和1.25分别为水对陶器的o粒子和3粒子的相对质量阻止本领,1.14为水对土壤的y射线相对质量阻止本领.F为地下水波动因子.在大多数情况下,地下水波动率为20%,因此取 F=0.8是比较合适的. Aitken 将陶器含水率在10%~20%、土壤含水率在20%~40%时α,和y年剂量降低的百分率及其误差列于表5(假定陶器和土壤的放射性含量在典型情况下). 在一般情况下,W和W'的平均值可以在遗址发掘时实地采集粘土样品测量.如果陶器早已出土,含水率只能估计.但是估计错一档,计算得到的年龄就会偏大或偏小.例如把W'=30%错误估计为W'=20%,年龄会偏轻10%,反之,年龄会偏老10%.由此可见,含水率对年代测定的影响非常大,所以在发掘遗址采集陶片时,,-一定保持陶片出土时的含水率,以便回实验室可以准确地将其测定.有不少考古工作者忽略这一点,把水分已经部分挥发或者完全干燥的陶片送到热释光实验室测定,就会给测得的年代造成很大的误差.当然,这个人为因素是可以避免的.但倘若陶片出土处的地下水位在历史上曾有过大起大落,那么含水率的严重影响仍不能排除.这是因为我们测量得到的含水率只能代表出土时状态,不能反映历史的真实情况,这也是影响热释光测定年代准确度的重要原因之一. 瓷器质地紧密,空隙度很小,含水率非常低,故水分对瓷器剂量率的衰减可以以略. 4.7 氡逃逸 氡从陶器和土壤中逃逸也是热释光测定年代中 一个比较复杂和难解决的问题.钍系、铀系和锕系三个系衰变到中途时有一个质子数为86的气体成员氡,在Th 系中为220Rn,早期的专用名词为钍射气;在两个U系中,分别为 2??Rn和219Rn,219Rn早期的专用名词为锕射气.因为是气体,所以有一些氡很容易从样品或者土壤中散发出来,特别在多孔样品中最容易逃逸.如果氡逃逸25%,不仅氡放射性比母体降低25%,而且后面所有成员的放射性都要降低25%.这三个氡气中最容易逃逸的是222Rn,因为它的半衰期长,达 3.8 d,有足够的时间可以从陶器中逃逸出来.其他两个220Rn 和219Rn只能在陶器内部散发,因为它们的半衰期比较短,分别为55秒和4秒,还来不及从陶器中逃逸出来,就分别衰变成下一个子体216Po和215Po,因为这两个子体都不是气态,所以没有办法从陶器中跑出来.尽管如此,有时候钍射气还是有逃逸的机会,特别在测量年剂量时,如果样品早已经被夹碎,或者被研磨成粉末,那么钍射气很可能已经逃逸. 还有一些其他情况也可能使放射系失去平衡.陶器埋藏在遗址中时,如果镭被地下水浸沥出来,这个衰变系后面所有的放射性成员都没有作用了.又如,在石笋方解石中,形成石笋时,铀可能被溶合进碳酸钙中,但是没有钍.因此,在年轻的石笋和钟乳石中,有铀而没有它的子体.然后随着时间的消逝,开始逐渐积累,这个积累取决于230Th 的半衰期75×10年,直到这个系衰变停止,这个过程就是铀系法测定年代的基础. 氡气从陶器中逃逸分两步.第一步,氡气从固体物质中逃逸,进入充满水或者空气的空隙中;第二步,氡沿着空隙扩散,然后从陶器中逃逸出去.当镭核发射一个α粒子形成氡核后,第一步就完成了.但是动量的保存需要有一个动能为 0.1 MeV 的对相核反冲,才能在陶器中达到 0.02 um 的射程.如果这个过程在 Water content (%)Da (%) PotterySoil DB(%) D,(%) D (%) Fine-grain Inclusion W=10. W'=20 12±3 10±2 18±5 13±3 13±3 W=10. W'=30 12±3 10±2 27±7 15±4 17±5 W=20. W'=40 24±6 20±5 36±9 25±6 26±7 一个空隙中结束,只要它没有经历放射性衰变,第二步仍发生在这个空隙里.这个放射性衰变链的下一个成员不是气体,这个子体将附着在这个空隙的壁上.由于氡的半衰期为3.8d,所以在一个充满水的空隙中,其衰变前的扩散路程已经达到几个厘米,那么在一个充满空气的空隙中,其扩散路程更长.另一个类似是情况发生在 Th 系中, 232 Th 放射系衰变到一半时也会产生一个放射性气体——钍射气220Rn, 它是222Rn 的同位素,因为它的半衰期比氡短得多,只有55 s,所以它的扩散路程比 222Rn小6000倍,其逃逸可以忽略..2235U 放射系衰变链中也有一个气体核素为锕射气219Rn,它也是氡的同位素,半衰期仅4 s,所以扩散路程更短,其逃逸程度完全可以忽略.由此可见,我们最关心的是222Rn 的逃逸. Tanner 研究表明,氡在干燥土壤中寿命长到可以运动1000 mm 左右[116].这种运动会产生两个结果,一个是陶器吸收环境y剂量的有效距离只有300 mm,所以这种迁移作用,使y剂量率发生变化;二是y剂量98%是氡和氡以后的同位素产生的,氡损失率可达15%.这两种情况都会使环境剂量率降低.但另一方面,土壤中的水会阻止氡的自由移动,使其减少到只有20mm,不过也有可能地下水会把会带到2000 mm的地方. 当遗址中土壤和陶器都有地下水渗透时,水对氡产生一个双重作用.一方面水的阻止本领比土壤和陶器大,使水更容易吸收辐射能,这样陶器吸收的y剂量减少;另一方面水会阻止氡的运动,使氡逃逸减少,这样陶器吸收的y剂量相对增多,当这一对正反作用恰好相互抵消时,则含水率和氡逃逸对y剂量都不需要修正.可惜这种完全抵消的可能性不大,而且也无法确定,所以一般只能作各自修正.不过就细粒测定年代技术来说,环境剂量中由U系提供的的y剂量仅占年剂量的7%,因此,氡逃逸引起的y年剂量对年代测定的影响不是很大. 陶瓷器内部α和β剂量受氡的影响比较小.因为在238U中,氡和氡以后产生的α剂量率只有 54%,β也只有62%.如果陶瓷器质地紧密(瓷器肯定紧密),或者空隙中有水分存在,都会减少氡的损失,因为氡可能被地下水输送,也可能被地下水和陶瓷致密性所阻止. 厚源α粒子计数在测量中也遇到了氡气问题,因为一个闪烁屏要达到低本底的计数率是非常困难的.首先要屏蔽来自空气中氡射气和钍射气发射的α粒子,还有建筑物内砖头和花岗石发出大量的氡射气和钍射气,如果闪烁屏暴露在空气中,就会大量吸收,使闪烁屏本底升高.但是它们随时间是衰退还是比较快的,例如212Pb的半衰期是10 h, 它发生在钍链的钍射气后面.只要在使用前将闪烁屏密封贮藏在一个容器里几天,基本上可以解决这个问题. 样品的总α粒子计数率一般都要测量 24 h 以上.为了探测样品中全部的α粒子,要把样品颗粒研磨得非常小,这样就造成样品中的氡气大量释放出来,积累在样品的测量盒中,使α粒子计数率大幅度上升,重新建聿放射系平衡.钍链按照 212Pb的10h半衰期重新建立平衡,铀链则按照氡的3.8d半衰期重新建立平衡.但是210Pb和 210Po仍然维持在器物里藏期间已经建立平衡的水平上,这是因为 210Pb 的半衰期比较长,达22年.早些时候,氡的积累是作为估算氡逃逸程度的一个方法,在测量一个样品时,用三个计数率来表示三个状态:cvo表示样品未密封的计数率;o表示样品盒密封后第一个24h内的平均计数率;α表示密封10d后的计数率.