
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转6127
仪器信息网认证电话,请放心拨打

法规探讨3 | 《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征
在前两期的推送中,我们分别对《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》的数据分析和皮肤要求(详情请点击链接查看)部分分别进行了详细深入的探讨,本期我们将继续讨论征求意见稿中关于部分实验条件的内容。体外释放试验(In Vitro Release Test,IVRT)和体外透皮试验( In Vitro Permeation Test ,IVPT)是外用制剂仿制药体外关键质量属性研究的重要内容,其中,IVRT是评估药物制剂在体外环境下释放行为的一种实验方法,而IVPT则是在体外条件下模拟药物的经皮渗透过程,所以接近生理环境的实验条件是IVRT与IVPT研究的基础。IVRT研究中接收介质的pH范围征求意见稿中对IVRT研究的实验条件进行了规定,指出IVRT研究中常用的接收介质包括pH5-7的缓冲液-醇二元混合体系,并要求在研究期间,接收介质的pH应保持恒定。征求意见稿截图我们知道,通常情况下,皮肤的pH在5~6,但是接收介质的pH应根据配方制剂的pH、原料药在不同pH条件下的溶解度以及药物作用部位的pH等来综合进行确定【1】。在实际IVPT实验中,接收介质较常见的是所选用的pH值7.2的缓冲液。翻看国外的相关法规指南,我们也可以发现,EMA指南中未明确说明接收介质的pH范围,但要求在整个释放测试期间,应使接收介质的 pH值保持恒定;FDA IVRT指南和USP对介质pH范围也没有明确要求;PMDA虽然要求采用pH值在5~7的缓冲液(离子强度约为0.05)作为接收介质,但同时又指出当采用标准制剂进行试验,如果24 h的平均释放量小于20%,则可改变离子强度,如添加表面活性剂、改变pH。另外,有研究人员【2】研究了两种药物在一系列pH缓冲溶液中的饱和溶解度,结果表明,pH值会影响药物的溶解性和稳定性,以及在这些测试中使用的膜的完整性。调整pH值可以优化药物释放速率,更准确地模拟生理条件。特别是对于对pH变化敏感的药物,进行pH调整是确保结果一致性和可靠性的必要措施。IVRT和IVPT研究中膜/接收介质温度征求意见稿中还提到,在IVRT和IVPT研究中膜表面/接收介质温度应保持在32℃±0.5℃ 。征求意见稿截图IVRT和IVPT研究中的膜表面/接收介质温度范围应该是根据人体皮肤表面的平均温度范围来模拟的实验条件。然而,人体皮肤表面的温度影响因素较多,环境温度、湿度、风速、活动状态、不同部位等因素都会影响皮肤温度。欧聪颖等人【3】在相对封闭的环境中研究了人体皮肤表面不同部位的温度,结果表明,环境温度24℃湿度70%时,人体局部皮肤温度在约31.7℃~34.2℃范围内波动;而且随着环境温度降越低,皮肤温度分布越不均匀。Weiwei Liu等人【4】对不同空气温度下人体不同部位的皮肤温度进行了研究,在 21℃、24℃、26℃ 和 29℃ 的空气温度下,温度增加和温度降低得到的人体局部皮肤温度结果如下图所示。可以发现在空气温度21℃时,人体不同部位皮肤温度在约25℃~35℃的范围内变动,变动范围较大。对比上述研究数据,征求意见稿中规定的IVRT和IVPT的实验温度32℃±0.5℃范围相比于人体皮肤表面不同部位的温度波动范围小了很多。另外,参考一下国外的法规指南,FDA IVRT指南、FDA IVPT指南、 USP<1724>等法规中均要求对于外用皮肤类产品,测试温度为32±1℃。在整个研究过程中皮肤切片表面的温度保持在32±1℃,皮肤表面的温度波动会影响药物扩散,并可能增加实验变异性,法规建议使用可进行温度调节的扩散池来控制膜表面的温度。VDC12 Plus透皮扩散仪合邦科仪自主研发的VDC12 Plus透皮扩散仪,采用干加热方式控温,红外测温监控系统,可实现温度精准控制,温度调整范围为室温+5~50℃,分辨率为±0.01℃,控温误差<± 0.5℃,可以很好地满足法规要求,是外用制剂透皮研究的首选。参考文献:[1]殷连珍,李慧敏,苏梅,肖群,潘宪伟.外用药物制剂体外释放试验技术要求概况[J].中南药学, 2024(003):022.[2] Kokate, A.; Li, X.; Singh, P.; Jasti, B.R. Effect of Thermodynamic Activities of the Unionized and Ionized Species on Drug Flux across Buccal Mucosa. J. Pharm. Sci. 2008, 97, 4294–4306.[3] 欧聪颖,刘何清,张强.室内静坐状态下影响皮肤温度的环境因素[J].湖南科技大学学报:自然科学版, 2021.[4] Liu W, Lian Z, Deng Q, et al. Evaluation of calculation methods of mean skin temperature for use in thermal comfort study[J]. Building and Environment, 2011, 46(2): 478-488.
企业动态
2024.08.05

法规探讨2 | 《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征
在上一期的推送中,我们对《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》中关于IVRT的数据处理部分进行了详细讨论(详情点击链接查看),本期我们将继续探讨征求意见稿中对IVPT实验皮肤要求的内容。体外透皮试验(In Vitro Permeation Test,简称IVPT),是使用人或动物的离体皮肤模拟经皮给药制剂在生理条件下的经皮渗透过程,通过动态监测给药后特定时间内制剂有效成分的累计药物量和渗透速率,从而考察药物及其制剂的体外经皮渗透行为,为评价制剂的安全性和有效性提供保证。皮肤模型对IVPT实验结果是否可靠起到了关键作用,离体皮肤的来源、部位、处理方式、储存方式、完整性等都可能会对实验结果造成影响,正因如此,USP<1724>、FDA、EMA等法规中对皮肤的要求进行了比较详细的阐述。《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》中也对皮肤模型给出了指导意见:征求意见稿截图(点击可放大查看)IVPT皮肤模型开发需要考虑的因素很多,如:皮肤来源、部位、层次、皮肤储存条件及时长,皮肤类型选择和皮肤制备技术等。皮肤的选择EMA在2018年发布的《Draft guideline on quality and equivalence of topical products》中仅推荐使用离体人类皮肤进行IVPT研究;FDA 阿昔洛韦指南和IVPT指南均推荐采用离体人类皮肤;USP 中规定,除采用离体人类皮肤外,因猪的皮肤与人类皮肤具有相对相似的形态,可作为研究的次要选择,然而由于啮齿类动物(如鼠、豪猪)的皮肤在形态学和角质层结构和厚度方面与人类皮肤显著不同,不鼓励使用;合成膜(包括模拟生物膜开发的合成膜)也不适用于IVPT方法,因为采用合成膜得到的结果不能反应透过皮肤的药物量和渗透速率,故用作临床预测的意义不大。在大多数情况下,由于人类的皮肤比较难获得,或者伦理的要求,限制了人体皮肤的使用,征求意见稿中将猪的皮肤作为首要选择,这与我国的国情相符。目前,用于经皮研究的猪品种有Gottingen小型猪、Yucatan小型猪(墨西哥无毛猪)、Yorkshire猪、Landrace猪及巴马香猪等,这些也是目前IVPT研究中,最为常用的皮肤模型。除了皮肤的种属,皮肤的不同组织部位、年龄和种族,甚至是同一供体的相同组织部位,其渗透性均存在差异。因此,实验所用的皮肤应有一致的来源,且为相同的解剖区域(如背部躯干;也可以使用其他生物膜,如直肠、阴道或角膜上皮组织,这取决于所评估药品的预期给药途径)。皮肤厚度的显著变化可能会增加IVPT结果的变异性,需采用相同的皮肤厚度,FDA建议皮肤(人类)的厚度在500±250μm;但USP要求,皮肤(人类)厚度通常在250~500 µm。猪皮肤的厚度与人类皮肤接近且稍厚于人类皮肤,通常IVPT实验中使用的猪皮肤厚度为1mm左右;对于难渗药物,可以选用厚度稍薄的猪皮肤,但是建议最低厚度不低于0.4mm。皮肤的处理和保存皮肤的制备可以采用电动取皮机设置一定的厚度进行取皮,去毛去油过程中注意保护皮肤完整性,处理好的皮肤低温保存(OCDE建议储存温度为-20℃)。实验中需规定每组供体皮肤的储存温度和时间,以及冻融循环次数。目前,对于皮肤的储存条件尚无统一要求,但是有研究表明新鲜皮肤的电阻较高、渗透性较低,不同的储存温度和时间对皮肤屏障渗透性和电阻抗特性会产生相应的影响。另外,在上样前皮肤的解冻过程中,采用接收介质浸泡的方式可能会破坏皮肤的屏障完整性,通常可将皮肤在室温条件下放置约10min即可,并避免反复冻融皮肤。皮肤屏障完整性测试《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》中也对皮肤屏障的完整性测试提出了要求:征求意见稿截图(点击可放大查看)征求意见稿指出在每次试验前后应检测皮肤屏障完整性。FDA、EMA 与 PMDA 的法规中均要求在试验前需进行皮肤屏障完整性测试;FDA 及 USP 均未明确说明试验后是否需要进行皮肤屏障完整性测试;但 EMA 要求,如果试验时间大于 24h,试验后需评估皮肤屏障完整性。虽然有法规要求,但是在通常情况下试验结束后测定皮肤屏障完整性不太容易实现,因为在皮肤上已经有药品存在,可能会干扰测定结果。我们可以在试验前对所有皮肤进行完整性检测,试验后对空白皮肤进行完整性检测。皮肤屏障完整性测试常用方法关于皮肤屏障完整性的测试方法,虽然征求意见稿中提到可采用经皮水分散失法、氚化水渗透法和电阻/电导值法等三种方法,但是考虑到氚元素具有放射性,用户一般较多选用经皮水分散失法和电阻/电导值法两种方法。征求意见稿对上述两种方法没有展开描述,FDA IVPT 工业指南中对皮肤屏障完整性测试的常用方法描述的比较详细,具体内容如下:应在IVPT方法开发期间建立皮肤屏障完整性测试方法。常用的皮肤屏障完整性测试方法还没有相应的药典收载或法定可接受标准。尽管如此,下面讨论常见类型屏障完整性测试的推荐参数。(1)经皮水分散失法测试皮肤屏障完整性TEWL皮肤屏障完整性测试是通过测定皮肤外表面附近的水分(蒸汽),评估水分从皮肤下侧通过皮肤屏障的速率。在测定时,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤下侧与接收室中的接收介质(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32℃±1℃。如果皮肤切片切割的足够大,能覆盖皮肤安装位点的扩散池法兰,可在皮肤表面温度平衡至32℃±1℃几小时后,在不破坏皮肤切片与其下部的扩散池法兰粘附的情况下,将供给室轻轻移去,此时可将TEWL探针直接放置在皮肤表面,而不用放置在供给室的顶部。通常,在测试结果稳定后,每个皮肤切片需至少重复测定三次,并记录相关结果。商业化可用于TEWL测定的设备,有不同的设计和操作原理。由于设计的差异,一些TEWL测试设备可能相对更容易受到环境条件的影响(如,开放式腔室与封闭式腔室相比)。因此,通常需要尽可能精确的控制环境的温度和湿度(比较理想的情况是,将温度控制在21°C ± 2°C、湿度控制在50% RH± 20%RH范围内)。更精确的控制相对湿度(如,控制在40%RH~50%RH范围内)可以降低一些设备测定TEWL结果的变异性。TEWL测试设备生产商提供的用于体外的一些测试探针和适配器,可能适用于TEWL测定。不一致的测量探针室直径、测量探针孔、体外适配器和待测的皮肤面积,以及由于供给室高度的不同导致的探测传感器与皮肤表面距离的差异,均会增加TEWL测定结果的变异。与供给室和/或皮肤切片相关的TEWL测试设备对准的差异也可能增加TEWL测量结果的变异。此外,有些TEWL测试设备可能相对更容易受到握持探针的手的热传递的影响。对于特定的测试设备,申请人应遵循生产商使用手册中的相关说明。对于人体躯干或大腿皮肤,采用TEWL屏障完整性测试法,合理的皮肤屏障完整性接受(截止)标准是每小时每平方米不大于约15g水(即,≤15g/m2/hr);如果选择其作为截止标准,TEWL大于15g/m2/hr的皮肤,则不具有皮肤完整性。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可用作为无剂量空白对照皮肤切片。但是,如果通过实验数据表明,所选的接受标准可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片,也可以采用更高的截止标准(如,≤20g/m2/hr)。然而,具有合格屏障完整性皮肤的TEWL测量值,可能会因TEWL测试设备、操作方式和环境条件的不同而变化(如,较高的环境湿度或距离皮肤表面较远的距离可能会降低TEWL的测量值)。对环境和设备/操作因素的精确控制可将TEWL测量的可变性将至最低。因此,在IVPT方法开发过程中应对TEWL的测定方法进行优化(或基于实验室之前进行该测试的优化结果)。此外,应对TEWL测试设备进行适当的校准。申请人可提供生产商制定的相关校准程序信息,与IVPT方法开发报告一起在ANDA中递交,用于支持申请人为TEWL测量制定的技术程序的合理性。在IVPT方法开发、初步研究、验证和/或正式研究的整个研究阶段,进行TEWL屏障完整性测试时,均应监测和报告实验室的环境温度和湿度。(2)电阻/电导值法测试皮肤屏障完整性电阻/电导法测定皮肤完整性有几种变量,以电阻、电导或相关的电概念报告测试结果,用于表征流过皮肤的电流量。与皮肤相关的跨膜电阻测试通常指跨膜电阻(TEER)皮肤屏障完整性测试。测试结果可以用电阻的倒数“电导”为单位进行描述。电阻/电导值法测试皮肤屏障完整性通常采用“旨在测量电子电路或电子元件的电感(L)、电容(C)和电阻(R)”的仪器;这些仪器通常被称为LCR仪表,具有可调节的不同设置(测试参数)。TEER皮肤屏障完整性测试的一个推荐方法是,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤的角质层暴露在空气中(即,朝向供给室),皮肤下侧与接收液(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32°C±1°C。将少量的离子溶液(足以覆盖皮肤切片的整个表面)短暂的施加到皮肤角质层上;然后,将LCR仪表的一端电极与接收室中溶液接触,而另一端电极与供给室中溶液接触。在测定通过皮肤的电阻后(如,以kΩ为单位,按面积归一化法,注意电阻与面积成反比),将供给室中的溶液移出,用吸收性低脂棉将皮肤表面轻轻吸干。然后,在自研外用制剂或RS上样前,先将皮肤与上面的干燥空气接触,并平衡足够长的时间,使角质层恢复到正常的水合状态。TEER皮肤屏障完整性测试的结果,可能因LCR仪表设置(如,频率)和所用的测试技术方法不同,产生很大变化。电阻/电导值法测试皮肤屏障完整性接受标准的合理性,可通过试验数据证明,即:所选的接受标准应当可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片。在下一期的推送中,我们将对《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》的实验条件进行探讨,欢迎关注合邦科仪微信公众号。
企业动态
2024.08.02

法规探讨 | 对于《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征
《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》(点击链接查看)已经于2024年7月15日发布,相信很多朋友们已经关注到并进行了详细解读。首先在这里我们需要感谢CDE为我们国家制药行业做的辛勤付出,作为一家实验室分析仪器行业的一员,我们当然很希望国家制药行业可以蓬勃发展。既然是征求意见稿,很多朋友一定看过之后有撰写回复意见的想法。在这里我们抛砖引玉一下,谈论一些自己的看法,也希望能帮助到持续关注我们的广大朋友们。本篇文章我们重点对《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》中关于IVRT的数据处理部分进行详细讨论。IVRT(体外释放试验,In Vitro Release Test)是用于评估药物制剂在体外环境下释放行为的一种实验方法。它在药物研发和药品审批过程中扮演了重要角色,具体包括以下几个方面:01药物释放特性评估IVRT提供了一种标准化的方法来评估药物制剂的释放特性,这对于确保药物的疗效和安全性至关重要。通过模拟体内条件,IVRT可以帮助预测药物在实际应用中的表现。02监管合规在药物审批过程中,IVRT数据是必不可少的。监管机构通常要求药品开发商提供IVRT数据,以确保药物制剂符合规定的释放要求。03产品质量控制IVRT实验用于药品生产过程中的质量控制。通过监测药物制剂的释放特性,可以确保产品在生产过程中的一致性和稳定性。04改进药物配方通过分析IVRT数据,制药公司可以优化药物配方,提高药物的释放效果。这对于提升药物的疗效和患者依从性具有重要意义。总的来说,IVRT实验是确保药物制剂质量和效果的重要工具,其数据处理和分析对于药品的研发、生产和审批具有重要意义。那么我们来看一下征求意见稿中,对IVRT数据处理的要求内容:综合分析我们发现法规中对数据结果斜率RSD的要求以及分析方法,主要参考FDA的相关资料。而在结果判定上,继续秉持着从严的态度选择了EMA的标准。那么我们根据征求意见稿中法规的要求进行了数据模拟(如对各法规数据处理方式有疑问,可扫码关注我们的视频号的直播回放):扫码关注视频号,观看直播回放因为我们的仪器通常RSD做的很小,无法做出RSD15%的情况,但是为了验证法规需要极端数据,所以我们进行了数据模拟。模拟数据一我们假设了一种极端情况,对同一参比制剂进行一次6+6的实验,若满足法规要求的RSD,我们这里将12组数据的RSD设置到了接近15%。具体如下:(实验数据)(整合之后的数据)我们按照法规要求的曼惠特尼检验方式,数据按照ABABAB来分组:以上比值做百分比转化后按照从小到大排序,第8个和第29个分别为74.9%和99.0%,超过了法规的90%-111%的要求。思考以上结果,我们发现导致这一结果的原因可能是因为我们的数据质量要求以及统计学处理方式是参考了FDA的标准,判定结果按照EMA的标准。如果我们将统计学处理方式改为EMA的要求,同时为了尽量遵从EMA要求我们再加一组6+6数据结果会如何呢?同样因为我们的仪器通常RSD做的很小,无法做出RSD15%的情况,但是为了验证法规需要极端数据,所以我们再次进行了数据模拟。模拟数据二为了满足以上需要,我们另外模拟了一组12+12的实验数据,RSD取比较极限的情况13.55%,我们还是假设这是同一种药物,具体模拟每组斜率数据如下:(实验数据)(整合之后的数据)我们按照EMA法规对数据进行了处理以及分析,因为没有对24个数据是否满足正态分布进行检验,我们直接按照复杂处理方式进行:根据法规要求将144个比值进行自然对数转化:按照公式:进行统计分析,查表t值介于1.66~1.653之间,我们按照1.66进行计算,上下限分别为0.090014和-0.09562,我们进行反对数转换后上下限为109.42%和90.88%。满足征求意见稿要求的90%-111%要求。当然如果做了这方面修改的话我们需要多做12组数据,对24组数据进行统计,实验难度实际上加大了很多。如果我们不增加样本量,依然按照法规的判定标准90%-111%进行判定的话,数据需要做到什么程度呢?实验数据这一次我们实际用自己仪器进行了实验,同样只选用一种参比制剂做6+6实验,各组释放斜率数据如下:我们按照曼惠特尼检验方法,此处不赘述处理步骤了。最终下限为92.2%上限为109.4%。在同一种参比情况下,我们对此半固体制剂进行IVRT实验需要12组数据达到RSD 8.15%才能满足法规要求的90%-111%的上下限。虽然我们的上下限数据距离法规要求还有一定的余地,然而在实际研发过程中我们对两种药品进行测试实际上很难达到这个标准。讨论在实际的实验过程当中,数据的RSD依然是越小越好。在IVRT和IVPT研究中影响RSD的因素是多方面的,扩散设备的性能、制剂样品的稳定性、人工膜与生物膜的选择、操作差异等都会对实验误差有影响,我们需要综合考虑并采取相应的措施来减少这些因素对实验结果的影响。透皮扩散仪在外用制剂研发和皮肤科学研究中扮演着重要角色,透皮扩散仪的性能直接影响实验数据的准确性和RSD。扩散池中转子转速快慢决定药物在介质中的分布情况,从而间接影响药物扩散速率;温度的变化会改变分子的热运动状态,从而影响其扩散能力,在透皮扩散实验中,保持恒定的温度对于获得可重复的实验结果至关重要;取样精度直接关系到实验结果的准确性和可靠性,在IVRT和IVPT实验中,取样过程需要精确控制,以避免因取样误差而导致的实验结果偏差。VDC-12Plus 透皮扩散仪VDC-12Plus透皮扩散仪独特的设计,完全可以满足USP,FDA,EMA,PMDA以及征求意见稿等法规和指导原则的要求:根据不同的实验要求,转子转速可调,转速误差范围可达±2%;采用干加热式控温系统,可实时监测温度变化,控温误差<± 0.5℃,有效保证扩散池温度均一稳定;一体式化设计,仪器占用空间更小,管路更短,减少了管路死体积,取样更精准;独特设计的扩散池上样盖,上样便利快捷。对于《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》的后续探讨欢迎继续关注合邦科仪公众号。
企业动态
2024.07.26

《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》发布
7月15日,国家药品监督管理局药品审评中心发布通知,对《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》进行公开征求意见。如有相关意见和建议,请及时反馈至国家药品监督管理局药品审评中心,同时也欢迎与我们联系,就征求意见稿进行深入探讨。通知和征求意见稿原文如下(相关附件下载链接见文后):为明确局部给药局部起效化学仿制药体外关键质量属性研究中体外释放(IVRT)与体外透皮(IVPT)的技术要求,更好地指导企业进行研究以及统一监管要求,我中心组织起草了《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》。我们诚挚地欢迎社会各界对征求意见稿提出宝贵意见和建议,并及时反馈给我们,以便后续完善。征求意见时限为自发布之日起1个月。请将您的反馈意见发到以下联系人的邮箱。联系人:姜典卓,刘孟斯邮箱:jiangdzh@cde.org.cn,liums@cde.org.cn感谢您的参与和大力支持。国家药品监督管理局药品审评中心 2024年7月15日 相关附件:1《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT) 研究技术指导原则(征求意见稿)》2《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT) 研究技术指导原则(征求意见稿)》起草说明3《局部起效化学仿制药体外释放(IVRT)与体外透皮(IVPT)研究技术指导原则(征求意见稿)》意见反馈表.docx可参考的国外指导原则: 1 FDA IVPT Studies for Topical Drug Products Submitted in ANDAs2 FDA IVRT Studies for Topical Drug Products Submitted in ANDAs
企业动态
2024.07.19
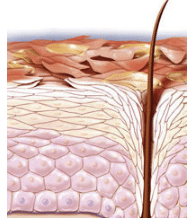
离体猪皮肤完整性随IVPT试验时间的动态变化探究
透皮扩散试验(IVPT)作为评价经皮给药制剂效能的重要试验之一,通过模拟真实环境,利用离体皮肤模型(典型是猪皮),动态监测给药后特定时间内透过皮肤的药物累积渗透量和渗透速率,从而考察药物及其制剂与参比药物的一致性,为评价制剂的安全性和有效性提供参考和保证。 确保离体皮肤各层次,尤其是角质层的完整无损,是获取准确、可靠IVPT数据的前提与基石。遵循USP<1724>半固体药品性能试验的严格标准,在上样前对生物膜的完整性进行确认是不可或缺的一环;OECD发布的皮肤吸收研究指导文件亦强调了在试验前后对皮肤完整性的细致确认;而《经皮给药制剂体外经皮渗透试验技术规范专家共识探讨》更明确指出,对离体皮肤屏障功能,即皮肤完整性的双重检测(试验前后),是确保实验结果有效性的关键步骤。经皮水分流失测量仪——皮肤完整性检测仪器通过测量TEWL值来反映皮肤屏障的完整性;用于体外透皮试验(IVPT)中离体皮肤的完整性评价(SIFT);可单独使用或与电脑连接一起使用;便携,无电线,可轻松测量。2022年10月FDA发布的中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且经皮水分流失测定法是推荐方法之一。皮肤电阻率仪——皮肤完整性检测仪器离体皮肤电阻测试仪用于体外透皮试验(IVPT)中离体皮肤的屏障完整性评价(SIFT),通过测量电阻进行离体皮肤的评估筛选,控制皮肤的质量,降低体外透皮试验的相对标准偏差。2022年10月FDA发布的中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且皮肤电阻率测定法是推荐方法之一。 鉴于目标化合物的特异性,IVPT试验时长与取样时间点需量身定制,以充分反映药物特性及制剂配方的影响。《经皮给药制剂体外经皮渗透试验技术规范专家共识探讨》中还提到,基于皮肤细胞的活性考量,通常建议将IVPT时长控制在24小时以内,以维护皮肤屏障功能的稳定与完整;若研究需求超越此时限,则需采取特别措施,确保在整个试验周期内,皮肤屏障的完整性与功能得以有效维持。 为进一步探究实验过程中皮肤完整性的动态变化,我们设计了一套验证性实验方案,旨在通过系统性监测,直观展现随着时间推移皮肤屏障状态的变化趋势,为IVPT试验的精准执行与结果解读提供强有力的支持。实验详情如下:01实验目的 探究离体猪皮肤在实验环境下皮肤完整性随时间的变化。02实验前准备仪器:透皮扩散仪(HB合邦科仪) 经皮失水仪(瑞士SKT) 皮肤电阻率仪(瑞士SKT)介质:生理盐水,庆大霉素样品:巴马香猪皮肤6片(来自不同供体)03实验方法1. 配制介质溶液,在0.9%氯化钠溶液中加入0.02%的硫酸庆大霉素,配置300ml介质溶液;2. 取自不同供体的巴马香猪皮肤6片,安装在透皮扩散池上,将组装好的透皮扩散池安装到透皮扩散仪上;3. 透皮扩散仪方法设定:温度32℃,转速600rpm;4. 分别在0 h、2 h、4 h、6h、8 h、24 h,测量并记录6组皮肤的经皮失水值和皮肤电阻数据。04测试结果图1. 猪皮肤24h经皮失水变化趋势图 6组皮肤的0-24h经皮失水数据变化趋势如上图所示,由图可知,在实验过程中,各组皮肤的经皮失水值(TEWL)呈现一定幅度的上下浮动,但这种波动始终维持在一个相对稳定的范围内,即总体波动区间限制在1.0至3.0g/m²/hr之间,且所有记录的最高值均未超过15g/m²/hr的阈值,验证结果表明在24小时内IVPT实验中皮肤经皮水分流失数值始终保持在合格且相对稳定的范围内。图2. 猪皮肤24h电阻变化趋势图 空白介质电阻为0.3KΩ,在为期24h的实验周期内,6组皮肤的电阻值均相对稳定,总体变化幅度较小,验证结果同样表明在24小时内IVPT实验中皮肤电阻数值始终保持在合格且相对稳定的范围内。05结论 基于上述详尽的数据分析,我们可以明确得出结论:在24h的实验周期内,尽管各组皮肤的经皮水分流失(TEWL)值均经历了细微的波动,但这些波动均严格控制在法规限定的安全阈值(≤15g/m²/hr)之内,充分证明了皮肤完整性在此过程中未发生显著变化,完全符合相关法规的严格要求。 同时,各组皮肤的电阻值变化模式也进一步印证了皮肤状态的稳定性。实验表明在温度32℃,转速600rpm的IVPT实验环境下,皮肤屏障功能在24小时内可以良好维持。 综上所述,在24小时以内的IVPT实验中,皮肤在电阻以及TEWL上的数值变化相对稳定且满足法规要求保持完整性合格状态。巴马小香猪皮肤品种:巴马小香猪猪龄:1~2月龄部位:全身皮肤(腹背部)皮肤类型:全层皮(角质+表皮+真皮)产品特点:巴马小香猪皮肤产品对完整性等关键质量参数严格把控;可按照客户需求定制。关于离体猪皮肤的评估,我们在往期的推送中进行过深入阐述,详情如下:体外渗透试验(IVPT)中生物膜的选择体外渗透试验(IVPT)中生物膜的控制皮肤屏障完整性测试:经皮水分散失法(TEWL)皮肤屏障完整性测试:电阻电导值法
企业动态
2024.07.15

透皮研究中离体猪皮活性的保存与挑战
离体猪皮因与人类皮肤具有高度相似性,近年来备受关注,成为外用制剂、化妆品等透皮扩散研究的关键实验材料【1】。离体猪皮模型通过保持皮肤的完整性和活性,能够在实验室条件下模拟皮肤屏障的功能,为评估化妆品和外用制剂等各类产品的安全性和有效性提供了强有力的支持。相较于传统的实验动物模型,离体猪皮模型在伦理接受度、成本效益及实验结果的可重复性方面展现出了显著优势。表. 人类和动物皮肤厚度测量数据【2】表. 毛囊的密度【3】根据表中数据,我们推荐在IVPT试验中使用猪皮。因为与人类皮肤相比,虽然猪皮的厚度虽然稍厚,大鼠的皮肤厚度更接近人类皮肤,但是猪皮的单位面积毛囊数量与人类皮肤更接近。然而,值得注意的是,离体的猪皮需要妥善保存,很多因素会导致离体猪皮发生变色现象,尤其是不当的储存条件,如暴露在空气中、遭遇高温、潮湿或强烈光照,均会加速猪皮浅层角质细胞中关键脂质成分(如神经酰胺、胆固醇及游离脂肪酸)的氧化过程,进而引发猪皮在较短时间内出现变色发黄现象【4】。关于发黄猪皮对实验的影响,后续我们将进行相关验证,敬请关注。为了最大限度地延缓这些不利变化,研究人员采取了诸多措施,其中,低温、密封及避光的储存环境被证明尤为有效【5】。另一方面,甲醛等有机溶剂在防止猪皮发黄、变质方面展现出了显著效果。它们通过溶解并清除皮肤表面的脂质层,实现了防腐、消毒及漂白的多重目的。然而,这一处理方式亦伴随着潜在风险,多项研究表明,甲醛等有机溶剂会通过交联胶原纤维改变皮肤细胞的结构,使蛋白质变性,导致皮肤机械性能发生变化,皮肤的硬度增加弹性降低,从而影响皮肤的渗透性【6】。经有机溶剂处理后的活体皮肤,其氚水经皮渗透量增加,类似的结果在离体皮肤实验中也能观察到【7】,即使完整性按法规要求依然合格,但是IVPT的实验结果也很难与未经有机溶剂浸泡的离体皮肤对应。鉴于上述,确保离体猪皮的活性对于透皮扩散试验(IVPT)的成功至关重要。那么,我们应该如何评估离体猪皮呢?在过往的推送中我们进行过深入阐述,详情如下:体外渗透试验(IVPT)中生物膜的选择体外渗透试验(IVPT)中生物膜的控制皮肤屏障完整性测试:经皮水分散失法(TEWL)皮肤屏障完整性测试:电阻电导值法参考文献:【1】Abd,Eman,Yousef,et al.Skin models for the testing of transdermal drugs.[J].Research & Reports in Transdermal Drug Delivery, 2016.DOI:10.2147/CPAA.S64788.)【2】Todo H. Transdermal Permeation of Drugs in Various Animal Species. Pharmaceutics. 2017 Sep 6;9(3):33. doi: 10.3390/pharmaceutics9030033. PMID: 28878145; PMCID: PMC5620574.【3】Mangelsdorf S, Vergou T, Sterry W, Lademann J, Patzelt A. Comparative study of hair follicle morphology in eight mammalian species and humans. Skin Res Technol. 2014 May;20(2):147-54. doi: 10.1111/srt.12098. Epub 2013 Jun 25. PMID: 23800212.【4】Gray G M , White R J , Williams R H ,et al.Lipid composition of the superficial stratum corneum cells of pig epidermis[J].British Journal of Dermatology, 1982, 106.DOI:10.1111/j.1365-2133.1982.tb00902.x.【5】Foutz T., Stone E. & Abrams C. Jr. Effects of freezing on mechanical properties of rat skin. Am. J. Vet. Res. 53, 788–792 (1992).【6】Ranamukhaarachchi SA, Lehnert S, Ranamukhaarachchi SL, Sprenger L, Schneider T, Mansoor L, Rai K, Häfeli UO, Stoeber B. A micromechanical comparison of human and porcine skin before and after preservation by freezing for medical device development. Sci Rep. 2016;6:32074.【7】王旭平,任道凤,金锡鹏,顾平.有机溶剂对活体皮肤屏障功能的影响[J].中华劳动卫生职业病杂志, 1998(3).
企业动态
2024.07.08

USP<1724>半固体药品性能试验 | 生物膜的关键质量要求
在外用制剂药品开发的过程中,如何精准模拟药物经过人体皮肤的渗透行为,是研发团队必须面临的重要挑战。而在这其中,生物膜无疑起到了举足轻重的作用。在过往的推送中,合邦兴业已对体外渗透试验或透皮试验(IVPT)中生物膜的关键质量要求进行了详尽的探讨和解析,期望能为相关领域的研究和实践提供一些参考。阐述的核心内容包括:体外渗透试验(IVPT)中生物膜的控制皮肤屏障完整性测试:经皮水分散失法(TEWL)皮肤屏障完整性测试:电阻电导值法体外渗透试验(IVPT)中生物膜的选择在今天的探讨中,合邦兴业精心摘录并系统整理了关于USP<1724>半固体药品性能试验中对生物膜的具体质量要求,旨在为各位行业专家及老师们提供一份权威的参考资料,助力药物渗透性能研究的精准与高效。法规原文下载链接见文后。以下是摘录整理的法规译文:(法规原文截图,点击可放大)体外渗透试验(IVPT)的目的是利用旨在模拟体内条件的方法参数,表征应用于生物膜表面的药物渗透进入和通过它的速率和程度。IVPT 方法可以采用多种生物膜,包括离体人类皮肤,这些生物膜应具备可表现出渗透性自然变化的性质,以反映体内的变化情况。这种渗透性的可变性很大;某一特定化合物(在同一配方制剂中)在人群中的不同个体之间或同一个体的不同解剖区域之间的皮肤渗透性,存在 10 倍的差异是很常见的。实验差异也可能是化合物的物理化学性质导致。在所有产品或试验组的对比研究中,应采用同一供体(或相同供体组)、同一解剖区域(如腹部、背部等)、同一来源(如选择性外科手术或尸体)和相同处理方式的重复皮肤切片,以减少变异。(法规原文截图,点击可放大)生物膜从尸体或选择性外科手术(如腹部整形和乳房缩小)获得的离体人类皮肤(皮片厚度通常在 250-500 µm 左右),由于其与人类药物产品开发的内在相关性,以及在实验过程中易于处理,通常是最合适的选择膜。将膜安装到扩散池前,应确认皮肤厚度,因为厚度的显著变化可能会增加 IVPT 结果的变异性。即使文献中采用了仅有角质层的皮肤样本(isolated stratum corneum preparations),在 IVPT 研究中也不建议使用,因为它们需要专门处理技术,并可能导致较高的实验变异性和/或产生难以解释的结果。由于这些原因,仅有角质层的皮肤样本不适用于 IVPT 研究。表皮样本(epidermal preparations,在真皮和表皮交界处从真皮层分离出来的表皮薄片)需要技术经验处理,与离体人类皮肤相比,其结构完整性较差,而且更容易撕裂或失去屏障的完整性。然而,表皮样本与离体人类皮肤相比,往往厚度比较一致,且一些化合物的渗透程度更大。因此,虽然在大多数情况下离体人类皮肤是一个很好的选择,但在某些情况下(例如,当感兴趣的药物渗透通过皮肤的量无法量化时),在 IVPT 方法开发过程中可以采用表皮样本进行评估。除监管指南另有规定外,猪的皮肤与人类皮肤具有相对相似的形态,可作为(开发)研究的次要选择。由于啮齿类动物的皮肤在形态学和角质层结构和厚度方面与人类皮肤显著不同,因此不鼓励使用。人工培养的皮肤组织(重构人类皮肤表皮)的使用情况并不理想,因为这类组织的渗透性目前还不能代表人类皮肤。合成膜(包括模拟生物膜开发的合成膜)也不适用于 IVPT 方法;因为采用合成膜得到的测试结果不能反应透过皮肤的药物量和渗透速率,可能会造成误导。鼠、猪和人皮肤的相关数据对比如下两个表格所示:另外,有人曾对鼠和猪来源的生物膜进行体外渗透试验的研究,研究结果表明:大鼠皮肤的体外渗透性高于体内实验;大鼠皮肤对所有受试物质的渗透性均高于人体皮肤,存在显著的种间差异。促渗剂筛选:可选择大鼠或裸鼠皮肤,渗透性较好,变异性小猪皮肤和人的皮肤表现出较好的一致性,两者表面脂质和厚度相似。参考文献:[1] OECD (2004), Guidance Document for the Conduct of Skin Absorption Studies, OECD Series on Testing and Assessment, No. 28, OECD Publishing, Paris, https://doi.org/10.1787/9789264078796-en. [2] 世界中医药学会联合会经皮给药专业委员会,汤秀珍.经皮给药制剂体外经皮渗透试验技术规范专家共识探讨[J].中国现代应用药学, 2022(008):039.使用人或猪的全层(non-dermatomed)皮肤,可能会对实验造成挑战,一般不建议。也可以使用其他生物膜,如直肠、阴道或角膜上皮组织,这取决于所评估药品的预期给药途径。在这种情况下,由于考虑模拟体内的相关条件,试验方法的几个参数可能与进行皮肤 IVPT 研究的参数不同。例如,膜的维持温度可能不同。巴马小香猪皮肤品种:巴马小香猪猪龄:1~2月龄部位:全身皮肤(腹背部)皮肤类型:全层皮(角质+表皮+真皮)产品特点:巴马小香猪皮肤产品对完整性等关键质量参数严格把控;可按照客户需求定制。(法规原文截图,点击可放大)膜温度在整个研究过程中,每个皮肤切片的表面温度应保持在32℃±1℃。皮肤表面温度的波动会影响药物扩散,并可能增加实验的变异性。建议使用可进行温度调节的扩散池来控制膜表面的温度。透皮扩散仪VDC 12Plus透皮扩散仪器是用于外用制剂关键质量属性研究的重要仪器。VDC-12PLUS的设计满足USP、FDA、EMA、PMDA等法规和指导原则的标准。一体化设计一体化设计使得仪器整体尺寸更小,占用空间更少;同时优化管路设计,减少了管路死体积,让实验数据可靠性获得有效提升;7×2设计可以两侧设计不同的实验参数,如温度、转速、取样时间。同时每组6+1的设计满足法规要求;一台仪器相当于两台,可以同时完成两组不同实验;空白位满足法规要求的空白位设计,在进行IVPT实验时,更方便设计非给药对照组,可排除皮肤基质及其他潜在杂质的干扰。(法规原文截图,点击可放大)膜完整性在上样前,应确认每个扩散池中皮肤屏障的完整性,可以采用诸如经皮水分散失值(TEWL)、皮肤的电阻/电导值或氚化水渗透等技术手段。只有满足屏障完整性接受标准的皮肤切片才可以适用。TEWL通常是首选方法,不同于其他方法,TEWL是在不受外部空气影响的条件下,测定水分通过皮肤屏障的量,相对快速和方便。点击下载UPS<1724>半固体药品性能试验法规原文。经皮水分流失测量仪——皮肤完整性检测仪器通过测量TEWL值来反映皮肤屏障的完整性;用于体外透皮试验(IVPT)中离体皮肤的完整性评价(SIFT);可单独使用或与电脑连接一起使用;便携,无电线,可轻松测量。2022年10月FDA发布的中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且经皮水分流失测定法是推荐方法之一。皮肤电阻率仪——皮肤完整性检测仪器离体皮肤电阻测试仪用于体外透皮试验(IVPT)中离体皮肤的屏障完整性评价(SIFT),通过测量电阻进行离体皮肤的评估筛选,控制皮肤的质量,降低体外透皮试验的相对标准偏差。2022年10月FDA发布的中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且皮肤电阻率测定法是推荐方法之一。
企业动态
2024.06.28

FDA IVPT资料 | 透皮外用制剂(IVPT)研究参考案例
在IVPT测试中,评估皮肤的完整性非常重要。FDA IVPT测试工业指南中要求,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,即皮肤屏障完整性测试,并对皮肤屏障完整性测试的验收标准进行了规定。以下文章来源于FDA官网,文章主要介绍了两种评估透皮外用制剂(IVPT)研究中皮肤完整性的方法,以及实验过程全部取样与部分取样的优势,而对于一些局部制剂可能会出现的非常规通量曲线,需要进一步研究与确认Jmax、AUC等关键质量参数。原文下载链接见文后。皮肤完整性测试Skin Integrity testingThere are two methodogies that are currently employed to asses skin integrity for IVPT studies. However, there is not standardized criteria that are available to assess viable skin membranes for use in IVPT.目前有两种方法用于评估透皮外用制剂(IVPT)研究中的皮肤完整性。然而,目前尚无可用于评估IVPT使用的活性皮肤膜完整性的标准。1. 透皮电阻(ER)测试The transepidermalelectrical resistance (ER)• The measured resistance is dependent on the device, ie., applied frequency, resulting current, ionic strength of the solution as well as the surface area of the skin sample. Actual readings, measured in kΩ, is difficult to correlate with the data that has been reported in various existing literature.- 测得的电阻取决于设备,即应用频率、产生的电流、溶液的离子强度以及皮肤样本的表面积。实际读数以kΩ为单位,测量结果很难与各种现有文献中报告的数据相关联。• It was determined that the skin sections that showed resistance values less than 3 times of baseline (without skin membrane, solution reading) had compromised barrier and rejected for the study. Alternatively, those skin section with resistance readings higher than 20 times of the baseline resistance, were with keratinized epithelium and also rejected from the study经确定,显示电阻值低于基准值(无皮肤膜,溶液读数)3倍的皮肤部分具有受损屏障并被排除在研究之外。相反,那些电阻读数高于基准电阻的20倍的皮肤部分,则具有角化上皮层,也被排除在研究之外。对于不同生物膜在不同装置中的电阻值我们进行了如下验证实验:将皮肤1在10ml玻璃池、40ml塑料池中进行透皮电阻(ER)测试,平行测量三次,取中位数进行统计,测试结果如下:皮肤1在不同扩散池中的透皮电阻测试结果(单位:kΩ)将皮肤2在10ml塑料池、10ml玻璃池中进行透皮电阻(ER)测试,平行测量三次,取中位数进行统计,测试结果如下:皮肤2在不同扩散池中的透皮电阻测试结果(单位:kΩ)由上述数据可知,相同的生物膜在不同扩散池中的绝对电阻值不同,但与空白相比,其电阻的倍数值可以重现,据此我们可以评估皮肤的完整性。同时,不同电阻对累计渗透量也有一定影响,我们曾经发表过不同厚度皮肤的电阻值对药物体外渗透实验的影响相关文章,供各位老师参考。2. 经皮水分流失(TEWL)测试The transepidermal water loss (TEWL)• Furthermore as the supporting data we measured the transepidermal water loss (TEWL) with a VapoMeter (Delfin Technologies Ltd., Finland), the standard limited value as suggested in various literatures -2 h-1 was used.- 此外,我们还使用经皮水分流失测量仪测量了经皮水分流失(TEWL),采用了各种文献建议的标准限值 -2 h-1。However the criteria that has been used for skin selection for the IVPT studies in many cases exhibited with quite different flux profiles within the same and different donors.然而,目前在许多情况下用于IVPT研究的皮肤选择标准在同一供体和不同供体之间显示出相当不同的通量曲线。More research is need to standardized skin integrity testing procedures in qualification of the viable skin membranes used IVPT studies.需要进行更多的研究来标准化皮肤完整性测试程序,以确定可用于IVPT研究的活性皮肤膜。皮肤电阻率仪——皮肤完整性检测仪器离体皮肤电阻测试仪用于体外透皮试验(IVPT)中离体皮肤的屏障完整性评价(SIFT),通过测量电阻进行离体皮肤的评估筛选,控制皮肤的质量,降低体外透皮试验的相对标准偏差。2022年10月FDA发布的中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且皮肤电阻率测定法是推荐方法之一。经皮水分流失测量仪——皮肤完整性检测仪器通过测量TEWL值来反映皮肤屏障的完整性;用于体外透皮试验(IVPT)中离体皮肤的完整性评价(SIFT);可单独使用或与电脑连接一起使用;便携,无电线,可轻松测量。2022年10月FDA发布的中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且经皮水分流失测定法是推荐方法之一。在每个时间点进行全部与部分取样/补液的优势The complete vs partial receptor volume removal/replacement at each time pointsAdvantages of complete volume removal/replacement• Simplify flux calculation• Close to In Vivo situation• Help solubilize a hydrophobic analyte and maintain sink• Avoiding the occurrence of negative flux values as sometimes occurs with slow/low penetrating compounds when aliquot sampling is used全部取样/补液的优势:• 简化通量计算• 接近体内情况• 有助于溶解亲脂性分析物并维持溶液• 避免使用等分采样时,有时会出现负通量值的情况,特别是对于慢速/低渗透化合物Adventives of partial volume removal/replacement• Easy to handle sample collections during manual operation• Giving opportunity to use automated diffusion cells systems• Sink condition is maintain• Cumulative amounts are detectable in low level drug presented at early time points samples部分取样/补液的优势:• 易于手动操作时处理样品收集• 提供使用自动扩散池系统的机会• 保持溶液状态• 在早期时间点的样品中,可以检测到低水平药物的累积量More research is required to compare skin absorption profiles obtained with complete and partial volume replacement in IVPT studies需要进行更多研究来比较在IVPT研究中使用完整和部分体积取样/补液所获得的皮肤吸收曲线。Fig1 Mean Flux (μg/cm2/hr) Results: Partial volume (0.5 mL) volume withdraw/ replacement at each time points. Across Donor Summary Percutaneous Absorption of low permeable anaytethrough ex vivo Human Torso Skin over 48 hours from a Single Application.(Mean ±SD, n=6 Donors)图1 均值通量(μg/cm2/hr)结果:在每个时间点进行部分体积(0.5 mL)的取样/补液。跨供体数据统计单次应用在48小时内,低渗透性药物透过人离体躯干皮肤的的通量曲线。(均值±标准偏差,n=6 供体)透皮扩散仪VDC 12Plus透皮扩散仪器是用于外用制剂关键质量属性研究的重要仪器。VDC-12PLUS的设计满足USP、FDA、EMA、PMDA等法规和指导原则的标准。一体化设计一体化设计使得仪器整体尺寸更小,占用空间更少;同时优化管路设计,减少了管路死体积,让实验数据可靠性获得有效提升;7×2设计可以两侧设计不同的实验参数,如温度、转速、取样时间。同时每组6+1的设计满足法规要求;一台仪器相当于两台,可以同时完成两组不同实验;空白位满足法规要求的空白位设计,在进行IVPT实验时,更方便设计非给药对照组,可排除皮肤基质及其他潜在杂质的干扰。非常规通量曲线Unconventional Flux ProfilesFor some of topical formulations we observed unconventional flux profiles when no maximum peak, Jmax was identify across multiple subsequent time points even 72hrs or 98hrs IVPT duration.对于一些局部制剂,我们观察到非常规通量曲线,即在多个连续时间点甚至在72小时或98小时的IVPT持续时间内,未发现最大峰值Jmax。Fig2 The Percutaneous Absorption of low permeable analyte through ex vivo Human Torso Skin over 96 hours from a single application on individual donors. Mean on of n=3 skin pieces for each donor is presented. a) donor 1, with 5 mg/cm2; b) donor 1, with 15 mg/cm2; c) donor 2, with 5 mg/cm2; d) donor 2, with 15 mg/cm2;图2 透过体外人体躯干皮肤的低渗透分析物的经皮吸收,从单次应用开始到96小时,对各个供体进行了研究。每个供体的n=3 皮肤片的均值呈现。a)供体1,5 mg/cm2;b)供体1,15 mg/cm2;c)供体2,5 mg/cm2;d)供体2,15 mg/cm2;There is a need to determine:- How such profiles can be used in pharmacokinetic endpoints calculation- If the dose duration method is applicable for such cases有必要确定:- 如何将这种曲线用于药代动力学终点的计算- 剂量持续时间方法是否适用于此类情况原文下载链接:https://www.fda.gov/media/138052/download原文在FDA网站搜索:“There are two methodogies that are currently employed to asses skin integrity for IVPT studies. However, there is not standardized criteria that are available to assess viable skin membranes for use in IVPT”也可以找到原文。巴马小香猪皮肤品种:巴马小香猪猪龄:1~2月龄部位:全身皮肤(腹背部)皮肤类型:全层皮(角质+表皮+真皮)产品特点:巴马小香猪皮肤产品对完整性等关键质量参数严格把控;可按照客户需求定制。
企业动态
2024.06.24

