








Cr(III)是人体必需的微量元素,Cr(VI)具有强烈毒性,对人体健康产生严重危害。本实验通过
快速原位分离,石墨炉原子吸收直接检测Cr(III)和Cr(VI),并讨论其存在形式
方案详情

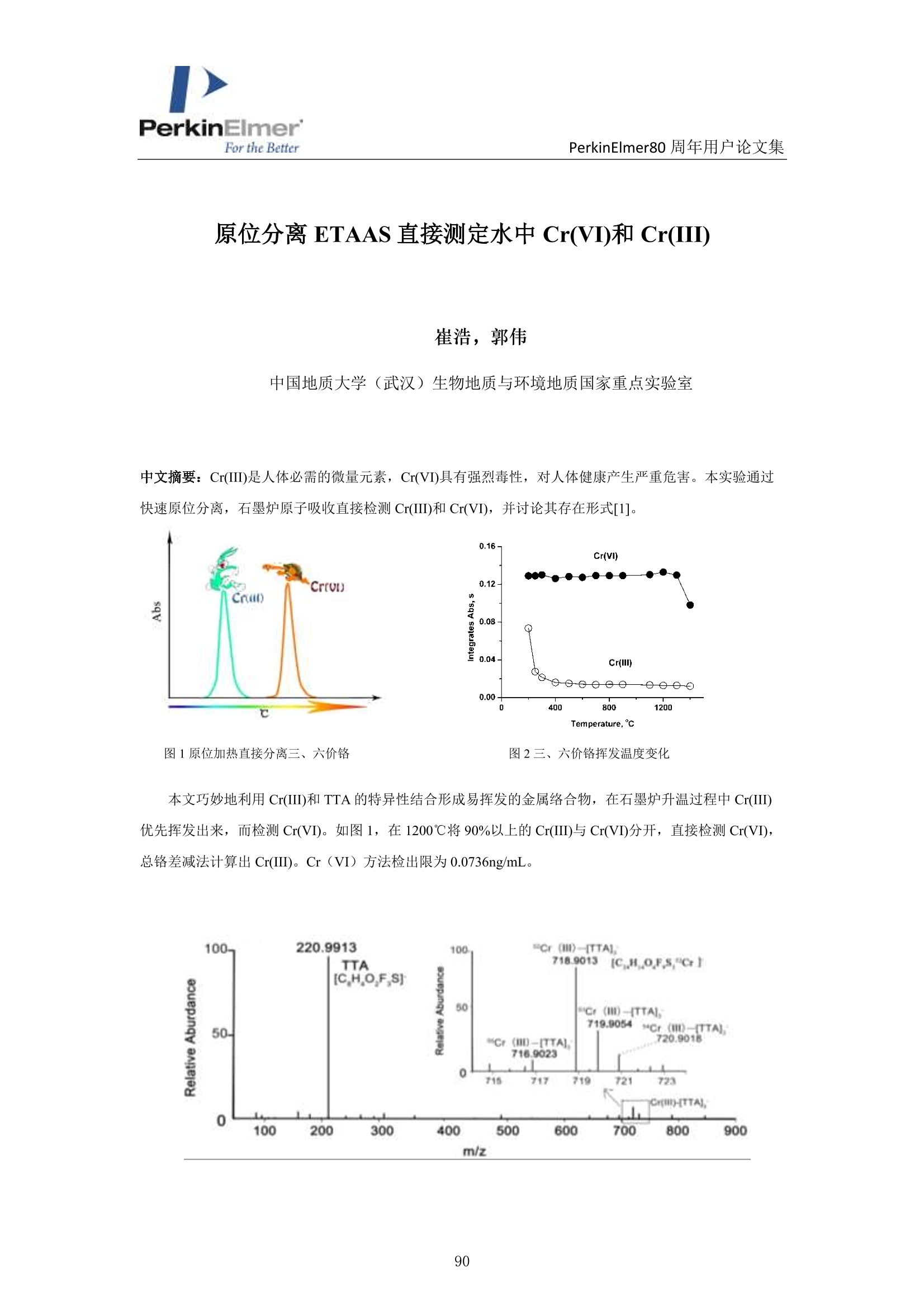
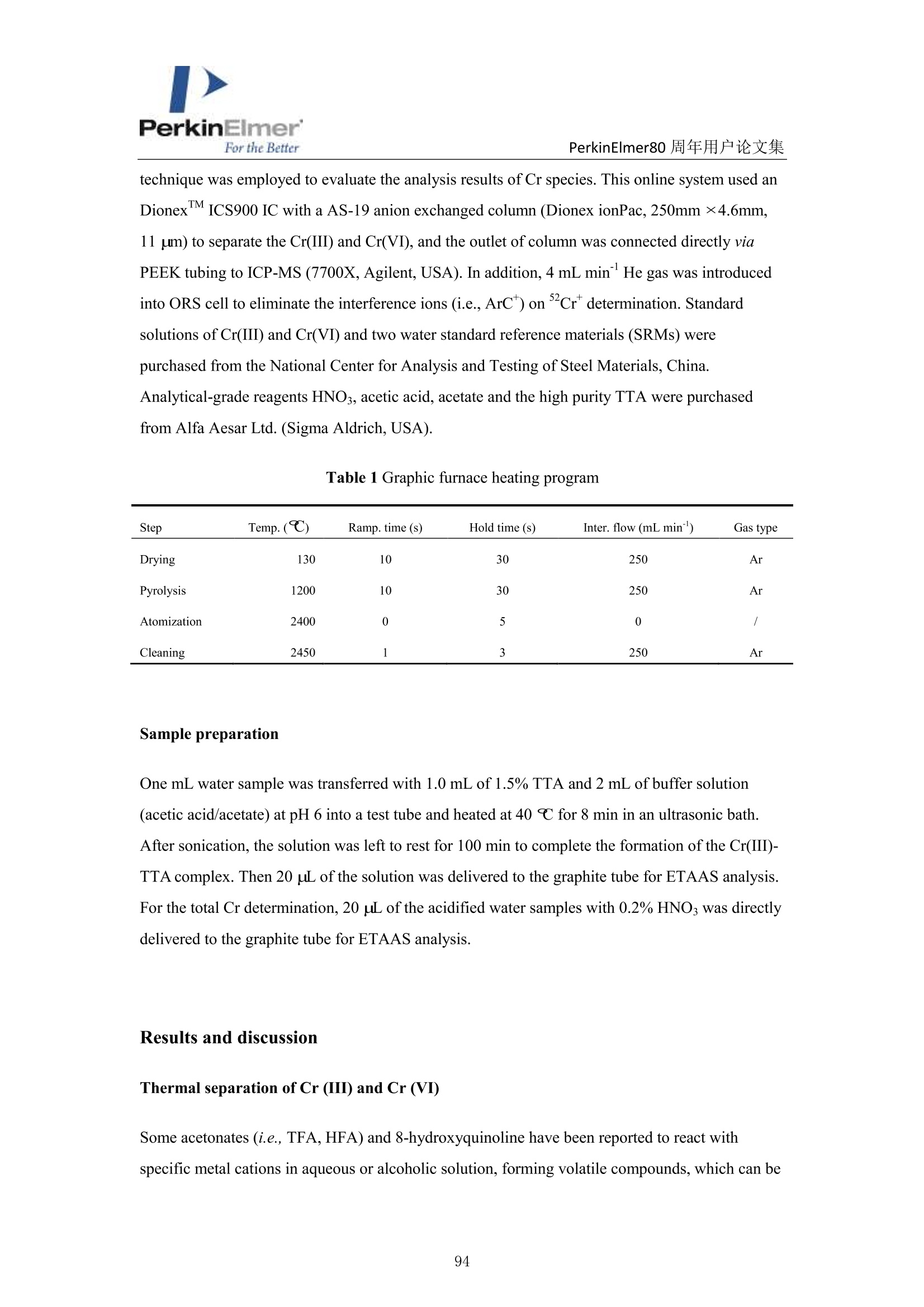
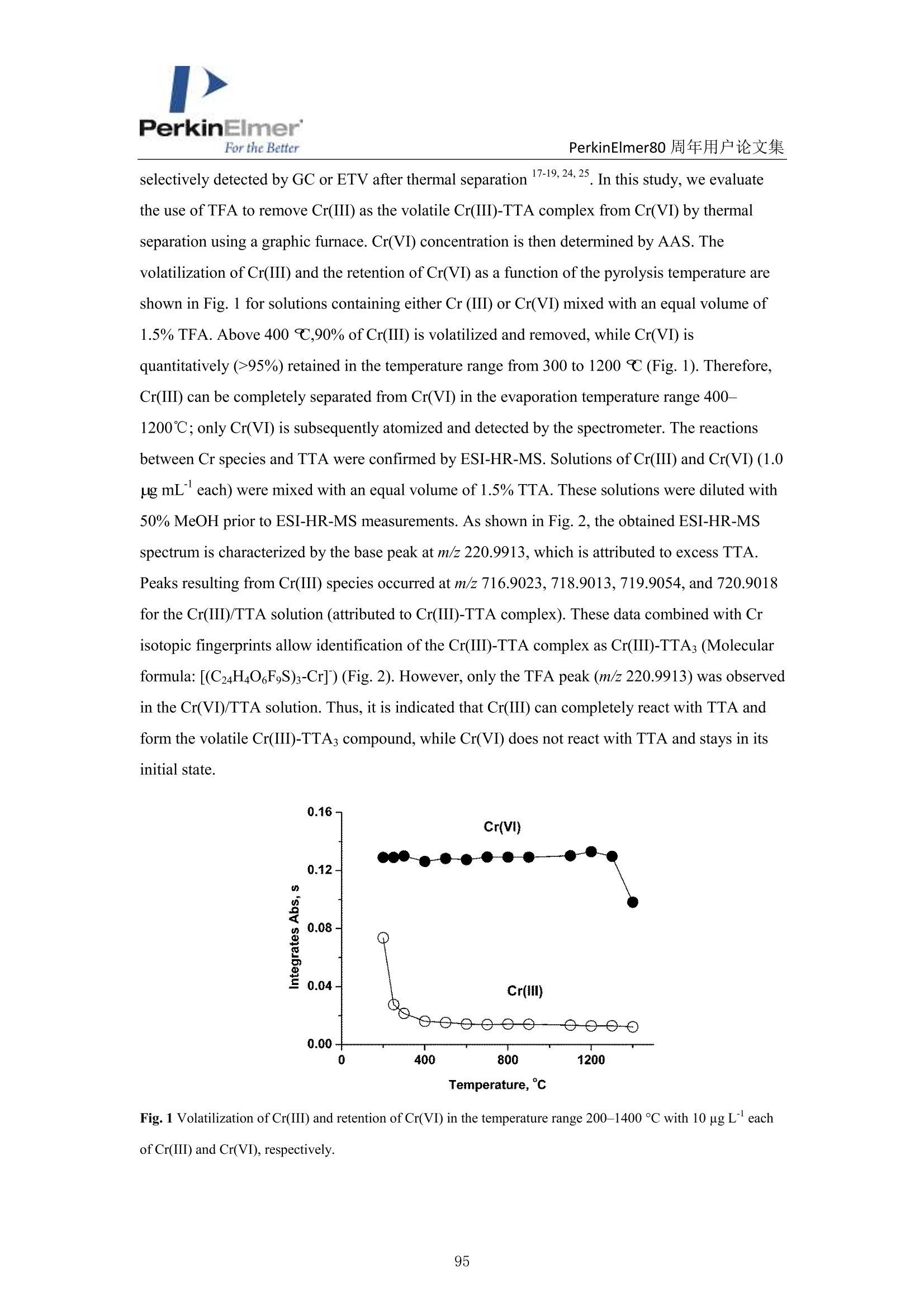
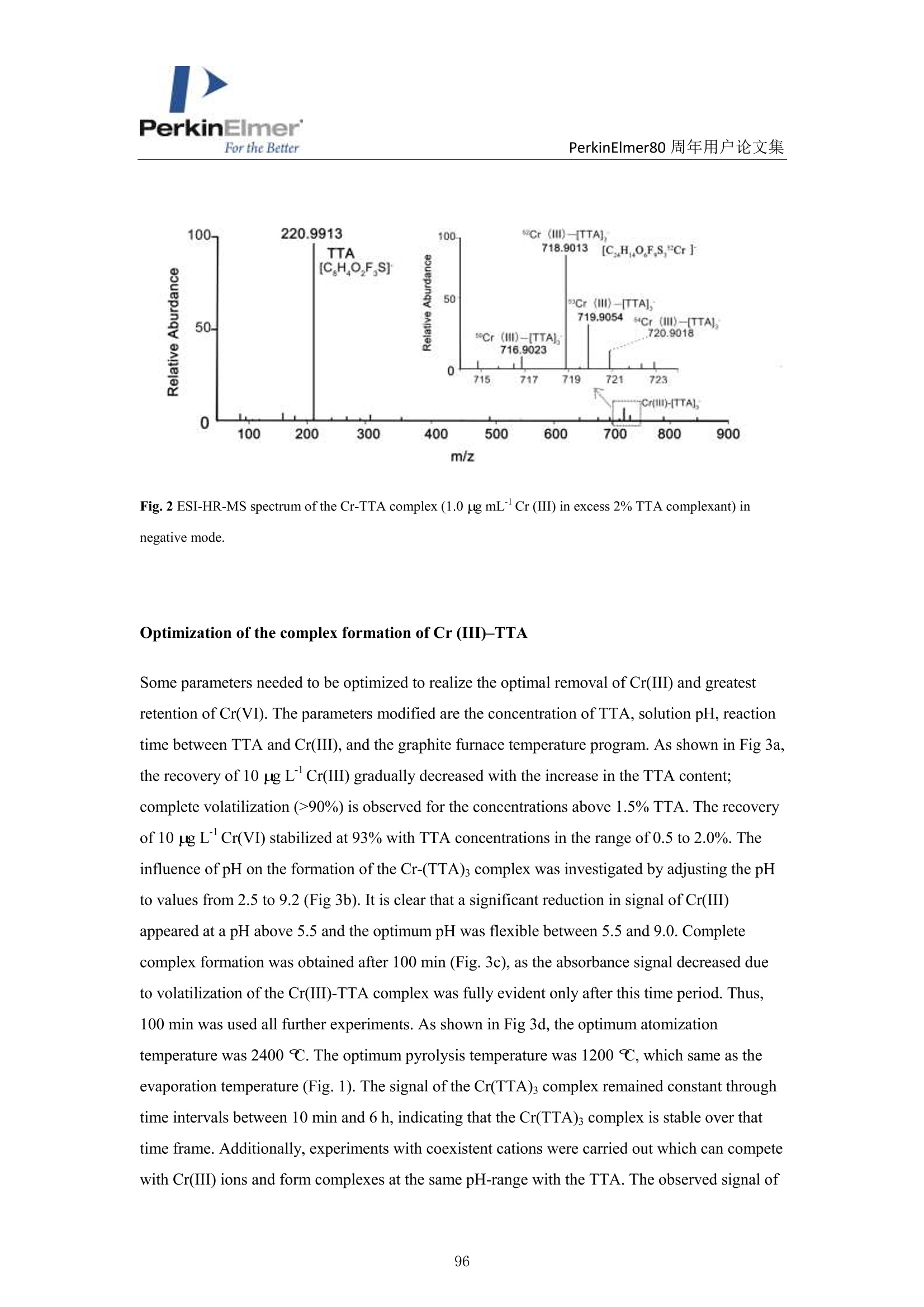
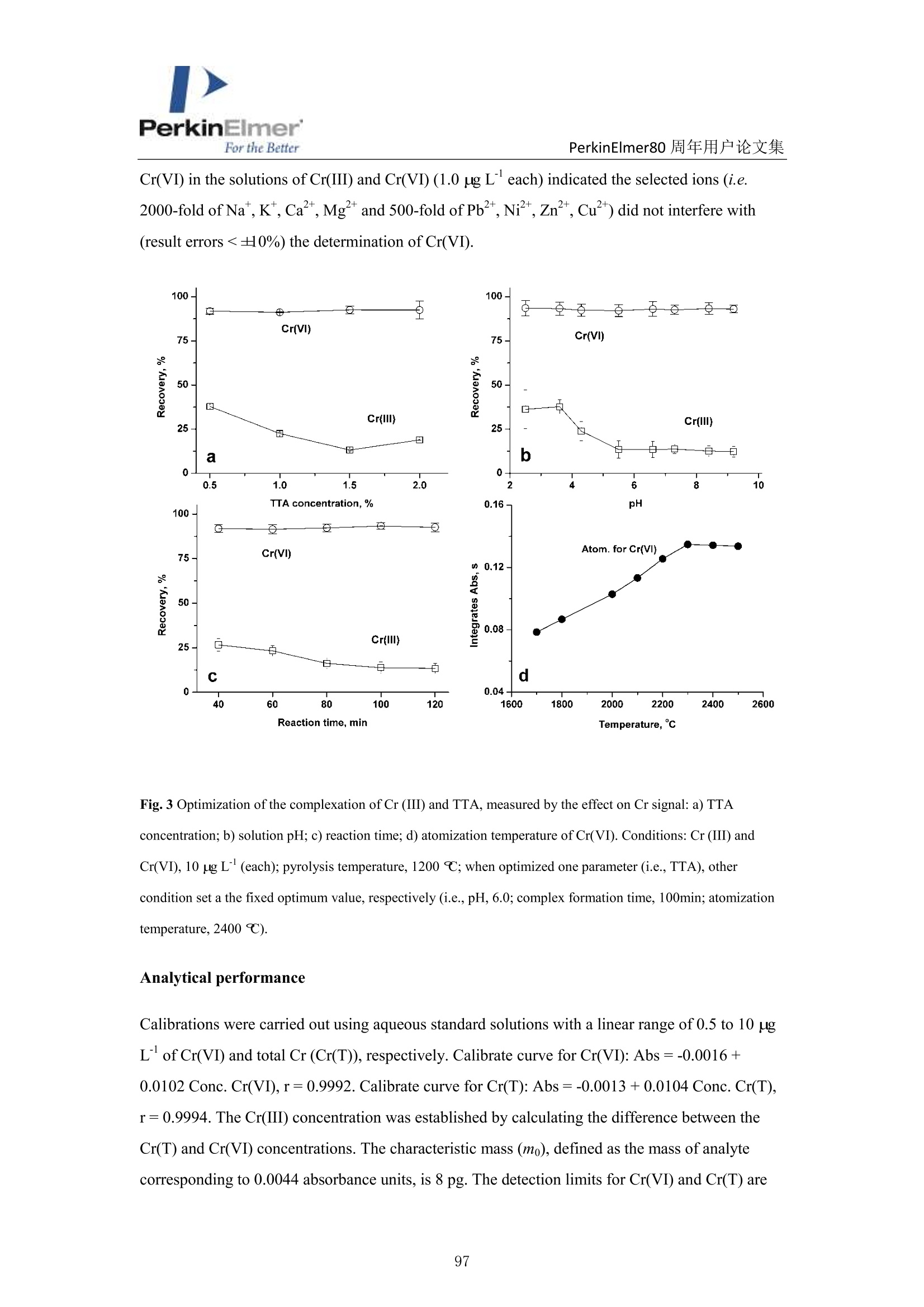
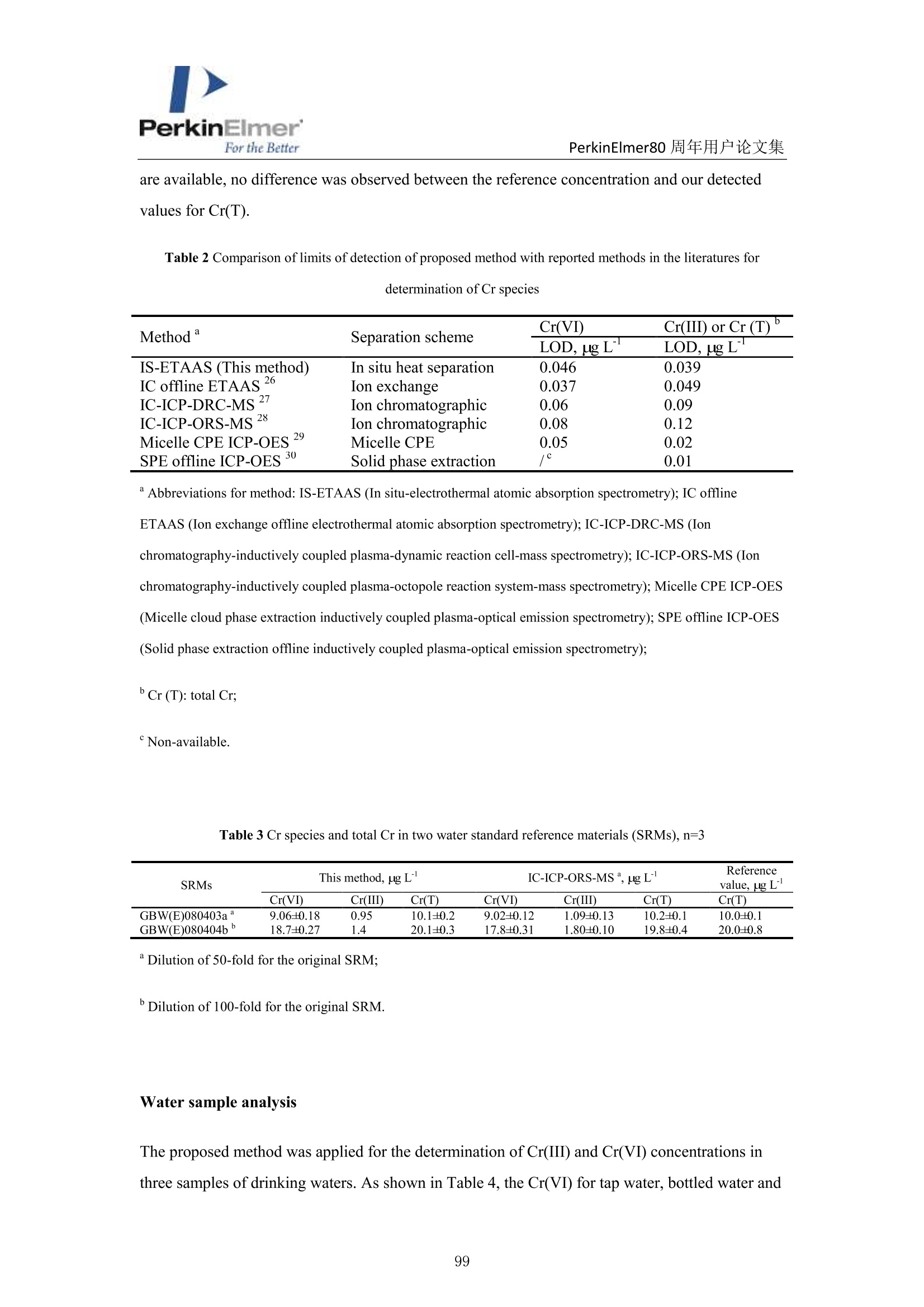
PerkinElmerFor the BetterPerkinElmer80 周年用户论文集 PerkinElmer80 周年用户论文集 原位分离 ETAAS 直接测定水中 Cr(VI)和 Cr(III) 崔浩,郭伟 中国地质大学(武汉)生物地质与环境地质国家重点实验室 中文摘要: Cr(III)是人体的需的微量元素, Cr(VI)具有强烈毒性,对人体健康产生严重危害。本实验通过快速原位分离,石墨炉原子吸收直接检测 Cr(ⅢII)和 Cr(VI), 并讨论其存在形式[1]。 图1原位加热直接分离三、六价铬 图2三、六价铬挥发温度变化 本文巧妙地利用 Cr(III)和 TTA 的特异性结合形成易挥发的金属络合物,在石墨炉升温过程中 Cr(III)优先挥发出来,而检测 Cr(VI)。如图1,在1200℃将90%以上的Cr(III)与 Cr(VI)分开, 直接检测 Cr(VI),总铬差减法计算出 Cr(III)。Cr(VI) 方法检出限为 0.0736ng/mL。 图3Cr(III)-TTA产物 利用高分辨质谱仪(ESI-HR-MS)验证其存在形式如图3所示,在质量数 m/=z 220.9913 出现明显TTA 的质谱峰,在m/z=716.9023,718.9013,719.9054 和720.9018 出现了一系列的含有铬同位素的 Cr(III))(TTA) 的质谱峰,猜测其化学式为[(C24H4O,FgS)3-Cr]-c 参考文献 [1] H. Cui, W. Guo, L.L. Jin et al. Anal.Methods,2017,9,1307-1312. Direct speciation of Cr in drinking water by in-situ thermalseparation ETAAS Hao Cui, Wei Guo* State Key Laboratory of Biogeology and Environmental Geology, School ofEarth Sciences, ChinaUniversity of Geosciences, Wuhan, 430074, P. R. China Abstract In situ speciation of Cr(VI) and Cr(III) based on thermal separation and electrothermal atomic absorptionspectrometry (ETAAS) detection has been developed. The complexant 2-thenoyltrifluoracetone (TTA) was used toselectively react with Cr(III); the product of the Cr(III)-TTA complex was vaporized at 400 ℃, while Cr(VI)quantitatively remains up to a temperature of 1200 ℃ and was subsequently detected by AAS. The complexationreaction products of the volatile Cr-TTA complex were verified using electro-spray ionization high resolutionquadrupole-Orbitrap-mass spectrometry (ESI-HR-MS). The Cr(III) concentration was established by calculatingthe difference between the total Cr (Cr(T)) and Cr(VI) concentrations. Under optimum experimental conditions,the detection limits for Cr(VI) and Cr(T) are 0.046 ug Land 0.039 pg L, with RSDs for 1.0 pg L"ofCr(VI)and 1.0 ug Lof Cr(T) of 3.1% and 2.8% (n=5), respectively. The accuracy of this method was validated bytesting two water standard reference materials (SRMs) for total chromium. The proposed method was employedfor analysis of drinking waters. The Cr(VI) concentrations for tap water, bottled water, and well water were found to be 1.41,0.53 and 0.68 pg L,respectively, which is in good agreement with the reference values measured byion chromatographic ICP-MS. This high throughput method has great potential for screening of Cr species at ultra-trace levels in drinking water. Introduction Cr(III) is essential for organisms, but Cr(VI) has strongly toxic and carcinogenic effects becauseof its high oxidizing potential and easy permeation of biological membranes 12. One of the mainsources of Cr(VI) for humans is drinking water, which may be contaminated