








方案详情
文
锂电池鼻祖J.-M. Tarascon《Issues and challenges facing rechargeable lithium batteries》
方案详情

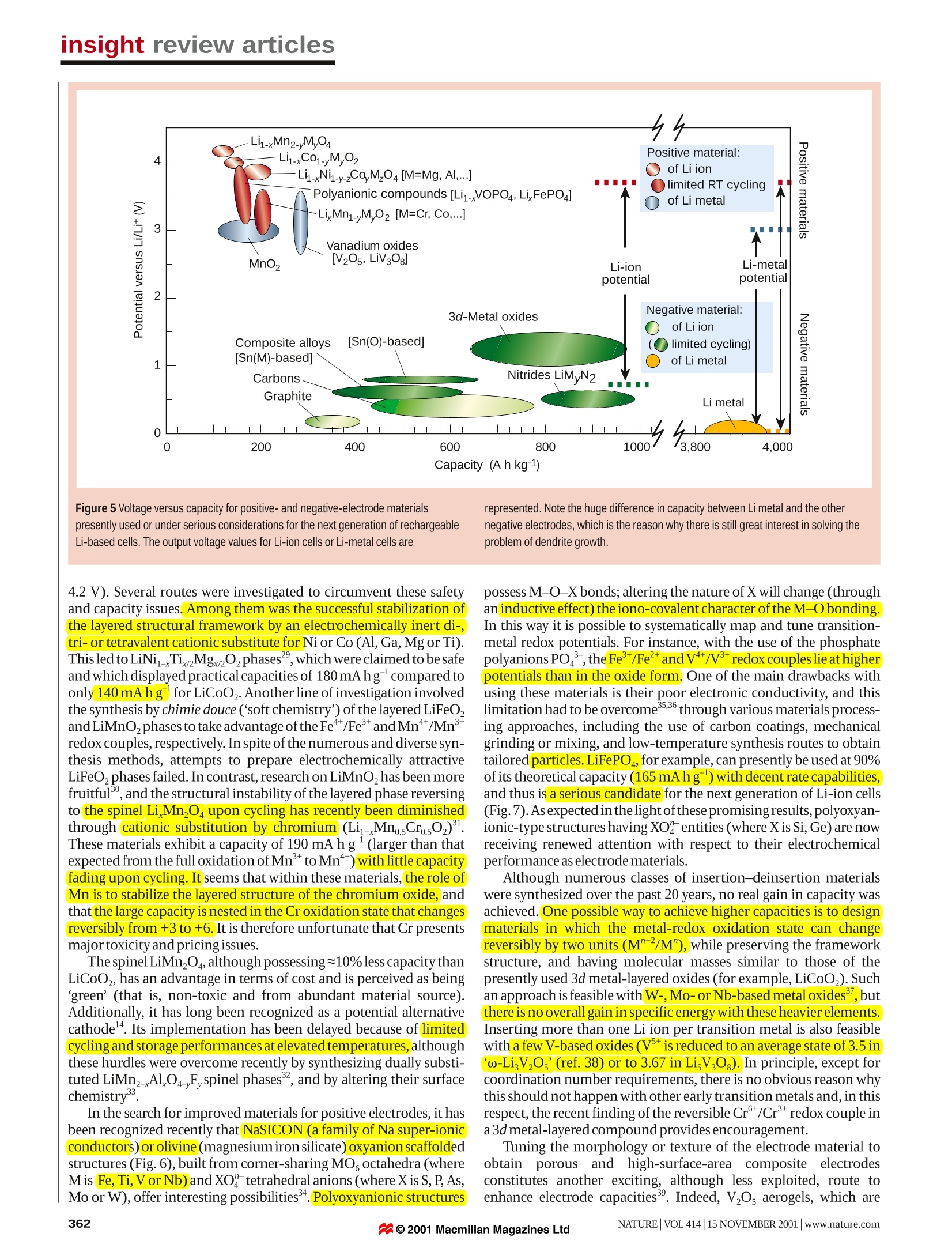
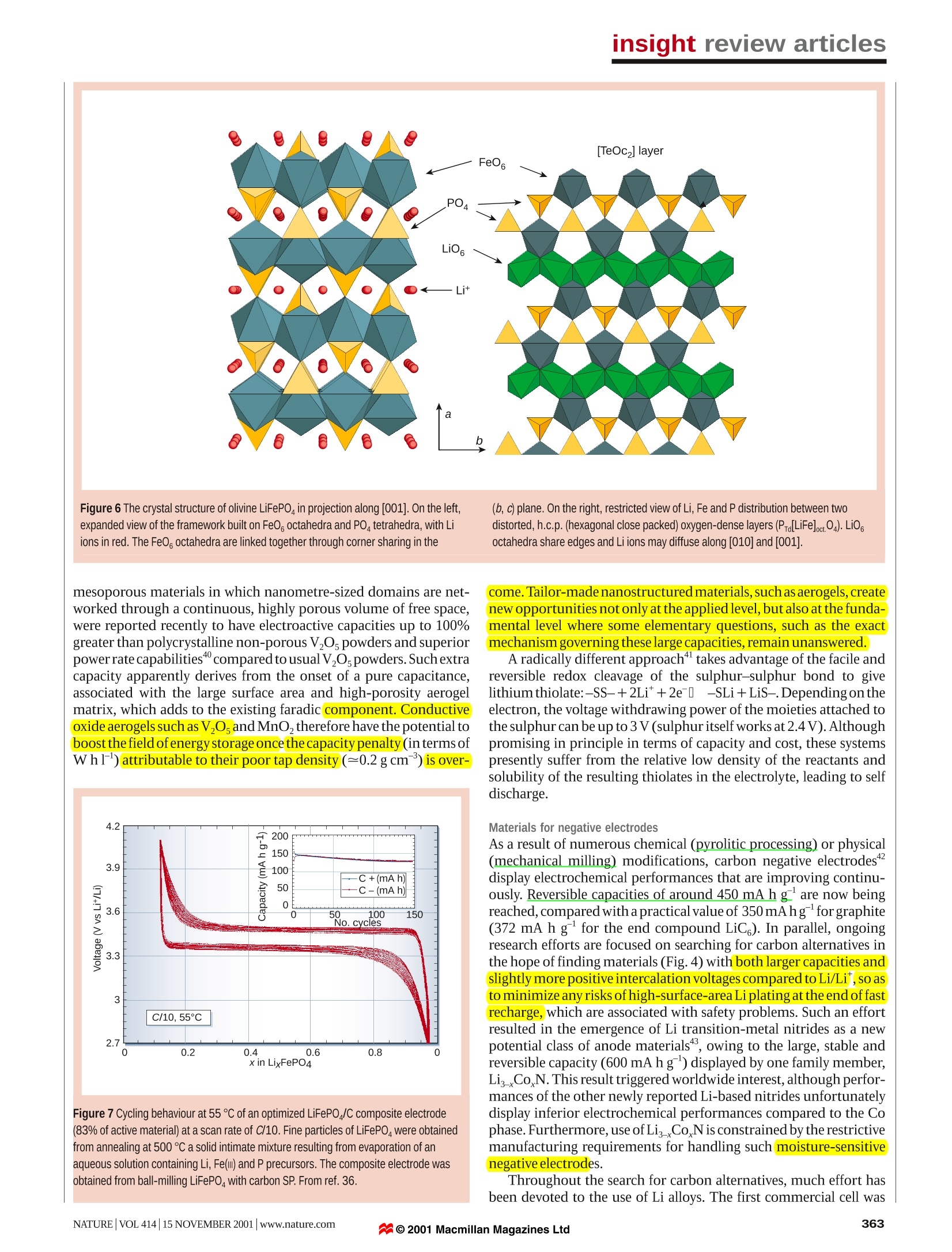
insight review articles J.-M. Tarascon*& M. Armandt *Universite de Picardie Jules Verne, Laboratoire de Reactivite et Chimie des Solides,UMR-6007, 33 rue Saint Leu, 80039, Amiens, FrancetDepartment of Chemistry, University ofMontreal, C.P. 6128 Succ. Centre Ville, Montreal, Quebec H3C 3J7, Canada Issues and challenges facingrechargeable lithium batteries demand for portable electronic devices. Lithium-ion batteries are the systems of choice, offering high energydensity, flexible and lightweight design, and longer lifespan than comparable battery technologies. We presenta brief historical review of the development of lithium-based rechargeable batteries, highlight ongoingresearch strategies, and discuss the challenges that remain regarding the synthesis, characterization,electrochemical performance and safety of these systems echargeable Li-ion cells are key components ofthe portable, entertainment, computing andRtelecommunication equipment required bytoday’s information-rich, mobile society.Despite the impressive growth in sales ofbatteries worldwide, the science underlying batterytechnology is often criticized for its slow advancement.This is true whatever the technology considered (forexample, nickel-cadmium, nickel-metal hydride or Liion). Certainly, when compared, energy storage cannotkeep pace with the rate of progress in the computerindustry (Moore’s law predicts a doubling of memorycapacity every two years), yet the past decade hasproduced spectacular advances in chemistry andengineering within the emerging technologies of Ni-MeHand Li-ion batteries. These cells are now supplanting thewell known Ni-Cd batteries. A battery is composed of several electrochemical cellsthat are connected in series and/or in parallel to provide therequired voltage and capacity, respectively.Eachcell consistsof a positive and a negative electrode (both sources ofchemical reactions) separated by an electrolyte solutioncontaining dissociated salts, which enable ion transferbetween the two electrodes. Once these electrodes are con-nected externally, the chemicalreactions proceed in tandemat both electrodes, thereby liberating electrons and enablingthecurrentto betapped bythe user.The amountofelectricalenergy, expressed either per unit of weight (Whkg) or perunit of volume (Whl), that a battery is able to deliver is afunction of the cell potential (V) and capacity (A h kg),both of which are linked directly to the chemistry of thesystem. Among the various existing technologies (Fig. 1),Li-based batteries - because of their high energy densityand design flexibility - currently outperform other sys-tems, accounting for 63% of worldwide sales values inportable batteries. This explains why they receive mostattention at both fundamental and appliedlevels. Historical developments in Li-battery research Before reviewing the present status of research and futurechallenges for Li-battery technologies, we present a briefhistorical account of developments over the past 30 years, aspersonally perceived. The motivation for using a battery technology based on Limetal as anode relied initially on the fact that Li is the mostelectropositive (-3.04V versus standard hydrogen electrode) Figure 1 Comparison of the different battery technologies in terms of volumetricand gravimetric energy density. The share of worldwide sales for Ni-Cd, Ni-MeHand Li-ion portable batteries is 23, 14 and 63%, respectively. The use of Pb-acidbatteries is restricted mainly to SLI (starting, lighting, ignition) in automobiles orstandby applications, whereas Ni-Cd batteries remain the most suitabletechnologies for high-power applications (for example, power tools). as wellas thelightest (equivalent weight M=6.94gmol,andspecific gravity p=0.53 g cm) metal, thus facilitating thedesign of storage systems with high energy density. Theadvantage in using Li metal was first demonstrated in the1970s with the assembly of primary (for example, non-rechargeable) Li cells. Owing to their high capacity andvariable discharge rate, they rapidly found applications aspower sources for watches, calculators or for implantablemedical devices. Over the same period, numerous inorganiccompounds were shown to react with alkali metals in areversible way. The discovery of such materials, which werelateridentified as intercalation compounds, was crucial in thedevelopment of high-energy rechargeable Li systems. Likemost innovations, development of the technology resultedfrom several contributions. By 1972, the concept ofelectro-chemical intercalation and its potential use were clearlydefined,althoughthe information was not widely dissemi-nated, being reported only in conferencee proceedings.Before this time, solid-state chemists had been accumulating Figure 2 Schematic representation and operating principles of Li batteries.a, Rechargeable Li-metal battery (the picture of the dendrite growth at the Li surfacewas obtained directly from in situ scanning electron microscopy measurements).b,Rechargeable Li-ion battery. structural data on the inorganic layered chalcogenides,and mergingbetween the two communities was immediate and fruitful. could give sufficient ion diffusion disappeared as a framework struc-ture (V,Oi3) proved to function perfectly. Later, Goodenough, withLiMO, (where M is Co, Ni or Mn)13,14, would propose the families ofcompounds that are still used almost exclusively in today's batteries. To circumvent the safety issues surrounding the use of Li metal,several alternative approaches were pursued in which either the elec-trolyte or the negative electrode was modified. The first approachinvolved substituting metallic Li for a second insertion material (Fig.2b).5The concept was first demonstrated in the laboratory by Murphyet al.and then by Scrosati et al.and led, at the end of the 1980s andearly 1990s, to the so-called Li-ion or rocking-chair technology. Theprinciple of rocking-chair batteries had been used previously inNi-MeH batteries18,19. Because of the presence of Li in its ionic ratherthan metallic state,Li-ion cells solve the dendrite problem and are, inprinciple, inherently safer than Li-metal cells. To compensate for theincrease in potential of the negative electrode, high-potential inser-tion compounds are needed for the positive electrode, and emphasisshifted from the layered-type transition-metal disulphides to layered-orthree-dimensional-typetransition-metaloxides13.Metal