如果o超过a表示有钍射气的积累,如果进一步增加到,则表示有氡的积累.由于“超计数”的出现,这个方法有些问题.例如计数率增加10%,一般就将其作为气体逃逸的警戒线,但也可能是由超计数引起的.不过密封计数率超过未密封计数率作为一个半定量的标志还是有用的,它能说明气体逃逸的严重程度.但是容器中氡的保持量往往估计过低,因为氡气非常容易从薄的塑料容器或者封口中逃逸. Pernicka等人[117,118]发现在厚源α粒子计数中,即使样品不密封,测量得到的计数率也比用其他方法根据钍和铀含量得到的结果高,这个现象称“超计数”.他们使用的样品是陶器、粘土和沙.另外,他们还有一个试验样品,是一个已知放射性含量的人工制备的α辐射体混合物.因为预期的计数率是根据钍和铀的平均含量来计算的,所以样品中如果含有放射性夹杂物,或者放射系失去平衡,可能出现计数率降低,而不是超计数.如果子体过剩太多,则出现超计数.但是放射性夹杂物的出现,将导致从o计数率得到的 剂量率大于从母核浓度的化学分析得到的剂量率. Aitken 设计一个测量兆逃逸的“气体密室”这是将厚源α粒子计数装置的样品盒改成一个容积比较大的能够放置整块陶片的密封容器.先用整块陶片测量,模拟陶片埋藏在遗址土壤中的状态,然后将其压碎,放在托盘上测量.这样,到达硫化锌屏的α粒子只有已经从样品中逃逸的氡和已经附着在托盘的壁和底部的疼气子体.因空气吸收使计数率降低,但是用已知发射率的放射源进行标定后,可以估算出氡的发射率.气体密室使用的所有材料,必须是低放射性的.例如承托硫化锌的玻璃用光学硅硫酸硼玻璃,气体密室的材料用黄铜. 密室放在光电倍增管的顶部,.5与用塑料样品容器测量厚源α粒子计数时相同.使用 Aitken 提出的合适计数一览表I119],就可以根据不同的半衰期对氡气和钍射气的逃逸量分别进行估算.当压碎样品测量得到的逃逸量超过整块样品的逃逸量,就应该考虑“丢失的计数”在这种情况下,需要测量io. 因为气体是从一个非常大的样品中聚集起来(由几克样品代替0.2 g气体直接提供α计数),所以气体密室是一个高灵敏度器具.一个放射性含量典型的陶器的氡逃逸量只要超过1%~2%就能够被探测到.虽然这个装置灵敏度很高,但是由于计数效率的不规则漂移,所以能够做到的还只有半定量. 氡逃逸的精确测量还是应该用传统的电离室. Per-nicka和Wagner介绍过测量氡逃逸的离子脉冲室171.大约10g的陶片放在一个密封的锥形烧瓶内,贮藏4天以上.然后将混合气体送到一个离子脉冲室,进行计数.德国海德堡大学环境物理研究所的Fischer又有了新发展,他们从海水中提取空气,测量了空气中的氡含量. 4.8 钍/铀比的变化 用厚源α粒子计数法测量钍和铀的年剂量时只记录这两个系发射的总α粒子数目,并假定样品中钍和铀的放射性是相同的.但事实上并非这样,对β年剂量来说,单位放射性比度铀链剂量比钍链高,而对v剂量来说,单位放射性比度钍链剂量比铀链高.如果不分析样品中的钍铀比,仅从总α计数率转换成β和y年剂量就会产生一定的误差.于是人们就利用厚 源α计数法中的“慢对”和“快对”技术,从总α计数率中分离出钍系系铀系各自的贡献.但是“慢对”和“快对”的计数率非常低,一个放射性比度为41 Bq/kg 的典型陶器样品,“对率”仅0.2计数/千秒,需要连续测量8个多星期,才能达到±3%的统计误差,这是不实用的.因此,在常规工作中,,一般都不测量钍铀比,只测量样品的总α计数率,并在计算年剂量时假定样品中的钍系和铀系的放射性是相同的,即钍铀重量比约为3.2.事实上许多样品的钍铀重量比并不等于3.当钍铀比在较大范围内变化时,从总α计数率换算得到的年剂量会产生一定的误差.我们计算了在任意钍、铀含量下和所有钍铀比覆盖范围内(即0≤Th/U≤oo)的总α率数率对α年剂量、β年剂量和y年剂量转换因子[120] 从文献[120]可以看出,当假定样品中钍铀重量比为3而事实上钍铀重量比为零或无穷大时,α年剂量转换因子的误差为5.2%,β年剂量转换因子的误差为19.8%,y年剂量转换因子的误差为20.8%.由此可见,即使钍铀比在这两个极端情况下,用总α计数率计算α年剂量时误差也不大,仅5%,但是对β和y年剂量影响较大,各为20%左右.这种极端情况多数出现在某些陆相和海相地质样品中,对陶瓷和砖瓦等考古样品并不多见.所以在测定陶瓷器样品时,如果取两个极端误差的一半,即当钍铀重量比在1.1~9.5范围内变化时,用总α计数率计算得到的α,β和y年剂量转换因子的误差分别为2.6%, 9.9%和10.4%.对细粒技术测定年代的影响为2.5%,对粗粒石英技术测定年代影响为4.1%,详见文献[120]. 4.9 地下水的化学作用 某些矿物和地下水中的离子交换使少量矿物产生溶解后再结晶过程.这个过程会使原来贮藏的热释光遭到破坏,有些石笋沉积物就利用这个现象来计算其生成年龄.陶器粘土中的方解石,在低温烧制时,与粘土矿物结合成石灰石,随后被水合成氢氧化钙,再慢慢地变成方解石.同样,方解石和另一些比较容易溶解的盐类,在一定的埋葬条件下,也有可能沉淀在多孔的陶器组织中.这种地下水对天然放射性同位素的浸出和沉淀作用,使陶器接受的内部剂量率发生变化.如果我们采用粗粒石英技术,因为这 种晶体是经过挑选分离出来,影响较小.如果采用细粒技术,则这种影响比较严重. 4.10 α热释光相对效率 如前所述,α热释光相对效率是热释光测定年代中最为复杂的-1—-个参数.常用三个符号表示,分别为K3.7,K 和a,虽然它们都是α热释光相对效率,但是含义不完全一样,如果用错的话,会给测量得到的年代造成很大的误差. 1)K3.7是“细粒混合矿物技术”创始人Zimmer-man161提出.也是细粒样品测定年代时,测量样品α外辐照效率必需要测量的值.但是计算年代时不能使用这个外辐照效率,因为陶器样品在陶器中接受的α辐照是“内辐照”,而内辐照效率K在一般情况下是没有办法测量的,所以 Zimmerman通过实验,得到了α内外辐照效率的转换系数0.85.如果用实验室α源辐照样品,测量得到的α效率一定要乘上这个转换系数,否则,会给年代测定带来一个系统误差. 2)α外辐照效率现在已经定位在α能量3.7 MeV,所以测定年代实验室的α辐照源辐照在样品上的能量一定要 3.7 MeV, 否则测量得到的α外辐照效率是错误的.但是对一般的热释光测定年代实验室来说,要使α源辐照在样品上的剂量正好是3.7 MeV能量是非常困难的,有的根本忽略这个问题,不考虑什么α能量,以至于测量得到的年代出现很大的误差既不知道什么缘故,也找不出什么原因. 3) 由于能量响应问题非常麻烦,不容易解决,所以 Aitken 提出用“a值系统”代替“K值系统”,解决了α能量响应问题.a是石英专用的α相对热释光效率,它的求法与K3.7完全相同,但是含义差别很大.要得到a值,先决条件是α源的强度不能用辐照剂量Gy 标定,必需用“S”标定,否则得到的α效率不是a值. 4)α剂量能量响应比较大,而且各种物质的α吸收效率不同.