by the leather tanninindustries, timber processing and waste-disposal companies, and via steel works,smelting, andelectroplating processes. The maximum allowable concentration (MAC) of Cr (VI) in drinkingwater is set at 6.0 ug Lin China . Therefore, the development of methods for the accuratedetermination of Cr(VI) concentration in drinking water is very important. Two main analytical strategies for Cr speciation have been published during the last threedecades. The classical analysis method involves two steps: 1) use of solid-phase extraction (SPE),liquid-liquid extraction (LLE), cloud-point extraction (CPE), or co-precipitation approach toseparate the two Cr species in the analysis solution; and 2) offline determination of Cr(III) or Cr(VI) concentrations by atomic spectrometry techniques (i.e. atomic absorption spectrometry(AAS), inductively coupled plasma-optical emission spectrometry (ICP-OES), and inductivelycoupled plasma- mass spectrometry (ICP-MS)) 5-8. Recent studies have implemented onlinecoupling of chromatographic methods (i.e., high performance liquid chromatography (HPLC), ionchromatography (IC), and capillary electrophoresis (CE)) to elemental detectors (atomicspectrometry techniques); especially HPLC-ICP-MS is an attractive tool for Cr speciation analysis9-13. Although the HPLC-ICP-MS combines high separation capability and high sensitivity forelement species analysis, it has some obvious disadvantages, including long retention times, therisk of contamination, unavailable standard hyphenated instruments, required sophisticatedoperation, and problems associated with spectral and/or non-spectral interferences caused by theuse of organic solvents 14,15. However, in some cases, it is enough to determine the concentrationof the most toxic species (i.e, Cr(VI)) as a screening parameter along with the total content of anelement or to distinguish between organic and inorganic compounds 1.Bermejo-Barrera et al.reported that some acetonates (i.e. trifluoroacetylacetonates TFA or hexafluoracetylacetonatesHFA) could react with appropriate metal cation in aqueous or alcoholic solution and form volatile compounds, which can be selectively detected by GC with thermal separation 18. Zhu et al. 1employed 8-hydroxyquinoline (8-Ox) to react with Cr(III) and the product ofCr(III)-8-Ox chelatecan be first volatilized at 1000℃ by electrothermal vaporization (ETV) and subsequentlydetected by ICP-OES; meanwhile, the stabilized Cr(VI) was measured at 2650℃ in the presenceof a polytetrafluoroethylene (PTFE) slurry chemical modifier. However, direct speciation analysisof Cr using a commercial ETAAS may be more attractive for the real samples, and the reactionmechanism ofCr(III) and the complexant is complicated and should be studied in detail. The present work aims to develop a reliable method for the direct determination of Crspecies in drinking water by in situ thermal separation by ETAAS using 2-thenoyltrifluoracetone(TTA) as a volatilizing chelating