oxides aremore oxidizing than disulphides (for example, they have a higherinsertion potential)-owing to the more pronounced ionic character ofM O’bonds compared with ‘M S’bonds.Nevertheless, it tookalmost ten years to implement the Li-ion concept. Delays were attrib-uted to the lack of suitable materials for the negative electrode (eitherLi alloys or insertion compounds) and the failure of electrolytes tomeet - besides safety measures - the costs and performancerequirements for a battery technology to succeed. Finally, capitalizingon earlier findings20.21, the discovery of the highly reversible, low-voltage Li intercalation-deintercalation process in carbonaceousmaterial" (providing that carefully selected electrolytes are used), ledto the creation ofthe C/LiCoO rocking-chair cell commercialized bySony Corporation in June 1991((ref.23). This type of Li-ion cell,having a potential exceeding 3.6 V (three times that of alkalinesystems) and gravimetric energy densities as high as 120-150 Whkg(two to three times those ofusual Ni-Cdbatteries), is found in mostoftoday's high-performance portable electronicdevices. In 1972, Exxon’embarked on a large project using TiS, as thepositive electrode, Li metal as the negative electrode and lithiumperchlorate in dioxolane as the electrolyte.TTiS, was the best intercala-tion compound available at the time,having a very favourable Thesecondapproachinvolved replacing theliquidelectrolyte by alayered-type structure. As the results were published in readily avail- dry polymer electrolyte (Fig. 3a), leading to the so-called Li solidable literature, this work convinced a wider audience. But in spite of polymerelectrolyte (Li-SPE) batteries. But this technology is restrictedthe impeccable operation of the positive electrode, the system was not tolarge systems (electric traction orbackup power)andnotto portableviable. It soon encountered the shortcomings of a Li-metal/liquid devices, as it requires temperatures up to 80 ℃. Shortly after this,electrolyte combination-uneven (dendritic) Li growth as the metal several groups tried to develop a Li hybrid polymer electrolytewas replated during each subsequent discharge-recharge cycle (Li-HPE) battery, hoping to benefit from the advantages of polymer(Fig.2a),which led to explosion hazards. Substituting Li metal for an electrolyte technology without the hazards associated with the use ofalloy with Al solved the dendrite problem but, as discussedlater, alloy Li metal. Hybrid'meant that the electrolyte included three compo-electrodes survived only a limited number of cycles owing to extreme nents: a polymer matrix (Fig.3b)swollen with liquid solventand a salt.changes in volume during operation. In the meantime, significant Companies such as Valence and Danionics were involved indevelopingadvances in intercalation materials had occurred with the realization these polymer batteries, but HPE systems never materialized at theat Bell Labs that oxides, besides their early interest for the heavier industrial scale because Li-metal dendrites were still a safety issue.chalcogenides1.11, were giving higher capacities and voltages. More-over, the previously held belief that only low-dimensional materials With theaim ofcombining the recentcommercial success enjoyedby liquid Li-ion batteries with the manufacturing advantages Figure 3 Schematic representations of polymerelectrolyte networks. a, Pure(dry) polymerconsisting of entangled chains, through whichthe Li ions (red points) move assisted by themotion of polymer chains. b, A hybrid (gel)network consisting of a semicrystallinepolymer, whose amorphous regions are swollenin a liquid electrolyte, while the crystallineregions enhance the mechanical stability.c, A poly-olefin membrane (Celgard forinstance) in which the liquid electrolyte is heldby capillaries. a Liquid electrolyte c Cell can - Separator- Li1+xMn204 Separator- Carbon < Cell can 3.8V Cu Al -Separator 1.5 Ah Carbon Li+Mn204 Separator b Liquid electrolyte Carbon Separator . Li+Mn204 Cell can Figure 4 Schematic drawing showing the shape and components of various Li-ionbattery configurations. a, Cylindrical; b, coin; c, prismatic; and d, thin and flat. Note the unique flexibility of the thin and flat plastic LilON configuration; in contrast to the otherconfigurations, the PLilON technology does not contain free electrolyte. presentedby the polymer technology, Bellcore researchersintroduced polymeric electrolytes in a liquid Li-ion system. Theydeveloped the first reliable and practical rechargeable Li-ion HPEbattery, called plastic Li ion (PLiON), which differs considerablyfrom the usual coin-, cylindrical- or prismatic-type cell configura-tions(Fig.4). Such a thin-film battery technology,which offers shapeversatility,flexibility andlightness, has been developed commerciallysince l1999, and has many potential advantages in the continuingtrendtowards electronic miniaturization. Finally,the‘nextgeneration'of bonded liquid-electrolyte Li-ion cells, derived fromthe plastic Li-ion concept, are beginning to enter the market place.Confusingly called Li-ion polymer batteries, these new cells use agel-coated, microporous poly-olefin separator bonded to theelectrodes (also gel-laden), rather than the P(VDF-HFP)-basedmembrane (that is, a copolymer of vinylidene difluoride with hexa-fluoropropylene)used in the plastic Li-ioncells. Having retraced almost 30 years of scientific venture leading tothe development of the rechargeable Li-ion battery, we now describesome ofthe significant issues and opportunities provided by the fieldby highlighting the various areas in need oftechnological advances. Present status and remaining challenges that might be placed on tomorrow’s portable devices, which in turnplaces different requirements on the active material chemistry. Forinstance, with respect to the lower operating voltages of emergingelectronics,much debate has focused on whether we should developa low-voltage active chemistry or rely entirely on electronics(d.c.