隆重上市 | 合邦科仪VDC12 Plus透皮扩散仪性能验证表现
体外释放实验(IVRT)是目前评价半固体制剂(如乳膏剂、软膏剂、凝胶剂等)处方工艺的重要手段,主要用于外用制剂的药学质量控制,是药物关键质量属性之一,可用于表征某些工艺、配方和/或生产的变更对药品的影响,也可用于药品开发过程中处方工艺的筛选研究。扩散池法是进行半固体制剂体外释放实验(IVRT)的可靠方法,该方法在美国药典 (USP) 半固体药品性能测试中有详细记载。合邦科仪现重磅推出新产品——VDC12 Plus透皮扩散仪,用于软膏、硬膏、涂抹剂、洗剂、薄膜、气雾剂等的体外释放测试,其设计满足USP<1724>,FDA、EMA、PMDA等法规和指导原则的标准。VDC12 Plus透皮扩散仪搭载先进的自动取样技术,可完成自动排出气泡、自动取样、自动采集样品、自动补液、自动清洗,使药物透皮释放实验更加准确高效。VDC12 Plus 透皮扩散仪VDC12 Plus 透皮扩散仪产品特点:一体化设计一体化设计使得仪器整体尺寸更小,占用空间更少;同时优化管路设计,减少了管路死体积,让实验数据可靠性获得有效提升;7×2 设计可以两侧设计不同的实验参数,如温度、转速、取样时间。同时每组6+1的设计满足法规要求;一台仪器相当于两台,可以同时完成两组不同实验;空白位满足法规要求的空白位设计,在进行IVPT实验时,更方便设计非给药对照组,可排除皮肤基质及其他潜在杂质的干扰。为了验证VDC12 Plus透皮扩散仪的性能,我们对利多卡因乳膏样品的体外释放速率进行了测试,实验详情如下:01实验目的通过测试样品,对透皮扩散仪在体外释放实验过程中的性能进行验证。02样品信息样品剂型:利多卡因乳膏03主要分析仪器1)VDC12 Plus 透皮扩散仪(HB合邦科仪)2)分析天平3)液相色谱-紫外检测器(HPLC-UV)04体外释放实验参数溶出装置:透皮扩散装置温度:32℃ ± 1℃标准池:12 ml取样量:10ml取样时间:分别在第0.5h、1h、2h、3h、4h、5h、6h时进行取样05液相色谱方法参数流动相:甲醇:0.3%磷酸氢二铵 67:33色谱柱:C18-150×4.6mm流速:1.5 ml/min进样量:20 μl检测波长:210 nm06测试结果6.1 累计释放曲线6.2 拟合曲线在0.5h、1h、2h、3h、4h、5h、6h时间点,以单位面积累计释放量(ug/cm2)(y轴)对时间(h)(x轴)做图,拟合线性回归方程(部分取样模式)如下:6.3 释放速率07结论在0.5h、1h、2h、3h、4h、5h、6h时间点对样品(同时7个扩散池)平行进行实验。0h时,扩散池中未检出目标物;在0.5h-6h的7个取样点分别对7个扩散池的累计释放量做线性考察,释放速率的平均值为430.9;释放速率RSD为3.42%。FDA IVRT测试工业指南中提到,根据每个扩散池的释放速率(斜率)计算的批内精密度,其变异系数(%CV)应不大于15%。在上述实验中,采用合邦科仪VDC12 Plus 透皮扩散仪对利多卡因乳膏进行的体外释放实验(IVRT),7个扩散池的释放速率(斜率)RSD为3.42%,远远小于FDA IVRT测试工业指南中提到的15%,这表明合邦科仪VDC12 Plus 透皮扩散仪的性能完全符合FDA IVRT测试工业指南的法规要求。
企业动态
2024.06.20

精彩回顾 | 合邦科仪水活度仪亮相制药工业微生物大会
2024年5月11日,位于杭州举办的第八届制药工业微生物大会圆满落幕。这次大会,我们向制药用户展示了水活度仪在微生物限度检测及药品稳定性分析中的技术及应用优势。制药工业微生物大会汇聚了业内众多专家、制药企业代表及科研人员,共同探讨制药微生物检测领域的最新技术和应用。水活度作为2025版药典新仪器,在本次会议中备受瞩目。 在展会上,我们详细展示了水活度仪器在微生物限度检测及药品稳定性分析中的应用。通过现场演示和讲解,与会者了解到该仪器能够准确测量样品中的水活度值,为制药企业提供关键的微生物控制参数,从而确保药品的安全性和稳定性。 水活度仪器在展会上受到了制药企业的广泛关注。他们表示,该仪器能够满足其微生物限度检测及药品稳定性分析的需求,有助于为企业降本增效。许多企业代表还主动与我们进行了深入交流和探讨,寻求合作机会。与会的行业专家也对水活度仪器给予了高度评价。他们认为,该仪器在制药微生物检测领域具有独特优势,能够为制药企业提供更加准确、高效的检测手段。我们深知技术创新是保持竞争力的关键。我们将继续加大研发投入力度,不断优化和升级水活度仪器的性能和功能,以满足客户不断变化的需求。 展望未来,我们将继续专注于医药仪器领域的研发与创新工作。我们将以客户需求为导向。同时,我们也期待与更多的制药企业和行业专家建立合作关系,共同推动我国制药行业的发展与进步。感谢广大制药用户、客户群体对我们的关注和支持!我们将继续努力,为您提供更优质的产品和服务。Aqualab水活度仪采用USP/EMA/PMDA/AOAC水活度检测金标准-镜面露点法设计美国药典USP优先推荐镜面露点法,该方法可高精度测量所有样品,测量时间小于5min。可以对生产过程中微生物限度进行全流程控制。是业内公认具有潜力的微生物检测新方法。快速精准-测量时间小于5分钟!产品特点:国际公认水活度金标准镜面冷凝露点法。全球用户量第一的品牌Aqualab水活度仪。★合邦科仪作为独家仪器厂商参与2025版新药典第四部水活度测定法标准制定课题——《水活度测定法的增订》。部分用户名单:
企业动态
2024.05.13

重要文献 | 人体各部位TEWL参考值
健康成人的经表皮水分流失:一项系统综述和汇总分析更新(翻译稿)M. Akdeniz*, S. Gabriel*, A. Lichterfeld-Kottner, U. Blume-Peytavi, J. Kottner,Charité-Universitätsmedizin Berlin, Department of Dermatology and Allergy, Clinical Research Center for Hair and Skin Science通讯作者:Merve Akdeniz关于经皮失水,我们已经了解了什么?1、经表皮失水量(TEWL)被认为是测量皮肤屏障完整性的参数之一。2、较高的TEWL通常与皮肤屏障损伤有关。3、“正常 ”的TEWL值仍然未知。这此研究补充了什么内容?1、提供了86个皮肤区域的TEWL参考估计数2、从头到脚,个体内部存在大量的变异。3、随着年龄的增长,TEWL似乎在下降。TEWL测量设备和测量方法之间的平均差异看起来很小。研究背景:经表皮失水量(TEWL)是最重要的皮肤屏障特征之一。较高的TEWL通常与皮肤屏障损伤有关,而较低的TEWL与健康的皮肤有关。研究目的: 目的是更新现有的数据和汇总分析,以提供成人健康皮肤的TEWL参考值。方法:本次更新包括了从1947年到2017年4月13日的两次搜索所确定的研究。在健康成年人中进行观察性和干预性研究,提供了TEWL测量的定量估测,包含了如明确的皮肤面积和年龄的分布测度。按照皮肤区域提取数据并进行统计汇总。结果:经过全文评估,在现有荟萃分析基础基础纳入45项研究的结果。在212项研究中,总共确定了86个皮肤区域的TEWL估计数。乳房皮肤的TEWL最低为2.3(95%CI1.9-2.7)g/m²/h,腋窝的TEWL最高为44.0(95% CI 39.8-48.2)g/m²/h,样本量从4个(前额左中侧)到4013(右前臂中侧)。不同测量设备的TEWL估计之间的差异的临床相关性似乎很小。老年人的TEWL与年轻组相似或更低。结论:提供的参考估计有助于临床研究计划和结果解释。TEWL高度依赖于皮肤面积,我们的研究结果进一步支持了左右测量部位之间的对称性。与年轻人相比,老年人的TEWL似乎普遍相似或下降,但现有的证据有限。TEWL的报告应得到改进:在未来的研究中,应始终报告均值和扩散参数。图1文献的识别与选择表1 通过OvidSP再Medline和Embase数据库搜索表2 所有研究TEWL测量值汇总表3 年轻人与老年人TEWL值范围的比较在文章后边有原文下载~介绍:经皮失水量(TEWL)被认为是测量皮肤屏障完整性的最重要的参数之一。第一次测量方法是在1911年描述的1,今天,这个参数被认为是各种皮肤学和皮肤研究环境中的标准参数2,4。TEWL被定义为水的通量密度,它从真皮层和表皮通过角质层扩散到皮肤表面。这种持续的水扩散不能直接观察到。在皮肤表面的一个特定区域上的蒸汽通量可以通过现有的测量仪器进行测量。有不同的TEWL测量仪器(封闭腔,开放腔系统),但不能确定哪种测量技术是最准确的5-7。TEWL的增加似乎与皮肤屏障功能障碍相关,而TEWL的降低则被认为是皮肤屏障完整或恢复的一个指标8-10。有证据似乎表明,TEWL在衰老过程中也会减少,这可能被误解为一种皮肤屏障的改善1,11。TEWL受到许多环境和个体因素的影响,包括年龄、性别、种族、解剖区域、皮肤温度和环境条件、季节、吸烟状况、测量技术和许多其他因素2,3,12。因此,一个TEWL值是否“正常 ”或是再病理相关性阈值范围内目前仍然是一个有争议的问题。然而,在2013年,首次尝试总结健康成年人TEWL的实证证据11。在截至2012年5月的搜索期间,我们总结了年轻和老年健康人类的50个皮肤区域的TEWL参考值11。研究证明这些参考值对设计临床和实验研究以及解释研究结果是有用的13-15。自2012年以来,已经发表了大量测量和报告TEWL的新研究。因此,这次系统审查的主要目的是更新现有的审查结果。第二个目标是进一步调查年轻人和老年人之间以及不同测量条件和设备之间可能存在的差异。方案审查方案已提前在普洛斯彼罗数据库(CRD42016037977)中注册。资格标准研究必须满足与之前的汇总分析相同的标准11。(1)提供TEWL测量的定量估计的初步实证研究,(2)皮肤面积的明确报告,(3)平均年龄的明确报告, (4)在体内对健康人体皮肤进行的TEWL测量, (5) TEWL的测量和报告,包括传播的测量,如标准差(6)发表语言:英语或德语。类型的研究和引用被排除:(1)皮肤病、皮肤切除,或其他疾病可能影响TEWL, (2)不明确的测量区域, (3)系统或局部治疗影响皮肤屏障功能, (4)人类18岁以下或没有报告的年龄, (5)体外或体外测量, (6)动物研究, (7)评论,意见。信息来源和搜索策略之前的系统回顾和荟萃分析涵盖了从1947年到2012年5月的搜索期。对于这次更新,通过OvidSP在数据库MEDLINE和Embase(2012年5月6日至2016年4月14日)中进行了并发搜索,最后一次更新是在2017年4月13日(表1)。这次更新涵盖了从1947年到4月13日的搜索期间2017.我们访问了所有TEWL探针制造商的网站,并访问了三个网站(英国Biox有限公司;芬兰Delfin技术公司;德国Courage & Khazaka)提供了出版清单。这些发表列表被筛选为其他的初级研究。对所有符合科学网络中纳入标准的研究进行了正向检索,以获取其他文献。研究的选择和数据的收集过程数据库搜索和发布列表的结果被导入引用管理器(Endnote X7)。根据标题和摘要对研究的资格进行了评估。结果由两位审稿人(MA、SG)独立筛选。任何不确定性都由第三位审稿人(AL,JK,UBP)进行了讨论和解决。数据项目提取以下变量:作者(s)、年份、国家、季节、样本量、性别、平均年龄、皮肤照片类型、种族来源、吸烟状况、健康状况、TEWL设备、皮肤面积、测量的平均TEWL和相应的扩散值[SD、标准误差(SE),95%置信区间(CI)]。如果只给出标准偏差(SD),我们根据公式SE=SD/√(样本量)将其转换为标准误差(SE)16。在新的研究中,我们还提取了以下变量:吸烟状况、室温、相对湿度(RH)、适应环境、仪器校准、避免使用空气湍流和测量的次数)。数据提取由两位审稿人独立进行。任何分歧都由第三位审稿人(AL, JK,UBP)解决。个体研究中的偏倚风险目前还没有对报告TEWL测量的研究进行偏倚风险评估,但已经发表了一些开展和报告指南2,4。Plessis等人的最新研究结果。20133被用来创建一个定制的偏差风险工具。我们认为以下项目对于准确和可重复性的测量很重要: (1)在测量前是否有至少15分钟的适应期(是/否)? (2)研究前是否对仪器进行了校准(是/否/不适用)? (3)如果使用了开放式腔室系统(是/否/不适用),则在测量过程中是否避免了不良的空气湍流? (4)是否在相同皮肤区域进行两次或三次测量,结果取平均值(是/否)?两名审稿员(MA、SG)独立评估了每个纳入参数的偏倚风险。我们通过共识或与第三位评论作者(AL,JK,UBP)协商来解决分歧。总结结果的措施每个皮肤面积的平均TEWL值作为每项研究的主要结果。采用随机效应模型,将所有新研究和以前研究的每个皮肤区域的TEWL测量值结合起来11纳入汇总分析的研究。每个特定地点的总结测量给出95%的置信区间。在这篇综述中,我们重点关注了TEWL值的描述性信息。各个皮肤区域的TEWL汇总测量的如果不在95%置信区间则被认定为有统计学意义的差异。我们还基于I2统计方法17使用SPSS统计版本22软件(IBM,美国)进行描述性信息汇总统计如平均年龄,并使用StatsDirect Version2.8.0( StatsDirect 有限公司,英国)计算汇总分析。其他分析采用亚组分析来确定18-64岁的65岁以上受试者之间可能存在的差异。为了确定不同的TEWL测量仪器和测量条件可能造成的影响,我们进一步进行亚组分析。因为右前臂掌侧是最常测量的皮肤区域,所以我们只选用此区域分析。95%置信区间不重叠被解释为有统计学差异。结果选择和特征研究研究的鉴定和选择过程如图1所示。.去除重复文献后,筛选了MEDLINE和Embase的366篇论文和其他来源的113篇论文。最终,对195项研究进行了全文评估,并纳入了45项研究。之前的原始综述包括167项研究,并新增45项研究结果添加到现有的汇总分析中11。根据所有212项研究,共确定了86个皮肤区域。对于所有被排除的研究,都记录了排除的原因(表S1)。排除全文的主要原因是缺少基准TEWL值以及缺少扩展参数。关于每个研究的TEWL估计数和皮肤区域的详细信息见表S2。将45个新研究的结果纳入到现有的数据集上1159项研究有时期信息;62项研究报告了种族起源,30项研究报告了皮肤照片。在几乎所有的研究中,TEWL的测量都是根据现有的标准,在类似的温度和湿度控制的房间中进行的2-4(室温在20~22±1 °C之间,RH低于60%)。在82项研究中使用了Evaporimeter(Servomed,瑞典),随后在76项研究中使用了Tewameter(Courage+Khazaka,Cologne)。分析45项新研究的结果,其中14项研究中至少进行了两种设备测量18-31在45项研究中,并且在这45项研究前对TEWL设备进行了校准31-34。45项研究中提到避免空气湍流影响报道的有五项18,19,31,34,35。偏差风险在表2中显示了关于偏差风险的信息(例如。适应,仪器校准),在大多数纳入的研究中没有提供。因此,不可能将评估结果纳入分析。结果的汇总汇总分析的主要结果如表2、表3和图1中所示。总的来说,提取了86个皮肤区域的TEWL估计值,并将样本量从n=4(前额左中)到右中掌前臂最大n = 4013。除了现有的结果外,还研究了36个新的皮肤区域。只有一份出版物报道了18个皮肤区域36。乳房皮肤的TEWL最低为2.3(95% CI 1.9-2.7)g/m²/h,TEWL最高为44.0(95% CI 39.8-48.2)腋窝为g/m²/h。TEWL似乎在面部皮肤区域的发病率通常较高。右脸颊13.9(95%CI12.914.9)g/m²/h,与手臂(例如右前臂中腹侧6.8(95%CI6.5-7.0)g/m²/h)、腿部(例如大腿5.1(95% CI 4.1-6.1)g/m²/h)(腹部5.9(95%CI4.5-7.4)g/m²/h).提取18至64岁受试者的82个皮肤区域TEWL数值进行评估计算。在较年轻的群体中,合并的平均年龄范围在21.8岁至44.8岁之间。对65岁以上受试者的27个皮肤区域的TEWL估计, 合并平均年龄为68.3-78.8岁。两个年龄组之间可以对24个皮肤区域进行比较。老年组的平均TEWL普遍低于中年组。13例患者的差异有统计学意义。表3附加分析大多数TEWL估计数集中在右前臂中掌侧(n = 4013)。因此,我们选择这个皮肤面积来比较每个测量仪器的汇总估计值(表S3)。H 4300型(日本)的最低TEWL为3.1(95% CI 1.0-5.2) , 而采用冷凝室测量法(英国)的最高TEWL为11.4(95%CI10.10.712.0)。大部分测量值使用Evaporimeter(Servomed,瑞典)和Tewameter210(C+K,Köln)。开放式Evaporimeter的TEWL为5.1(95% CI 4.8-5.4),封闭式Vapometer的TEWL为7.5(95% CI 6.6-8.3)。讨论结果汇总本次回顾和汇总分析旨在提供有关健康成人TEWL值的现有实证证据。我们能够提取并计算出人体的86个皮肤区域。总的来说,更新后的估计数似乎与之前的汇总分析相似。因此, 目前的结果可以被认为是健康皮肤的广义TEWL估计。敏感性分析支持了不同的测量仪器和条件提供的结果略有不同的事实,但这些差异可能是次要的临床相关性。总的来说,对TEWL的临床相关性的解释具有挑战性。回顾结果清楚地表明,研究结果之间存在很大的差异性,但健康成年人的皮肤也存在广泛的个体内差异性。众所周知,在皮肤科研究中测量的实际TEWL值受到许多已知因素的影响,如年龄、 白天、季节、地理区域,但也有未知因素37。最有可能的是,绝对的TEWL值不是重要的,更重要的是随着时间的相对变化或治疗之间的差异1。另一方面,有新的证据表明,基准TEWL数值可能对儿童的特应性皮炎有预测价值38,39。目前还正在讨论健康成年人的基准TEWL是否也能预测皮肤情况40或可用作皮肤甚至皮下疾病的早期(亚临床)标志41,42。最近的一项研究确定了TEWL与屏障功能遗传之间的联系43。无论如何,我们的研究结果强烈表明,这些预测高度依赖于皮肤区域。不同皮肤区域的TEWL差异与不同的角质细胞大小和新城代谢速率有关41,42。我们的结果显示,面部区域的TEWL值普遍高于手臂、腿部或腹部(表2和3)。我们的研究结果与 Plewig 和 Marples 的研究结果一致44,相对于前臂、腹部和大腿,面部区域的角质层细胞更小,细胞层更少;因此,渗透路径长度更短,TEWL值更高41。细胞间脂质双分子层的数量和组成也可能影响由内而外的水扩散45。回顾结果表明,关于皮肤老化对TEWL值可能造成的影响。老年人TEWL降低的原因是一个有争议的问题1,11。表皮细胞增殖的减缓导致角质细胞体积增大, 自然保湿因子的减少和细胞间脂质的变化可能起到一定作用46,47。内在和外部老化皮肤之间的皮肤屏障特征的差异不能表明29,48而且目前的汇总分析都不支持。老年皮肤中皮肤屏障功能的降低最近被提出可以通过改善紧密连接功能来补偿49。然而,皮肤老化是一个高度复杂的过程,依赖于各种内在和外在的过程,因此这些结果在皮肤屏障能力方面的临床相关性仍不清楚50。这项研究最重要的发现之一是不同测量设备之间的比较。开室式测量腔很容易受到外部空气湍流的影响,这需要控制环境因素。然而,它们能够进行更长时间和连续的测量。封闭腔室装置不能用于连续测量,每次测量后都必须进行平衡,但是它们不受外部空气湍流的影响。关于开室系统的大部分基于Evaporimeter,而闭室系统则是基于Vapometer。汇总分析表明,封闭系统和开放系统之间不存在系统性差异(表S3)。开放式测量(TEWL 5.1(95% CI 4.8-5.4))和封闭式测量(TEWL 7.5 (95% CI 6.6- 8.3)之间的差异的临床相关性)似乎是最小的。最高的TEWL估计数是用AquaFlux(UK)测量的,这是一种封闭的冷凝器室测量装置。最低的TEWL估计值是用H4300模型测量的,该模型使用了一种不通风的闭室方法, 目前已不再生产。这些设备在功能上彼此不同,也与开室测量设备不同51。但似乎没有证据表明有一种可用的设备优于其他设备。TEWL可能会受到其他几个参数的影响,如温度和RH、校准、适应和测量频率。因此,需要进行高度的标准化2-4。我们的研究结果进一步支持了身体左右两侧TEWL值的对称模式(例如在左右两侧的前额和脸颊之间,表2和表3)。TEWL值在前额中央区分布较高,而向外侧区分布最小。此外,我们的结果显示,从右前臂的近端(5.3(95 % CI 4.3-6.3))到远端(8.8(95 % CI 7.6-10.0)), TEWL增加明显且具有统计学意义。在对皮肤区域进行个体间随机化时,必须考虑到这一点。我们综述的另一个重要发现是TEWL估计数的报告质量较差。由于缺失扩散参数以及更清晰的报告内容,我们排除了22项报告95个皮肤区域TEWL值的研究。一个明确清晰的报告才可以用来比较不同研究的结果。根据国际准则2-4以及我们的调查结果,我们建议报告以下最低信息:测量对象(不包括对象)基准值TEWL值(不是与基准值的差异)TEWL扩展参数测量条件(温度和相对湿度)测量装置的类型(包括根据制造商提供的探头)测量对象(不包括测量对象)的样本特征确切的解剖区域限制我们应用了一种广泛的搜索策略,筛选了数百项研究,包括由TEWL设备制造商创建的出版物列表,并在科学网络上进行了正向搜索。然而,可能有一些出版物没有被确定,特别是因为TEWL测量通常是大型研究的一小部分。关注英语或德语的出版物可能会导致对潜在相关研究的额外排除。然而,可能的语言偏差不太可能存在,因为描述性(基准值)TEWL报告的系统差异是不可信的。由于对不同研究设计的TEWL估计被合并,因此也不可能进行正式的发表偏倚风险评估。一些皮肤区域的样本量很小,在实证研究设计中评估TEWL估计的偏倚评估风险的公认方法是未知的。因此,概括性可能受到限制。皮肤区域之间的差别会使得测量结果有区别。众所周知,皮肤屏障的特征甚至在紧密相邻的皮肤区域之间也有所不同。33,52因此,我们决定尽可能详细地将这些内容分开。另一个限制可能是在许多被纳入的研究中常用的术语“健康志愿者 ”。也许这些受试者在过去可能患有皮肤病,这可能对皮肤屏障功能有更持久的影响。结论我们提供了86个皮肤区域TEWL的参考值。这些参考估计对研究计划和结果解释很有用。TEWL高度依赖于皮肤面积,我们的研究结果进一步支持了左右身体部位之间的对称性。与年轻人相比,老年人的TEWL似乎普遍相似或降低,但其临床相关性尚不清楚。应改进TEWL的报告,基准值在未来的研究中应始终报告均值和扩散参数下载原文:Transepidermal water loss in healthy adults: a systematic review and meta-analysis update
应用实例
2024.05.10

相约杭州 | 合邦科仪邀您参加制药工业微生物技术大会
药品是关乎人民身体健康的特殊性商品,其质量是对于整个社会而言意义重大。随着药品制造与监管科学的发展,国内外对药品生产过程中微生物污染的风险控制高度关注。药品从前期的研发、中期的临床到后期的商业生产,每一个周期内都有微生物相关的质量活动。为保障药品质量与患者用药安全,提升药品微生物污染控制水平,第八届制药工业微生物技术大会于2024年5月10-11日在杭州举办。本次会议合邦科仪携Aqualab多款水活度仪在现场展示,其中包含国际金标准镜面露点法以及独家用于测量挥发性样品的激光法水活度仪。另外在现场也有合邦科仪透皮扩散仪,生物均质机等多款其他仪器的展示。欢迎您莅临展位,同时有精美礼品相送~
企业动态
2024.05.10

水活度对口服固体制剂化学和物理稳定性的影响
水活度对口服固体制剂化学和物理稳定性的影响 在整个产品生命周期内,需要对口服固体制剂的多种化学和物理特性,进行测试评估。本文将介绍三个案例研究,以表明通过水分活度评估水分对效价和溶解的影响。 国际药物开发创新与质量联合会(IQ)报告,“自十年前ICH Q1A稳定性方案首次发表以来,稳定性方面研究已经得到完善和发展”。例如《加速预测稳定性(APS):基础知识和制药行业实践》一书,其中包括总共22章详细的技术细节供开发和执行稳定性研究的科学家参考。此书中大量描述的概念之一就是水活度(Aw)。 最初稳定性试验通常在敞口烧杯或小瓶中进行,将口服固体制剂暴露在高温和一定的相对湿度(RH)下,且没有保护性包装,在一定时间后从稳定性试验箱中取出样品分析化学和物理稳定性。 虽然每种药物都会吸附水分,但每种药物水分吸收速率以及药物在稳定性试验箱中达到平衡湿度所需的时间有很大差异。 通过最初测量产品的水活度,以及测量记录水活度在暴露和吸湿期间随时间的变化,可以确定口服固体制剂的游离水与环境湿度达到平衡的速度。 在平衡状态下,稳定性实验箱的相对湿度相当于当时状态下产品的水活度。绘制的曲线是水活度对温度的函数,水活度这一数值代表了药物产品中游离水所占的比例;或者换句话说,游离的水可以参与我们不想发生的反应,因此产品稳定性可以根据水活度来定义,将水活度测试添加到稳定性试验中是一个重要的质量控制方法。 除了定义产品化学稳定性外,即使化学稳定性在允许的范围内,也可以通过水活度识别水分对口服固体制剂物理稳定性的影响。本文描述了三个案例研究,其中高水活度对不同产品的效价和/或溶出度产生了负面影响。尽管这些研究根据相对湿度条件进行描述的,但我们也可以通过直接测量产品水分活度的方法来表述。 水活度会影响降解速率指数对水分敏感的药物会与水发生化学反应,破坏原料药中的化学键。这既降低了效价,又增加了杂质含量。多项研究表明,在低湿度条件下本具有长达几年稳定性终点的化合物在RH从40%变成75%储存时有明显变化。他们的稳定性终点从几年减少到了几个月。 在下面的例子中,研究人员定量测定了温度和湿度对硝西泮固体制剂分解的影响。硝西泮在双分子酸碱催化反应中降解,游离水分的多少会显著影响这一反应。因此,水活度值的大小决定了两种主要分解产物所占的比例。强制破坏试验的结果如图1所示,样品在四种温度(40、55、70和85℃)和六种相对湿度条件(30、40、50、60、70和80%相对湿度)下储存在开放的杯子中。图1A描述了硝西泮降解常数(K)与温度和相对湿度之间的明显指数相关性;在对数标度上绘制时,观察到线性相关性(图1B)。在高相对湿度(80%相对湿度及更高)下,分解化合物变得不稳定,因此无法轻易建立降解曲线。如前所述,平衡相对湿度相当于产品的水活度——因此,以下数据可以直接与水活度相关,硝西泮的后续水活度测量将决定产品的稳定性。 图1 硝西泮在不同相对湿度和温度下的降解速率A)指数关系 B)对数关系 水活度会影响溶出速率在进行化学稳定性测试的同时还应进行物理稳定性测试,以确定产品是否保持了其他关键质量属性。即使原料药本身对水分不敏感,高水分活度也会导致产品某些配方的溶解速率、硬度或易碎性发生改变。在下面的例子中,研究人员通过研究体外溶出度和效价(化学稳定性),评估了不同商品化环丙沙星(CIP)片剂在加速变性条件下(40℃/75%相对湿度)初级包装的稳定性。结果显示,配方VII未通过体外溶出度试验和效价试验;60分钟后,只有30.8%的药物溶解,储存6个月后,只有79.4%的原料药残留(表1)。仅6个月后,配方I未通过溶出试验,而效价仍在规范范围内。 表1 环丙沙星的加速稳定性试验 在另一项已发表的研究中,研究人员使用热分析和X射线分析来研究盐酸贝那普利的溶解稳定性。结果显示,当片剂在40℃/75%相对湿度下储存3个月时,即使原料药本身也没有变化,制剂物理结构也会发生改变。当含水量增加到临界水平以上时,溶解速率呈线性下降。崩解剂的“预激活”导致片剂的结构变化,是溶出速率降低的根本原因(表2)。 表2 盐酸贝那普利的加速稳定性试验 通过这两个溶解稳定性示例,我们发现实验结果可直接与平衡相对湿度(即水活度)相关。一旦溶解和水活度之间的相关性已知,产品水活度测量也可用于确定产品溶解质量。通过测定水活度来确定干燥药品的稳定性和质量非常重要,美国药典USP-水活度通则的导言指出,“一些水分子可能与API或药物辅料紧密结合,无法参与化学、生物化学或物理化学反应(例如,作为水合物),而有些水分子可以自由地参与水解等反应,或者参与到支持微生物生长的中。确定可用(游离)水所占比例很重要,而水活度(aw)的测定可以为我们提供这一信息。”USP中描述的水活度方法之一是可调谐二极管激光吸收光谱法,这是唯一能够准确测量含有挥发性物质样品水活度的技术。Aqualab TDL激光法测定水活度 Aqualab 4TE水活度仪采用USP/EMA/PMDA/AOAC水活度检测金标准-镜面露点法设计。美国药典USP优先推荐镜面露点法,该方法可高精度测量所有样品,测量时间小于5min。可以对生产过程中微生物限度进行全流程控制。是业内公认具有潜力的微生物检测新方法。 Aqualab 4TE 镜面冷凝露点法水活度仪 产品特点:·国际公认水活度金标准镜面冷凝露点法。·全球用户量第一的品牌Aqualab水活度仪。
应用实例
2024.05.07

直播预告 | 1小时解决您IVPT数据处理与分析问题
新的一期直播来啦!首先预祝各位伙伴劳动节快乐~节后我们会为大家献上新的一期直播。这次我们来解答您外用制剂关键质量属性的核心问题!时间:5月8日,15:00-16:00直播内容概述:体外渗透(IVPT)是外用制剂关键质量属性之一,该实验数据处理复杂,且有众多因素影响实验结果。本次我们为您详细讲解在不使用软件的情况下如何进行IVPT数据处理,并结合案例为您分析影响实验数据的因素。赶快扫码预约吧!~
企业动态
2024.04.30

美国USP建议新方法 | 水活度≥0.6aw产品须做洋葱伯克霍尔德菌群(BBC)测试
美国药典委员会(USP)致力于确保非无菌药品的微生物达到最高质量水平,为此,USP建议采用新的药典方法来实施"通则非无菌产品的微生物检验:洋葱伯克霍尔德菌群(BBC)的测试"。降低与微生物污染相关的风险是保障药品质量的一个重要方面。BCC具有在各种环境中存活的能力,而且与严重不良事件有关,因此备受关注。美国食品药品管理局(FDA)强调了对水基非无菌产品中的BCC进行严格检测的重要性1。为了加强我们对此类风险的共同防范,USP正计划修订几个通则,以明确该测试的应用。新提议的药典方法新提议的药典方法强调在协调的章节中收载必要的本地(未协调)文本,确保BCC检测适用于吸入用产品或口腔、口腔黏膜、皮肤或鼻腔用水性制剂。提议的更新方法强调了确保某些剂型不含 BCC以保护患者安全和产品完整性的重要性。USP正在分享新提议的药典方法,用于建立洋葱伯克霍尔德菌群微生物检测的整体框架。该方法包括以下步骤:修订通则在通则特定微生物的测试(Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms)加入一项要求,即水活度为0.6aw或更高的产品必须按照通则的指示进行测试,以证明不含BCC。这将为何时需要进行BCC测试提供更明确的指导。更新相关参考性通则a.调整通则非无菌产品微生物检查法: 药物制剂及药用原料的微生物限度标准(Microbiological Examination of Nonsterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use),以响应这些要求,确保处理BCC测试的应用是一致的。b. 修订通则水活度测定在非无菌制剂中的应用(Application of Water Activity Determination to Nonsterile Pharmaceutical Products),以保持一致性。药典协调小组(PDG)合作与PDG密切协作,在全球范围内统一标准。作为第一步,USP打算将本地(未协调)文本纳入通则和通则中。更新各论根据需要审查和修订各论。USP正在考虑对高风险产品直接引用通则。建议修订的初期示例以下示例包括拟纳入通则的文本,以及对通则表1的提议修订。USP期待与监管机构和全球药典机构进行合作讨论,以协调和加强微生物测试标准。建议参与评议人员药物供应商和制造商、制药商、合同生产和检测机构、监管机构以及质量保证/质量控制专家。欢迎提交评议提交评议的截止日期: 2024年6月27日USP欢迎业界提出反馈意见,请通过发送电子邮件提交评议至: microbiology@usp.org,如需了解更多信息,请联系USP通则高级科学家II Leslie Leslie Furr(Furleslie.furr@usp.org或microbiology@usp.org) 。参考文献:1. FDA advises drug manufacturers that Burkholderia cepacia complex poses a contamination risk in non-sterile, water-based drug products | FDA原文链接:实施“通则非无菌产品的微生物检验: 洋葱伯克霍尔德菌群的测试”的提议药典方法Aqualab水活度仪采用USP/EMA/PMDA/AOAC水活度检测金标准-镜面露点法设计。美国药典USP优先推荐镜面露点法,该方法可高精度测量所有样品,测量时间小于5min。可以对生产过程中微生物限度进行全流程控制。是业内公认具有潜力的微生物检测新方法。快速精准-测量时间小于5分钟!产品特点:国际公认水活度金标准镜面冷凝露点法。全球用户量第一的品牌Aqualab水活度仪。★合邦科仪作为独家仪器厂商参与2025版新药典第四部水活度测定法标准制定课题——《水活度测定法的增订》。部分用户名单:
应用实例
2024.04.24

新闻资讯|合邦科仪亮相2024CBC全产业链资源大会
新闻资讯|合邦科仪亮相2024CBC全产业链资源大会2024 CBC中国医药全产业链新资源大会圆满落幕。本次展会是合邦科仪在行业中的一次重要展示,我们在这次展会中向专家、客户和合作伙伴展示了我们公司的最新技术和产品成果,吸引了广泛关注和认可。合邦科仪是一家技术创新型仪器科技公司,本次展会合邦科仪携多款新品与大家见面,产品广受好评,直击行业痛点。合邦科仪的仪器产品为行业提供了顶尖技术,解决很多卡脖子问题大会第一天下午,合邦科仪总经理汤宏敏于药典新仪器分会场,进行了关于镜面露点法水活度仪的报告。汤宏敏总经理精彩地介绍了我们公司的镜面露点法水活度仪,展示了该仪器在测量水活度方面的准确性和可靠性,水活度仪是国内试行指导原则中用于微生物限度检测的新方法。合邦代理的Aqualab水活度仪可以为客户提供了高质量的水活度测量解决方案。报告结束后,汤宏敏总经理带领药典委专家以及用户,在展位现场进行了仪器实际使用展示。专家对合邦科仪的镜面露点法水活度仪表示了高度肯定和认可,这种专业级别的认可将进一步增强合邦在行业内的信誉和地位。第一天会议尾声,汤宏敏总经理在圆桌会议上分享了对透皮行业未来发展的深刻见解,引发了圆桌会议其他专家的新思路。汤总经理的发言展示了我们公司的前瞻性思维和战略规划,受到了与会者的广泛关注。会议第二天世界中医药联合会经皮给药专业委员会会长梁秉文教授亲临合邦科仪展位。梁秉文教授对合邦科仪的透皮扩散仪新颖的设计思路以及人性化的应用设计高度赞赏,这种专业认可将进一步提升合邦的产品影响力和市场竞争力。同时梁秉文教授以及上海皮肤病医院专家朱全刚教授也考察了我们公司的皮肤完整性检测相关仪器。经皮失水和皮肤电阻是FDA皮肤外用制剂体外渗透指导原则中提到的皮肤完整性评价方法,合邦科仪代理的瑞士SKT经皮失水和皮肤电阻测量仪器测量数据准、重复性强,可以极大降低评价难度。梁老现场也对合邦科仪的高质量猪皮表示了赞赏和认可,这种对产品质量的肯定将进一步提升合邦在市场上的品牌价值和信誉度。大会第二天下午,合邦科仪市场总监李天宇介绍了一款公司重磅新产品,一种皮下采样探针技术-微灌注OFM,该技术有望改变行业格局,打通透皮外用制剂制剂最后一公里。作为透皮行业可以用于活体检测的新技术,李天宇经理的报告吸引了众多行业专家以及相关用户的关注。李经理详细介绍了开放式微灌流的技术原理以及应用案例,该技术为行业内用户提供了更好的取样方式和实验方案,受到了与会者的积极反响和关注。报告结束后,李天宇经理在展会现场进行了体外渗透实验全流程操作演示,包括皮肤的实验前处理,皮肤完整性评价,体外渗透实验上样,以及实验后回收率测试。结合合邦科仪自有或代理的仪器,该场实操演示吸引了众多观众的关注和参与,也展示了合邦在技术应用方面的专业实力。合邦科仪作为透皮行业专业的实验室仪器以及解决方案服务商,总经理汤宏敏也被大会主办方特别安排进行了一次采访。采访中汤总就国内透皮行业研发现状,站在研发仪器的角度抒发了自己的见解。采访内容包含未来法规方向,各仪器所检测的数据重要性以及仪器偏差可能对结果的影响,汤总的讲述专业中肯为研发用户指明了新思路。展会最后一天,汤宏敏总经理于药典新仪器现场精彩地介绍了我们公司的透皮扩散仪,该仪器是外用制剂关键质量属性研究的必用仪器,在透皮制药研究中具有重要意义。合邦科仪透皮扩散仪可以为客户提供准确可靠的实验数据和结果,受到了与会者的高度赞誉。通过本次展会,专家、客户和合作伙伴对我们的产品和服务表示了极高的兴趣和认可,为公司未来的发展打下了坚实基础。展望未来,合邦今后会一直持续保持钻研创新的精神,不断推出新品,致力于为国内医药产业升级和科学仪器国产替代,为行业的快速发展做出更大的贡献,为人类的健康事业添砖加瓦。最后,我们衷心感谢所有参与展会的专家、客户和合作伙伴的支持和信任,期待未来与大家携手共进,共创美好明天!
企业动态
2024.04.22

TEER皮肤电阻测量仪介绍
皮肤电阻测量仪皮肤电阻测量仪是用于评估皮肤屏障功能和皮肤健康状况的重要工具之一。其原理是通过电流施加到皮肤,测量通过皮肤的电流强度,并根据欧姆定律计算出皮肤的电阻值。通过电阻测量也可以辅助我们分析皮肤的水分含量、皮脂分泌、表皮层厚度等参数,从而为美容皮肤分析、皮肤疾病诊断和治疗监测提供重要数据支持。 在FDA IVPT指导原则中,皮肤电阻率是用于皮肤完整性评价建议方法之一。法规指出,在实验前后应对实验用皮肤进行完整性评价,以确保整个IVPT实验以及实验结果的可靠性。同时也有相关文献指出,IVPT的透过率在一定程度上与皮肤电阻值有相关性。 将皮肤安装到扩散池中,使皮肤的角质层暴露在空气中,皮肤下侧与接收液(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32°C±1°C。将少量的离子溶液施加到皮肤角质层上;然后,将Ag电极与接收室中溶液接触,AgCl电极与供给室中溶液接触。在测定通过皮肤的电阻后,将供给室中的溶液移出,用吸收性低脂棉将皮肤表面轻轻吸干。在上样前,先将皮肤与干燥空气中平衡足够长的时间,使角质层恢复到正常的水合状态。 型号ER2310测量优势精度高,便捷操作,1秒内显示测量结果。数据存储有数据记录功能,能够提供准确的测量结果历史存储数据。档位切换无需手动调整电阻档位,直接测量自动换挡出读数。环境要求低不受温湿度环境影响,确保测量结果可靠性。 应用:在医疗美容、皮肤科医学和生物医学研究等领域,皮肤电阻测量仪器被广泛应用,为行业研究和临床实践提供重要支持。医美行业:皮肤水分含量评估:皮肤电阻率可以用于评估皮肤的水分含量。医美机构可以利用皮肤电阻率测量结果为客户提供个性化的皮肤护理方案,包括补水保湿、皮肤调理等。皮肤科医学:皮肤疾病诊断和治疗监测:皮肤电阻率可以用于评估不同皮肤疾病患者的皮肤屏障功能和病变程度。医生可以通过测量皮肤电阻率来辅助诊断和监测皮肤疾病的发展和治疗效果。 生物医学研究:皮肤生理学研究:皮肤电阻率可以用于研究不同生理状态下皮肤的电性质和屏障功能。研究人员可以通过测量皮肤电阻率来探索皮肤对外界刺激的响应和适应机制。药物递送系统研究:皮肤电阻率可以用于评估不同药物递送系统在皮肤上的渗透性能和递送效率。研究人员可以利用皮肤电阻率测量结果指导药物递送系统的设计和优化。相关法规: In Vitro Permeation Test Studies for Topical Drug Products Submitted in ANDAs.pdf 相关文献:【1】Musazzi UM, Casiraghi A, Franzé S, Cilurzo F, Minghetti P. Data on the determination of human epidermis integrity in skin permeation experiments by electrical resistance. Data Brief. 2018 Oct 26;21:1258-1262. doi: 10.1016/j.dib.2018.10.098. PMID: 30456241; PMCID: PMC6231038. 【2】Morin M, Ruzgas T, Svedenhag P, Anderson CD, Ollmar S, Engblom J, Björklund S. Skin hydration dynamics investigated by electrical impedance techniques in vivo and in vitro. Sci Rep. 2020 Oct 14;10(1):17218. doi: 10.1038/s41598-020-73684-y. PMID: 33057021; PMCID: PMC7557913. 【3】Zhu XM, Li Y, Xu F, Gu W, Yan GJ, Dong J, Chen J. Skin Electrical Resistance Measurement of Oxygen-Containing Terpenes as Penetration Enhancers: Role of Stratum Corneum Lipids. Molecules. 2019 Jan 31;24(3):523. doi: 10.3390/molecules24030523. PMID: 30709044; PMCID: PMC6384980. 【4】Knoth K, Zäh RK, Veldung B, Burgio D, Wiegand B, Smola H, Bock U, Lehr CM, Hittinger M, Groß H. Development and evaluation of a quality control system based on transdermal electrical resistance for skin barrier function in vitro. Skin Res Technol. 2021 Sep;27(5):668-675. doi: 10.1111/srt.12998. Epub 2021 Jan 6. PMID: 33404151. 【5】Rachakonda VK, Yerramsetty KM, Madihally SV, Robinson RL Jr, Gasem KA. Screening of chemical penetration enhancers for transdermal drug delivery using electrical resistance of skin. Pharm Res. 2008 Nov;25(11):2697-704. doi: 10.1007/s11095-008-9696-y. Epub 2008 Aug 6. PMID: 18683029. 【6】Schwingenschuh S, Scharfetter H, Martinsen ØG, Boulgaropoulos B, Augustin T, Tiffner KI, Dragatin C, Raml R, Hoefferer C, Prandl EC, Sinner F, Hajnsek M. Assessment of skin permeability to topically applied drugs by skin impedance and admittance. Physiol Meas. 2017 Nov 2;38(11):N138-N150. doi: 10.1088/1361-6579/aa904e. PMID: 28967873. 【7】Arpaia P, Cesaro U, Moccaldi N. Noninvasive measurement of transdermal drug delivery by impedance spectroscopy. Sci Rep. 2017 Mar 24;7:44647. doi: 10.1038/srep44647. PMID: 28338008; PMCID: PMC5364508.
应用实例
2024.04.12

