








本文集针对于兽药残留中的抗生素、磺胺类、驱肠虫类、镇静剂类药物等项目建立了相关液相色谱解决方案,从而为有效监控动物源性食品中兽药残留提供了强有力的检测手段,对发展畜牧业的产品质量和稳定食品安全大环境产生重大意义。
方案详情

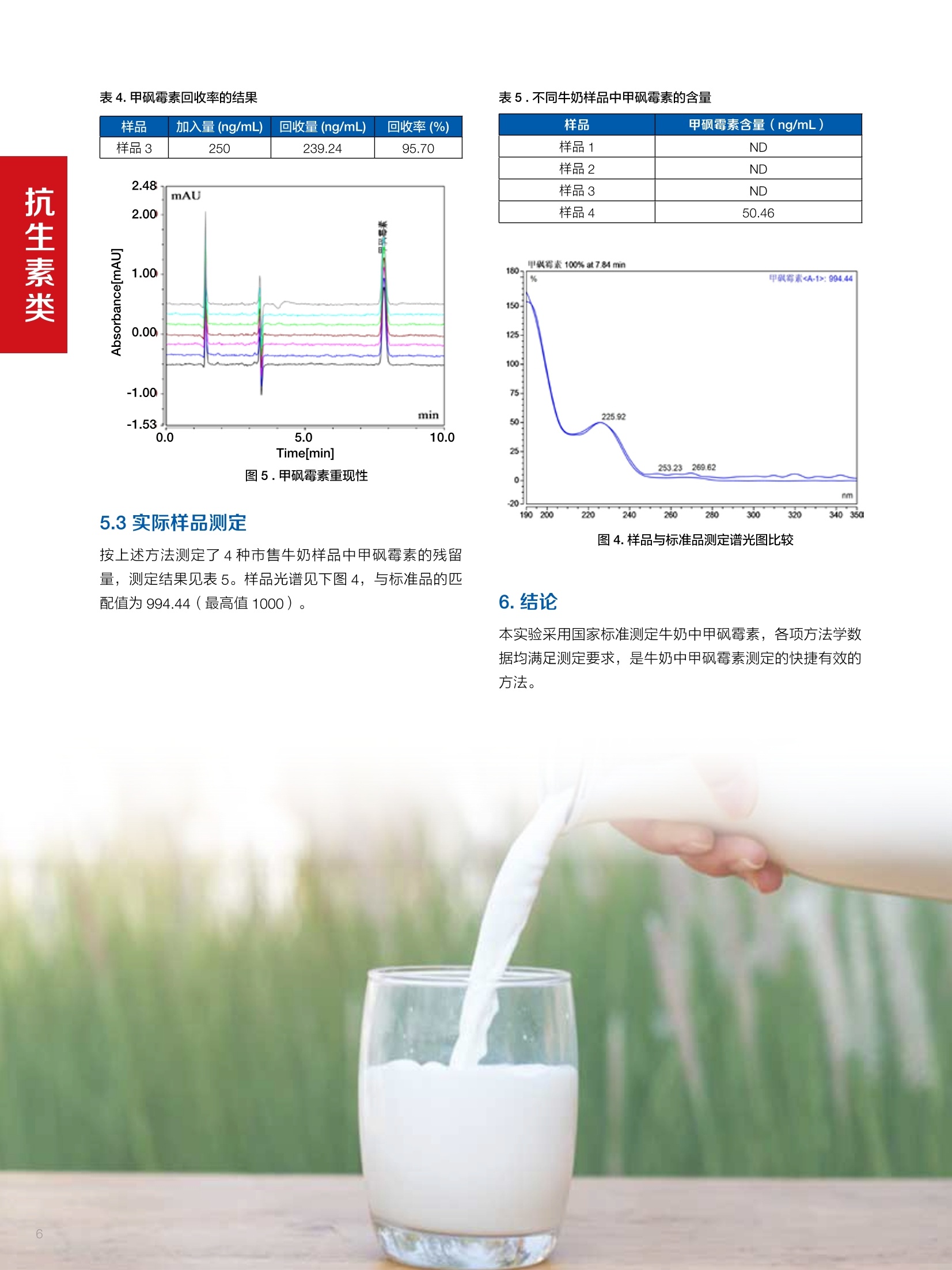
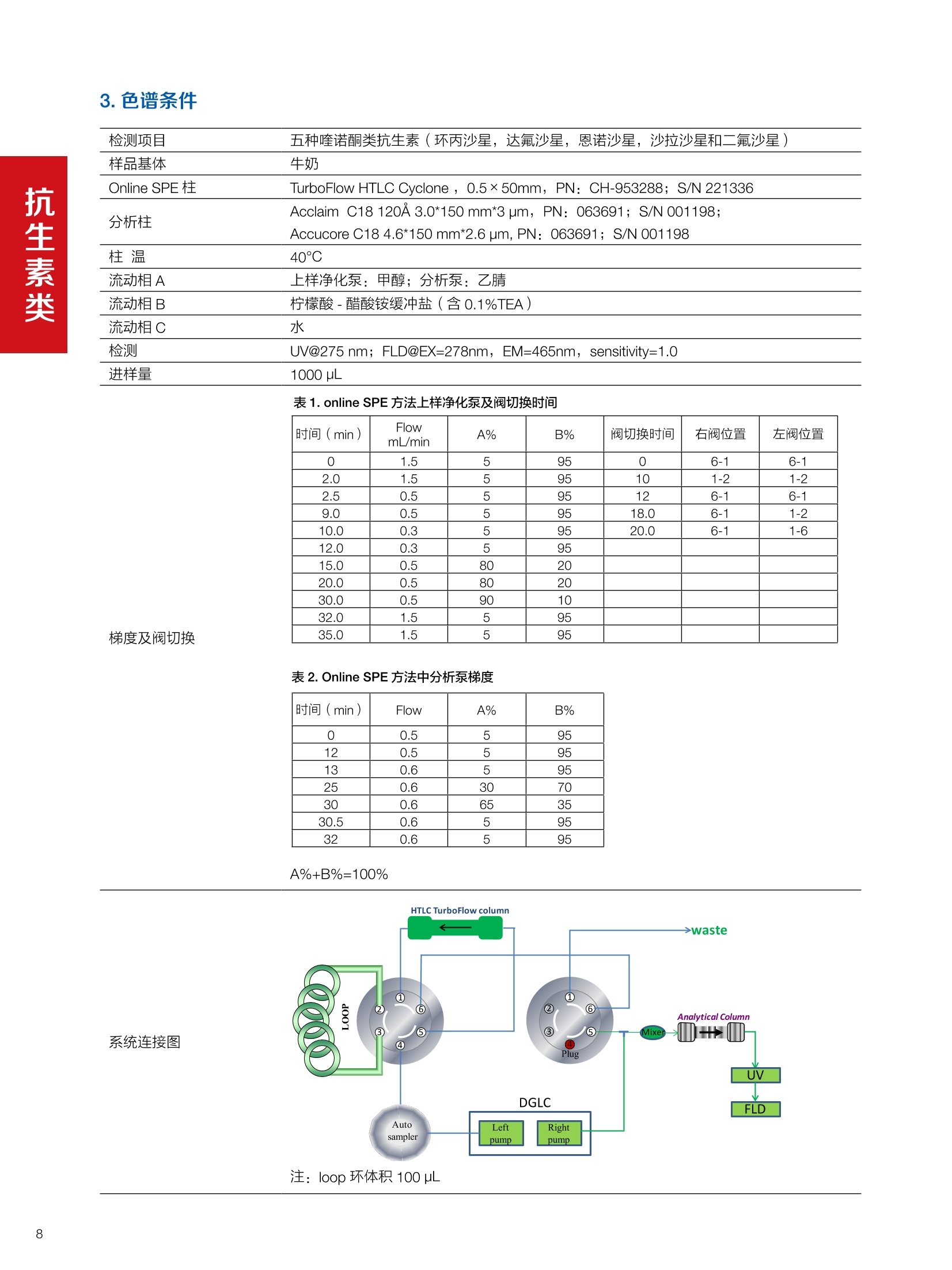
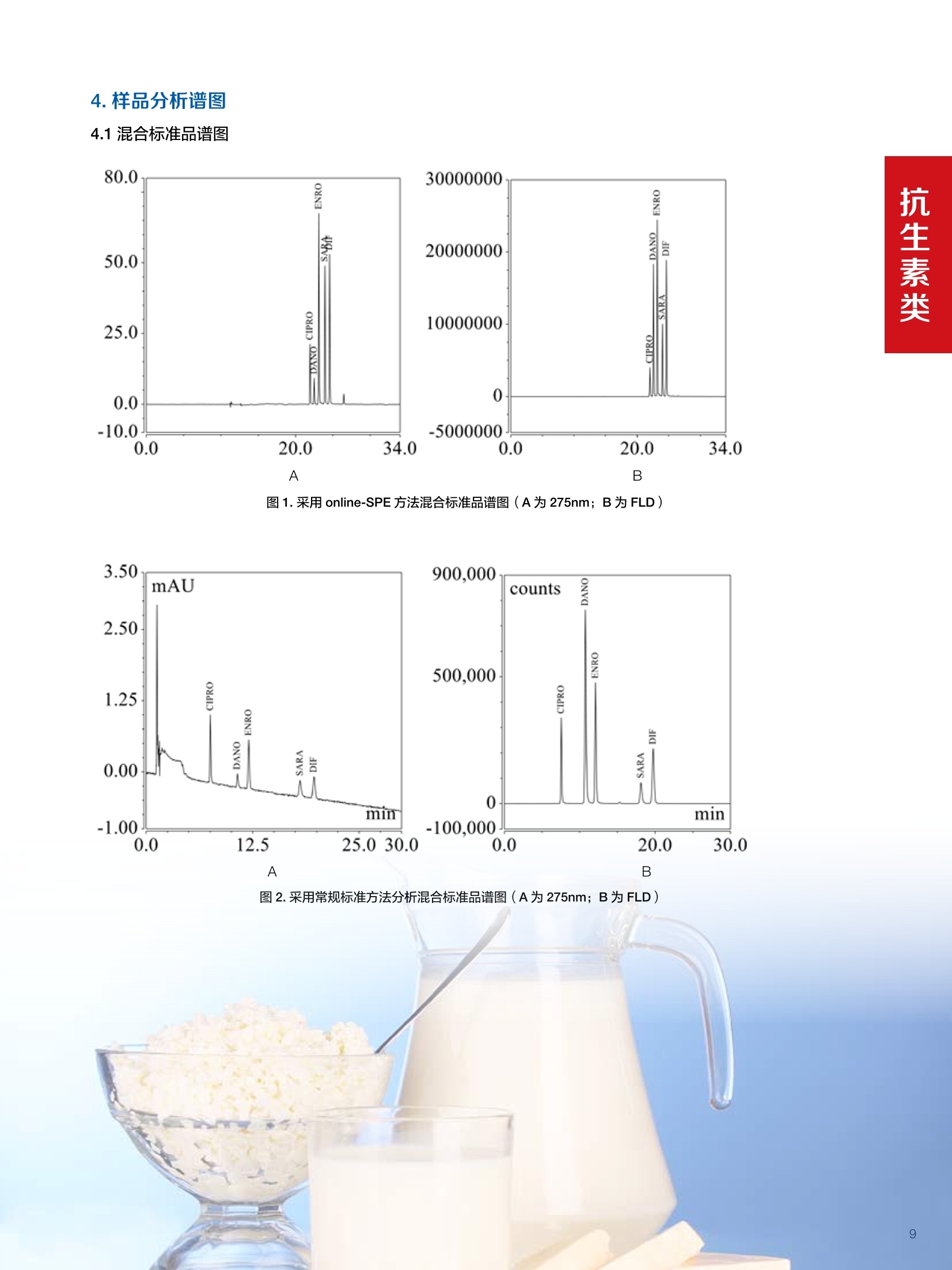
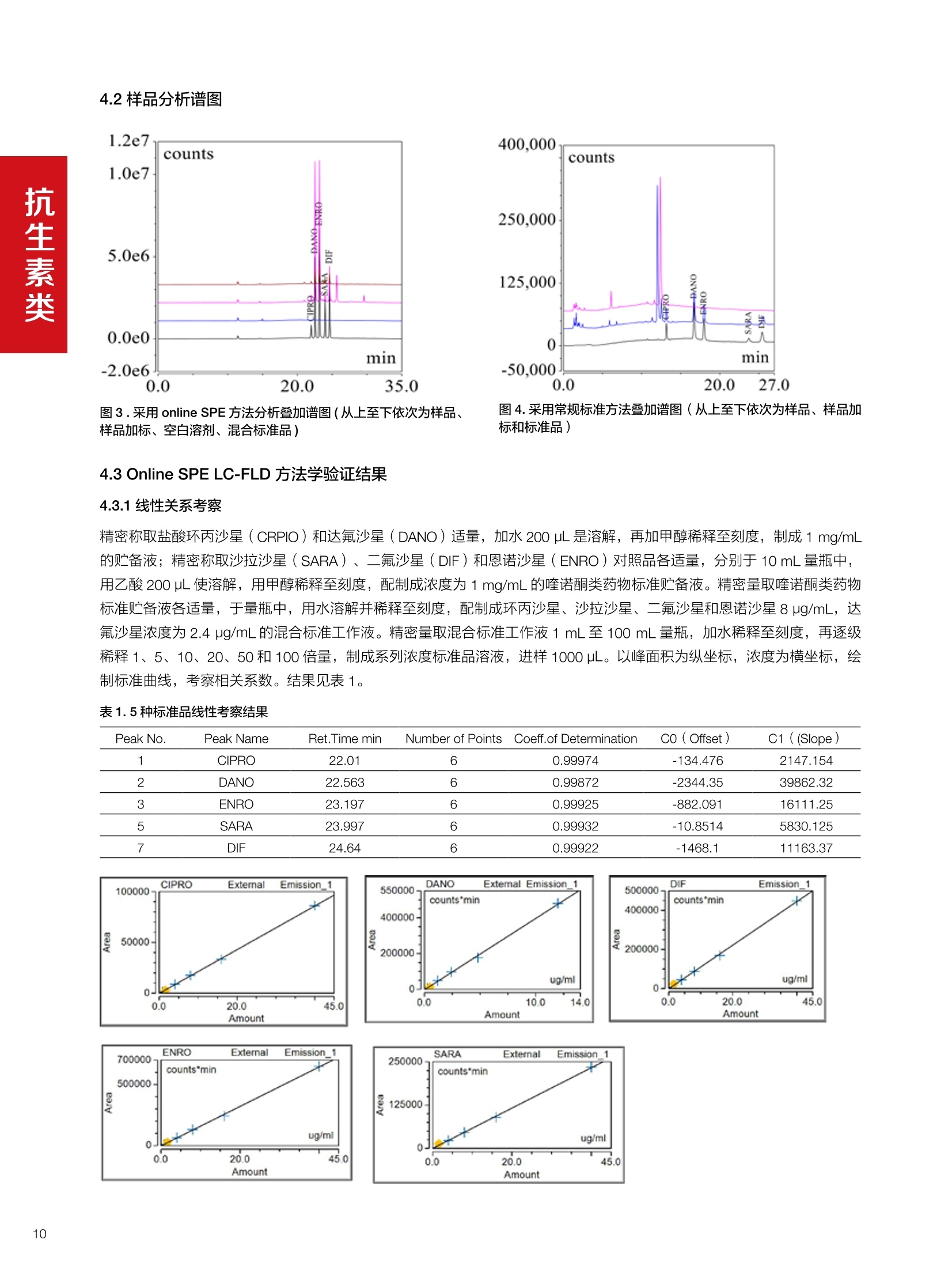
水产品中甲氧苄啶的测定 兽药残留分析应用文集 目录 1.抗生素类 HPLC-UV检测牛奶中甲砜霉素残留. 4 Online SPE-HPLC 法快速测定牛奶中五种喹诺酮类抗生素的残留量 2.磺胺类 水产品中甲氧苄啶的测定.mmmimmmm 14 3.驱肠虫药类 液相色谱紫外检测器测定牛奶中左旋咪唑. 17 鸡可食性组织中尼卡巴嗪残留量的测定 19 动物性食品中常山酮残留量的测定. 22 牛奶中阿维菌素类药物多残留的测定. 24 .水产品中阿维菌素和伊维菌素多残留的测定. .29 鸡可食性组织中地克珠利残留量的测定....... .33 4.镇静剂类 HPLC-UV动物性食品中氮哌酮及代谢物多残留的测定 .35 5.其他兽药类 猪肉中对乙酰氨基酚残留分析. 38 前言 随着人们生活水平的不断提高和现代养殖业日益趋向于规模化、集约化,人们对动物源食品由需求型向质量型发生转变,特别是食品安全性是大家关注的焦点。包括抗菌药、抗寄生虫药、消毒药和各种生长促进剂等在内的兽药应用已逐渐成为保障养殖业发展必不可少的环节之一,但是由于养殖人员对科学用药知识的缺乏,同时有些不法人员一味追求经济利益,导致滥用兽药的现象在当前畜牧业中普遍存在。滥用兽药容易造成动物源性食品中有害物质的残留,一方面兽药在动物源性的食品中残留可导致人体毒性反应、变态反应、致畸、致突变作用等诸多方面不利影响;另一方面兽药无论是用于饲料添加还是直接用于治疗,最终都会以原形或者代谢物的形式随粪、尿等排泄物进入生态环境,造成兽药在生态环境中的残留,当动物体内排出的这些化学物质超过环境的自净能力时,就会对人类生活的环境和生态系统产生不利影响。 在动物源食品中较易引起兽药残留超标的主要有抗生素类、磺胺类、激素类、抗寄生虫类药物。抗生素类药物可使动物机体中的耐药致病菌很容易感染人类,而且抗生素药物残留可使人体中细菌产生耐药性,扰乱人体微生态而产生各种毒副作用;磺胺类药物主要通过输液、口服、创伤外用等用药方式或作为饲料添加剂而残留在动物源食品中,在近15年~20年,动物源食品中磺胺类药物残留量超标现象十分严重,多在猪、禽、牛等动物中发生;同时养殖业中常见的激素和β-兴奋剂类主要有性激素类、皮质激素类和盐酸克仑特罗等,许多研究已经表明盐酸克仑特罗、已烯雌酚等激素类药物在动物源食品中的残留超标可极大危害人类健康。 针对兽药滥用造成动物源性食品中残留的情况,农业部在2003年(265)号公告中明文规定,不得使用《食品动物禁用的兽药及其他化合物清单》所列21类药物及未经农业部批准的兽药,不得使用进口国明令禁用的兽药,畜禽产品中不得检出禁用药物。在此基础上,农业部和卫计委在2013年联联合发布《食品安全国家标准牛奶中左旋咪唑残留量的测定高效液相色谱法》等29项兽药残留检测方法标准,明确了各种兽药残留检测方法和残留限度,保证人们食用动物源性食品的安全。 本文集针对于兽药残留中的抗生素、磺胺类、驱肠虫类、镇静剂类药物等项目建立了相关液相色谱解决方案,从而为有效监控动物源性食品中兽药药留提供了强有力的检测手段,对发展畜牧业的产品质量和稳定食品安全大环境产生重大意义。 HPLC-UV 检测牛奶中甲砜霉素残留 甲砜霉素,英文/拉丁名称: Thiamphenicol,又名“硫霉素” “甲砜氯霉素”或“赛芬妥霉素”,是一种酰胺醇类抗生素。它是氯霉素的甲基磺酰基相似体,有着相似的反应,强度却小2.5-5倍。与氯霉素相似,甲砜霉素不溶于水,但却极易溶于脂类。与氯霉素相比,甲砜霉素的主要优点是不会引起再生不良性贫血。许多国家主要将甲砜霉素用于兽药,但在中国及意大利该抗生素还可以用于人类疾病的治疗。在巴西,甲砜霉素的应用也十分广泛,特别是用于治疗性传播疾病及盆腔炎。但甲砜霉素对人类也具有毒害,引起血液系统不良反应等,乳制品中可能存在甲砜霉素残留,给消费者带来潜在的危害。本方法参考国家标准 GB 29689-2013《食品安全国家标准牛奶中甲砜霉素残留量的测定高效液相色谱法》。 图1.甲砜霉素分子结构式 2.样品前处理 2.1标准溶液的制备 甲砜霉素标准储备液:精密密取甲砜霉素对照品10mg,于10mL容量瓶中,用乙腈溶解并稀释至刻度尺,配置成浓度为1 mg/mL的标准储备液。 甲砜霉素标准曲线:将甲砜霉素标准储备液,用流动相(20%ACN)稀释,配制成20,50,125, 250,500 ng/mL的系列标准溶液。 2.2样品前处理 选取市售的4种牛奶,参考中华人民共和国国家标准GB29689-2013牛奶中甲砜霉素残留检测高效液相色谱法,称取样品 5 g, 于50mL离心管中,加乙酸乙酯20 mL,振辰 10 min, 4 000 r/min 离心5min, 取上清液于鸡心瓶中,残渣中加乙酸乙酯20 mL, 重复提取一次,合并两次提取液,于45℃旋转蒸发至近干,加水5mL于鸡心瓶中,超声5 min 使充分溶解,转至50mL离心管中。再加水5mL于鸡心瓶中重复溶解后转至同一离心管中。加20mL 正己烷,振荡5 min, 4 000 r/min离心2 min, 取下层液备用。 C18 柱(200 mg/3 mL, Thermo Scientific, 货号:60108-303 HyperSep C18 SPE 柱, 200 mg/3mL)依次用乙腈5mL和水5mL活化。取备用液过柱,控制流速1mL/min, 挤干。用乙腈5mL洗脱,收集洗脱液,于45℃氮气吹干,用流动相1.0mL溶解残余物,滤膜过滤,供高效液相色谱测定。 检测项目 甲砜霉素 样品基体 牛奶 Thermo Ultimate 3000: 仪器型号 泵: DGP-3600SD 自动进样器: WPS-3000TSL 柱温箱: TCC-3000SD 检测器:DAD-3000SD 色谱软件: Chromeleon Chromatography Data System 7.2 色谱柱类型尺 Acclaim C18, 120A(250×4.6 mm, 寸、S/N号及 5pm, P/N:059149, S/N:004594) ; 柱温 30℃ 检测器类型、 工作参数及 DAD, 225 nm S/N 号 流动相组成及 流动相A:水;流动相B:乙腈 流速 80:20(v/v) 进样体积 20pL 4.样品分析谱图 4.1标准品色谱图 4.2供试品溶液测定谱图 图2.甲砜霉素标准溶液 UV色谱图 图3.样品4溶液UV测定谱图 5.测定结果 5.1线性、检出限及 RSD 5.2回收率和精密度 由低浓度至高浓度依次进样检测,以质量浓度与峰面积作标准曲线线性回归。结果表明,在20~500 ng/mL范围内显良好线性关系(2=0.9968)。通过溶液逐步稀释法,测得最低定量限为10 ng/mL。外标法定量。标准曲线如下: 取牛奶样品,添加一定浓度的标准品,精密度数据结果见表3,如表4回收率为87.2%。 表3.甲砜霉素精密度的结果 浓度范围 峰面积(mAU*min) 第一针 0.1973 第二针 0.2031 第三针 0.1999 第四针 0.2026 第五针 0.2027 第六针 0.2050 第七针 0.1996 RSD% 1.30% 图4.甲砜霉素标准曲线 表2.甲砜霉素标准曲线方程 Ret.Time (min) Peak Name Cal.Type Coeff.of Determination co (Offset) C1 (Slope) C2 (Curve) 7.837 甲砜霉素 Lin, WithOffset 0.99915 0.0062 0.0008 0 表4.甲砜霉素回收率的结果 样品 加入量(ng/mL)回收量(ng/mL) 回收率(%) 图5.甲砜霉素重现性 5.3实际样品测定 按上述方法测定了4种市售牛奶样品中甲砜霉素的残留量,测定结果见表5。样品光谱见下图4,与标准品的匹配值为 994.44(最高值1000)。 表5.不同牛奶样品中甲砜霉素的含量 样品 甲砜霉素含量(ng/mL) 样品1 ND 样品2 ND 样品3 ND 样品4 50.46 图4.样品与标准品测定谱光图比较 本实验采用国家标准测定牛奶中甲砜霉素,各项方法学数据均满足测定要求,是牛奶中甲砜霉素测定的快捷有效的方法。 Online SPE-HPLC 法快速测定牛奶中五种喹诺酮类抗生素的残留量 1.引言 氟喹诺酮(Fluoroquinolone, FQs)类药物是近20年来喹 诺酮类药物迅速发展起来的一类十分重要的广谱抗生素。因其有抗菌谱广、吸收好、血液浓度高、能迅速分解到各组织、半衰期长、能制成各种剂型等特点而得到迅速推广。不仅在临床中得到了广泛应用,随着畜牧业发展的需要,氟哇诺酮类药物在治疗和预防动物疾病、促进动物生长、提高饲料转化率上应用相当广泛。这类药物的原形及其代谢产物可蓄积或贮存于动物的细胞、组织、器官或可食性产品(如蛋、奶)中,这便形成药物残留。FQs在动物源性食品中的残留可直接对人体健康造成危害,长期食用药物残留超标的牛奶,可能会引起皮疹、过敏性体克等不良反应,并会抑制人体肠道中正常的敏感菌群生长,低浓度的残留药物也可能诱导对喹诺酮类药物敏感的致病菌产生耐药性,间接危害人类健康。基于食品安全考虑,一些国家像中国、澳大利亚、加拿大、美国和欧盟成员国已禁止在家畜饲料中添加一些抗生素类药物。同时为保护消费者健康,并减少潜在危害,欧盟(Commission Decision37/2010/CE) (European Commission Regulation,2010)和美国FDA制定了抗生素类药物的最大残留限量。作为牛奶,达氟沙星的最大限度为30ug/kg, 恩诺沙星及其代谢物环丙沙星残留总量不超过100 pg/kg, 因此,建立灵敏度较高的动物性食品中喹诺酮类抗生素残留量的测 定方法,以符合法规财测的要求,具有重要的意义。 文献关于食品中氟喹诺酮类药物的残留测定方法较多,通常在提取、净化和检测原理不同。基于液相色谱结合不同检测技术的方法较为常见。例如UVe、电化学检测、荧光检测4-6)、质谱17-9]和化学发光检测小10。方法建立的主要难点是样品的前处理,文献中也报道了很多关于样品净化和富集的方法,如液相萃取④、QuEChERS 方法7.11、基于分子印迹技术的SPE 法去、SPE法I12、基于离子液体的液液微萃取法等。与离线的样品处理方法比较,在线方法具有自动化程度高、准确度和重现性较好,且溶剂消耗少、样品分析效率高等优点, Lina Kantianil13采用在线固相的方法,利用 Spark Holland 的专用 online SPE 仪器,以 oasis HLB 作为 SPE 柱, MS/MS 的检测对15种喹诺 酮类抗生素进行了分析,进样300微升,最低检出限做到0.01~ 1.93ppb,但该方法需要专用仪器,且 SPE 柱不可多次重复使用,检测成本较高。涡流色谱技术(Turbulentflow chromatography, TFC) 是利用大粒径填料使流动相在高流速下产生涡流状态,在这种情况下,大分子的基质成分如蛋白质等,还未能扩散进入填料颗粒内部就已被洗出柱外,而小分子的待测物则可以保留下来,与基质分离,从而对生物样品进行净化与富集。