reagent. In the graphite furnace (GF), TTA reacts with Cr(III)forming a volatile compound whereas Cr(VI) remains unreacted. It should be possible to separatethese two oxidation states by application of a suitable GF temperature program. The reactionbetween Cr(III) and TTA was studied using a electro-spray ionization high resolution quadrupole-Orbitrap-mass spectrometry (ESI-HR-MS). Experimental Instrumentation and reagents All measurements were performed with a PinAAcleM 900T atomic absorption spectrometer(PerkinElmer, Inc., Shelton, USA), which has been described elsewhere in detail 20-22. It equippedwith a longitudinal Zeeman-effect background correction and transversely heated graphiteatomizer (THGA). Standard THGA tubes with integrated pyrolytically coated platforms and a Crhollow cathode lamp (Wavelengh, 357.87 nm; spectral bandwidth, 0.7 nm; cathode lamp intensity,9 mA) were used. The peak area measurement mode ad 5 s integration time were employedthroughout in this work. All aqueous solutions (Injection volume, 20 pL) were delivered into thegraphite tube by means of an AS autosampler (PerkinElmer, Inc., Shelton, USA). The optimizedgraphic furnace heating program was listed in Table 1. An ESI-HR-MS (QExactive hybridQuadrupole-Orbitrap MS, Thermo Scientific, Germany) was used to investigate the complexationbetween the Cr species and TTA. The operating conditions employed were similar to those for thedetermination of organic iodine in our previous work 2. An ion exchange chromatographic-inductively coupled plasma-octopole reaction system-mass spectrometry (IC-ICP-ORS-MS) technique was employed to evaluate the analysis results of Cr species. This online system used anDionexTM ICS900 IC with a AS-19 anion exchanged column (Dionex ionPac, 250mm ×4.6mm,11 um) to separate the Cr(III) and Cr(VI), and the outlet of column was connected directly viaPEEK tubing to ICP-MS (7700X, Agilent, USA). In addition, 4 mL min He gas was introducedinto ORS cell to eliminate the interference ions (i.e.,ArC*) on cr determination. Standardsolutions of Cr(III) and Cr(VI) and two water standard reference materials (SRMs) werepurchased from the National Center for Analysis and Testing of Steel Materials, China.Analytical-grade reagents HNO3, acetic acid, acetate and the high purity TTA were purchasedfrom Alfa Aesar Ltd. (Sigma Aldrich, USA). Table 1 Graphic furnace heating program Step Temp.(℃) Ramp. time (S) Hold time (s) Inter. flow (mL min) Gas type Drying 130 10 30 250 Ar Pyrolysis 1200 10 30 250 Ar Atomization 2400 0 5 0 Cleaning 2450 1 3 250 Ar Sample preparation One mL water sample was transferred with 1.0 mL of 1.5% TTA and 2 mL of buffer solution(acetic acid/acetate) at pH 6 into a test tube and heated at 40℃ for 8 min in an ultrasonic bath.After sonication, the solution was left to rest for 100 min to complete the formation of the Cr(III)-TTA complex. Then 20 uL of the solution was delivered to the graphite tube for ETAAS analysis.For the total Cr determination, 20 uL