如果一个α源用某一个样品或者某一种人造磷光体来标定其辐照吸收剂量(Gy),那么用这个标定好的源的辐照剂量率来辐照其他样品时,其他样品吸收的α剂量并不是你已经标定好的这个剂量,而是根据样品不同而异.所以用辐照剂量来标签-个α源实际上是错误的.现在有了α源原S值,就可以 用S值计算不同辐照样品的吸收剂量率和α效率,因为一个o源的S值可以独立存在,与被辐照的样品无关.用S值计算各种样品的吸收剂量率见文献[121].S值还有一个非常大的用处,就是它可以通过热释光将已经标定好的α源的S值传递给另外一个被标定的α源,解决了以往o源标定的难题. 5 一些失败的典型例子 从失败的例子中可以看到古陶瓷测定年代的复杂性.我们在上一节中已经分析了影响古剂量、年剂量和年代准确测定的大量的复杂因素和存在的问题.这些问题有的得到了解决,有的部分解决,有的根本没有解决.这些错综复杂的因素往往相互交叉影响,同时出现.如果在一个样品的年代测定中,有几个复杂因素同时出现,而且分不出主次,那就非常麻烦,得到的年代往往是错误的.如果一件珍贵的古代陶瓷器,其真伪被我们测错,不仅会造成巨大的经济损失,还可能牵涉到法律,或者在名誉和信誉上造成不良的影响. 5.1 瓷器年代测定失败实例 瓷器年代测定的失败和成功例子已经列举很多,可见参考文献[51,59].其中最典型的例子就是两件“青花云龙纹象耳瓶”的热释光年代.如果用国际上流行的标准前剂量技术测定,其中一件的瓷器的热释光年代就为距今800年,这就是一件(或者两件)元代青花珍品,价值连城.但是用前剂量饱和指数法测定,得到的这两件瓷器的热热光年代都小于距今50年,那么这两件瓷器就为现代仿制品.两个结果大相径庭,到底孰是孰非,经过考古界和科学界反复讨论,-致认为这两件青花云龙纹象耳瓶不是元代真品.为什么同样用热释光技术,同样用前剂量方法,会得到截然相反的结果,这就是热释光测定年代的复杂性和存在的问题,其原因已经有文章报道,可以参阅参考文献[59],这里不再赘述. 另外,用前剂量饱和指数法测定瓷器年代还存在一些失败的例子,其中最典型的是S,偏高.图16是一件宋代紫定窑白瓷碗样品的TAC曲线.这件瓷器的热激活和辐照熄灭特性非常好,热释光灵敏度很高,熄灭率也很大,按理说是前剂量测定年代最理想 图 16 一件宋代紫定窑白瓷瓷样品的 TAC曲线 的样品,但是从图中可以明显地看到, So远大于 Sy,这是违反常规和不合理的,因为这样得到的古剂量和年代都是负值.我们采用包括预热在内的各种处理手段都没有办法使So降低.这样的瓷器就不能测定年代,这个问题至今没有办法解决. 5.2 陶器年代测定失败实例 陶器热释光测定年代相对地比瓷器测定年代简单, -般地说,大多数热释光测定年代实验室对陶器的年代测定和真伪鉴定已没有什么问题,但是唐三彩就不是这样.从事热释光测定年代的人都发现,不论是细粒混合矿物技术还是石英粗颗粒技术都很难测定唐三彩的烧制年代,有不少测定结果以失败而告终. 图17是一件编号 C1469 的民国时代的三彩陶器细粒样品的热释光曲线,图18是编号T031的一件唐代的三彩陶器细粒样品的热释光曲线.两个图中三条曲线分别为自然热释光N,自然加β剂量热释光N+β和自然加2β剂量热释光 N+23. 两个图中所有热释光曲线的本底已经被减去.两件陶器的热释光曲线形状倒是很想象,但是都没有“坪”.如果任意从高温部分截取热释光计算古剂量,那么从不同温度范围截取的热释光计算的古剂量各不相同,这样,得到年代也各种各样,到底哪一个年代是正确的,无法确定.图17测量的样品是取自一位收藏家送来的一件三彩陶马,物主称这件器物是唐代真品.我们根据大多数陶器的“坪”的位置,在高温部分截取一组热释光测量,采用的标定剂量为 6.53 Gy,按图2作线性回归,或者按(2)式计算,得到 P=7.664 Gy, D=8.69 mGy/a(唐三 彩细粒样品的平均年剂量),于是年代 A=880±202(±1o)aBP. 这个年代在±1o误差范围内与唐代(1101~1390 aBP)基本符合.所以测量者和物主都非常高兴.但实际上这个测量是失败的,如果我们再截取另一个温度范围的热释光来计算古剂量,就会得到另一个热释光年代 A=560 a. 到底是哪-个年代是正确的没有办法确定,因为没有坪的热释光是不稳定的,所以测得的年代也是不可靠的. 为了得到这件三彩陶器的真实年代,我们采用前剂量饱和指数法测定,结果年代为距今100左右,这与民国时代符合.前剂量饱和指数法的具体测量方法见文献[26]. 同样道理,如果图18中编号T031这件真的的唐三彩也用上述方法测量,得到的年代也是错误的.我们在图18中取300~330℃的热释光积分值,则 N=6298,N+C=14449, N+2=26806,单位cps(计数/秒),使用上述例子的同样方法,得到等效剂量Q-554 mGy,采用唐三彩细粒样品平均年剂量计算年代, A=64 aBP. 图17-一件明代三彩陶马细粒样品的热释光曲线 图18 一件唐代三彩陶器细粒样品的热释光曲线 如果再取另一个温度范围的热释光积分值,又得到一个年代 A=139 aBP. 我们再采用前剂量饱和指数法测定,得到的年代A=1200±280 aBP, 与唐代吻合.当然也有不少陶器不能用前剂量饱和指数法测定年代,主要是这些陶器前剂量灵敏度很低,无规律,致使得到的年代与已知年代相差甚远. 随着热释光测定古陶瓷年代知识的普及,一个新的问题也出现了.古陶瓷仿制者已经懂得只要将现代制造的仿古陶瓷,用y射线辐照一定的人工剂量,用热释光测定年代时,就会得出这件陶瓷器是“古代真品”的假象.因此,如何识别从一件陶瓷器中测量得到的热释光是自然辐照还是人工辐照产生的,将成为古陶瓷热释光测定年代的一个新课题.另外,热释光测定年代都要在器物上取样,这在珍贵文物上 是不允许的,因此,不需要取样的热释光无损检测又将成为一个新的和热门的研究课题. 另外,由加拿大西蒙·弗雷泽大学 Huntley 等在1985年发展起来的一项新技术-—“光释光”(Optically-stimulated luminescence, OSL)测定年代[122]已在国际上广泛流行,它在沉积物年代测定上发挥了独特的作用.因为就测定年代来说,晶格缺陷可以分为两类,一类是热敏陷阱,只有用加热才能排空陷阱中所有的俘获电子,这就是热释光.另一类是光敏陷阱,只要用光照就可以排空陷阱中的俘获电子,这就是光释光.由风形成的沉积物如黄土正是靠阳光来排空光敏陷阱中的电子,所以它对测定黄土沉积年龄非常有用.