该技术可以在线处理生物样品,速度快、选择性好、灵敏度高,易于实现自动化,近年来在生物领域尤其是体内药物分析中得到了广泛的应用114,在牛奶和动物可食组织的喹诺酮残留测定中也有应用[15.16],两个方法采用聚合物基质的 Cyclone柱作为TFC柱,以 MS/MS 检测同时测定了环丙沙星等氟喹诺酮的残留量,具有快速、灵敏度较高,且TFC柱耐用性较好等优点,但方法也有不足之处,如需要专用仪器或多个液相泵的配置和复杂连接,采用串联质谱进行检测时,随样品基质不同存在基质效应等,使该方法不具有普适性。本文采用双三元液相色谱(DGLC) Online-TFC-LC-FLD法,结合在线稀释,建立了快速测定牛奶中环丙沙星、达氟沙星、恩诺沙星、沙拉沙星和二氟沙星残留的分析方法。 方法一:称取牛奶样品(2±0.05)g, 于50mL离心管中,加85%磷酸100uL, 乙腈4mL,涡旋混匀,振荡 5 min,10000 r/min 离心5 min, 取上清液于另一离心管中,加正已烷5 mL,涡旋1min,静置,取下层清液于25mL鸡心瓶中。残渣中加乙腈4mL,重复提取一次,上清液经同一份正已烷分配,合并两次提取液,于50℃氮吹至干。用流动相1.0mL溶解残余物,滤膜过滤,供高效液相色谱法去定。方法二:称称牛奶样品(1±0.02)g,于15mL聚丙烯离心管中,加乙腈-0.2%三氯乙酸(10:90)溶液5mL,涡旋混匀,超声5 min, 于10000 r/min 离心5min, 取上清液,直接进样测定。 3.色谱条件 检测项目 五种喹诺酮类抗生素(环丙沙星,达氟沙星,恩诺沙星,沙拉沙星和二氟沙星) 样品基体 牛奶 Online SPE 柱 TurboFlow HTLC Cyclone, 0.5×50mm, PN: CH-953288; S/N 221336 分析柱 Acclaim C18 120A 3.0*150 mm*3 um, PN: 063691; S/N 001198; Accucore C18 4.6*150 mm*2.6 um, PN: 063691; S/N 001198 柱温 40℃ 流动相A 上样净化泵:甲醇;分析泵:乙腈 流动相B 柠檬酸-醋酸铵缓冲盐(含0.1%TEA) 流动相C 水 检测 UV@275 nm; FLD@EX=278nm, EM=465nm, sensitivity=1.0 进样量 1000 pL 表1. online SPE 方法上样净化泵及阀切换时间 梯度及阀切换 时间 (min) Flow mL/min A% B% 阀切换时间 右阀位置 左阀位置 0 1.5 5 95 0 6-1 6-1 2.0 1.5 5 95 10 1-2 1-2 2.5 0.5 5 95 12 6-1 6-1 9.0 0.5 5 95 18.0 6-1 1-2 10.0 0.3 5 95 20.0 6-1 1-6 12.0 0.3 5 95 15.0 0.5 80 20 20.0 0.5 80 20 30.0 0.5 90 10 32.0 1.5 5 95 35.0 1.5 5 95 表2. Online SPE 方法中分析泵梯度 时间(min) Flow A% B% 0 0.5 5 95 12 0.5 5 95 13 0.6 5 95 25 0.6 30 70 30 0.6 65 35 30.5 0.6 5 95 32 0.6 5 95 A%+B%=100% 注: loop环体积100 pL 4.1混合标准品谱图 A B 图1.采用 online-SPE方法混合标准品普图(A为275nm;B为FLD) 图2.采用常规标准方法分析混合标准品谱图(A为 275nm; B为FLD) 图3.采用online SPE 方法分析叠加谱图(从上至下依次为样品、样品加标、空白溶剂、混合标准品) 图4.采用常规标准方法叠加谱图(从上至下依次为样品、样品加标和标准品) 4.3 Online SPE LC-FLD方法学验证结果 4.3.1线性关系考察 精密称取盐酸环丙沙星(CRPIO)和达氟沙星(DANO)适量,加水200 pL是溶解,再加甲醇稀释至刻度,制成1 mg/mL的贮备液;精密称取沙拉沙星(SARA)、二氟沙星(DIF)和恩诺沙星(ENRO)对照品各适量,分别于10mL量瓶中,用乙酸200 pL使溶解,用甲醇稀释至刻度,配制成浓度为 1 mg/mL 的喹诺酮类药物标准贮备液。精密量取喹诺酮类药物标准贮备液各适量,于量瓶中,用水溶解并稀释至刻度,配制成环丙沙星、沙拉沙星、:二氟沙星和恩诺沙星 8 pg/mL,达氟沙星浓度为2.4 pg/mL的混合标准工作液。精密量取混合标准工作液1mL 至100 mL瓶瓶,加水稀释至刻度,再逐级稀释1、5、10、20、50和100倍量,制成系列浓度标准品溶液,进样1000uL。以峰面积为纵坐标,浓度为横坐标,绘制标准曲线,考察相关系数。结果见表1。 表1.5种标准品线性考察结果 Peak No. Peak Name Ret.Time min Number of Points Coeff.of Determination C0(Offset) C1( (Slope) 1 CIPRO 22.01 6 0.99974 -134.476 2147.154 2 DANO 22.563 6 0.99872 -2344.35 39862.32 3 ENRO 23.197 6 0.99925 -882.091 16111.25 5 SARA 23.997 6 0.99932 -10.8514 5830.125 7 DIF 24.64 6 0.99922 -1468.1 11163.37 Amount 4.3.2重复性考察结果 取混合标准品溶液,连续进样6次,计算峰面积的RSD,结果CIPRO、DANO、ENRO、SARA、DIF 的 RSD 分另为 1.64%、1.93%、1.54%、1.1%和1.02%。叠加谱图见图 6. 图6.连续进样叠加谱图 4.3.3回收率实验 取某品牌的市售纯牛奶4份,每份1g, 其中两份按照40 pig/kg (达氟沙星 12 uig/kg)加入混合标准品溶液,另外两份按照80 pg/kg(达氟沙星24 ug/kg) 加入混合标准品溶液,并按照方法二制备样品溶液,按照本法测定各目标物的总量,每个样品连续进样2次,计算平均回收率。 表2.回收率实验结果 名称 样品中含量(ug) 加标量(ug/kg) 测得量(ug/kg) 回收率(%) 平均值(%) 环丙沙星 0 40 38.6 96.40 95.68 40 38.4 96.10 80 76.4 95.55 80 75.7 94.68 达氟沙星 0 12 11.3 94.00 92.60 12 11.2 93.33 24 22.2 92.58 24 21.7 90.48 恩诺沙星 0.09 40 32.6 81.53 83.11 40 33.4 83.51 80 67.7 84.64 80 66.2 82.75 沙拉沙星 0 40 33.4 83.56 85.11 40 33.2 83.03 80 69.2 86.55 80 69.8 87.29 二氟沙星 0 40 30.1 75.23 75.87 40 30.3 75.75 80 61.5 76.93 80 60.5 75.57 取某品牌奶粉2份,每份2g, 按照 80ug/kg 加入混合标准品溶液,按照方法一进行样品溶液制备和含量测定,结果CIPRO、DANO、ENRO、SARA、DIF的平均回收率为102%、84.14%、85.94%、82.97%和85.38%。 5.1本文利用 DGLC 在线固相萃取技术建立了牛奶中5种氟喹诺酮残留的测定方法,并经过了系统的方法学验证,结果表明环丙沙星、恩诺沙星、沙拉沙星和二氟沙星在0.8~40 ug/L范围内,达氟沙星在0.24~12 pg/L范围内,相关系数均>0.999,表明线性相关性较好;连续进样6次各目标物峰面积的RSD均<2%,表明精密度较好;加标样品的回收率均>75%,表明采用本方法测定,结果准确度较高;按照 S/N=3计算,环丙沙星、达氟沙星、恩诺沙星、沙拉沙星和二氟沙星的检出限分别为0.13、0.01、0.04、0.12和0.05 pg/L.。 5.2参考标准方法,实验采用 Cyclone HTLC 柱作为 SPE柱,增加了方法耐用性;与以往 onlline SPE 连接方式不同 (如图7所示),本实验利用预先填充在 Loop 环中80:20(甲醇:水相)作为洗脱溶剂,由于减少了系统延迟体积(GDV)的影响,可达到快速转移目标分析物至分析柱中的目的。实验采用一个三通加一个六通阀的方式实现了在线稀释功能,达到了“峰聚焦”(Peak Focusing)作用,改善了目标物峰形和分离度,由表3结果可知,峰对称因子得到了明显改善。 5.3交叉污染问题:实验由于采用荧光检测,且经过在线富集,因此对交叉污染要求较高。在仪器方法中加入进样前后清洗,另外为避免右侧阀非三通间的残留,实验首先将目标物的转移窗口时间由 1min 变成 2min, 另外尽量减少了此段距离,从而大大减小了交叉污染。 图7.典型的在线固相萃取连接图(其中A为反向冲洗方式;B为顺向冲洗方式) 表3.分别采用 A\B 两种连接方式时不对称因子 (asymmetry) 比较结果 名称 FLD UV Peak name 以图7-A方式连接 本法连接方式 以图7-A方式连接 本法连接方式 CIPRO 1.53 1.17 1.67 1.03 DANO 1.55 1.07 1.32 1.07 ENRO 1.4 1.05 1.93 1.01 SARA 1.36 1.04 1.54 0.98 DIF 1.39 1.01 1.38 0.92 本研究利用双三元液相色谱在线固相萃取技术建立了快速测定牛奶中5种氟喹诺酮残留量的测定方法,经过系统的方法学验证,表明方法简便、快速、准确,从SPE柱富集、净化,到完成分析,整个过程只需35 min, 且通过大体体进样,方法检出限可做到 0.01 ~0.13 pg/L。在系统连接上,采用更加普遍的2位置六通阀加三通的方式实现了在线稀释过程,改善了目标分析物的峰形和分离度,结合荧光检测,使该方法更具有普适性。方法充分利用涡流色谱技术特点,有效的去除了大分子基质成分干扰,有文献报道121,使用Turboflow 柱可连续进样500~2000次,且样品在进样前已经过初步的蛋白沉淀处理,使其耐用性更好,单次检测成本更低,适合牛奶样品中兽残的快速筛查检测。 ( USDA International Maximum Residue Level Database,2012. USDA Veterinary D rug MRL.Retrieved from USDA h ome page: http://www.mrldatabase.com/default.cfm?selectvetdrug=1 (02.02.12). ) ( [2] Gao, S., Jin, H ., You, J., Ding, Y ., Zhang, N., Wang, Y.,2011. l onic liquid-based homogeneous liquid-liquid microextraction f o r the d e termination of an t ibiotics in milk by high-performance liquid chromatography.Journal of Chromatography A 1218,7254-7263. ) ( 3 Rodri guez Ca’ c e res, M . I. , Guiberteau Ca b anillas,A., Galeano Di'az, T., Martinez Can~ as, M.A.,2010. S imultaneous determination of q uinolonesfor veterinary use by high-performance liquidchromatography w i th electrochemical detection.Journal of Chromatography B 878,398-402. ) ( 4 Cho, H.J., Y i, H., Cho, S .M., Lee, D . G., C h o, K. , Abd El-Aty, A.M., S him, J.H., L ee, S .H., Jeong, J .Y.,Shin, H.C., 2 010. S i ngle-step extraction followed byLC determination of (fluoro)quinolone drug residuesin muscle, eggs, and m ilk. Journal of Separation Science 23, 1034-1043. ) ( 5 Rambla-Alegre, M., C ollado-Sa'nchez, M . A., Esteve-Romero, J ., Carda-Broch, S., 2011.Quinolonescontrol in milk and eggs samples by liquicchromatography using a surfactant-mediated mobilephase. Analytical and B ioanalytical Chemistry 400, 1303-1313. ) ( 6 Zhen, M.M . , G o ng, R., Z hao, X. , Feng, Y. , 2010.Selective sample pretreatment by m olecularly imprinted p olymer m o nolith for the analysis offluoroquinolones from milk samples. Journal of Chromatography A 1217,2075-2081. ) ( 7 曲斌,朱志谦,陆桂萍,蒋天梅,耿士伟,郭良雪.QuEChERS-UPLC-MS/MS快速测定生鲜牛乳中的氟 喹诺酮类药物残留[].中国兽牧兽医,2013(40): 50~54. ) ( Bogialli, S., D'Ascenzo, G., Di Corcia, A., Lagana`, A., T ramontana, G ., 2009. S imple assay for monitoringseven quinolone antibacterials in eggs: e x tractionwith hot water and liquid chromatography coupledto tandem mass s p ectrometry Laboratory validation ) ( in the line with t h e European Union CommissionDecision 657/2002/EC. Journal o f C hromatography A 1216, 794-800 ) ( 9] K rebber, R ., Hoffend, F.J . , Ruttmann, F.,2009. Simpleand r apid d etermination of enrofloxacin i n edibletissues by turbulent flow chromatography/tandemmass spectrometry. Analytica Chimica Acta 6 3 7, 208-213. ) ( [10] L i, Y., Zhang, Z . , Li, J., L i , H., C hen, Y. , Liu, Z.,2011. Simple, s t able and sensitive electrogeneratedchemiluminescence d e tector f o r high-performanceliquid chromatography and its application i n d irectdetermination o f multiple fluoroquinolone residues inmilk. Talanta 84, 690-695. ) ( [11] Lombardo-Agu"i, M. , Ga’miz-Gracia, L., Cruces-Blanco, C., Garc i a-Campan~ a, A.M., 2 011.Comparison of different sample treatments for theanalysis of quinolones in milk by c a pillary-liquidchromatography with laser induced fluorescence detection. Journal of Chromatography A1218, 4966- 4971. ) ( [12] Herrera-Herrera, A.V., Herna ndez-Borges, J.,Rodri guez-Delgado, M.A., 2009. Fluoroquinoloneantibiotic determination in bovine, ovine and caprinemilk by using s olid-phase extraction and h i gh-performance liquid chromatographyfluorescencedetection with i onic liquids as mobile phase additives.Journal of Chromatography A 1216,7281-7287. ) ( [13] K rebber, R ., Hoffend, F .J., Ruttmann, F., 2009. Simpleand r apid d etermination o f e n rofloxacin in e d ibletissues by turbulent flow chromatography/tandemmass spectrometry. Analytica Chimica Acta 637,208-213. ) ( [14] Lewis Couchman., T urbulent flow chromatographyin bioanalysis: a revie w . Biomed. Chromatogr.2012;26:892-905 ) ( [15] K rebber, R ., Hoffend, F .J., Ruttmann, F., 2009. Simpleand r a pid determination of en r ofloxacin in edibletissues by turbulent flow chromatography/tandemmass s pectrometry. Analytica Chimica Acta 637, 208-213. ) 甲氧苄啶(CAS No.: 738-70-5),即 5-[(3,4,5-三甲氧基苯基)甲基]-2,4-嘧啶二胺,作为广谱抗菌药,被广泛应用于养殖业的病害防治,其结构式见图1。但是养殖户使用不当,就会在水产品体内产生药物残留,通过食物链的富集,影响人类健康。基于甲氧苄啶对人体健康的危害,香港已对甲氧苄啶进行管制,我国农业行业标准 NY5070《无公害食品水产品中渔药残留限量》中规定甲氧苄啶的限量为50 pg/kg。因此,建立一种快速、灵敏测定水产品中甲氧苄啶的方法是十分必要的。 目前测定甲氧苄啶的主要方法有高效液相色谱法、紫外分光光度法、示差折光法等。分光光度法的检出线较高,而高效液相色谱法具有灵敏度高、快速简便的特点,被广泛应用。本文拟采用高效液相色谱法,通过优化实验条件,建立一种快速、准确测定水产品中甲氧苄啶的方法。 图1.甲氧苄啶的结构式 2样品前处理 2.1标准溶液配制: 准确称取0.01g甲氧苄啶标准品于 10 mL容量瓶中,并用甲醇定容至10 mL, 得到浓度为1000 mg/L的标准储备液1。将标准储备液1用甲醇进行逐级稀释,得到浓度为0.05,0.1,0.2,0.5,1,2,5,10,20,50,100,200, 500, 1000 mg/L 的标准工作溶液。 2.2流动相水相(0.5%高氯酸溶液)的配制:取高氯酸5mL,用水溶解并稀释至1000 mL。 2.3样品前处理所需溶液的配制: 0.1 moL/L 硫酸溶液:取硫酸0.54 mL,用水溶解并稀释至100 mL; 2 moL/L氢氧化钾:取氢氧化钾11.22 g, 用水溶解并稀释至100mL;5%乙酸溶液:取乙酸5mL,用水溶解并稀释至100 mL;5%氨水甲醇溶液:取氨水5mL,用甲醇溶解并稀释100mL。 2.4.样品前处理: 分别称取两种小黄鱼、虾和鳕鱼三种不同的水产品试样,于50mL具塞离心管中,加三氯甲烷 15 mL、甲醇14mL、0.1 mol/L硫酸溶液6mL, 旋涡混合2 min, 4 000 r/min 离心3 min, 取上清液于 150 mL分液漏斗中。残渣中加甲醇14mL 和0.1 mol/L硫酸溶液6mL,重复提取一次,合并两次上清液于分液漏斗中,加2 mol/L 氢氧化钾溶液2mL、二氯甲烷30 mL, 振摇2 min, 静置分层,取下层液于茄形瓶中,分液漏斗中加二氯甲烷30 mL重复提取一次,合并两次下层液,于40℃旋转蒸发至近干,用5%乙酸溶液6mL溶解残余物,备用。为考察方法的可靠性,本文进行了加标实验以考察其方法的回收率,过程如下:称取两份5g小黄鱼试样,分别加入0.6mL浓度为 10 mg/L 非标准单位和 500 mg/L的甲氧苄啶标准品,按照样品前处理步骤与样品同步处理,样品的理论加标浓度分别为 1 mg/L和50 mg/L。 检测项目 甲氧苄啶 样品基体 水产品 仪器型号 Ultimate 3000 系列: 泵:DGP-3600RS P/N: 5040.0066 自动进样器: WPS-3000TRS P/N: 5840.0020 柱温箱: TCC-3000RS P/N: 5730.0000 检测器: DAD-3000RS P/N: 5082.0020 色谱软件: Chromeleon Chromatography Data System 色谱柱类型尺寸、 Acclaim RSLC 120, C18, 3pm, 3x150 mm; P/N 063691, S/ S/N号及柱温 N:001249;35℃ 检测器类型、工作 检测波长:230 nm; 参数及 S/N 号 流动相组成及流速 甲醇:0.5%高氯酸溶液(45:55, v/v),0.3 mL/min 进样体积 1pL 4.1标准溶液分析谱图 4.2方法重现性分析结果 为考察方法的重现性,通过连续进样9次浓度为 10 mg/L的标准溶液,得到甲氧苄啶的保留时间和峰面积的相对标准偏差(RSD)分别是 0.094%和0.498%,见图4. 图2.甲氧苄啶的标样谱图(10mg/L) 图4.连续进样甲氧苄啶标准品的叠加色谱图(n=9,浓度为10mg/L) 图3.甲氧苄啶的标准曲线 以测得峰面积为纵坐标(Y),对应的标准溶液浓度(X),绘制标准曲线。得到甲氧苄啶的回归方程分别:为Y=0.1931X+0.1277,相关系数R=0.9995. 4.3仪器检出限 分别以3倍S/N和10倍S/N计算目标化合物的检出限(LOD))和定量限(LOQ),得到其LOD 和 LOQ分别为11 pg/L和37pg/L。 4.4样品加标分析结果 为考察方法的可靠性,本文对实际样品进行了1 mg/L 和50 mg/L 两个浓度的加标,得到的回收率分别为78%和85%,说明该方法具有良好的回收率,适用于水产品中甲氧苄啶的测定,见图5. 