








数据非依赖性采集(DIA)是随着定量蛋白质组学而建立的质谱扫描技术。 DIA 能够获得扫描范围内所有母离子及二级子离子信息,不会造成低丰度离子信息的丢失,同时突破了高分辨质谱二级定量的通量限制。 本研究基于静电场轨道阱 Q-qIT-OT 三合一质谱,发展了经典 DIA 方法以及 WiSIM-DIA 和 Full MS-DIA两种全新 DIA 方法,并对 Hela 细胞全蛋白中添加的 10 条低浓度肽段进行定量分析,考察方法的线性、重现性和灵敏度。 结果表明,3 种方法的定量限均低至 amol (14 ~435 amol),并展示出良好的线性和定性确证可靠性。 其中,WiSIM-DIA 基于超高分辨一级监测定量,与经典 DIA 优势互补;Full MS鄄DIA 的选择窗口仅 3 amu,
能够直接进行搜库鉴定,实现了数据依赖性采集(DDA)和 DIA 的统一,摆脱了 DIA 依赖于 DDA 建立谱图库的局限性。
方案详情

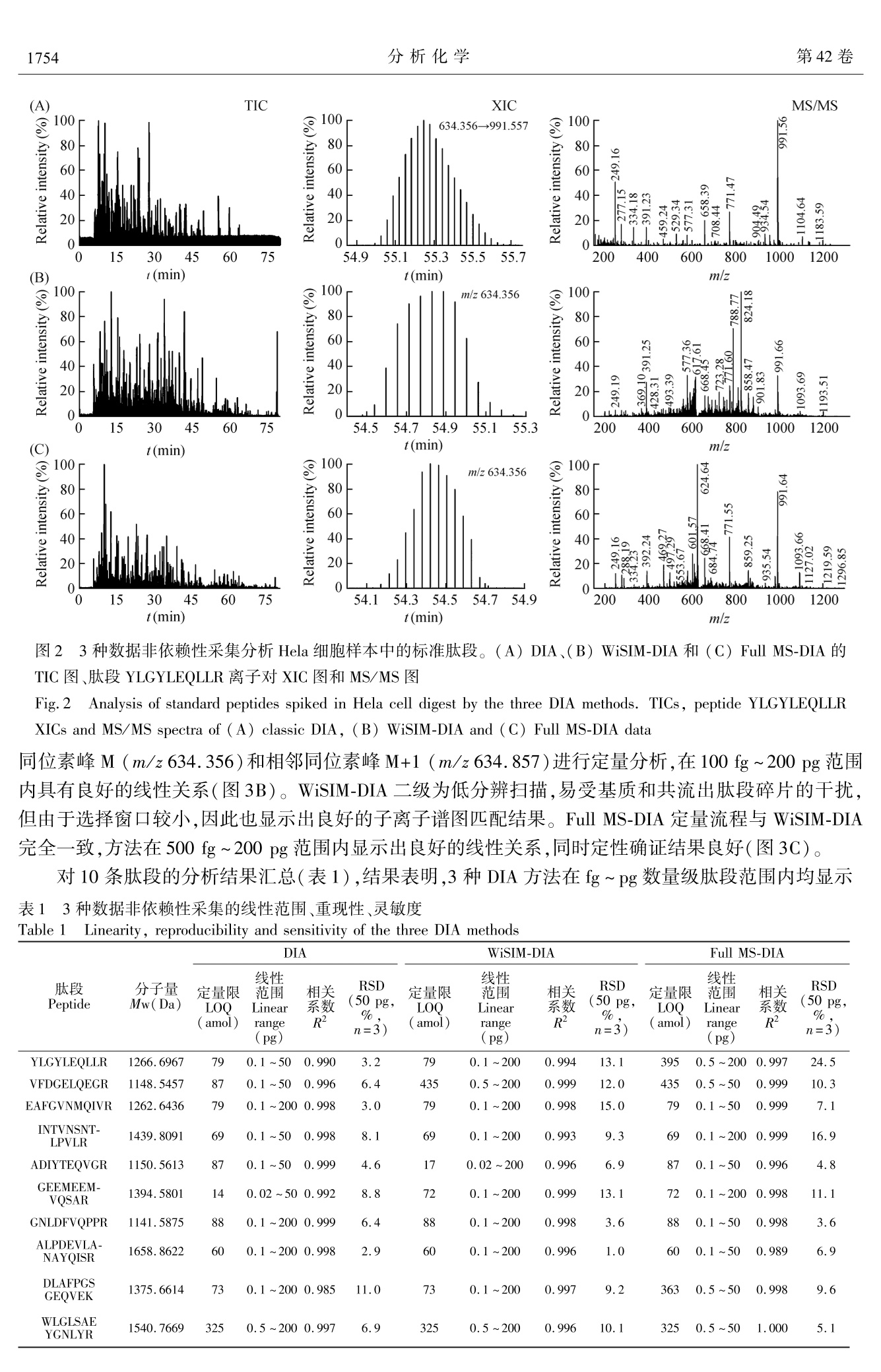
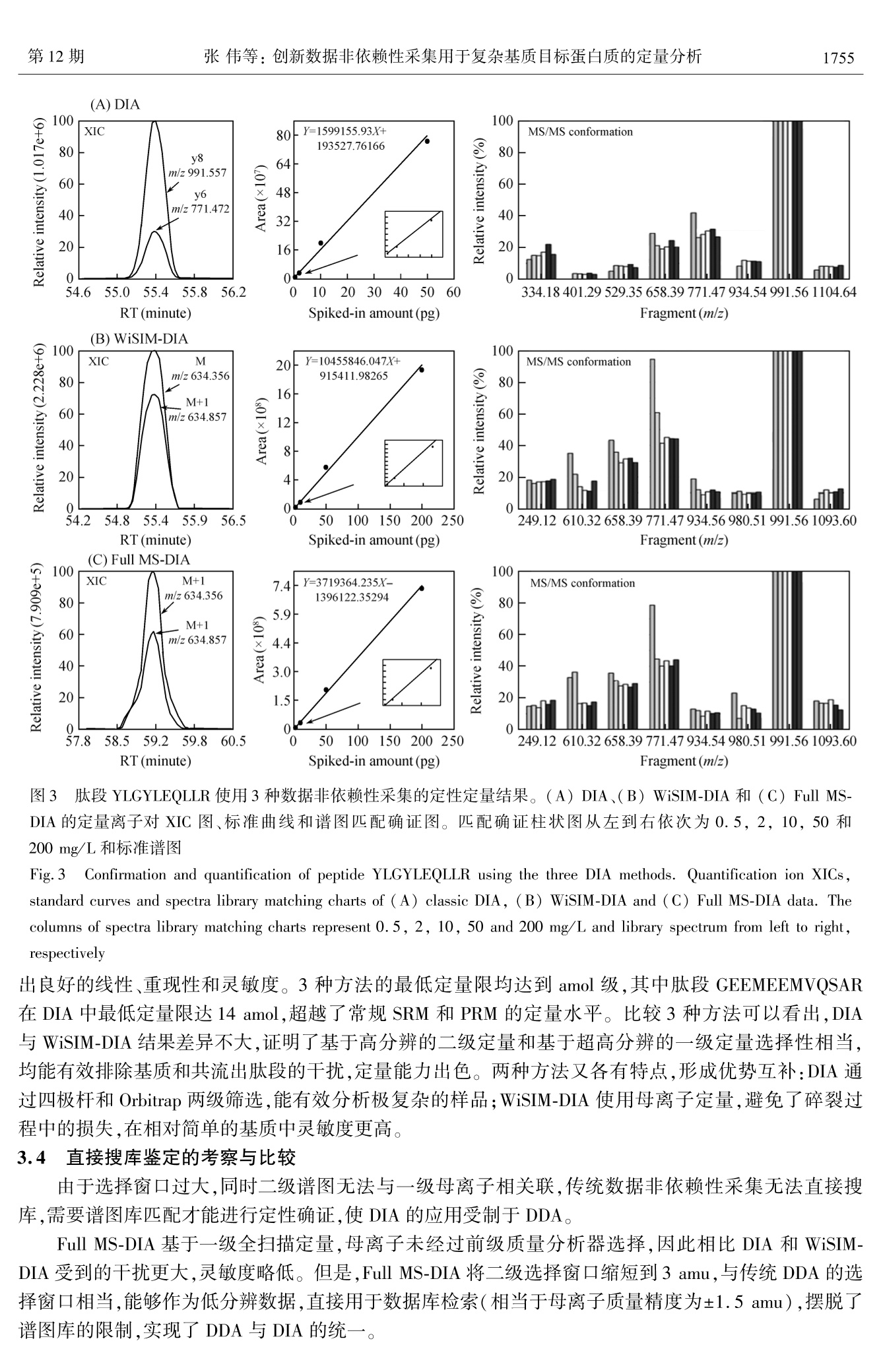
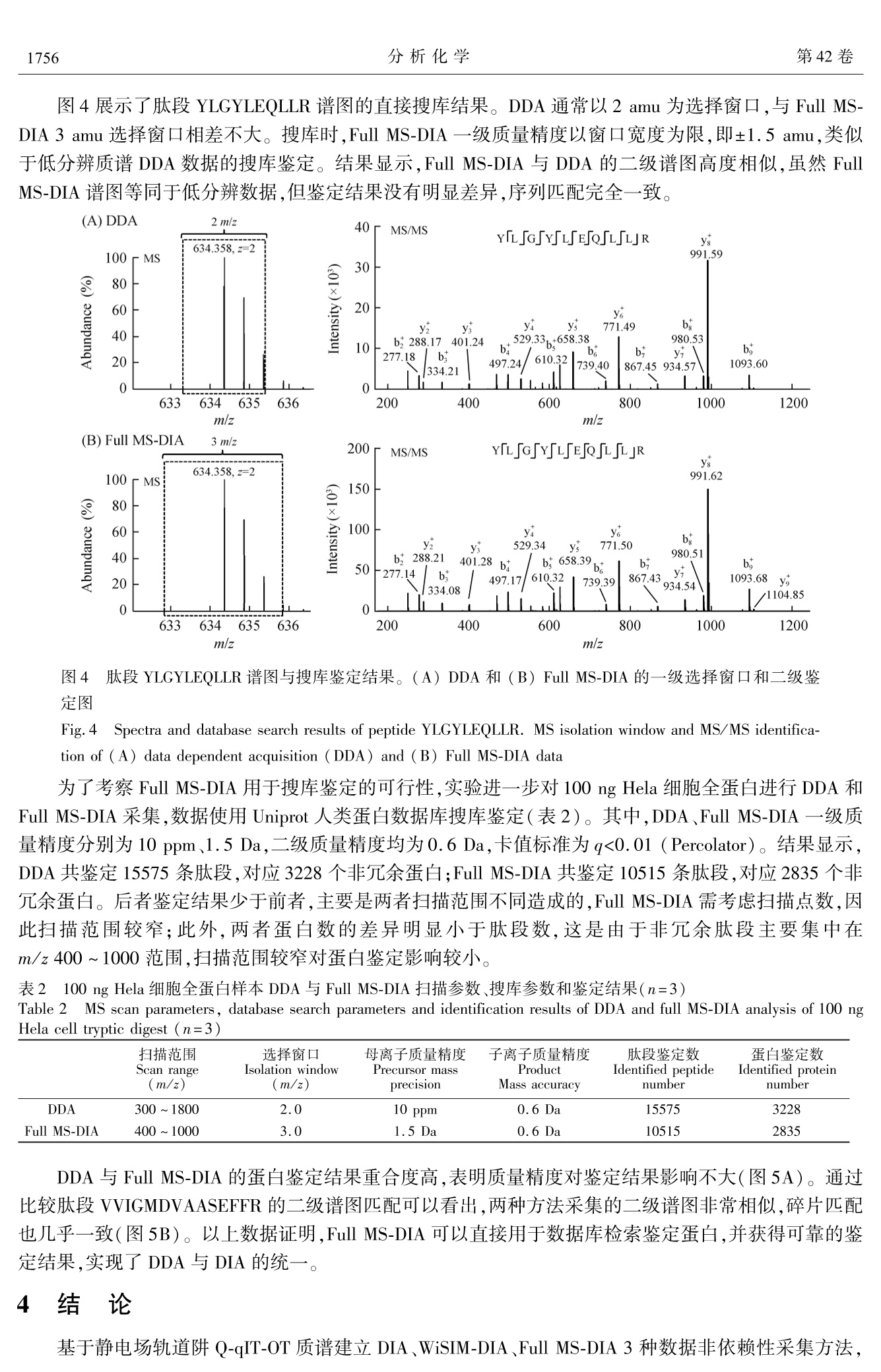
分析化学 (FENXI HUAXUE))研究报告Chinese Journal of Analytical Chemistry第12期1750~1758第42卷2014年12月 张伟等:创新数据非依赖性采集用于复杂基质目标蛋白质的定量分析第12期1751 DOI:10.11895/j.issn.0253-3820.140365 创新数据非依赖性采集用于复杂基质目标蛋白质的定量分析 张伟 Reiko Kiyonami 江峥*1 陈伟 (赛默飞世尔科技(中国)有限公司,上海201206) (ThermoFisher Scientific, San Jose, CA, USA) 摘 要 数据非依赖性采集(DIA)是随着定量蛋白质组学而建立的质谱扫描技术。DIA 能够获得扫描范围内所有母离子及二级子离子信息,不会造成低丰度离子信息的丢失,同时突破了高分辨质谱二级定量的通量限制。本研究基于静电场轨道阱 Q-qIT-OT 三合一质谱,发展了经典 DIA 方法以及 WiSIM-DIA 和 Full MS-DIA两种全新 DIA 方法,并对 Hela 细胞全蛋白中添加的10条低浓度肽段进行定量分析,考察方法的线性、重现性和灵敏度。结果表明,3种方法的定量限均低至 amol (14~435 amol),并展示出良好的线性和定性确证可靠性。其中, WiSIM-DIA 基于超高分辨一级监测定量,与经典 DIA优势互补;Full MS-DIA 的选择窗口仅3 amu,能够直接进行搜库鉴定,实现了数据依赖性采集(DDA)和 DIA 的统一,摆脱了 DIA 依赖于 DDA 建立谱图库的局限性。 关键词 静电场轨道阱;数据非依赖性采集;蛋白质组学;绝对定量 引 言 数据依赖性采集( Data dependent acquisition, DDA)是串联质谱非目标化合物分析的主要手段。蛋白质组学的经典策略-鸟枪法(Shotgun)即基于 DDA 发展而来,利用一级全扫描检测肽段母离子,然后按信号强度排列,将前若干位的母离子依次选择、碎裂,并扫描二级碎片离子。同时,动态排除、价态排除、中性丢失/诊断离子触发等技术,使 DDA 尽可能多地采集有效肽段谱图,实现鉴定结果最大化11。基于鸟枪法,蛋白质组学已经实现酵母蛋白质组接近完全覆盖,人类蛋白质组也已达到50%以上的基因组覆盖和7个数量级的动态范围[2.3]。然而,DDA 的局限性也逐渐显现:(1)先强后弱的采集方式易造成低丰度肽段信息丢失;(2)母离子选择有一定的随机性,造成重现性不佳;(3)每个循环获得的谱图数量不一,造成扫描点数不均匀,影响定量分析准确性。 目标蛋白质组学针对目标蛋白/肽段离子实时监测和采集,避免了 DDA 的信息丢失和重现性问题。主要采集方法包括选择离子监测(Selected ion monitoring, SIM)、基于三重四极杆的选择反应监测(Selectedreaction monitoring, SRM)和基于高分辨质谱的平行反应监测(Parallel reaction monitoring, PRM),是目标蛋白验证和绝对定量的有效手段[4.5]。但是目标性的采集方法需要指定目标肽段,对于未知肽段无法采集;通量限制也使得一次实验只能监测数量有限肽段或离子对,难以满足大规模蛋白分析的需要。 数据非依赖性采集(Data independent acquisition, DIA)使用25 amu 或更大间隔将整个质量范围等分为若干窗口,每个窗口依次选择、碎裂、扫描。DIA 能够获得质量范围内所有母离子的全部碎片离子信息,通量无上限,循环时间固定,同时数据可以回溯,有效解决了 DDA 和目标采集方法存在的问题161。