光释光测定古陶瓷年代目前只是初步试验一下,可能今后会有比较大的发展. ( 1( Grogler N , Houtermans F G, Stauffer H. Ue b er die datierung von keramik und z i egel d urch t hermolumineszenz. Helv Phys A C TA, 1960,33:595 —5 96 ) ( 2 Kennedy G C, K nopff L . Dating by thermoluminescence. Archaeology,1960,1 3 :14 7— 148 ) ( 3 Fleming S J, Moss H M, Joseph A . Thermoluminescence aut h enticity testing of some“Six Dynasties”figures. Archaeometry, 1970, 12: 57—65 ) ( 4 F l eming S J, Sampson E H . The a uthe n ticity of figur i nes, anim a ls and potte r y facsimiles of br o nzes in the Hu i Hs i ne s t yle. A r -chaeometry, 1972,14(2):237— 2 44 ) ( 5 Fleming S J. Thermoluminescence and glaze studies o f a group of Tang Dynasty ceramic s . Archaeometry,1973 , 15(1):31 —5 2 ) ( 6 Mc-Dougall DJ. Thermoluminescence of Geological Mate r ials. New York: Academic Press, 1968 ) ( 7 Aitken M J, Tite M S, Reid J . Thermoluminescent dating of ancient c eramics. N ature, 1 964,202: 1032-1033 ) ( 8 Ichikawa Y. Dating of ancient ceramics by t h ermoluminescence. Bull Inst of Chem Res Kyoto Univ, 1965, 43: 1—6 ) ( 9 Mazess R B, Zimmerman D W . Pot t ery da t ing from th e rmoluminescence. Science, 19 6 6, 152: 347—348 ) ( 10 Ralph E K , H an M C . D a ting of pottery by thermoluminescence. N ature,1966, 2 10:245—247 ) ( 11 Aitken M J, Zimmerman D W, Fle m ing S J. T h ermoluminescent dat i ng of ancient pottery. N ature. 1 968, 219: 442 —4 44 ) ( 12 Mejdahl V. Thermoluminescence dating of ancient danish ceramics. Archaeometry, 1969, 11: 9 9— 104 ) ( 13 F l eming S J. Thermoluminescence Techniques in Archaeology. Oxford: Clarendon Press, 1979 ) ( 14 Zimmerman D W . Introduction to basic procedures for s ample preparation and T L measurement of ceramics. PACT, 1978, 2: 1—6 ) ( 15 Aitken M J. Thermoluminescence Da t ing. London: Academic Press, 198 5 ) ( 16 Zimmerman D W. Thermoluminescent dating using fine grains f r om pottery. A rchaeometry, 1971, 13:29—52 ) ( 17 Fleming S J. Study o f thermoluminescence of crystalline extracts f rom pottery. A rchaeometry, 1966,9: 17 0 —173 ) ( 18 王维达,朱连芳.热释光测定古陶瓷年代.自然杂志,1980,11,3:844—8 4 7 ) ( 19 王维达. 古 代陶器的热释光年代.考古,1979,1:82一88 ) ( 20 王维达.陶器细粒热释光断代的原理和实验.上海博物馆馆刊,1981,1(1):123- —138 ) ( 21 Oberhofe M, Scharmann A. 应用热释光剂量学.北京 : 中国计量出版社,1988.290 ) ( 22 Aitken M J. Physics and Archaeology. 2nd e d. Oxford: C larendon Press,1974 ) ( 23 Wang W D, Xi a J D. A c onvenient method for preparation of fine-grain samples. Ancient TL, 1989, 7(3): 47—53 ) ( 24 夏君定.热释光法快速鉴定唐三彩真伪.核技术,1993,16(4):247— 2 50 ) ( 25 Huxtable J. Fine grain dating . PACT, 1978, 2: 7- 1 1 ) ( 26 王维达,夏君定,周智新.热释光前剂量饱和指数法测定中国古瓷器年代.中国科学E辑:技术科学,2006,36(5):525—540 ) ( 27 王维达,周智新,夏君 定 .内外照射效率的转换系数.核技术,199 7 ,20(8): 450- -454 ) ( 28 Wang W D,Zh o u Z X, XiaJ D, et al. The conversion factor of Ker to K. in thermoluminescence da t ing. J Nucl Sci T echnol, 1 998,9(4):227—230 ) ( 29 F leming SJ. Supralinearity c o rrections i n f i ne-grain thermoluminescence dating: A reappraisal. Archa e ometry, 1975, 1 7: 12 2 — 1 2 9 ) ( 30 王维达.河姆渡和禾皮岩陶片热释光年代的测定——兼论粗粒石英断代技术.考古学集刊,1984,4:321—327 ) ( 31 Bell W T . Alpha d ose attenuation in quartz grain s for thermoluminescence dating. Ancient TL, 1980, 1 2:4-8 ) ( 32 王维达,梁宝鎏,周智新,等.