图5.空白、标样、样品、样品加标(加标浓度:上 图-1 mg/L和下图-50 mg/L)的色谱图 为考察甲氧苄啶在水产品中的使用情况,本文对小黄鱼、虾米、鳕鱼三种不同的水产品进行测试,见图6.由图6可以看出甲氧苄啶在所测试的三个水产品中均未检出,符合我国农业行业标准 NY5070《无公害食品水产品中渔药残留限量》。 图6.1-标样(10mg/L)、2-空白和样品(3-虾米、4-小黄鱼、5-鳕鱼)的色谱图 5.结论 本文通过高效液相色谱-紫外检测法建立了一种快速、准确测定水产品种甲氧苄啶的方法,为规范水产品中甲氧苄啶的使用提供技术支持。 液相色谱紫外检测器测定牛奶中左旋咪唑 1.引言 左旋咪唑,化学名为2,3,5,6四氢-6-苯基咪唑噻唑,分子结构式见图1,是一种广谱驱肠虫药物,主要用于控制牛、羊、马、猪等动物胃肠道、肺中的线虫。不合理地使用左旋咪唑会造成动物产品中残留,而残留的左旋咪唑会对人体造成危害。为此,为保证动物源性产品食用的安全性,各国都制定了严格的限量标准。美国食品和药物管理局(FDA) 规定其允许残留量是100 pg/kg, 我国最新发布的食品安全国家标准 GB 29681-2013《牛奶中左旋咪唑残留量的测定高效液相色谱法》对其做了分析规定,本方法主要参照了该标准进行了相关测定。 图1.左旋咪唑分子结构式 2.样品前处理 标准溶液配制:左旋咪唑标准物质购自德国 Dr.Ehrenstorfer公司,准确配制成 10 pg/mL 左旋咪唑标准储备液,再用流动相稀释,配制成浓度为10、20、50、100、200、400和800 pg/L的系列标准工作溶液,供高效液相色谱测定。 样品前处理:称取试料(5±0.05)g,于离心管中,加碳酸盐缓冲液5mL,加乙酸乙酯10mL, 混匀,6000r/min 离心 10 min, 取上清液于茄形瓶中,再加乙酸乙酯10mL萃取一次,合并两次上清液,于50℃水浴旋转蒸发至干,加碳酸盐缓冲液5mL溶解残余物,再净化。先将C18 SPE柱(型号:Thermo Hypersil C18 3mg)依次用水3mL、甲醇3 mL和碳酸盐缓冲液3mL活化,取备用液过柱,用水3mL淋洗,用甲醇5mL洗脱,收集洗脱液,于50℃水浴氮气吹干,用流动相1.0mL溶解残余物,滤膜过滤,供高效液相色谱测定。 检测项目 左旋咪唑 样品基体 水产品 仪器型号 Ultimate 3000系列: 泵: LPG-3400SD P/N: 5040.0031 自动进样器:WPS-3000SL P/N: 5822.0010 柱温箱: TCC-3000RS P/N: 5730.0000 检测器: DAD-3000 P/N:5082.0010 色谱软件: Chromeleon Chromatography Data System 色谱柱类型尺寸、 Acclaim 120 C18, 150×4.6mm, 5pm,P/N:059148,S/N:001211; S/N 号及柱温 30℃ 检测器类型、工作 VWD, 220nm 参数及 S/N号 流动相组成及流速 流动相:0.02 mol/L 磷酸二氢钠二 乙胺缓冲溶液+甲醇(70+30,v/v) 进样体积 50 pL 4样品分析谱图 4.1标准溶液分析普图 Time [min] 图2.左旋咪唑标准溶液谱图 No. Peak Name RetentionTime (min) Cal.Type #Points Coeff.Det. % Offset Slope 1 左旋咪唑 7.90 LOff 6 99.9 -0.0594 0.0026 左旋咪唑标准曲线图 以测得峰面积为纵坐标(Y),对应的标准溶液浓度为横坐标 (X),绘制标准曲线。求回归方程得: Y=0.0026X-0.0594,相关系数r=99.9%,线性较好。 4.2左旋咪唑标准溶液重现性分析结果 表2.标准溶液重现性分析结果(浓度为100 pg/L,n=5) Sample No. Sample Name Ret.Time(min) 1 左旋咪唑 7.950 2 左旋咪唑 7.952 3 左旋咪唑 7.945 4 左旋咪唑 7.950 5 左旋咪唑 7.942 Average: 7.95 Rel.Std.Dev: 0.07% 从以上表可以看出,对浓度为100 pg/L 的标准溶液连续进样5次,重现性较好,保留时间RSD 值为 0.07%。 4.3仪器检出限 对最低浓度 10.0 pg/L进行了分析,根据信噪比进行计算,仪器最低检出限(以S/N=3计)为 1.0 pg/L. 称取了一份牛奶样品,同时进行加标分析,谱图如下所示: 图4.样品分析谱图(上面-蓝色-样品,下面-黑色-为标准溶液) 样品加标回收率实验:对3号样品进行基质加标实验,加入浓度值100 pg/L, 测得值91.0 pg/L,通过计算得回收率为91.0%,回收率较好。 4.5五份不同品牌牛奶样品分析谱图与结果 图5.实际样品分析图(从上至下分别为1、2、3、4、5号样品) 从以上谱图,及外标法计算得,该5份样品均未检出左旋咪唑,结果为 N.D。 5 结论 该方法可用于对牛奶中的左旋咪唑的测定,同时需要对前处理步骤进行严格的操作,以保证回收率值在标准规定范围内。 尼卡巴嗪为二硝基均二苯脲和羟基二甲基嘧啶复合物。为黄色或黄绿色粉末;本品在二甲基甲酰胺中微溶,在水、乙醇、乙酸乙酯、氯仿、乙醚中不溶。用于预防鸡和火鸡球虫病,具有高效、低毒、性能稳定、抗药性小等优点。然而尼卡巴嗪残留对人体存在潜在的威胁,需对其进行残留监控。尼卡巴嗪是由两种单一成分:4,4-二硝基苯脲(DNC)和2-羟基-4,6-二甲基嘧定(HDP)组成的复合物。结构式见图1、图2。由于尼卡巴嗪复合物中的 HDP成分在动物体内可迅速经尿排除体外,而抗球虫活性成分 DNC 通过粪便排泄较缓慢。因此国际上和我国均规定尼卡巴嗪在鸡组织中的残留标识物为 DNC, 我国农业部235号公告限定其在鸡的所有组织中的残留限量(MRL)为 0.2 mg/kg(200 ug/kg)。我国最新发布的食品安全国家标准 GB 29691-2013《鸡可食性组织中尼卡巴嗪残留量的测定高效液相色谱法》对其做了分析规定,本方法主要参照了该标准进行了相关测定。 图1.尼卡巴嗪结构式 图2.4,4-二硝基均二苯脲结构式 2样品前处理 2.1标准溶液的制备 1 mg/mL4,4'-二硝基均二苯脲标准贮备液:精密称取4,4'-二硝基均二苯脲对照品 25 mg, 于25mL的容量瓶中,用二甲基甲酰胺溶解并稀释至刻度,配制成浓度为1mg/mL的4,4'-二硝基均二苯脲标准贮备液。-20℃以下保存,有效期6个月。 100 pg/mL 4, 4-二二硝基均二苯脲标准工作液:精密量取1 mg/mL 4,4'-二硝基均二苯脲标准贮备液 1.0 mL于10 mL瓶瓶中,用甲醇溶解并稀释至刻度,配制成浓度为100 pg/mL 的4,4'-二硝基均二苯脲标准工作液。2-4℃以下保存,有效期一周。 2.2样品前处理 称取匀质后的试料(2.0±0.02)g, 于50mL离心管中,加乙腈10mL, 超声 5min, 4000 r/min 离心12 min, 取上清液,于50mL鸡心瓶中。残渣中加乙腈10 mL, 重复提取一次,合并两次上清液,加正丙醇3mL, 60℃旋转蒸至近干。加乙腈0.5 mL、正己烷1 mL, 涡旋 3 min,溶解,移至10 mL的具塞离心管中。用乙腈0.5 mL和正己烷1mL重复涡旋溶解一次。合并两次溶液至10 mL的具塞离心管中,加入正己烷2mL,漩涡混合3 min,4000 r/min 离心5 min, 正上层正己烷液。再口正己烷2mL, 重复提取一次,4000 r/min 离心 5 min, 取下层液,备用。 C18 SPE 柱用乙腈 10 mL活化,取备用液过柱,自然流干,收集滤液。用洗脱液4mL洗脱,收集洗脱液,合并滤液和洗脱液,于60℃旋转蒸发近干,用流动相2.0 mL溶解残余物,滤膜过滤,供高效液相色谱测定。 3.色谱条件 检测项目 尼卡巴嗪 样品基体 鸡胸肉、鸡肝 Thermo UItimate 3000: 仪器型号 泵:LGP-3400SD 自动进样器:WPS-3000TSL 柱温箱: TCC-3000SD 检测器: DAD-3000RS 色谱软件: Chromeleon Chromatography Data System 6.8 SR11 分析柱 Acclaim 120 C18(250×4.6mm, 5um) (PN059149 SN004806) 流动相 乙:水=58:42 柱温 30°℃ UV检测波长 340 nm 进样量 20uL 4样品分析谱图 4.1标准品色谱图 图3.标准溶液色谱图 图5.鸡肝样品测定谱图 4.2供试品溶液测定谱图 4.3空白试验谱图 图4.鸡胸肉样品测定谱图 图6.空白试验测定谱图 5.测定结果 5.1精密度实验结果 取 100 pg/L和1000 ug/L的对照品溶液,分别连续进样6次,计算4,4'-二硝基均二苯脲的保留时间和峰面积的RSD 值。 表1.100 pg/L 对照品精密度测定结果 100 pg/L 1 2 3 4 5 6 RSD% 保留时间(min) 7.180 7.193 7.193 7.190 7.190 7.173 0.11 峰面积(mAU*min) 0.1225 0.1222 0.1189 0.1226 0.1201 0.1212 1.23 表2.1000 pg/L 对照品精密度测定结果 1000 pg/L 1 2 3 4 5 6 RSD% 保留时间(min) 7.180 7.177 7.180 7.180 7.180 7.180 0.02 峰面积(mAU*min) 1.0353 1.0358 1.0372 1.0338 1.0336 1.0314 0.20 5.2检出限与定量限 表3.检出限与定量限测定结果 S/N 浓度(ug/L) 样品含量(ug/kg) 检出限 3.0 5 5 定量限 10.1 20 20 5.3线性与相关系数 精密量取 100 ug/mL 4,4'-二硝基均二苯脲标准工作液适量,用流动相稀释,配制成浓度为20、50、100、200、500、1000、4000和8000 pg/L的系列标准溶液,供高效液相色谱测定。以测得峰面积为纵坐标,对应的标准溶液浓度为横坐标,绘制标准曲线。 回归方程y=0.065x-0.046相关系数R=0.9998 5.4加标回收率 取空白鸡胸肉样品,按照125 pg/kg 和400 ug/kg 两个浓度加标,按供试品溶液制备方法进行样品制备。实验结果表明各组分的加标回收率均在73-80%之间,符合分析检测的要求。 表4.加标回收率测定结果 加标量(pg) 加标测定值(ug) 回收率% 125 pg/kg 0.25 0.196 78.34 125 pg/kg 0.25 0.192 78.62 400 pg/kg 0.80 0.589 73.65 400 pg/kg 0.80 0.602 75.19 5.5实际样品测定 称取匀质后的鸡胸肉和鸡肝试料,按照供试品溶液制备方法制备供试溶液,进液相色谱仪进行测定。 表5.实际样品含量测定结果 称样量(g) 含量(ug/kg) 鸡胸肉1 1.994 未检出 鸡胸肉2 1.991 鸡肝1 2.008 62.50 鸡肝2 2.004 未检出 本实验采用 U3000 液相色谱仪和 Acclaim 120 C18 色谱柱测定鸡可食性组织中尼卡巴嗪残留物4,4'-二硝基均二苯脲的含量,按照 GB 29691-2013进行试验,方法学各项指标符合要求,检出限和定量限比国标更低,满足对鸡可食性组织中该组分残留的测定要求。 动物性食品中常山酮残留量的测定 1.引言 常山酮( Halofuginone),又名卤夫酮,为新型广谱抗球虫药,对多种鸡球虫均有较强的抑杀作用;常作为一种饲料添加剂广泛用于防治家禽的球虫病。