目前,已发展了多种基于飞行时间(Q-TOF)、静电场轨道阱( Orbitrap)和离子阱的 DIA方法’。 Gillet 等使用32个连续的25 amu 窗口,基于 Q-TOF (TripleTOF 5600)发展了 SWATH 技术,并证明该技术的定量能力与 SRM 相当[8]。 Egertson 等利用 Q-Orbitrap (Q Exactive )独有的多重扫描功能(Multiplexing,MSX)发展了 MSX-DIA 技术,将选择窗口缩小到4 amu,最大程度减少了共流出肽段和杂质的干扰。 然而,传统数据非依赖性采集仍存在诸多局限:(1)由于扫描速度的限制, DIA 难以使用超高分辨率扫描;(2) DIA 的大窗口选择引入较大干扰,虽然 MSX-DIA 缩小了选择窗口,但需要特定的算法解析数据,增加了工作量;(3)DIA依赖于 DDA 建立的谱图库进行匹配,实现定性确证和定量离子选择,因 ( 2014-04-24 收稿;2014-07-04接受 ) ( *E-mail: zheng. jiang@ thermofisher. com ) 此 DDA 鉴定不到的蛋白,DIA 也无法分析。 本研究基于四极杆-静电场轨道阱-线性离子阱(Q-0T-qIT)三合一质谱,利用添加10条低浓度肽段的 Hela样本,发展并考察了3种数据非依赖性采集方法,包括经典的 DIA、全新的宽窗口 SIM 扫描 DIA(WiSIM-DIA)和全扫描 DIA (Full MS-DIA),定量限均达到 amol 水平,线性、重现性良好。其中, WiSIM-DIA 和 Full MS-DIA 基于24万超高分辨率,利用一级精确质量数定量、二级离子阱定性确证,进一步缩小了选择窗口,提高了检测特异性。此外, Full-MS DIA 可以直接搜库,实现了 DDA 与 DIA 的统一,蛋白鉴定数量与 DDA 相当,摆脱了谱图库的限制。基于 Q-OT-qIT的数据非依赖性采集方法灵活多样、流程简单有效,在目标蛋白质组学领域具有广阔的应用前景。 2 实验部分 2.1 仪器与试剂 Orbitrap Fusion 三合一质谱仪、EASY-nLC 1000 纳流超高效液相色谱(Thermo Fisher Scientific)。标准肽段由生工生物(上海)股份有限公司合成;乙腈、司酸(Thermo Fisher Scientific);其它试剂均购自Sigma-Aldrich 公司。 2.2 Hela 细胞全蛋白酶解液制备 取适量 Hela 细胞沉淀,加入含7 mol/L 尿素、2 mol/L 硫脲、1 mmol/L苯甲基磺酰氟(PMSF)和50 mmol/L 二硫苏糖醇(DTT)的蛋白提取液,在超声细胞破碎仪中超声3次各5s。冰上放置20 min后,4℃离心(15000 g) 30 min, 取上清液,Bradford 法确定总蛋白浓度。向蛋白提取液中加入 DTT (终浓度10 mmol/L),于56℃振荡30 min。加入碘乙酰胺(终浓度50 mmol/L),室温避光振荡 40 min。反应后加加6倍体积的预冷丙酮,-20℃放置3h,4℃离心(15000 g)30 min, 弃上清液。将蛋白沉淀溶于50 mmol/L NH HCO,溶液,加入胰蛋白酶 Trypsin(酶:蛋白为1:40,w/w),37℃酶解过夜。将全蛋白酶解液稀释至终浓度为100 mg/L。 2.3 标准肽段添加 等重称取10条标准肽段粉末,混合后使用0.1%甲酸溶液充分溶解为 1 g/L 肽段混合溶液(总浓度10 g/L)。肽段混合溶液进一步使用100 mg/L Hela 酶解液逐级稀释为7种浓度梯度(200,50,10,2,0.5,0.1 和0.02 mg/L)溶液。另取少量标准肽段溶液用水稀释至500 mg/L, 用作 PRM 扫描,构建谱图库。 2.4 纳流液相色谱方法 色谱柱:自制纳流 Cig色谱柱(3 pm, 100 A, 75 pum x 15 cm);上样量:1uL;流速:300 nL/min;A相:0.1%甲酸溶液;B相:0.1%甲酸-乙腈溶液;分析梯度:0~4 min,3%~10% B;4~74 min, 10%~35% B;74~78 min,35%~90% B;78~90 min,90% B。 2.5 质谱方法 离子源:nano-Flex 纳喷雾离子源;扫描模式:正离子;喷雾电压:2.2kV;离子传输管温度:275℃;RF-lens:60%。 DDA 扫描:一级扫描模式:全扫描;一级扫描范围:m/z300~1800;一级检测:Orbitrap(分辨率240 K);选择模式:四极杆;选择窗口:2 amu;碎裂模式:CID;碎裂能量:30%;二级检测:离子阱;动态排除:60s;循环时间:3 s。 PRM 扫描:母离子列表:10条标准肽段的精确质量数;选择模式:四极杆;选择窗口:2amu;碎裂模式:HCD;碎裂能量:30%;二级检测:Orbitrap(分辨率30K);循环模式:按母离子列表依次扫描。 