of the acidified water samples with 0.2%HNOs was directlydelivered to the graphite tube for ETAAS analysis. Results and discussion Thermal separation of Cr (III) and Cr (VI) Some acetonates (i.e.,TFA,HFA) and 8-hydroxyquinoline have been reported to react withspecific metal cations in aqueous or alcoholic solution, forming volatile compounds, which can be selectively detected by GC or ETV after thermal separation 17-19,24,25. In this study, we evaluatethe use ofTFA to remove Cr(III) as the volatile Cr(III)-TTA complex from Cr(VI) by thermalseparation using a graphic furnace. Cr(VI) concentration is then determined by AAS. Thevolatilization of Cr(III) and the retention of Cr(VI) as a function of the pyrolysis temperature areshown in Fig. 1 for solutions containing either Cr (III) or Cr(VI) mixed with an equal volume of1.5% TFA. Above 400 ℃,90% of Cr(III) is volatilized and removed, while Cr(VI) isquantitatively (>95%) retained in the temperature range from 300 to 1200℃ (Fig.1). Therefore,Cr(III) can be completely separated from Cr(VI) in the evaporation temperature range 400-1200℃; only Cr(VI) is subsequently atomized and detected by the spectrometer. The reactionsbetween Cr species and TTA were confirmed by ESI-HR-MS. Solutions of Cr(III) and Cr(VI) (1.0ug mLeach) were mixed with an equal volume of 1.5% TTA. These solutions were diluted with50% MeOH prior to ESI-HR-MS measurements. As shown in Fig.2, the obtained ESI-HR-MSspectrum is characterized by the base peak at m/z 220.9913, which is attributed to excess TTA.Peaks resulting from Cr(III) species occurred at m/z 716.9023, 718.9013, 719.9054, and 720.9018for the Cr(III)/TTA solution (attributed to Cr(III)-TTA complex). These data combined with Crisotopic fingerprints allow identification of the Cr(III)-TTA complex as Cr(III)-TTA(Molecularformula: [(C24H4O6F9S)3-Cr]) (Fig. 2). However, only the TFA peak (m/z 220.9913) was observedin the Cr(VI)/TTA solution. Thus, it is indicated that Cr(III) can completely react with TTA andform the volatile Cr(III)-TTA3compound, while Cr(VI) does not react with TTA and stays in itsinitial state. Fig. 1Volatilization of Cr(III) and retention of Cr(VI) in the temperature range 200-1400 C with 10 ug L’eachof Cr(III) and Cr(VI), respectively. Fig. 2 ESI-HR-MS spectrum of the Cr-TTA complex(1.0 ug mLCr(III) in excess 2% TTA complexant) innegative mode. Optimization of the complex formation of Cr (III)-TTA Some parameters needed to be optimized to realize the optimal removal of Cr(III) and greatestretention of Cr(VI). The parameters modified are the concentration of TTA, solution pH,reactiontime between TTA and Cr(III), and the graphite furnace temperature program. As shown in Fig 3a,the recovery of 10 ug L’Cr(III) gradually decreased with the increase in the TTA content;complete volatilization (>90%) is observed for the concentrations above 1.5% TTA. The recoveryof 10 ug L’Cr(VI) stabilized at 93% with TTA concentrations in the range of 0.5 to 2.0%. Theinfluence of pH