TEWL经皮水分流失测量仪介绍
经皮水分流失测量仪 研究表明,离体皮肤的质量评价受皮肤新鲜度、厚度、质量和皮肤屏障完整性等因素的影响。皮肤屏障完整性是皮肤质量评价的重要参考属性。 经皮失水(TEWL)是衡量皮肤屏障完整性的一个重要标准,同时也是FDA IVPT工业指南中建议的皮肤屏障完整性评价方法之一,TEWL是通过测量皮肤表面的水蒸气分压的变化来推断出来的。TEWL的单位是g/h/m2(以每小时每平方米的失水克数表示)TEWL数值受环境条件的影响,如环境湿度、温度和气流,应在控制相应条件下进行测量。TEWL在不同的部位数值有显著差异,它的大小取决于汗腺、皮肤温度和角质层厚度等因素。 TE-007的特点:测量:对于离体皮肤的TEWL测量,常用的方法包括开放式仪器和封闭式仪器。开放式仪器通常通过放置TEWL传感器在皮肤表面,并在环境中保持恒定的湿度和温度来测量TEWL。而封闭式仪器则将皮肤样品与一个密封的环境接触,并测量一定时间内该环境中水汽的增加量,从而计算TEWL的数值。 在实际应用中,选择合适的TEWL测量方法取决于具体的实验需求和研究目的。封闭式仪器通常提供更严密的控制和更准确的结果,但可能需要更复杂的操作步骤和设备。而开放式仪器则更加灵活,并且适用于不同类型的实验条件和样品。TE-007属于封闭式测量方式,在一定条件下可以在封闭腔室内忽略温度湿度以及气流的影响。在测定时,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤下侧与接收室中的接收介质(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32℃±1℃半小时后,将TEWL探头直接放置在皮肤表面进行测量。通常,在测试结果稳定后,每个皮肤切片需至少重复测定3次,并记录相关结果。 应用医药行业:皮肤药物透过性评价:TEWL作为评价皮肤屏障完整性的重要指标,可用于评估不同药物在皮肤上的透过性能。药物传递系统研发:通过测量TEWL,可以评估不同药物传递系统的透皮输送效率和皮肤刺激性。皮肤疾病研究:TEWL可以用于评估不同皮肤疾病患者的皮肤屏障功能,如湿疹、银屑病等。 美容行业:皮肤保湿性能评价:TEWL可以作为评估不同护肤品的保湿效果的指标,用于确定护肤品的保湿性能和持久度。皮肤护理产品测试:通过测量TEWL,可以评估不同护肤产品对皮肤屏障功能的影响,包括清洁剂、化妆品和抗衰老产品等。 皮肤科:皮肤疾病诊断和治疗监测:TEWL可以用于评估皮肤疾病患者的皮肤屏障功能,并监测治疗效果的变化。皮肤创面愈合评估:TEWL可用于评估不同创面治疗方法对皮肤屏障恢复的影响,以指导创面愈合过程的管理和治疗。 生物医学研究:皮肤生理学研究:TEWL可以用于研究不同生理状态下皮肤屏障功能的变化,如年龄、性别和种族等因素对TEWL的影响。药物递送系统研究:TEWL可用于评估不同药物递送系统在皮肤上的渗透性能和递送效率,以指导药物递送系统的设计和优化。 相关法规: In Vitro Permeation Test Studies for Topical Drug Products Submitted in ANDAs.pdf 相关文献:【1】Schoenfelder H, Liu Y, Lunter DJ. Systematic investigation of factors, such as the impact of emulsifiers, which influence the measurement of skin barrier integrity by in-vitro trans-epidermal water loss (TEWL). Int J Pharm. 2023 May 10;638:122930. doi: 10.1016/j.ijpharm.2023.122930. Epub 2023 Apr 6. PMID: 37028576.【2】Verdier-Sévrain S, Bonté F. Skin hydration: a review on its molecular mechanisms. J Cosmet Dermatol. 2007 Jun;6(2):75-82. doi: 10.1111/j.1473-2165.2007.00300.x. PMID: 17524122.【3】Alexander H, Brown S, Danby S, Flohr C. Research Techniques Made Simple: Transepidermal Water Loss Measurement as a Research Tool. J Invest Dermatol. 2018 Nov;138(11):2295-2300.e1. doi: 10.1016/j.jid.2018.09.001. PMID: 30348333.【4】Peer RP, Burli A, Maibach HI. Unbearable transepidermal water loss (TEWL) experimental variability: why? Arch Dermatol Res. 2022 Mar;314(2):99-119. doi: 10.1007/s00403-021-02198-y. Epub 2021 Feb 26. PMID: 33638033.【5】Barel, A., Clarys, P., Gabard, B. (2017). Transepidermal Water Loss. In: Humbert, P., Fanian, F., Maibach, H., Agache, P. (eds) Agache’s Measuring the Skin. Springer, Cham. https://doi.org/10.1007/978-3-319-26594-0_142-2【6】Vemkata Vamsi K Venuganti and Omathanu P Perumal Effect of poly(amidoamine) (PAMAM) dendrimer on skin permeation of 5-fluorouracil. International Journal of Pharmaceutics 361:230-238, 2008. 【7】Dini V, Salibra F, Brilli C and Romanelli M Instrumental evaluation of the protective effects of a barrier film on surrounding skin in chronic wounds. Wounds 2008;20(9):254-257【8T.Amano.T.Takeda,H.YanoandT.Tamura Olopatadinehydrochlorideacceleratestherecoveryofskinbarrierfunctioninmice.BritishJournalofDermatology2007156:906:912.【9】Retroperitoneoscopic lumbar sympathectomy for the treatment of plantar hyperhidrosis: technique and preliminary findings Rieger R. and Pedevilla S., Surg Endosc, Jan;21(1):129-35, 2007.【10】Shampooing with Pyrithione Zinc Reduces Transepiderman Water Loss in Scalp of Dandruff-involved Patients Billhimer W, Erb J, Bacon R, Ertel K, Coopas M. Poster presentation at AAD 2006, Cape Town Efficacy of dietary hempseed oil in patients with atopic dermatitis Callaway J., Schwab U., Harvima I., Halonen P., Mykkänen O., Hyvönen P. and Järvinen T.Dermatology Treat, 16: 87-94, 2005.
应用实例
2024.04.12