-d.c.converters) andlpersist in searching for high-voltage activeLichemistry.Findingthe best-performingcombination ofelectrode-electrolyte-electrode can be achieved only through theselective use of existing and new materials as negative and positiveelectrodes, and of the right electrolyte combination, so as to mini-mize detrimental reactions associated with the electrode-electrolyteinterface-the criticalphase ofany electrochemical system. Materials for positive electrodes The choice of the positive electrode depends on whether we are deal-ing with rechargeable Li-metal or Li-ion batteries (Fig. 5)2. Forrechargeable Li batteries, owing to the use of metallic Li as thenegative electrode,the positive electrode does not need to be lithiatedbefore cell assembly. In contrast, for Li-ionbatteries, because the car-bon negative electrode is empty (no Li), the positive one must act as asource of Li, thus requiring use of air-stable Li-based intercalationcompounds to facilitate the cell assembly. Although rechargeableLi-SPE cells mainly use Li-free V0s or its derivatives as the positiveelectrode,LiCoO,is most widely used in commercial Li-ion batteries,deintercalating and intercalating Li around 4V. Initially, the use of layered LiNiO, was considered, as this dis-played favourable specific capacity compared with LiCoO. Butexpectations were dismissed for safety reasons after exothermic oxi-dation of the organic electrolyte with the collapsing delithiatedLiNiO,structure.Delithiated LiCoO,was found to be more ther-mally stable than its Li NiO, counterpart. Thus, substitution of CoforNiinLiNi_Co.02was adopted to provide apartial solution to thesafety concerns surrounding LiNiO,. Whatever the considered battery technology, measures of its perfor-mance (for example, cell potential, capacity or energy density) arerelated to the intrinsic property ofthe materials that form thepositiveand negative electrodes. The cycle-life and lifetime are dependent onthe nature of the interfaces between the electrodes and electrolyte,whereas safety is a function of the stability of the electrode materialsand interfaces.Compared with mature batteries technologies, suchas lead-acid or Ni-Cd, rechargeable Li-based battery technologiesare still in their infancy, leaving much hope for improvementover thenext decade. Such improvements should arise from changes in bat-tery chemistry and cellengineering. Advances in active chemistry areleft to the solid-state chemists'creativity andinnovation in the designand elaboration of new intercalation electrodes. At the same time,they must bear in mind that it is impossible to predict the demands Although the reversible delithiation of LiCoO, beyond 0.5 Li isfeasible, delithiation for commercial applications has been limitedtothat value for safety reasonss (charged cut-off limited to around 4.2V). Several routes were investigated to circumvent these safetyand capacity issues. Among them was the successful stabilization ofthe layered structural framework by an electrochemically inert di-,tri- or tetravalent cationic substitute forlNi or Co (Al, Ga, Mg orTi).This led to LiNiTiMg202phases,which were claimed to be safeandwhich displayed practical capacities of 180mAhg compared toonly 140 mAhgfor LiCoO,. Another line of investigation involvedthe synthesis by chimie douce (‘soft chemistry’) ofthe layered LiFeO,andLiMnO,phases to take advantage oftheFeω/FeandMn*/Mn’+redoxcouples,respectively. In spite ofthe numerous and diverse syn-thesis methods, attempts to prepare electrochemically attractiveLiFeO phases failed. In contrast, research on LiMnO,has beenmorefruitful, and the structural instability ofthe layered phase reversingto the spinel Li Mnz04 upon cycling has recently been diminishedthroughcationic substitution by chromium MnsCrs0)3.These materials exhibit a capacity of 190 mA h1g(larger than thatexpected from the full oxidation ofMn’to Mn) with little capacityfading upon cycling. It seems that within these materials, the role ofMn is to stabilize the layered structure of the chromium oxide, andthat the large capacity is nested in the Cr oxidation state that changesreversibly from +3 to +6. It is therefore unfortunate that Cr presentsmajor toxicity andpricing issues. possess M-O-X bonds; altering the nature of X will change (throughan inductive effect) the iono-covalent character ofthe M-O bonding.In this way it is possible to systematically map and tune transition-metal redox potentials. For instance, with the use of the phosphatepolyanions PO,theFe/FeandV*/Vredox couples lie at higherpotentials than in the oxide form. iOne of the main drawbacks withusing these materials is their poor electronic conductivity, and thislimitation had to be overcome35.36 through various materials process-ing approaches, including the use of carbon coatings, mechanicalgrinding or mixing, and low-temperature synthesis routes to obtaintailoredparticles. LiFePO4, for example, can presently be used at 90%ofits theoretical capacity (165 mAhg) with decent rate capabilities,and thus is a serious candidate for the next generation of Li-ion cells(Fig.7). As expected in the light ofthese promising results, polyoxyan-ionic-type structures having XO entities (where X is Si, Ge) are nowreceiving renewed attention with respect to their electrochemicalperformance as electrode materials. The spinelLiMn,O4,although possessing~10%less capacity thanLiCoo, has an advantage in terms of cost and is perceived as beinggreen’ (that is, non-toxic and from abundant material source).Additionally, it has long been recognized as a potential alternativecathode . Its implementation has been delayed because of limitedcycling and storage performances at elevated temperatures,althoughthese hurdles were overcome recently by synthesizing dually substi-tuted LiMnAl.04F,spinel phases,and by altering their surfacechemistry. Although numerous classes of insertion-deinsertion materialswere synthesized over the past 20 years, no real gain in capacity wasachieved. One possible way to achieve higher capacities is to designmaterials in which the metal-redox oxidation state can changereversibly by two units (M"*/M"), while preserving the frameworkstructure, and having molecular masses similar to those of thepresently used 3d metal-layered oxides (for example, LiCoO,). Suchan approach is feasible with W-,Mo-orNb-based metal oxides’,butthere is no overall gain in specific energy with these heavier elements.Inserting more than one Li ion per transition metal is also feasiblewith a few V-based oxides (V’is reduced to an average state of3.5 inω-Li V 0,(ref. 38) or to 3.67 in Li, V,O;). In principle, except forcoordination number requirements, there is no obvious reason whythis should not happen with other early transition metals and, in thisrespect, the recent finding of the reversible Crt/Crredox couple ina3d metal-layered compound provides encouragement. In the search for improved materials for positive electrodes, it hasbeen recognized recently that NaSICON (a family of Na super-ionicconductors)orolivine (magnesiumiron silicate)oxyanion scaffoldedstructures (Fig. 6), built from corner-sharing MO, octahedra (whereM is