on the formation of the Cr-(TTA)3 complex was investigated by adjusting the pHto values from 2.5 to 9.2 (Fig 3b). It is clear that a significant reduction in signal of Cr(III)appeared at a pH above 5.5 and the optimum pH was flexible between 5.5 and 9.0. Completecomplex formation was obtained after 100 min (Fig. 3c), as the absorbance signal decreased dueto volatilization of the Cr(III)-TTA complex was fully evident only after this time period. Thus,100 min was used all further experiments. As shown in Fig 3d, the optimum atomizationtemperature was 2400 ℃C. The optimum pyrolysis temperature was 1200℃, which same as theevaporation temperature (Fig.1). The signal of the Cr(TTA)3 complex remained constant throughtime intervals between 10 min and 6 h, indicating that the Cr(TTA)3 complex is stable over thattime frame. Additionally, experiments with coexistent cations were carried out which can competewith Cr(III) ions and form complexes at the same pH-range with the TTA. The observed signal of Cr(VI) in the solutions of Cr(III) and Cr(VI) (1.0 pg L" each) indicated the selected ions (i.e.2000-fold ofNa , K, Ca ,Mg’and 500-fold of Pb , Ni , Zn, Cu) did not interfere with(result errors <+10%) the determination of Cr(VI). Fig. 3 Optimization of the complexation of Cr (III) and TTA, measured by the effect on Cr signal: a) TTAconcentration; b) solution pH;c) reaction time; d) atomization temperature ofCr(VI). Conditions: Cr (III) andCr(VI), 10 pg L"(each); pyrolysis temperature, 1200 ℃; when optimized one parameter (i.e., TTA), othercondition set a the fixed optimum value, respectively (i.e.,pH, 6.0; complex formation time, 100min; atomizationtemperature,2400℃). Analytical performance Calibrations were carried out using aqueous standard solutions with a linear range of 0.5 to 10 ugL"of Cr(VI) and total Cr (Cr(T)), respectively. Calibrate curve for Cr(VI): Abs =-0.0016+0.0102 Conc.Cr(VI),r=0.9992.Calibrate curve for Cr(T): Abs=-0.0013+0.0104 Conc. Cr(T),r=0.9994. The Cr(III) concentration was established by calculating the difference between theCr(T) and Cr(VI) concentrations. The characteristic mass (mo), defined as the mass of analytecorresponding to 0.0044 absorbance units, is 8 pg. The detection limits for Cr(VI) and Cr(T) are 0.046 ug L’ and 0.039 ug L, which were calculated as the three times of the relative deviation ofthe 11 consecutive measurements of the blank solutions (0.2% HNO3 for Cr(T) and 1.5% TTA forCr(VI)) and the blank values are 0.045 and 0.031 ug L for Cr(VI) and total Cr, respectively. Thelimit of quantitation (LOQ) for Cr(VI) and Cr(T) are 0.15 ug L and 0.013 ug L, which werecalculated as the ten times of the relative deviation of the 11 consecutive measurements of theblank solutions (0.2% HNO3 for Cr(T) and 1.5% TTA for Cr(VI)).The relative standarddeviations (RSDs) for 1.0 g L"of Cr(VI) and Cr(T) of 3.1% and 2.8% (n=5), respectively. Table2 also shows that the LODs of Cr species are comparable with the results of the method ofchromatographic