法规译文|ANDA申请中递交的外用药物产品的体外渗透(IVPT)研究
ANDA申请中递交的外用药物产品的体外渗透(IVPT)研究Guidance for Industry 工业指南 DRAFT GUIDANCE 指南草案This guidance document is being distributed for comment purposes only. 本草案仅用于征求意见Comments and suggestions regarding this draft document should be submitted within 60 days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit electronic comments to https://www.regulations.gov. Submit written comments to the Dockets Management Staff (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register.For questions regarding this draft document, contact (CDER) Susan Levine at 240-402-7936.请于草案公布后60日内反馈建议或意见。网上提交请点击http://www.regulations.gov。书面资料请邮寄至FDA文件管理中心(HFA-305),地址为5630 Fishers Lane, rm. 1061, Rockville, MD 20852。反馈意见时请注明草案内容对应的行号。如有疑问请拨打240-4020-7936联系(CDER)Susan Levine。U.S. Department of Health and Human Services 美国健康福利部Food and Drug Administration 食品和药品管理局Center for Drug Evaluation and Research (CDER) 医药审评和研究中心(CDER)October 2022 2022年10月Generic Drugs 仿制药Additional copies are available from: 其他副本可从Office of Communications, Division of Drug Information 药品信息通讯厅Center for Drug Evaluation and Research 医药审评和研究中心Food and Drug Administration 食品和药品管理局10001 New Hampshire Ave., Hillandale Bldg., 4 th Floor Silver Spring, MD 20993-0002Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353Email: druginfo@fda.hhs.govhttps://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugsIn Vitro Permeation Test Studies for Topical Drug Products Submitted in ANDAsANDA申请中递交的外用药物产品的体外渗透(IVPT)研究Guidance for Industry(注释1:指南由食品药品管理局(FDA)的医药审评与研究中心( CDER)仿制药办公室( OGD)起草制订。)This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA or Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff responsible for this guidance as listed on the title page.该指南代表了 FDA 对该主题目前的看法。它并不会赋予任何人任何权利,也不会约束 FDA 或公众,如果有替代的方法能够满足法律法规的要求,可以使用替代的方法。如果想探讨替代的方法,请联系该指南首页中 FDA负责执行该指南的工作人员。I. Introduction 绪论This guidance is intended to assist applicants who are submitting abbreviated new drug applications (ANDAs) for liquid-based and/or other semisolid products applied to the skin, including integumentary and mucosal (e.g., vaginal) membranes, which are hereinafter called “topical products.”(注释2)Because of the complex route of delivery associated with these products, which are typically locally acting, and the potential complexity of certain formulations, topical products (other than topical solutions) are classified as complex products.(注释3)This guidance provides recommendations for in vitro permeation test (IVPT) studies comparing a proposed generic (test) topical product and its reference standard (RS) for the purpose of supporting a demonstration of bioequivalence (BE) to the reference listed drug (RLD). The reference standard ordinarily is the RLD.(注释4)本指南旨在帮助申请人提交用于皮肤的液体和/或其他半固体产品的仿制药申请(ANDA),包括皮肤和粘膜(如阴道),以下简称“外用制剂”(注释2)。由于这些制剂通常局部起效,具有复杂递送途径和配方,外用制剂(外用溶液除外)通常称为复杂制剂(注释3)。本指南为拟申报的仿制药与其对照制剂(RS)的体外渗透试验(IVPT)研究提供建议,以论证与参比制剂(RLD)的生物等效性(BE)。参比制剂通常简称为RLD。(注释4)注释2:在ANDA申请中,本指南所述的外用制剂包括软膏剂、乳膏剂、洗剂lotions、乳剂emulsions、糊剂pastes、洗发剂shampoos、凝胶剂、混悬剂、喷雾剂、气雾剂、泡沫剂foams、溶液和其他半固体和/或液体剂型,这些剂型具有特定的物质排列结构(可能包括多个相态)。注释3:根据仿制药使用者付费法案(GDUFA)重新授权绩效目标和2023-2027财年强化计划(常称为GDUFA III承诺函)(参见网址https://www.fda.gov/media/153631/download)中所述的定义,除另有规定外,复杂制剂产品包括具有复杂配方(如:胶体)和复杂递送途径(如:具体起效的皮肤用产品)的产品。注释4:根据21 CFR 314.3(b)定义,参比制剂是指作为ANDA申报中参照的FDA指定的已批准药物;对照制剂(RS)被定义为“寻求ANDA的批准必须在所需的体内生物等效性研究中使用的对照药品”。推荐采用对照制剂(RS)进行体外试验。在有些情况下(如RLD已经退市),RS可以不是RLD。关于RLD和RS的更多信息,见“ANDA申请参照药品行业指南(2020年10月)”,我们会对指南定期进行更新,关于指南的最新版本,详见网址:https://www.fda.gov/regulatory-information/search-fda-guidance-documents.This guidance does not address drug products that are administered via ophthalmic, otic, nasal, inhalation, oral, or injection-based routes, or that are transdermal or topical delivery systems (including products known as patches, topical patches, or extended release films).本指南不适用于通过眼、耳、鼻、吸入、口服或注射途径的药品,亦不适用于经皮或局部给药系统(包括贴剂、局部贴剂或缓释膜剂)。It is beyond the scope of this guidance to discuss specific topical products to which this guidance applies. FDA recommends that applicants consult this guidance and any relevant product-specific guidances (PSGs)(注释5)and any other relevant guidances for industry,(注释6)when considering the design and conduct of IVPT studies that, in conjunction with other studies, as deemed necessary, may be appropriate to support a demonstration that a proposed generic topical product and its RLD are bioequivalent. FDA also recommends that applicants routinely refer to FDA’s guidance web pages, because additional guidances may become available that could assist in the development of a generic topical product.本文不是讨论适用于该指南范围的特定外用制剂产品的指南。FDA建议申请人,在进行IVPT研究的设计和实施时,查阅特定产品开发指南(PSGs)(注释5)和其他行业指南(注释6),同时结合其他必要的研究,以支持拟申请的仿制药与RLD的生物等效性。此外,FDA建议申请人定期查阅FDA指南网站,及时获取最新的相关指南,以促进仿制药的开发。注释5:Generic drug product-specific guidances are available at FDA’s Product-Specific Guidances for Generic Drug Development web page at https://www.fda.gov/drugs/guidances-drugs/product-specific-guidances-generic-drugdevelopment. FDA仿制药特定产品开发指南,详见网址。注释6:其他相关行业指南包括:ANDA申请中递交的外用药物产品的体外释放研究指南(2022年10月)和ANDA 申请中递交的外用药物产品的物理化学和结构(Q3)特性(2022年10月)。该指南代表了FDA对该主题目前的看法。In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.一般而言,FDA 指南性文件并非具有强制执行的法律职能。实际上,指南陈述了管理部门对某一个问题当前的看法,并且仅作为建议,除非当具体的法规或法令要求被引用时。在指南中用到的词语“应该”,是指建议,并非要求的意思。II. Background 背景This guidance has been developed as part of FDA’s “Drug Competition Action Plan,”(注释7)which, in coordination with the Generic Drug User Fee Amendments (GDUFA)(注释8) program and other FDA activities, is intended to increase competition in the market place for prescription drugs, facilitate the entry of high-quality and affordable generic drugs, and improve public health.本指南已发展为FDA“药品竞争行动计划”(注释7)的一部分,配合GDUFA(注释8)项目和其他FDA举措,旨在促进处方药市场的竞争,促进优质和实惠的药品进入市场,提高公众医疗水平。注释7:See FDA Drug Competition Action Plan (describing the FDA’s Drug Competition Action Plan, implemented in 2017 and designed to, among other things, further encourage robust and timely market competition for generic drugs), available at https://www.fda.gov/drugs/guidance-compliance-regulatory-information/fda-drug-competitionaction-plan.注释8:In this guidance, GDUFA refers to the generic drug user fee program codified in the Generic Drug User Fee Amendments of 2012, Title III, Food and Drug Administration Safety and Innovation Act (Public Law 112-144), the Generic Drug User Fee Amendments of 2017, Title III, FDA Reauthorization Act of 2017 (Public Law 115-52), and the Generic Drug User Fee Amendments of 2022, Title III of Division F (the FDA User Fee Reauthorization Act of 2022) of the Continuing Appropriations and Ukraine Supplemental Appropriations Act, 2023 (Public Law 117-180).The Federal Food, Drug, and Cosmetic Act (FD&C Act) generally requires an ANDA to contain, among other things, information to show that the proposed generic drug product 1) is the same as the RLD with respect to the active ingredient(s), conditions of use, route of administration, dosage form, strength, and labeling (with certain permissible differences) and 2) is bioequivalent to the RLD.(注释9)Thus, an ANDA will not be approved if the information submitted in the ANDA is insufficient to show that the test product is bioequivalent to the RLD.(注释10)《联邦食品、药品和化妆品法案》(FD&C法案)要求ANDA申请中应包含合适的信息,说明:1)仿制药与RLD相比,具有相同的活性成分、使用条件、给药途径、剂型、规格和说明书(允许存在一些差异);2)与RLD生物等效。(注释9)因此,如果ANDA中提交的信息不足以证明自制制剂与RLD具有生物等效性,则ANDA将不予批准(注释10)。注释9:See sections 505(j)(2)(A), (j)(2)(C), and (j)(4) of the FD&C Act (21 U.S.C. 355(j)(2)(A), (j)(2)(C), (j)(4)); see also 21 CFR 314.94.注释10:21 CFR 314.127(a)(4), (6).An IVPT study may be used to assess the rate and extent to which a drug (i.e., an active ingredient) from a topical product becomes available at or near a site of action in the skin, and may be used to characterize and compare the rate and extent of bioavailability for a drug from a test topical product and RS. The IVPT flux profiles resemble pharmacokinetic profiles and can be analyzed using unique IVPT endpoints that are somewhat analogous to the pharmacokinetic endpoints of maximum concentration (Cmax) and the area under the concentration-time curve (AUC). Yet, IVPT studies characterize the rate and extent of absorption, not the distribution, metabolism and excretion that occurs in vivo. Therefore, while it is relevant to characterize the kinetics of topical drug bioavailability monitored by IVPT studies, the use in this guidance of the term “cutaneous pharmacokinetics” should not be construed to embody all aspects of pharmacokinetics—only those related to the absorption component that directly controls the rate and extent to which a topically applied drug becomes available locally at the site of action. This guidance focuses on general considerations and recommendations for the method development, method validation, and conduct of IVPT studies that are submitted in ANDAs and intended to support a demonstration of BE.(注释11)IVPT研究可用于评估外用药物产品中药物(即活性成分)到达皮肤作用部位或附近的速率和程度,表征和评估自研外用制剂与对照制剂(RS)中药物的生物利用度和程度。IVPT通量曲线类似于药代动力学曲线,可以使用独特的IVPT终点进行分析,该终点与最大浓度(Cmax)的药代动力学终点和浓度-时间曲线下面积(AUC)相似。然而,IVPT研究表征的是药物吸收的速率和程度,而不是体内的分布、代谢和排泄。因此,IVPT研究虽然可用于外用药物制剂中活性成分生物利用度的动力学表征,但本指南中的俗语“皮肤药代动力学”不应被解释为药代动力学的所有方面,其表征的仅仅是“外用制剂中活性成分达到作用部位的速率和程度”。本指南旨在说明,在ANDA申请中提交、拟用于BE论证的IVPT研究的方法开发和验证的一般考虑和建议。(注释11)注释11:A demonstration of no significant difference in the rate and extent of drug permeation into and through the skin of the test topical product and RS using an appropriately validated IVPT method can be used to support a demonstration of BE along with other data in the application (which may be specified in a PSG), as part of a comparative product characterization-based approach.在ANDA申请中,通过采用合适且经过验证的IVPT方法,说明自研外用制剂与对照制剂(RS)中药物的渗透速率和通过皮肤的量无明显差异,作为产品特性比较方法的一部分,并结合其他数据用于支持BE的论证(可能在“特定产品开发指南”中有规定)。III. IVPT method developmen IVPT方法开发The development of an IVPT method that is suitable to support a demonstration of BE for a specific topical product routinely involves a systematic series of exploratory studies. Inappropriate or insufficient efforts to develop an IVPT method that is suitable for its intended purpose increases the likelihood that the subsequent IVPT validation, pilot, and pivotal studies will ultimately be inadequate to support a demonstration of BE. By contrast, appropriate and systematic IVPT method development studies help to identify IVPT study designs and protocol (method) parameters which reliably produce flux profiles that can facilitate a comparison of the cutaneous pharmacokinetics of a drug delivered topically to the skin from test topical products and RSs.适用于支持特定外用制剂BE论证的IVPT方法的开发,通常需要进行一系列系统的探索性研究。不合适或不充分的IVPT方法开发可能会导致,后续的IVPT验证、初步和正式研究不足于支持BE论证。相反,合理且系统的IVPT方法开发研究,有助于明确IVPT研究设计和方案(方法)参数,产生可靠的通量曲线,以便于自研制剂和RSs的皮肤药代动力学比较。A detailed and well-organized IVPT method development report should be submitted in an ANDA to show how the IVPT method was optimized, and to support a demonstration that the method parameters selected for the IVPT are appropriate or necessary, particularly in situations where the method parameters are different from the methods recommended in this guidance). The Agency’s interest in reviewing the method development report is to understand why specific IVPT method parameters were selected and whether the resulting IVPT method is suitably sensitive and reproducible. This method development report should clearly indicate/distinguish the method parameters used for each set of data, illustrate the efforts made to optimize the IVPT method, and demonstrate that the method parameters selected for the IVPT are appropriate.应在ANDA中提交详细且结构清晰的IVPT方法开发报告,说明IVPT方法是如何优化的,所选择的IVPT方法参数是合理性或必要的,尤其是方法参数与本指南中建议的方法不同时。监管机构在审核IVPT方法开发的过程中,比较关注IVPT方法参数选择的合理性,以及确定的IVPT方法是否具有合适的灵敏度和重现性,因此,需要说明在IVPT方法优化过程中所作的努力,并证明所选择的IVPT方法参数是合适的。Applicants are encouraged to use the recommendations in this guidance, and if an applicant elects to use methods that are different from those recommended in this guidance, the IVPT method development report should demonstrate why it is scientifically justified to use an alternative approach than what is recommended in this guidance to optimize the IVPT method.(注释12)Some examples of recommended procedures are described in subsequent sections, to help applicants identify circumstances when information should be submitted in the ANDA to explain why a different procedure was utilized.鼓励申请人采用本指南中推荐的方法,如果申请人选择其他方法,应在IVPT方法开发报告中说明,采用替代的方法优化IVPT方法的科学合理性(注释12)。以下部分描述了本指南中推荐方法的一些示例,以帮助申请人识别在ANDA申请中何时需要递交相关信息,解释为什么采用了不同的方法。注释12:Applicants may choose to use an approach different from the approach recommended in this guidance. However, the alternative approach must comply with relevant statutes and regulations. See 21 CFR 10.115(d). 申请人可以采用指南中没有描述的其它方法。然而,所选用的替代方法必须遵守相关法律法规要求。A. IVPT Method Parameters IVPT方法参数All relevant parameters of the final IVPT method should be summarized (e.g., in a table) and submitted in the ANDA. Also, information should be provided to briefly explain the choice of the final IVPT method parameters like the equipment (e.g., a vertical diffusion cell (VDC)), skin source (e.g., cadaver), skin type (e.g., posterior torso), skin preparation (e.g., dermatomed), skin barrier integrity test (e.g., trans-epidermal water loss (TEWL) measurement), skin barrier integrity test acceptance criteria (e.g., 应对最终确定的IVPT方法的相关参数进行总结(如,以表格形式),并在ANDA申报资料中递交。此外,还应提供信息,简要说明最终确定IVPT方法参数选择的理由,譬如说:设备(如,立式扩散池(VDC))、皮肤来源(如,尸体)、皮肤类型(如,背部躯干)、皮肤处理方法(如,离体皮肤/皮节)、皮肤屏障完整性测试(如,经皮水分散射法(TEWL))、皮肤屏障完整性接受标准(如,<15克/平方米/小时(g/m2/h))、上样量(如,15毫克/平方厘米(15mg/cm2))、剂量维持时间(如,6小时)、研究持续时间(如,24小时、48小时等)、接受液取样时间点(如,1、2、4、6、8、12、16和24小时)等。B. IVPT Method Considerations IVPT方法考虑因素The choice of some IVPT method parameters like the equipment, skin source, skin type, skin preparation, and skin barrier integrity test procedures may be based upon investigator experience or convenience, like the availability of specific equipment or instrumentation in a laboratory, established tissue supply agreements, or other logistical considerations. However, if the chosen IVPT method parameters do not appear to be well-suited for a specific IVPT method, it is the applicant’s responsibility to systematically evaluate alternative method parameters, and ultimately, to validate that the IVPT method parameters chosen are suitable for the intended purpose. The recommended procedures for IVPT method validation are detailed in section IV of this guidance.IVPT方法参数的选择,如设备、皮肤来源、皮肤类型、皮肤处理方法和皮肤屏障完整性测试方法等,可能取决于研究人员的经验或便利性,譬如说实验室已有的设备或仪器、既定的组织供应协议或其他后勤保障等因素。但是,如果所选的IVPT方法参数似乎并不适合特定的IVPT方法,申请人有责任系统的评估替代的方法参数,确认所选的IVPT方法参数适用于既定的目的。本指南第四节详细介绍IVPT方法验证的推荐程序。The choice of other IVPT method parameters like the topical product dose amount, dose duration, study duration (which may be longer than the dose duration), sampling schedule, sampling procedures, receptor solution composition, and sample analytical method may be different for each IVPT method, and such parameters of IVPT methods should be systematically developed, optimized, and/or validated for the relevant topical product, as appropriate. The IVPT method development studies should characterize how differences in these method parameters influence the resulting IVPT flux profile so that optimal study conditions can be objectively selected from among those evaluated.IVPT其他方法参数的选择,如外用制剂的上样量、剂量维持时间、研究持续时间(可能比剂量维持时间长)、取样计划、取样方法、接收介质组成和样品分析方法,不同IVPT方法可能存在差异,因此需要对这些参数进行系统的开发、优化和/或验证,以确保其适用于相关的外用制剂产品。应在IVPT方法开发研究中,描述这些方法参数的差异如何影响最终的IVPT通量曲线,以便从这些评估中客观地选择最佳的研究条件。The selection of the dose amount used in the study should be assessed for each IVPT method based upon studies performed during IVPT method development. Different dose amounts may be compared in parallel on replicate skin sections from the same set of donors to optimize the dose amount for the IVPT study. Considerations for selecting an optimal dose amount may include (1) the consistency with which the dose can be applied (potentially using different dispensing and/or spreading techniques), (2) the reproducibility of the flux profiles, (3) the influence of dose amount and dose duration on the shape of the flux profile, and (4) the approximate range of drug concentrations in receptor solution samples at different time points (relative to the sample analytical method limits of quantification).应根据IVPT方法开发期间进行的研究,对每个IVPT方法中所选的上样量进行评估。可通过采用相同皮肤供体的重复皮肤切片进行不同剂量的平行对比研究,优化IVPT研究中所需的上样量。优化上样量需要考虑的因素包括:(1)采用相同的上样方式(可采用的上样技术包括:直接分配和/或涂覆);(2)通量曲线的重现性;(3)上样量和剂量维持时间对通量曲线的影响;和(4)接收液中不同取样时间点的药品浓度大致范围(与样品分析方法的定量限度相关)。The selected sampling schedule and study duration should be sufficient to characterize the cutaneous pharmacokinetics of the drug, which ideally includes a sufficiently complete flux profile to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points. A dose that remains on the skin for the duration of the study may continue to deliver the drug for a sustained period and may not necessarily exhibit a suitable decline in the flux at later time points. In such instances, it may be appropriate to develop an IVPT method that involves wiping off the applied dose after a suitable duration on the skin and continuing to monitor the receptor solution for an extended period thereafter, during which the decline in the flux profile can be characterized. The sampling frequency should be selected to provide a suitable resolution for the flux profile, and a minimum of eight non-zero sampling time points is recommended across the study duration (e.g., 48 hours).所选择的取样计划(即:取样时间点)和研究持续时间应足以表征药物的皮肤药代动力学,理想情况下应包括一个足够完整的通量分布,以识别最大(峰值)通量和此后多个时间点的通量下降情况。在研究期间,如果药品持续保留在皮肤上面,其可能会持续递送药物,在最大(峰值)通量之后时间点可能不会通量下降的现象。此时,在IVPT方法开发时,可在上样一段时间后将样品擦去,然后持续监控接收液一定时间,使之呈现出通量下降的情况。所选的取样频率应足以为通量曲线提供合适的分辨率,在整个研究期间(如,48小时),推荐至少采用8个非零取样点。C. IVPT Method Procedures and Controls IVPT方法程序和控制Suitable technical procedures and control parameters should be established during method development. These may include procedures for preparing and mounting the skin on the diffusion cell in a consistent manner, determining the instrument settings that regulate the skin surface temperature within the specified range, performing the barrier integrity test appropriately, controlling the accuracy and precision of the dose amount dispensed on each skin section.在方法开发过程中,应制定合适的技术规程和控制参数,包括:采用一致的方式准备皮肤并将其安装到扩散池上;确定仪器参数设置,使皮肤表面温度在既定的范围内;进行合适的屏障完整性测试;控制皮肤切片上上样量的准确度和精密度。For example, a dosing procedure may be developed that uses a positive displacement pipette to dispense a volumetrically controlled amount of a topical product, targeting the deposition on the skin of a certain mass (e.g., 15 mg/cm2) of topical product. If the inner diameter of the orifice in the dosing compartment of the diffusion cell is 15 millimeters (mm), and the effective dose area is ~1.77 cm2, this would indicate a target dose of ~26.5 mg of topical product per diffusion cell. Experiments during method development may establish that, based upon the density of the topical product, a specific volumetric setting on a specific model of positive displacement pipette with a specific pipette tip repeatedly dispenses ~27.5 mg of topical product (e.g., characterized by multiple replicate pipette dispensations into a weigh boat on a fine balance). This pipette setting may be optimal for a dosing procedure where the dose spreading instrument, like the flat bottom of a high performance liquid chromatography (HPLC) glass vial, or the rounded end of a glass rod or capillary tube, is subsequently used to spread the dispensed dose evenly upon the skin section mounted in the diffusion cell, and where repeatedly weighing the dose-spreading instrument before and after the dose spreading indicates that the residual topical product remaining on the bottom of the glass vial after the dose spreading reproducibly amounts to ~1.0 mg of topical product (indicating that ~26.5 mg of the topical product would reproducibly be dosed to each skin section). Such characterizations of the technical procedures and control parameters for the IVPT method, like the reproducibility of the dosing procedure, should be established during method development and may not need to be demonstrated thereafter each time the same procedure is used.例如,在上样方式的研究中,可以采用外置活塞式移液器(即:移液枪)把一定体积的外用药物制剂分配到皮肤上,使皮肤上具有一定重量的药物制剂(如,15mg/cm2);如扩散池供给室的孔口内径是15mm,则其有效面积约为1.77cm2,意味着每个扩散池中制剂的目标剂量约为26.5mg。在方法开发过程中,采用外置活塞式移液器及配套的枪头,根据外用制剂的密度,将外置活塞式移液器设定为相应的体积,依次重复分配外用药物制剂使其约为27.5mg(如,通过“可重复分配相应重量的外用药物制剂到天平称量盘上”进行论证)。这种移液器是上样分配样品的最佳选择,分配样品后可采用高效液相色谱(HPLC)玻璃瓶的平底、玻璃棒或毛细管的圆端,将分配到皮肤上的药品涂覆均匀;同时,在涂覆前后应分别称量涂覆用的工具,确保涂覆工具在涂覆前后重量相差约为1.0mg(表明可将约26.5mg的外用制剂重复均匀的涂覆到每个皮肤切片上)。IVPT方法的技术程序和控制参数等特性,如上样方法的可重现性,应在方法开发期间进行确定,但之后就不需要对这些特性再次论证。D. IVPT Skin Barrier Integrity Testing: Common Methods IVPT皮肤屏障完整性测试:常用方法The technical procedures for the skin barrier integrity test should be established during IVPT method development. Three types of barrier integrity tests are common, however, there are currently no applicable compendial standard protocols or acceptance criteria for any of these three types of human skin barrier integrity tests. Nonetheless, recommended parameters for the three common types of barrier integrity tests are discussed below.应在IVPT方法开发期间建立皮肤屏障完整性测试方法。常用的皮肤屏障完整性测试方法有三种,但还没有相应的药典收载或法定可接受标准。尽管如此,下面讨论三种常见类型屏障完整性测试的推荐参数。1. Trans-Epidermal Water Loss Skin Barrier Integrity Test 经皮水分散失法测试皮肤屏障完整性A TEWL skin barrier integrity test involves a measurement near the outer surface of the skin of the rate at which water (vapor) is fluxing through the skin barrier from the underside of the skin section. For the test, the skin section is mounted in a diffusion cell (e.g., clamped in place between the donor and receptor compartments), with the underside of the skin in contact with the receptor solution in the receptor compartment (e.g., phosphate buffered saline, pH 7.4), and equilibrated to a skin surface temperature of 32°C ± 1°C. If skin sections are cut large enough to cover the flange of the diffusion cell in which they are mounted, then after they have equilibrated for several hours at a skin surface temperature of 32°C ± 1°C, it may be feasible to gently remove the donor compartment without disrupting a skin section’s adherence to the lower flange of the diffusion cell, thereby allowing the TEWL probe to be placed directly on the skin surface, instead of being placed atop the donor compartment. Typically, a minimum of three replicate measurements are made on each skin section, which are recorded after the measurements have stabilized.TEWL皮肤屏障完整性测试是通过测定皮肤外表面附近的水分(蒸汽),评估水分从皮肤下侧通过皮肤屏障的速率。在测定时,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤下侧与接收室中的接收介质(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32℃±1℃。如果皮肤切片切割的足够大,能覆盖皮肤安装位点的扩散池法兰,可在皮肤表面温度平衡至32℃±1℃几小时后,在不破坏皮肤切片与其下部的扩散池法兰粘附的情况下,将供给室轻轻移去,此时可将TEWL探针直接放置在皮肤表面,而不用放置在供给室的顶部。通常,在测试结果稳定后,每个皮肤切片需至少重复测定三次,并记录相关结果。Commercially available devices to measure TEWL may differ in design and operational principles. The TEWL measured by devices with certain designs (e.g., an open chamber versus a closed chamber) may be relatively more susceptible to the influence of environmental conditions. Therefore, environmental temperature and humidity are typically controlled as precisely as possible (e.g., a temperature range of 21°C ± 2°C and a humidity range of 50% ± 20% relative humidity are ideal, if feasible). More precise control of the relative humidity (e.g., in the range of 40% – 50%) may reduce the variability of TEWL measurements for devices with certain designs. Certain designs of measurement probes and adapters for in vitro use are available by the manufacturers of TEWL devices, and may be appropriate to use. Inconsistency in the diameters for the measurement probe chamber, the measurement probe orifice, the in vitro adapters, and the skin area being measured, as well as variation in the distance of the probe sensor(s) from the skin surface, potentially because of the (variable) height of donor compartments (when applicable), could increase the variability of TEWL measurements. Inconsistent control of the alignment of the TEWL measurement device in relation to the donor compartment and/or the skin section may also increase the variability of TEWL measurements. Also, the TEWL measured by devices with certain designs may be relatively more susceptible to the influence of heat transfer from the hand that holds the probe. Applicants should follow relevant instructions in the manufacturer’s user manual for the specific TEWL measurement device used.商业化可用于TEWL测定的设备,有不同的设计和操作原理。由于设计的差异,一些TEWL测试设备可能相对更容易受到环境条件的影响(如,开放式腔室与封闭式腔室相比)。因此,通常需要尽可能精确的控制环境的温度和湿度(比较理想的情况是,将温度控制在21°C ± 2°C、湿度控制在50% RH± 20%RH范围内)。更精确的控制相对湿度(如,控制在40%RH~50%RH范围内)可以降低一些设备测定TEWL结果的变异性。TEWL测试设备生产商提供的用于体外的一些测试探针和适配器,可能适用于TEWL测定。不一致的测量探针室直径、测量探针孔、体外适配器和待测的皮肤面积,以及由于供给室高度的不同导致的探测传感器与皮肤表面距离的差异,均会增加TEWL测定结果的变异。与供给室和/或皮肤切片相关的TEWL测试设备对准的差异也可能增加TEWL测量结果的变异。此外,有些TEWL测试设备可能相对更容易受到握持探针的手的热传递的影响。对于特定的测试设备,申请人应遵循生产商使用手册中的相关说明。No more than approximately 15 grams of water per square meter per hour (i.e., ≤15g/m2/hr)could be a reasoable skin barrier integrity acceptance (cutoff) criterion for a TEWL barrier integrity test on human torso or thigh skin; if this was selected as the cutoff criterion, skin sections with a TEWL> 15g/m2/hr would fail the test. Skin sections that fail a barrier integrity test should not be dosed, but may serve as non-dosed control skin sections. A higher cutoff (e.g., ≤20 g/m2/hr) may also be reasonable if justified by experimental data demonstrating that the selected acceptance criterion appropriately discriminates skin sections with a compromised barrier integrity from those with a competent barrier integrity.对于人体躯干或大腿皮肤,采用TEWL屏障完整性测试法,合理的皮肤屏障完整性接受(截止)标准是每小时每平方米不大于约15g水(即,≤15g/m2/hr);如果选择其作为截止标准,TEWL大于15g/m2/hr的皮肤,则不具有皮肤完整性。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可用作为无剂量空白对照皮肤切片。但是,如果通过实验数据表明,所选的接受标准可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片,也可以采用更高的截止标准(如,≤20g/m2/hr)。However, TEWL measurements for skin sections with a competent barrier integrity can vary depending upon the TEWL measurement device, the manner in which it is operated, and the environmental conditions (e.g., higher ambient humidity or greater distance from the skin surface may decrease the value of the TEWL measurement). Precise control of environmental and device/operational factors can minimize variability in TEWL measurements. Therefore, the technical procedures for measuring TEWL should be optimized during IVPT method development (or based upon prior optimization in the laboratory performing the test). Also, the TEWL measurement device should be appropriately calibrated (by the manufacturer, and for some devices, also before each set of tests). Applicants may provide information about the relevant calibration procedures specified by the manufacturer for the specific TEWL device used; this can be submitted in the ANDA along with the IVPT method development report, to support the appropriateness of the technical procedures established by the laboratory for TEWL measurements. When a TEWL barrier integrity test is used in any study phase (IVPT method development, pilot study, validation, and/or pivotal study) the ambient laboratory temperature and humidity during the TEWL barrier integrity test should be monitored and reported.然而,具有合格屏障完整性皮肤的TEWL测量值,可能会因TEWL测试设备、操作方式和环境条件的不同而变化(如,较高的环境湿度或距离皮肤表面较远的距离可能会降低TEWL的测量值)。对环境和设备/操作因素的精确控制可将TEWL测量的可变性将至最低。因此,在IVPT方法开发过程中应对TEWL的测定方法进行优化(或基于实验室之前进行该测试的优化结果)。此外,应对TEWL测试设备进行适当的校准(由生产商进行;对于某些设备,在每次实验前都需要进行校准)。申请人可提供生产商制定的相关校准程序信息,与IVPT方法开发报告一起在ANDA中递交,用于支持申请人为TEWL测量制定的技术程序的合理性。在IVPT方法开发、初步研究、验证和/或正式研究的整个研究阶段,进行TEWL屏障完整性测试时,均应监测和报告实验室的环境温度和湿度。2. Tritiated Water Skin Barrier Integrity Test 氚化水渗透法测试皮肤屏障完整性An example of a recommended approach to a tritiated water skin barrier integrity test would be to mount the skin in a diffusion cell (e.g., clamped in place between the donor and receptor compartments) and allow it to equilibrate to a skin surface temperature of 32°C ± 1°C with the stratum corneum exposed to the air in the donor compartment and the underside of the skin in contact with the receptor solution (e.g., phosphate buffered saline, pH 7.4).氚化水渗透法测试皮肤屏障完整性的一个推荐方法是,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤的角质层暴露在空气中(即,朝向供给室),皮肤下侧与接收液(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32°C ± 1°C。A small amount of tritiated water (sufficient to cover the entire surface of the skin section) would be briefly dosed on the stratum corneum. This dose of tritiated water would be left on the surface for a precisely controlled and relatively brief period (e.g., 5 minutes) after which it would be removed from the skin surface (e.g., using a pipette to remove the bulk volume and then an absorbent low lint laboratory tissue to gently blot dry). The receptor solution would then be sampled at a precise duration after the removal of the tritiated water from the skin surface (e.g., 30 minutes after the removal of the 5-minute dose of tritiated water from the skin surface).将少量的氚化水(足以覆盖皮肤切片的整个表面)上样到皮肤角质层,精确控制氚化水留在皮肤表面的时间(如,5分钟),到特定时长后立即将其从皮肤表面移除(如,先用吸管去除大量的氚化水,然后用吸收性低脂棉轻轻吸干)。在氚化水从皮肤表面移除后的特定时间点(如,将氚化水上样到皮肤表面5分钟后,再将其移除之后的30分钟)从接收液中取样。While the entire volume of the receptor compartment may be removed and replenished, typically only an aliquot of the receptor solution (e.g., phosphate buffered saline, pH 7.4) is transferred to a suitable volume of scintillation fluid for counting. The volume of the aliquot typically depends upon the type of scintillation fluid used and the maximum amount of aqueous fluid that is suitable to mix with the scintillation fluid. A scintillation counter is then used to quantify the amount of radioactivity in the aliquot sampled, which can be used to calculate the amount of tritiated water that permeated into the larger (entire) volume of receptor solution; the calculation is performed using the specific activity of the tritiated water to equate a given amount of radioactivity to the equivalent volume of tritiated water that permeated per square centimeter of skin surface area.虽然可以完全移取整个接收室的接收液(如PBS,pH7.4),通常情况下,移取一部分接收液转移至适当体积的闪烁液中用于计数即可。移取的接收液体积通常取决于闪烁液的类型及可与闪烁液混合的最大含水量。然后,采用闪烁计数器对取出的接收液的放射性进行定量,之后可进一步推导出整个接收室的接收液中含有的氚化水量;该计算是根据氚化水的比活度进行的,即:给定量的放射性与每平方厘米皮肤渗透的当量体积的氚化水相当。Approximately 1.5 equivalent (eq.) microliter (μL) of tritiated water per cm2 (i.e., ~1.5 eq. μL/cm2 or ~1.5 eq. mg/cm2) would be a reasonable skin barrier integrity acceptance (cutoff) criterion for a tritiated water barrier integrity test that involves a 5-minute dose followed by a 30minute sampling duration (i.e., sampling 30 minutes after dose removal) on human torso or thigh skin. Skin sections with a tritiated water test result of > 1.5 eq. mg/cm2 would fail the test and be excluded from the population of skin sections dosed with the topical product; skin sections that fail a barrier integrity test should not be dosed, but may serve as non-dosed control skin sections. Other acceptance criteria may also be reasonable if justified by experimental data demonstrating that the selected acceptance criterion appropriately discriminates skin sections with a compromised barrier integrity from those with a competent barrier integrity.对于人体躯干或大腿皮肤,采用氚化水屏障完整性测试法,将氚化水上样到皮肤表面5分钟后移除,在移除30分钟后取样(即:将氚化水移除后30分钟后取样),合理的皮肤屏障完整性接受(截止)标准是每平方厘米约1.5当量微升的氚化水(即:1.5 eq.μL/cm2 or ~1.5 eq.mg/cm2)。采用氚化水屏障完整性测试法,如果测定结果大于1.5 eq.mg/cm2,则不具有皮肤完整性。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可用作为无剂量空白对照皮肤切片。但是,如果通过实验数据表明,所选的接受标准可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片,也可以采用其他标准。When calculating the results for a tritiated water barrier integrity test, it may be important to account for the surface area dosed. For example, if using an acceptance criterion of 1.5 eq. mg/cm2 with a diffusion cell that has an orifice diameter of 15 mm and a skin surface area of 1.77 cm2, the mass of tritiated water that would be calculated to have permeated into the receptor compartment would be ~2.7 eq. mg/cm2 of tritiated water.对于氚化水屏障完整性测试法,在结果计算时,需要重点关注上样表面积。例如,当扩散池的直径是15mm时,相应的皮肤表面积为1.77 cm2,如果采用1.5 eq.mg/cm2的接受标准,则渗透进入接收室的氚化水的量应不超过2.7 eq. mg/cm2。3. Electrical Based Skin Barrier Integrity Tests 电阻/电导值法测试皮肤屏障完整性There are several variations of electrical based skin barrier integrity tests that report the test result as a measure of the resistance, conductance, or a related electrical concept that characterizes the bulk flow of electrical current across the skin. Transepithelial electrical resistance tests involving the skin may be referred to more specifically as Trans-Epidermal Electrical Resistance (TEER) skin barrier integrity tests. The test results may be described in units of conductance, which is the reciprocal of resistance. Electrical based skin barrier integrity tests often use instruments that are designed to measure the inductance (L), capacitance (C), and resistance (R) of electronic circuits or electrical components; these instruments are commonly known as LCR meters and have different settings (test parameters) that can be adjusted.电阻/电导法测定皮肤完整性有几种变量,以电阻、电导或相关的电概念报告测试结果,用于表征流过皮肤的电流量。与皮肤相关的跨膜电阻测试通常指跨膜电阻(TEER)皮肤屏障完整性测试。测试结果可以用电阻的倒数“电导”为单位进行描述。电阻/电导值法测试皮肤屏障完整性通常采用“旨在测量电子电路或电子元件的电感(L)、电容(C)和电阻(R)”的仪器;这些仪器通常被称为LCR仪表,具有可调节的不同设置(测试参数)。An example of a recommended approach to a TEER skin barrier integrity test would be to mount the skin in a diffusion cell (e.g., clamped in place between the donor and receptor compartments) and allow it to equilibrate to a skin surface temperature of 32°C ± 1°C with the stratum corneum exposed to the air in the donor compartment and the underside of the skin in contact with an ionic solution (e.g., phosphate buffered saline, pH 7.4).TEER皮肤屏障完整性测试的一个推荐方法是,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤的角质层暴露在空气中(即,朝向供给室),皮肤下侧与接收液(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32°C±1°C。A small amount of the ionic solution (sufficient to cover the entire surface of the skin section) would be briefly dosed on the stratum corneum. Then, one lead/electrode from an LCR meter would be placed in contact with the solution in the receptor compartment while the other lead/electrode would be placed in contact with the solution in the donor compartment. After measuring the resistance across the skin (e.g., in kΩ, normalized for area, noting that resistance is inversely proportional to area) the solution in the donor compartment would be removed and the skin surface would be gently blotted dry with an absorbent low lint laboratory tissue. The skin (still mounted in the diffusion cell) would then be allowed to equilibrate with the dry air above for a sufficient duration to normalize the hydration state of the stratum corneum before being dosed with the test topical product or RS.将少量的离子溶液(足以覆盖皮肤切片的整个表面)短暂的施加到皮肤角质层上;然后,将LCR仪表的一端电极与接收室中溶液接触,而另一端电极与供给室中溶液接触。在测定通过皮肤的电阻后(如,以kΩ为单位,按面积归一化法,注意电阻与面积成反比),将供给室中的溶液移出,用吸收性低脂棉将皮肤表面轻轻吸干。然后,在自研外用制剂或RS上样前,先将皮肤与上面的干燥空气接触,并平衡足够长的时间,使角质层恢复到正常的水合状态。The results for a TEER skin barrier integrity test can vary substantially depending on the LCR meter settings (e.g., frequency) and the technical procedures used for the test. The acceptance criterion for a specific electrical based skin barrier integrity test method may be justified by experimental data demonstrating that the selected acceptance criterion appropriately discriminates skin sections with a compromised barrier integrity from those with a competent barrier integrity.TEER皮肤屏障完整性测试的结果,可能因LCR仪表设置(如,频率)和所用的测试技术方法不同,产生很大变化。电阻/电导值法测试皮肤屏障完整性接受标准的合理性,可通过试验数据证明,即:所选的接受标准应当可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片。E. IVPT Skin Barrier Integrity Testing: General Considerations IVPT皮肤完整性测试:注意事项There are three general considerations for the development or adoption of technical procedures for any skin barrier integrity test method during IVPT method development:在方法开发过程中,皮肤屏障完整性的任一测试方法的开发或选择,都应注意以下三个方面:The technical procedures should not irreversibly alter the skin barrier. It may be acceptable to temporarily alter the hydration state of the stratum corneum by briefly depositing an aqueous solution on the surface of the skin, as long as sufficient time is afforded for the hydration of the stratum corneum to normalize before dosing of the topical product. The procedure described above for a brief (e.g., 5-minute) exposure of the skin surface to tritiated water followed by a 30-minute duration during which the hydration state of the stratum corneum is re-equilibrating would likely be appropriate. By contrast, a 30-minute exposure of the skin surface to an aqueous solution for an electrical-based test method, followed within 5 minutes by dosing of the topical product, may not be appropriate without further characterization of the influence of the hydration state of the stratum corneum on the discrimination sensitivity of the skin to differences in topical bioavailability. Similarly, if a portable lamp were placed close to the skin to improve visibility while study procedures were being performed, the heat from the lamp may alter the local (micro)environment of the skin in a manner that is not representative of the ambient environmental conditions in the laboratory; this should be avoided.测定方法不应不可逆的改变皮肤完整性。可以接受将水溶液上样到皮肤表面后,短暂的改变角质层水合状态的情况,但在外用制剂上样前,经过足够长的时间后,角质层应可以恢复到正常的水合状态。如前面描述的情况,将皮肤表面短暂的暴露于氚化水一段时间(如,5分钟)后移除,经过30分钟的重新平衡,角质层的水合状态可以恢复到合适水平。相比之下,对于电阻/电导值测试法,将皮肤暴露于水溶液中30分钟后移除,而后,在5分钟后上样是不合适的,因为没有进一步评估皮肤角质层水合状态对不同外用制剂生物利用度区分灵敏度的影响。类似情况是,在测试过程中,如果将携带式手提灯放置在皮肤附近以提高能见度,从灯中散发出的热可能会改变皮肤的局部(微)环境,并不能代表实验室中的环境条件,应当避免。The acceptance criterion should be a cutoff value for the test result, at which a skin section fails the test. Skin sections that fail a barrier integrity test should not be dosed but may serve as non-dosed control skin sections. Skin sections with a passing barrier integrity test result may be considered to have a competent barrier integrity and may be dosed. This acceptance criterion should be selected based upon an understanding of the distribution of test results (among multiple replicate skin sections from multiple donors) for the specific barrier integrity test procedure performed with the specific type and preparation of skin under conditions relevant to the IVPT pivotal studies submitted in the ANDA. The intention of the barrier integrity test is to identify (and exclude) skin sections whose barrier integrity (intactness) is compromised. The intent is not to reduce the inherent variability in barrier function (permeability) in human skin that is representative of real variation in the human population. Also, the relative permeability of the skin to a drug from a topical product may not necessarily correlate with the permeability of the skin to water, and therefore, constraining the variability of the skin permeability to water (using a stricter acceptance criterion that excludes a larger number of skin sections) may not necessarily reduce the variability in the IVPT study results.接受标准应是皮肤未通过屏障完整性测试的截止值。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可作为无剂量空白对照皮肤切片。通过皮肤完整性测试的皮肤切片,可被视为具有合格的屏障完整性,可以进行上样试验。接受标准的选择可以依据,在ANDA申请中递交的IVPT正式研究中,采用特定类型和制备工艺皮肤进行的屏障完整性测试,对获得的测试结果(采用多个供体的多个重复皮肤切片进行)分布的理解进行选择。皮肤屏障完整性测试的目的是识别(和排除)屏障完整性(完整无缺)受损的皮肤切片。其目的并不是减少人体皮肤屏障功能(渗透性)的内在变异性,人体皮肤的内在变异性是其固有的特性。此外,皮肤对外用制剂中药物的相对渗透性,可能与皮肤对水分的渗透性无关;因此,限制皮肤对水分渗透的可变性(使用更严格的接受标准,排除更多的皮肤切片)可能不一定会减少IVPT研究结果的可变性。The acceptance criterion should be able to discriminate skin sections with a compromised barrier integrity. This may be demonstrated by measuring the barrier integrity of skin sections mounted and equilibrated in a diffusion cell before and after deliberately compromising the skin barrier (e.g., by repeatedly using adhesive tape to strip away increasing amounts of the stratum corneum, piercing the skin several times with a 30 gauge needle, or using other physical or chemical insults to damage the skin barrier). Based upon the acceptance criterion selected, the test result for skin sections that pass the test before being damaged should fail the test after the damage.接受标准应能够区分屏障完整性受损的皮肤切片。对于接受标准的论证,可通过“在特意破坏皮肤屏障的前后(如,通过反复使用胶布剥离越来越多的角质层、用30号针多次刺穿皮肤,或使用其他物理或化学手段破坏皮肤屏障)”,将其分别安装到扩散池并平衡后,根据屏障完整性的测定结果来评估。根据选择的接受标准,在破坏前通过测试的皮肤切片,对其破坏后应不能通过测试。F. Differences Between IVPT Method Development and Validation IVPT方法开发和验证的区别1. Optimization of an IVPT Method Prior to Advancing to IVPT Method Validation 在IVPT方法验证前进行IVPT方法的优化Different study designs and method parameters may be evaluated during the IVPT method development phase. For example, if the selected study parameters initially involve a dose duration of 48 hours and a study duration of 48 hours, and the flux profile is measurable, but it is not feasible to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points, then it may be appropriate to evaluate other study parameters as part of the IVPT method development. For example, a different target dose of the topical product and/or a longer sampling duration may be evaluated. An alternate study design may involve a shorter dose duration (e.g., 4–6 hours) after which the applied dose is removed from the skin, and the receptor solution continues to be sampled across a study duration that is sufficient to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points. While shorter dose durations can help to improve the shape of IVPT flux profiles, the removal of the topical product dose from the skin surface can be challenging and often requires its own method development and optimization. Also, the design of sensitivity studies for such an IVPT study design may require a more sophisticated understanding of IVPT studies. While reasonable efforts should be made to develop an IVPT method that produces a well-defined maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points, this may not be feasible for certain topical products even with study durations of 96 hours, or, at least, may not be feasible to produce reliably in all donors. In such circumstances, the IVPT method development report should detail the systematic efforts made to optimize the IVPT method.在IVPT方法开发期间可以对不同的研究设计和方法参数进行评估。例如,如果最初选择的研究参数是剂量维持时间和研究总体时长均为48小时,但测定结果发现,在该条件下不能识别出最大(峰值)通量和之后多个取样时间的通量下降情况。此时,采用较短的剂量持续时间(如,4~6小时)可能有助于改善IVPT通量分布曲线,但将外用制剂从皮肤表面移除是具有挑战性的,通常需要单独进行方法开发和优化。此外,对于IVPT研究中灵敏度的研究设计,可能需要对IVPT研究有更全面清晰的理解。虽然应尽力进行IVPT方法开发,以便可以识别出最大(峰值)通量和此后多个时间点的通量下降情况,但某些外用制剂即使研究持续时长达到96小时,也不能产生理想的通量分布曲线,或者,不能在所有的供体中产生可重复的结果。在这种情况下,应在IVPT方法开发报告中详细说明为IVPT方法优化所做出的努力。2. Use of a Validated Sample Analytical Method for IVPT Method Validation 采用已验证的分析方法进行IVPT方法验证The IVPT method development studies, being exploratory in nature, are often performed using a sample analytical method that is not validated (e.g., an HPLC or ultrahigh performance liquid chromatography (UPLC) method, often involving mass spectrometry (MS)); also, IVPT method development studies are often conducted in a manner that is not compatible with a quality management system which would otherwise make the evidence generated suitable to support valid conclusions. Such method development studies would not be suitable to demonstrate the validity of an IVPT method, or associated results. Therefore, although it may appear to be redundant, certain experiments performed during IVRT method development may need to be repeated during IVPT method validation, using appropriate controls, like a validated analytical method and procedures that are compatible with a suitable quality management system.IVPT方法开发研究,本质上具有探索性,通常是采用未经验证的样品分析方法(如,HPLC或UPLC,通常需要进行质谱分析(MS))进行的;此外,IVPT方法开发研究通常与质量管理体系不兼容,产生的数据不足以支撑可靠的结论。因此,方法开发研究不足以支撑IVPT方法验证或相关的结果。尽管看起来似乎是多余的,但在IVPT方法开发期间进行某些试验可能需要在IVPT方法验证期间重复进行考察,进行适当的控制,如与适当的质量管理体系相兼容的已验证的分析方法和程序。It is important to clearly segregate and consistently identify those experiments and results that were part of IVPT method development separately from those that were part of IVPT method validation. It is also important to consistently identify all relevant method parameters and experimental conditions/controls for each set of IVPT results. Information in the method development report should clearly identify/distinguish when the results for apparently similar sets of experiments may have been obtained using different method parameters. Method development reports should clarify which sets of diffusion cells were run in parallel or separately (e.g., on separate days). In addition, the sample analytical method parameters used to analyze the samples from each set of IVPT experiments should be specified, and the report should indicate whether or not the sample analytical method was validated (either at the time of sample analysis or subsequently).需要注意的是,应将IVPT方法开发和IVPT方法验证的试验结果分开来看,并采用一致的评价标准;对于每组IVPT结果,所有相关的方法参数和试验条件/对照都应保持一致,这点非常重要。当采用不同的方法参数可以获得明显相似的试验结果时,应在方法开发报告中清楚的说明。在方法开发报告中,也应阐明扩散池是平行还是单独运行(如,在不同的日期)的。此外,应说明在IVPT试验中,用于样品分析的具体方法参数,并说明分析方法是否经过了验证(在样品分析时或样品分析后)。IV. IVPT method validation IVPT方法验证When all the relevant parameters of the IVPT method have been established, a pilot study should be performed using the final IVPT method and using a validated sample analytical method. The purpose of the pilot study is to validate the suitability of the selected IVPT method parameters by demonstrating that the performance characteristics of the IVPT method are appropriate to compare the cutaneous pharmacokinetics of a drug delivered topically from a test product and RS. The results from the pilot study, thereby, support the systematic validation of the IVPT method, which proceeds as a distinct study phase following IVPT method development.在所有相关的IVPT方法参数确定之后,应采用最终确定的IVPT方法和已验证的样品分析方法进行初始研究。初步研究的目的是,通过论证IVPT方法的性能特性适用于自研制剂和对照制剂(RS)中药物的皮肤药代动力学比较,确认所选IVPT方法参数的适用性。因此,初步研究的结果支持IVPT方法的系统验证,这是IVPT方法开发之后的一个特有的研究阶段。The results from this IVPT pilot study can help to estimate the number of donors that may be needed to adequately power the IVPT pivotal study. In addition to the test topical product and RS evaluated in the pilot study, a parallel assessment should be performed with a third topical product or formulation that is known or designed to be different from the RS, to validate the selectivity of the IVPT method to discriminate differences in bioavailability. The IVPT pilot study results should be plotted with error bars, comparing the permeation profiles for the three treatment groups in the pilot study. Separate plots should be prepared for average flux results and average cumulative permeation results. These data can be used to support specific IVPT method validation parameters (e.g., permeation profile and range).IVPT的初步研究结果有助于评估在IVPT正式研究中所需的供体数量。在IVPT的初步研究中,除了对自研外用制剂和对照制剂(RS)评估外,还应对已知或设计与RS不同的另一种外用产品或配方进行平行评估,以确认IVPT方法在区分生物利用度差异方面的选择性。应用误差线绘制IVPT的初步研究结果,对三个试验组的渗透曲线进行比较。这些数据可用于支持特定的IVPT方法验证参数(如,渗透概况和范围)。A pilot IVPT study performed with multiple skin donors (e.g., 4–6 skin donors) and a minimum of four replicate skin sections per donor per treatment group is recommended. As skin from an increasing number of donors is evaluated in the pilot study, the accuracy of the estimated number of donors needed to adequately power the IVPT pivotal study may improve. While skin from the same donors evaluated in the pilot study may also be used in the IVPT pivotal study, the results from the pilot study should not be combined with the results from the IVPT pivotal study for the purpose of statistical analysis.在IVPT初步研究中,建议采用多个皮肤供体(如,4~6个皮肤供体),且每个试验组的每个供体至少4个重复皮肤切片。随着IVPT初步研究中评估的供体数量的增加,可逐步提高IVPT正式研究中所需的供体数量准确性。虽然在IVPT初步研究评估中用的相同供体皮肤也可用于IVPT正式研究,但初步研究的结果不能与IVPT正式研究中获得的结果合并进行统计分析。The equipment, methodologies, and study conditions used in the IVPT pilot study (and the eventual IVPT pivotal study) should be appropriately validated or qualified. If an applicant elects to use equipment, methodologies, or study conditions that are different from those recommended in this guidance, the applicant should demonstrate why it was necessary and scientifically justified to do so. Detailed protocols and well-controlled study procedures are recommended to ensure the precise control of dosing, sampling, and other IVPT study parameters, as well as potential sources of experimental bias.IVPT初步研究(和最终的IVPT正式研究)中所用的设备、方法和研究条件应进行合适的验证或确认。如果申请人使用的设备、方法或研究条件与本指南中推荐的不一致,申请人应说明这种选择的必要性,并证明其科学合理性。建议建立详细的方案和良好的研究控制程序,以精确控制上样、取样和其它IVPT研究参数,以及潜在的试验偏差来源。The validation of the IVPT method should incorporate specific qualifications and controls (described below), performed using a validated sample analytical method, as applicable. The qualification of an IVPT method parameter refers to the process of defining what attributes make it suitable to perform its function in the IVPT method. For example, when repeated measurements of the temperature at the surface of skin mounted in a diffusion cell demonstrate that an IVPT equipment can maintain the skin surface temperature in the range of 32°C ± 1°C, the results can support a demonstration that the equipment is qualified to perform its function in an IVPT method for which a method parameter is the control of skin surface temperature in the range of 32°C ± 1°C across the relevant study duration.IVPT方法验证应包括下述的确认和控制内容,如适用,使用已验证样品分析方法。IVPT方法参数确认是表征IVPT方法的属性是否适合于执行其功能的过程。例如,当重复测定安装在扩散池中的皮肤表面温度时,IVPT设备可以维持皮肤表面温度在32℃±1℃范围内,这个结果可以用于证明,在整个IVPT方法研究期间,该设备可以执行其功能,将皮肤表面温度控制在32℃±1℃范围内。A. Equipment Qualification 设备确认Suitable equipment for the IVPT method includes various models of VDCs and flow-through diffusion cells. The operating principles and specific test procedures differ among the various equipment; relevant procedures from the manufacturer may be used for installation, operational, and performance qualifications. The laboratory qualification of each diffusion cell should, at minimum, include 1) measurements of the diffusional area of the orifices of the donor and receptor compartments between which the skin is mounted, 2) the empirically measured volume of the receptor solution compartment in each VDC or the empirically measured outflow tube length for each flow-through diffusion cell, 3) the stability of the temperature measured at the skin surface (e.g., 32°C ± 1°C) across a relevant duration (e.g., 48 hours), and 4) the rate of stirring or agitation in VDCs, or the flow rate for flow-through diffusion cells, as applicable.可用于IVPT方法的设备有VDCs(立式扩散池)和流通池。每种类型设备的操作原理和相关测定方法不同;可根据生产商提供的相关规程进行安装、操作和性能确认。各种型号的扩散池确认至少包括:1)测定供给室和接收室之间皮肤安装位置的孔口扩散面积;2)测量VDC接收室的容积,或流通池输出管的长度;3)在整个研究期间(如,48小时),测定皮肤表面温度(如,32℃±1℃)的稳定性;和4)如适用,测定VDC的搅拌速率,或流通池的流速。If information related to the diffusional area of the orifices and the volume of the receptor solution compartment for each diffusion cell is available from the manufacturer, that information should be provided for each relevant diffusion cell, in addition to the empirical measurements made by the laboratory performing the IVPT studies. The equipment should control the diffusion cell temperature so that the skin surface temperature is verified to be stable (e.g., 32°C ± 1°C) for each diffusion cell before dosing (e.g., measured by a calibrated infrared thermometer), and monitored periodically throughout the duration of the experiment by repeatedly measuring the skin surface temperature of a non-dosed control diffusion cell that is run in parallel with the other replicate dosed diffusion cells and connected to the same water bath or thermoregulation system.如果可以从生产商处获取每个扩散池的孔口扩散面积和接收室的容积信息,除了提供实验室进行IVPT研究时测定的相关结果,还应提供生产商提供的信息。设备应可以控制扩散池的温度,确保在上样前,皮肤表面温度稳定(如,通过校准的红外温度计测定在32℃±1℃范围内);并在整个试验期间通过重复测定,连接到相同的水浴或温度调节系统、与上样剂量组平行运行的非剂量控制组的皮肤表面温度进行定期监测。B. Membrane (Skin) Qualification 膜 (皮肤) 确认Excised human skin is recommended as the membrane for the IVPT study. The validity of each skin section dosed in the study should be qualified using an appropriate test procedure to evaluate the stratum corneum barrier integrity. Acceptable barrier integrity tests may be based upon tritiated water permeation, TEWL, or electrical impedance/conductance measured across the skin. The test parameters and acceptance criteria used for the skin barrier integrity test should be justified for the specific method and instrumentation that is used during the study. The skin from all donors whose skin is included in the study should be prepared in a consistent manner and dermatomed to a relatively consistent thickness, within limits specified in the study protocol. The skin thickness should be measured and reported for each skin section included in the study. The assignment of replicate skin sections from a donor to each treatment group should be randomized, as feasible. It is acceptable to balance the distribution of skin thicknesses in each treatment group (test topical product or RS) by a procedure specified in the study protocol建议采用离体人类皮肤作为IVPT研究用膜。应采用合适的方法评估角质层的屏障完整性,以对上样用皮肤切片的有效性进行验证。可接受的屏障完整性测试方法包括氚化水渗透、TEWL或电阻/电导值法。研究期间所用皮肤屏障完整性测试的参数和可接受标准,应根据所用的方法和仪器,说明其合理性。研究中所用的供体皮肤应采用相同的方式进行处理,且皮肤厚度应相对一致,以满足研究方案中拟定的限度要求。如适用,应将供体皮肤的重复皮肤切片随机分配给每个试验组。可以根据研究方案中规定的程序来平衡每个试验组(自研外用制剂和RS)的皮肤厚度分布。C. Receptor Solution Qualification 接收介质确认The composition and pH of the receptor solution used for the IVPT study should be qualified in relation to its compatibility with the skin as well as the stability and solubility of the drug in that receptor solution. The stability of the drug in the receptor solution samples should be validated as part of the receptor sample analytical method validation. The solubility of the drug in the IVPT receptor solution should be empirically determined in triplicate, to illustrate that the solubility of the drug in the receptor solution exceeds the highest sample concentration in the IVPT pivotal study, ideally by an order of magnitude. The solubility of the drug in the IVPT receptor solution should be sufficient to characterize the higher amounts of drug permeating from the increased drug delivery condition evaluated in the IVPT sensitivity assessment during IVPT method validation.IVPT研究中所用接收介质的组成和pH应根据其与皮肤的兼容性以及药物在接收介质中的稳定性和溶解度进行确认。药物在接收介质中的稳定性应作为接收液中样品分析方法验证的一部分。药物在接收介质中的溶解度,应经过三次重复检测经验确定,确保其超过IVPT正式研究中最高样品浓度,理想情况下是一个数量级。在IVPT方法验证期间的灵敏度评估中,药物在接收介质中的溶解度应足以表征,增加药物递送条件时药物渗透的最高量。The inclusion of 0.1% polyoxyethylene[20]oleyl ether (also known as Oleth-20, Volpo-20, or Brij-20; CAS number 9004-98-2) is recommended to enhance the solubility of physiological buffer based (aqueous) receptor solutions for hydrophobic drugs. If additional solubility is needed, small increases in the concentration of polyoxyethylene[20]oleyl ether (e.g., from 0.1% or 0.2%, which is typically adequate for most hydrophobic drugs, to higher concentrations) are recommended, but should not exceed 6% polyoxyethylene[20]oleyl ether. Other strategies to improve the solubility of the drug in the receptor solution that may have the potential to alter the permeability of the skin (e.g., inclusion of organic solvents and alcohols in the receptor solution) are not recommended and may invalidate the IVPT method.对于疏水性药物,推荐在基于生理缓冲盐的接收介质中添加0.1%(w/v)聚氧乙烯20油醚(别名Oleth-20, Volpo-20, or Brij-20;CAS:9004-98-2)提高溶解度。如有必要,可在接收介质中略微增加聚氧乙烯20油醚的浓度[例如,从0.1%(w/v)到0.2%(w/v)],通常足以满足大多数疏水药物溶解度的要求,但不应超过6%。其他改善药物在接收介质中溶解度的策略可能会改变皮肤的渗透性(例如,在接收介质中加入有机溶剂和醇),使IVPT方法失效,不建议使用。The inclusion of an anti-microbial agent in the receptor solution (e.g., ~0.1% sodium azide or ~ 0.01% gentamicin sulfate) is recommended to mitigate potential bacterial decomposition of the dermis and/or epidermis in the diffusion cell, regardless of the study duration. Other antimicrobial agents may also be acceptable, and if used, information should be included in the ANDA to explain the reason for their selection (and for the concentration at which they were used).无论研究时间的长短,建议在接收介质中加入一种抗微生物剂(例如,~0.1%叠氮化钠或~0.01%硫酸庆大霉素),以减轻在整个研究期间扩散池中细菌对真皮和/或表皮的潜在分解。如果采用其它抗微生物剂,应在ANDA中阐述选择的理由(以及使用的浓度)。D. Receptor Solution Sampling Qualification 接收液取样确认The accuracy and precision of receptor solution sample collection at each time point should be appropriately qualified. Evidence to qualify a sampling procedure should illustrate that the sampling technique can reliably collect a consistent volume of the sample from the well-mixed volume of the receptor compartment at each sampling event, and that no artifacts are likely to be created by the sampling technique. Information should be included describing the equipment manufacturer’s specification for the accuracy and precision of receptor solution sampling, when available.应对接收液中每个时间点取样的准确性和精密度进行适当的确认。在取样程序的确认过程中应证明,所用取样技术可以从混合良好的接收室中始终一致的收集到相同体积的接收液,且不会因取样技术的原因引起误差。如适用,应描述设备生产商关于接收液取样准确度和精密度的规范信息。For IVPT studies using a flow-through diffusion cell, it may be appropriate to qualify the lengths of tubing, and their associated dead volumes, to accurately calculate the lag time before a sample elutes through the tubing and is collected. For IVPT studies using a VDC, removal of the entire receptor solution volume and full volume replacement of the receptor solution at each time point may provide optimal solubility sink conditions. The sampling of small aliquots of the receptor solution for an IVPT study may introduce anomalous measurements of apparently negative flux in certain regions of the IVPT study and produce flux profiles that are difficult to interpret.对于采用流通扩散池进行IVPT研究的情况,应对流通扩散池每个管路的长度,以及相关的死体积进行确认,以准确计算样品通过管路洗脱和收集前的延迟时间。对于采用VDC进行IVPT研究的情况,在每个时间点移出所有接收液,并补充相应体积的新鲜接收介质,这种方法可以提供最佳的溶解度漏槽条件。在IVPT研究中,移取较少量的接收液体积,可能会导致某些时间点出现明显异常的负通量结果,产生难以解释通量曲线。E. Environmental Control 环境控制Ambient laboratory temperature and humidity during the study should be monitored and reported. An environmentally controlled temperature range of 21°C ± 2°C is recommended, and a humidity range of 50% ± 20% relative humidity is recommended, if feasible.在研究期间,应监控和报告实验室环境的温度和湿度。如适用,建议将温度控制在21℃±2℃、湿度控制在50%RH±20%RH之间。F. Permeation Profile and Range 渗透曲线和范围The flux profile and cumulative permeation profile for the IVPT pilot study should be plotted across a range of sampling times, which corresponds to the IVPT pivotal study duration. The calculation of flux and cumulative total permeation is discussed in more detail below. The results of the IVPT pilot study should validate that the selected study parameters are suitable to adequately characterize the permeation profile (the cutaneous pharmacokinetics) of the drug within the selected study duration (the range of sampling time points).在IVPT初步研究和正式研究中,应在整个取样时间范围内分别绘制通量分布曲线和累积渗透分布曲线。下文有关于通量和累积渗透量计算方式的详细讨论。应根据IVPT初步研究的结果确认,在所选择的研究期间(取样时间点范围内),所选择的研究参数是否足以表征渗透概况(皮肤药代动力学)。A sufficiently complete flux profile should be adequate to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points in the IVPT pilot study. The results of the IVPT pilot study should also validate that the sampling frequency provides suitable resolution to adequately characterize the permeation profile (particularly the flux profile).在IVPT初步研究中,一个充分完整的通量分布应足以识别出最大(峰值)通量和此后多个时间点的通量下降情况。应根据IVPT初步研究结果,确定取样频率,确保为表征渗透分布(特别是通量分布)提供合适的分辨率。G. Precision and Reproducibility 精密度和重现性The flux and cumulative permeation results from the IVPT pilot study (and the eventual IVPT pivotal study) should be calculated, tabulated, and reported for each diffusion cell at each time point, with summary statistics to describe the intra-donor average, standard deviation, and percent coefficient of variation (%CV) among replicates, as well as the inter-donor average, standard error, and %CV. Complete results for all data values used in the calculations should be reported in a clear and organized manner, to facilitate the reconstruction of the flux and cumulative permeation results. The design of the study should be detailed and clear, and data values should be clearly associated with specific donors, replicates, treatment groups, time points, etc.在IVPT初步研究(和最终的IVPT正式研究)中,应计算和报告各扩散池在每个时间点的通量和累计渗透结果,并以表格的形式汇总统计以下信息:重复测定的批内均值、标准偏差和变异系数(%CV);以及批间均值、标准偏差和变异系数。计算中所用数据的完整结果应以清晰和有组织的方式进行报告,以便通量和累计渗透结果的重现。研究设计应是详细和清晰,将数据结果与相关供体、重复数、试验组和时间点等相关联。H. Dose Depletion 剂量消耗The recovery of permeated drug in the receptor solution should be characterized in each diffusion cell as the cumulative total permeation of the drug in the receptor solution over the IVPT duration. This may be expressed as a percentage of the nominal amount of drug in the applied dose (which may be estimated based upon the nominal strength of the drug in the topical product and the approximate mass of topical product dosed on the skin).在IVPT研究期间,每个扩散池单元中药物渗透进入接收液中的回收率可以表征为“接收液中药物的总累积渗透量”,以上样量中药物标示量的百分比表达(根据外用制剂中药物的标识浓度和外用制剂在皮肤上的大概质量进行估计)。For example, if 10 mg of a topical product containing 5% drug was dosed on the membrane, the amount of drug in the applied dose may be estimated to be 0.5 mg (or 500 μg). If a cumulative total of 10 μg of drug diffused into the receptor solution across a 48-hour duration of the IVPT, it would be possible to estimate that the 500 μg dose would have been depleted by approximately 10 μg, amounting to an approximately 2% dose depletion. The average percentage dose depletion may thereby be estimated (not accounting for skin content) and should be reported.例如,如果含有5% 药物的外用制剂上样为10mg,则在上样中药物的量大约为0.5mg(或500μg)。如果在IVPT研究的整个48小时内,药物渗透进入接收液的总量为10μg,则估计500μg的剂量消耗为10μg,相当于约2%的剂量消耗。应估算和报告平均剂量消耗百分比(不考虑皮肤含量)。I. Discrimination Sensitivity and Selectivity 区分力—灵敏度和选择性The discrimination ability of the IVPT method may be described using two concepts: sensitivity and selectivity. The IVPT sensitivity studies are necessarily performed during IVPT method development to establish IVPT method parameters like the dose amount, dose duration, study duration, etc. However, the analysis of the results from these studies is qualitative in nature, and they need not be repeated during the IVPT method validation phase.可以用以下两个概念描述IVPT方法的区分力:灵敏度和选择性。在IVPT方法开发期间,必须进行IVPT灵敏度研究,以确定IVPT方法参数,如:上样量、剂量维持时间等。但这些研究结果本质上为定性研究,在IVPT方法验证阶段无需重复进行。The IVPT sensitivity studies are typically performed toward the end of the IVPT method development phase, and a key purpose of these studies is to incorporate the final IVPT method parameters for the target dose and dose duration to be used in the pivotal study so that the IVPT sensitivity studies can support a demonstration of the validity of the final IVPT method. Therefore, IVPT sensitivity studies are described within this section of the guidance in the context of IVPT validation (rather than method development) to avoid dissociating the discussions of IVPT sensitivity (which is performed to establish the suitability of the final IVPT method parameters) and IVPT selectivity (which is performed once the final IVPT method parameters are established, and which is based upon the IVPT pilot study that is performed as part of the IVPT method validation). With the exception of the alternative dose amounts or dose durations used in the IVPT sensitivity study, it is important that the IVPT method parameters are consistent across the IVPT sensitivity, pilot, and pivotal studies (including the anatomical region specified in the study protocol (e.g., posterior torso), the skin source, and skin preparation).IVPT灵敏度研究通常在IVPT方法开发阶段末进行,这些研究的一个关键目的是为了确定“在正式研究中所用的最终IVPT方法参数”,包括目标上样量和剂量维持时间,因此,IVPT灵敏度研究可以为最终IVPT方法的有效性提供合理性证明。因此,本文在IVPT方法验证(而非“方法开发”)模块描述IVPT灵敏度研究,是为了避免将IVPT灵敏度(以确定最终IVPT方法参数的适用性)和IVPT选择性(基于IVPT初步研究,在最终IVPT方法参数确定之后进行,是IVPT方法验证的一部分)分开讨论。除了在IVPT灵敏度研究中的上样量或剂量维持时间不同外,在IVPT灵敏度、初步研究和正式研究的所有阶段,IVPT方法参数均应保持一致(包括:研究方案中指定的解剖区域(如,背部躯干)、皮肤来源和处理方法)。1. IVPT Sensitivit 灵敏度IVPT sensitivity is the ability of the IVPT method to detect changes in the cutaneous pharmacokinetics of the drug as a function of differences in drug delivery. If the IVPT method consistently demonstrates higher and lower flux profiles (i.e., higher and lower values for IVPT endpoints) in response to increased and decreased drug delivery, respectively (or in response to other conditions expected to increase and decrease drug delivery, respectively), the IVPT method may be considered sensitive.IVPT灵敏度是IVPT方法检测药物皮肤药代动力学变化的能力,以反映药物递送的差异。如果随着药物递送的增加和降低(或预期会增加和降低药物递送的其他条件),IVPT方法能分别给出较高或较低通量分布曲线(即,较高和较低的IVPT终点),则认为IVPT方法是灵敏度。There are a few potential approaches by which to produce the differences in drug delivery that can be differentiated by a suitably discriminating IVPT method. Regardless of the approach used, the differences in the IVPT permeation profiles are not necessarily expected to be specifically proportional to differences in the dose amount, dose duration, or product strength. For example, three-fold differences in the dose amount (even if outside the recommended target dose range) may provide distinct flux curves but may not result in three-fold differences in the IVPT endpoints because the skin barrier may be rate-limiting both in vitro and in vivo.有几种潜在具有合适区分力的IVPT方法设计,去表征药物的不同递送行为。但无论采用何种方式,均不能期望IVPT渗透曲线的差异与药物上样量、剂量持续时间或产品规格的差异成比例关系。例如,上样量的三倍差异(即使超出了建议的目标剂量范围)可产生不同的通量曲线,但不会出现IVPT终点的三倍差异,因为皮肤屏障限制体外和体内的(渗透)速率。In other words, if the target dose for the pivotal IVPT study was 10 mg/cm2, a 3-fold lower dose would be ~3 mg/cm2and a 3-fold higher dose would be 30 mg/cm2; thus, an IVPT sensitivity study might compare the flux profiles from 3, 10, and 30 mg/cm2doses of the topical product. Similarly, if the target dose for the pivotal IVPT study was 15 mg/cm2, a 3-fold lower dose would be 5 mg/cm2and a 3-fold higher dose would be 45 mg/cm2; thus, an IVPT sensitivity study might compare the flux profiles from 5, 15, and 45 mg/cm2doses of the topical product. An IVPT sensitivity study performed with multiple skin donors (e.g., 4–6 skin donors) and a minimum of four replicate skin sections per donor per treatment group is recommended.换言之,如果正式IVPT研究的目标剂量是10 mg/cm2,则三倍的较低剂量为3 mg/cm2,三倍的较高剂量为30 mg/cm2;因此,可通过比较3、10和30 mg/cm2外用制剂上样量的通量曲线,确认IVPT方法的灵敏度。同理,如果正式IVPT研究的目标剂量是15 mg/cm2,则三倍的较低剂量为5 mg/cm2,三倍的较高剂量为45 mg/cm2;因此,可通过比较5、15和45 mg/cm2外用制剂上样量的通量曲线,确认IVPT方法的灵敏度。在IVPT灵敏度研究中,建议采用多个皮肤供体(如,4~6个皮肤供体),且每个试验组的每个供体至少4个重复皮肤切片。Modulation of Dose Amount: An IVPT method development study with different dose amounts may provide supportive evidence that the IVPT methodology is sensitive to differences in drug delivery.This approach is well suited to topical products that contain volatile components that evaporate from the formulation following dose application to the skin. Modulating the dose amount for such topical products effectively alters the thickness of the applied dose. The majority of volatile components from a thinner dose will tend to evaporate more rapidly (compared to a thicker dose), and a thinner dose will tend to deliver less drug into the skin (and/or for a shorter duration) compared to a thicker dose.调整上样量:可以采用不同的上样量进行IVPT方法开发研究,为IVPT方法对药物递送的差异具有灵敏度提供支持性证据。该方法适用于含有挥发性成分的外用制剂,这些挥发性成分在药物制剂上样到皮肤后会挥发。调整这类制剂的上样量可以有效改变上样的厚度。采用较薄的上样量,与较厚的上样量相比,大部分挥发性成分会挥发,因此,较薄的上样量(和/或较短的维持时间)递送进入皮肤的药物量较少。Modulating the dose amount can be an effective technique to modulate differences in drug delivery for formulations with volatile components, like gels, lotions, and many creams. However, modulating the dose amount may not necessarily produce perceptible differences in drug delivery for topical products like petrolatum-based ointments, or other types of topical products that do not evaporate on the skin, or that may not experience dose-dependent differences in metamorphosis that can alter the rate and extent of drug delivery.对于含有挥发性组分的配方,如凝胶、洗剂和很多乳膏剂,调整上样量是改变药物递送行为的有效技术手段。然而,对于基于凡士林的软膏剂、或不会在皮肤上挥发的外用制剂、或可改变药物递送速率和程度的形变过程不具有剂量依赖性,通过改变这些制剂的上样量不一定会产生药物递送行为的明显差异。Modulation of Dose Duration: For many topical products, it may be more effective to modulate the dose duration, instead of the dose amount, to produce differences in drug delivery and associated changes in the cutaneous pharmacokinetics of the drug.调整剂量维持时间:对于很多外用制剂,相比于调整上样量,通过调整剂量维持时间可更有效地评估药物递送的差异和与药物皮肤药代动力学相关的变化。An IVPT method development study with a controlled dose amount (e.g., 15 mg/cm2) dosed for different durations (e.g., 2 hours, 6 hours, and 12 hours) may be well suited to provide supportive evidence that the IVPT methodology is sensitive to differences in drug delivery from many topical products. The scenario described in this example would support an IVPT study design where a topical product dose of 15 mg/cm2 is dosed for 6 hours (the target duration for the IVPT study) and then wiped off. The applied dose may be removed with a series of cotton-tipped swabs, one or more of which may be dry and one or more of which may be moistened (e.g., with a soap solution or water). The initial (dry) swab typically removes the bulk of the dose and subsequent swabs are used to remove the residual dose (i.e., the residue of the topical product which may otherwise continue to deliver drug into the skin) and/or to rinse the skin.在IVPT方法开发研究中,通过将一定量的外用制剂(如,15 mg/cm2)在皮肤上维持不同的时间(如,2h、6h和12h)可能是提供支持性证据的比较合适方式,以证明IVPT方法对很多外用制剂中药物递送的差异具有敏感性。本例描述了用于支持IVPT研究设计的场景,将上样量为15 mg/cm2的外用制剂在皮肤上维持6h(研究的目标维持时间),然后将其擦除。可以采用一些棉签移除样品,包括一个或多个干燥的棉签,和一个或多个浸湿的棉签。首先用干燥的棉签移除大部分样品,然后用浸湿的棉签移除残留的样品(即,残留的样品可能会继续递送药品进入皮肤)和/或清洗皮肤。To support a demonstration of the sensitivity of the IVPT study, the permeation profile produced by the target dose duration for the IVPT study (e.g., 6 hours) should be compared with a shorter dose duration (e.g., 2 hours) that is expected to perceptibly decrease the drug delivery, and also be compared with a longer dose duration (e.g., 12 hours) that is expected to perceptibly increase the drug delivery. Thereby, the three dose durations compared in the IVPT sensitivity study are designed to produce perceptible changes in the cutaneous pharmacokinetics of the drug as a function of differences in drug delivery, and thereby support a demonstration of the sensitivity of the IVPT method.为了证明IVPT研究的灵敏度,将IVPT研究中目标剂量持续时间(如,6小时)的渗透曲线分别与较短剂量维持时间(如,2小时)和较长剂量维持时间(如,12小时)的渗透曲线进行比较,较短和较长剂量维持时间应分别有明显较低和较高的剂量递送。因此,在IVPT灵敏度研究中,通过设计比较三种不同剂量维持时间,可产生明显不同的“能反映药物递送差异的”药物皮肤药代动力学,支持IVPT方法的灵敏度论证。The specific dose durations may be selected based upon an initial exploratory IVPT study performed during IVPT method development that characterizes the permeation profile when the dose is left on the skin for a longer duration (e.g., 24 or 48 hours). An important feature of the results from such an IVPT study is the duration of the initial phase of the permeation profile, when the flux is increasing at a relatively rapid rate.在IVPT方法开发期间,可以根据初始的探索性IVPT研究中,剂量在皮肤上保留较长时间(如,24或48小时)的渗透曲线,选择特定的剂量维持时间。这种研究结果的一个重要特征是,在渗透曲线的初始阶段,通量以一个相对较快的速率增加。For example, if such an exploratory study indicates that the flux increases on a steep slope until approximately 12 hours, and then continues to deliver the drug at a gradually increasing rate thereafter, it may suggest that the permeation profile for a dose duration of longer than 12 hours (e.g., 24 hours) may not be perceptibly different from that of the 12 hour dose duration, especially when compared in a relatively small number of donors and replicates (e.g., four donors with four replicates each per dose duration). It may also suggest that a 12-hour dose duration may be a good choice for the longest of the three dose durations in the IVPT sensitivity study.例如,如果探索性研究结果表明,在12小时前通量快速增加,之后随着剂量维持时间的增加,通量与12小时无明显的差异;这种结果表明,超过12小时(如24小时)的剂量维持时间,获得的通量分布曲线可能与12小时无明显差异,尤其是在供体和重复数量较少的情况下(如,每个剂量维持时间4组供体皮肤,每组供体皮肤4个重复皮肤切片)。这也可能表明,在IVPT灵敏度研究中,12小时的剂量持续时间可能是一个较好的选择,也是三个剂量维持时间中最长时间。The target dose duration should be selected based upon considerations like the sensitivity of the sample analytical method, the ability to produce a permeation profile that can be perceptibly discriminated from that produced by the longer (12 hour) dose duration, and/or the labeled use of the topical product (which may indicate that the topical product should be reapplied every 4–6 hours).目标剂量维持时间的选择应基于以下考虑因素,如:样品分析方法的灵敏度、产生能明显区别于最长(12小时)剂量维持时间所产生的渗透曲线的能力、和/或外用制剂的说明书(其中可能说明,每4~6小时需要重复进行给药)。The shortest of the three dose durations in the IVPT sensitivity study should be selected based upon the sensitivity of the sample analytical method and its ability to produce a permeation profile that can be perceptibly discriminated from that produced by the target (6 hour) dose duration.在IVPT灵敏度研究中,三个剂量维持时间中最短时间的选择应基于样品分析方法的灵敏度,以及其产生能明显区别于目标剂量维持时间(6小时)所产生的渗透曲线的能力。Modulation of Product Strength: To validate the sensitivity, specificity, and selectivity of an in vitro release test (IVRT) method, altered strength formulations are routinely prepared. While it may seem convenient to use these altered strength formulations in an attempt to demonstrate the sensitivity and selectivity of an IVPT method, doing so may not produce the desired outcomes. There may be circumstances when this strategy may produce perceptibly different permeation profiles, however, in many instances, the resulting permeation profiles may not be perceptibly different when compared in a relatively small number of donors and replicates (e.g., four donors with four replicates each per topical product strength). In general, the modulation of topical product strength to support a demonstration of IVPT sensitivity is not recommended because it may not consistently produce the expected increase or decrease in drug delivery; however, in certain situations, higher and lower strength formulations (relative to the nominal strength of the RS) may suitably increase and decrease the drug delivery and cutaneous pharmacokinetics relative to that from the nominal strength topical product.调整产品规格:对于IVRT方法的灵敏度、专属性和选择性验证而言,常见的方式是改变配方制剂的规格。虽然在IVPT方法的灵敏度和选择性验证中,采用改变配方制剂规格的方式似乎也比较方便,但这种方式可能不会产生预期的结果。在某些情况下,这种策略可能会产生明显不同的渗透曲线;然而,在很多情况下,由于在对比研究中采用的供体组和重复数较少(如,每种外用制剂规格采用4个供体组,每组供体4个重复),并不能产生具有明显不同的渗透曲线。通常,不建议通过调整外用制剂规格的方式去论证IVPT的灵敏度,因为它可能不会持续产生预期的药物递送增加或降低;然而,在某些情况下,相对于外用制剂的目标规格,较高和较低规格的配方制剂(相对于对照制剂的规格)可以适当的增加和降低药物递送和皮肤药代动力学。2. IVPT Selectivity 选择性IVPT selectivity is the ability of the IVPT method to discriminate the cutaneous pharmacokinetics of the drug between the RS and a topical product or formulation that exhibits differences in drug delivery relative to the RS. The IVPT pilot study with the parallel assessment of the RS, the test topical product, and a third topical product or formulation that is known or designed to be different from the RS may provide supportive evidence that the IVPT methodology is selective for differences in drug delivery. Topical product batch information for all topical product lots used in IVPT method development, validation and pilot studies, as applicable, should be submitted in the study reports. The topical product information should include, but not be limited to, information about the batch formula, manufacturing date, batch size, altered manufacturing processes (if applicable) and, if available, potency and content uniformity. The evaluation of inequivalence may be based upon a qualitative or quantitative comparison of the permeation profiles and/or the IVPT endpoints.IVPT选择性是IVPT方法区分“对照制剂(RS)”和“与RS在药物递送方面存在差异的外用制剂或配方”在药物皮肤药代动力学差异的能力。在IVPT初步研究中,通过将RS、自研外用制剂和“已知或设计不同于RS”的第三种外用制剂或配方进行平行评估,提供支持性证据,说明IVPT方法对药物递送的差异具有选择性。如适用,应在研究报告中递交,IVPT方法开发、验证和初步研究中用到的所有外用制剂的相关批信息,包括但不限于:批配方、生产日期、批量、变更的生产工艺(如适用),以及效价和含量均匀度(如有)。可基于渗透曲线和/或IVPT终点的定性或定量比较进行不等效性评估。J. Robustness 耐用性A primary assumption related to robustness testing is that the test system performs consistently when all system variables (e.g., temperature, stirring rate) are at nominal settings. A value of robustness testing is that it can verify whether the system continues to provide a consistent output when specific variables are slightly altered, thereby qualifying operational ranges for those variables. However, the variability inherent in the permeability of human skin, whether in vitro or in vivo, may not be compatible with the primary assumption related to the consistency of the test system.与耐用性测试相关的一个主要假设是,当所有系统变量(如,温度、搅拌速率)为标准设置时,测试系统的性能一致。耐用性测试的意义在于,它可以确认当特定变量稍微改变时,系统是否可以提供一致的输出结果,从而确定这些变量的操作范围。然而,无论是在体外还是在体内,人体皮肤渗透性的固有可变性可能与测试系统一致性的初始假设不相容。Nonetheless, results from studies during IVPT method development that appear to support the robustness of the IVPT method or system should be reported, if relevant. For example, an IVPT method may be robust to substantial variations in the stirring rate of the receptor compartment. Similarly, the permeation profile of a drug into and through human skin may appear to be robust to certain differences in the topical product strength. Ultimately, because it may not always be feasible to validate the robustness of IVPT method parameters, IVPT study procedures should be controlled as precisely as possible.尽管如此,如果相关,应报告在IVPT方法开发期间获得的可以支持IVPT方法或系统耐用性的研究结果。例如,IVPT方法对接收室中搅拌速率的显著变化具有耐用性。类似地,药物进入并通过人体皮肤的渗透曲线可能对外用制剂规格的改变具有耐用性。因此,对IVPT方法参数的耐用性验证并不总是可行的,在IVPT研究中应尽可能精准的控制IVPT研究程序。V. Sample analytical method validation 样品分析方法验证While exploratory studies performed during IVPT method development may use an unvalidated sample analytical method, it is essential that all studies conducted as part of the IVPT method validation use a validated sample analytical method. A validated IVPT method should use a validated sample analytical method (e.g., HPLC/MS or UPLC/MS). Therefore, a discussion of the sample analytical method for the IVPT method is included in this guidance under this section on IVPT method validation.虽然在IVPT方法开发的探索性研究中,可以采用未验证的样品分析方法,但作为IVPT方法验证的一部分,在IVPT方法验证时,必须采用已验证的样品分析方法。已验证的IVPT方法应当使用已验证的样品分析方法(如,HPLC/MS或UPLC/MS)。因此,该部分讨论用于IVPT方法的样品分析方法。However, the study protocols and reports related to the IVPT method are distinct from those for the sample analytical method that is used to quantify drug concentrations in IVPT receptor solution samples. The validation of a sample analytical method, in and of itself, does not demonstrate the validity of an IVPT method. Separate and specific reports should be submitted for the validation of the sample analytical method (e.g., HPLC/MS or UPLC/MS) and for the validation of the IVPT method.然而,与IVPT方法相关的研究方案和报告与用于IVPT接收液中样品定量的分析方法不同。样品分析方法验证,就其本身而言,并不能证明IVPT方法的有效性。因此,应分别递交样品分析方法(如,HPLC/MS或UPLC/MS)和IVPT方法的验证报告。Any results from studies of the IVPT method that are performed during method development using a different sample analytical method than that which is ultimately validated, cannot support a demonstration of the validity of the IVPT method. Information should be provided in the IVPT method validation report referencing the (separate) sample analytical method validation, and clearly indicate that all relevant results in the IVPT method validation report were obtained using a validated sample analytical method (as opposed to a sample analytical method with different parameters than those which were validated).如果,在方法开发期间使用的样品分析方法与最终验证的样品分析方法不同,与其相关的IVPT方法研究结果不能用于支持论证IVPT方法的有效性。在IVPT方法验证报告中,应提供参考的样品分析方法验证信息,并明确表明IVPT方法验证报告中的所有相关结果均是采用经验证的样品分析方法(而不是与经验证的分析方法具有不同参数的样品分析方法)获得的。The receptor sample analysis procedures (e.g., typically involving an HPLC/MS or UPLC/MS system) should be performed using chromatography software (e.g., a chromatography data system) with audit trails, and should include a multi-point (6–8 concentration) calibration curve with suitable quality control samples, and should be validated in a manner compatible with the FDA guidance for industry Bioanalytical Method Validation (May 2018).接收液中样品的分析方法(如,通常为HPLC/MS或UPLC/MS系统)应使用具有审计追踪的色谱软件(如,色谱数据系统),并应包括具有适当质量控制样品的多点(6~8个)校准曲线,同时,应符合FDA生物分析方法验证行业指南要求。The validation of the receptor sample analytical method should include relevant qualifications of dilution integrity, if applicable, as well as stability assessments with the highest relevant temperature in the receptor solution for the longest relevant duration; the highest relevant temperature may be warmer than 32°C because the temperature of the receptor solution is often higher than the temperature at the surface of the skin, and the longest relevant duration may be the longest interval between sampling time points for methods in which the entire receptor solution is replaced at each sampling time point, or it could be longer in scenarios with only partial sampling of the receptor solution (e.g., 34°C for 48 hours).如适用,接收液中样品分析方法的验证应包括相关的稀释完整性确认,以及样品在最高相关温度的接收液中放置最长相关时间的稳定性评估;最高相关温度可以略高于32℃,因为接收液的温度通常高于皮肤表面温度。对于在每个取样时间点替换整个接收液的情况,最长相关时间为各相邻取样时间点中,间隔最长时间;但对于接收液部分取样的情况,时间可能更长(如,34℃放置48h)。If the samples are processed in specific ways for analysis (e.g., by drying and reconstituting the receptor samples in a smaller volume to concentrate the sample and increase the effective analytical sensitivity, or by dilution of receptor solution samples into the validated curve range of the sample analytical method) those procedures should be validated (e.g., by qualifying the dilution integrity during the sample analytical method validation). The stability of the drug in the receptor solution sample should be validated in a receptor solution matrix that has been exposed to the underside of the skin in a diffusion cell under conditions relevant to the IVPT pivotal study.如果在测定前,需要用特定的方式对样品进行处理(如:将样品干燥后重新用小体积溶剂配制以提高分析灵敏度;或通过稀释接收液样品,使其浓度到已验证的样品分析方法曲线范围内),应对该处理方法进行确认(如,在样品分析方法验证期间确认稀释完整性)。药物在接收液中的稳定性,应在IVPT正式研究相关的条件下,与扩散池中皮肤下侧接触的接受液基质中进行。VI. IVPT Pivotal study IVPT正式研究The IVPT pivotal study protocol should incorporate considerations relevant to BE studies, in general.IVPT正式研究方案通常应包含与BE研究相关的考虑因素。A. Handling and Retention of Samples 样品的处理和保存Refer to 21 CFR 320.38, 320.63 and the FDA guidances for industry Handling and Retention of BA and BE Testing Samples (May 2004) and Compliance Policy for the Quantity of Bioavailability and Bioequivalence Samples Retained Under 21 CFR 320.38(c) (August 2020), as applicable, regarding considerations for retention of study drug samples and to 21 CFR 320.36 for requirements for maintenance of records of BE testing. Retention samples should be randomly selected from the drug supplies received before allocating topical product units for use in an IVPT study in which the test topical product and RS are compared.参考21 CFR 320.38,320.63和FDA行业指南《生物利用度(BA)和生物等效性(BE)研究中试验样品的处理和保存》(2004年5月)和《21CFR 320.38(c) BE样品留存的数量及生物利用度》(2020年8月),如适用,关于保留研究药物样品的考虑以及21 CFR 320.36关于保存BE检测记录的要求。在采用自研外用制剂和RS进行IVPT对比研究前,应从收到的药品中随机选择样品进行留样。B. Control of Study Procedures 研究程序的控制Study procedures that have the potential to influence the results of the study should be appropriately controlled. Also, experimental observations that may have the potential to influence the interpretation of the study results, as well as any protocol or standard operating procedure (SOP) deviations, should be reported.应对可能影响研究结果的研究程序进行适当控制。另外,应报告可能影响研究结果解释的试验观察,以及任何与方案或标准操作规程(SOP)发生的偏离。Control of procedures related to the skin include the consistent control across the study of the skin preparation (e.g., dermatoming of skin sections) and the thickness of skin sections mounted on diffusion cells, as well as the skin storage conditions, including the duration for which the skin was frozen and the number of freeze-thaw cycles to which the skin was exposed. Skin from the same anatomical location should be used from all donors, and the demographics (age, race, sex) should be reported for all donors. Also, the IVPT sensitivity, pilot, and pivotal studies should use skin from the same anatomical site; otherwise, if skin from different anatomical sites is used across the different study phases, it may not be possible for the results of the IVPT sensitivity and pilot studies to support a demonstration of the discrimination ability of the IVPT method used for the pivotal study because the method parameters would not be aligned across the respective studies. Similarly, if a non-rate-limiting support membrane is used beneath the skin section (e.g., a filter membrane used in a validated IVRT method for the same topical product) then it should be used in a consistent manner for the IVPT sensitivity, pilot, and pivotal studies.与皮肤相关的控制程序,包括:采用相同的皮肤处理方法(如,皮肤的剥离)和安装在扩散池上皮肤切片的厚度的一致性,以及皮肤存储条件,如皮肤冷冻存储时间和取出后的冻融循环次数。所有供体皮肤应采用相同的解剖区域,并报告所有供体的人口统计信息(年龄、种族、性别)。另外,在IVPT灵敏度、初步试验和正式研究中,应采用相同的解剖区域;否则,如果在不同的研究阶段,采用的皮肤解剖区域不同,在IVPT灵敏度和初步研究中获得结果可能不足以支持IVPT正式研究中区分力的论证,因为各研究阶段采用的方法参数不一致。同样,如果在皮肤下面使用非速率限制支撑膜(如,同品种IVRT验证研究中所用的滤膜),则IVPT灵敏度、初步和正式研究中应保持一致。Control of procedures related to the dose include the control of the area of dose application, the dose amount, the dosing technique, the dose duration, and the blinding and randomization consistent manner for all diffusion cells in the study. Differences in dosing technique may alter the metamorphosis of the dosage form on the skin, and inconsistencies in the diameter of the area dosed on each diffusion cell may significantly influence the dosed area and contribute to errors in the calculation of flux.与剂量或上样相关的控制程序,包括:对上样面积、上样量、上样技术、剂量维持时间的控制,以及在研究中每个扩散池采用的盲法和随机化方法的一致性。不同的上样技术可能改变上样到皮肤上剂型的形态,扩散池孔口扩散面积的不一致可能会显著影响上样面积,进而导致通量计算的偏差。Control of procedures related to sampling include the control of sampling time precision, the sampling technique, the duration of sampling and replacement of receptor solution, the sample volume or flow rate, and sample handling and storage.与取样相关的控制程序,包括:取样时间精准度、取样技术、取样和替代接收液的时间间隔、取样体积或流速,以及样品的处理和储存。Control of procedures related to the pivotal study should include a non-dosed control skin section from each skin donor, which should be mounted in a diffusion cell and otherwise treated identically to the dosed skin sections, including sampling of the receptor solution at all time points to ensure that drug concentrations monitored in the receptor solution are associated with the dose applied in the IVPT pivotal study, and not drug contamination in the skin from that donor that might permeate into the receptor solution across the duration of the study. A pre-dose “zero” sample collected from each diffusion cell is also recommended, which may identify potential contamination associated with each skin section and/or each diffusion cell.在正式研究期间的相关控制程序,包括:对每组皮肤供体的非剂量控制皮肤切片,按照与上样皮肤切片一致的方式将其安装到扩散池中并进行后续处理,在所有时间点对接收液进行取样,以确保IVPT正式中,在接收液中检测到的药物为实际渗透进入的量,而不是研究期间由于皮肤的药物污染渗透进入。建议从每个扩散池中采集“零”浓度样品,用于识别每个皮肤切片和/或每个扩散池的潜在污染。In addition, investigators should perform the IVPT validation and pivotal studies within a quality management system that includes, but is not limited to, documented procedures for:此外,研究人员应在一定的质量管理体系条件下,进行IVPT验证和正式研究,包括但不限于:Study personnel identification, training, qualification, and responsibilities 研究人员的识别、培训、资质和职责Study management and study management personnel responsibilities 研究管理和研究管理人员职责Quality control (QC) and QC personnel responsibilities 质量控制(QC)和QC人员职责Quality assurance (QA) and QA personnel responsibilities 质量保证(QA)和QA人员职责Use of SOPs 操作规程(SOP)的制定Use of study protocols 研究方案的制定Use of study reports 研究报告的制定Maintenance and control of the study facility environment and systems 研究设施环境和系统的维护和控制Qualification and calibration of instruments and computerized systems 仪器和计算机系统的确认和校准Good documentation practices including, but not limited to, contemporaneous documentation of study procedures and recording of experimental observations or deviations from procedures specified in the study protocol or in relevant SOPs 良好的文件规范,包括但不限于:及时记录研究过程、试验观察、以及与研究方案及相关SOPs中规格程序的偏离Maintenance of suitable records that facilitate the reconstruction of study events and procedures, including study sample handling and storage records (e.g., sample tracking logs), audit trails for sample analysis procedures, control of study materials and reagents, and electronic data control 对记录进行适当的维护,以便研究过程可以重现,包括:研究样品的处理和保存记录(如,样品跟踪日志)、样品分析过程的审计跟踪、研究物料和试剂的控制以及电子数据控制Archival of study records 研究记录归档C. Blinding Procedure 盲法程序A detailed description of the blinding procedure should be provided in the study protocol and final report. The packaging of the test topical product and RS should be similar in appearance to maintain adequate blinding of the investigator and any experimental operators.应在研究方案和最终报告中递交详细的盲法程序信息。自研外用制剂和RS的包装应有相似的外观,以确保对研究人员和任何试验操作人员充分致盲。D. Randomization 随机化The method of randomization should be described in the protocol of the IVPT study and the randomization schedule provided, preferably in a SAS data set in .xpt format (created using the SAS XPORT procedure). It is recommended that an independent third party generate and hold the randomization code throughout the conduct of the study to minimize bias. The applicant may generate the randomization code if not involved in the packaging and labeling of the test topical product and RS dosed in the study. A sealed copy of the randomization scheme should be retained at the study site and should be available to FDA investigators at the time of site inspection to allow for verification of the treatment identity of each skin section.应在IVPT研究方案中描述随机化方法,并推荐以.xpt格式的SAS数据集形式(使用SAS XPORT程序创建)提供随机化计划表。在整个研究过程中,建议由独立的第三方生成并保存随机化代码,以减少偏差。如果不参与自研外用制剂和RS的包装和标签,申请人可以自行生成随机化代码。随机化方案的密封副本应保留在研究现场,以便在现场核查期间可随时提供给FDA审评员,用于确认每个皮肤切片的具体信息。E. Dosing 上样In the IVPT pivotal study, the test topical product and RS should be dosed in an alternating pattern on successive diffusion cells (skin sections) from each donor. One of two dosing sequences (illustrated below) may be randomly assigned for each donor: a. ABABAB… b. BABABA…在IVPT正式研究中,对于每个供体组,可以在扩散池(皮肤切片)上以交替给药方式依次放置自研外用制剂和RS。对于每个供体组,可采用下述两个方式中的一个进行随机放样: a. ABABAB… b. BABABA…F. Study Design 研究设计The IVPT pivotal study should compare the cutaneous pharmacokinetics of the drug from the test topical product versus that from the RS using excised human skin with a competent skin barrier mounted on a qualified diffusion cell system. The IVPT pivotal study should use a design that directly compares the test topical product and RS on skin from the same set of donors, each with the same number of replicate skin sections per donor per treatment group (dosed with either test topical product or RS topical), using the same IVPT method parameters.在IVPT正式研究中,应采用皮肤屏障完整性良好的离体人类皮肤和经确认的扩散池系统,对自研外用制剂和RS的皮肤药代动力学进行对比研究。在自研外用制剂和RS的对比研究中,每个试验组应采用相同的皮肤供体、相同的皮肤切片重复数,并采用相同的IVPT方法参数。The IVPT pivotal study design, methodology, and diffusion cell equipment considerations relating to sampling precision should be controlled as precisely as possible. For example, it may be appropriate to stagger the dose application on successive diffusion cells and to synchronize the sampling time points with the dosing time for that diffusion cell, to ensure consistent durations between dosing and sampling of all diffusion cells.应尽可能精准的控制IVPT正式研究的设计、方法和扩散池设备的取样精密度。例如,对于连续的扩散池,可以错开上样时间,根据上样时间的差异同步取样时间,以确保所有扩散池的上样和取样时间的间隔一致。G. Inclusion Criteria 纳入标准In general, the following inclusion criteria should apply: healthy, normal, barrier-competent skin from male and/or female donors of at least 18 years of age. Inclusion criteria related to donor demographics (e.g., age, race, sex) should be specified in the study protocol and demographic information should be reported for each donor. Additional criteria may be added by the applicant.通常,应采用以下纳入标准:至少18周岁的健康、正常、具有屏障完整性的雄性和/或雌性供体。应在研究方案中规定符合纳入标准的人口统计信息(如,年龄、种族、性别),并报告每个供体的个人信息。申请人可以增加其它纳入标准。The skin may be harvested following excision from patients undergoing a surgical procedure or excised from cadavers. A consistent source is recommended for all the skin used. The anatomical region specified in the study protocol (e.g., posterior torso) should be consistent for all donors whose skin is included in the study.建议使用人体离体皮肤,可以从接受外科手术的病人或尸体获取。所用的所有皮肤应有一致的来源,且所有的供体应为相同的解剖区域(如,背部躯干),并在研究方案中进行明确规定。The study protocol should specify the inclusion (acceptance) criteria for skin sections based upon the barrier integrity test result, which should be reported for each skin section. The study protocol should specify inclusion criteria related to the temperature and duration of skin storage as well as the number of freeze-thaw cycles, all of which should be reported for each donor’s skin. The study protocol should specify the inclusion criteria related to the skin harvesting/processing procedures and skin thickness (e.g., dermatomed skin of 500 μm ± 250 μm thickness) used in the IVPT study.研究方案中应规定皮肤切片的屏障完整性测试纳入(接受)标准;并规定每组供体皮肤的储存温度和时间,以及冻融循环次数;同时规定IVPT研究中所用皮肤的获取/处理方法和厚度(如,厚度为500 μm ± 250 μm)。H. Exclusion Criteria 排除标准In general, the following exclusion criteria should apply. Skin from subjects with a known (history of) dermatological disease should be excluded from the study. Skin with tattoos, stretch marks, or any visible sign of abnormality should be excluded from the study. Skin exhibiting a significant density of terminal hair is not recommended and should be excluded from the study. Additional criteria may be added by the applicant.通常,应采用以下排除标准。包括:患有已知皮肤疾病(病史)的供体皮肤;带有纹身、妊娠纹或任何异常迹象的皮肤;表现出明显浓密的末端毛发的皮肤等。申请人可以增加其它排除标准。While gentle washing or rinsing of the skin surface is appropriate, submerging the skin in an aqueous solution for more than a few minutes may damage the skin barrier and should be avoided; such skin sections should be excluded from the study. Also, skin that has been subjected to shaving with a blade; abrasive polishing; tape-stripping; or cleansing with alcohols, solvents, or other strong solutions that could damage the skin barrier should be excluded from the study.虽然可以采用温和的清洗或冲洗方式处理皮肤表面,但是,如果将皮肤浸泡在水溶液中几分钟,就有可能破坏皮肤屏障,应避免该处理方式。此外,也不建议采用刀片剃须、研磨抛光、胶带剥离,或采用乙醇或其它强极性溶剂处理皮肤,因为这些处理方式可能破坏皮肤屏障。Skin from donors with significant background levels of the drug or other compounds that may interfere with the quantification of the drug in receptor solution samples should be excluded from the study. Skin from donors exhibiting a high barrier integrity test failure rate among replicate skin sections may be excluded from the study, and skin from an alternative donor may be used instead.含有显著的药物背景水平或含有的其他化合物可能干扰接收液中药物定量测定的皮肤供体,应排除在研究之外。重复皮肤切片中屏障完整性测试失败率高的供体皮肤应排除在研究之外,采用其他的供体皮肤进行替代。I. IVPT Endpoints IVPT终点The endpoints for the IVPT pivotal study are based upon parameters that characterize the rate and extent to which the drug permeates into and through the skin and becomes available in the receptor solution. Specifically, the rate of drug permeation is characterized by the flux (J) and the extent of drug permeation is characterized by the total cumulative amount (AMT) of drug permeated into the receptor solution across the study duration.IVPT关键研究的皮肤药代动力学终点是基于表征药物渗透和通过皮肤进入接收液的速率和程度的参数。具体来说,药物的渗透速率采用通量(J)进行表征,而药物的渗透程度采用整个研究期间渗透到接收液的累积总量进行表征。The flux (rate of drug permeation) should be plotted as J on the Y-axis in units of mass/area/time (e.g., nanograms (ng)/cm2/hr) versus time on the X-axis. Flux profiles commonly resemble plasma pharmacokinetic profiles, however, it is important to distinguish that the flux is a rate, rather than a concentration. The extent of drug permeation should also be plotted, as the total cumulative amount (AMT) of drug permeated on the Y-axis in units of mass/area (e.g., ng/cm2) versus time on the X-axis.以通量(药物渗透速率)为纵坐标Y-轴,时间为横坐标X-轴作图;其中Y-轴用“J”标识,以质量/面积/时间(如,纳克/平方厘米/小时)为单位。通量分布模型通常类似于血浆药代动力学分布模型,尽管通量是一个速率单位,并不代表浓度。也应对药物的渗透程度作图,以药物累积渗透量(单位为质量/面积,如纳克/平方厘米)为纵坐标Y-轴,时间为横坐标X-轴。The flux should be calculated based upon: the receptor sample concentration (e.g., 2.0 ng/mL) at each time point; the precise, empirically measured volume of that specific diffusion cell (e.g., 6.0 mL) which may vary between individual cells; the area of dose application (e.g., 1 cm2); and the duration for which the receptor volume was accepting the drug. For example, if the sample exemplified here represented a 2-hour period following dosing, then J would be calculated based upon the values above as:通量的计算应基于:每个时间点的接收液中样品的浓度(如,2.0 ng/mL);精确的、经验测量的扩散池的体积(如,6.0 mL),每个扩散池的体积可能不同;上样区域的面积(如,1 cm2);和整个研究的持续时间。例如,如果此处所示样本代表给药后2小时内的情况,则J值按下式进行计算:J = [(2.0 ng/mL) x (6.0 mL)]/(1 cm2)/(2 hrs) = 6 ng/cm2/hrThis flux should be calculated and reported for each diffusion cell for each sampling interval and plotted across the entire study duration to generate the flux profile for each diffusion cell. The rate calculated above may be plotted at the 2-hour time point, or at the midpoint between 0 and 2 hours (i.e., 1 hour).In addition, the AMT should be calculated and reported for each diffusion cell. This cumulative amount of drug that has permeated (in total across the entire study) should be reported as the AMT endpoint, rather than using a trapezoid rule to calculate the area under the flux curve.应计算并报告各扩散池的所有相邻采样点之间的通量,并为整个研究期间的所有扩散池绘制通量曲线。上述计算的速率可以对应于2小时的实际时间点进行绘制,也可以采用0和2小时之间的中间点(即,1小时)。此外,应计算并报告每个扩散池的总累计渗透量(AMT)。AMT终点为药物渗透的累计量(整个研究期间的渗透总量),而不是采用梯形法则计算的通量曲线下的面积。The maximum flux (Jmax) at the peak of the drug flux profile and the AMT should both be compared for locally-acting test topical products and RSs. This is somewhat analogous to the comparison of the Cmax and AUC for systemically-acting test products and RSs, inasmuch as the pair of endpoints in each case facilitates a comparison of the rate and extent to which the drug from each type of product (locally-acting or systemically-acting) becomes available at the site of action.A confidence interval (CI) should be calculated for each IVPT endpoint: a. the natural log-transformed maximum flux (Jmax) b. the natural log-transformed total cumulative amount (AMT) permeated应将局部起效的自研制剂和RSs的最大通量(Jmax,药物通量曲线的最高峰)和AMT进行对比。这类似于全身起效的自研制剂和RSs的Cmax和AUC的比较,因为这些终点可分别用于表征各剂型(局部起效或全身起效)药物到达作用部位的速率和程度。应计算每个IVPT终点的置信区间(CI): a. 自然对数换算后的最大通量(Jmax) b. 自然对数换算后的总累积渗透量(AUC)It is the responsibility of the applicant to determine the number of donors required to adequately power the IVPT pivotal study, however, a minimum of four dosed replicates per donor per treatment group (test product or RS) is recommended.At the completion of the study, if the number of skin replicates is the same for all donors in the test topical product and RS treatment groups in the IVPT study, a statistical analysis for a balanced design is recommended. If skin sections or diffusion cells are excluded from the final statistical analysis because of experimental loss/issues, and the resulting data set is unbalanced, a statistical analysis for an unbalanced design is recommended.申请人有责任确定足以支持IVPT关键研究所需的供体数量,然而,建议每个试验组(自研制剂或RS)每个供体至少4个重复剂量。在IVPT研究结束时,如果自研外用制剂和RS试验组中所有供体的皮肤切片重复数均一致,建议采用平衡设计法进行统计分析。如果因试验问题,需将皮肤切片或扩散池从最终的统计分析中排除,则结果数据集不平衡/对称,建议采用非平衡设计法进行统计分析。Approaches to statistical analysis of the pivotal study are described in section VIII of this guidance. Appendix I provides example SAS code for determining BE with both a balanced dataset and an unbalanced dataset. Appendix II provides numerical examples with simulated data sets. Appendix III provides example R code for determining BE.在本指南的第VIII部分,对正式研究的统计分析方法进行了描述。附录I提供了用于测定平衡数据集和非平衡数据集BE的SAS代码示例。附录II提供了模拟数据集的具体示例。附录III提供了测定BE的R代码示例。VII. Submitting information on IVPT studies in an ANDA ANDA中递交的IVPT研究信息For IVPT studies with topical products submitted in ANDAs that are intended to support a demonstration of BE, detailed study protocols, relevant SOPs, and detailed reports should be submitted for the IVPT method validation (including the IVPT pilot study) and the IVPT pivotal study. In addition, a detailed report describing the IVPT method development should be submitted. These protocols, SOPs, and reports should be submitted in module 5.3.1.2 of the electronic Common Technical Document (eCTD) and should describe experimental procedures, study controls, quality management procedures, and data analyses.对于支持外用制剂产品BE论证的IVPT研究,在ANDA申请中应递交IVPT方法验证(包括IVPT初步研究)和IVPT正式研究的详细研究方案、相关SOPs和报告。此外,应递交描述IVPT方法开发的详细报告。这些方案、SOPs和报告应在电子通用技术文件(eCTD)的模块5.3.1.2中递交,并对相关的试验方法、研究控制、质量管理程序和数据分析进行描述。Note that the study protocols, SOPs, and reports related to the IVPT method are distinct from those for the sample analytical method that is used to quantify drug concentrations in IVPT receptor solution samples (e.g., an HPLC/MS or UPLC/MS method). Separate protocols and SOPs should be submitted for the sample analytical method validation. Sample analytical method development and validation reports, pilot and pivotal IVPT study sample analysis reports, as well as associated SOPs and protocols relevant to the sample analysis of an IVPT study with human skin should be submitted in Module 5.3.1.4 of the eCTD.注意:用于IVPT接收液中样品浓度的定量分析方法(如,HPLC/MS或UPLC/MS方法)与IVPT方法相关的研究方案、SOPs和报告不同。应单独递交样品分析方法验证的方案和SOPs。样品分析方法开发和验证报告、初步和正式IVPT研究样品分析报告,以及与采用人体皮肤进行IVPT研究相关的样品分析SOPs和方案,应在eCTD的模块5.3.1.4中递交。VIII. IVPT pivotal study statistical analysis IVPT正式研究统计分析
应用实例
2024.04.12