Fe, Ti, VorNb) andXO"tetrahedral anions (where X is S,P,As,Mo or W), offer interesting possibilities’Polyoxyanionic structures Tuning the morphology or texture of the electrode material toobtain porous and high-surface-areal composite electrodesconstitutes another exciting, although less exploited, route toenhance electrode capacities. Indeed, VO, aerogels, which are mesoporous materials in which nanometre-sized domains are net-worked through a continuous, highly porous volume of free space,were reported recently to have electroactive capacities up to 100%greater than polycrystalline non-porous V,0s powders and superiorpower ratecapabilities"compared tousualV,0,powders. Suchextracapacity apparently derives from the onset of a pure capacitance,associated with the large surface area and high-porosity aerogelmatrix, which adds to the existing faradic component. Conductiveoxide aerogels such as V,O,andMnO,therefore have the potential toboostthe field ofenergy storage once the capacity penalty (interms ofWh1)) attributable to their poor tap density (=0.2gcm)is over- Figure 7 Cycling behaviour at 55℃ of an optimized LiFePO /C composite electrode(83% of active material) at a scan rate of C/10. Fine particles of LiFePO were obtainedfrom annealing at 500 ℃ a solid intimate mixture resulting from evaporation of anaqueous solution containing Li, Fe(ll) and P precursors. The composite electrode wasobtained from ball-milling LiFePOwith carbon SP. From ref. 36. come. Tailor-made nanostructured materials, such as aerogels, createnew opportunities not only at the applied level, but also at the funda-mental level where some elementary questions, such as the exactmechanism governing these large capacities, remain unanswered. A radically different approachtakes advantage of the facile andreversible redox cleavage of the sulphur-sulphur bond to givelithium thiolate:-SS-+2Li*+2e→-SLi+LiS-.Depending on theelectron, the voltage withdrawing power of the moieties attached tothe sulphur can be up to 3V (sulphur itselfworks at2.4V). Althoughpromising in principle in terms of capacity and cost, these systemspresently suffer from the relative low density of the reactants andsolubility of the resulting thiolates in the electrolyte, leading to selfdischarge. Materials for negative electrodes As a result of numerous chemical (pyrolitic processing) or physical(mechanical milling) modifications, carbon negative electrodes"display electrochemical performances that are improving continu-ously.Reversible capacities of around 450 mAhg are now beingreached, compared with a practical value of 350 mAhg forgraphite(372 mA h g for the end compound LiC). In parallel, ongoingresearch efforts are focused on searching for carbon alternatives inthe hope of finding materials (Fig. 4) with both larger capacities andslightly more positive intercalation voltages compared to Li/Li ,so asto minimize any risks ofhigh-surface-area Li plating at the end offastrecharge, which are associated with safety problems. Such an effortresulted in the emergence of Li transition-metal nitrides as a newpotential class of anode materials", owing to the large, stable andreversible capacity (600 mAhg) displayed by one family member,LisCoN. This result triggered worldwide interest, although perfor-mances ofthe other newly reported Li-based nitrides unfortunatelydisplay inferior electrochemical performances compared to the Cophase.Furthermore,use ofLis_CoNis constrained by the restrictivemanufacturing requirements for handling such(moisture-sensitivenegative electrodes. Throughout the search for carbon alternatives, much effort hasbeen devoted to the use of Li alloys. The first commercial cell was introduced in the 1980s by Matsushita; this was based on Wood'smetal (a low-melting alloy of Bi, Pb, Sn and Cd), whose cyclingperformances were found to deteriorate with increased depth ofdischarge. While attractive in terms of gravimetric capacity, Li alloyssuffer from cyclability issues resulting from large Li-driven volumeswings (up to 200%), which cause disintegration and hence a loss ofelectrical contacts between particles. Although a reduction in alloyparticle size clearly benefits the cyclability by increasing tolerance tostress cracking, so far the gains are not sufficient". However, itbecame clear that any physical or chemical means ofovercoming theproblem of reactant expansion should be beneficial, hence the use ofcomposite negative electrodes. The basis behind this concept is theuse of a ‘buffer matrix' to compensate for the expansion of the reac-tants, sopreserving the electrical pathway?4.Initially, such a bufferaction was achieved by mixing two alloys that reacted at differentpotentials so that the electrochemically active phase was imbedded inanon-electrochemicallyactive matrix. A similar approach held considerable promise in 1997, when Fujiannounced the commercialization of a new Li-ion technology(STALION) using an amorphous tin composite oxide (ATCO) asnegative electrode. This reacts reversibly with Li at about 0.5 V,andhas a specific capacity twice that of graphite. In situ X-ray diffrac-tion studies of the ATCO electrode led to the conclusion that the Lireactivity mechanism in these composites was based on oxidedecomposition by Li through an initial irreversible process to formintimately mixed Li,O and metallic Sn, followed by a Li alloying reac-tion to form nanodomains of LinSn embedded within the Li,Omatrix4. However, the STALION cell was never commercialized, inspite ofits announcement at the end of1998. This was most likely dueto poor long-term cyclability, the huge and irreversible capacity lossduring the first cycle, which was reported by many groups, and thenecessity of finding a convenient source for the two initial Li ionsneeded for the SnO reduction process. did not react with Li according to the classical processes of Liinsertion-deinsertion or Li alloying, the catchwords of the past20 years. For instance, MO-type compounds (where M is Co, Ni, Fe,Cu or Mn), having a rocksalt structure and containing metalelements (M) that do not alloy with Li, exhibited capacities two tothree times those of carbon with 100% capacity retention for up to100 cycles. The mechanism of Li reactivity in such materials differsfrom the classical processes, and is nested in the electrochemicallydriven, in situ formation of metal nanoparticles during the firstdischarge, which enables the formation and decomposition of Li,Oupon subsequent cycling.Remaining issues relate to a problem ofsurface, with the chemical reactivity being enhanced as the particlesize becomes