DIA 方法:母离子列表:m/z510~890, 间隔20 amu;选择模式:四极杆;选择窗口:20 amu;碎裂模式:HCD;碎裂能量:30%;二级检测:Orbitrap(分辨率30K);二级扫描范围:m/z150~2000;循环模式:按母离子列表依次扫描。 WiSIM-DIA 方法:一级扫描模式:SIM;一级选择模式:离子阱;一级扫描范围:m/z 400~600, m/z600~800, m/z 800~1000;一级检测:Orbitrap(分辨率240K);母离子列表:m/z 406~598, m/z 606~798, m/z 806~998间隔12 amu;二级选择模式:四极杆;选择窗口:12 amu;碎裂模式:CID;碎裂能量: 30%;二级检测:离子阱;二级扫描范围:m/z150~2000;循环模式:3个 SIM+3个母离子列表依次扫描。Full MS-DIA 方法:一级扫描模式:全扫描;一级扫描范围: m/z 400~1000;一级检测:Orbitrap(分辨率240K);母离子列表:m/z401.5~518.5, m/z 521.5~638.5, m/z 641.5~758.5,m/z 761.5~878.5,m/z 881.5~998.5,间隔3 amu;选择模式:四极杆;选择窗口:3 amu;碎裂模式:CID;碎裂能量:30%;二级检测:离子阱;二级扫描范围:m/z150~2000;循环模式:5个一级全扫描+5个母离子列表依次扫描。 2.6 数据处理 标准肽段 PRM数据使用 Proteome Discoverer 1. 4软件进行搜库鉴定,母离子质量精度:10 ppm,子离子质量精度:0.02 Da。鉴定结果作为谱图库进行 DIA 的定性确证和定量离子挑选。DIA 数据使用Pinpoint 1.4软件进行谱图库离子筛选、定性确证、定量分析和标准曲线绘制。质量精度设置同上,母离子定量选择单同位素峰和第一个同位素峰,子离子定量选择强度最高的2个y离子,定性确证选择强度最高的8个子离子。100 ng Hela 细胞全蛋白 DDA 和 Full MS-DIA 数据使用 Proteome Discoverer 1.4 软件进行 Uniprot 人类蛋白数据库搜库鉴定与比较,质量精度设置同上(Full MS-DIA 母离子质量精度设置为1.5 Da),可变修饰:甲硫氨酸氧化(M+15.995 Da),固定修饰:半斗氨酸脲甲基化(C+57.021 Da)。鉴定结果使用 Percolator 进行严格卡值(q<0.01)。 3 结果与讨论 3.1 基于Q-qIT-OT的创新数据非依赖性采集策略的建立 数据非依赖性采集依次扫描整个质量范围的所有母离子及二级子离子,循环时间长,因此对质谱扫描速度要求高。目前,主要的 DIA 方法的质谱扫描速度均在10 Hz 左右,例如 SWATH 等,因此采用较大选择窗口(25~50 amu),以缩短循环时间(约2.5~3.2s)。Q-qIT-OT三合一质谱集四极杆、线性离子阱和 Orbitrap 3 种质量分析器为一体,Orbitrap 扫描速度达到15 Hz,线性离子阱达到20 Hz,为缩短传统 DIA 的选择窗口、降低干扰、提高灵敏度提供了可能10]。 本研究首先基于四极杆和 Orbitrap,建立了经典 DIA 方法(图1A):使用20 amu 选择窗口,利用Orbitrap 30K 高分辨率和 HCD 高能碎裂,依次选择、碎裂、扫描整个质量范围(m/z500-900),循环时间仅为1.9s,数据处理时挑选强度最高的子离子组成母子离子对进行色谱峰提取和定量。此外,利用线性离子阱的高灵敏度和高扫描速度,建立了两种基于一级定量的新方法:宽窗口 SIM 扫描 DIA (WiSIM-DIA)和全扫描 DIA (Full MS-DIA)。 WiSIM-DIA(图1B)利用200 amu 宽窗口筛选母离子,在 m/z 400~1000 范围依次使用 Orbitrap 进行240K超高分辨离子监测扫描,最大程度排除基质干扰,基于一级精确质量数进行定量。在每个 SIM扫描之后,利用12 amu 窄窗择选择母离子,在质量范围内依次进行线性离子阱二级碎片离子扫描,基于二级谱图实现定性确证。 Full MS-DIA(图1C)在 WiSIM-DIA 基础上将12 amu 选择窗口缩短到3 amu,达到与 DDA 相当的选择窗口,但扫描窗口数量增加了4倍。为了避免循环时间过长对定量造成的影响,在 m/z 400~1000的质量范围内插入5个 Orbitrap 240 K 超高分辨一级全扫描以保证扫描点数。Full MS-DIA一级扫描间隔仅2.6s,保证了定量结果的可靠性。 肽段纳流液相分离的出峰时间一般在30s以上,因此在合适的循环时间内,3种 DIA 方法可以根据样本复杂程度灵活调节质量范围、分辨率、选择窗口等参数,达到最佳分析效果。