ANDA申请中递交的外用药物产品的体外渗透(IVPT)研究
ANDA申请中递交的外用药物产品的体外渗透(IVPT)研究Guidance for Industry 工业指南 DRAFT GUIDANCE 指南草案This guidance document is being distributed for comment purposes only. 本草案仅用于征求意见Comments and suggestions regarding this draft document should be submitted within 60 days of publication in the Federal Register of the notice announcing the availability of the draft guidance. Submit electronic comments to https://www.regulations.gov. Submit written comments to the Dockets Management Staff (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register.For questions regarding this draft document, contact (CDER) Susan Levine at 240-402-7936.请于草案公布后60日内反馈建议或意见。网上提交请点击http://www.regulations.gov。书面资料请邮寄至FDA文件管理中心(HFA-305),地址为5630 Fishers Lane, rm. 1061, Rockville, MD 20852。反馈意见时请注明草案内容对应的行号。如有疑问请拨打240-4020-7936联系(CDER)Susan Levine。U.S. Department of Health and Human Services 美国健康福利部Food and Drug Administration 食品和药品管理局Center for Drug Evaluation and Research (CDER) 医药审评和研究中心(CDER)October 2022 2022年10月Generic Drugs 仿制药Additional copies are available from: 其他副本可从Office of Communications, Division of Drug Information 药品信息通讯厅Center for Drug Evaluation and Research 医药审评和研究中心Food and Drug Administration 食品和药品管理局10001 New Hampshire Ave., Hillandale Bldg., 4 th Floor Silver Spring, MD 20993-0002Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353Email: druginfo@fda.hhs.govhttps://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugsIn Vitro Permeation Test Studies for Topical Drug Products Submitted in ANDAsANDA申请中递交的外用药物产品的体外渗透(IVPT)研究Guidance for Industry(注释1:指南由食品药品管理局(FDA)的医药审评与研究中心( CDER)仿制药办公室( OGD)起草制订。)This draft guidance, when finalized, will represent the current thinking of the Food and Drug Administration (FDA or Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations. To discuss an alternative approach, contact the FDA staff responsible for this guidance as listed on the title page.该指南代表了 FDA 对该主题目前的看法。它并不会赋予任何人任何权利,也不会约束 FDA 或公众,如果有替代的方法能够满足法律法规的要求,可以使用替代的方法。如果想探讨替代的方法,请联系该指南首页中 FDA负责执行该指南的工作人员。I. Introduction 绪论This guidance is intended to assist applicants who are submitting abbreviated new drug applications (ANDAs) for liquid-based and/or other semisolid products applied to the skin, including integumentary and mucosal (e.g., vaginal) membranes, which are hereinafter called “topical products.”(注释2)Because of the complex route of delivery associated with these products, which are typically locally acting, and the potential complexity of certain formulations, topical products (other than topical solutions) are classified as complex products.(注释3)This guidance provides recommendations for in vitro permeation test (IVPT) studies comparing a proposed generic (test) topical product and its reference standard (RS) for the purpose of supporting a demonstration of bioequivalence (BE) to the reference listed drug (RLD). The reference standard ordinarily is the RLD.(注释4)本指南旨在帮助申请人提交用于皮肤的液体和/或其他半固体产品的仿制药申请(ANDA),包括皮肤和粘膜(如阴道),以下简称“外用制剂”(注释2)。由于这些制剂通常局部起效,具有复杂递送途径和配方,外用制剂(外用溶液除外)通常称为复杂制剂(注释3)。本指南为拟申报的仿制药与其对照制剂(RS)的体外渗透试验(IVPT)研究提供建议,以论证与参比制剂(RLD)的生物等效性(BE)。参比制剂通常简称为RLD。(注释4)注释2:在ANDA申请中,本指南所述的外用制剂包括软膏剂、乳膏剂、洗剂lotions、乳剂emulsions、糊剂pastes、洗发剂shampoos、凝胶剂、混悬剂、喷雾剂、气雾剂、泡沫剂foams、溶液和其他半固体和/或液体剂型,这些剂型具有特定的物质排列结构(可能包括多个相态)。注释3:根据仿制药使用者付费法案(GDUFA)重新授权绩效目标和2023-2027财年强化计划(常称为GDUFA III承诺函)(参见网址https://www.fda.gov/media/153631/download)中所述的定义,除另有规定外,复杂制剂产品包括具有复杂配方(如:胶体)和复杂递送途径(如:具体起效的皮肤用产品)的产品。注释4:根据21 CFR 314.3(b)定义,参比制剂是指作为ANDA申报中参照的FDA指定的已批准药物;对照制剂(RS)被定义为“寻求ANDA的批准必须在所需的体内生物等效性研究中使用的对照药品”。推荐采用对照制剂(RS)进行体外试验。在有些情况下(如RLD已经退市),RS可以不是RLD。关于RLD和RS的更多信息,见“ANDA申请参照药品行业指南(2020年10月)”,我们会对指南定期进行更新,关于指南的最新版本,详见网址:https://www.fda.gov/regulatory-information/search-fda-guidance-documents.This guidance does not address drug products that are administered via ophthalmic, otic, nasal, inhalation, oral, or injection-based routes, or that are transdermal or topical delivery systems (including products known as patches, topical patches, or extended release films).本指南不适用于通过眼、耳、鼻、吸入、口服或注射途径的药品,亦不适用于经皮或局部给药系统(包括贴剂、局部贴剂或缓释膜剂)。It is beyond the scope of this guidance to discuss specific topical products to which this guidance applies. FDA recommends that applicants consult this guidance and any relevant product-specific guidances (PSGs)(注释5)and any other relevant guidances for industry,(注释6)when considering the design and conduct of IVPT studies that, in conjunction with other studies, as deemed necessary, may be appropriate to support a demonstration that a proposed generic topical product and its RLD are bioequivalent. FDA also recommends that applicants routinely refer to FDA’s guidance web pages, because additional guidances may become available that could assist in the development of a generic topical product.本文不是讨论适用于该指南范围的特定外用制剂产品的指南。FDA建议申请人,在进行IVPT研究的设计和实施时,查阅特定产品开发指南(PSGs)(注释5)和其他行业指南(注释6),同时结合其他必要的研究,以支持拟申请的仿制药与RLD的生物等效性。此外,FDA建议申请人定期查阅FDA指南网站,及时获取最新的相关指南,以促进仿制药的开发。注释5:Generic drug product-specific guidances are available at FDA’s Product-Specific Guidances for Generic Drug Development web page at https://www.fda.gov/drugs/guidances-drugs/product-specific-guidances-generic-drugdevelopment. FDA仿制药特定产品开发指南,详见网址。注释6:其他相关行业指南包括:ANDA申请中递交的外用药物产品的体外释放研究指南(2022年10月)和ANDA 申请中递交的外用药物产品的物理化学和结构(Q3)特性(2022年10月)。该指南代表了FDA对该主题目前的看法。In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.一般而言,FDA 指南性文件并非具有强制执行的法律职能。实际上,指南陈述了管理部门对某一个问题当前的看法,并且仅作为建议,除非当具体的法规或法令要求被引用时。在指南中用到的词语“应该”,是指建议,并非要求的意思。II. Background 背景This guidance has been developed as part of FDA’s “Drug Competition Action Plan,”(注释7)which, in coordination with the Generic Drug User Fee Amendments (GDUFA)(注释8) program and other FDA activities, is intended to increase competition in the market place for prescription drugs, facilitate the entry of high-quality and affordable generic drugs, and improve public health.本指南已发展为FDA“药品竞争行动计划”(注释7)的一部分,配合GDUFA(注释8)项目和其他FDA举措,旨在促进处方药市场的竞争,促进优质和实惠的药品进入市场,提高公众医疗水平。注释7:See FDA Drug Competition Action Plan (describing the FDA’s Drug Competition Action Plan, implemented in 2017 and designed to, among other things, further encourage robust and timely market competition for generic drugs), available at https://www.fda.gov/drugs/guidance-compliance-regulatory-information/fda-drug-competitionaction-plan.注释8:In this guidance, GDUFA refers to the generic drug user fee program codified in the Generic Drug User Fee Amendments of 2012, Title III, Food and Drug Administration Safety and Innovation Act (Public Law 112-144), the Generic Drug User Fee Amendments of 2017, Title III, FDA Reauthorization Act of 2017 (Public Law 115-52), and the Generic Drug User Fee Amendments of 2022, Title III of Division F (the FDA User Fee Reauthorization Act of 2022) of the Continuing Appropriations and Ukraine Supplemental Appropriations Act, 2023 (Public Law 117-180).The Federal Food, Drug, and Cosmetic Act (FD&C Act) generally requires an ANDA to contain, among other things, information to show that the proposed generic drug product 1) is the same as the RLD with respect to the active ingredient(s), conditions of use, route of administration, dosage form, strength, and labeling (with certain permissible differences) and 2) is bioequivalent to the RLD.(注释9)Thus, an ANDA will not be approved if the information submitted in the ANDA is insufficient to show that the test product is bioequivalent to the RLD.(注释10)《联邦食品、药品和化妆品法案》(FD&C法案)要求ANDA申请中应包含合适的信息,说明:1)仿制药与RLD相比,具有相同的活性成分、使用条件、给药途径、剂型、规格和说明书(允许存在一些差异);2)与RLD生物等效。(注释9)因此,如果ANDA中提交的信息不足以证明自制制剂与RLD具有生物等效性,则ANDA将不予批准(注释10)。注释9:See sections 505(j)(2)(A), (j)(2)(C), and (j)(4) of the FD&C Act (21 U.S.C. 355(j)(2)(A), (j)(2)(C), (j)(4)); see also 21 CFR 314.94.注释10:21 CFR 314.127(a)(4), (6).An IVPT study may be used to assess the rate and extent to which a drug (i.e., an active ingredient) from a topical product becomes available at or near a site of action in the skin, and may be used to characterize and compare the rate and extent of bioavailability for a drug from a test topical product and RS. The IVPT flux profiles resemble pharmacokinetic profiles and can be analyzed using unique IVPT endpoints that are somewhat analogous to the pharmacokinetic endpoints of maximum concentration (Cmax) and the area under the concentration-time curve (AUC). Yet, IVPT studies characterize the rate and extent of absorption, not the distribution, metabolism and excretion that occurs in vivo. Therefore, while it is relevant to characterize the kinetics of topical drug bioavailability monitored by IVPT studies, the use in this guidance of the term “cutaneous pharmacokinetics” should not be construed to embody all aspects of pharmacokinetics—only those related to the absorption component that directly controls the rate and extent to which a topically applied drug becomes available locally at the site of action. This guidance focuses on general considerations and recommendations for the method development, method validation, and conduct of IVPT studies that are submitted in ANDAs and intended to support a demonstration of BE.(注释11)IVPT研究可用于评估外用药物产品中药物(即活性成分)到达皮肤作用部位或附近的速率和程度,表征和评估自研外用制剂与对照制剂(RS)中药物的生物利用度和程度。IVPT通量曲线类似于药代动力学曲线,可以使用独特的IVPT终点进行分析,该终点与最大浓度(Cmax)的药代动力学终点和浓度-时间曲线下面积(AUC)相似。然而,IVPT研究表征的是药物吸收的速率和程度,而不是体内的分布、代谢和排泄。因此,IVPT研究虽然可用于外用药物制剂中活性成分生物利用度的动力学表征,但本指南中的俗语“皮肤药代动力学”不应被解释为药代动力学的所有方面,其表征的仅仅是“外用制剂中活性成分达到作用部位的速率和程度”。本指南旨在说明,在ANDA申请中提交、拟用于BE论证的IVPT研究的方法开发和验证的一般考虑和建议。(注释11)注释11:A demonstration of no significant difference in the rate and extent of drug permeation into and through the skin of the test topical product and RS using an appropriately validated IVPT method can be used to support a demonstration of BE along with other data in the application (which may be specified in a PSG), as part of a comparative product characterization-based approach.在ANDA申请中,通过采用合适且经过验证的IVPT方法,说明自研外用制剂与对照制剂(RS)中药物的渗透速率和通过皮肤的量无明显差异,作为产品特性比较方法的一部分,并结合其他数据用于支持BE的论证(可能在“特定产品开发指南”中有规定)。III. IVPT method developmen IVPT方法开发The development of an IVPT method that is suitable to support a demonstration of BE for a specific topical product routinely involves a systematic series of exploratory studies. Inappropriate or insufficient efforts to develop an IVPT method that is suitable for its intended purpose increases the likelihood that the subsequent IVPT validation, pilot, and pivotal studies will ultimately be inadequate to support a demonstration of BE. By contrast, appropriate and systematic IVPT method development studies help to identify IVPT study designs and protocol (method) parameters which reliably produce flux profiles that can facilitate a comparison of the cutaneous pharmacokinetics of a drug delivered topically to the skin from test topical products and RSs.适用于支持特定外用制剂BE论证的IVPT方法的开发,通常需要进行一系列系统的探索性研究。不合适或不充分的IVPT方法开发可能会导致,后续的IVPT验证、初步和正式研究不足于支持BE论证。相反,合理且系统的IVPT方法开发研究,有助于明确IVPT研究设计和方案(方法)参数,产生可靠的通量曲线,以便于自研制剂和RSs的皮肤药代动力学比较。A detailed and well-organized IVPT method development report should be submitted in an ANDA to show how the IVPT method was optimized, and to support a demonstration that the method parameters selected for the IVPT are appropriate or necessary, particularly in situations where the method parameters are different from the methods recommended in this guidance). The Agency’s interest in reviewing the method development report is to understand why specific IVPT method parameters were selected and whether the resulting IVPT method is suitably sensitive and reproducible. This method development report should clearly indicate/distinguish the method parameters used for each set of data, illustrate the efforts made to optimize the IVPT method, and demonstrate that the method parameters selected for the IVPT are appropriate.应在ANDA中提交详细且结构清晰的IVPT方法开发报告,说明IVPT方法是如何优化的,所选择的IVPT方法参数是合理性或必要的,尤其是方法参数与本指南中建议的方法不同时。监管机构在审核IVPT方法开发的过程中,比较关注IVPT方法参数选择的合理性,以及确定的IVPT方法是否具有合适的灵敏度和重现性,因此,需要说明在IVPT方法优化过程中所作的努力,并证明所选择的IVPT方法参数是合适的。Applicants are encouraged to use the recommendations in this guidance, and if an applicant elects to use methods that are different from those recommended in this guidance, the IVPT method development report should demonstrate why it is scientifically justified to use an alternative approach than what is recommended in this guidance to optimize the IVPT method.(注释12)Some examples of recommended procedures are described in subsequent sections, to help applicants identify circumstances when information should be submitted in the ANDA to explain why a different procedure was utilized.鼓励申请人采用本指南中推荐的方法,如果申请人选择其他方法,应在IVPT方法开发报告中说明,采用替代的方法优化IVPT方法的科学合理性(注释12)。以下部分描述了本指南中推荐方法的一些示例,以帮助申请人识别在ANDA申请中何时需要递交相关信息,解释为什么采用了不同的方法。注释12:Applicants may choose to use an approach different from the approach recommended in this guidance. However, the alternative approach must comply with relevant statutes and regulations. See 21 CFR 10.115(d). 申请人可以采用指南中没有描述的其它方法。然而,所选用的替代方法必须遵守相关法律法规要求。A. IVPT Method Parameters IVPT方法参数All relevant parameters of the final IVPT method should be summarized (e.g., in a table) and submitted in the ANDA. Also, information should be provided to briefly explain the choice of the final IVPT method parameters like the equipment (e.g., a vertical diffusion cell (VDC)), skin source (e.g., cadaver), skin type (e.g., posterior torso), skin preparation (e.g., dermatomed), skin barrier integrity test (e.g., trans-epidermal water loss (TEWL) measurement), skin barrier integrity test acceptance criteria (e.g., 应对最终确定的IVPT方法的相关参数进行总结(如,以表格形式),并在ANDA申报资料中递交。此外,还应提供信息,简要说明最终确定IVPT方法参数选择的理由,譬如说:设备(如,立式扩散池(VDC))、皮肤来源(如,尸体)、皮肤类型(如,背部躯干)、皮肤处理方法(如,离体皮肤/皮节)、皮肤屏障完整性测试(如,经皮水分散射法(TEWL))、皮肤屏障完整性接受标准(如,<15克/平方米/小时(g/m2/h))、上样量(如,15毫克/平方厘米(15mg/cm2))、剂量维持时间(如,6小时)、研究持续时间(如,24小时、48小时等)、接受液取样时间点(如,1、2、4、6、8、12、16和24小时)等。B. IVPT Method Considerations IVPT方法考虑因素The choice of some IVPT method parameters like the equipment, skin source, skin type, skin preparation, and skin barrier integrity test procedures may be based upon investigator experience or convenience, like the availability of specific equipment or instrumentation in a laboratory, established tissue supply agreements, or other logistical considerations. However, if the chosen IVPT method parameters do not appear to be well-suited for a specific IVPT method, it is the applicant’s responsibility to systematically evaluate alternative method parameters, and ultimately, to validate that the IVPT method parameters chosen are suitable for the intended purpose. The recommended procedures for IVPT method validation are detailed in section IV of this guidance.IVPT方法参数的选择,如设备、皮肤来源、皮肤类型、皮肤处理方法和皮肤屏障完整性测试方法等,可能取决于研究人员的经验或便利性,譬如说实验室已有的设备或仪器、既定的组织供应协议或其他后勤保障等因素。但是,如果所选的IVPT方法参数似乎并不适合特定的IVPT方法,申请人有责任系统的评估替代的方法参数,确认所选的IVPT方法参数适用于既定的目的。本指南第四节详细介绍IVPT方法验证的推荐程序。The choice of other IVPT method parameters like the topical product dose amount, dose duration, study duration (which may be longer than the dose duration), sampling schedule, sampling procedures, receptor solution composition, and sample analytical method may be different for each IVPT method, and such parameters of IVPT methods should be systematically developed, optimized, and/or validated for the relevant topical product, as appropriate. The IVPT method development studies should characterize how differences in these method parameters influence the resulting IVPT flux profile so that optimal study conditions can be objectively selected from among those evaluated.IVPT其他方法参数的选择,如外用制剂的上样量、剂量维持时间、研究持续时间(可能比剂量维持时间长)、取样计划、取样方法、接收介质组成和样品分析方法,不同IVPT方法可能存在差异,因此需要对这些参数进行系统的开发、优化和/或验证,以确保其适用于相关的外用制剂产品。应在IVPT方法开发研究中,描述这些方法参数的差异如何影响最终的IVPT通量曲线,以便从这些评估中客观地选择最佳的研究条件。The selection of the dose amount used in the study should be assessed for each IVPT method based upon studies performed during IVPT method development. Different dose amounts may be compared in parallel on replicate skin sections from the same set of donors to optimize the dose amount for the IVPT study. Considerations for selecting an optimal dose amount may include (1) the consistency with which the dose can be applied (potentially using different dispensing and/or spreading techniques), (2) the reproducibility of the flux profiles, (3) the influence of dose amount and dose duration on the shape of the flux profile, and (4) the approximate range of drug concentrations in receptor solution samples at different time points (relative to the sample analytical method limits of quantification).应根据IVPT方法开发期间进行的研究,对每个IVPT方法中所选的上样量进行评估。可通过采用相同皮肤供体的重复皮肤切片进行不同剂量的平行对比研究,优化IVPT研究中所需的上样量。优化上样量需要考虑的因素包括:(1)采用相同的上样方式(可采用的上样技术包括:直接分配和/或涂覆);(2)通量曲线的重现性;(3)上样量和剂量维持时间对通量曲线的影响;和(4)接收液中不同取样时间点的药品浓度大致范围(与样品分析方法的定量限度相关)。The selected sampling schedule and study duration should be sufficient to characterize the cutaneous pharmacokinetics of the drug, which ideally includes a sufficiently complete flux profile to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points. A dose that remains on the skin for the duration of the study may continue to deliver the drug for a sustained period and may not necessarily exhibit a suitable decline in the flux at later time points. In such instances, it may be appropriate to develop an IVPT method that involves wiping off the applied dose after a suitable duration on the skin and continuing to monitor the receptor solution for an extended period thereafter, during which the decline in the flux profile can be characterized. The sampling frequency should be selected to provide a suitable resolution for the flux profile, and a minimum of eight non-zero sampling time points is recommended across the study duration (e.g., 48 hours).所选择的取样计划(即:取样时间点)和研究持续时间应足以表征药物的皮肤药代动力学,理想情况下应包括一个足够完整的通量分布,以识别最大(峰值)通量和此后多个时间点的通量下降情况。在研究期间,如果药品持续保留在皮肤上面,其可能会持续递送药物,在最大(峰值)通量之后时间点可能不会通量下降的现象。此时,在IVPT方法开发时,可在上样一段时间后将样品擦去,然后持续监控接收液一定时间,使之呈现出通量下降的情况。所选的取样频率应足以为通量曲线提供合适的分辨率,在整个研究期间(如,48小时),推荐至少采用8个非零取样点。C. IVPT Method Procedures and Controls IVPT方法程序和控制Suitable technical procedures and control parameters should be established during method development. These may include procedures for preparing and mounting the skin on the diffusion cell in a consistent manner, determining the instrument settings that regulate the skin surface temperature within the specified range, performing the barrier integrity test appropriately, controlling the accuracy and precision of the dose amount dispensed on each skin section.在方法开发过程中,应制定合适的技术规程和控制参数,包括:采用一致的方式准备皮肤并将其安装到扩散池上;确定仪器参数设置,使皮肤表面温度在既定的范围内;进行合适的屏障完整性测试;控制皮肤切片上上样量的准确度和精密度。For example, a dosing procedure may be developed that uses a positive displacement pipette to dispense a volumetrically controlled amount of a topical product, targeting the deposition on the skin of a certain mass (e.g., 15 mg/cm2) of topical product. If the inner diameter of the orifice in the dosing compartment of the diffusion cell is 15 millimeters (mm), and the effective dose area is ~1.77 cm2, this would indicate a target dose of ~26.5 mg of topical product per diffusion cell. Experiments during method development may establish that, based upon the density of the topical product, a specific volumetric setting on a specific model of positive displacement pipette with a specific pipette tip repeatedly dispenses ~27.5 mg of topical product (e.g., characterized by multiple replicate pipette dispensations into a weigh boat on a fine balance). This pipette setting may be optimal for a dosing procedure where the dose spreading instrument, like the flat bottom of a high performance liquid chromatography (HPLC) glass vial, or the rounded end of a glass rod or capillary tube, is subsequently used to spread the dispensed dose evenly upon the skin section mounted in the diffusion cell, and where repeatedly weighing the dose-spreading instrument before and after the dose spreading indicates that the residual topical product remaining on the bottom of the glass vial after the dose spreading reproducibly amounts to ~1.0 mg of topical product (indicating that ~26.5 mg of the topical product would reproducibly be dosed to each skin section). Such characterizations of the technical procedures and control parameters for the IVPT method, like the reproducibility of the dosing procedure, should be established during method development and may not need to be demonstrated thereafter each time the same procedure is used.例如,在上样方式的研究中,可以采用外置活塞式移液器(即:移液枪)把一定体积的外用药物制剂分配到皮肤上,使皮肤上具有一定重量的药物制剂(如,15mg/cm2);如扩散池供给室的孔口内径是15mm,则其有效面积约为1.77cm2,意味着每个扩散池中制剂的目标剂量约为26.5mg。在方法开发过程中,采用外置活塞式移液器及配套的枪头,根据外用制剂的密度,将外置活塞式移液器设定为相应的体积,依次重复分配外用药物制剂使其约为27.5mg(如,通过“可重复分配相应重量的外用药物制剂到天平称量盘上”进行论证)。这种移液器是上样分配样品的最佳选择,分配样品后可采用高效液相色谱(HPLC)玻璃瓶的平底、玻璃棒或毛细管的圆端,将分配到皮肤上的药品涂覆均匀;同时,在涂覆前后应分别称量涂覆用的工具,确保涂覆工具在涂覆前后重量相差约为1.0mg(表明可将约26.5mg的外用制剂重复均匀的涂覆到每个皮肤切片上)。IVPT方法的技术程序和控制参数等特性,如上样方法的可重现性,应在方法开发期间进行确定,但之后就不需要对这些特性再次论证。D. IVPT Skin Barrier Integrity Testing: Common Methods IVPT皮肤屏障完整性测试:常用方法The technical procedures for the skin barrier integrity test should be established during IVPT method development. Three types of barrier integrity tests are common, however, there are currently no applicable compendial standard protocols or acceptance criteria for any of these three types of human skin barrier integrity tests. Nonetheless, recommended parameters for the three common types of barrier integrity tests are discussed below.应在IVPT方法开发期间建立皮肤屏障完整性测试方法。常用的皮肤屏障完整性测试方法有三种,但还没有相应的药典收载或法定可接受标准。尽管如此,下面讨论三种常见类型屏障完整性测试的推荐参数。1. Trans-Epidermal Water Loss Skin Barrier Integrity Test 经皮水分散失法测试皮肤屏障完整性A TEWL skin barrier integrity test involves a measurement near the outer surface of the skin of the rate at which water (vapor) is fluxing through the skin barrier from the underside of the skin section. For the test, the skin section is mounted in a diffusion cell (e.g., clamped in place between the donor and receptor compartments), with the underside of the skin in contact with the receptor solution in the receptor compartment (e.g., phosphate buffered saline, pH 7.4), and equilibrated to a skin surface temperature of 32°C ± 1°C. If skin sections are cut large enough to cover the flange of the diffusion cell in which they are mounted, then after they have equilibrated for several hours at a skin surface temperature of 32°C ± 1°C, it may be feasible to gently remove the donor compartment without disrupting a skin section’s adherence to the lower flange of the diffusion cell, thereby allowing the TEWL probe to be placed directly on the skin surface, instead of being placed atop the donor compartment. Typically, a minimum of three replicate measurements are made on each skin section, which are recorded after the measurements have stabilized.TEWL皮肤屏障完整性测试是通过测定皮肤外表面附近的水分(蒸汽),评估水分从皮肤下侧通过皮肤屏障的速率。在测定时,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤下侧与接收室中的接收介质(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32℃±1℃。如果皮肤切片切割的足够大,能覆盖皮肤安装位点的扩散池法兰,可在皮肤表面温度平衡至32℃±1℃几小时后,在不破坏皮肤切片与其下部的扩散池法兰粘附的情况下,将供给室轻轻移去,此时可将TEWL探针直接放置在皮肤表面,而不用放置在供给室的顶部。通常,在测试结果稳定后,每个皮肤切片需至少重复测定三次,并记录相关结果。Commercially available devices to measure TEWL may differ in design and operational principles. The TEWL measured by devices with certain designs (e.g., an open chamber versus a closed chamber) may be relatively more susceptible to the influence of environmental conditions. Therefore, environmental temperature and humidity are typically controlled as precisely as possible (e.g., a temperature range of 21°C ± 2°C and a humidity range of 50% ± 20% relative humidity are ideal, if feasible). More precise control of the relative humidity (e.g., in the range of 40% – 50%) may reduce the variability of TEWL measurements for devices with certain designs. Certain designs of measurement probes and adapters for in vitro use are available by the manufacturers of TEWL devices, and may be appropriate to use. Inconsistency in the diameters for the measurement probe chamber, the measurement probe orifice, the in vitro adapters, and the skin area being measured, as well as variation in the distance of the probe sensor(s) from the skin surface, potentially because of the (variable) height of donor compartments (when applicable), could increase the variability of TEWL measurements. Inconsistent control of the alignment of the TEWL measurement device in relation to the donor compartment and/or the skin section may also increase the variability of TEWL measurements. Also, the TEWL measured by devices with certain designs may be relatively more susceptible to the influence of heat transfer from the hand that holds the probe. Applicants should follow relevant instructions in the manufacturer’s user manual for the specific TEWL measurement device used.商业化可用于TEWL测定的设备,有不同的设计和操作原理。由于设计的差异,一些TEWL测试设备可能相对更容易受到环境条件的影响(如,开放式腔室与封闭式腔室相比)。因此,通常需要尽可能精确的控制环境的温度和湿度(比较理想的情况是,将温度控制在21°C ± 2°C、湿度控制在50% RH± 20%RH范围内)。更精确的控制相对湿度(如,控制在40%RH~50%RH范围内)可以降低一些设备测定TEWL结果的变异性。TEWL测试设备生产商提供的用于体外的一些测试探针和适配器,可能适用于TEWL测定。不一致的测量探针室直径、测量探针孔、体外适配器和待测的皮肤面积,以及由于供给室高度的不同导致的探测传感器与皮肤表面距离的差异,均会增加TEWL测定结果的变异。与供给室和/或皮肤切片相关的TEWL测试设备对准的差异也可能增加TEWL测量结果的变异。此外,有些TEWL测试设备可能相对更容易受到握持探针的手的热传递的影响。对于特定的测试设备,申请人应遵循生产商使用手册中的相关说明。No more than approximately 15 grams of water per square meter per hour (i.e., ≤15g/m2/hr)could be a reasoable skin barrier integrity acceptance (cutoff) criterion for a TEWL barrier integrity test on human torso or thigh skin; if this was selected as the cutoff criterion, skin sections with a TEWL> 15g/m2/hr would fail the test. Skin sections that fail a barrier integrity test should not be dosed, but may serve as non-dosed control skin sections. A higher cutoff (e.g., ≤20 g/m2/hr) may also be reasonable if justified by experimental data demonstrating that the selected acceptance criterion appropriately discriminates skin sections with a compromised barrier integrity from those with a competent barrier integrity.对于人体躯干或大腿皮肤,采用TEWL屏障完整性测试法,合理的皮肤屏障完整性接受(截止)标准是每小时每平方米不大于约15g水(即,≤15g/m2/hr);如果选择其作为截止标准,TEWL大于15g/m2/hr的皮肤,则不具有皮肤完整性。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可用作为无剂量空白对照皮肤切片。但是,如果通过实验数据表明,所选的接受标准可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片,也可以采用更高的截止标准(如,≤20g/m2/hr)。However, TEWL measurements for skin sections with a competent barrier integrity can vary depending upon the TEWL measurement device, the manner in which it is operated, and the environmental conditions (e.g., higher ambient humidity or greater distance from the skin surface may decrease the value of the TEWL measurement). Precise control of environmental and device/operational factors can minimize variability in TEWL measurements. Therefore, the technical procedures for measuring TEWL should be optimized during IVPT method development (or based upon prior optimization in the laboratory performing the test). Also, the TEWL measurement device should be appropriately calibrated (by the manufacturer, and for some devices, also before each set of tests). Applicants may provide information about the relevant calibration procedures specified by the manufacturer for the specific TEWL device used; this can be submitted in the ANDA along with the IVPT method development report, to support the appropriateness of the technical procedures established by the laboratory for TEWL measurements. When a TEWL barrier integrity test is used in any study phase (IVPT method development, pilot study, validation, and/or pivotal study) the ambient laboratory temperature and humidity during the TEWL barrier integrity test should be monitored and reported.然而,具有合格屏障完整性皮肤的TEWL测量值,可能会因TEWL测试设备、操作方式和环境条件的不同而变化(如,较高的环境湿度或距离皮肤表面较远的距离可能会降低TEWL的测量值)。对环境和设备/操作因素的精确控制可将TEWL测量的可变性将至最低。因此,在IVPT方法开发过程中应对TEWL的测定方法进行优化(或基于实验室之前进行该测试的优化结果)。此外,应对TEWL测试设备进行适当的校准(由生产商进行;对于某些设备,在每次实验前都需要进行校准)。申请人可提供生产商制定的相关校准程序信息,与IVPT方法开发报告一起在ANDA中递交,用于支持申请人为TEWL测量制定的技术程序的合理性。在IVPT方法开发、初步研究、验证和/或正式研究的整个研究阶段,进行TEWL屏障完整性测试时,均应监测和报告实验室的环境温度和湿度。2. Tritiated Water Skin Barrier Integrity Test 氚化水渗透法测试皮肤屏障完整性An example of a recommended approach to a tritiated water skin barrier integrity test would be to mount the skin in a diffusion cell (e.g., clamped in place between the donor and receptor compartments) and allow it to equilibrate to a skin surface temperature of 32°C ± 1°C with the stratum corneum exposed to the air in the donor compartment and the underside of the skin in contact with the receptor solution (e.g., phosphate buffered saline, pH 7.4).氚化水渗透法测试皮肤屏障完整性的一个推荐方法是,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤的角质层暴露在空气中(即,朝向供给室),皮肤下侧与接收液(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32°C ± 1°C。A small amount of tritiated water (sufficient to cover the entire surface of the skin section) would be briefly dosed on the stratum corneum. This dose of tritiated water would be left on the surface for a precisely controlled and relatively brief period (e.g., 5 minutes) after which it would be removed from the skin surface (e.g., using a pipette to remove the bulk volume and then an absorbent low lint laboratory tissue to gently blot dry). The receptor solution would then be sampled at a precise duration after the removal of the tritiated water from the skin surface (e.g., 30 minutes after the removal of the 5-minute dose of tritiated water from the skin surface).将少量的氚化水(足以覆盖皮肤切片的整个表面)上样到皮肤角质层,精确控制氚化水留在皮肤表面的时间(如,5分钟),到特定时长后立即将其从皮肤表面移除(如,先用吸管去除大量的氚化水,然后用吸收性低脂棉轻轻吸干)。在氚化水从皮肤表面移除后的特定时间点(如,将氚化水上样到皮肤表面5分钟后,再将其移除之后的30分钟)从接收液中取样。While the entire volume of the receptor compartment may be removed and replenished, typically only an aliquot of the receptor solution (e.g., phosphate buffered saline, pH 7.4) is transferred to a suitable volume of scintillation fluid for counting. The volume of the aliquot typically depends upon the type of scintillation fluid used and the maximum amount of aqueous fluid that is suitable to mix with the scintillation fluid. A scintillation counter is then used to quantify the amount of radioactivity in the aliquot sampled, which can be used to calculate the amount of tritiated water that permeated into the larger (entire) volume of receptor solution; the calculation is performed using the specific activity of the tritiated water to equate a given amount of radioactivity to the equivalent volume of tritiated water that permeated per square centimeter of skin surface area.虽然可以完全移取整个接收室的接收液(如PBS,pH7.4),通常情况下,移取一部分接收液转移至适当体积的闪烁液中用于计数即可。移取的接收液体积通常取决于闪烁液的类型及可与闪烁液混合的最大含水量。然后,采用闪烁计数器对取出的接收液的放射性进行定量,之后可进一步推导出整个接收室的接收液中含有的氚化水量;该计算是根据氚化水的比活度进行的,即:给定量的放射性与每平方厘米皮肤渗透的当量体积的氚化水相当。Approximately 1.5 equivalent (eq.) microliter (μL) of tritiated water per cm2 (i.e., ~1.5 eq. μL/cm2 or ~1.5 eq. mg/cm2) would be a reasonable skin barrier integrity acceptance (cutoff) criterion for a tritiated water barrier integrity test that involves a 5-minute dose followed by a 30minute sampling duration (i.e., sampling 30 minutes after dose removal) on human torso or thigh skin. Skin sections with a tritiated water test result of > 1.5 eq. mg/cm2 would fail the test and be excluded from the population of skin sections dosed with the topical product; skin sections that fail a barrier integrity test should not be dosed, but may serve as non-dosed control skin sections. Other acceptance criteria may also be reasonable if justified by experimental data demonstrating that the selected acceptance criterion appropriately discriminates skin sections with a compromised barrier integrity from those with a competent barrier integrity.对于人体躯干或大腿皮肤,采用氚化水屏障完整性测试法,将氚化水上样到皮肤表面5分钟后移除,在移除30分钟后取样(即:将氚化水移除后30分钟后取样),合理的皮肤屏障完整性接受(截止)标准是每平方厘米约1.5当量微升的氚化水(即:1.5 eq.μL/cm2 or ~1.5 eq.mg/cm2)。采用氚化水屏障完整性测试法,如果测定结果大于1.5 eq.mg/cm2,则不具有皮肤完整性。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可用作为无剂量空白对照皮肤切片。但是,如果通过实验数据表明,所选的接受标准可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片,也可以采用其他标准。When calculating the results for a tritiated water barrier integrity test, it may be important to account for the surface area dosed. For example, if using an acceptance criterion of 1.5 eq. mg/cm2 with a diffusion cell that has an orifice diameter of 15 mm and a skin surface area of 1.77 cm2, the mass of tritiated water that would be calculated to have permeated into the receptor compartment would be ~2.7 eq. mg/cm2 of tritiated water.对于氚化水屏障完整性测试法,在结果计算时,需要重点关注上样表面积。例如,当扩散池的直径是15mm时,相应的皮肤表面积为1.77 cm2,如果采用1.5 eq.mg/cm2的接受标准,则渗透进入接收室的氚化水的量应不超过2.7 eq. mg/cm2。3. Electrical Based Skin Barrier Integrity Tests 电阻/电导值法测试皮肤屏障完整性There are several variations of electrical based skin barrier integrity tests that report the test result as a measure of the resistance, conductance, or a related electrical concept that characterizes the bulk flow of electrical current across the skin. Transepithelial electrical resistance tests involving the skin may be referred to more specifically as Trans-Epidermal Electrical Resistance (TEER) skin barrier integrity tests. The test results may be described in units of conductance, which is the reciprocal of resistance. Electrical based skin barrier integrity tests often use instruments that are designed to measure the inductance (L), capacitance (C), and resistance (R) of electronic circuits or electrical components; these instruments are commonly known as LCR meters and have different settings (test parameters) that can be adjusted.电阻/电导法测定皮肤完整性有几种变量,以电阻、电导或相关的电概念报告测试结果,用于表征流过皮肤的电流量。与皮肤相关的跨膜电阻测试通常指跨膜电阻(TEER)皮肤屏障完整性测试。测试结果可以用电阻的倒数“电导”为单位进行描述。电阻/电导值法测试皮肤屏障完整性通常采用“旨在测量电子电路或电子元件的电感(L)、电容(C)和电阻(R)”的仪器;这些仪器通常被称为LCR仪表,具有可调节的不同设置(测试参数)。An example of a recommended approach to a TEER skin barrier integrity test would be to mount the skin in a diffusion cell (e.g., clamped in place between the donor and receptor compartments) and allow it to equilibrate to a skin surface temperature of 32°C ± 1°C with the stratum corneum exposed to the air in the donor compartment and the underside of the skin in contact with an ionic solution (e.g., phosphate buffered saline, pH 7.4).TEER皮肤屏障完整性测试的一个推荐方法是,将皮肤安装到扩散池中(如,夹在供给室和接收室之间),使皮肤的角质层暴露在空气中(即,朝向供给室),皮肤下侧与接收液(如PBS,pH7.4)接触,然后将皮肤表面温度平衡至32°C±1°C。A small amount of the ionic solution (sufficient to cover the entire surface of the skin section) would be briefly dosed on the stratum corneum. Then, one lead/electrode from an LCR meter would be placed in contact with the solution in the receptor compartment while the other lead/electrode would be placed in contact with the solution in the donor compartment. After measuring the resistance across the skin (e.g., in kΩ, normalized for area, noting that resistance is inversely proportional to area) the solution in the donor compartment would be removed and the skin surface would be gently blotted dry with an absorbent low lint laboratory tissue. The skin (still mounted in the diffusion cell) would then be allowed to equilibrate with the dry air above for a sufficient duration to normalize the hydration state of the stratum corneum before being dosed with the test topical product or RS.将少量的离子溶液(足以覆盖皮肤切片的整个表面)短暂的施加到皮肤角质层上;然后,将LCR仪表的一端电极与接收室中溶液接触,而另一端电极与供给室中溶液接触。在测定通过皮肤的电阻后(如,以kΩ为单位,按面积归一化法,注意电阻与面积成反比),将供给室中的溶液移出,用吸收性低脂棉将皮肤表面轻轻吸干。然后,在自研外用制剂或RS上样前,先将皮肤与上面的干燥空气接触,并平衡足够长的时间,使角质层恢复到正常的水合状态。The results for a TEER skin barrier integrity test can vary substantially depending on the LCR meter settings (e.g., frequency) and the technical procedures used for the test. The acceptance criterion for a specific electrical based skin barrier integrity test method may be justified by experimental data demonstrating that the selected acceptance criterion appropriately discriminates skin sections with a compromised barrier integrity from those with a competent barrier integrity.TEER皮肤屏障完整性测试的结果,可能因LCR仪表设置(如,频率)和所用的测试技术方法不同,产生很大变化。电阻/电导值法测试皮肤屏障完整性接受标准的合理性,可通过试验数据证明,即:所选的接受标准应当可以适当地区分屏障完整性受损的皮肤切片与屏障完整性合格的皮肤切片。E. IVPT Skin Barrier Integrity Testing: General Considerations IVPT皮肤完整性测试:注意事项There are three general considerations for the development or adoption of technical procedures for any skin barrier integrity test method during IVPT method development:在方法开发过程中,皮肤屏障完整性的任一测试方法的开发或选择,都应注意以下三个方面:The technical procedures should not irreversibly alter the skin barrier. It may be acceptable to temporarily alter the hydration state of the stratum corneum by briefly depositing an aqueous solution on the surface of the skin, as long as sufficient time is afforded for the hydration of the stratum corneum to normalize before dosing of the topical product. The procedure described above for a brief (e.g., 5-minute) exposure of the skin surface to tritiated water followed by a 30-minute duration during which the hydration state of the stratum corneum is re-equilibrating would likely be appropriate. By contrast, a 30-minute exposure of the skin surface to an aqueous solution for an electrical-based test method, followed within 5 minutes by dosing of the topical product, may not be appropriate without further characterization of the influence of the hydration state of the stratum corneum on the discrimination sensitivity of the skin to differences in topical bioavailability. Similarly, if a portable lamp were placed close to the skin to improve visibility while study procedures were being performed, the heat from the lamp may alter the local (micro)environment of the skin in a manner that is not representative of the ambient environmental conditions in the laboratory; this should be avoided.测定方法不应不可逆的改变皮肤完整性。可以接受将水溶液上样到皮肤表面后,短暂的改变角质层水合状态的情况,但在外用制剂上样前,经过足够长的时间后,角质层应可以恢复到正常的水合状态。如前面描述的情况,将皮肤表面短暂的暴露于氚化水一段时间(如,5分钟)后移除,经过30分钟的重新平衡,角质层的水合状态可以恢复到合适水平。相比之下,对于电阻/电导值测试法,将皮肤暴露于水溶液中30分钟后移除,而后,在5分钟后上样是不合适的,因为没有进一步评估皮肤角质层水合状态对不同外用制剂生物利用度区分灵敏度的影响。类似情况是,在测试过程中,如果将携带式手提灯放置在皮肤附近以提高能见度,从灯中散发出的热可能会改变皮肤的局部(微)环境,并不能代表实验室中的环境条件,应当避免。The acceptance criterion should be a cutoff value for the test result, at which a skin section fails the test. Skin sections that fail a barrier integrity test should not be dosed but may serve as non-dosed control skin sections. Skin sections with a passing barrier integrity test result may be considered to have a competent barrier integrity and may be dosed. This acceptance criterion should be selected based upon an understanding of the distribution of test results (among multiple replicate skin sections from multiple donors) for the specific barrier integrity test procedure performed with the specific type and preparation of skin under conditions relevant to the IVPT pivotal studies submitted in the ANDA. The intention of the barrier integrity test is to identify (and exclude) skin sections whose barrier integrity (intactness) is compromised. The intent is not to reduce the inherent variability in barrier function (permeability) in human skin that is representative of real variation in the human population. Also, the relative permeability of the skin to a drug from a topical product may not necessarily correlate with the permeability of the skin to water, and therefore, constraining the variability of the skin permeability to water (using a stricter acceptance criterion that excludes a larger number of skin sections) may not necessarily reduce the variability in the IVPT study results.接受标准应是皮肤未通过屏障完整性测试的截止值。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可作为无剂量空白对照皮肤切片。通过皮肤完整性测试的皮肤切片,可被视为具有合格的屏障完整性,可以进行上样试验。接受标准的选择可以依据,在ANDA申请中递交的IVPT正式研究中,采用特定类型和制备工艺皮肤进行的屏障完整性测试,对获得的测试结果(采用多个供体的多个重复皮肤切片进行)分布的理解进行选择。皮肤屏障完整性测试的目的是识别(和排除)屏障完整性(完整无缺)受损的皮肤切片。其目的并不是减少人体皮肤屏障功能(渗透性)的内在变异性,人体皮肤的内在变异性是其固有的特性。此外,皮肤对外用制剂中药物的相对渗透性,可能与皮肤对水分的渗透性无关;因此,限制皮肤对水分渗透的可变性(使用更严格的接受标准,排除更多的皮肤切片)可能不一定会减少IVPT研究结果的可变性。The acceptance criterion should be able to discriminate skin sections with a compromised barrier integrity. This may be demonstrated by measuring the barrier integrity of skin sections mounted and equilibrated in a diffusion cell before and after deliberately compromising the skin barrier (e.g., by repeatedly using adhesive tape to strip away increasing amounts of the stratum corneum, piercing the skin several times with a 30 gauge needle, or using other physical or chemical insults to damage the skin barrier). Based upon the acceptance criterion selected, the test result for skin sections that pass the test before being damaged should fail the test after the damage.接受标准应能够区分屏障完整性受损的皮肤切片。对于接受标准的论证,可通过“在特意破坏皮肤屏障的前后(如,通过反复使用胶布剥离越来越多的角质层、用30号针多次刺穿皮肤,或使用其他物理或化学手段破坏皮肤屏障)”,将其分别安装到扩散池并平衡后,根据屏障完整性的测定结果来评估。根据选择的接受标准,在破坏前通过测试的皮肤切片,对其破坏后应不能通过测试。F. Differences Between IVPT Method Development and Validation IVPT方法开发和验证的区别1. Optimization of an IVPT Method Prior to Advancing to IVPT Method Validation 在IVPT方法验证前进行IVPT方法的优化Different study designs and method parameters may be evaluated during the IVPT method development phase. For example, if the selected study parameters initially involve a dose duration of 48 hours and a study duration of 48 hours, and the flux profile is measurable, but it is not feasible to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points, then it may be appropriate to evaluate other study parameters as part of the IVPT method development. For example, a different target dose of the topical product and/or a longer sampling duration may be evaluated. An alternate study design may involve a shorter dose duration (e.g., 4–6 hours) after which the applied dose is removed from the skin, and the receptor solution continues to be sampled across a study duration that is sufficient to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points. While shorter dose durations can help to improve the shape of IVPT flux profiles, the removal of the topical product dose from the skin surface can be challenging and often requires its own method development and optimization. Also, the design of sensitivity studies for such an IVPT study design may require a more sophisticated understanding of IVPT studies. While reasonable efforts should be made to develop an IVPT method that produces a well-defined maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points, this may not be feasible for certain topical products even with study durations of 96 hours, or, at least, may not be feasible to produce reliably in all donors. In such circumstances, the IVPT method development report should detail the systematic efforts made to optimize the IVPT method.在IVPT方法开发期间可以对不同的研究设计和方法参数进行评估。例如,如果最初选择的研究参数是剂量维持时间和研究总体时长均为48小时,但测定结果发现,在该条件下不能识别出最大(峰值)通量和之后多个取样时间的通量下降情况。此时,采用较短的剂量持续时间(如,4~6小时)可能有助于改善IVPT通量分布曲线,但将外用制剂从皮肤表面移除是具有挑战性的,通常需要单独进行方法开发和优化。此外,对于IVPT研究中灵敏度的研究设计,可能需要对IVPT研究有更全面清晰的理解。虽然应尽力进行IVPT方法开发,以便可以识别出最大(峰值)通量和此后多个时间点的通量下降情况,但某些外用制剂即使研究持续时长达到96小时,也不能产生理想的通量分布曲线,或者,不能在所有的供体中产生可重复的结果。在这种情况下,应在IVPT方法开发报告中详细说明为IVPT方法优化所做出的努力。2. Use of a Validated Sample Analytical Method for IVPT Method Validation 采用已验证的分析方法进行IVPT方法验证The IVPT method development studies, being exploratory in nature, are often performed using a sample analytical method that is not validated (e.g., an HPLC or ultrahigh performance liquid chromatography (UPLC) method, often involving mass spectrometry (MS)); also, IVPT method development studies are often conducted in a manner that is not compatible with a quality management system which would otherwise make the evidence generated suitable to support valid conclusions. Such method development studies would not be suitable to demonstrate the validity of an IVPT method, or associated results. Therefore, although it may appear to be redundant, certain experiments performed during IVRT method development may need to be repeated during IVPT method validation, using appropriate controls, like a validated analytical method and procedures that are compatible with a suitable quality management system.IVPT方法开发研究,本质上具有探索性,通常是采用未经验证的样品分析方法(如,HPLC或UPLC,通常需要进行质谱分析(MS))进行的;此外,IVPT方法开发研究通常与质量管理体系不兼容,产生的数据不足以支撑可靠的结论。因此,方法开发研究不足以支撑IVPT方法验证或相关的结果。尽管看起来似乎是多余的,但在IVPT方法开发期间进行某些试验可能需要在IVPT方法验证期间重复进行考察,进行适当的控制,如与适当的质量管理体系相兼容的已验证的分析方法和程序。It is important to clearly segregate and consistently identify those experiments and results that were part of IVPT method development separately from those that were part of IVPT method validation. It is also important to consistently identify all relevant method parameters and experimental conditions/controls for each set of IVPT results. Information in the method development report should clearly identify/distinguish when the results for apparently similar sets of experiments may have been obtained using different method parameters. Method development reports should clarify which sets of diffusion cells were run in parallel or separately (e.g., on separate days). In addition, the sample analytical method parameters used to analyze the samples from each set of IVPT experiments should be specified, and the report should indicate whether or not the sample analytical method was validated (either at the time of sample analysis or subsequently).需要注意的是,应将IVPT方法开发和IVPT方法验证的试验结果分开来看,并采用一致的评价标准;对于每组IVPT结果,所有相关的方法参数和试验条件/对照都应保持一致,这点非常重要。当采用不同的方法参数可以获得明显相似的试验结果时,应在方法开发报告中清楚的说明。在方法开发报告中,也应阐明扩散池是平行还是单独运行(如,在不同的日期)的。此外,应说明在IVPT试验中,用于样品分析的具体方法参数,并说明分析方法是否经过了验证(在样品分析时或样品分析后)。IV. IVPT method validation IVPT方法验证When all the relevant parameters of the IVPT method have been established, a pilot study should be performed using the final IVPT method and using a validated sample analytical method. The purpose of the pilot study is to validate the suitability of the selected IVPT method parameters by demonstrating that the performance characteristics of the IVPT method are appropriate to compare the cutaneous pharmacokinetics of a drug delivered topically from a test product and RS. The results from the pilot study, thereby, support the systematic validation of the IVPT method, which proceeds as a distinct study phase following IVPT method development.在所有相关的IVPT方法参数确定之后,应采用最终确定的IVPT方法和已验证的样品分析方法进行初始研究。初步研究的目的是,通过论证IVPT方法的性能特性适用于自研制剂和对照制剂(RS)中药物的皮肤药代动力学比较,确认所选IVPT方法参数的适用性。因此,初步研究的结果支持IVPT方法的系统验证,这是IVPT方法开发之后的一个特有的研究阶段。The results from this IVPT pilot study can help to estimate the number of donors that may be needed to adequately power the IVPT pivotal study. In addition to the test topical product and RS evaluated in the pilot study, a parallel assessment should be performed with a third topical product or formulation that is known or designed to be different from the RS, to validate the selectivity of the IVPT method to discriminate differences in bioavailability. The IVPT pilot study results should be plotted with error bars, comparing the permeation profiles for the three treatment groups in the pilot study. Separate plots should be prepared for average flux results and average cumulative permeation results. These data can be used to support specific IVPT method validation parameters (e.g., permeation profile and range).IVPT的初步研究结果有助于评估在IVPT正式研究中所需的供体数量。在IVPT的初步研究中,除了对自研外用制剂和对照制剂(RS)评估外,还应对已知或设计与RS不同的另一种外用产品或配方进行平行评估,以确认IVPT方法在区分生物利用度差异方面的选择性。应用误差线绘制IVPT的初步研究结果,对三个试验组的渗透曲线进行比较。这些数据可用于支持特定的IVPT方法验证参数(如,渗透概况和范围)。A pilot IVPT study performed with multiple skin donors (e.g., 4–6 skin donors) and a minimum of four replicate skin sections per donor per treatment group is recommended. As skin from an increasing number of donors is evaluated in the pilot study, the accuracy of the estimated number of donors needed to adequately power the IVPT pivotal study may improve. While skin from the same donors evaluated in the pilot study may also be used in the IVPT pivotal study, the results from the pilot study should not be combined with the results from the IVPT pivotal study for the purpose of statistical analysis.在IVPT初步研究中,建议采用多个皮肤供体(如,4~6个皮肤供体),且每个试验组的每个供体至少4个重复皮肤切片。随着IVPT初步研究中评估的供体数量的增加,可逐步提高IVPT正式研究中所需的供体数量准确性。虽然在IVPT初步研究评估中用的相同供体皮肤也可用于IVPT正式研究,但初步研究的结果不能与IVPT正式研究中获得的结果合并进行统计分析。The equipment, methodologies, and study conditions used in the IVPT pilot study (and the eventual IVPT pivotal study) should be appropriately validated or qualified. If an applicant elects to use equipment, methodologies, or study conditions that are different from those recommended in this guidance, the applicant should demonstrate why it was necessary and scientifically justified to do so. Detailed protocols and well-controlled study procedures are recommended to ensure the precise control of dosing, sampling, and other IVPT study parameters, as well as potential sources of experimental bias.IVPT初步研究(和最终的IVPT正式研究)中所用的设备、方法和研究条件应进行合适的验证或确认。如果申请人使用的设备、方法或研究条件与本指南中推荐的不一致,申请人应说明这种选择的必要性,并证明其科学合理性。建议建立详细的方案和良好的研究控制程序,以精确控制上样、取样和其它IVPT研究参数,以及潜在的试验偏差来源。The validation of the IVPT method should incorporate specific qualifications and controls (described below), performed using a validated sample analytical method, as applicable. The qualification of an IVPT method parameter refers to the process of defining what attributes make it suitable to perform its function in the IVPT method. For example, when repeated measurements of the temperature at the surface of skin mounted in a diffusion cell demonstrate that an IVPT equipment can maintain the skin surface temperature in the range of 32°C ± 1°C, the results can support a demonstration that the equipment is qualified to perform its function in an IVPT method for which a method parameter is the control of skin surface temperature in the range of 32°C ± 1°C across the relevant study duration.IVPT方法验证应包括下述的确认和控制内容,如适用,使用已验证样品分析方法。IVPT方法参数确认是表征IVPT方法的属性是否适合于执行其功能的过程。例如,当重复测定安装在扩散池中的皮肤表面温度时,IVPT设备可以维持皮肤表面温度在32℃±1℃范围内,这个结果可以用于证明,在整个IVPT方法研究期间,该设备可以执行其功能,将皮肤表面温度控制在32℃±1℃范围内。A. Equipment Qualification 设备确认Suitable equipment for the IVPT method includes various models of VDCs and flow-through diffusion cells. The operating principles and specific test procedures differ among the various equipment; relevant procedures from the manufacturer may be used for installation, operational, and performance qualifications. The laboratory qualification of each diffusion cell should, at minimum, include 1) measurements of the diffusional area of the orifices of the donor and receptor compartments between which the skin is mounted, 2) the empirically measured volume of the receptor solution compartment in each VDC or the empirically measured outflow tube length for each flow-through diffusion cell, 3) the stability of the temperature measured at the skin surface (e.g., 32°C ± 1°C) across a relevant duration (e.g., 48 hours), and 4) the rate of stirring or agitation in VDCs, or the flow rate for flow-through diffusion cells, as applicable.可用于IVPT方法的设备有VDCs(立式扩散池)和流通池。每种类型设备的操作原理和相关测定方法不同;可根据生产商提供的相关规程进行安装、操作和性能确认。各种型号的扩散池确认至少包括:1)测定供给室和接收室之间皮肤安装位置的孔口扩散面积;2)测量VDC接收室的容积,或流通池输出管的长度;3)在整个研究期间(如,48小时),测定皮肤表面温度(如,32℃±1℃)的稳定性;和4)如适用,测定VDC的搅拌速率,或流通池的流速。If information related to the diffusional area of the orifices and the volume of the receptor solution compartment for each diffusion cell is available from the manufacturer, that information should be provided for each relevant diffusion cell, in addition to the empirical measurements made by the laboratory performing the IVPT studies. The equipment should control the diffusion cell temperature so that the skin surface temperature is verified to be stable (e.g., 32°C ± 1°C) for each diffusion cell before dosing (e.g., measured by a calibrated infrared thermometer), and monitored periodically throughout the duration of the experiment by repeatedly measuring the skin surface temperature of a non-dosed control diffusion cell that is run in parallel with the other replicate dosed diffusion cells and connected to the same water bath or thermoregulation system.如果可以从生产商处获取每个扩散池的孔口扩散面积和接收室的容积信息,除了提供实验室进行IVPT研究时测定的相关结果,还应提供生产商提供的信息。设备应可以控制扩散池的温度,确保在上样前,皮肤表面温度稳定(如,通过校准的红外温度计测定在32℃±1℃范围内);并在整个试验期间通过重复测定,连接到相同的水浴或温度调节系统、与上样剂量组平行运行的非剂量控制组的皮肤表面温度进行定期监测。B. Membrane (Skin) Qualification 膜 (皮肤) 确认Excised human skin is recommended as the membrane for the IVPT study. The validity of each skin section dosed in the study should be qualified using an appropriate test procedure to evaluate the stratum corneum barrier integrity. Acceptable barrier integrity tests may be based upon tritiated water permeation, TEWL, or electrical impedance/conductance measured across the skin. The test parameters and acceptance criteria used for the skin barrier integrity test should be justified for the specific method and instrumentation that is used during the study. The skin from all donors whose skin is included in the study should be prepared in a consistent manner and dermatomed to a relatively consistent thickness, within limits specified in the study protocol. The skin thickness should be measured and reported for each skin section included in the study. The assignment of replicate skin sections from a donor to each treatment group should be randomized, as feasible. It is acceptable to balance the distribution of skin thicknesses in each treatment group (test topical product or RS) by a procedure specified in the study protocol建议采用离体人类皮肤作为IVPT研究用膜。应采用合适的方法评估角质层的屏障完整性,以对上样用皮肤切片的有效性进行验证。可接受的屏障完整性测试方法包括氚化水渗透、TEWL或电阻/电导值法。研究期间所用皮肤屏障完整性测试的参数和可接受标准,应根据所用的方法和仪器,说明其合理性。研究中所用的供体皮肤应采用相同的方式进行处理,且皮肤厚度应相对一致,以满足研究方案中拟定的限度要求。如适用,应将供体皮肤的重复皮肤切片随机分配给每个试验组。可以根据研究方案中规定的程序来平衡每个试验组(自研外用制剂和RS)的皮肤厚度分布。C. Receptor Solution Qualification 接收介质确认The composition and pH of the receptor solution used for the IVPT study should be qualified in relation to its compatibility with the skin as well as the stability and solubility of the drug in that receptor solution. The stability of the drug in the receptor solution samples should be validated as part of the receptor sample analytical method validation. The solubility of the drug in the IVPT receptor solution should be empirically determined in triplicate, to illustrate that the solubility of the drug in the receptor solution exceeds the highest sample concentration in the IVPT pivotal study, ideally by an order of magnitude. The solubility of the drug in the IVPT receptor solution should be sufficient to characterize the higher amounts of drug permeating from the increased drug delivery condition evaluated in the IVPT sensitivity assessment during IVPT method validation.IVPT研究中所用接收介质的组成和pH应根据其与皮肤的兼容性以及药物在接收介质中的稳定性和溶解度进行确认。药物在接收介质中的稳定性应作为接收液中样品分析方法验证的一部分。药物在接收介质中的溶解度,应经过三次重复检测经验确定,确保其超过IVPT正式研究中最高样品浓度,理想情况下是一个数量级。在IVPT方法验证期间的灵敏度评估中,药物在接收介质中的溶解度应足以表征,增加药物递送条件时药物渗透的最高量。The inclusion of 0.1% polyoxyethylene[20]oleyl ether (also known as Oleth-20, Volpo-20, or Brij-20; CAS number 9004-98-2) is recommended to enhance the solubility of physiological buffer based (aqueous) receptor solutions for hydrophobic drugs. If additional solubility is needed, small increases in the concentration of polyoxyethylene[20]oleyl ether (e.g., from 0.1% or 0.2%, which is typically adequate for most hydrophobic drugs, to higher concentrations) are recommended, but should not exceed 6% polyoxyethylene[20]oleyl ether. Other strategies to improve the solubility of the drug in the receptor solution that may have the potential to alter the permeability of the skin (e.g., inclusion of organic solvents and alcohols in the receptor solution) are not recommended and may invalidate the IVPT method.对于疏水性药物,推荐在基于生理缓冲盐的接收介质中添加0.1%(w/v)聚氧乙烯20油醚(别名Oleth-20, Volpo-20, or Brij-20;CAS:9004-98-2)提高溶解度。如有必要,可在接收介质中略微增加聚氧乙烯20油醚的浓度[例如,从0.1%(w/v)到0.2%(w/v)],通常足以满足大多数疏水药物溶解度的要求,但不应超过6%。其他改善药物在接收介质中溶解度的策略可能会改变皮肤的渗透性(例如,在接收介质中加入有机溶剂和醇),使IVPT方法失效,不建议使用。The inclusion of an anti-microbial agent in the receptor solution (e.g., ~0.1% sodium azide or ~ 0.01% gentamicin sulfate) is recommended to mitigate potential bacterial decomposition of the dermis and/or epidermis in the diffusion cell, regardless of the study duration. Other antimicrobial agents may also be acceptable, and if used, information should be included in the ANDA to explain the reason for their selection (and for the concentration at which they were used).无论研究时间的长短,建议在接收介质中加入一种抗微生物剂(例如,~0.1%叠氮化钠或~0.01%硫酸庆大霉素),以减轻在整个研究期间扩散池中细菌对真皮和/或表皮的潜在分解。如果采用其它抗微生物剂,应在ANDA中阐述选择的理由(以及使用的浓度)。D. Receptor Solution Sampling Qualification 接收液取样确认The accuracy and precision of receptor solution sample collection at each time point should be appropriately qualified. Evidence to qualify a sampling procedure should illustrate that the sampling technique can reliably collect a consistent volume of the sample from the well-mixed volume of the receptor compartment at each sampling event, and that no artifacts are likely to be created by the sampling technique. Information should be included describing the equipment manufacturer’s specification for the accuracy and precision of receptor solution sampling, when available.应对接收液中每个时间点取样的准确性和精密度进行适当的确认。在取样程序的确认过程中应证明,所用取样技术可以从混合良好的接收室中始终一致的收集到相同体积的接收液,且不会因取样技术的原因引起误差。如适用,应描述设备生产商关于接收液取样准确度和精密度的规范信息。For IVPT studies using a flow-through diffusion cell, it may be appropriate to qualify the lengths of tubing, and their associated dead volumes, to accurately calculate the lag time before a sample elutes through the tubing and is collected. For IVPT studies using a VDC, removal of the entire receptor solution volume and full volume replacement of the receptor solution at each time point may provide optimal solubility sink conditions. The sampling of small aliquots of the receptor solution for an IVPT study may introduce anomalous measurements of apparently negative flux in certain regions of the IVPT study and produce flux profiles that are difficult to interpret.对于采用流通扩散池进行IVPT研究的情况,应对流通扩散池每个管路的长度,以及相关的死体积进行确认,以准确计算样品通过管路洗脱和收集前的延迟时间。对于采用VDC进行IVPT研究的情况,在每个时间点移出所有接收液,并补充相应体积的新鲜接收介质,这种方法可以提供最佳的溶解度漏槽条件。在IVPT研究中,移取较少量的接收液体积,可能会导致某些时间点出现明显异常的负通量结果,产生难以解释通量曲线。E. Environmental Control 环境控制Ambient laboratory temperature and humidity during the study should be monitored and reported. An environmentally controlled temperature range of 21°C ± 2°C is recommended, and a humidity range of 50% ± 20% relative humidity is recommended, if feasible.在研究期间,应监控和报告实验室环境的温度和湿度。如适用,建议将温度控制在21℃±2℃、湿度控制在50%RH±20%RH之间。F. Permeation Profile and Range 渗透曲线和范围The flux profile and cumulative permeation profile for the IVPT pilot study should be plotted across a range of sampling times, which corresponds to the IVPT pivotal study duration. The calculation of flux and cumulative total permeation is discussed in more detail below. The results of the IVPT pilot study should validate that the selected study parameters are suitable to adequately characterize the permeation profile (the cutaneous pharmacokinetics) of the drug within the selected study duration (the range of sampling time points).在IVPT初步研究和正式研究中,应在整个取样时间范围内分别绘制通量分布曲线和累积渗透分布曲线。下文有关于通量和累积渗透量计算方式的详细讨论。应根据IVPT初步研究的结果确认,在所选择的研究期间(取样时间点范围内),所选择的研究参数是否足以表征渗透概况(皮肤药代动力学)。A sufficiently complete flux profile should be adequate to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points in the IVPT pilot study. The results of the IVPT pilot study should also validate that the sampling frequency provides suitable resolution to adequately characterize the permeation profile (particularly the flux profile).在IVPT初步研究中,一个充分完整的通量分布应足以识别出最大(峰值)通量和此后多个时间点的通量下降情况。应根据IVPT初步研究结果,确定取样频率,确保为表征渗透分布(特别是通量分布)提供合适的分辨率。G. Precision and Reproducibility 精密度和重现性The flux and cumulative permeation results from the IVPT pilot study (and the eventual IVPT pivotal study) should be calculated, tabulated, and reported for each diffusion cell at each time point, with summary statistics to describe the intra-donor average, standard deviation, and percent coefficient of variation (%CV) among replicates, as well as the inter-donor average, standard error, and %CV. Complete results for all data values used in the calculations should be reported in a clear and organized manner, to facilitate the reconstruction of the flux and cumulative permeation results. The design of the study should be detailed and clear, and data values should be clearly associated with specific donors, replicates, treatment groups, time points, etc.在IVPT初步研究(和最终的IVPT正式研究)中,应计算和报告各扩散池在每个时间点的通量和累计渗透结果,并以表格的形式汇总统计以下信息:重复测定的批内均值、标准偏差和变异系数(%CV);以及批间均值、标准偏差和变异系数。计算中所用数据的完整结果应以清晰和有组织的方式进行报告,以便通量和累计渗透结果的重现。研究设计应是详细和清晰,将数据结果与相关供体、重复数、试验组和时间点等相关联。H. Dose Depletion 剂量消耗The recovery of permeated drug in the receptor solution should be characterized in each diffusion cell as the cumulative total permeation of the drug in the receptor solution over the IVPT duration. This may be expressed as a percentage of the nominal amount of drug in the applied dose (which may be estimated based upon the nominal strength of the drug in the topical product and the approximate mass of topical product dosed on the skin).在IVPT研究期间,每个扩散池单元中药物渗透进入接收液中的回收率可以表征为“接收液中药物的总累积渗透量”,以上样量中药物标示量的百分比表达(根据外用制剂中药物的标识浓度和外用制剂在皮肤上的大概质量进行估计)。For example, if 10 mg of a topical product containing 5% drug was dosed on the membrane, the amount of drug in the applied dose may be estimated to be 0.5 mg (or 500 μg). If a cumulative total of 10 μg of drug diffused into the receptor solution across a 48-hour duration of the IVPT, it would be possible to estimate that the 500 μg dose would have been depleted by approximately 10 μg, amounting to an approximately 2% dose depletion. The average percentage dose depletion may thereby be estimated (not accounting for skin content) and should be reported.例如,如果含有5% 药物的外用制剂上样为10mg,则在上样中药物的量大约为0.5mg(或500μg)。如果在IVPT研究的整个48小时内,药物渗透进入接收液的总量为10μg,则估计500μg的剂量消耗为10μg,相当于约2%的剂量消耗。应估算和报告平均剂量消耗百分比(不考虑皮肤含量)。I. Discrimination Sensitivity and Selectivity 区分力—灵敏度和选择性The discrimination ability of the IVPT method may be described using two concepts: sensitivity and selectivity. The IVPT sensitivity studies are necessarily performed during IVPT method development to establish IVPT method parameters like the dose amount, dose duration, study duration, etc. However, the analysis of the results from these studies is qualitative in nature, and they need not be repeated during the IVPT method validation phase.可以用以下两个概念描述IVPT方法的区分力:灵敏度和选择性。在IVPT方法开发期间,必须进行IVPT灵敏度研究,以确定IVPT方法参数,如:上样量、剂量维持时间等。但这些研究结果本质上为定性研究,在IVPT方法验证阶段无需重复进行。The IVPT sensitivity studies are typically performed toward the end of the IVPT method development phase, and a key purpose of these studies is to incorporate the final IVPT method parameters for the target dose and dose duration to be used in the pivotal study so that the IVPT sensitivity studies can support a demonstration of the validity of the final IVPT method. Therefore, IVPT sensitivity studies are described within this section of the guidance in the context of IVPT validation (rather than method development) to avoid dissociating the discussions of IVPT sensitivity (which is performed to establish the suitability of the final IVPT method parameters) and IVPT selectivity (which is performed once the final IVPT method parameters are established, and which is based upon the IVPT pilot study that is performed as part of the IVPT method validation). With the exception of the alternative dose amounts or dose durations used in the IVPT sensitivity study, it is important that the IVPT method parameters are consistent across the IVPT sensitivity, pilot, and pivotal studies (including the anatomical region specified in the study protocol (e.g., posterior torso), the skin source, and skin preparation).IVPT灵敏度研究通常在IVPT方法开发阶段末进行,这些研究的一个关键目的是为了确定“在正式研究中所用的最终IVPT方法参数”,包括目标上样量和剂量维持时间,因此,IVPT灵敏度研究可以为最终IVPT方法的有效性提供合理性证明。因此,本文在IVPT方法验证(而非“方法开发”)模块描述IVPT灵敏度研究,是为了避免将IVPT灵敏度(以确定最终IVPT方法参数的适用性)和IVPT选择性(基于IVPT初步研究,在最终IVPT方法参数确定之后进行,是IVPT方法验证的一部分)分开讨论。除了在IVPT灵敏度研究中的上样量或剂量维持时间不同外,在IVPT灵敏度、初步研究和正式研究的所有阶段,IVPT方法参数均应保持一致(包括:研究方案中指定的解剖区域(如,背部躯干)、皮肤来源和处理方法)。1. IVPT Sensitivit 灵敏度IVPT sensitivity is the ability of the IVPT method to detect changes in the cutaneous pharmacokinetics of the drug as a function of differences in drug delivery. If the IVPT method consistently demonstrates higher and lower flux profiles (i.e., higher and lower values for IVPT endpoints) in response to increased and decreased drug delivery, respectively (or in response to other conditions expected to increase and decrease drug delivery, respectively), the IVPT method may be considered sensitive.IVPT灵敏度是IVPT方法检测药物皮肤药代动力学变化的能力,以反映药物递送的差异。如果随着药物递送的增加和降低(或预期会增加和降低药物递送的其他条件),IVPT方法能分别给出较高或较低通量分布曲线(即,较高和较低的IVPT终点),则认为IVPT方法是灵敏度。There are a few potential approaches by which to produce the differences in drug delivery that can be differentiated by a suitably discriminating IVPT method. Regardless of the approach used, the differences in the IVPT permeation profiles are not necessarily expected to be specifically proportional to differences in the dose amount, dose duration, or product strength. For example, three-fold differences in the dose amount (even if outside the recommended target dose range) may provide distinct flux curves but may not result in three-fold differences in the IVPT endpoints because the skin barrier may be rate-limiting both in vitro and in vivo.有几种潜在具有合适区分力的IVPT方法设计,去表征药物的不同递送行为。但无论采用何种方式,均不能期望IVPT渗透曲线的差异与药物上样量、剂量持续时间或产品规格的差异成比例关系。例如,上样量的三倍差异(即使超出了建议的目标剂量范围)可产生不同的通量曲线,但不会出现IVPT终点的三倍差异,因为皮肤屏障限制体外和体内的(渗透)速率。In other words, if the target dose for the pivotal IVPT study was 10 mg/cm2, a 3-fold lower dose would be ~3 mg/cm2and a 3-fold higher dose would be 30 mg/cm2; thus, an IVPT sensitivity study might compare the flux profiles from 3, 10, and 30 mg/cm2doses of the topical product. Similarly, if the target dose for the pivotal IVPT study was 15 mg/cm2, a 3-fold lower dose would be 5 mg/cm2and a 3-fold higher dose would be 45 mg/cm2; thus, an IVPT sensitivity study might compare the flux profiles from 5, 15, and 45 mg/cm2doses of the topical product. An IVPT sensitivity study performed with multiple skin donors (e.g., 4–6 skin donors) and a minimum of four replicate skin sections per donor per treatment group is recommended.换言之,如果正式IVPT研究的目标剂量是10 mg/cm2,则三倍的较低剂量为3 mg/cm2,三倍的较高剂量为30 mg/cm2;因此,可通过比较3、10和30 mg/cm2外用制剂上样量的通量曲线,确认IVPT方法的灵敏度。同理,如果正式IVPT研究的目标剂量是15 mg/cm2,则三倍的较低剂量为5 mg/cm2,三倍的较高剂量为45 mg/cm2;因此,可通过比较5、15和45 mg/cm2外用制剂上样量的通量曲线,确认IVPT方法的灵敏度。在IVPT灵敏度研究中,建议采用多个皮肤供体(如,4~6个皮肤供体),且每个试验组的每个供体至少4个重复皮肤切片。Modulation of Dose Amount: An IVPT method development study with different dose amounts may provide supportive evidence that the IVPT methodology is sensitive to differences in drug delivery.This approach is well suited to topical products that contain volatile components that evaporate from the formulation following dose application to the skin. Modulating the dose amount for such topical products effectively alters the thickness of the applied dose. The majority of volatile components from a thinner dose will tend to evaporate more rapidly (compared to a thicker dose), and a thinner dose will tend to deliver less drug into the skin (and/or for a shorter duration) compared to a thicker dose.调整上样量:可以采用不同的上样量进行IVPT方法开发研究,为IVPT方法对药物递送的差异具有灵敏度提供支持性证据。该方法适用于含有挥发性成分的外用制剂,这些挥发性成分在药物制剂上样到皮肤后会挥发。调整这类制剂的上样量可以有效改变上样的厚度。采用较薄的上样量,与较厚的上样量相比,大部分挥发性成分会挥发,因此,较薄的上样量(和/或较短的维持时间)递送进入皮肤的药物量较少。Modulating the dose amount can be an effective technique to modulate differences in drug delivery for formulations with volatile components, like gels, lotions, and many creams. However, modulating the dose amount may not necessarily produce perceptible differences in drug delivery for topical products like petrolatum-based ointments, or other types of topical products that do not evaporate on the skin, or that may not experience dose-dependent differences in metamorphosis that can alter the rate and extent of drug delivery.对于含有挥发性组分的配方,如凝胶、洗剂和很多乳膏剂,调整上样量是改变药物递送行为的有效技术手段。然而,对于基于凡士林的软膏剂、或不会在皮肤上挥发的外用制剂、或可改变药物递送速率和程度的形变过程不具有剂量依赖性,通过改变这些制剂的上样量不一定会产生药物递送行为的明显差异。Modulation of Dose Duration: For many topical products, it may be more effective to modulate the dose duration, instead of the dose amount, to produce differences in drug delivery and associated changes in the cutaneous pharmacokinetics of the drug.调整剂量维持时间:对于很多外用制剂,相比于调整上样量,通过调整剂量维持时间可更有效地评估药物递送的差异和与药物皮肤药代动力学相关的变化。An IVPT method development study with a controlled dose amount (e.g., 15 mg/cm2) dosed for different durations (e.g., 2 hours, 6 hours, and 12 hours) may be well suited to provide supportive evidence that the IVPT methodology is sensitive to differences in drug delivery from many topical products. The scenario described in this example would support an IVPT study design where a topical product dose of 15 mg/cm2 is dosed for 6 hours (the target duration for the IVPT study) and then wiped off. The applied dose may be removed with a series of cotton-tipped swabs, one or more of which may be dry and one or more of which may be moistened (e.g., with a soap solution or water). The initial (dry) swab typically removes the bulk of the dose and subsequent swabs are used to remove the residual dose (i.e., the residue of the topical product which may otherwise continue to deliver drug into the skin) and/or to rinse the skin.在IVPT方法开发研究中,通过将一定量的外用制剂(如,15 mg/cm2)在皮肤上维持不同的时间(如,2h、6h和12h)可能是提供支持性证据的比较合适方式,以证明IVPT方法对很多外用制剂中药物递送的差异具有敏感性。本例描述了用于支持IVPT研究设计的场景,将上样量为15 mg/cm2的外用制剂在皮肤上维持6h(研究的目标维持时间),然后将其擦除。可以采用一些棉签移除样品,包括一个或多个干燥的棉签,和一个或多个浸湿的棉签。首先用干燥的棉签移除大部分样品,然后用浸湿的棉签移除残留的样品(即,残留的样品可能会继续递送药品进入皮肤)和/或清洗皮肤。To support a demonstration of the sensitivity of the IVPT study, the permeation profile produced by the target dose duration for the IVPT study (e.g., 6 hours) should be compared with a shorter dose duration (e.g., 2 hours) that is expected to perceptibly decrease the drug delivery, and also be compared with a longer dose duration (e.g., 12 hours) that is expected to perceptibly increase the drug delivery. Thereby, the three dose durations compared in the IVPT sensitivity study are designed to produce perceptible changes in the cutaneous pharmacokinetics of the drug as a function of differences in drug delivery, and thereby support a demonstration of the sensitivity of the IVPT method.为了证明IVPT研究的灵敏度,将IVPT研究中目标剂量持续时间(如,6小时)的渗透曲线分别与较短剂量维持时间(如,2小时)和较长剂量维持时间(如,12小时)的渗透曲线进行比较,较短和较长剂量维持时间应分别有明显较低和较高的剂量递送。因此,在IVPT灵敏度研究中,通过设计比较三种不同剂量维持时间,可产生明显不同的“能反映药物递送差异的”药物皮肤药代动力学,支持IVPT方法的灵敏度论证。The specific dose durations may be selected based upon an initial exploratory IVPT study performed during IVPT method development that characterizes the permeation profile when the dose is left on the skin for a longer duration (e.g., 24 or 48 hours). An important feature of the results from such an IVPT study is the duration of the initial phase of the permeation profile, when the flux is increasing at a relatively rapid rate.在IVPT方法开发期间,可以根据初始的探索性IVPT研究中,剂量在皮肤上保留较长时间(如,24或48小时)的渗透曲线,选择特定的剂量维持时间。这种研究结果的一个重要特征是,在渗透曲线的初始阶段,通量以一个相对较快的速率增加。For example, if such an exploratory study indicates that the flux increases on a steep slope until approximately 12 hours, and then continues to deliver the drug at a gradually increasing rate thereafter, it may suggest that the permeation profile for a dose duration of longer than 12 hours (e.g., 24 hours) may not be perceptibly different from that of the 12 hour dose duration, especially when compared in a relatively small number of donors and replicates (e.g., four donors with four replicates each per dose duration). It may also suggest that a 12-hour dose duration may be a good choice for the longest of the three dose durations in the IVPT sensitivity study.例如,如果探索性研究结果表明,在12小时前通量快速增加,之后随着剂量维持时间的增加,通量与12小时无明显的差异;这种结果表明,超过12小时(如24小时)的剂量维持时间,获得的通量分布曲线可能与12小时无明显差异,尤其是在供体和重复数量较少的情况下(如,每个剂量维持时间4组供体皮肤,每组供体皮肤4个重复皮肤切片)。这也可能表明,在IVPT灵敏度研究中,12小时的剂量持续时间可能是一个较好的选择,也是三个剂量维持时间中最长时间。The target dose duration should be selected based upon considerations like the sensitivity of the sample analytical method, the ability to produce a permeation profile that can be perceptibly discriminated from that produced by the longer (12 hour) dose duration, and/or the labeled use of the topical product (which may indicate that the topical product should be reapplied every 4–6 hours).目标剂量维持时间的选择应基于以下考虑因素,如:样品分析方法的灵敏度、产生能明显区别于最长(12小时)剂量维持时间所产生的渗透曲线的能力、和/或外用制剂的说明书(其中可能说明,每4~6小时需要重复进行给药)。The shortest of the three dose durations in the IVPT sensitivity study should be selected based upon the sensitivity of the sample analytical method and its ability to produce a permeation profile that can be perceptibly discriminated from that produced by the target (6 hour) dose duration.在IVPT灵敏度研究中,三个剂量维持时间中最短时间的选择应基于样品分析方法的灵敏度,以及其产生能明显区别于目标剂量维持时间(6小时)所产生的渗透曲线的能力。Modulation of Product Strength: To validate the sensitivity, specificity, and selectivity of an in vitro release test (IVRT) method, altered strength formulations are routinely prepared. While it may seem convenient to use these altered strength formulations in an attempt to demonstrate the sensitivity and selectivity of an IVPT method, doing so may not produce the desired outcomes. There may be circumstances when this strategy may produce perceptibly different permeation profiles, however, in many instances, the resulting permeation profiles may not be perceptibly different when compared in a relatively small number of donors and replicates (e.g., four donors with four replicates each per topical product strength). In general, the modulation of topical product strength to support a demonstration of IVPT sensitivity is not recommended because it may not consistently produce the expected increase or decrease in drug delivery; however, in certain situations, higher and lower strength formulations (relative to the nominal strength of the RS) may suitably increase and decrease the drug delivery and cutaneous pharmacokinetics relative to that from the nominal strength topical product.调整产品规格:对于IVRT方法的灵敏度、专属性和选择性验证而言,常见的方式是改变配方制剂的规格。虽然在IVPT方法的灵敏度和选择性验证中,采用改变配方制剂规格的方式似乎也比较方便,但这种方式可能不会产生预期的结果。在某些情况下,这种策略可能会产生明显不同的渗透曲线;然而,在很多情况下,由于在对比研究中采用的供体组和重复数较少(如,每种外用制剂规格采用4个供体组,每组供体4个重复),并不能产生具有明显不同的渗透曲线。通常,不建议通过调整外用制剂规格的方式去论证IVPT的灵敏度,因为它可能不会持续产生预期的药物递送增加或降低;然而,在某些情况下,相对于外用制剂的目标规格,较高和较低规格的配方制剂(相对于对照制剂的规格)可以适当的增加和降低药物递送和皮肤药代动力学。2. IVPT Selectivity 选择性IVPT selectivity is the ability of the IVPT method to discriminate the cutaneous pharmacokinetics of the drug between the RS and a topical product or formulation that exhibits differences in drug delivery relative to the RS. The IVPT pilot study with the parallel assessment of the RS, the test topical product, and a third topical product or formulation that is known or designed to be different from the RS may provide supportive evidence that the IVPT methodology is selective for differences in drug delivery. Topical product batch information for all topical product lots used in IVPT method development, validation and pilot studies, as applicable, should be submitted in the study reports. The topical product information should include, but not be limited to, information about the batch formula, manufacturing date, batch size, altered manufacturing processes (if applicable) and, if available, potency and content uniformity. The evaluation of inequivalence may be based upon a qualitative or quantitative comparison of the permeation profiles and/or the IVPT endpoints.IVPT选择性是IVPT方法区分“对照制剂(RS)”和“与RS在药物递送方面存在差异的外用制剂或配方”在药物皮肤药代动力学差异的能力。在IVPT初步研究中,通过将RS、自研外用制剂和“已知或设计不同于RS”的第三种外用制剂或配方进行平行评估,提供支持性证据,说明IVPT方法对药物递送的差异具有选择性。如适用,应在研究报告中递交,IVPT方法开发、验证和初步研究中用到的所有外用制剂的相关批信息,包括但不限于:批配方、生产日期、批量、变更的生产工艺(如适用),以及效价和含量均匀度(如有)。可基于渗透曲线和/或IVPT终点的定性或定量比较进行不等效性评估。J. Robustness 耐用性A primary assumption related to robustness testing is that the test system performs consistently when all system variables (e.g., temperature, stirring rate) are at nominal settings. A value of robustness testing is that it can verify whether the system continues to provide a consistent output when specific variables are slightly altered, thereby qualifying operational ranges for those variables. However, the variability inherent in the permeability of human skin, whether in vitro or in vivo, may not be compatible with the primary assumption related to the consistency of the test system.与耐用性测试相关的一个主要假设是,当所有系统变量(如,温度、搅拌速率)为标准设置时,测试系统的性能一致。耐用性测试的意义在于,它可以确认当特定变量稍微改变时,系统是否可以提供一致的输出结果,从而确定这些变量的操作范围。然而,无论是在体外还是在体内,人体皮肤渗透性的固有可变性可能与测试系统一致性的初始假设不相容。Nonetheless, results from studies during IVPT method development that appear to support the robustness of the IVPT method or system should be reported, if relevant. For example, an IVPT method may be robust to substantial variations in the stirring rate of the receptor compartment. Similarly, the permeation profile of a drug into and through human skin may appear to be robust to certain differences in the topical product strength. Ultimately, because it may not always be feasible to validate the robustness of IVPT method parameters, IVPT study procedures should be controlled as precisely as possible.尽管如此,如果相关,应报告在IVPT方法开发期间获得的可以支持IVPT方法或系统耐用性的研究结果。例如,IVPT方法对接收室中搅拌速率的显著变化具有耐用性。类似地,药物进入并通过人体皮肤的渗透曲线可能对外用制剂规格的改变具有耐用性。因此,对IVPT方法参数的耐用性验证并不总是可行的,在IVPT研究中应尽可能精准的控制IVPT研究程序。V. Sample analytical method validation 样品分析方法验证While exploratory studies performed during IVPT method development may use an unvalidated sample analytical method, it is essential that all studies conducted as part of the IVPT method validation use a validated sample analytical method. A validated IVPT method should use a validated sample analytical method (e.g., HPLC/MS or UPLC/MS). Therefore, a discussion of the sample analytical method for the IVPT method is included in this guidance under this section on IVPT method validation.虽然在IVPT方法开发的探索性研究中,可以采用未验证的样品分析方法,但作为IVPT方法验证的一部分,在IVPT方法验证时,必须采用已验证的样品分析方法。已验证的IVPT方法应当使用已验证的样品分析方法(如,HPLC/MS或UPLC/MS)。因此,该部分讨论用于IVPT方法的样品分析方法。However, the study protocols and reports related to the IVPT method are distinct from those for the sample analytical method that is used to quantify drug concentrations in IVPT receptor solution samples. The validation of a sample analytical method, in and of itself, does not demonstrate the validity of an IVPT method. Separate and specific reports should be submitted for the validation of the sample analytical method (e.g., HPLC/MS or UPLC/MS) and for the validation of the IVPT method.然而,与IVPT方法相关的研究方案和报告与用于IVPT接收液中样品定量的分析方法不同。样品分析方法验证,就其本身而言,并不能证明IVPT方法的有效性。因此,应分别递交样品分析方法(如,HPLC/MS或UPLC/MS)和IVPT方法的验证报告。Any results from studies of the IVPT method that are performed during method development using a different sample analytical method than that which is ultimately validated, cannot support a demonstration of the validity of the IVPT method. Information should be provided in the IVPT method validation report referencing the (separate) sample analytical method validation, and clearly indicate that all relevant results in the IVPT method validation report were obtained using a validated sample analytical method (as opposed to a sample analytical method with different parameters than those which were validated).如果,在方法开发期间使用的样品分析方法与最终验证的样品分析方法不同,与其相关的IVPT方法研究结果不能用于支持论证IVPT方法的有效性。在IVPT方法验证报告中,应提供参考的样品分析方法验证信息,并明确表明IVPT方法验证报告中的所有相关结果均是采用经验证的样品分析方法(而不是与经验证的分析方法具有不同参数的样品分析方法)获得的。The receptor sample analysis procedures (e.g., typically involving an HPLC/MS or UPLC/MS system) should be performed using chromatography software (e.g., a chromatography data system) with audit trails, and should include a multi-point (6–8 concentration) calibration curve with suitable quality control samples, and should be validated in a manner compatible with the FDA guidance for industry Bioanalytical Method Validation (May 2018).接收液中样品的分析方法(如,通常为HPLC/MS或UPLC/MS系统)应使用具有审计追踪的色谱软件(如,色谱数据系统),并应包括具有适当质量控制样品的多点(6~8个)校准曲线,同时,应符合FDA生物分析方法验证行业指南要求。The validation of the receptor sample analytical method should include relevant qualifications of dilution integrity, if applicable, as well as stability assessments with the highest relevant temperature in the receptor solution for the longest relevant duration; the highest relevant temperature may be warmer than 32°C because the temperature of the receptor solution is often higher than the temperature at the surface of the skin, and the longest relevant duration may be the longest interval between sampling time points for methods in which the entire receptor solution is replaced at each sampling time point, or it could be longer in scenarios with only partial sampling of the receptor solution (e.g., 34°C for 48 hours).如适用,接收液中样品分析方法的验证应包括相关的稀释完整性确认,以及样品在最高相关温度的接收液中放置最长相关时间的稳定性评估;最高相关温度可以略高于32℃,因为接收液的温度通常高于皮肤表面温度。对于在每个取样时间点替换整个接收液的情况,最长相关时间为各相邻取样时间点中,间隔最长时间;但对于接收液部分取样的情况,时间可能更长(如,34℃放置48h)。If the samples are processed in specific ways for analysis (e.g., by drying and reconstituting the receptor samples in a smaller volume to concentrate the sample and increase the effective analytical sensitivity, or by dilution of receptor solution samples into the validated curve range of the sample analytical method) those procedures should be validated (e.g., by qualifying the dilution integrity during the sample analytical method validation). The stability of the drug in the receptor solution sample should be validated in a receptor solution matrix that has been exposed to the underside of the skin in a diffusion cell under conditions relevant to the IVPT pivotal study.如果在测定前,需要用特定的方式对样品进行处理(如:将样品干燥后重新用小体积溶剂配制以提高分析灵敏度;或通过稀释接收液样品,使其浓度到已验证的样品分析方法曲线范围内),应对该处理方法进行确认(如,在样品分析方法验证期间确认稀释完整性)。药物在接收液中的稳定性,应在IVPT正式研究相关的条件下,与扩散池中皮肤下侧接触的接受液基质中进行。VI. IVPT Pivotal study IVPT正式研究The IVPT pivotal study protocol should incorporate considerations relevant to BE studies, in general.IVPT正式研究方案通常应包含与BE研究相关的考虑因素。A. Handling and Retention of Samples 样品的处理和保存Refer to 21 CFR 320.38, 320.63 and the FDA guidances for industry Handling and Retention of BA and BE Testing Samples (May 2004) and Compliance Policy for the Quantity of Bioavailability and Bioequivalence Samples Retained Under 21 CFR 320.38(c) (August 2020), as applicable, regarding considerations for retention of study drug samples and to 21 CFR 320.36 for requirements for maintenance of records of BE testing. Retention samples should be randomly selected from the drug supplies received before allocating topical product units for use in an IVPT study in which the test topical product and RS are compared.参考21 CFR 320.38,320.63和FDA行业指南《生物利用度(BA)和生物等效性(BE)研究中试验样品的处理和保存》(2004年5月)和《21CFR 320.38(c) BE样品留存的数量及生物利用度》(2020年8月),如适用,关于保留研究药物样品的考虑以及21 CFR 320.36关于保存BE检测记录的要求。在采用自研外用制剂和RS进行IVPT对比研究前,应从收到的药品中随机选择样品进行留样。B. Control of Study Procedures 研究程序的控制Study procedures that have the potential to influence the results of the study should be appropriately controlled. Also, experimental observations that may have the potential to influence the interpretation of the study results, as well as any protocol or standard operating procedure (SOP) deviations, should be reported.应对可能影响研究结果的研究程序进行适当控制。另外,应报告可能影响研究结果解释的试验观察,以及任何与方案或标准操作规程(SOP)发生的偏离。Control of procedures related to the skin include the consistent control across the study of the skin preparation (e.g., dermatoming of skin sections) and the thickness of skin sections mounted on diffusion cells, as well as the skin storage conditions, including the duration for which the skin was frozen and the number of freeze-thaw cycles to which the skin was exposed. Skin from the same anatomical location should be used from all donors, and the demographics (age, race, sex) should be reported for all donors. Also, the IVPT sensitivity, pilot, and pivotal studies should use skin from the same anatomical site; otherwise, if skin from different anatomical sites is used across the different study phases, it may not be possible for the results of the IVPT sensitivity and pilot studies to support a demonstration of the discrimination ability of the IVPT method used for the pivotal study because the method parameters would not be aligned across the respective studies. Similarly, if a non-rate-limiting support membrane is used beneath the skin section (e.g., a filter membrane used in a validated IVRT method for the same topical product) then it should be used in a consistent manner for the IVPT sensitivity, pilot, and pivotal studies.与皮肤相关的控制程序,包括:采用相同的皮肤处理方法(如,皮肤的剥离)和安装在扩散池上皮肤切片的厚度的一致性,以及皮肤存储条件,如皮肤冷冻存储时间和取出后的冻融循环次数。所有供体皮肤应采用相同的解剖区域,并报告所有供体的人口统计信息(年龄、种族、性别)。另外,在IVPT灵敏度、初步试验和正式研究中,应采用相同的解剖区域;否则,如果在不同的研究阶段,采用的皮肤解剖区域不同,在IVPT灵敏度和初步研究中获得结果可能不足以支持IVPT正式研究中区分力的论证,因为各研究阶段采用的方法参数不一致。同样,如果在皮肤下面使用非速率限制支撑膜(如,同品种IVRT验证研究中所用的滤膜),则IVPT灵敏度、初步和正式研究中应保持一致。Control of procedures related to the dose include the control of the area of dose application, the dose amount, the dosing technique, the dose duration, and the blinding and randomization consistent manner for all diffusion cells in the study. Differences in dosing technique may alter the metamorphosis of the dosage form on the skin, and inconsistencies in the diameter of the area dosed on each diffusion cell may significantly influence the dosed area and contribute to errors in the calculation of flux.与剂量或上样相关的控制程序,包括:对上样面积、上样量、上样技术、剂量维持时间的控制,以及在研究中每个扩散池采用的盲法和随机化方法的一致性。不同的上样技术可能改变上样到皮肤上剂型的形态,扩散池孔口扩散面积的不一致可能会显著影响上样面积,进而导致通量计算的偏差。Control of procedures related to sampling include the control of sampling time precision, the sampling technique, the duration of sampling and replacement of receptor solution, the sample volume or flow rate, and sample handling and storage.与取样相关的控制程序,包括:取样时间精准度、取样技术、取样和替代接收液的时间间隔、取样体积或流速,以及样品的处理和储存。Control of procedures related to the pivotal study should include a non-dosed control skin section from each skin donor, which should be mounted in a diffusion cell and otherwise treated identically to the dosed skin sections, including sampling of the receptor solution at all time points to ensure that drug concentrations monitored in the receptor solution are associated with the dose applied in the IVPT pivotal study, and not drug contamination in the skin from that donor that might permeate into the receptor solution across the duration of the study. A pre-dose “zero” sample collected from each diffusion cell is also recommended, which may identify potential contamination associated with each skin section and/or each diffusion cell.在正式研究期间的相关控制程序,包括:对每组皮肤供体的非剂量控制皮肤切片,按照与上样皮肤切片一致的方式将其安装到扩散池中并进行后续处理,在所有时间点对接收液进行取样,以确保IVPT正式中,在接收液中检测到的药物为实际渗透进入的量,而不是研究期间由于皮肤的药物污染渗透进入。建议从每个扩散池中采集“零”浓度样品,用于识别每个皮肤切片和/或每个扩散池的潜在污染。In addition, investigators should perform the IVPT validation and pivotal studies within a quality management system that includes, but is not limited to, documented procedures for:此外,研究人员应在一定的质量管理体系条件下,进行IVPT验证和正式研究,包括但不限于:Study personnel identification, training, qualification, and responsibilities 研究人员的识别、培训、资质和职责Study management and study management personnel responsibilities 研究管理和研究管理人员职责Quality control (QC) and QC personnel responsibilities 质量控制(QC)和QC人员职责Quality assurance (QA) and QA personnel responsibilities 质量保证(QA)和QA人员职责Use of SOPs 操作规程(SOP)的制定Use of study protocols 研究方案的制定Use of study reports 研究报告的制定Maintenance and control of the study facility environment and systems 研究设施环境和系统的维护和控制Qualification and calibration of instruments and computerized systems 仪器和计算机系统的确认和校准Good documentation practices including, but not limited to, contemporaneous documentation of study procedures and recording of experimental observations or deviations from procedures specified in the study protocol or in relevant SOPs 良好的文件规范,包括但不限于:及时记录研究过程、试验观察、以及与研究方案及相关SOPs中规格程序的偏离Maintenance of suitable records that facilitate the reconstruction of study events and procedures, including study sample handling and storage records (e.g., sample tracking logs), audit trails for sample analysis procedures, control of study materials and reagents, and electronic data control 对记录进行适当的维护,以便研究过程可以重现,包括:研究样品的处理和保存记录(如,样品跟踪日志)、样品分析过程的审计跟踪、研究物料和试剂的控制以及电子数据控制Archival of study records 研究记录归档C. Blinding Procedure 盲法程序A detailed description of the blinding procedure should be provided in the study protocol and final report. The packaging of the test topical product and RS should be similar in appearance to maintain adequate blinding of the investigator and any experimental operators.应在研究方案和最终报告中递交详细的盲法程序信息。自研外用制剂和RS的包装应有相似的外观,以确保对研究人员和任何试验操作人员充分致盲。D. Randomization 随机化The method of randomization should be described in the protocol of the IVPT study and the randomization schedule provided, preferably in a SAS data set in .xpt format (created using the SAS XPORT procedure). It is recommended that an independent third party generate and hold the randomization code throughout the conduct of the study to minimize bias. The applicant may generate the randomization code if not involved in the packaging and labeling of the test topical product and RS dosed in the study. A sealed copy of the randomization scheme should be retained at the study site and should be available to FDA investigators at the time of site inspection to allow for verification of the treatment identity of each skin section.应在IVPT研究方案中描述随机化方法,并推荐以.xpt格式的SAS数据集形式(使用SAS XPORT程序创建)提供随机化计划表。在整个研究过程中,建议由独立的第三方生成并保存随机化代码,以减少偏差。如果不参与自研外用制剂和RS的包装和标签,申请人可以自行生成随机化代码。随机化方案的密封副本应保留在研究现场,以便在现场核查期间可随时提供给FDA审评员,用于确认每个皮肤切片的具体信息。E. Dosing 上样In the IVPT pivotal study, the test topical product and RS should be dosed in an alternating pattern on successive diffusion cells (skin sections) from each donor. One of two dosing sequences (illustrated below) may be randomly assigned for each donor: a. ABABAB… b. BABABA…在IVPT正式研究中,对于每个供体组,可以在扩散池(皮肤切片)上以交替给药方式依次放置自研外用制剂和RS。对于每个供体组,可采用下述两个方式中的一个进行随机放样: a. ABABAB… b. BABABA…F. Study Design 研究设计The IVPT pivotal study should compare the cutaneous pharmacokinetics of the drug from the test topical product versus that from the RS using excised human skin with a competent skin barrier mounted on a qualified diffusion cell system. The IVPT pivotal study should use a design that directly compares the test topical product and RS on skin from the same set of donors, each with the same number of replicate skin sections per donor per treatment group (dosed with either test topical product or RS topical), using the same IVPT method parameters.在IVPT正式研究中,应采用皮肤屏障完整性良好的离体人类皮肤和经确认的扩散池系统,对自研外用制剂和RS的皮肤药代动力学进行对比研究。在自研外用制剂和RS的对比研究中,每个试验组应采用相同的皮肤供体、相同的皮肤切片重复数,并采用相同的IVPT方法参数。The IVPT pivotal study design, methodology, and diffusion cell equipment considerations relating to sampling precision should be controlled as precisely as possible. For example, it may be appropriate to stagger the dose application on successive diffusion cells and to synchronize the sampling time points with the dosing time for that diffusion cell, to ensure consistent durations between dosing and sampling of all diffusion cells.应尽可能精准的控制IVPT正式研究的设计、方法和扩散池设备的取样精密度。例如,对于连续的扩散池,可以错开上样时间,根据上样时间的差异同步取样时间,以确保所有扩散池的上样和取样时间的间隔一致。G. Inclusion Criteria 纳入标准In general, the following inclusion criteria should apply: healthy, normal, barrier-competent skin from male and/or female donors of at least 18 years of age. Inclusion criteria related to donor demographics (e.g., age, race, sex) should be specified in the study protocol and demographic information should be reported for each donor. Additional criteria may be added by the applicant.通常,应采用以下纳入标准:至少18周岁的健康、正常、具有屏障完整性的雄性和/或雌性供体。应在研究方案中规定符合纳入标准的人口统计信息(如,年龄、种族、性别),并报告每个供体的个人信息。申请人可以增加其它纳入标准。The skin may be harvested following excision from patients undergoing a surgical procedure or excised from cadavers. A consistent source is recommended for all the skin used. The anatomical region specified in the study protocol (e.g., posterior torso) should be consistent for all donors whose skin is included in the study.建议使用人体离体皮肤,可以从接受外科手术的病人或尸体获取。所用的所有皮肤应有一致的来源,且所有的供体应为相同的解剖区域(如,背部躯干),并在研究方案中进行明确规定。The study protocol should specify the inclusion (acceptance) criteria for skin sections based upon the barrier integrity test result, which should be reported for each skin section. The study protocol should specify inclusion criteria related to the temperature and duration of skin storage as well as the number of freeze-thaw cycles, all of which should be reported for each donor’s skin. The study protocol should specify the inclusion criteria related to the skin harvesting/processing procedures and skin thickness (e.g., dermatomed skin of 500 μm ± 250 μm thickness) used in the IVPT study.研究方案中应规定皮肤切片的屏障完整性测试纳入(接受)标准;并规定每组供体皮肤的储存温度和时间,以及冻融循环次数;同时规定IVPT研究中所用皮肤的获取/处理方法和厚度(如,厚度为500 μm ± 250 μm)。H. Exclusion Criteria 排除标准In general, the following exclusion criteria should apply. Skin from subjects with a known (history of) dermatological disease should be excluded from the study. Skin with tattoos, stretch marks, or any visible sign of abnormality should be excluded from the study. Skin exhibiting a significant density of terminal hair is not recommended and should be excluded from the study. Additional criteria may be added by the applicant.通常,应采用以下排除标准。包括:患有已知皮肤疾病(病史)的供体皮肤;带有纹身、妊娠纹或任何异常迹象的皮肤;表现出明显浓密的末端毛发的皮肤等。申请人可以增加其它排除标准。While gentle washing or rinsing of the skin surface is appropriate, submerging the skin in an aqueous solution for more than a few minutes may damage the skin barrier and should be avoided; such skin sections should be excluded from the study. Also, skin that has been subjected to shaving with a blade; abrasive polishing; tape-stripping; or cleansing with alcohols, solvents, or other strong solutions that could damage the skin barrier should be excluded from the study.虽然可以采用温和的清洗或冲洗方式处理皮肤表面,但是,如果将皮肤浸泡在水溶液中几分钟,就有可能破坏皮肤屏障,应避免该处理方式。此外,也不建议采用刀片剃须、研磨抛光、胶带剥离,或采用乙醇或其它强极性溶剂处理皮肤,因为这些处理方式可能破坏皮肤屏障。Skin from donors with significant background levels of the drug or other compounds that may interfere with the quantification of the drug in receptor solution samples should be excluded from the study. Skin from donors exhibiting a high barrier integrity test failure rate among replicate skin sections may be excluded from the study, and skin from an alternative donor may be used instead.含有显著的药物背景水平或含有的其他化合物可能干扰接收液中药物定量测定的皮肤供体,应排除在研究之外。重复皮肤切片中屏障完整性测试失败率高的供体皮肤应排除在研究之外,采用其他的供体皮肤进行替代。I. IVPT Endpoints IVPT终点The endpoints for the IVPT pivotal study are based upon parameters that characterize the rate and extent to which the drug permeates into and through the skin and becomes available in the receptor solution. Specifically, the rate of drug permeation is characterized by the flux (J) and the extent of drug permeation is characterized by the total cumulative amount (AMT) of drug permeated into the receptor solution across the study duration.IVPT关键研究的皮肤药代动力学终点是基于表征药物渗透和通过皮肤进入接收液的速率和程度的参数。具体来说,药物的渗透速率采用通量(J)进行表征,而药物的渗透程度采用整个研究期间渗透到接收液的累积总量进行表征。The flux (rate of drug permeation) should be plotted as J on the Y-axis in units of mass/area/time (e.g., nanograms (ng)/cm2/hr) versus time on the X-axis. Flux profiles commonly resemble plasma pharmacokinetic profiles, however, it is important to distinguish that the flux is a rate, rather than a concentration. The extent of drug permeation should also be plotted, as the total cumulative amount (AMT) of drug permeated on the Y-axis in units of mass/area (e.g., ng/cm2) versus time on the X-axis.以通量(药物渗透速率)为纵坐标Y-轴,时间为横坐标X-轴作图;其中Y-轴用“J”标识,以质量/面积/时间(如,纳克/平方厘米/小时)为单位。通量分布模型通常类似于血浆药代动力学分布模型,尽管通量是一个速率单位,并不代表浓度。也应对药物的渗透程度作图,以药物累积渗透量(单位为质量/面积,如纳克/平方厘米)为纵坐标Y-轴,时间为横坐标X-轴。The flux should be calculated based upon: the receptor sample concentration (e.g., 2.0 ng/mL) at each time point; the precise, empirically measured volume of that specific diffusion cell (e.g., 6.0 mL) which may vary between individual cells; the area of dose application (e.g., 1 cm2); and the duration for which the receptor volume was accepting the drug. For example, if the sample exemplified here represented a 2-hour period following dosing, then J would be calculated based upon the values above as:通量的计算应基于:每个时间点的接收液中样品的浓度(如,2.0 ng/mL);精确的、经验测量的扩散池的体积(如,6.0 mL),每个扩散池的体积可能不同;上样区域的面积(如,1 cm2);和整个研究的持续时间。例如,如果此处所示样本代表给药后2小时内的情况,则J值按下式进行计算:J = [(2.0 ng/mL) x (6.0 mL)]/(1 cm2)/(2 hrs) = 6 ng/cm2/hrThis flux should be calculated and reported for each diffusion cell for each sampling interval and plotted across the entire study duration to generate the flux profile for each diffusion cell. The rate calculated above may be plotted at the 2-hour time point, or at the midpoint between 0 and 2 hours (i.e., 1 hour).In addition, the AMT should be calculated and reported for each diffusion cell. This cumulative amount of drug that has permeated (in total across the entire study) should be reported as the AMT endpoint, rather than using a trapezoid rule to calculate the area under the flux curve.应计算并报告各扩散池的所有相邻采样点之间的通量,并为整个研究期间的所有扩散池绘制通量曲线。上述计算的速率可以对应于2小时的实际时间点进行绘制,也可以采用0和2小时之间的中间点(即,1小时)。此外,应计算并报告每个扩散池的总累计渗透量(AMT)。AMT终点为药物渗透的累计量(整个研究期间的渗透总量),而不是采用梯形法则计算的通量曲线下的面积。The maximum flux (Jmax) at the peak of the drug flux profile and the AMT should both be compared for locally-acting test topical products and RSs. This is somewhat analogous to the comparison of the Cmax and AUC for systemically-acting test products and RSs, inasmuch as the pair of endpoints in each case facilitates a comparison of the rate and extent to which the drug from each type of product (locally-acting or systemically-acting) becomes available at the site of action.A confidence interval (CI) should be calculated for each IVPT endpoint: a. the natural log-transformed maximum flux (Jmax) b. the natural log-transformed total cumulative amount (AMT) permeated应将局部起效的自研制剂和RSs的最大通量(Jmax,药物通量曲线的最高峰)和AMT进行对比。这类似于全身起效的自研制剂和RSs的Cmax和AUC的比较,因为这些终点可分别用于表征各剂型(局部起效或全身起效)药物到达作用部位的速率和程度。应计算每个IVPT终点的置信区间(CI): a. 自然对数换算后的最大通量(Jmax) b. 自然对数换算后的总累积渗透量(AUC)It is the responsibility of the applicant to determine the number of donors required to adequately power the IVPT pivotal study, however, a minimum of four dosed replicates per donor per treatment group (test product or RS) is recommended.At the completion of the study, if the number of skin replicates is the same for all donors in the test topical product and RS treatment groups in the IVPT study, a statistical analysis for a balanced design is recommended. If skin sections or diffusion cells are excluded from the final statistical analysis because of experimental loss/issues, and the resulting data set is unbalanced, a statistical analysis for an unbalanced design is recommended.申请人有责任确定足以支持IVPT关键研究所需的供体数量,然而,建议每个试验组(自研制剂或RS)每个供体至少4个重复剂量。在IVPT研究结束时,如果自研外用制剂和RS试验组中所有供体的皮肤切片重复数均一致,建议采用平衡设计法进行统计分析。如果因试验问题,需将皮肤切片或扩散池从最终的统计分析中排除,则结果数据集不平衡/对称,建议采用非平衡设计法进行统计分析。Approaches to statistical analysis of the pivotal study are described in section VIII of this guidance. Appendix I provides example SAS code for determining BE with both a balanced dataset and an unbalanced dataset. Appendix II provides numerical examples with simulated data sets. Appendix III provides example R code for determining BE.在本指南的第VIII部分,对正式研究的统计分析方法进行了描述。附录I提供了用于测定平衡数据集和非平衡数据集BE的SAS代码示例。附录II提供了模拟数据集的具体示例。附录III提供了测定BE的R代码示例。VII. Submitting information on IVPT studies in an ANDA ANDA中递交的IVPT研究信息For IVPT studies with topical products submitted in ANDAs that are intended to support a demonstration of BE, detailed study protocols, relevant SOPs, and detailed reports should be submitted for the IVPT method validation (including the IVPT pilot study) and the IVPT pivotal study. In addition, a detailed report describing the IVPT method development should be submitted. These protocols, SOPs, and reports should be submitted in module 5.3.1.2 of the electronic Common Technical Document (eCTD) and should describe experimental procedures, study controls, quality management procedures, and data analyses.对于支持外用制剂产品BE论证的IVPT研究,在ANDA申请中应递交IVPT方法验证(包括IVPT初步研究)和IVPT正式研究的详细研究方案、相关SOPs和报告。此外,应递交描述IVPT方法开发的详细报告。这些方案、SOPs和报告应在电子通用技术文件(eCTD)的模块5.3.1.2中递交,并对相关的试验方法、研究控制、质量管理程序和数据分析进行描述。Note that the study protocols, SOPs, and reports related to the IVPT method are distinct from those for the sample analytical method that is used to quantify drug concentrations in IVPT receptor solution samples (e.g., an HPLC/MS or UPLC/MS method). Separate protocols and SOPs should be submitted for the sample analytical method validation. Sample analytical method development and validation reports, pilot and pivotal IVPT study sample analysis reports, as well as associated SOPs and protocols relevant to the sample analysis of an IVPT study with human skin should be submitted in Module 5.3.1.4 of the eCTD.注意:用于IVPT接收液中样品浓度的定量分析方法(如,HPLC/MS或UPLC/MS方法)与IVPT方法相关的研究方案、SOPs和报告不同。应单独递交样品分析方法验证的方案和SOPs。样品分析方法开发和验证报告、初步和正式IVPT研究样品分析报告,以及与采用人体皮肤进行IVPT研究相关的样品分析SOPs和方案,应在eCTD的模块5.3.1.4中递交。VIII. IVPT pivotal study statistical analysis IVPT正式研究统计分析
应用实例
2024.04.08