在我国,其在饲料中的最大允许添加浓度为3 mg/kg, 其休药期为4天。如果食用了含常山酮残留浓度较高的禽肉组织,会出现身体不适等中毒症状,对人体有一定的毒害作用。目前,许多国家建立了常山酮的残留检测方法并对其残留限量作了规定。本实验参照国标《GB 29693-2013动物性食品中常山酮残留的测定高效液相色谱法》,建立了高效液相色谱法测定鸡肉组织中常山酮的残留量的测定方法,灵敏度高、回收率好、重复性好,可供相关检测工作者参考。 2.样品前处理 标准溶液配制:准确称取常山酮标准品5mg、精确到0.1mg,置于50mL容量瓶中,用0.125M 乙酸铵缓冲液溶解并定容至刻度,配置成浓度为 0.1 mg/mL的标准储备液。 常山酮标准工作液:分别移取适量体积的上述常山酮标准储备液,加入适量的流动相,配置成常山酮浓度为 0.05、0.1、0.2、0.5、1 pg/mL 的标准工作溶液。 样品前处理:提取:称称4.0g鸡肉组织匀浆,加入到100 mL离心管中,加入胰蛋白酶 50 mg 和10mL水,涡动1 min;接着用10%碳酸钠溶液调节 pH至8,40℃恒温酶解3h;取出,放置室温,加入2 mL 10%碳酸钠溶液,涡动1 min;加入20 mL 乙酸乙酯,震荡3 min, 以5000 r/min 离心5 min; 移取上清液后同样步骤提取一次,合并乙酸乙酯层。加入 0.125 M 乙酸铵溶液 15mL, 震荡3min,静置,收集下层水相;再加0.125M 乙酸铵溶液重复萃取一次,合并水相层。转至旋蒸瓶中,于38℃旋转蒸发除去残余乙酸乙酯,备用。 净化:精确量取3mL提取液至已活化的固相萃取小柱中(HLB固相萃取柱,依次用3mL甲醇、3mL水和3mL0.125M乙酸铵溶液淋洗活化),用甲苯2mL洗涤,挤干;用甲醇3mL洗脱,收集洗脱液,于38℃氮气吹干;用流动相 1.0 mL溶解残余物,微孔滤膜过滤,供高效液相色谱仪测定。 3.色谱条件 检测项目 常山酮残留 样品基体 动物性食品 仪器型号 Ultimate 3000: 泵: LPG-3400SD 自动进样器:WPS-3000SL 柱温箱: TCC-3000SD 检测器: VWD-3400RS 色谱软件: Chromeleon Chromatography Data System 7.2 分析柱 Acclaim C18; 250×4.6mm, 5 um; P/N 059149, S/N 004771; 柱温 30°℃ ACN : 0.05M 乙酸铵(pH=4.3) 流动相组成及流速 =25:75 流速:1mL/min 进样方式及进样体 自动进样器,进样量:25pL 积 固相萃取小柱 HLB 固相萃取柱 4.1标准品测试图谱 取标准品溶液,重复进样7次,计算保留时间和峰面积的RSD,结果常山酮保留时间和峰面积的RSD分别为0.04%,0.35%。 图2.重复性谱图(n=7) 4.3线性及检出限 取系列浓度标准品溶液,进样25uL。以峰面积为纵坐标,浓度为横坐标,绘制标准曲线,考察相关系数。结果见表1和图3、图4。以仪器信噪比 S/N=10计算,常山酮的最低定量限约为 0.02 pg/mL. 图3.各浓度标准品叠加普图 表1.线性数据 图4.标准曲线 4.4回收率及实际样品 取鸡肉组织样品,匀浆,加入一定量的常山酮标准溶液,按照样品前处理方法制备成供试品溶液,测定图谱见图5,测定结果见表2。按上述方法测定从菜市场随机采购的鸡组织样品,样品中均未检出常山酮残留。 图5.样品、样品加标图谱(上面-蓝色-样品加标,下面-黑色-为品溶液)。 表2.回收率数据 名称 实际样品含量(ng/g) 加标量 (ng/g) 测得量 (ng/g) 加标回收率(%) 鸡肉样品 未检出 500 435 87.0% No. Ret.Time (min) Peak Name Cal.Type Point Offset (CO) Slope (C1) Range ( pg/mL) Corr.Coeff% 1 8.4 常山酮 LOff 5 0.0096 1.9472 (0.05~1) 99.98 6.结论 本实验采用国家标准 GB 29693-2013测定动物性食品中常山酮,各项方法学数据均满足测定要求,可用于动物性食品中常山酮残留的测定。值得注意的是,本实验前处理比较繁琐复杂,需要操作者对各项前处理过程严格操作,以保证方法回收率符合规定。 阿维菌素类(Avermectins)药物是由链霉素产生的一组大环内酯类物质,包含结构相似的8种组分。该类化合物对线虫和节肢动物有及强的驱杀作用,从而使抗蠕虫药物用药剂量由 mg/kg 级下降到 pg/kg 级。目前在这类药物中已商品化的有阿维菌素、伊维菌素、多拉菌素、依普菌素,其中伊维菌素是阿维菌素由加氢还原获得“。这类药物是目前兽医临床上用量最大的抗寄生虫药,对各种寄生线型动物和节肢动物具有很强的抑杀作用,其效果比常用农药高5-50倍,并且药效持久。但是这些药物的广泛使用不仅造成牛组织中药物残留超标,还会造成牛奶中药物污染12-3。JECFA(联合食品添加剂专家委员会)规定牛奶中EP、DOR和ⅣM 的最高残留限量值分别为20、15和10 pg/kg2。我国农业部规定牛奶中ⅣM的最高残留限量为20 ug/kg, 且泌乳期禁用 AVM、DOR和EP。 近年来阿维菌素类(AVMs)药物的浅留分析技术有较大的发展,但有关牛奶中该类药物的残留检测较少[4-10],阿维菌素类药物多残留检测方法集中于对猪、牛、羊等动物组织样品中该类药物的残留研。卢平等报道了牛奶和山羊奶中伊维菌素类药物的 HPLC 检测方法。Perez 等报道了牛奶中伊维菌素的 HPLC 检测方法。Heller 等研究了牛奶中伊维菌素的LC-MS确证检测方法。本文依据GB29696-2013方法1对牛奶中四种阿维菌素类药物在Ultimate3000 液相色谱仪上同时进行测定,且检测限低于标准方法,线性范围和回收率等参数均满足国标要求,可对实际样品进行准确定量分析。 2.1配制各种溶液 洗涤液:取乙腈30 mL、水70 mL 和三乙胺20L, 混匀; 衍生化试剂A液:取N-甲基咪唑1mL、乙腈1mL,混匀, 现配现用; 衍生化试剂B液:取三氟乙酸酐1mL、乙腈2mL, 混匀,现配现用。 2.2标准曲线的制备 精密量取10 mg/L阿维菌素类药物混合标准贮备液适量,用乙乙稀释,配制成浓度为 0.0005、0.001、0.002、0.005、0.01、0.05、0.1、0.5和1.0 mg/L的系列标准溶液,各取1.0mL于5mL试管中,于60℃水浴氮气吹干,依次加衍生化试剂A液100pL和衍生化试剂B液150pL,密闭,涡动10s, 依次加冰醋酸和三乙胺各50 pL, 涡动 10s, 与样品溶液同步密闭反应 30 min,加650 uL甲醇混匀,供高效液相色谱测定。以测得峰面积为纵坐标,对应的标准溶液浓度为横坐标,绘制标准曲线。求回归方程和相关系数。 2.3样品前处理过程: ( 提取:称取取料(5±0.05)g于50mL离心管中,加乙 腈8mL,涡动1 min, 4500 r/min 离心 10 min, 取上清液。残渣中加乙腈8mL,重复提取一次,合并两次上清液,加水 20 mL、三乙胺50pL,混匀,备用。 ) 净化: HyperSep C18 柱 (Thermofisher)依次用乙腈5 mL 和洗涤液5mL活化,取备用液过柱,自然流干,抽干5 min, 加异辛烷3mL洗涤,抽干5 min, 乙腈5mL洗脱,收集洗脱液于10mL试管中,于60℃水浴氮气吹干,备用。 ( 衍生化:于备用试管中依次加衍生化试剂A液100 uL、衍生化试剂 B液150uL , 密 闭 ,涡动10s,依次加冰醋酸50 pL和三乙胺50 pL, 涡动10s, 于室温密闭反应 30 min, 加甲醇650 pL混匀。用0.45 pm 有机滤膜过滤, 直接上机分析。 ) 检测项目 阿维菌素、埃普利诺菌素、伊维菌素、多拉菌素 样品基体 鲜牛奶、酸奶等 仪器型号及配置 Dionex Ultimate 3000 系统: 泵: DGP-3600SD 自动进样器:WPS-3000TSL 柱温箱: TCC-3000SD 检测器: FLD-3400RS 色谱软件: Chromeleon Chromatography Data System7.2 色谱柱类型尺寸、S/N、P/N号 Acclaim C18, 120A, 3×150mm, 3 um(S/N001201, P/N063691); 保护住类型尺寸、S/N、P/N号及柱温 温度:30℃ 检测器类型、工作参数及 S/N 号 FLD:激发波长:365nm;发射波长:475nm; 流动相组成及流速 乙腈: H,O=92:8;流速:0.5 ml/min; 进样方式及进样体积 自动进样,10L 4样品分析谱图 4.1空白图谱和阿维菌素类药物标准品谱图 图1.空白色谱图 图2.四种阿维菌素类药物标准品图谱 4.2精密度实验结果 取阿维菌素等四种菌素的对照品照液 0.1 mg/L,分别连续进样7次,计算各自保留时间和峰面积的RSD 值, 结果见表1, RSD 均小于0.8%,重现性结果较好。 表1.重复性实验数据 保留时间 (min) 峰面积(counts*min) 埃谱利诺菌素 阿维菌素 多拉菌素 伊维菌素 埃谱利诺菌素 阿维菌素 多拉菌素 伊维菌素 1 9.443 12.067 16.053 22.243 698014.2 749644.7 733974.1 618226.2 2 9.443 12.063 16.047 22.233 703733.4 756923.3 741471.9 622479.3 3 9.440 12.060 16.050 22.223 705057.5 760688.4 741764.6 623729.7 4 9.433 12.047 16.030 22.197 706779.7 760297.4 744779.6 624321.8 5 9.427 12.037 16.013 22.170 708746.8 761955.5 745516.9 627050.1 6 9.403 12.003 15.967 22.113 711256.4 764493.1 747307.8 627064.2 7 9.397 11.993 15.950 22.093 711406.9 767922.9 748235.1 632692.2 RSD/% 0.20 0.24 0.26 0.27 0.67 0.77 0.65 0.72 4.3检出限与定量限 四种菌素的最低检测限为 0.0002 mg/L, 远低于标准的要求(最低检测限0.001 mg/kg, 定量限0.002 mg/L)。 4.4线性与相关系数 取不同浓度对照品溶液依次进样检测,以质量浓度(X)与峰面积(Y)作标准曲线线性回归,结果表明表0.0005-0.5 mg/L 的浓度范围内,线性相关系数均为0.9999. 图3.重现性图谱 表2.标准曲线方程 No. Ret.Time (min) Peak Name Cal.Type Offset(CO) Slope (C1) Range(mg/L) Corr.Coeff% 1 9.443 埃谱利诺菌素 LOff -5652.8 11575015.3 0.0005-0.5 0.9999 2 12.067 阿维菌素 LOff -2149.7 12400775.6 0.0005-0.5 0.9999 3 16.053 多拉菌素 LOff -4526.5 12157589.9 0.0005-0.5 0.9999 4 22.243 伊维菌素 LOff -155.0 10201248.5 0.0005-0.5 0.9999 图4.埃普利诺菌素标准曲线 图5.阿维菌素标准曲线 图6.多拉菌素标准曲线 图7.伊维菌素标准曲线 4.5样品加标分析结果 counts min 图8.样品1色谱图图 图9.样品2色谱图 图10.样品3色谱图 图11.