例如,分析分子量较大的肽段时,质量范围可以调整为 m/z 400~1200 或更宽。 3.2 复杂基质加标样本的数据采集与分析 为了考察与比较3种 DIA 方法的分析效果,实验对 Hela 细胞全蛋白酶解液中添加的10条低浓度肽段进行了数据采集。10条合成的标准肽段均来自于非人源的植物蛋白,分子量在1100~1700 Da 之间,具有高度特异性。标准肽段溶液使用 100 mg/L Hela 细胞酶解液逐级稀释为7种浓度梯度,并逐一使用3种 DIA 方法采集(上样量1p.L),考察方法的选择性、线性、重现性和灵敏度。 图2展示了50 mg/L 浓度肽段样本的总离子流图(TIC)、目标肽段 YLGYLEQLLR (m/z 634.356, 图1 基于 Q-qIT-OT的3种数据非依赖性采集原理。(A)经典数据非依赖性采集 DIA;(B)宽窗口 SIM 扫描DIA(WiSIM-DIA);(C)全扫描 DIA (Full MS-DIA) Fig. 1 Schematic illustrating quadrupole-ion-orbitrap (Q-qIT-OT) based three data-independent acquisition (DIA)methods. (A) Classic DIA, (B) Wide isolation window SIM scan DIA (WiSIM-DIA), (C) Full scan DIA (Full MS-DIA) 2+)的提取离子流图(XIC)和子离子谱图(MS/MS)。由于 Hela 细胞样本高度复杂,标准肽段的色谱峰在 TIC 中被完全掩盖。另外,相比 WiSIM-DIA 和 Full MS-DIA, DIA 只有二级扫描没有一级扫描,因此TIC 均由二级谱图构成,信噪比相对较低。进一步提取该肽段的 XIC 图,其中 DIA 基于子离子定量,选择最强子离子 m/z 991.557 形成634.356~991.557 母子离子对提取色谱峰; WiSIM-DIA 和 Full MS-DIA基于母离子定量,利用母离子精确质量数 m/z 634.356提取色谱峰。通过比较 XIC 可以看出,在复杂Hela 细胞基质中,3种方法均能有效排除干扰。 肽段 YLGYLEQLLR 的出峰时间约为 35 s,3 种 DIA 在出峰时间内扫描点数均达到10以上,分别为22,14,14,具有良好的色谱峰型和可靠的定量峰面积。 MS/MS 谱图显示了3种选择窗口下二级碎片离子的扫描结果。DIA 的选择窗口最大(20 amu),质谱峰最多,但 Orbitrap 高分辨扫描能够有效区分碎片离子,因此谱图具有良好的信噪比,可以有效用于定性与定量分析。 WiSIM-DIA 窗口缩短为12 amu,但属于线性离子阱低分辨扫描,因此质谱峰的区分能力相对较弱,造成一定干扰。Full MS-DIA 窗口仅3 amu,与 DDA 相当,把基质干扰降到了最低,提供了良好的定性确证信息。 3.3 方法的线性、灵敏度的考察与比较 数据进一步使用 Pinpoint 1.4 软件处理,考察方法在100 ng 复杂 Hela 细胞基质下,20~200 pg低浓度肽段范围内的线性、重现性和灵敏度。并以10条标准肽段的 PRM 数据鉴定结果作为谱图库,进行谱图匹配和定性确证。DIA 选取标准谱图中强度最高的8个子离子作为定性确证依据,并提取其中最好的2个y离子进行定量分析。WiSIM-DIA 和 Full MS-DIA 同样选取最强的8个子离子进行定性确证,定量分析则使用一级母离子的单同位素峰和相邻的第一个同位素峰。 以肽段 YLGYLEQLLR (m/z 634.356,2+)为例,DIA 数据选取最强的 y8 (m/z 991.557) 和y6 (m/z 771.472)作为定量离子对,绘制标准曲线(图3A)。曲线在100 fg~ 50 pg范围内显示出良好的线性,同时,8个定性确证离子与谱图库中的标准谱图高度匹配,强度比例一致。WiSIM-DIA 选取单 TIC XIC MS/MS 图23种数据非依赖性采集分析 Hela 细胞样本中的标准肽段。(A) DIA、(B) WiSIM-DIA 和(C) Full MS-DIA 的TIC 图、肽段 YLGYLEQLLR 离子对 XIC 图和 MS/MS图 Fig.2Analysis of standard peptides spiked in Hela cell digest by the three DIA methods. TICs, peptide YLGYLEQLLRXICs and MS/MS spectra of (A) classic DIA, (B)WiSIM-DIA and (C) Full MS-DIA data 同位素峰 M (m/z 634.356)和相邻同位素峰 M+1 (m/z 634.857)进行定量分析,在100 fg~ 200 pg范围内具有良好的线性关系(图3B)。