前剂量 饱 和指数法研究.核技术,1999,22(10):578- 582 ) ( 33 王维达,梁宝鎏,夏君定.前剂量饱和指数法测定中国瓷器年代.核技术,2001, 24(12):996—1001 ) ( 34 王维达,梁宝鎏,夏君定.前剂 量 中热激活和辐照熄灭研究.核技术,2005,28(5):350— 3 55 ) ( 35 Fleming S J. The acquisition of radioluminescence by ancient ceramics.Thesis of Doctor. Oxford: Oxford U niversity, 1969 ) ( 36 Fleming SJ, T hompson J. Quartz as a h e at resistant dosimeter. Health Phys, 1970, 18:567 -568 ) ( 37 Thompson J. T h e influence of p r evious i r radiation on t hermoluminescence sensitivity. Thesis of Doctor. Oxford: Oxford U niversity,1970 ) ( 38 Zimmerman J. The radiation-i n duced in c rease of t h ermoluminescence sensitivity of fired quartz. Solid State Phys, 1 971, 4 : 3277 —3 291 ) ( 39 Fleming S J. The pre-dose technique: a new thermoluminescence d a ting method. A r chaeometry, 1973, 12 : 13 3 —14 7 ) ( 40 Aitken M J. Pre-dose dating predictions from the model. PACT, 1979, 3: 319—324 ) ( 41 M cKeever S W S. Mechanisms of thermoluminescence production: Some problems and a few an s wers. Nucl T r acks R a diat M eas, 1991, 18:5—12 ) ( 42 H a skell E H . R etrospective accident dosimetry using environmental m aterials. Radiat Prot Dosim, 1993, 47(1-4) : 297— 30 3 ) ( 43 Bailiff I K. Pre-dose dating: High S sherds. PACT, 1979, 3: 3 4 5—35 5 ) ( 44 Bailiff I K. Pre-dose dating: Sensitization of R traps. PACT, 1983, 9:208—214 ) ( 45 Bailiff I K. Pre-dose a nd i n clusion dating: A comparison usi n g iron age and early medieval potte r y. Nucl T racks Radiat M eas, 1 9 85,10(4-6):771—777 ) ( 46 Haskell E H, Kaipa P L, Wrenn M E. Pre-dose TL characteristics of quartz inclusion removed from bricks exposed t o fallout radiationfrom atmospheric testing at the Nevada Test S i te. R adiat M eas, 1988, 1 4 :114 — 120 ) ( 47 Bailiff I K. Per-dose dating. In : Gokus H Y , O berhofer M, Regulla D, eds. Science Da t ing Method. CEC: Advan c ed Scientific Tech-niques, Kluwer Academic Publisher. 1991 ) ( 48 Bailiff I K. Per-dose technique. Radiat Meas, 1994, 23(2-3):471—479 ) ( Chen R. Saturation s ensitization of 110℃ TL peak in quartz and i ts applicatio n in pre-dose technique. PACT, 1978, 3 : 325 -335 ) ( 梁宝鎏, Stokes MJ, 王维 达 ,等.细粒石英前剂量方法.核技术,1995,18(8):463—466 ) ( 51 王维达,梁宝鎏,夏君定.前剂量中热激活和辐照熄灭研究.核技术,2005,28(5):350—355 ) ( 52 Chen R, Y a n g X H,McKeever S W S . The strongly supe r linear dose dep e ndence of thermoluminescence in synthetic quar t z. J P h ys DAppl Phys, 1988, 21 : 1452 —14 57 ) ( 53 Chen R , Leung L P. Modeling the pre-dose effect in thermoluminescence. Radiat Prot Dosim, 1999, 84: 43—46 ) ( 54 Chen R, L eung L P. A m odel for d ose- r ate dependence of thermoluminescence. J Phys D Appl Phys, 2000, 33: 846—850 ) ( 55 Bailiff I K. Pre-dose dating. In: Gokus H Y, Oberhofer M , Regull D , eds. S cience D ating Method: A d vanced Scientific Techniques. CEC: Eurocourses, Kluwer A cademic Publishers, 1991. 