体外渗透试验(IVPT)中生物膜的控制 一、皮肤的纳入标准
体外渗透试验(IVPT)中生物膜的控制一、皮肤的纳入标准 所用的皮肤应有一致的来源,且为相同的解剖区域(如背部躯干;也可以使用其他生物膜,如直肠、阴道或角膜上皮组织,这取决于所评估药品的预期给药途径);同时,需规定每组供体皮肤的储存温度和时间,以及冻融循环次数。此外,FDA建议皮肤的厚度在(500±250)μm;但USP要求,厚度通常在250~500 µm。在研究中,需采用相同的皮肤厚度,因为厚度的显著变化可能会增加IVPT结果的变异性。 朱慧勇等[1]研究表明将皮肤的新鲜度(将裸鼠皮肤于-20℃放置6个月,其皮肤屏障性能无明显差异)、厚度(RSD≤15%)、质量(RSD≤15%)、经皮水分散失(TEWL)达到稳态等因素作为皮肤模型的质量控制手段,可进一步增加研究结果的可靠性和准确度。 但是,MORIN M等[2]研究表明将猪耳皮肤在不同的温度条件下放置不同的时间,新鲜皮肤相对其它条件具有较高的电阻且对亲水性化合物阿昔洛韦和疏水性水杨酸甲酯的渗透性均较低;在不同温度条件下放置不同时间的皮肤对阿昔洛韦在皮肤中的组织分布有显著影响;但根据水杨酸甲酯的研究结果可知,在不同的放置条件下,皮肤中均保存一定的酯酶活性。 目前,关于皮肤的储存条件尚未有统一的要求,但通常情况下,新鲜的皮肤渗透性较低,因此,在厚度、质量及解剖区域一致的情况下,不同时间测得的结果亦会有不同的差异,这也侧面说明,对受试制剂和参比制剂平行测定的重要性。 二、皮肤的处理和保存 可以采用电动取皮机设置一定的厚度进行取皮,然后使用纯水或生理盐水冲洗皮肤外表面,并用镊子和刀片等小心刮去残留的皮下脂肪和血管等其他组织,然后反复冲洗干净,低温保存(建议储存于-20℃或-80℃冰箱中)。 虽然可以采用温和的清洗或冲洗方式处理皮肤表面,但是,如果将皮肤浸泡在水溶液中几分钟,就有可能破坏皮肤屏障,应避免该处理方式。此外,也不建议采用刀片剃须、研磨抛光、胶带剥离,或采用乙醇或其它强极性溶剂处理皮肤,因为这些处理方式可能破坏皮肤屏障。 因此,在上样前皮肤的解冻过程中,应尽可能的避免采用接收介质浸泡的方式,以免破坏皮肤的屏障完整性;实际操作中,通常可将皮肤在室温条件下放置约30 min即可。 三、采用人体皮肤存在的挑战除来源、理论因素外,人体皮肤的存放也存在一定的挑战。将皮肤存放在-22℃,或模拟表皮与真皮的剥离程序,将其放置在60℃进行加热处理,均会对皮肤的组织活力造成损伤[3]。经济合作与发展组织(OECD)指南规定,在试验中,必须对皮肤的屏障特性进行评估,但并没有给出有关最佳储存条件的具体要求。该指南指出,新鲜的离体皮肤应在24小时内使用,但可接受的储存期限可能因储存温度和酶系统的新陈代谢而异(OECD,2004年)。Nielsen等人[4]采用在80℃条件下储存三周的人体皮肤进行体外经皮渗透研究,结果发现,由于皮肤的上层及其深层的结构变化,药物的渗透速率和总渗透量明显增加。目前,对经皮吸收的评估而言,欧洲替代方法验证中心(ECVAM)建议将皮肤储存在20℃。在世界卫生组织发布的《环境健康标准:皮肤吸收》中指出,基于Bronaugh等人[5]的研究成果,人体皮肤可以在20℃下储存长达一年(1986)。无论储存条件如何,都必须证明皮肤屏障功能的完整性。这可以通过进行初步的渗透研究,并将测定结果与预定的可接受标准进行比较来实现,如采用氚化水渗透和TEWL。 参考文献:文献1:朱慧勇,武余波,卢望丁等. 用于经皮给药系统研究的皮肤模型与关键质量控制[J].中国医药工业杂志, 2022, 53(5): 592-600. 文献2:MORIN M, RUNNSJö A, RUZGAS T, et al.Effects of storage conditions on permeability and electrical impedance properties of the skin barrier[J]. Int J Pharm, 2023.122891.文献3:Wester RC, Christoffel J, et al. Human Cadaver Skin Viability for In Vitro Percutaneous Absorption: Storage and Detrimental Effects of Heat- Separation and Freezing[J]. Pharm Res, 1998, 15(1): 82– 84.文献4:Nielsen JB, Plasencia I, Sørensen JA, et al. Storage Conditions of Skin Affect Tissue Structure and Subsequent In Vitro Percutaneous Penetration. Skin Pharmacol Physiol[J]. 2011, 24(2): 93– 102.文献5:Bronaugh RL, Stewart RF, et al. Methods for In Vitro Percutaneous Absorption Studies. I. Comparison with In Vivo Results. Toxicol Appl Pharmacol[J]. 1982, 62(3): 474– 480.
企业动态
2024.03.26