样品3及加标色普图 选择了鲜奶和酸奶等三种牛奶样品,样品中未检出四种阿维菌素类药物(见表3),其中样品3进行加标测定,加标回收率在93.0%-109.0%之间(表4),满足标准测定要求。 表3.样品中阿维菌素类药物多残留含量 成分 埃谱利诺菌素 阿维菌素 多拉菌素 伊维菌素 测量值\mg/L) 实际含量(mg/kg) 测量值(mg/L) 实际含量(mg/kg) 测量值(mg/L) 实际含量(mg/kg ) 测量值(mg/L) 实际含量(mg/kg) 样品1 <0.0005 <0.0001 <0.0005 < 0.0001 <0.0005 <0.0001 <0.0005 <0.0001 样品2 <0.0005 < 0.0001 <0.0005 <0.0001 <0.0005 < 0.0001 <0.0005 <0.0001 样品3 <0.0005 <0.0001 <0.0005 <0.0001 <0.0005 <0.0001 <0.0005 <0.0001 表4.样品中阿维菌素类药物多残留含量 成分 测量值(mg/L) 加标值(mg/L) 加标后测量值(mg/L) 回收率(%) 实际样品中含量(mg/kg) 埃谱利诺菌素 <0.0005 0.01 0.0109 109.0 <0.0001 阿维菌素 <0.0005 0.01 0.0093 93.0 <0.0001 多拉菌素 <0.0005 0.01 0.0109 109.0 <0.0001 伊维菌素 <0.0005 0.01 0.0108 108.0 <0.0001 本方法参考国标 GB29696-2013标准测定牛奶样品中四种阿维菌素类药物残留,阿维菌素、埃普利诺菌素、伊维菌素、多拉菌素的标准品经过N-甲基咪唑和三氟乙酸酐衍生反应,牛奶样品经过HyperSep C18柱净化后衍生反应,选择 Acclaim C18色谱柱,在乙腈和水的等度条件下得到很好的分离,结果各项方法学数据均满足测定要求。 ( 参考文献 : ) ( [1] CAMPBELL W C . Ivermectin and abamecti n [ M].New York: S p ringer-Verlag New York Inc. 1 989. ) ( 2] CAC/MRL. 02-2009 Maximum R e sidue Limits for Veterinary Drugs in Foods Updated a s a t the 32nd S ession o f the Codex Alimentarius Commission[S].2009 ) ( [3] 中华人民共和国农业部.第235号公告动物性食品中兽药最高残留限量 [S].2002 ) ( [4] 扈洪波,朱蓓蕾,李俊锁.阿维菌素类药物的研究进 展[J].畜牧兽医学报,2000,31:5200-5209 ) ( [5] 卢平,古丽曼,宫秀杰,等.伊维菌素在牛奶山羊 奶中药物残留检测方法的的核研究[].动物医学进 展,2002,23(6):104-105 ) ( [6] Perez L ,Palma C,Villegas R , et al.Analytical method-ology f o r the detection of ivermectin residues in milk samples from dairy farms in the ) ( province of Nuble,Chile[J].Archivos de MedicinaVeterinaria,2006,38(2) : 143-150 ) ( [7] Heller D N,Schenck F J . Particle b eam liquidchromatography/mass spectrometry with negative ion chemical ionization for the confirmation of ivermectin r esiduein b ovine milk and liver[J].Bio MassSpectrom,1993,22:184-193 ) ( [8] Helena D,Darinka Z D,Ksenija S G , et a l .Residues o f certain veterinary drugs i n raw milk i n Slovenia in the2002 period[J].International Journal of Environmentand P ollution,2007,31(1/2):155-166 ) ( [9] Sheridan R ,Desjardins L.Determination of abamectin,doramectin, e mamectin, eprinomectin, ivermectin,and m oxidectin i n milk by liquid c hromatographyelectrospray tandem mass specrometry[J].J AOAC Int,2006,89(4):1088-1094 ) ( [10] Kolberg DI S ,Presta M A,Wickert C , et al.Rapid a nd accurate simultaneous d etermination of abamectin and ivermectin in b ovine milk b y high performanceliquid chromatography with fluorescence detection[J].J Braz Chem Soc,2009(1):1-7 ) ( [11] GB29696-2013 食品安全国家标准牛奶中阿维菌素 类药物残留检测高效液相色谱法 ) 阿维菌素类是由放线菌产生的一组大环内酯类抗生素.包括伊维菌素,阿维菌素,多拉菌素,莫能菌素,伊普菌素等,广泛用于畜禽体内外抗寄生虫药。近年来,该药也被广泛应用于水产养殖中,对水产品的寄生虫防治有较好的疗效。但在动物体内的残留时间较长,世界卫生组织仍将其列为高毒化合物,,各国也都制定了食品中阿维菌素残留的相关限值。 已有报道的阿维菌素检测方法包括高效液相色谱法[1],液质联用法[2],液相色谱串联质谱法[3]等。液质联用是确证方法,但仪器昂贵,普及程度较低,所以高效液相色谱荧光检测法仍为目前常用的检测方法。本文依据 GB29695-2013水产品中阿维菌素和伊维菌素多残留的测定,对龙利鱼片和鳕鱼片样品进行了测定,结果满足标准要求。 2.样品前处理 2.1标准溶液配制 100 pg/mL 阿维菌素和伊维菌素混合标准贮备液:精密称取阿维菌素和伊维菌素标准品各 10 mg, 于100 mL量瓶中,用甲醇溶解并稀释至刻度,配制成浓度为 100 pg/mL的阿维菌素和伊维菌素混合标准贮备液。-18℃以下保存,有效期6个月。 10 pg/mL 阿维菌素和伊维菌素混合标准工作液:精密量取100 pg/mL 阿维菌素和伊维菌素混合标准贮备液10mL,于100 mL容量瓶中,用甲醇稀释至刻度,配制成浓度为10 pg/mL 阿维菌素和伊维菌素混合标准工作液。2~8℃保存,有效期3个月。 精密量取10 pg/mL 阿维菌素和伊维菌素混合标准工作液适量,用甲醇稀释,配制成浓度为0.1、0.2、0.5、1和2 ug/mL的系列标准溶液,各取0.1mL于5mL离心管中,于45~55℃水浴氮气吹干,按衍生化步骤处理。供高效液相色谱测定。以测得峰面积为纵坐标,对应的标准溶液浓度为横坐标,绘制标准曲线。求回归方程和相关系数。 2.2样品前处理 提取:称取试料(5±0.05)g,于50mL聚丙烯离心管中,加乙腈15mL, 旋涡混合1min, 超声30 min,4000 r/min 离心5 min, 取上清液于另一50mL聚丙烯离心管中。残渣中加入乙腈10 mL,重复提取一次,合并两次提取液。在提取液中加正己烷10 mL, 充分涡旋混合1 min, 4000 r/min 离心 5 min, 弃上层正己烷层。提取液中再加正己烷10 mL,重复提取一次,弃正己烷层,备用。 净化:取备用液,依次过用乙腈10 mL活化的无水硫酸钠柱和碱性氧化铝固相萃取柱,收集滤液。待滤液流至将尽,用乙腈10mL冲洗离心管,转至无水硫酸钠柱和碱性氧化铝固相萃取柱,吹干,合并滤液于茄形瓶中,于40~50℃旋转蒸发至干。用乙腈3mL分2次溶解残余物,并转至5mL具塞玻璃离心管中,45~55℃水浴氮气吹干。 衍生化:于上述5mL离心管中加衍生化试剂 A100 uL,盖紧塞子,旋涡混合30s, 再加衍生化试剂B150uL,盖紧塞子,旋涡混合30s, 密封避光,室温下衍生15 min,加甲醇750 uL,旋涡混合30 s,过滤,供高效液相色谱测定。 检测项目 阿维菌素和伊维菌素 样品基体 龙利鱼片,鳕鱼片 仪器型号及配置 Dionex Ultimate 3000RS 系列 泵: DGP-3600RS(S/N:8048214) 自动进样器: WPS-3000TRS(S/N: 8048519) 柱温箱: TCC-3000RS(S/N: 8047332) 检测器: DAD-3000RS(S/N: 8024592) 色谱软件: Chromeleon Chromatography Data System7.2 Acclaim C18, 120A, 4.6×250mm, 5 pm (S/N005562, P/N059149); 色谱柱类型尺寸、S/N、 P/N号及柱温 温度:35℃ 流动相组成及流速 时间(min) 甲醇% 乙腈% 0.4%乙酸 0 39 55 6 13 39 55 6 15 45 55 0 25 39 55 6 4.样品分析谱图 4.3龙利鱼片普图 4.1阿维菌素和伊维菌素标准品谱图 Time [min] 图3.龙利鱼片谱图 图1.标准溶液UV色谱图(0.5pg/mL) 4.4鳕鱼片谱图 4.2重复性谱图(5针) 图2.标准溶液重复性色谱图 图4.鳕鱼片谱图 4.5龙利鱼片及加标谱图 4.6龙利鱼片加标重现性谱图 图5.龙利鱼片及加标谱图 图6龙利鱼片加标重现性谱图 表2.重复性实验数据 保留时间(min) 峰面积(mAU*mi) 峰高(min) 阿维菌素 伊维菌素 阿维菌素 伊维菌素 阿维菌素 伊维菌素 1 10.487 16.890 10226.23 11876.85 40788.10 62208.35 2 10.490 16.893 10407.36 11872.34 41464.69 61687.38 3 10.483 16.897 10338.77 12025.02 41032.76 61799.40 4 10.480 16.897 10295.62 11833.41 41096.35 62214.01 5 10.493 16.900 10365.03 11880.53 41158.64 62418.61 RSD/% 0.05% 0.02% 0.67% 0.62% 0.59% 0.50% 表3.龙利鱼片中阿维菌素和伊维菌素含量实验数据 样品 实际样品中含量(ug/kg) 阿维菌素 伊维菌素 阿维菌素 伊维菌素 龙利鱼片 <0.1 <0.1 <2 <2 鳕鱼片 <0.1 <0.1 <2 <2 实际样品中含量(pg/kg)=测量值(ng/mL)>*0.10 mL/量值(g) 表4.龙利鱼片中阿维菌素和伊维菌素加标回收率及精密度实验数据 添加浓度(ug/kg) 4 20 阿维菌素 伊维菌素 阿维菌素 伊维菌素 加标后浓度(mg/kg) 3.604 3.502 18.736 16.634 回收率(%) 90.1 87.5 93.7 83.2 RSD(%) 3.94 2.46 1.06 1.21 6.