155 —1 73 ) ( 56 Bailiff I K. The pre-dose technique . Radiat Meas , 1994,23(2-3):417 —47 9 ) ( 57 Leung P L, Yang B, S t okes M J. A re g ression method for ev a luation of paleodose in the pre-dose technique. Ancient TL , 19 9 7, 15 : 1—5 ) ( 58 Leung P L, Stokes M J, Wang W D, e t al. Study on fi n e quartz pre-dose technique. J Nucl Sc i Technol, 1996,7:85—89 ) ( 59 王维达. 热 激活和辐照熄灭特性及其在瓷器前剂量测定年代中的应用.中国科学E辑:技术科学,2009, 39(1):94 -102 ) ( 60 Aitken M J. Appendix G: Annual D o se Evaluation: S u mmary of Radioactivity D a ta, i n Thermoluminescence Dating. London: Aca- demic Press , 1985.28 2— 286 ) ( 61 王维达,周智新,夏君定.从TL测定年代比对看α源的标定.核技术,1999,22(10):627—632 ) ( 62 王维达,周智新,夏君定.α内外辐照效率转换因子.核技术,1997,20(8):450—454 ) ( 63 王维达.热释光断代中直接从α源强度S求α效率K3.7.文物保护与考古科学,2002,12(增刊): 258- 265 ) ( 64 王维达,夏君定.用厚源α计数测定 TL 和ESR年代中的钍、铀年剂量.核技术,1991,14(2):101—1 0 8 ) ( 65 Przibram K . Irradiation Colours and Luminescence. London: Pergamon Press, 1956 ) ( 66 Cameron J R , Z i mmerman D, K e nney G, et al. Thermoluminescent radiati o n dosimetry utilizing LiF. Heal t h P hys, 1 964, 10: 25一29 ) ( 67 Schulman J H . Survey of luminescence dosimetry. Luminescence dosimetry. USA Conf,1 9 67,650637:3—33 ) ( 68 Aitken M J. L ow-level e n vironmental radiation m e asurement usi n g natural calcium flu o ride. In: Proceedings of 2nd Int e rnationalConference on L uminescence D o simetry. G a tlinburg: USAEC Conf, 1968. 28 1 --292 ) ( 69 Aitken M J. Thermoluminescent dosimetry of e nvironmental radiation on archaeological sites. Archaeometry, 19 6 9,11: 1 09—114 ) ( 70 Y amashita T, Nada N, Onishi H, et al. Calcium sulfate phosphor activated by rare earth. In: Proceedings of nd I n ternational C on-ference on Luminescence Dosimetry. Gatlinburg: USAEC Conf, 1968.4— 17 ) ( 71 Mejdahl V. Measurement of en v iro n mental radiation at a rchaeological excavation sites. Archaeometry, 1970,12:281 -283 ) ( Mejdahl V. Dosimetry techniques in thermoluminescence dating. Riso Report No.261, Riso N atio n al Laboratory, Roskilde. 1972 ) ( Mejdahl V . Measurement of enviro n mental rad i ation at ar c haeological ex c avation si t es b y m e ans of TL dos i meters. PAC T , 1978, 2:70—83 ) ( 74 Burgkhardt B, H errera R, P iesch E . L ong-term f a ding experiment w i th d i fferent T LD system. In: S c harmann, ed . Proceedings of 5thInternational Conference on L uminescence D osimetry. Giessen: Physikalisches Institut, 1977.7 5— 83 ) ( 75 Metha S K, Sengupta S. Annealing characteristics and nature of traps in Al, O ,thermoluminescent phosphor. P hys Med Biol, 1977, 22: 863—872 ) ( 76 Bhasin B D, S a sidharan R , Sunta C M. Preparation and the r moluminescent characteristics of t e rb i um doped magnesium orthosilicatephosphor. Health Phys,1976,30:139—142 ) ( 77 Yamashita T. A s e ries of s ulfate phosphors having different e ffective atomic numbers f o r evaluating radiation e nergy, In: Niewiadom-sky T, ed. Proceedings of the 4th Internationa l Conference on Luminescence Dosimetry. K r akow: In s titute of nuclear physics, 1974. 