separation. Aparna et al. 26 used a sulphate-form of Dowex-1 as the ion exchangeresin to selectively adsorb Cr(VI) selectively, eluting out 99% Cr(III) for seawater determination.The absorbed Cr(VI) is eluted using 2M HNO3 and the residual Cr(III) is preconcentrated bycoprecipitation with Fe(OH)3 for ET-AAS determination 2. Wolf et al." used a reversed-phaseion-pairing liquid chromatography to separate the Cr species and ICP-MS determination for Crspecies in soil leachates.Cr(III) is complexed with ethylenediaminetetraacetic acid (EDTA) priorto separation by mixing samples with the mobile phase containing 2.0 mM tetrabutylammoniumhydroxide (TBAOH), 0.5 mM EDTA (dipotassium salt), and 5%(vol/vol) methanol, adjusted topH 7.6." Cuello et al. 28 used an Agilent 1100 HPLC with a PLRP-S 100A polymeric reversedphase column (150 mm ×4.6mm,3 um) to separate the Cr(VI) and Cr(ⅢII) in cigarette smokecondensate, and the outlet of column was connected directly via PEEK tubing to ICP-MS.Meeravali et al. 2 used Triton X-114 micelle and cetylpyridinium bromide-Triton X-114 mixed-micelle to extract sequentially the hydrophobic Cr(III)-trifluoropentanedione and hydrophilicCr(VI),respectively, and then ICP-OES was used for its determination. This micelle cloud phaseextraction and ICP-OES offline detection method successfully used to analyzed wastewater andnatural water 2. Chen et al. 30 developed a method for the speciation of Cr(III) and Cr(VI) inenvironmental water samples by solid phase extraction (SPE) using a micro-column (20 mm ×4.0mm, i.d.) packed with chitosan chemically modified mesoporous silica and determination by ICP-OES. Only Cr(III) can be selectively adsorbed on the micro-column in the pH range of 5-9, whileCr(VI) cannot be retained and passes through the column directly.3Cr(VI) concentrations wereobtained as the respective differences between total Cr and Cr(III). Table 3 shows the analysisresults for Cr species of the two water SRMs. Results for Cr(VI) for GBW(E)080403a andGBW(E)080404b are 9.06 and 18.7 ug L,respectively, which are in good agreement with thereference values measured by IC-ICP-ORS-MS method. Although no reference values for Cr(VI) are available, no difference was observed between the reference concentration and our detectedvalues for Cr(T). Table 2 Comparison of limits of detection of proposed method with reported methods in the literatures fordetermination of Cr species Method“a Separation scheme Cr(VI) Cr(III) or Cr (T)" LOD, ug L -1 LOD, ug L -1 IS-ETAAS (This method) In situ heat separation 0.046 0.039 IC offline ETAAS‘ 26 Ion exchange 0.037 0.049 IC-ICP-DRC-MS 27 Ion chromatographic 0.06 0.09 IC-ICP-ORS-MS 28 Ion chromatographic 0.08 0.12 Micelle CPE ICP-OES 29 Micelle CPE 0.05 0.02 SPE offline ICP-OES 30 Solid phase extraction / 0.01 Abbreviations for method: IS-ETAAS (In situ-electrothermal atomic absorption spectrometry); IC offlineETAAS (Ion exchange offline electrothermal atomic absorption spectrometry); IC-ICP-DRC-MS (Ionchromatography-inductively coupled plasma-dynamic reaction cell-mass spectrometry); IC-ICP-ORS-MS (Ionchromatography-inductively coupled plasma-octopole reaction system-mass spectrometry); Micelle CPE ICP-OES(Micelle cloud phase extraction inductively coupled plasma-optical emission spectrometry); SPE offline ICP-OES(Solid phase extraction offline inductively coupled plasma-optical emission spectrometry); Cr (T): total Cr; Non-available. Table 3 Cr species and total Cr in two water standard reference materials (SRMs), n=3 SRMs This method, ugL ReferenceIC-ICP-ORS-MS , ugL value, ugL Cr(VI) Cr(III) Cr(T) Cr(VI) Cr(III) Cr(T) Cr(T) GBW(E)080403a 9.06=0.18 0.95 10.1=0.2 9.02+0.12 1.09+0.13 10.2+0.1 10.0=0.1 GBW(E)080404b° 18.7±0.27 1.4 20.1+0.3 17.8±0.31 1.80=0.10 19.8=0.4 20.0=0.8 Dilution of 50-fold for the original SRM; 'Dilution of 100-fold for the original SRM. Water sample analysis The proposed method was applied for the determination of Cr(III) and Cr(VI) concentrations inthree samples of drinking waters. As shown in Table 4, the Cr(VI) for tap water, bottled water and well water are 1.41, 0.53, and 0.68 ug L, respectively. They are in good agreement with thereference values measured by ion exchange-inductively coupled plasma-octopole reaction system-mass spectrometry (IC-ICP-ORS-MS). Our results for Cr(VI) (0.53-1.41 ug L) and Cr(T) (1.37-2.21 ug L) are within the range of results previously published by other researchers for drinkingwater from US, Japan, Slovakia, Poland, and China (0-2.7 ug L"for Cr(VI) and 1.06-4.60 pg Lfor Cr(T)) 3,19,31,33. Table 3 Cr species and total Cr in two water standard reference materials (SRMs), n=3 SRMs This method, ug L ReferenceIC-ICP-ORS-MS, ug Lvalue, ugL Cr(VI) Cr(III) Cr(T) Cr(VI) Cr(III) Cr(T) Cr(T) GBW(E)080403a 9.06±0.18 0.95 10.10.2 9.02+0.12 1.09±0.13 10.2±0.1 10.0=0.1 GBW(E)080404b° 18.7=0.27 1.4 20.1+0.3 17.8+0.31 1.80=0.10 19.8=0.4 20.0=0.8 Dilution of 50-fold for the original SRM; 'Dilution of 100-fold for the original SRM. Conclusions TTA was used to selectively react with Cr(III), and the product of Cr(III)-TTA complex wasremoved from the commercial graphite furnace at 400℃, while Cr(VI) quantitatively remains (>93%) up to a temperature of 1200℃, for subsequent detection by AAS. The simple andeconomical in-situ method is suitable for the direct screening of trace levels of Cr species in watersamples with no chemical separation; and high throughput, and good sensitivity, precision andaccuracy. References 1 A. Zhitkovich, Chem. Res. Toxicol.,2005,18,3-11. ( 2Y.S. Fung and W. C. S h am, Analyst, 1994, 31, 1029-1032. ) 3 R. Karosi, V. Andruch, J. Posta and J. Balogh, Microchem.J., 2006,82,61-65. 4 People’s Republic of China Ministry of health, GB 5749-2006 Standards for drinking water quality,revised in 2006. ( 5 N. N. Meeravali , K. Madhavi and S. J. Kumar, J. Anal. At. Spectrom., 2016,31,1582-1589. ) 6 S. M. Yousefi and F. Shemirani,J. Hazard. Mater,2013,254-255,134-140. 