smaller. These findings open new avenues of researchaimed at capitalizing on the beneficial effect that particle-sizeconfinement could have within the field of electrochemistry. Theseand related nanocluster systems under development hold muchpromise for future developments. Besides ATCO, other investigations, such as those pursued byDahnetal.48onthe-Sn-Fe-Csystem, have also revealed an appealinglow-voltage reversible reactivity in composite materials developed asnegative electrodes (in spite of initial irreversibility and short-livedcapacities). The best experimental proof of the beneficial aspect ofthe buffer matrix arises from the ability to obtain several hundredcycles on a composite made by precipitating Sn metal at the grainboundaries ofelectrochemically inactive SnFe,C grains. However,the cycling performance was improved at the expense ofthe electrodeelectrochemical capacity. A new approach to alleviate the problems of alloy expansion,proposed by Thackeray et al., involved selecting intermetallic alloyssuch as CuSns, InSb and CuSb that show a strong structuralrelationship to their lithiated products,Li,CuSn and Li, Sb for the Snand Sb compounds, respectively.InSb and Cu,Sb electrodesi areparticularly attractive candidates because theyoperate through areversible process of lithium insertion and metal extrusion, with aninvariant face-centred-cubic Sb array (that is, this array provides astable host framework for both the incoming and extruded metalatoms). In the ternary Li InSb system (04V versus Li/Li) positive electrodematerial for Li-ion batteries requires electrolyte combinations thatoperate well outside their window of thermodynamic stability(3.5V).This is the reason why early workers in the field ignored verypositive cathode materials. But fortunately this electrolyte stability iskinetically controlled, enabling the use of non-aqueous electrolytesat potentials as high as 5.5 V(ref. 55). Similarly, the use of a polymerrather than a liquid electrolyte adds further selection criteria linkedto the electrochemical stability ofthe polymer. There arenumerousliquid solvents available, each with different dielectric constants andviscosity, and we can select specific solvents to favour the ionic Based on the peculiar behaviour (that is, large capacity at lowpotential) of the transition-metal vanadates M-V-O, first proposedbyy Fuji Co.and later studied by several groups, Poizot et al.reinvestigated the reactivity of Li-metal oxide.Surprisingly, theyfound a Li electrochemical activity for well known oxides.lbut these Figure 8 Arrhenius plot of conductivity for various solid electrolytes. 1,First-generation PEO-LiCF,SO; 2, new solutes with high-dissociation PEO-Li[(CF,SO2)2N];3,low-7, combination polymer; 4, plasticized polymer electrolytePEO-Li[(CF,SO2)2N]+25% w/w PEG-dimethylether (molecular weight, 250); liquidcrystalline polymer electrolytes; 5, heating curves; 6, cooling curve;7, gel-typepolymer (X-linked PEO-dimethacrylate-Li[(CF,SO2)2N]-PC 70%); 8, liquid electrolytePC/DME LiCF,SO ; 9, liquid electrolyte EC/DMC-LiPF, at low temperature; 10, gelelectrolyte P(VDF-HFP)/EC/DMC-LiPF,(ref.61). Polymer and liquid electrolytes conductivity of the electrolyte. In contrast, there are only a fewLi-based salts or polymers to choose from, the most commonly usedones being based on polyethylene oxidee ((PEO). The results fromresearch efforts aimed at counterbalancing this deficit have led to thepresent level ofresearch and development on electrolytes. Guided by general concepts of viscosity and dielectric constants,optimizing the ionic conductivity of a liquid electrolyte almostbecomes a field-trial approach with the hope of finding the keyingredients. For instance,only ethylene carbonate can provide theadhoc protective layer on the surface of graphite that prevents furtherreaction (continuouselectrolyte reduction and self-discharge).Ethylene carbonate is therefore present in almost all commercialcompositions, thinned with other solvents owing to its high meltingpoint. Why the homologous propylene carbonate is unsuitable forthis protective layer remains an open question, reminding us thatchemistry has its secrets. In contrast, achieving high ionic conductivity in Li-basedpolymer electrolytes requires a better understanding of thefundamentals of ion dissociation and transport. Both the nature ofthe polymer-salt interaction and the precise structure of highlyconcentrated electrolyte solutions have always resisted rationaliza-tion. Nevertheless, a principal goal has been to search for new, highlyconductive salts with a large electrochemical window, which form aeutectic composition with PEO that melts at the lowest possible tem-perature. The concept of non-coordinating anions with extensivecharge delocalization was achieved with the perfluorosulphonimideLi*[CF,SO,NSO,CF,] salt (abbreviated as LiTFSI)58. Figure 8 showsthe marked improvement when passing, with simple PEO, from a‘conventional'LiCF,SO, salt (curve 1) to the imide salt (curve 2),where an order of magnitude is gained, not ignoring the larger elas-tomeric domaintowards low temperature. The polymer architecturehas a role independently of dissociation. Attaching the side chains ofthe solvating group to the polymer increases the degrees of freedomas a result of dangling chain ends; this improves conductivity (Fig. 8,curve3), but compromises the mechanical properties. Although efforts aimed at enhancing the ionic conductivity ofpolymer electrolytes have been insufficient to permit operation atroom temperature, they have benefited liquid-based electrolytesystems in terms of cost and safety, so that battery manufacturers ofLi-ion cells are eager to see the further developmentoforganic anion-based salts able to operate at voltages greater than 4.5 V. LiTFSI is anexample of this cross-fertilization. Although extremely resistant tooxidation itself. the electrochemical use of such a salt is limited to 4 Vin presence of an Al collector, because a stable and soluble Al salt canbe formed as a consequence of the robustness of the anion bonds.With the less stable coordination anions (LiPF), decompositionoccurs immediately and is accompanied by formation of protectiveAlF. However, owing to its high conductivity in any medium, itssafety and lack oftoxicity, LiTFSI is being used increasingly in Li-ionbatteries, the corrosion problem having being solved by simpleaddition of a passivating coordination-type salt. A wide range ofanion-forming systems now exists, especially in the imide family,and these are viewed as candidates for high conductivity and