467—479 ) ( Becker K. S olid State Dosimetry. Cleveland Ohio: CRC P r ess, 1973 ) ( S chayes R, Brooke C, Kozlowitz I,et al. Thermoluminescent prop e rties of n a tu r al calcium fluo r ide, Lum i nescence disimetry, USAECConf-650637, US Atomic Energy Commission, 1967,138—1 57 ) ( 80 Storm E , Israel H I . P hoton Cross Sections fr o m 0.0 0 1 to 1 00 MeV for E l eme n ts 1 throug h 100. Los Ala m os: Universit y of California, 1967 ) ( 81 F r ancois H, M cKerrell H, M e jdahl V . Thermoluminescence dating of ceramics from Gloze. In: Scharmann A, e d . Proc e edings of 5th I nternational C onference on Lu m inescence Dosimetry. Giessen: Physikalisches Institu e , Justus - Liebig-Universitat, 1977. 469 479 ) ( 82 Mejdahl V . Dosimetry techniques in thermoluminescence dating, Riso Report No.261,Riso N ational Laboratory,Roskilde, 1972 ) ( 83 Aitken M J, F leming S J, Doell R , et al. Etna and other historic flows : Preliminary resul t s. I n: McDouga l l D J,ed. Thermolumines-cence of Geological Material s . New York: Academic Press, 1968.35 9—3 36 ) ( 84 Fleming S J . D evelopment and app l ication of c alcium fluoride for evaluation of dos a ge with a radi o active powe r matri x . In: Auxier J A,Becker K , Robinson E M , eds. Proceedings of 2nd International Conference on L uminescence Dosimetry. N T IS: Springfield, 1 968. 465 ) ( Fleming S J. Thermoluminescent d a ting: Refinement of the quartz inclusion method. A rchaeometry, 1 9 70,12 ( 2): 1 3 3—134 ) ( Bailliff L K, Aitken M J. Use of thermoluminescence dosimetry for evaluation of internal beta dose-rate in archaeological dating. NuclInstru Methods, 1980, 173(2):423 —4 29 ) ( 87 MejdahlV. T hermoluminescence dating: A thermoluminescence te c hnique for beta-ray dosimetry. PACT, 1978, 2:35-4 7 ) ( 88 Wang W D . The ultrathin T L D system for beta-ray dosimetry. Chin Sci B u ll,1984,29 ( 1):128—132 ) 89 王维达.陶片中β剂量的厚度响应.核技术,1993,16(4):228—231 ( 90 周智新,黄刚.第一次辐射剂量防护会议论文选编.北京:原子能出版社,1982.82 ) ( 91 Wang W D . Ult r athin TLD measurement of alpha dose-rate and com p arison with alph a counting. Ancient TL, 1983 , 1(1): 2—4 ) ( 92 王维达,周智新.热释光测定年代中用 TLD 测量α年剂量研究.科学通报,1983,28(13):807一80 9 ) ( 93 Wang W D , Zhou Z X. Thermoluminescence dating of Chinese pottery. Archaeometry, 1983,25(2):99—106 ) ( 94 Wa n g W D, Xi a J D, Zh o u Z X . Comparison betwe e n fine-g r ain and ulrat h in TLD i n mea s urement of alph a dose-rate. Ancien t TL,1989,7(2):38- -42 ) ( 95 周智新,黄刚.超薄型CaSO :Tm 热释光剂量元件的制作及应用.核技术,1 9 80,3(5):43-46 ) ( Chen R. On t h e calculation o f activation energie s an d frequenc y factors from glo w curves. JAppl Phys, 1969,40: 570— 58 5 ) ( C hen R . M e thods for kinetic analysis of thermally st imulated processes. J M a ter Sci, 1 9 76, 1 1 : 1 521—1541 ) ( Shenker D, Chen R . Numerical fitting of general order kinetics peaks. J Phys D Appl Phys , 1971 , 4: 287—291 ) ( Shalgaonkar C S, N arlikar A V. Recent methods f o r determining trap depth from glow c u rves. J M ater Sci, 1972, 7: 1465—14 7 1 ) ( 100 Ganguly