WiSIM-DIA二级为低分辨扫描,易受基质和共流出肽段碎片的干扰,但由于选择窗口较小,因此也显示出良好的子离子谱图匹配结果。Full MS-DIA 定量流程与 WiSIM-DIA完全一致,方法在500 fg~200 pg 范围内显示出良好的线性关系,同时定性确证结果良好(图3C)。 对10条肽段的分析结果汇总(表1),结果表明,3种 DIA 方法在fg~pg 数量级肽段范围内均显示表1 3种数据非依赖性采集的线性范围、重现性、灵敏度 Table 1 Linearity, reproducibility and sensitivity of the three DIA methods 肽段 Peptide DIA WiSIM-DIA Full MS-DIA 线性 RSD 分子量 Mw(Da) 定量限 LoQ (amol) 线性 范围 相关 Linear 系数 range R’ (Pg) RSD (50 pg, % n=3) 定量限 LOQ (amol) 线性 范围 Linear range (pg) 相关 系数 R RSD (50 pg, %. n=3) 定量限 LOQ (amol) 范围 Linear range (pg) 相关 系数 R (50pg, % n=3) YLGYLEQLLR 1266.6967 79 0.1~500.990 3.2 79 0.1~200 0.994 13.1 395 0.5~200 0.997 24.5 VFDGELQEGR 1148.5457 87 0.1~500.996 6.4 435 0.5~200 0.999 12.0 435 0.5~50 0.999 10.3 EAFGVNMQIVR 1262.6436 79 0.1~200 0.998 3.0 79 0.1~200 0.998 15.0 79 0.1~50 0.999 7.1 INTVNSNT- LPVLR 1439.8091 69 0.1~500.998 8.1 69 0.1~200 0.993 9.3 69 0.1~200 0.999 16.9 ADIYTEQVGR 1150.5613 87 0.1~500.999 4.6 17 0.02~200 0.996 6.9 87 0.1~50 0.996 4.8 GEEMEEM- VQSAR 1394.5801 14 0.02~50 0.992 8.8 72 0.1~200 0.999 13.1 72 0.1~2000.998 11.1 GNLDFVQPPR 1141.5875 88 0.1~200 0.999 6.4 88 0.1~200 0.998 3.6 88 0.1~50 0.998 3.6 ALPDEVLA- NAYQISR 1658.8622 60 0.1~200 0.998 2.9 60 0.1~200 0.996 1.0 60 0.1~50 0.989 6.9 DLAFPGS GEQVEK 1375.6614 73 0.1~2000.985 11.0 73 0.1~200 0.997 9.2 363 0.5~50 0.998 9.6 WLGLSAE YGNLYR 1540.7669 325 0.5~2000.997 6.9 325 0.5~200 0.996 10.1 325 0.5~50 1.000 5.1 图3 肽段 YLGYLEQLLR 使用3种数据非依赖性采集的定性定量结果。(A)DIA、(B)WiSIM-DIA 和(C) Full MS-DIA 的定量离子对 XIC 图、标准曲线和谱图匹配确证图。匹配确证柱状图从左到右依次为0.5,2,10,50和200 mg/L和标准谱图 Fig.3 Confirmation and quantification of peptide YLGYLEQLLR using the three DIA methods. Quantification ion XICs,standard curves and spectra library matching charts of (A) classic DIA, (B) WiSIM-DIA and(C) Full MS-DIA data. Thecolumns of spectra library matching charts represent 0.5, 2, 10,50 and 200 mg/L and library spectrum from left to right,respectively 出良好的线性、重现性和灵敏度。3种方法的最低定量限均达到 amol 级,其中肽段 GEEMEEMVQSAR在 DIA中最低定量限达14 amol,超越了常规 SRM 和 PRM 的定量水平。