可行性建议 本方法参考 GB 29695-2013水产品中阿维菌素和伊维菌素的液相测定方法,水产品样品经过乙腈提取,,7无水硫酸钠和碱性氧化铝萃取柱净化,再衍生过滤后直接进样分析,荧光检测的激发波长365 nm, 发射波长475 nm, 杂质干扰较少,阿维菌素和伊维菌素含量测定、回收率和重现性与标准的结果一致。选择Acclaim C18色谱柱,在甲醇,乙腈和0.4%乙酸溶液的等度条件下得到很好的分离,测定低限均为2 pg/kg。 附录:标准曲线 No. Ret.Time (min) Peak Name Cal.Type Offset(C0) Slope(C1) Range (pg/mL) Corr.Coeff% 1 10.483 阿维菌素 Lin 52074.277 0.1-2 0.9973 2 16.893 伊维菌素 Lin 0 60885.351 0.1-2 0.9962 ( 参考文献: ) [1]徐英江,任传博,刘慧慧,宫向红,张秀珍,邹荣婕,李佳蔚,于召强。液相色谱荧光法测定水产品中伊维菌素,阿维菌素,莫能菌素,埃普里诺菌素和多拉菌素含量,中国渔业质量与标准,2011,1(1):70-74 ( [2 ] HFowells L , Sauer MJ. Multi-residue analysis of avermectins and moxidectin by ion-trap LC-MSn, Analyst. 2 001,126(2): 155-60 ) ( [3] Sherdan R, D esjardins L . Determination of abamectin do-ramectin emamectin,Eprinomectin and m o xidectin i n milkbyliquid chromatography electrospray tandem mass spectrometry. J AOAC Int, 2006,89(4):1088-1094 ) 鸡可食性组织中地克珠利残留量的测定 1.引言 地克珠利(Diclazuril),分子式为C7H,CI,N4O2,分子量为407.63,属三嗪苯乙腈类化合物,为类白色或淡黄色粉末;几乎无臭;在二甲基甲酰胺中略溶,在四氢呋喃中微溶,在水、乙醇中几乎不溶。地克珠利作为一种新型、高效、低毒抗球虫药,已经被广泛应用于治疗和预防禽类及哺乳动物的球虫感染。为了确保动物性食品卫生安全,我国农业部第2002-235号公告对地克珠利最高残留限量做了规定。本实验参照国标《GB 29701-2013鸡可食性组织中地克珠利残留量的测定高效液相色谱法》,建立了高效液相色谱法测定鸡可食性组织中地克珠利的残留量的测定方法,结果良好,具有一定的参考价值。 2.样品前处理 2.1标准溶液配制 地克珠利标准储备液:精确称取地克珠利对照品 10 mg,置于50 mL棕色容量瓶中,加入50 mL 四氢呋喃溶解并定容至刻度。即的 500 pg/mL 的标准储备溶液。 地克珠利标准曲线:移取地克珠利标准储备液,采用逐步稀释法,分别加入适量流动相,配制成100,500,1000,5000, 10000 ng/mL 的系列标准溶液。 2.2样品前处理 准确称取试料2g, 剪碎,置于50 mL聚丙烯离心管中;加乙乙10mL, 均质1 min,10000 r/min 离心5 min,收集乙腈提取液于50mL茄形瓶中,残渣再重复提取一次,合并两次提取液,加入环已烷5mL,置摇床震摇 15min, 静置5 min, 弃环已烷层液,加正丙醇5mL,于50℃减压蒸干,用流动相1.0mL溶解残余物,微孔滤膜过滤,供高效液相色谱仪测定。 3.色谱条件 检测项目 地克珠利 样品基体 鸡可食性组织 仪器型号 Ultimate 3000: 泵: LPG-3400SD 自动进样器: WPS-3000SL 柱温箱: TCC-3000SD 检测器: DAD-3000RS 色谱软件:Chromeleon Chromatography Data System 7.2 分析柱 Syncronis C18; 250x4.6mm, 5pm; P/N97105-254130, S/N 0314616P3 流动相 乙腈+0.2%磷酸(57+43,v/v); 流速:1mL/min 柱温 30°C UV检测波长 278 nm 进样量 20 pL 4.样品分析谱图 4.1标准品谱图 图1.地克珠利标准溶液UV色谱图 4.2供试品溶液及加标测定谱图 Time [min] 图2.样品溶液测定谱图 图3.样品溶液加标测定谱图 5.测定结果 5.1线性、检出限 由低浓度至高浓度依次进样检测,以质量浓度与峰面积作标准曲线线性回归。结果表明,在100~10000ng/mL范围内显良好线性关系(r=0.9995)。通过溶液逐步稀释法,以 S/N=10计算,最低定量限为50 pg/kg。外标法定量。标准曲线如下: 图4.地克珠利标准曲线 表1.地克珠利标准曲线方程 Ret.Time (min)) Peak Number Coeff. of Co C1 Name Of points Determination (Offset) (Slope) 12.775 地克珠利 5 0.9995 -0.0278 0.0005 5.2重复性考察结果 取标准品溶液,连续进样7次,计算峰面积的RSD, RSD为1.01%。 5.3回收率 取鸡肉组织样品,添加一定浓度的标准品,静置30 min,按照样品前处理方法制备样品加标溶液,测定计算回收率,如表2所示。 表2.回收率结果 名称 实际含量 加入量 (ng/mL) 回收量 (ng/mL) 平均回收率 (%) 样品1 未检出 2000 1672 83.6 5.4实际样品测定 按上述方法测定从菜市场随机采购的鸡组织样品,样品中均未检出地克珠利残留。 本实验采用国家标准 GB 29701-2013测定鸡可食性组织中地克珠利残留,各项方法学数据均满足测定要求,可用于鸡可食性组织中地克珠利残留的测定。值得注意的是,本实验前处理第二步萃取净化溶剂选择的是实验室现有的环己烷,环己烷密度跟乙腈接近,导致与乙腈层分层不完全,可能会损失回收率。后续试验建议采用标准方法使用的正己烷作为第二步萃取净化溶剂,有助于提高萃取回收率. HPLC-UV 动物性食品中氮哌酮及代谢物多残留的测定 氮哌酮(Azaperone) 是一种丁酰苯类神经安定药。动物经肌肉注射此药后可消除紧张感,活动力下降,对环境淡漠并长期处于安静状态。有助于避免动物因“聚居应激”和在混群时发生互相攻击和打斗的现象,降低因应激及创伤引起的死亡率,因此在长途运输中常常给猪等动物使用该药,但不合理地使用本品可能造成氮哌酮及其代谢物氮哌醇在动物器官组织中残留,为保证动物源性产品食用的安全性,我国最新发布的食品安全国家标准 GB29709-2013《动物性食品中氮哌酮及代谢物多残留的测定高效液相色谱法》对其做了分析规定,本方法主要参照了该标准进行了相关测定。 图1.氮哌酮分子式 图2.氮哌醇分子式 标准工作溶液:精密称取氮哌酮和氮哌醇标准品品10mg,用甲醇溶解定容至10 mL, 配成浓度为1 mg/mL的标准贮备液。精密量取氮哌酮和氮哌醇标准贮备液适量,以50%乙腈稀释,配制成浓度为 10、20、50、100、200、500和1000 pg/L的系列混合标准溶液。 样品前处理:选取市售的猪肉、猪肝、猪肾,参考《中华人民共和国国家标准》GB 29709-2013“动物性食品中氮哌酮及代谢物残留的测定高效液相色谱法”,称取试料(5±0.05) g, 于50mL离心管中,加乙腈10mL, 均质混合1min, 5000 r/min 离心5 min, 取上清液,残渣中加乙腈10 mL 重复提取一次,合并两次上清液,于40℃水浴旋转蒸干,用2%甲酸水溶液5 mL 溶解残余物,加正己烷4mL,混匀,5000 r/min 离心 5 min,弃正己烷 层液,备用。 固相萃取柱*依次用甲醇3mL、水3mL和2%甲酸水溶液3mL活化,取备用液过柱,控制流速小于1mL/min,用2%甲酸水3mL和甲醇3mL淋洗,抽干,用5%氨化甲醇4mL 洗脱。收集洗脱液,于40℃氮气吹干,用流动相1.0mL溶解残余物,旋涡混匀,0.22 pm有机滤膜过滤,供高效液相色谱法测定。 *GB29707-2013中规定使用的固相萃取柱型号为MCX(混合阳离子基质),规格为60 mg/3 mL,本试验选择的固相萃取柱为 Hypersep Retain C X (60 mg, 3 ml)(PN60107-303),填料型号和规格与之完全一致。 3.色谱条件 检测项目 氮哌酮及代谢物(氮哌醇) 样品基体 动物性食品(猪肉、猪肝、猪肾) 仪器型号 Thermo Ultimate 3000: 泵: DGP-3600RS 自动进样器: WPS-3000TRS 柱温箱: TCC-3000RS 检测器: DAD-3000RS 色谱软件: Chromeleon Chromatography Data System 7.1 色谱柱类型 Acclaim 120 C18,250×4.6mm, 尺寸、S/N号 5 um, P/N: 059149,S/N:004738; 及柱温 30℃ UV检测波长 250 nm 流动相 5mM醋酸铵+乙腈(50+50,v/v) 进样量 50 pL 4.1标准溶液分析谱图 时间[min] Ret. Time (min) Peak Name Cal. Type Coeff. of Determination Co (Offset) C1 (Slope) C2 (Curve) 7.5 氮呱醇 Lin, WithOffset 1.00000 0.0007 0.0014 0 11.6 氮呱酮 Lin, WithOffset 0.99998 0.0005 0.0026 0 mAUmin ulL 250-200150100-0.50-000J 图2.氮哌醇标准曲线图 图3.氮哌酮标准曲线图 取不同浓度对照品溶液依次进样检测,以质量浓度(X)与峰面积(Y)作标准曲线线性回归,结果表明在10~1000ug/L浓度范围,氮哌酮及其代谢物氮哌醇均显良好线性关系(r>0.9999)。 4.2标准溶液重现性分析结果 4.4样品加标分析结果 取混合对照品溶液(20 ug/L),连续进样7针,峰面积精密度考察结果见表3。 取样品(1#)5g,精密加入50 pg/L的标准品1mL,照样品前处理方法制备供试品溶液,进样50uL测定,测定结果如下。 表3.精密度考察结果 序号 氮哌醇 氮哌酮 1 0.0273 0.0511 2 0.0279 0.0519 3 0.0278 0.0506 4 0.0279 0.0505 5 0.0279 0.0506 6 0.0274 0.0516 0.0270 0.0510 RSD % 1.33 1.05 从以上图表可以看出,氮哌醇和和哌酮的峰面积值RSD均小于2%,说明重现性较好。 4.3仪器检出限 对最低浓度10.0 pg/L进行了分析,根据信噪比进行计算,氮哌酮及其代谢物氮哌醇的仪器检出限(以S/N=3计)均约为3ug/L,根据供试品取样量计,方法检出限均约为0.6 pg/kg (GB29707-2013规定检测限为 2 pg/kg)。 图4.样品及加标样品溶液UV色谱图(上-蓝色-样品,,下-黑色-加标样品) 待则物 加入量(ug/kg) 回收量(ug/kg) 回收率(%) GB29709-2013 回收率限值(%) 氮哌醇 10.0 8.33 83.3 60~110 氮呱酮 10.0 7.04 70.4 60~110 由测试结果,方法回收率在 GB29709-2013规定的范围以内。 4.5不同样品的分析结果 表5.样品测定结果 样品 氮哌醇(ug/L) 氮哌酮(ug/L) 1#(猪肉) 未检出 未检出 2#(猪肝) 未检出 未检出 3#(猪肾) 未检出 未检出 5.