7 H. Y. Peng, N. Zhang, M. He, B. B. Chen and B. Hu, Talanta, 2015,131,288-272. 8M. Soylak and N. Kizil, Atom. Spectrosc.,2013,36,216-220. 9 E. Leese, J. Morton, P. H. E. Gardiner and V. A. Carolan,J. Anal. At. Spectrom., 2016,31,924-933.10 C. Winter and A. Seubert, J. Anal. At. Spectrom.,2016,31,1262-1268.11 H. Y. Cheng, P. Li, J.H. Liu and M. Y. Ye, J. Anal. At. Spectrom., 2016, DIO: 10.1039/C6JA00206D. 12 B. Markiewicz, I Komorowicz, A. Sajnog, M. Belter and D. Baralkiewicz, Talanta, 2015, 132,814-828. 13 F. Hernandez, F. Seby, S. Millour, L. Noel, T. Guerin, Food Chem., 2017, 214,339-346. 14 A. Gonzalvez, M. L. Cervera, S. Armenta, M. de la Guardia, Anal. Chim. Acta, 2009, 636,129-157. 15 M. A. Vieira, P. Grinberg, C. R. R. Bobeda, M. N.M. Reyes, R. C. Campos, Spectrochim. Acta B,2009,64,459-476. 16 A. Gonzalvez, S. Armenta, M. L. Cervera and M. de la Guardia, TRAC-Trend. Anal. Chem., 2010, 29,260-268. ( 17 P. B ermejo-Barrera, M . C.Barciela-Alonso, B. Perez-Fernandez and A . Bermejo-Barrera, Spectrochim Acta Part B, 2003, 58, 167-173. ) ( 18 E. W. Berg and J. T. Truemper, Anal.Chim. Acta, 1 9 65, 32,245-252. ) ( 19X.S. Zhu, B . Hu, Z. C. Jiang, Y. L. Wu and S. Xiong, Anal. Chim. Acta, 2002,471,121-126.20 W. Guo, P . Zhang, L. L. Jin and S . H . H u , J. Anal. At. Spectrom., 2014, 2 9 ,19 4 9-1954. ) ( 21 H . Cui , W . G uo, M. T . Cheng, P . Zhang, L . L. Jin, Q. H. Guo and S. H . H u, Anal. Methods, 2015, 7 , 8970-8976. ) ( 22 Y. E. Peng, W. Guo, P. Zhang and L. L. Jin, J. Food Compos. Anal.,2015, 4 2, 78 - 83. ) ( 23 Y. Yang, Y. Peng, Q. Chang, C. Dan, W. Guo and Y. Wang, Anal. Chem.,2016,88,1275-1280. 24 S. Arpadjan andV. Krivan, Anal. Chem., 1986,58,2811- 2 814. ) ( 25 E . B eceiro-Gonzalez, J. B arciela-Garcia, P. Bermejo-Barrera a nd A. Bermejo-Barrera, F r es. J. A nal. Chem., 1 992,344,301-305. ) ( 26 A. A parna, S. S umithra, G. V enkateswarlu, A. C . S ahayam, S. C. Chaurasia a n d T. Mukherjee, Atom.Spectrosc.,2006,27,123-127. ) ( 27 R. E. Wolf, J. M. Morrison and M. B. Goldhaber, J. Anal. At. Spectrom.,2007,22,1 0 51-1 06 0. ) ( 28 S . Cuello, J. Entwisle, J . Benning, C. Liu, S. Cobur n , K. G . McAdam, J. Braybrook and H. Goenaga- Infante,J. Anal. At. Spectrom., 2016, DOI: 10.1039/C5JA00442J. ) ( 29 N. N . Meeravali, K . M a dhavi and S. J . Ku m ar, J. Anal. At. Spectrom., 2011, 2 6 , 2 14- 2 19.30 D. H. Chen, M . H e, C. Z. Huang and B. H u, Atom. Spectrosc.,2008,2 9 ,165-17 1 . ) ( 31 S. I. Ohira , K . Nakamura, C. P. Shelor, P . K . Dasgupta and K. Tod a , Ana l . Chem., 201 5 ,87 , 11575- 11580. ) ( 32 B. Markiewicz, I. Komorowicz and D. B a ralkiewicz, T a lanta, 2002,152,489-497. ) 本文巧妙地利用Cr(III)和TTA 的特异性结合形成易挥发的金属络合物,在石墨炉升温过程中Cr(III)优先挥发出来,而检测Cr(VI)。如图1,在1200℃将90%以上的Cr(III)与Cr(VI)分开,直接检测Cr(VI),总铬差减法计算出Cr(III)。Cr(VI)方法检出限为0.0736ng/mL。
确定






还剩10页未读,是否继续阅读?

产品配置单

珀金埃尔默企业管理(上海)有限公司为您提供《水中Cr(VI)和Cr(III)检测方案(原子吸收光谱)》,该方案主要用于饮用水中(类)金属及其化合物检测,参考标准--,《水中Cr(VI)和Cr(III)检测方案(原子吸收光谱)》用到的仪器有珀金埃尔默原子吸收光谱仪PerkinElmer PinAAcle 900
推荐专场





相关方案
更多
该厂商其他方案
更多