比较3种方法可以看出, DIA与 WiSIM-DIA 结果差异不大,证明了基于高分辨的二级定量和基于超高分辨的一级定量选择性相当,均能有效排除基质和共流出肽段的干扰,定量能力出色。两种方法又各有特点,形成优势互补:DIA 通过四极杆和 Orbitrap 两级筛选,能有效分析极复杂的样品;WiSIM-DIA 使用母离子定量,避免了碎裂过程中的损失,在相对简单的基质中灵敏度更高。 3.4 直接搜库鉴定的考察与比较 由于选择窗口过大,同时二级谱图无法与一级母离子相关联,传统数据非依赖性采集无法直接搜库,需要谱图库匹配才能进行定性确证,使 DIA 的应用受制于 DDA。 Full MS-DIA 基于一级全扫描定量,母离子未经过前级质量分析器选择,因此相比 DIA 和 WiSIM-DIA 受到的干扰更大,灵敏度略低。但是,Full MS-DIA 将二级选择窗口缩短到3 amu,与传统DDA 的选择窗口相当,能够作为低分辨数据,直接用于数据库检索(相当于母离子质量精度为±1.5 amu),摆脱了谱图库的限制,实现了 DDA 与DIA 的统一。 图4展示了肽段 YLGYLEQLLR 谱图的直接搜库结果。DDA 通常以2 amu 为选择窗口,与Full MS-DIA 3 amu 选择窗口相差不大。搜库时,Full MS-DIA一级质量精度以窗口宽度为限,即±1.5 amu,类似于低分辨质谱 DDA 数据的搜库鉴定。结果显示,Full MS-DIA 与 DDA 的二级谱图高度相似,虽然FullMS-DIA 谱图等同于低分辨数据,但鉴定结果没有明显差异,序列匹配完全一致。 图4 肽段 YLGYLEQLLR 谱图与搜库鉴定结果。(A) DDA 和(B) Full MS-DIA 的一级选择窗口和二级鉴定图 Fig.4 Spectra and database search results of peptide YLGYLEQLLR. MS isolation window and MS/MS identifica-tion of (A) data dependent acquisition (DDA) and (B) Ful MS-DIA data 为了考察 Full MS-DIA 用于搜库鉴定的可行性,实验进一步对100 ng Hela 细胞全蛋白进行 DDA 和Full MS-DIA 采集,数据使用 Uniprot 人类蛋白数据库搜库鉴定(表2)。其中,DDA、Full MS-DIA 一级质量精度分别为 10 ppm、1.5 Da,二级质量精度均为0.6 Da,卡值标准为 q<0.01 (Percolator)。结果显示,DDA 共鉴定15575 条肽段,对应3228个非冗余蛋白;Full MS-DIA 共鉴定10515条肽段,对应2835个非冗余蛋白。后者鉴定结果少于前者,主要是两者扫描范围不同造成的,Full MS-DIA需考虑扫描点数,因此扫描范围较窄;此外,两者蛋白数的差异明显小于肽段数,这是由于非冗余肽段主要集中在m/z 400~1000范围,扫描范围较窄对蛋白鉴定影响较小。 表2 100 ng Hela 细胞全蛋白样本 DDA 与 Full MS-DIA 扫描参数、搜库参数和鉴定结果(n=3) Table 2 MS scan parameters, database search parameters and identification results of DDA and full MS-DIA analysis of 100 ngHela cell tryptic digest (n=3) 扫描范围 选择窗口 母离子质量精度 子离子质量精度 肽段鉴定数 蛋白鉴定数 Scan range Isolation window Precursor mass Product Identified peptide Identified protein (m/z) (m/z) precision Mass accuracy number number DDA 300~1800 2.0 10 ppm 0.6 Da 15575 3228 Full MS-DIA 400~1000 3.0 1.5 Da 0.6 Da 10515 2835 DDA 与 Full MS-DIA 的蛋白鉴定结果重合度高,表明质量精度对鉴定结果影响不大(图5A)。通过比较肽段 VVIGMDVAASEFFR 的二级谱图匹配可以看出,两种方法采集的二级谱图非常相似,碎片匹配也几乎一一(图5B)。以上数据证明,Full MS-DIA可以直接用于数据库检索鉴定蛋白,并获得可靠的鉴定结果,实现了 DDA 与 DIA 的统一。 4 结 论 基于静电场轨道阱Q-qIT-OT 质谱建立 DIA、WiSIM-DIA、Full MS-DIA 3 种数据非依赖性采集方法, (A) 图5 100 ng Hela 细胞全蛋白样本 DDA 与 Full MS-DIA 鉴定结果比较。