性能测试在半固体制剂生物等效性评价中的作用
性能测试在半固体制剂生物等效性评价中的作用第一章|前言“外用药物制剂具有复杂的药物递送途径,通常局部起效,辅料成分复杂,处方组成和制备工艺参数的微小差异就有可能导致产品不同的质量特性,从而影响药物的安全性和有效性。外用药物制剂是一类发挥局部或全身治疗作用的制剂,剂型包括软膏剂、乳膏剂、凝胶剂、散剂、乳剂、糊剂、混悬剂、喷雾剂、气雾剂、溶液和其他半固体和/或液体剂型。该类制剂的质量测试分为两类:一是评估产品质量属性的测试,包括物理化学和结构特性(Q3),如外观、混悬药物的晶型、粒度分布、液滴粒径、流变特性、pH值、黏度、含量/含量均匀度、微生物限度、有关物质、抑菌剂含量、抗氧剂含量和无菌等;另一是评估产品性能的测试,包括体外释放试验(IVRT)和体外透皮试验(IVPT)。外用药物制剂在处方组成(Q1)和用量(Q2)一致性的基础上,对Q3特性的评估是生物等效性豁免的重要基础。“Part 02第二章|IVPT和IVRT的作用“根据2021年03月《皮肤外用化学仿制药研究技术指导原则(试行)》所述,半固体药物在体外释放的程度和速度是制剂性能的综合体现,主要用于外用产品的药学质量控制,也可用于药品开发过程中处方工艺的筛选研究。体外透皮试验的设计目的是模拟外用药物在生理条件下的透皮过程,以反映外用制剂的质量。 IVRT是一个重要且有用的工具,因为:(1)它为合适的临床候选处方的选择和剂型的表征提供了科学依据;(2)它可用于评估药物生产和质量的一致性;(3)它为不同企业之间药物的比较提供一致的标准;(4)它为临床中预测该剂型的性能提供了可测量的指标;(5)在上市后的变更(如生产地点、组分和生产工艺的变更)中,它可用于论证变更前后的“相似性”;(6)它有助于识别和评估关键的生产变量。 跟 USP《1724》、FDA IVRT 和 IVPT 指南,二者分别可用于以下情况:IVRT 体外释放试验1. 表征一个产品批次的稳态药物释放速率。当采用一个经过验证的IVRT分析方法,对支持产品安全性和/或有效性的产品批次进行性能(释放速率)表征时,获得的该外用制剂的性能(释放速率)特征可作为未来产品性能的参考基础。2. 通过比较变更后和变更前产品的稳态药物释放速率,表征某些工艺、配方和/或生产的变更对药品的影响,在某些情况下,可用于论证产品批量放大或已获批产品批准后变更的等效性。3. 在某些情况下,通过比较仿制药和参比制剂的稳态药物释放速率,表征特定工艺、配方和/或生产变化对药品的影响,用于论证药物的生物等效性。IVPT 体外渗透试验1. 表征一个产品批次中药物渗透进入和透过皮肤的速率和程度。2.通过比较药物渗透进入和透过皮肤的速率和程度,表征特定工艺、配方和/或生产变更对药物产品的影响。在进行外用药物产品的配方对比,以及变更后和变更前产品的对比中,可能需要进行IVPT测试。3. 通过比较仿制药和参比制剂中药物渗透进入和透过皮肤的速率和程度,表征特定工艺、配方和/或生产变化对药物产品的影响,用于论证药物的生物等效性。 根据EMA指南《外用制剂质量和等效性指南草案》,亦可知 IVRT 和 IVPT 的重要性,如该指南给出以下信息: 1. IVRT:尽管该测定不能模拟体内性能情况,但释放速率仍是一项关键质量属性,应订入产品放行和货架期质量标准;对于简单的配方制剂(例如单相的溶液剂、凝胶剂和软膏剂),可通过证明质量等效性(包括Q3特性、体外释放),用于支持与参照药物相比的治疗等效性,以代替等效性临床试验2. IVPT:对于比较复杂的制剂(如乳剂),或含有的辅料可能直接影响生物利用度或产品性能的制剂,可采用合适等效性测试方法,例如:IVPT、角质层取样(胶带剥离)或药代动力学,代替临床治疗研究此外,在药物获批上市后,IVRT和IVPT试验可用于生产放大和处方工艺变更的评估,如根据PMDA指南《皮肤局部适用制剂(半固体制剂和贴剂)处方变更生物等效性研究指南》所述:可通过IVRT和IVPT试验对已上市半固体制剂和贴剂的A、B、C水平处方变更进行论证。综述可知,IVRT和IVPT研究在半固体制剂研究中的重要性。“Part 03第三章|IVRT和IVPT研究的难点“虽然FDA、EMA和PMDA均有发布关于IVRT和IVPT研究的相关指南,但目前国内暂无统一的技术要求规范,且各国技术要求亦不统一。 在IVRT研究中有很多技术难点,如合成膜的选择及处理、接收介质的筛选、上样方式的控制,如何开发具有区分力的试验方法等。 IVPT试验结果受多种因素的影响,如扩散池的选择,样品分析方法,生物膜的来源、处理和贮存,试验设计(如上样量、上样技术,接收介质的种类、pH和体积,设备平衡,试验时长和采样)等,常导致各扩散池间、操作者间及实验室间重现性差,试验结果不可靠。 合邦兴业 将针对上述研究中存在的难点,后期分别从以下主题,详细对 IVRT 和 IVPT 的试验设计进行详细阐述,敬请期待: 1. IVR方法开发:阐述分析方法的开发、合成膜的选择、接收介质的种类、上样方式、设备平衡、重复次数等等2. IVRT数据统计分析:依次举例阐述FDA、EMA和PMDA对统计分析的要求3. IVRT方法确认及验证:包括分析方法验证和IVRT试验方法验证两部分4. IVPT生物膜选择:包括皮肤选择、纳入/拒绝标准、处理保存、屏障完整性5.IVPT方法开发:包括上样方式(如何减少上样导致的变异)、接收介质、温度控制、药物组织分析等6.IVPT分析方法选择:皮上样品处理、组织分布研究、接收液分析方法IVPT统计分析:依次举例阐述FDA、EMA和PMDA对统计分析的要求
企业动态
2024.03.26