讨论 5.1对照品溶液配制方法修改说明: GB29709-2013采用纯甲醇配制对照品溶液,测定过程中发现,由于对照品溶液极性(100%甲醇)大于流动相溶液极性(50%乙腈),当使用进样器和色谱柱之间死体积较小的HPLC仪时,容易产生溶剂效应,导致色谱峰变形(见图5),若采用与流动相极性一致的溶剂(如50%乙腈)配制对照品溶液则可解决该问题(见图1)。 5.2样品溶液配制方法修改说明 GB29709-2013采用旋涡混合结合超声方式提取样品,实际操作中发现,该方式对粘度较大的样品(如肉馅)提取效率较低,加标回收率小于60%,而采用均质提取可在一定程度上提高提取效率和加标回收率。 原标准方法在最后一步采用直接溶解后即进样测定的方式;实际操作时发现,溶解后样品可能存在一定的浑浊,为保护色谱柱,采用微孔滤膜过滤后进样的方式。 6.结论 本实验参照 GB29709-2013,对检测方法进行适当改进后,对动物性食品中“氮哌酮及代谢物残留”进行测定,结果各项方法学数据均满足测定要求。 对乙酰氨基酚为非甾体抗炎药,具有抗炎、解热镇痛等作用,常用于感冒发烧、关节痛、神经痛等疾病。除了用于人类疾病治疗外,也用于禽畜类的感染治疗。但禽畜体内的对乙酰氨基酚代谢较慢,会大量蓄积在动物的肌肉和肝脏等组织中,当人食用该类动物组织时,残留的对乙酰氨基酚则会转移到人体内而引起肌肉或肝脏的损伤,因此监测动物源食品中的对乙酰氨基酚残留量对食品安全有着重要意义。本研究参照国标 GB29683-2013(动物性食品中对乙酰氨基酚残留量的测定)对猪肉中的对乙酰氨基酚残留进行了检测,结果表明本方法专属性强,灵敏度高,可用于动物组织中对乙酰氨基酚残留分析。 图1.对乙酰氨基酚 2.样品前处理 标准品溶液制备:准确称取对乙酰氨基基对照品10mg并转移至10mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,制成浓度为1.0 mg/mL标准品储备液。密封并于4℃条件下避光保存。精密量取1mL标准品储备备至100mL量瓶中,用流动相稀释制成浓度为 10 pg/mL的标准溶液,摇匀,作为工作溶液。密封并于4℃条件下避光保存。 样品提取:称取5g猪肉样品,置于50mL具塞离心管中,加乙酸乙酯20mL涡旋2分钟,6000转/分离心15分钟,转移上层清液至另一离心管中,残渣中再加乙酸乙酯10mL涡旋2分钟,6000转/分离心10分钟,合并两次提取的上清液,于45℃下氮气吹干,用甲醇3mL溶解残余物,冰水浴中放置10分钟,将上层(甲醇溶液)液体转移至20 mL离心管中,加入正己烷5 mL,振摇混匀,6000转/分离心10分钟,弃去上层液,取下层液(甲醇溶液)于45℃水浴中氮气吹干,用水6mL溶解残渣, 10000转/分离心10分钟,取上清液备用。 样品纯化:纯化柱依次用甲醇3mL和水3mL活化。取样品备用液过柱(流速:<2mL/min)。用水3mL和5%甲醇3mL淋洗,甲醇4mL洗脱,收集洗脱液,于45℃水浴氮气吹干,用流动相1 mL溶解残余物,用微孔滤膜(0.22um)过滤,用于色谱分析。 加标样品制备:取洁净离心管(50mL),向其中添加浓 度分别为10,50,100 pg/L的标准品甲醇溶液,于45℃ 氮气吹干溶剂,再加入5g猪肉样沐,依次按照2.3和2.4 项下进行加标样品制备。 3.色谱条件 检测项目 对乙酰氨基酚 样品基体 猪肉 UltiMate3000: 仪器型号 HPG-3400 RS, SN: 8085552 WPS-3000TSL RS, SN: 8088671 TCC-3000 RS, SN:8061791 VWD-3100 RS, SN:8682235 变色龙(Chromeleon) 色谱管理软件(V.7.2) 色谱柱类型、S/N Acclaim 120 C18,4.6×250 mm, 5um, PN:059149, SN:004604; 号及柱温 柱温:30℃ 检测器类型、工作 检测波长: UV@250nm, 参数及S/N号 采集频率:5Hz 流动相组成及流速 流动相:甲醇+50mM乙酸铵 (20+80, v/v), 1.0mL/min 进样体积 50 pL 4.样品分析谱图 4.1标准溶液分析图谱 以测得峰面积为纵坐标(Y),对应的标准溶液浓度为横坐标(X),绘制标准曲线。对乙酰氨基酚回归方程:Y=3.3364 X+0.063,相关系数r=99.8%;将标准品溶液逐步稀释,测得最低检测限(S/N≥3) 为2.5 pg/L。 a)重现性分析结果 取对照品溶液,连续进样6针,计算保留时间和峰面积RSD%。结果见表1。 表1.重复性试验 序号 保留时间(min) 面积(min*mAU) 1 7.367 0.1758 2 7.373 0.1764 3 7.370 0.1750 4 7.371 0.1770 5 7.372 0.1761 6 7.369 0.1774 平均值 7.370 0.1763 RSD% 0.03 0.48 b)过滤影响测试 取对照品溶液,用微孔滤膜(尼龙材质,0.22 um)过滤,弃去初始滤液 0.2 mL, 取续滤液进样分析,测试其峰面积,并与未过滤对照品溶液进行比较(表1),结果见表2。 表4.过滤影响测试 序号 面积(min*mAU) 百分比(%) 1 0.1762 99.9 2 0.1752 99.3 3 0.1755 99.5 4.4样品测定与加样回收试验 按照色谱条件分析样品与加标样品,并计算其含量,结果见图2与表3 图2.样品加标分析图(黑色-加标,红色-未加标) 表3.样品测试与加标分析结果(n=2) 名称 含量 (ug/L) 加标量 (ug/L) 总含量 (ug/L) 回收率 (%) 样品 4.3 未检出 4.3 未检出 加标1 4.3 10 9.7 54.2 加标2 4.3 50 36.3 64.0 加标3 4.3 100 74.6 70.3 由于样品中目标组分为痕量成分,且样品前处理过程比较复杂,会产生样品损失,因此样品回收率较低,且随着加入标准品浓度升高,回收率会有所提高。 5.讨论 通过对前述方法学进行了考察,结果表明本方法有良好的专属性、重复性和线性范围,但由于样品前处理方法较复杂,目标组分损失较大,因此方法的准确度较差,可通过后期对样品前处理方法进行改善来提高方法准确度。 赛默飞世尔科技 上海 南京 上海市浦东新区新金桥路27号3,6,7号楼 南京市中央路201号南京国际广场南楼1103室 邮编201206 邮编210000 电话021-68654588*2570 电话021-68654588*2901 生命科学产品和服务业务 上海市长宁区仙霞路99号21-22楼 邮编200051 西安 电话021-61453628/021-61453637 西安市高新区科技路38号林凯国际大厦 1006-08单元 邮编710075 北京 电话029-84500588*3801 北京市东城区北三环东路36号环球贸易 中心C座7层/8层 邮编100013 昆明 电话+86 10 8794 6888 成都成都市临江西路1号锦江国际大厦1406室邮编610041电话 028-65545388*5300沈阳沈阳市沈河区惠工街10号卓越大厦3109室邮编110013电话 024-31096388*3901武汉武汉市东湖高新技术开发区高新大道生物园路生物医药园C8栋5楼邮编430075电话027-59744988*5401 云南省昆明市五华区三市街6号柏联广场写字 楼908单元 邮编650021 电话0871-63118338*7001 广州 广州国际生物岛寰宇三路36、38号合景 星辉广场北塔204-206单元 邮编510000 电话 020-82401600 欲了解更多信息,请扫描二维码关注我们的微信公众账号 赛默飞世尔科技在全国有共21个办事处。本资料中的信息,说明和技术指标如有变更,恕不另行通知。 热线8008105118 赛默飞 赛默飞色谱 电话4006505118 官方微信 口 与质谱中国 www.thermofisher.com The world leader in serving science 目录抗生素类HPLC-UV 检测牛奶中甲砜霉素残留 ...........................................................................................4Online SPE-HPLC 法快速测定牛奶中五种喹诺酮类抗生素的残留量 ..........................................72. 磺胺类水产品中甲氧苄啶的测定 ...........................................................................................................143. 驱肠虫药类液相色谱紫外检测器测定牛奶中左旋咪唑 ..................................................................................17鸡可食性组织中尼卡巴嗪残留量的测定 .....................................................................................19动物性食品中常山酮残留量的测定 .............................................................................................22牛奶中阿维菌素类药物多残留的测定 .........................................................................................24水产品中阿维菌素和伊维菌素多残留的测定 ..............................................................................29鸡可食性组织中地克珠利残留量的测定 .....................................................................................334. 镇静剂类HPLC-UV 动物性食品中氮哌酮及代谢物多残留的测定 ..............................................................355. 其他兽药类猪肉中对乙酰氨基酚残留分析 ....................................................................................................38
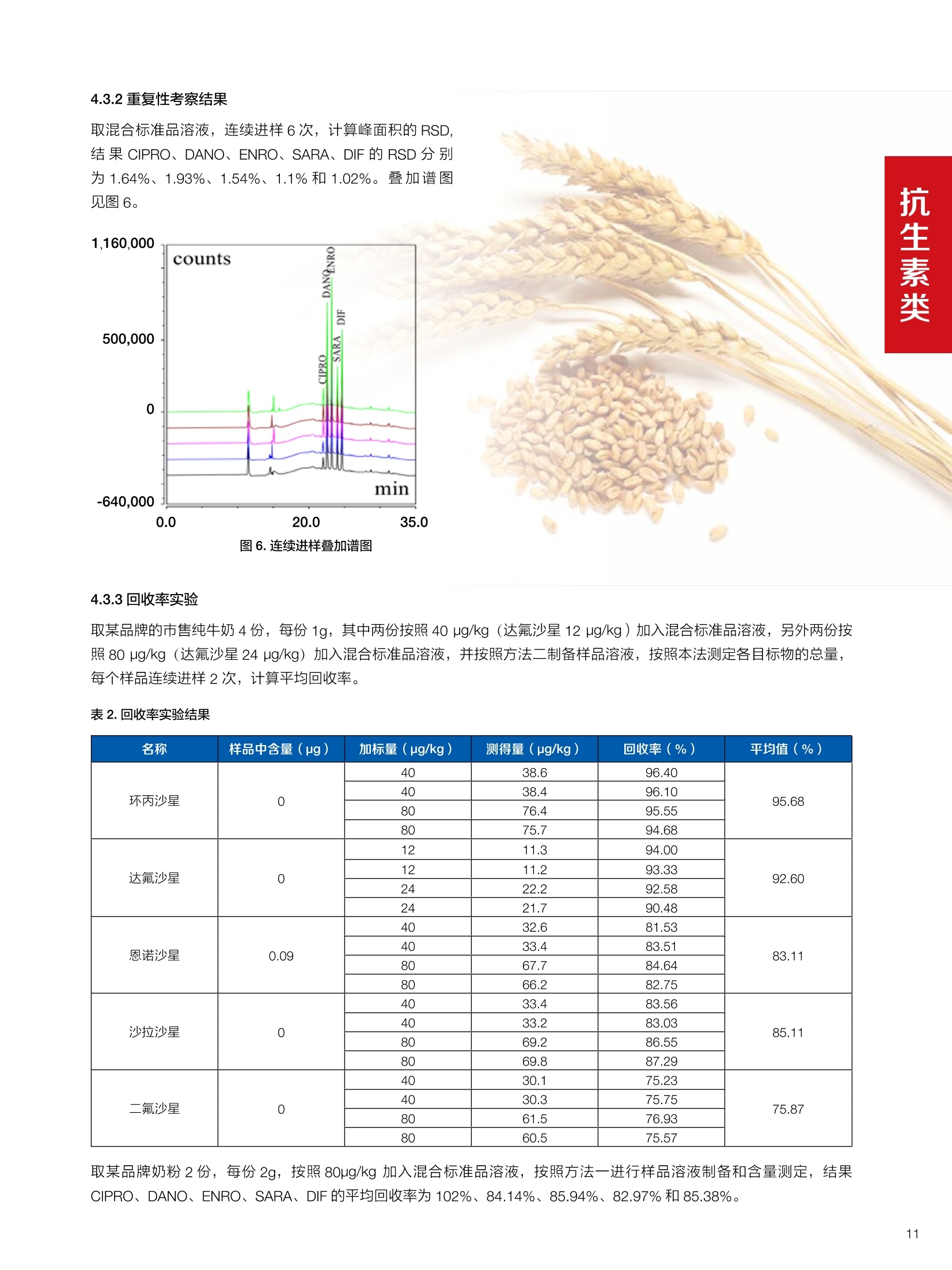
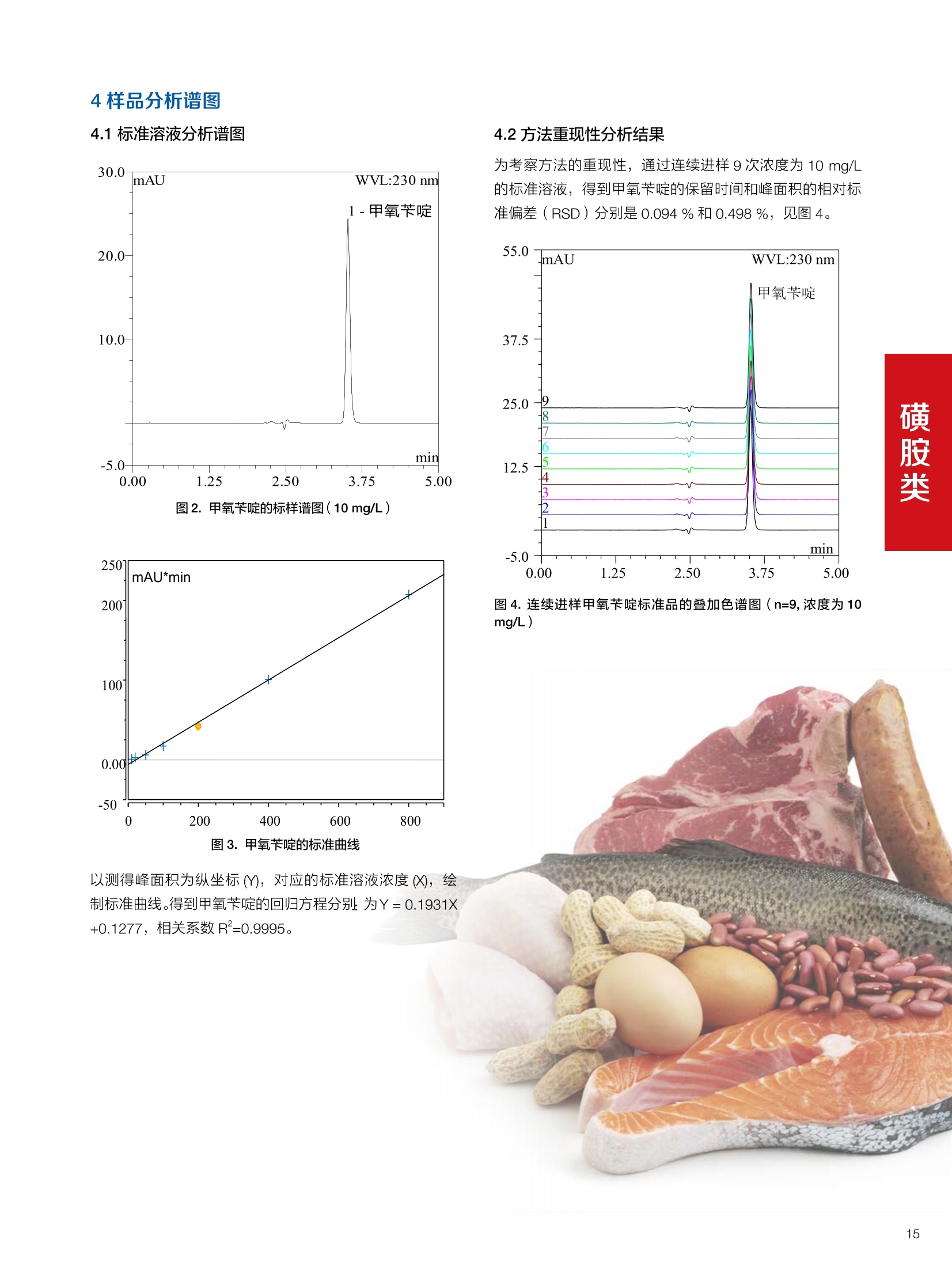
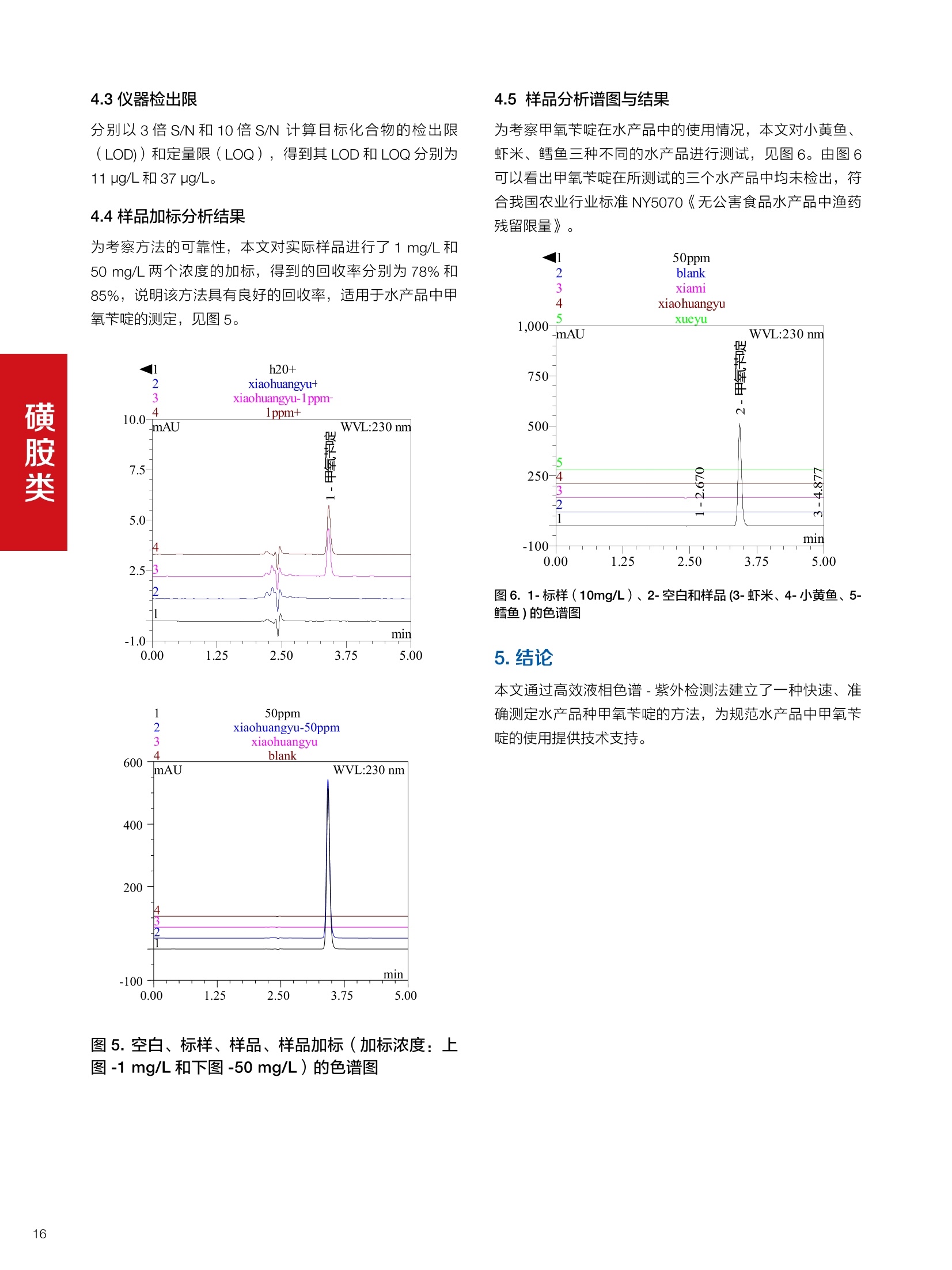
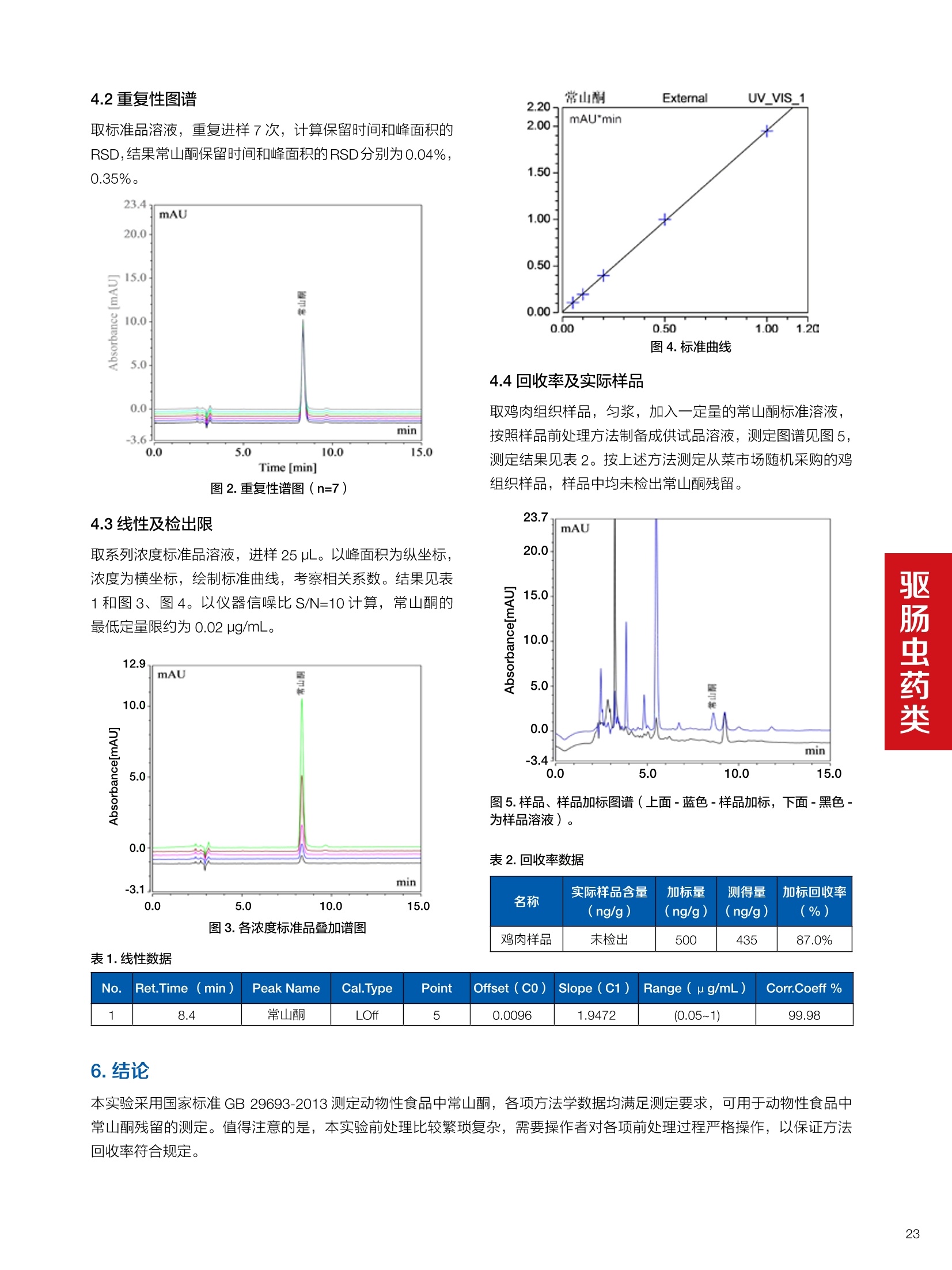
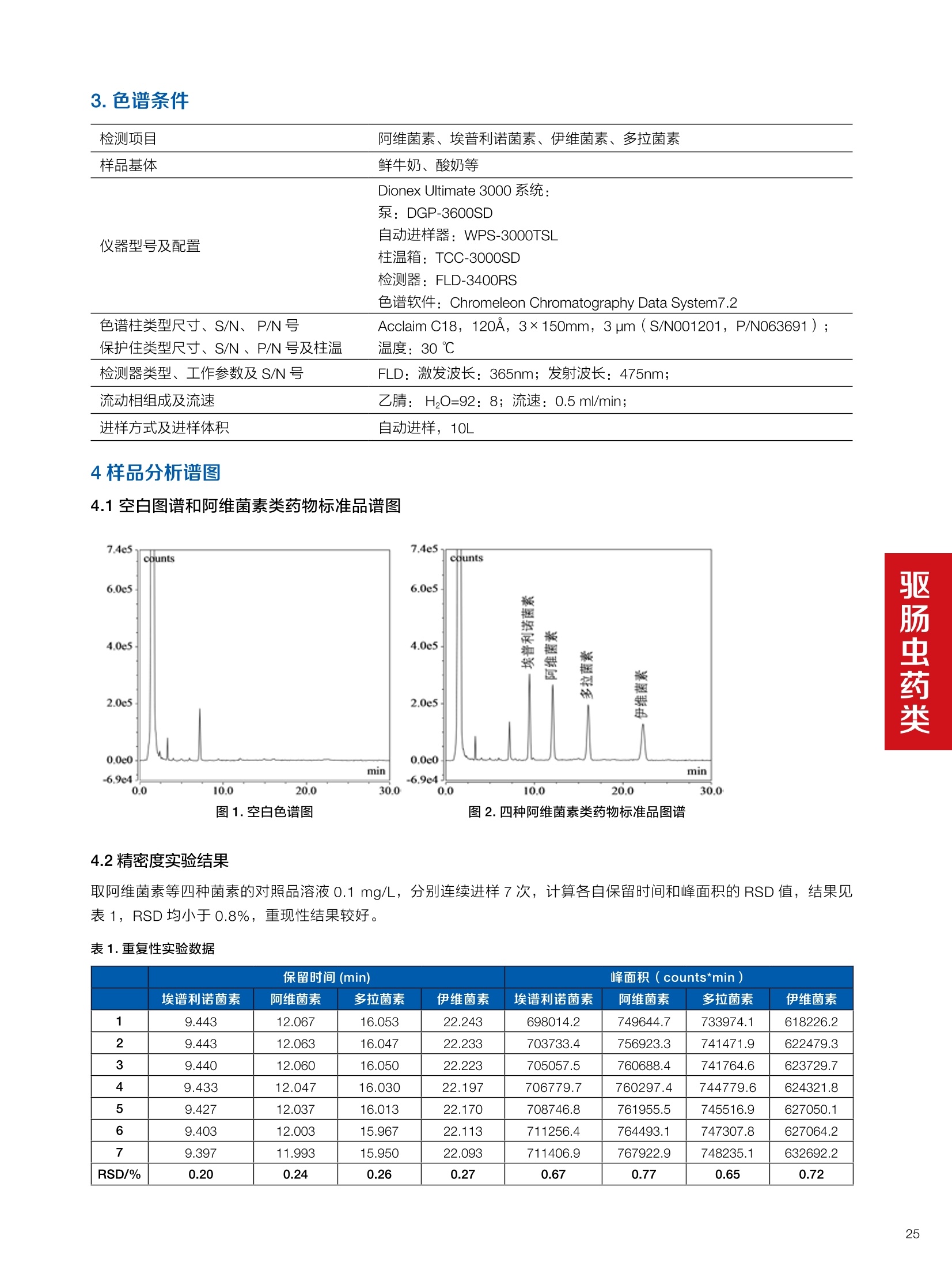
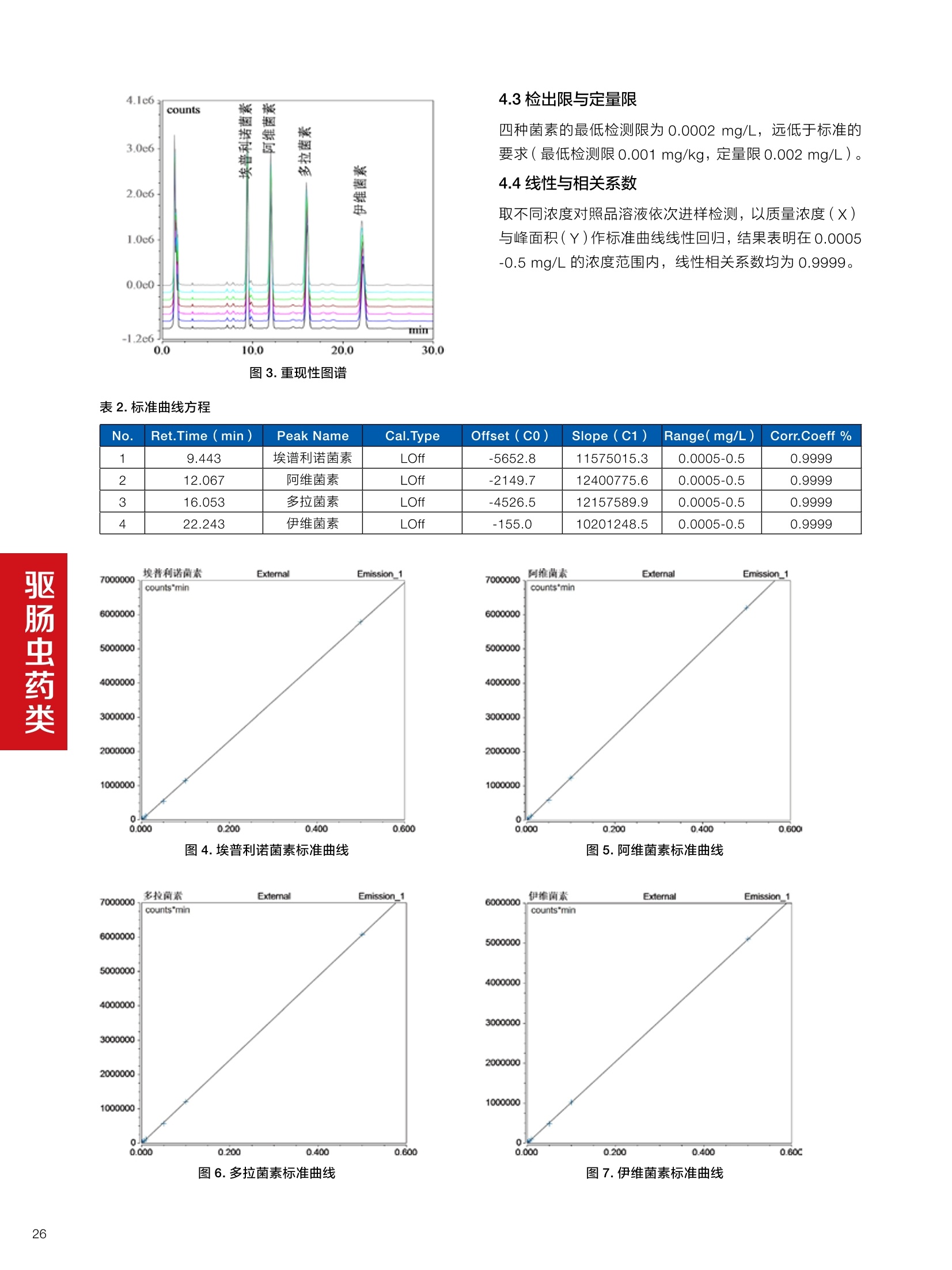
确定



























还剩38页未读,是否继续阅读?

产品配置单

赛默飞色谱与质谱为您提供《牛奶、鸡可食性组织中农兽残检测方案(液相色谱仪)》,该方案主要用于液体乳中兽药残留检测,参考标准《GB 29689-2013 食品安全国家标准 牛奶中甲砜霉素残留量的测定 高效液相色谱法》,《牛奶、鸡可食性组织中农兽残检测方案(液相色谱仪)》用到的仪器有赛默飞优谱佳UHPLC+高效液相色谱系统
推荐专场






相关方案
更多
该厂商其他方案
更多