(A)两种方法蛋白鉴定结果比较;(B)两种方法二级谱图匹配比较(肽段 VVIGMDVAASEFFR) Fig.5 Comparison of identification results of 100 ng Hela cell digest by DDA and Full MS-DIA methods. (A)Comparison of identified number of proteins, (B) Comparison of identified MS/MS spectra (peptide VVIGMD-VAASEFFR) 并使用添加10条低浓度肽段的 Hela 细胞全蛋白样本对方法进行考察。结果表明,3种方法的定量限均在14~435 amol 范围内,线性关系与重现性良好,定性确证可靠性高。其中, WiSIM-DIA 基于超高分辨SIM 定量,与经典 DIA 二级定量具有一定的互补性;而 Full MS-DIA 将二级选择窗口缩短到3 amu,实现了DIA数据直接搜库鉴定,共从 100 ng Hela 细胞全蛋白样本鉴定到2835 个非冗余蛋白,与 DDA 鉴定结果重合度高。基于 Q-qIT-OT的创新数据非依赖性采集方法为定量蛋白质组学提供了全新视角与策略。 ( References ) ( Kalli A, Smith G T, Sweredoski M J, Hess S. J. Proteome Res. , 2013, 1 2(7):3071-3086 ) ( 2 Nagaraj N, Kulak N A, C ox J , N euhauser N , Mayr K, H o erning O, V o rm O, Ma n n M. M ol. Cell. Pro t eomics, 20 1 2, 11(3):M111.013722 ) ( 3 Geiger T, Wehner A, Schaab C, Cox J , Mann M. Mol . Cel l . Proteomi c s, 20 1 2,11(3):M111.014050 ) ( 4 Gallien S, Duriez E, Crone C, Kellmann M, M oehring T, D omon B. Mol. Cell. P r oteomics, 2 012,11 ( 12):1709 - 1723 ) ( 5 Gallien S, Duriez E, Demeure K, Domon B. J. Proteomics, 2013,81:148-158 ) ( 6 Chapman J D, Goodlett D R, M a sselon C D. Ma s s Spectrom. Rev., 2013: 10.1002/mas. 21400 ) ( 7 Law K P, L im Y P. E xpert Rev. Proteomics, 2013, 10(6):551-566 ) ( 8 Gillet L C, Navarro P, Tate S, Rost H, S elevsek N , R e iter L, Bonner R, Ae b ersold R. Mo l . Cell. Pr o t eomics, 2012 , 11(6): 0 111.016717 ) ( 9 Egertson J D , Kuehn A , Merrihew G E, Bateman N W , M a cLean B X, Ting Y S , C a nterbury J D , M arsh D M, K e llmann M, Zabrouskov V, Wu C C, MacCoss M J. Nat. Methods,2013,1 0 (8):74 4 -746 ) ( 10 Senko M W , R e mes P M , Canterbury J D, Mathur R, Song Q, Eliuk S M, M ullen C, Earley L, H ar dm an M, B l eth r ow J D , Bui H, Specht A, L ange O , Denisov E , M a karov A , H o rning S, Z a brouskov V. Anal. Chem., 20 1 3, 85(24): 11710-11714 ) Quantification Analysis of Targeted Proteins in ComplexSample by Novel Data Independent Acquisition ZHANG Wei', Reiko Kiyonami, JIANG Zheng*1, CHEN Wei' (Thermo Fisher Scientific, Shanghai 201206, China)(Thermo Fisher Scientific, San Jose, CA, USA) Abstract