S, K aul I K . A nalysis of thermoluminescence glow p e aks f r om natural calcium fluoride. Mod Geol,19 8 4, 8: 1 5 5- -162 ) ( 101 Wi n tle A G. Thermal q uenching of thermoluminescence in quartz. Geophys J R oyal A str Soc, 1975, 5 : 165—167 ) ( 102 Visocekas R, Ceva T, Marti C, et a l. Tunnelling processes in afterglow of calcite. Phys Status Solidi A, 1976, 35:315 —3 27 ) 103 Chen R, Kirsch Y. Analysis of Thermally Stimulated Processes. London: Pergamon Press, 1981 ( 104 Wi n tle A G, Aitken, M J, H u xtable J. A b normal thermoluminescence fadin g characteristics. In: Proceeding of 3rd Inte r national Con-ference on L uminescence Dosimetry. Denmark: R iso, 1971: 105—131 ) ( 105 Wi n tle A G. Abnormal fading of thermoluminescence in m ineral sample. Nature, 1973,245:143—144 ) ( 106 Wintle A G. Detailed study of a thermoluminescent mineral exhibiting anomalous fading. J Lum i n, 1977,15:385—393 ) ( 107 Bailiff I K . Use of phototransfer for anomalous fading of thermoluminescence. N a ture, 1976, 264:531— 53 3 ) ( 108 Mejdahl V . F eldspar inclusion dating of ceramics and burnt stones . PACT, 1983, 9: 351 —3 64 ) ( 109 王维达.热释光测定年代研究.核电子学与探测技术,1983,3(4):1—11 ) ( 110 Visocekas R . M i scellaneous as p ects of artificial TL ca l cite: emission spectra, a t hermal de t rapping and anomalous fading. PACT,1979(3):258—265 ) ( 111 Visocekas R , Ceva T, Marti C, et al. Tunnelling processes in afterglow of calcite. P h ys Status Solidi A , 1976, 35:315—327 ) ( 112 Fleming S J. Supralinearity corrections in fine-grain thermolumine s cence dating:A reappraisal. A rchaeometry, 1975, 17:12 2 —129 ) ( 113 Chen R, Bowman S G E. Superlinear growth of thermoluminescence due to competition during irradiation . PACT, 1978, 2:216—230 ) ( 114 Fain J, Sanzelle S, Miallier D, et al. A TL model b ased on deep traps competition. Ra d iat Me a s, 1994, 23(2-3):287-291 ) ( 115 Jayaprakash R, Ratnam VV , Durr a ni S A. Optical bleaching of radiation-induced colour centres in fluorapatite. Nucl Tracks RadiatMeas, 1985, 10(4-6):601—604 ) ( 116 Tanner A B. Radon migration in t h e ground. I n : Adams J A S, Lowder WM, eds. The Natural Radiation Environment. Chicago: Uni-versity of Chicago Press,1964 ) ( 117 Pernicka E , Wagner G A . R a dioactive e q uilibrium and dose-rate d etermination in T L dating. PACT,19 8 2, 6:132—144 ) ( 118 Murray A S. S t udies of the s t ability of radioisotope concentrations and th e ir dependence on grain size. PAC T , 1982, 6: 21 6 —223 ) ( 119 Aitken M J . R adon loss e valuation b y alpha counting. PACT, 1 9 78,2:10 4 117 ) 120 王维达.未知钍一铀比的误差分析.核技术, 1993, 16(4):240—243 ( 121 王维达.热释光断代中直接从α强度S求α效率K3.7.文物保护与考古科学,2002,14(增刊):258—265 ) ( 122 H u ntley D J, Godfrey- S mith D I, Thewalt M L W. Op t ical dating studies of sediments. N ature, 19 85,313: 1 0 5 )
确定



























还剩31页未读,是否继续阅读?

产品配置单

上海茗准科学仪器有限公司为您提供《古陶瓷热释光测定年代的研究和进展》,该方案主要用于其他中--检测,参考标准--,《古陶瓷热释光测定年代的研究和进展》用到的仪器有丹麦Risø热释光/光释光测年仪
相关方案
更多