Alpassivation. Having exploited most of the possibilities offered by ‘dry’polymers to improve conductivity (ability, amorphous state andlowest possible glass-transition temperature Tcontrolling the ionmobility), a remaining option was to use additives, known inpolymer science as plasticizers, to act as chain lubricants, so leadingto the development of ‘hybrid’ polymer electrolytes°. Indeed,suitableplasticizers are chosen between the same polar solvents as forliquid electrolytes, such as propylene carbonate, y-butyrolactoneor polyethylene glycol ethers, or are formed from short-chain PEO(4-25 monomer units). A lightly plasticized material (10-25%additive) improves conductivity by an order of magnitude (Fig. 8,curve4). Gels, on the other hand, contain 60-95% liquid electrolyte,andare only 2-5times less conductive than their liquid counterpart (Fig. 8, curves 7-10). Interestingly, when the gelling agent is apolyether, most of the solvation still takes place through the polymerchains rather than the carbonate solvents, the latter being less proneto donate electron pairs. Understandably, the lightly plasticizedsystems can be used in a Li-metal configuration, as much of theresilience ofthe pristine polymer is retained, whereas the much softergels require a Li-ion configuration. It is surprising that in spite of the direct link between their ionicconductivity and their degree of amorphicity, very little is knownabout thestructural chemistry of polymer electrolytes. In contrast tothe well established dynamic view of ionic conductivity on thesematerials, Bruce et al.recently proposed a structural view,highlighting the importance of aligning or organizing the polymerchains in order to enhance the levels ofionic conductivity. Similarly,Wright and co-workers3 and Ingram4 focused on the liquidcrystalline state to force the solvating polymer into a conformationthat was dictated by the liquid crystal part. The result is a partialdecoupling of the conductivity from the glass-transition tempera-ture ofthe polymer. The conductivity of such liquid crystalline chainpolymers is low at room temperature, but reaches liquid-like valuesathigh temperature or when keptunder polarization, and remains soupon cooling to room temperature (Fig. 8, curves 5 and 6), withoutappreciable activation energy. As these new perspectives generaterenewed interest in the design of polymer electrolytes, it is hoped thatsolutions may eventually be found to the problems of ionicconductivity afflicting this class of materials at ambient orsubambient temperature. The addition of nanoparticle fillers (10% w/w), such as Al O orTiO,to simple PEO compounds increases the conductivity several-foldat60-80℃,andprevents crystallization for at least several weeksat room temperature. Two important advantages of these systemsare an increase in the apparent Li transport number, from a low of~0.3 (common to polymer, liquid and gels) to ~0.6, and the forma-tion of a stable, low-resistance interface in contact with Li. Becausethese materials obey different conduction mechanisms, they arepresently the focus of many studies, both practical and theoretical. Technologies based on either solid polymer or ‘hybrid'polymerelectrolytes offer great advantages that will be necessary to meet theflexible, shape-effective requirements dictated by today's electronicminiaturization, while at the same time providing a larger autonomy.Current Li technologies rely on liquid-jellyroll or prismatic-cellconfigurations. Neither fits well in a multiple-cell configuration.This is in marked contrast with the recent thin, plate-like plastic(PLiON) technology that enables excellent packing efficiency, asmultiple plates can be densely packaged in parallel within one cellwhile preserving the flexibility ofthe overall package.Future technol-ogy improvements should focus on better chemical engineering ofthe bonded laminates, so as to obtain even thinner cells. Similarattributes can be provided by the solid Li-polymer technology, whichin addition exhibits extra capacity and is free of electrolyte leakage.This currently operates at 80 ℃. Although warm temperatures maybe an advantage for the large batteries required by the transportationsector, problems of conductivity have to be solved for electronicapplications, as emphasized earlier. The electrode-electrolyte interface The Li-ion cell density can be improved through a selective use ofappropriate existing or new materials for negative and positiveelectrodes.However, optimizing an electrode material is only the firststep in the process leading to its implementation in a practical cell.Indeed, while the capacity of a cell is nested in the structural orelectronic behaviour of its electrode, poor cell lifetimes are rootedmainly in side reactions occurring at the electrode-electrolyte inter-face. Thus, mastering the chemical stability of any new electrodematerial with respect to its operating liquid or polymer electrolytemedium, which requires a control of the electrode-electrolyteinterface through surface chemistry, is as important as designing new materials. Tackling interfacial issues is both tedious and complex. Weshould remember that, despite many years of research devoted to themechanism by which the solid electrolyte interphase forms on Li orcarbonaceous materials, its composition and nature are still the sub-)-ject of much controversy. In contrast, the positive electrode interfacehas received little attention over the years, despite its equally crucialrole. Its importance is amplified with the Li-ion technology, wherehigh voltages exceed the electrochemical resistance of the electrolyte Consumers are in constant demand for thinner, lighter, space-oxidation, and even favour its catalytically driven decomposition. effective and shape-flexible batteries with larger autonomy. SuchThus, it is critical to control the electrode surface so as to modify its demand will continue to generate much research activity towards thecatalytic activity towards electrolyte decomposition. The strategy development of new cell configurations and new chemistries. In thisdeveloped to address this issue uses coatings that encapsulate, review we hope to have conveyed the message that the field of energythrough chemical or physical means, the electrode grains with either storage is advancing faster than it perhaps has ever done in the past.an inorganic or an organic phase. This concept, successfully applied The benefits, in terms ofweight, size and design flexibility provided byto the spinelLiMn,O4, is based on minimizing the surface area ofthe today's