Data independent acquisition (DIA)is a novel MS scan mode for quantitative proteomics,acquiring all precursors as well as fragments without any loss of low abundant ions, and breaks the throughputlimitation of product ion quantification by high-resolution MS. Here we developed three DIA methods onquadrupole-linear ion trap-Orbitrap (Q-qIT-OT) Tribrid MS, classic DIA, as well as novel wide isolationwindow SIM scan (WiSIM)-DIA and full scan-DIA (Full MS-DIA). Quantitative analysis of 10 low abundantpeptides spiked in Hela cell digest was performed by the three methods for linearity, reproducibility andsensitivity evaluation. The results showed that the LOQs reached amol level(14-435 amol)) wwiith goodlinearity and effective lMS/MS confirmation. WiSIM-DIAutilizes ultra-highh resolution SSIM scan forquantification complementary with classic DIA. The isolation window of Full MS-DIA was down to 3 amu, andthe data could be directly used for database searching, thus realizing the integration of data dependentacquisition (DDA) and DIA, and avoiding the limitation of using spectra library. Keywords Orbitrap; Data independent acquisition; Proteomics; Absolute quantification ( (Received 24 A pril 2014; accepted 4 J uly 2014) ) 中国化学会分析化学委员会关于申请“分析化学基础研究梁树权奖”的通知 分析化学基础研究梁树权奖是以我国著名分析化学家梁树权先生命名的分析化学领域的最高奖项。本奖项旨在鼓励我国中、青年分析化学工作者献身于分析化学学科的基础研究和教育工作,培养优秀人才,促进和推动我国分析化学学科的发展。 本奖项为国内分析化学基础研究成果个人奖。凡年龄在50周岁以下,具备下列条件之一者均可申报参加评选:(1)在分析化学基础研究中确有创新,观点明确、数据完整、结论可靠,并已在国内外刊物上发表或在全国性学术会议上宣读获得好评者;(2)在解决分析化学基础研究中某一技术难题有独创和革新,并经鉴定确认对国民经济建设有较大经济效益或社会效益者。 中国化学会分析化学委员会将于2014年12月1日至2015年3月10日受理第八届“分析化学基础研究梁树权奖”的申请。申请材料包括申请表、单位推荐书、成果技术资料(论文目录- 包括题目、发表时间及刊物,5~10篇有代表性的论文及对所取得的成就的综述1000~2000字),国内外的反映和引用期刊(最好附SCI检索),获奖成果及成果推广和应用情况,社会或经济效益,以及有关证明材料。全部申请材料均需打印,一式五份寄长春市人民大街5625号中国科学院长春应用化学研究所陈杭亭收(邮编130022)。材料概不退回,请自留底稿。 申请表格式登录网站 http://www. analchem. cn 本刊公告栏查询。 ( 中国化学会分析化学委员会 ) 2014年11月
确定






还剩7页未读,是否继续阅读?

产品配置单


赛默飞色谱与质谱为您提供《蛋白质中蛋白质的定量检测方案(液质联用仪)》,该方案主要用于其他中蛋白质的定量检测,参考标准--,《蛋白质中蛋白质的定量检测方案(液质联用仪)》用到的仪器有赛默飞Orbitrap Fusion三合一质谱仪、赛默飞EASY-nLC 1200纳升级UHPLC
推荐专场






相关方案
更多
该厂商其他方案
更多