state-of-the-artLi-ion configurations, which owe much to theactive material in direct contact with the electrolyte’P. The coating designengineers'striving to develop efficient, economical microtech-must allow easy diffusion of Li ions and, although insulating in nologies, are a good illustration. The Li-based battery chemistry isnature, must be thin enough to allow the electrons to tunnel through. relatively young, and as such is a source of aspirations as well asEqually relevant is the unexplained role of filler additives in polymer numerous exciting challenges. The latter are not limited to solid-stateelectrolytes", which markedly reduce the interfacial iimpedance in chemists. The effort should be highly multidisciplinary with strongcontact with Li. roots in the fields oforganic and inorganic chemistry,physics, surfacescience and corrosion. Through materials design we can expectsignificant improvements in energy density. And although designingnew materials can be intuitive or based on chemical concepts,coupling these efforts with those oftheorists who are able to performband-structure calculations on envisioned compounds will prove tobe highly beneficial. Ofequal importance is a better understanding ofthe electrode electrolyte interface to facilitate design of newinterfaces. Here the goal is well defined, although we must divergefrom the empirical approach used so far, and make full use of therecent progress achieved by in situ characterization. As Li-recharge-ablebatteries enter their teenage years,scientists andengineerspredictaneven brighter future lies ahead. 口 Thirty years after its initial observation, the key issue of Li den-drite growth, which was thought to be governed mainly by currentdensities,remains highly topical, especially in light ofrecentpromis-ing results obtained by Aurbarch’s and Bates’ groups. RevisitingExxon’s solvent 1-3-dioxolane, Aurbarch and co-workers showedthat the use ofLiAsF, salt led to a completely different Li morphologyfrom that obtained from an ethylenecarbonate-dimethyl carbonate(EC-DMC) electrolyte. They explained this in terms ofthe reactivityofdioxolane with lithium,which forms an elastomeric coatingendowing the Li surface with plasticity and flexibility, thereby reduc-ing dendrite growth. These findings were implemented in Li/MnO,commercial Tadiran cells that, under well defined cycling conditions,are claimed to be safe. The bulk polymerization of the cyclic ether,initiated at the positive electrode on overcharge, acts as a thermalshutdown. Even more spectacular are recent reports by Bates et al.who succeeded in cycling LiCoO,/Li thin-film batteries for morethan 50,000 cycles using a glassy electrolyte in ~1-um-thick filmsobtained by sputtering techniques. Byyccontrolling the uniform Listripping-plating mechanism, the same authors demonstrated thefeasibility of a Li-free, rechargeable, thin-film battery- that is, cellsconstructed in the discharged state with no Li metal initiallypresent. Such findings, whether resulting from low-current densityor the use of solid electrolyte, show that the problem of dendritegrowth can be solved, at least with special cell configurations. Visco70and co-workersrecently showed that a glassy nanometric layerdeposited on Li metal completely insulates it from its environment,even in the presence ofliquids, and that this coating can be applied ata high production rate. With further work devoted to the implemen-tation ofthese findings to large-size Li batteries, the development ofaLi-free rechargeable battery remains a realistic goal for the future. The principal challenge for Li-based rechargeable batteries, orindeed for any battery, lies in gaining better understanding andcontrol ofthe electrode-electrolyte interface in the hope ofdesigningnew solid-solid or solid-liquid interfaces. Forexample, the nature ofthe secondary reactions occurring at high temperature, which causecell failure, remains an unanswered question that must be addressedto ensure the practical success of these technologies. In this case,however, the main difficulty stems from a lack ofavailable techniquesto probe the evolution ofthe electrode-electrolyte interface at a locallevel. We have so far relied (with the exception of X-ray diffraction)on post-mortem rather than in situ studies to determine how theelectrodes or interfaces age with time either under cycling or storageconditions, thereby missing key information. Butintroduction oftheplastic Li-ion-type technology has created new opportunities toperform a wide variety of in situ characterization techniques. Theseinclude X-ray absorption near-edge structure, nuclear magnetic resonance and Mossbauer spectroscopies, or even scanning electronmicroscopy observations that allow real-time visualization ofdendrite growth at an interface. Efforts aimed at developing newcharacterization tools must be vigorously pursued so as to create acomprehensive database on the electrode-electrolyte interface. . Conclusion 1. Takeshita, H. Portable Li-ion,worldwide. Proc. Conf. Power 2000, San Diego, 25 September 2000. 2 Ikeda, H., Saito, T. & Tamura, H. in Proc. Manganese Dioxide Symp. Vol.1 (eds Kozawa, A. & Brodd,R.H.) (IC sample Office, Cleveland,OH, 1975). 3. Steele, B. C. H. in Fast Ion Transport in Solids (ed. Van Gool, W.) 103-109 (North-HollandAmsterdam, 1973). 4. Armand, M. B. in Fast Ion Transport in Solids (ed. Van Gool, W.) 665-673 (North-HollandAmsterdam, 1973). 5. Rouxel, J.,Danot, M., & Bichon, M. Les composites intercalaires Na, TiS,. Etude generale des phasesNa TiS, et K TiS,. Bull. Soc. Chim. 11,3930-3936 (1971). 6. Di Salvo, F.J., Schwall,R., Geballe, T. H., Gamble, F. R. & Osieki, J. H. Precursor effects of superconductivity up to 35°K in layered compounds. Phys. Rev. Lett. 27,310-313(1971). 7. Whittingham, M.S. Electrochemical energy storage and intercalation chemistry. Science 192, 1226(1976). 8. Whittingham, M. S. Chalcogenide battery. US Patent 4009052. 9.Rao,B. M. L., Francis, R. W. & Christopher, H. A. Lithium-aluminium electrodes.J. Electrochem. Soc.124,1490-1492(1977). 10.Broahead, J. & Butherus, A.D. Rechargeable non-aqueous battery. US Patent 3791867. 11.Broadhead,J.,DiSalvo,F. J. & Trumbore,F. A. Non-aqueous battery using chalcogenide electrode.US Patent 3864167. ( 1 2. Murphy, D. W.& Christian, P. A. Solid state electrodes for high energy bat t eries. Science 205,651-656(1979). 13 .M izushima, K., Jones,P. C. , Wiseman , P. J. & Goodenough,J.B . Li C oO , (0 < x≤1):a new cathode m aterial f o r batteries of high energy den s i t y. M at . Res . B ull. 1 5,783- 7 8 9 ( 1980). 14 . T hackeray, M . M . , Da v id,W. I. F. , Br u ce ,P . G. & Goodenough,J. B . L i thium in s ertion into mangane s e s pinels. M at. R es . B ull. 18,461- 4 72(1983). ) ( 1 5. A rmand,M. B. in Ma t eria l s for Advanced Batteries (Proc. N A T O Sy m p. Ma t erials Ad v . B a tteries) (eds M urphy, D. W . , Broadhead, J.& Steele, B. C. H . ) 145-161 (P l enum, New Y o rk, 1980). ) ( 1 6. Murphy,D. W., DiSalvo, F. J., C a r ides,J. N. & Waszczak,J.V. Topochemical reactions o f rutil e related s tructures w i th lithium. Mat. R e s. Bull. 13,139 5- 140 2 (1978). ) ( 17. ". L azzari, M. & Scros a ti, B. A cyclabl e lith i um organic electrolyte c ell based on two intercalation e lectrodes . J . E lect r o c h em. S oc . 12 7 ,7 7 3- 774(1980 ) . ) ( 1 8. Will, F. G. Hermetically sealed secondary battery with lanthanum nickel anode. US p atent 3874958( 1 975). ) 19.Percheron-Guegan, A., Achard, J.C., Sarradin,J.& Bronoel, G. Alliages a base de Lanthane et de ( N ickel et leurs applications electrochimiques. French patent 7516160 (1975). ) ( 2 0. Guerard, D . & H erold, A. New method for the preparation of l i thium insertion compounds ingraphite. C . R. Acad. Sci. C 275,571- 5 72(1 9 72). ) ( 2 1. B asu, S. A mbient temperature rechargeable battery. US p a tent 4,423,125 (filing date, 13 September1982; publication date, 27 Dec 1983) ) ( 22. Mohri, M. et al. Rechargeable lithium b attery based on pyrolytic carbon as a negative electrode.J. . Power Sources 26, 545 - 551(1 9 89). ) 23.Nagaura, T. & Tozawa, K. Lithium ion rechargeable battery. Prog. Batteries Solar Cells 9, 209 (1990). ( 2 4 . Armand, M., Chaba g no,J.M. & Du c lot, M. J. in Fast Ion Transport in Solids Electrodes and Electrolytes ( eds Vashishta,P., Mundy,J.-N. & Shenoy, G.K.) 1 3 1-1 3 6 (North-Ho l land, Amsterdam, 1979).2 5 .Kelly, I . E. , O w en,J. R. & St e el, B . H. Po l y ( ethyl e neoxide ) electrolyt e s f or o p eration at ne a r room ) temperature.J. Power Sources 14,13-21(1985);Interfacial Electrochem. 168,467(1984) 26.Tarascon,J.-M.,Gozdz, A. S., Schmutz, C., Shokoohi, F. & Warren, P. C. Performance ofBellcore’s plastic rechargeable Li-ion batteries. Solid State Ionics 86-88,49-54(1996). 27.Guyomard, D. in New Trends in Electrochemical Technology: Energy Storage Systems for Electronics Vol. 9 (eds Osaka, T.& Datta, M.)253-350(Gordon & Breach Science Publishers, 2000). ( 2 8.Dahn,J.R., Von Sacken, U., Juzkow, M. W. & A l - Janaby, H. Rechargeable LiNiO,/carbon cells.J.Ele c trochem. Soc. 138,2207-2211 (1 9 91). ) ( 2 9. Yuan Gao, Yakovleva, M . V. & Ebner, W. B. Novel L iNi.Tix2Mgx20,c o mpounds as cathode materials f or safer lithium-ion b atteries. Ele c tro c hem. Solid Stat e L ett . 1, 11 7-1 1 9 ( 1 998) . ) ( 30.Armstrong, A . R. & B r uce, P . G. S y nthesis of layered LiMnO, as an electrode for rechargeable lithium b atteries. Natur e 381,499- 5 00(1 9 96). ) ( 3 1.Ammundsen, B. e t al. in Proc. I nt . S y mp. El e ctroche m . Soc . Vo l . 99-24, 57-67 (ECS, Penn i ngton,NJ.2000) ). ) ( 3 2.Amatucci, G.G . , Pereira, N. , Zheng, T.& Tarascon, J .-M. Failure mechanism and improvement oftheelevated temperaturecycling ofLiMn , Ocompounds through the use of th e LiAl MnO4F , solidsolution. J. E lectrochem . Soc. 14 8 ,A171 -A1 82(2001) ) . ) ( 3 3.Amatucci, G. G . , d u Pasquier, A., Blyr, A., Zheng, T. & Tarascon, J.-M. T he elevated t emperature p erformance of the LiMn,O/C s y s tem: failure and solutions. Electrochem. Acta 45,255- 27 1(19 9 9). ) ( 3 4 . Padhi, A. K .,Nanjundaswamy, K. S ., Masquelier,C., O kada S. & Goodenough, J. B. Eff e ct of structure on t he Fe /F er ed ox couple in ir o n phosphates.J. E lectrochem. S o c. 144,1 6 09-16 1 3(1997). ) ( 3 5.Ravet, N. e t al. Improved iron-based cathode material. Abstr. N o. 12 7 , ECS Fall meeting, Hawaii,1999. ) ( 3 6.M o rcrette, M.,Wurm, C., Gaubicher, J . & M a s quelier, C. Poly a nionic structures as alternativematerials for lithium batteries. Abst r . No. 93 , Electrode Materials Meeti n g, Bordeaux Arcachon,27 M a y - 1 June 2001. ) ( . 37. C ava, R . J . , Murphy, D. W. & Zahurak, S. M . L ithium insertion in Wadsley-Roth phases based on N iobium oxide.J. Electrochem. Soc. 3 0,2345-2351(1983). ) ( 3 8.D e lmas,C., Brethes, S. & Menetrier, M. LiV s -ω, un n o uveau matèriau d’electrode pour a ccumulateur au lithium. C R Acad. S c i . 310, 1425-14 3 0(1990). ) ( 3 9.Le, D. B. e t al. High surface area V , O a e rogel i n tercalation electrodes.J. Electrochem. Soc. 143,2099-2104(1996). ) ( 4 0.Dong, W., Rolison, D . R. & D unn, B. El e ctrochemical properties of high surface area vanadium oxides aerogels. E l ectrochem. S olid State Lett . 3,457-459(2000). ) 41.Visco, S. J.& de Jonghe, L. C. in Handbook of Solid-State Batteries and Capacitors (ed. Munshi, M. Z.A.) 515 (World Scientific, Singapore, 1995). ( 42.Dahn, J. R. et al. Carbon and graphites as substitutes for the lithium anode. Industrial ChemistryLibrary V ol. 5 (ed.Pistoia, G.) (1994). ) 43.Shodai, T.,Okada, S., Tobishima, S. & Yamabi, I. Study of LiMN (M=Co, Ni or Cu) system for useas anode in lithium rechargeable cells. Solid State Ionics 86-88, 785-789 (1996). ( 4 4. Wi n ter, M . & Besenhard,J. O. E le c trochemical lithiation of tin and t i n-based intermetallics a n dcomposites. E lectrochem. Acta 45, 3 1-5 0 (1999) ) 45. Anani, A., Crouch-Baker, S. & Huggins, R. A. Kinetics and thermodynamic parameters of severalbinary alloys negative electrode materials at ambient temperature.J. Electrochem. Soc. 134,3098-3102(1987). 46.Idota, Y., Kubota, T., Matsufuji, A.,Maekawa, Y. & Miyasaka, T. Tin-based amorphous oxide: a high capacity lithium-ion storage material. Science 276,1395-1397 (1997) 47. Courtney, I. A. &Dahn,J. R. Electrochemical and in situ X-ray diffraction studies ofthe reaction of lithium with tin oxide composites.J. Electrochem. Soc. 144,2045-2052(1997). 48.Mao,O.,Dunlap, R. A. & Dahn, J. R. Mechanically alloyed Sn-Fe(-C) powders as anode materials for ( L i-ion batteries. I . The Sn,Fe-C s ystem.J. Electrochem. S o c. 146,40 5 -41 3 (199 9 ). ) 49. Beaulieu,L. Y., Larcher, B., Dunlap, R. A. & Dahn,J. R. Reaction ofLi with grain-boundary atoms innano structured compounds.J. Electrochem. Soc. 147,3206-3212(2000). ( 50. Kepler, K . D., Vaughey,J. T. & Thackeray,M. M. L i ,CugSn, (0
确定







还剩7页未读,是否继续阅读?

产品配置单



武汉科思特仪器股份有限公司为您提供《Issues and challenges facing rechargeable lithium batteries》,该方案主要用于其他中--检测,参考标准--,《Issues and challenges facing rechargeable lithium batteries》用到的仪器有CS2350H双单元电化学工作站(双恒电位仪)、CS350M电化学工作站/电化学测试系统、CS150M电化学工作站/测试系统
推荐专场






相关方案
更多
该厂商其他方案
更多