流变学评价在皮肤局部外用半固体制剂处方开发中的应用进展
流变学评价在皮肤局部外用半固体制剂处方开发中的应用进展本文介绍:流变学评价在皮肤局部外用半固体制剂处方开发中的应用进展作者:黄乐乐,马晋隆,王嘉明,徐 驿,徐盛超,朱慧勇,史家骏,倪 睿,罗华菲中国医药工业研究总院药物制剂国家工程研究中心期刊:中国医药工业杂志 Chinese Journal of Pharmaceuticals 2022, 53(5)摘要:皮肤局部外用半固体制剂的微观结构决定了制剂的流变学特性,流变行为可能会影响产品的生产制造、外观性状、稳定性、感官特性和体内性能等。通过流变学评价可表征皮肤局部外用半固体制剂的微观结构特征,对制剂处方开发具有较高的应用价值。流变学参数可作为皮肤局部半固体制剂的关键质量属性,并可用于预测产品稳定性和患者顺应性。本研究介绍了皮肤局部外用半固体制剂常用的流变学测试方法,及其在处方工艺开发、提高产品稳定性和改善患者顺应性方面的应用情况。市售皮肤局部外用半固体制剂以乳膏、软膏和凝胶剂型为主。这些剂型可以是单相体系(如矿物油、蜡质等脂质制成的软膏或高分子聚合物凝胶等),也可以是包含连续相和分散相的多相体系(如含有不溶性液滴或固体颗粒的软膏、乳液型凝胶及各种类型的乳膏和乳剂)。单相体系半固体制剂的微观结构取决于主要组分的分子结构,如凡士林或卡波姆;多相体系的微观结构主要受连续相与分散相相互作用的影响,如相互结合的作用力( 氢键、弱范德华力等)、分散相体积、乳滴大小、颗粒粒径和密度等。微观结构的内聚力使制剂得以维持其半固体状态并具有相应的抗变形能力和黏度,通常用流变学方法进行表征。当微观结构发生变化,相应流变行为的变化也可通过流变学测试进行量化,并结合其他物理方法( 如偏振光显微镜、差示扫描量热分析等) 获知结构变化的程度及原因。近年来出于质量源于设计的理念与仿制药质量和疗效一致性评价的要求,皮肤局部外用半固体制剂的流变学表征日益得到重视,在产品处方工艺开发、质量控制、贮存稳定性、使用时感官特性和患者顺应性等方面得到越来越多的应用。本研究将介绍皮肤局部外用半固体制剂的流变学研究方法及其在处方工艺开发中的应用。1 研究方法流体在较小剪切力作用下即可发生形变而具有流动性,但由于内部结构的相互作用会对流动产生一定阻力,表现为具有一定黏度;根据黏度与所施加剪切速率的关系,可分为牛顿流体和非牛顿流体。牛顿流体的黏度为固定值,与所受到的剪切力无关;非牛顿流体的黏度随剪切速率而变化,根据变化趋势又可分为剪切变稀和剪切变稠流体,前者表现为黏度随剪切速率增加而降低,后者则相反。皮肤外用半固体制剂通常为剪切变稀型非牛顿流体,在不受外力作用( 贮存) 时保持类固体形态,在外力作用下( 使用时) 能流动而易于涂布。流变学研究可确定流体类型及其流变学性质。半固体制剂的流变学研究包括 2 种主要方式(见表1),一种是通过施加周期性动态作用力来考察半固体的弹性形变,这通常要在半固体结构保持完整的静态下进行;另一种是在一定时间内持续施加作用力以考察半固体从静止到持续流动的行为。对应的测量模式为流动测量和黏弹性测量。表1 皮肤外用半固体制剂流变学研究方法1.1 流动测量 (flow measurements)1.1.1 稳态流动 (steady-state flow)局部半固体制剂通常为非牛顿流体,在剪切速率()恒定时,流变响应即剪切应力(σ)取决于剪切的持续时间,其表观粘度是时间的函数,因此也称为时间依赖性流体。测量非牛顿流体的黏度需基于稳态假设[1],如图1 所示,施加恒定剪切速率一定时间后材料的流动行为不再随时间变化,即材料结构的变化达到动态平衡,此时黏度可达到一个常数值( 图1中虚线所示),可作为相应剪切速率下的黏度。图1 在恒定剪切速率下黏度对时间的函数曲线Fig.1 Viscosity as Function of Time at a Constant Shear Rate通常为了全面考察制剂的流动性质,需在一定的剪切速率范围内测量黏度的变化,将黏度绘制为对剪切速率的曲线,即得到流动曲线(flow curve)。通过在稳态流动状态下逐步增加剪切速率,所得的流动曲线可称为稳态流动曲线。在低或高剪切速率下,黏度的变化幅度很小,流动曲线呈现平台段,出现平台段的流动曲线可被认为是完整反映了制剂的流动行为。在低剪切速率平台处,黏度曲线趋近于零的剪切黏度可看作是制剂在静止状态下( 如处于容器内) 的黏度,它反映了制剂微观结构的坚固性,是产品稳定性的指标之一。但黏度较小的体系可能在最低可测量剪切速率下也无法观察到平台段,此时可采用控制应力的模式来测量[2]。在处方开发阶段,可通过稳态流动曲线确定物料静止和使用时的黏度。低至中等剪切速率下的黏度决定了物料能否顺畅地从容器中挤出,中至高剪切速率下的黏度则决定产品是否易于涂布于皮肤。1.1.2 非稳态流动流体在剪切作用下,剪切应力或黏度随时间变化的性质称为“触变性”。触变流体受到一定程度的剪切作用时内部结构会被破坏,去除剪切作用后结构则会恢复,但在恢复完成之前有一定的时间滞后。触变性的测量通常是在非稳态流动下控制一段时间内的剪切速率先增大后减小,剪切应力响应时会产生包含上升和下降的流动曲线。上升曲线在下降曲线之上,说明结构的恢复是不完全的;上升曲线和下降曲线重合,说明结构能迅速恢复;上升曲线和下降曲线有交叉,则提示发生了重构。因上升和下降曲线不重叠而形成的滞后回环被称为“触变环”,它反映了剪切过程中材料结构的破坏和恢复情况。触变环面积通常被认为是触变性的度量[3],面积越小表明结构可越快恢复,面积越大则表明结构的破坏程度越高,但同时也提示产品具有较好的涂展性。因此,触变环通常被用来评价半固体制剂的稳定性,以及使用时的涂布性能[4]。1.2 黏弹性测量(viscoelastic measurements)上述持续剪切作用下的考察通常伴随着结构的破坏或分散,为了更全面地考察半固体制剂的黏弹性,需采用非破坏性的测试方法,即只施加小的作用力、不显著破坏制剂的静态结构,如蠕变和振荡测试。1.2.1 蠕变测试 (creep test)蠕变描述了半固体制剂在固定应力作用下的缓慢形变。蠕变测试是研究半固体黏弹性的最简单方法之一。测量时,突然对制剂施加一个应力,并在随后一段时间内保持不变,柔量(J =应变/应力)随时间变化的曲线称为蠕变曲线,应力消除后一段时间内的柔量-时间曲线称为蠕变恢复曲线。如果应变处于LVR(线性黏弹区 linear viscoelastic region)范围,则应力与应变的比值仅是时间的函数。典型的蠕变曲线如图2所示,曲线可分为3个部分,曲线AB段表示结构发生瞬时弹性形变,表现为柔量瞬时增大;BC段代表黏弹性流动,表现为柔量随时间延长而缓慢增加;CD段为黏性流动,柔量与时间成线性关系,代表结构在该应力下达到稳态[5]。在蠕变恢复过程中,AB段和BC段可完全或部分恢复,CD段则无法恢复。D点之后为蠕变恢复曲线。图2 蠕变 / 恢复曲线Fig.2 Creep/Recovery Curve蠕变曲线直接描述了制剂黏弹性与微观结构的关系,J 值增量越小,则结构的弹性越好、结构越稳定。PAL 测定了分散相比例不同的乳剂的蠕变曲线,发现分散相比例越大,J 值越小;在蠕变恢复阶段,J 值恢复比例随分散相比例增加而增加,因此分散相比例在一定范围内增加可增加乳剂的稳定性[6]。1.2.2 振荡测试 (oscillatory test)振荡试验被广泛应用于黏弹性体系及其内部结构弹性和完整性的表征和量化。振荡测试可采用多种形式进行,如应变或扭矩( 应力) 扫描、频率扫描、温度扫描和时间扫描。所测定的黏弹性参数见表2。表2 振荡测试的黏弹性参数Tab.2 Viscoelastic Parameters of Oscillatory Test在应变扫描(strain sweep)试验中,固定振荡频率(通常为0.1或1Hz),再测量黏弹性参数作为应变振幅的函数。在临界应变以下,|η*|、G'和G''与时间( 或频率) 的函数成线性关系。这个区域通常被称为 LVR,此区域内对微观结构的任何扰动都可以瞬间恢复(可逆过程)。在 LVR 内,通常 G'大于 G'',如果 G'比 G''高出1个数量级以上,可认为结构的弹性大于黏性[7]。用含有微晶结构的半固体辅料( 如白色软石蜡、羊毛醇等) 制备的简单软膏在剪切作用下易破碎,因此 LVR 较窄;而用具有较长分子链的交联网状结构辅料(如聚合物)制备的制剂则 LVR 较宽[8]。频率扫描(frequency sweep)试验是在频率不断增加的情况下考察G'的变化,用以表明材料在应力作用下抵抗结构变化的能力,可获得结构稳定性方面的信息[9]。如ANDRITOIU等用频率扫描试验考察了以植物油脂为油相的软膏的稳定性,观察到随频率增加(0.01~100 rad/s),G'均大于G'',说明软膏结构较稳定[10]。温度扫描(temperature sweep)试验是在固定剪切应变(LVR内)和频率(通常为1Hz)的情况下,逐步改变温度以考察黏弹性参数的变化。通常用于考察生产、贮存、运输或使用过程中可能遇到的温度变化对结构的影响。如 SHALVIRI 等通过温度扫描考察了黄原胶凝胶的热稳定性,观察到当温度升高到50℃时,凝胶发生了不可逆的结构变化[11]。LAUTERBACH 等以 tanδ 为响应,采用循环温度扫描考察了处方组成对乳剂和软膏微观结构的影响,观察到表面活性剂比例越高,乳剂弹性受温度的影响越大[12]。此外,考虑到皮肤外用半固体制剂的实际使用情况,例如在皮肤上揉擦软膏时施加的剪切应变和应力相对较大,对结构的破坏是不可逆的,因此若模拟实际使用时的应变进行测量,此时黏弹性参数的响应是非线性的[8]。如 LVR 临界处,G' 的变化达到2个数量级可认为制剂的涂抹性能较好。1.3 屈服应力 (yield stress)屈服应力是指制剂开始流动的临界应力。在该值以下,制剂主要呈现弹性性质,高于该值时制剂呈塑性流动。屈服应力决定了制剂的贮存稳定性和患者使用时的感官特性。半固体制剂的屈服应力应足够大,以避免因自身质量而流动,但同时也不应过大而难以在皮肤上涂抹均匀;屈服应力值若大于 800 Pa 则会过于黏稠而难以从软管中挤出[13]。屈服应力可通过不同的测量方式或模型拟合计算得到,常见于文献报道的方式见表3。表3 屈服应力的测量方式Tab.3 Measurement Methods of Yield Stress屈服应力是半固体制剂的一个重要流变学参数,较高时表明产品具有较好的稳定性,结构不易被破坏[14]。PARK 等考察了凡士林及用其制备的软膏和乳膏的屈服应力,观察到由于凡士林具有三维网状结构以及由微晶组成的胶态凝胶型结构,各样品均具有较大的屈服应力,可抵抗剪切[15]。KITAGAWA 等研究了不同工艺过程前后白凡士林的黏度和屈服应力变化,观察到经过不同的搅拌工艺,白凡士林的屈服应力有不同程度降低,且与其来源有关[16]。1.4 测量影响因素通常使用流变仪来测量各流变学参数。测量时,将样品小心置于下方的夹具,调整上下夹具的间距,再进行测试。夹具类型、制样和测定条件等因素都会对测量结果造成影响。1.4.1 夹具类型流变仪通常配备不同形状、尺寸和材质的测量夹具。一般有平行板、锥形板和同心圆筒3 种形状的夹具可供选择。目前测量中多采用平行板和锥形板夹具。一般来说,平行板夹具在使用上具有一些优点,如每次测量后易于清洁;与锥板夹具相比,两板之间的间隙较大,因此间隙误差相对较小;可轻松地改变板间的间隙,并可对同一样品进行一系列试验[15,17]。锥形板夹具则可减少上样量。此外,若测试材料和夹具之间有滑移现象,可采用砂质板面的夹具[15]。1.4.2 制样和测定条件加载样品时,需特别注意尽量减少对结构的扰动。每次加载完样品并调整夹具间距至测量间距后,应静置足够的时间,使样品结构充分恢复。对于刚性较强的样品,如凡士林,恢复时间可能长达数十分钟。可通过时间扫描模式施加极小应变以监测样品结构是否达到稳态。但对于具有挥发性的样品应考虑缩短恢复时间,防止挥发造成样品损耗,同时可在夹具上加装防挥发装置;水性样品也可用低黏度硅油封边。夹具间距应根据考察目的设定,间距小代表接近使用时状态,间距大代表接近贮存时状态[10,18—19]。此外,需考虑新制样品的老化问题,乳剂的老化可能长达数月,老化前后样品的流变学数据相差较大[20]。1.5 方法学验证目前流变学测定方法尚无标准化验证程序。SIMÕES 等用 1%氢化可的松乳膏为模型产品,尝试系统性验证试验装置和测量方法,包括精密度、区分力和耐用性[21]。具体如下:①流变仪验证,在 25 和 32 ℃下测定参考标准物(牛顿流体) 的黏度分布,RSD 应小于15% ;②日间精密度,每项试验在不同的 3 日内进行 12 次流变测量, RSD 应小于15% ;③区分力,交叉比较含不同比例关键辅料以及不同工艺制备的制剂的流变学性质,如有显著差异,则方法具有区分力;④耐用性,比较不同温度(±2 ℃)、不同上样方式(注射器或刮刀)及不同模具(锥板或平行板)的数据,与标称方法测得的平均值的偏差小于15%时可认为方法具有耐用性。考虑到流变学测试对结构变化比较敏感,我们认为后两项验证的意义还有待商榷,区分力的问题通常在于过度而不是不足,而耐用性的考察范围不应包括温度这种本身是测定变量的参数,且上样方式和模具也要根据具体样品和测定方法进行选择,不适宜作为耐用性指标。因此,流变学测试可能较难形成通用的方法学验证程序,需针对具体样品来开发具体的测定方法并进行验证。2 流变学性质考察的具体应用半固体制剂的流变学性质对微观结构的变化非常敏感。制剂开发过程中,处方和工艺因素对微观结构的影响以及贮存期间微观结构的变化均可通过流变学性质的变化表征出来。此外,制剂在使用时是否易从容器( 如软管) 中被挤出和涂布到皮肤上也是由其流变性质所决定的。因此,更好地了解影响流变特性的各种因素并加以控制,有助于半固体制剂处方工艺的开发及产品质量性能的提高。2.1 处方工艺开发2.1.1 考察辅料属性对制剂的影响辅料的流变学性质对于半固体制剂的处方开发具有重要的指导意义。应着重考虑对体系黏弹性贡献较大的主要辅料的流变学性质,以及加入其他辅料产生的影响。例如,凡士林是不同碳链长度的液态和微晶态烷烃类组成的混合物,具有三维网状结构,刚性较大,其流变学性质对以其为主要基质或油相组分的软膏或 w/o 型乳膏的结构有很大影响。PENA 等研究了一种由白凡士林、矿物油和微晶蜡组成的模型软膏,观察到软膏的流变学性质主要由白凡士林和矿物油决定,而微晶蜡有助于结构的稳定[22]。BAO 等报道不同来源凡士林的流变学性质有较大差异,当凡士林在软膏中所占比例足够大(>98% ) 时,软膏的流变学参数与其同步变化[23]。因此凡士林的流变学性质被认为是相关产品开发中最重要的参数之一,应作为关键物料属性进行控制。LARREA-WACHTENDORFF 等观察到相同工艺条件下,2 种淀粉( 玉米淀粉和大米淀粉) 形成的凝胶黏度和黏弹性参数不同,且甘油对其影响程度也不同:随甘油比例增加,玉米淀粉凝胶的黏度降低,而大米淀粉凝胶的黏度变化不明显,但 2 种凝胶黏弹性的变化均较小[24]。2.1.2 处方设计半固体制剂通常为多相体系,分散相的性质及其在连续相中的状态是流变学性质的主要影响因素。2.1.2.1 不溶性固体或液滴的影响当活性药物成分以固体颗粒形式混悬或以液滴形式分散于基质中时,其粒径大小和处方比例对基质的流变学性质将产生一定影响。XU 等报道在凡士林中加入的石蜡和不溶性药物颗粒的比例与软膏的 G' 呈负相关[25]。SIEMIRADZKA 等研制了一种促肾上腺皮质激素的皮肤外用软膏以代替注射给药,观察到水相的加入及加入比例对软膏的流变学性质均有影响[26]。药物溶于少量水相并通过乳化分散于亲脂性软膏基质时,能降低软膏的黏度和弹性,但同时能提高涂抹性。将黏度较低的载体如乳剂、醇质体、传递体、囊泡或脂质纳米粒加入到黏度较高的基质如凝胶或软膏中,可提高前者的局部给药性能,通过流变性测试可评估体系间的相容性。SOUTO 等通过流变学测试评估了水凝胶在加入固体脂质纳米粒(SLN)或纳米结构脂质载体(NLC) 前后的理化性质,结果表明与加入 NLC 相比,含有脂质含量较高的 SLN 的水凝胶 G' 值较高,黏度也较高,且不同凝胶基质(黄原胶、羟乙基纤维素、卡波姆和壳聚糖)制品均出现此现象[7]。CRISTIANO 等测定了泊洛沙姆 407 水凝胶在加入不同类型载体时的流变学特性和凝胶化温度的变化,结果表明醇质体等载体的加入对泊洛沙姆 407 水凝胶的三维网络结构和流变学特性没有不利影响,二者实现了协同作用,增加了这些载体在皮肤上的持续时间[27]。2.1.2.2 多相体系中乳化剂和分散相的影响乳膏和乳剂的流变学性能主要受乳化剂的性质和浓度影响,此外与分散相的体积分数、液滴的尺寸和分布、连续相的流变学性能、乳化剂膜的界面流变性等因素也有关。KORHONEN 等考察了4种山梨醇酐单酯乳化剂(Span-20、Span-40、Span-60 和Span-80) 的分子结构对简单三组分 o/w 型乳膏形成的影响,观察到乳化剂的饱和烃链越长,形成的乳膏黏弹性越好;临界胶束浓度小及烃链比油相烃链长的乳化剂均可增加黏弹性[28]。RIBEIRO等考察了在脂肪醇和非离子乳化剂形成的 o/w 乳膏中加入阳离子聚合物( 聚季铵盐) 和阿拉伯胶后制品流变学性质的变化,观察到前者的加入使乳膏中的液滴粒径增大、黏度降低、弹性变差( J 值增大、G' 值降低),而阿拉伯胶则可增加乳膏的稳定性,2 种物质分别通过削弱和加强乳化剂的双分子层网络结构而改变乳膏的流变性质[29]。THORGEIRSDÓTTIR 等研究了油相比例对卡波姆处方流变性能的影响,观察到增加油相比例可提高卡波姆乳膏的结构稳定性[9]。LAUTERBACH 等制备了一系列含不同浓度表面活性剂( 聚氧乙烯脂肪醇醚Brij) 的乳剂及不同比例液体和固体石蜡的软膏,测定其经历数个温度扫描循环(25 ℃→ 40 ℃→25 ℃→ 5 ℃→ 25 ℃ ) 的流变学参数( tanδ 及其极值的跨度),通过多元数据分析方法中的主成分分析确定了这些乳剂和软膏的关键物料属性分别是 Brij 的浓度和液体石蜡的比例[12]。2.1.3 工艺因素半固体制剂的制备工艺参数(如搅拌方式、搅拌速率、加热温度和时间、冷却温度和速率等)对制剂的微观结构有决定性影响,通过流变学考察有助于确定关键工艺参数及其范围。TAMBURIC 等考察了加热过程对凡士林结构的影响,观察到白凡士林在不经搅拌的情况下加热至70 ℃再自然冷却,其黏度( τ=50 Pa) 和屈服应力均有较大程度增加,触变环变小,说明单纯的熔融冷却过程使白凡士林建立了更紧密的微观结构[30]。通过适宜的搅拌可降低白凡士林结构的刚性,有助于形成柔软、延展性好的膏体;但如果搅拌过于剧烈则会增大对网络结构的破坏。他们还考察了工艺过程中的加热温度对以白凡士林为油相的乳膏结构的影响,观察到低温(25 ℃ ) 下制备的乳膏的黏弹性参数优于高温(70 ℃ ) 下的制品,因此在以凡士林为油相的乳膏制备中温度是关键工艺参数。BAO等考察了室温直接搅拌与熔融搅拌再冷却工艺的影响[23]。由于室温下凡士林仍处于半固体状态,直接搅拌工艺中需施加较大的剪切力才能混匀,对微观结构的破坏较大,与熔融搅拌再冷却法制备的软膏相比,G' 值和屈服应力显著降低,说明熔融混合对微观结构的影响较小。VAN HEUGTEN 等在对混悬型软膏制备工艺的筛选中观察到,冷却时的混合速率和灌装时的温度对软膏屈服应力的影响较大[13]。NISHIKAWA 等采用湿式气流粉碎工艺制备亲水性软膏,观察到软膏黏度随压力的增大而增大,触变环面积和屈服应力也相应增加,推测可能由于该工艺方式使油滴粒径减小、密度增大,其分子间的范德华力增强,因而膏体结构更稳固[3]。在凝胶的制备过程中,高分子聚合物胶凝剂需进行充分溶胀,普通的溶胀需较长时间,效率较慢,采用高速剪切或高压处理可加快溶胀过程,但条件较为剧烈;通过流变学参数确定处理的强度和时间,可避免对结构产生负面影响。LARREA WACHTENDORFF 等考察了高压处理过程(high pressure process,HPP) 对淀粉基水凝胶流变学性质的影响,观察到颗粒小的淀粉比表面积较大,在高压作用下形成的水凝胶黏度较高,结构更稳定。随 HPP 压力和处理时间的增加,G' 值增大,淀粉溶液中分子间的相互作用增强,得到了更强的水凝胶结构[24]。2.2 贮存稳定性由于流变学性质对微观结构的变化较敏感,因此适用于预测和评价半固体制剂在不同贮存条件和时间下的稳定性。多相体系由于结构复杂,易受环境条件影响而产生稳定性问题,如 o/w 型乳剂由于黏度较低,贮存期间易出现分散相的沉降或凝聚从而影响产品质量。TADROS 对乳剂的沉降、絮凝和凝聚进行了流变学表征,观察到用蠕变和振荡测试来预测稳定性较有效,而由于乳剂黏度本身偏低,黏度测试对乳剂稳定性变化不够敏感[31]。KRISHNAIAH 等考察了阿昔洛韦乳膏的热流变特性,观察到 G' 在30℃以上时会发生不可逆变化,因此应避免贮存温度超过30℃ [32]。LAUTERBACH 等观察到乳剂的流变测试温度扫描参数与40℃下贮存12周后经激光衍射法测得的液滴粒径具有良好相关性,即温度扫描参数数值变化幅度越大,40℃贮存的乳剂粒径增加越大,因此温度扫描可以预测乳液体系的稳定性趋势[12]。2.3 使用性能与患者的顺应性对患者来说,皮肤外用半固体制剂的感官特性尤为重要。易于挤出和涂抹、具有柔滑的质感可提高患者的顺应性,尤其是用于治疗慢性皮肤病的产品。虽然挤出和涂抹过程对应制剂在剪切力作用下的形变能力,但将具体流变参数与使用感官直接联系起来仍具有一定挑战性。有学者尝试将流变学参数与感官评价相结合,如 BRUMMER 等测定了具有良好和较差皮肤触感的乳膏的流变学参数,用以确定与使用时肤感相关的流变学参数范围[33]。首先,一组试用者对乳膏产品肤感进行评分,再测定高分值产品的非稳态流动曲线上最大黏度和其对应的应力,得到数值范围;再通过测定低分值产品的相同参数验证该范围的合理性。该研究认为在剪切速率为500 s–1 下,良好肤感对应的黏度和应力范围分别是 1350~3500 Pa·s 和 8.5~15 Pa,可为同类乳膏的开发提供参考。BEKKER 等采用相同方法确定了凡士林的上述流变学参数范围,分别为 35000~80000 Pa·s 和 16~41 Pa[34]。ALI 等采用了流变学和其他方法( 如猪皮触觉摩擦法和皮肤水分测量) 来评价外用 o/w 型乳膏的使用性能[4]。结果显示,流变学测试可用于确定相关的使用性能,如涂抹性与屈服应力呈负相关,即具有高屈服应力值(129 Pa) 的膏体较难涂抹;湿润感与G'、G'' 和黏度呈负相关,即低黏度[ 剪切速率为 0.25 s–1 时黏度为 (288.7±50.3)Pa·s] 的柔软膏体会让受试者感觉更湿润。GEH 等制备了一种以羧甲纤维素钠(CMC) 为胶凝剂的免疫球蛋白可喷雾凝胶,要求凝胶使用时易于喷射,喷洒至皮肤后可恢复黏性状态。他们考察了不同浓度CMC 凝胶的触变性,结果显示当 CMC 浓度在 0.5%~ 1.5% 时触变环面积随浓度增加而增大,高于1.5%时触变环面积则变小且出现交叉,说明高于此浓度的凝胶由于黏度较大而触变性降低,不利于喷射,因此1.5%是较适宜的浓度[35]。2.4 体外透皮行为局部外用制剂应用于皮肤上时,经涂抹形成较薄的制剂层与皮肤紧密接触,此时制剂的微观结构通常不是药物向皮肤渗透的限速因素,因此制剂的流变学性质与其所载药物的透皮行为相关性不明显。但是对于需要在皮肤表面持续发挥作用的制剂,如烧伤治疗药物、局部麻醉类药物或局部镇痛类药物的外用半固体制剂,应用时剂量较大,流变学性质会对体外透皮行为产生影响。ILIĆ 等比较了使用不同乳化剂制备的醋氯芬酸乳膏的理化性质和体外透皮行为 (猪耳皮肤,给药量 0.5 g/cm2),观察到黏度较大的乳膏药物透皮释放量较低,原因可能是黏稠膏体阻滞了混悬于其中的药物与皮肤的接触[36]。WELIN-BERGER 等考察了一种局部麻醉药物不同剂型(o/w 型乳剂、由o/w 型乳剂和增稠剂卡波姆组成的乳膏和w/o 型乳剂) 的流变学性质、体外释放和透皮行为,以及在体疗效。结果显示,2种乳剂的黏度和屈服应力值接近;而乳膏的黏度和屈服应力值较高,体外释放和透皮速率较低,从而导致其疗效( 豚鼠针刺法) 较低,原因在于聚合物网络阻碍了乳剂液滴的运动,进而对制剂中药物的迁移产生不利影响[37]。3 总结与展望皮肤局部外用半固体制剂由于微观结构的复杂性,通常处方工艺开发难度较大,且半固体的物理形态易受外力和环境的影响,从而产生稳定性问题。理想的半固体制剂应当在贮存时保持稳定,使用时具有良好的涂抹性和黏附性。由于流变学性质与微观结构的关联性,流变学表征已被证明是有效的质量和稳定性评价手段,并且可作为预测患者顺应性的手段,在皮肤局部外用半固体制剂开发中具有较高的应用价值。此外,流变学测试还可与其他评价方法( 如体外释放、体外透皮等) 相关联,从而构建整体质量评价体系,以用于皮肤局部给药系统的设计与开发。
企业动态
2024.03.26

外用药物制剂体外释放试验技术要求概况
作者:殷连珍1,李慧敏2*,苏梅1,肖群1,潘宪伟1*(1. 江苏柯菲平医药股份有限公司,南京 210016;2. 南京正大天晴制药有限公司,南京210046)* 代表通讯作者期刊:中南药学;首发日期:2023年11月03日 摘要:外用药物制剂具有复杂的药物递送途径,通常局部起效,辅料成分复杂,处方组成和制备工艺参数的微小差异就有可能导致产品不同的质量特性,从而影响药物的安全性和有效性。体外释放试验(in vitro release test,IVRT)可用于表征一个产品批次的稳态药物释放速率,表征某些工艺、配方和/或生产的变更对药品的影响,在某些情况下,可用于论证产品批量放大或上市后变更的等效性。尽管IVRT不能模拟体内性能情况,但其仍是一项关键质量属性,应订入产品放行和货架期质量标准。本文参考国内外相关技术指导原则及文献,对皮肤外用制剂的IVRT研究技术要求概况进行综述,期望为业内人士提供参考。关键词:外用药物制剂;经皮给药;体外释放试验(IVRT);一致性评价 外用药物制剂相较于口服、注射剂等给药方式具有许多优势:①避免肝脏的首过效应;②药物吸收不受胃肠道内pH 值、食物及其他药物等因素的影响;③具有控释效果;④维持恒定的有效血药浓度,避免因吸收过快产生血药浓度过高引起不良反应;⑤给药方便,安全性高,患者顺应性好[1]。已成为现代医学中重要的组成部分,同时具有较好的发展前景。 外用药物制剂是一类发挥局部或全身治疗作用的制剂,剂型包括软膏剂、乳膏剂、凝胶剂、散剂、乳剂、糊剂、混悬剂、喷雾剂、气雾剂、溶液和其他半固体和/或液体剂型[2-3]。其中,乳膏剂、软膏剂及凝胶剂处方组成复杂,多为半固体制剂,涉及油相、水相及油包水/水包油等热力学不稳定体系,使用多种不同性质的辅料(油相、水相、表面活性剂、矫味剂、清凉剂、抑菌剂等)。相对于传统的注射剂和片剂等剂型,其制备和稳定性过程中的动力学和热力学影响因素众多,在设计、制造、贮存和运输等多个环节均存在较大的挑战。 对于该类制剂,药物在体外释放的速率和程度是其性能的综合体现,主要用于产品的药学质量控制,也可用于药品开发过程中处方工艺的筛选研究。虽然不能根据IVRT释放速率的差异预测体内生物利用度的变化[4],但其具有潜在反应制剂中药物生物学特性变化的能力,而这种特性的变化可能与以下因素有关,如处方中的活性或非活性组分、制剂的物理化学特性、生产工艺的变更、运输和储存条件、产品的放置时间长短和其它对终产品质量特性具有关键作用的因素等[5]。 基于制剂的定性(Q1)、定量(Q2)以及物理化学和结构特性(Q3),SHAH VP等[6] 2015年提出了外用药物分类系统(topical drug classification system,TCS),该系统将皮肤外用制剂分为4类。如果仿制药和参比制剂(RLD)之间的Q1、Q2和Q3都相同,则仿制药可能实现生物等效性豁免(TCS 1类)。在TCS理念中,某些辅料可能在药物传递、发挥作用和随之产生的疗效中起到积极作用。但总体来看,许多辅料是惰性的。如果仿制药和RLD之间Q1、Q2不同,但在充分评估辅料的安全性和功效后,可进行IVRT试验,若仿制药和RLD一致,则可以实现生物等效性豁免(TCS 3类)。 欧洲药品管理局(EMA)允许通过证明Q3及IVRT的等效性,用于支持单相的溶液剂、凝胶剂、软膏剂等简单的配方制剂与参比制剂相比的治疗等效性,以代替等效性临床试验[7]。美国食品药品监督管理局(FDA)要求,对于外用半固体制剂,如果仿制药的Q3与对照标准制剂相同,仅需提供有限的其他证据(体外、in silico和/或体内),即可支持生物等效性论证[8],如根据2022年10月之后FDA更新的特定药品指南,在Q1、Q2和Q3一致的基础上,阿达帕林凝胶[9]、乳酸铵乳膏[10]、盐酸布替萘芬乳膏[11]、磷酸克林霉素凝胶[12]和酒石酸溴莫尼定凝胶[13]等仅需要论证IVRT等效,即可豁免临床等效性研究。日本医药食品局(PMDA)可通过IVRT试验对已上市半固体制剂和贴剂的B水平处方变更进行论证[14-15]。 综上可知,IVRT研究在外用药物制剂处方开发、生物等效性论证以及上市后变更中的重要性,因此对IVRT试验进行合理设计,确保其结果准确可靠十分重要。虽然FDA、EMA和PMDA均有发布关于IVRT研究的相关指南,但目前国内暂无统一的技术要求规范,且各国技术要求亦不统一,因此本文对IVRT研究相关技术要求概况进行总结,期望为业内人士提供参考。 1 国内外政策法规 在2018年国家药品监督管理局药品审评中心(CDE)发布的《新注册分类的皮肤外用仿制药的技术评价要求(征求意见稿)》中阐述可参照FDA SUPAC-SS指南[16]进行IVRT研究[17],但在2021年发布的《皮肤外用化学仿制药研究技术指导原则(试行)》中,建议参考FDA、EMA和PMDA相关指导原则开展相关研究工作。 在1997年FDA发布SUPAC-SS指南中首次对 IVRT 研究的相关参数及数据统计进行了阐述,包括上样量(300mg)、上样方式(闭环)、接收介质(水性缓冲液或水醇混合溶液)、合成膜、试验时长和取样点(至少5个取样点)等。2016年发布的《阿昔洛韦指南草案》[18]是第一份关于IVRT方法验证及接受标准的指南,但其部分参数(如线性与范围、专属性)的接受标准相对2022年10月FDA发布IVRT指南[3]较为宽松,目前已被FDA IVRT指南替代。2022年5月美国药典(USP)发布的《PF 48(3) Semisolid drug products-performance tests》(以下简称USP)是现有国内外药典或指南中对IVRT的研究设计最为全面的法规指南[4],而FDA IVRT指南是专门阐述IVRT方法验证的相关指南。 2010年PMDA发布的《皮肤局部适用制剂(半固体制剂和贴剂)处方变更生物等效性研究指南》中对IVRT的研究设计和数据统计进行了阐述,但未说明IVRT各参数的可接受标准,亦未对方法验证内容进行说明[14]。 2018年EMA发布的《外用制剂质量和等效性指南草案》中亦对IVRT研究设计的相关参数进行了阐述,但未明确膜惰性的可接受标准、接收介质的组成,且在方法验证模块仅要求对区分力、精密度和耐用性进行评估[7]。USP、FDA、EMA和PMDA对IVRT研究设计的技术要点如表1所示。 表1 USP、FDA、EMA及PMDA对外用半固体制剂IVRT试验的技术要求考察项目USP和FDAEMAPMDA设备立式扩散池、浸没池扩散池桨碟法,扩散池法膜合成膜(如混合纤维素酯膜、尼龙膜、聚丙烯膜、聚醚砜膜),不允许采用生物膜或模拟生物膜开发的合成膜;惰性(回收率95%~105%)不应限制活性成分释放,不与活性成分结合非匀速渗透分析方法高效液相色谱仪,采用多点校正曲线法(6~8个)//上样量及上样方式伪无限剂量(通常大于300mg,剂量消耗低于30%)、闭环上样伪无限剂量,确保上样量在目标上样的±5%范围内根据释放和渗透的药物量变化率确定接收介质水和醇或有机溶剂混合液,要求药物在接收介质中的溶解度应超过IVRT研究中最高样品浓度,理想情况下是一个数量级;同时需证明,采用150%规格的配方制剂,也可达到稳态药物释放接收介质中活性物质的最大浓度不应超过在该接收介质中活性物质最大溶解度的30%;应尽量减少接收介质的反向扩散,以避免药物产品的转化。在整个释放测试期间,应使接收介质的 pH值保持恒定pH值5~7的缓冲液(离子强度约为0.05);如果24 h的平均释放量小于20%,可以改变离子强度,如添加表面活性剂、改变pH。在不影响制剂形变的情况下,可以采用水和醇或有机溶剂混合液试验时长和取样试验时长通常为4~6小时,每小时至少取样一次(至少5个取样点);实际采样时间应在预定采样时间的±15 min 或 ±2%,取较窄的时间误差范围试验时长应足以表征释放曲线,理想情况下,释放量至少为活性物质的70%。在药物释放曲线的线性部分至少选取6个时间点,包括扩散达到稳态后的第一个取样时间点试验时长应确保标准制剂的释放曲线达到平台期或释放量超过70%,但最长应不超过24小时;从试验结束时间点回溯4个取样点,即包括最终取样时间点在内的至少5个取样点,取样时间点的个数应足以表征释放曲线重复次数 第一阶段6个剂量单位;第二阶段12个剂量单位在首次进行方法验证或等效性评估时,最少需要12个剂量单位。放行检测时每个批次至少6个剂量单位至少12个剂量单位膜温度32 ℃±1 ℃(内用制剂为37 ℃±1 ℃,如直肠和阴道用品)32 ℃±1 ℃32 ℃±0.5 ℃ 2 研究设计 2.1 设备 在IVRT研究中,可用的设备包括立式扩散池(VDC)、浸没池、USP装置4(流通池)、USP装置5(桨碟法)和桨式提取法[19]。其中,USP收载3种型号的VDC和2种型号的浸没池,PMDA可以采用VDC和桨碟法进行试验,EMA则没有明确说明。 采用桨碟法时,不需要使用膜,因此需评估配方制剂溶解于接收介质的可能性[20]。桨式提取法是将欧洲药典(Ph.Eur)通则2.9.4中的提取池[21]与USP装置2组合使用(见图1)。虽然,也可以采用浸没池和流通池,目前国内外最常用的设备仍是VDC。 对于VDC,应根据实验室的风险承受能力和生产商的建议,在6~12个月的周期内对其进行再确认,确认内容包括但不限于:(1)确认供给室和接收室的孔口直径在规定值的±5%;(2)将所有组件组装后测定接收室的体积(精确到0.01 mL),在实际计算过程中,采用测定的体积替代标称体积;(3)温度达到平衡后,确认膜表面温度的稳定性(在目标温度的±1 ℃范围内);(4)搅拌速率(通常在每分钟400~600转之间)。 在研究期间,应监控和报告实验室环境的温度和湿度。如适用,建议将温度控制在21 ℃±2 ℃、湿度控制在50% RH±20% RH之间,避免将设备置于空调的通风口之下及暴露在直射阳光中。工作台要保持水平,建议倾斜度≤1°[22]。 图1 (A)提取池,(B)将提取池组装到USP装置2中 2.2 合成膜 除桨碟法和桨式提取法外,其它类型的扩散池都需要采用一种膜来防止接收介质的搅拌对样品的机械扰动。在IVRT研究中,应选择具有适当的惰性和商业化的人工膜(如聚丙烯膜、聚醚砜膜、醋酸纤维素/尼龙混合酯膜、聚四氟乙烯膜、尼龙膜、聚砜膜等),但不宜采用生物膜或模拟生物膜开发的合成膜,因为这些膜可能会不恰当的影响药物释放的表观速率[4, 16]。 EMA要求膜不应限制活性成分释放,PMDA要求呈非匀速渗透。通常情况下,商业化的合成膜有不同的厚度、孔径和物理性质,在IVRT研究中常用的膜厚度为70 μm、孔径为0.45 μm。对于脂溶性药物和亲脂性基质,可采用较大孔径的膜(如1.2 μm),此外可能还需要将膜预先在适当的溶剂(如肉豆蔻酸异丙酯)中浸泡,以赋予膜亲脂性,提高药物从亲脂性基质中扩散的速率。 纤维素膜在制造时通常会添加增塑剂(如甘油),以保持膜的柔软性,使用前需去除增塑剂,如可在接受介质中浸泡30 min[23]。在测试亲脂性基质的软膏时,使用预先浸泡的膜可以确保恒定的体外测试条件。此外,接收介质和配方制剂不应影响膜的物理完整性,Realdon N等[24]使用不同类型的软膏,在进行IVRT研究期间和之后采用“气泡点(bubble point)”测试来评估膜的完整性,这个测试也可以帮助决定是否需要对膜进行浸泡处理。 膜的非匀速渗透,可通过采用不同规格制剂(如50%、100%和150%规格)可获得不同的释放速率进行评估。对于FDA和EMA要求的惰性(不与活性成分结合)评估方法为(1)在IVRT的整个研究期间,将膜放置到相应温度的接收液中进行孵化(例如,采用接收介质配制成待分析物的预期最高和最低浓度水平,在32℃±1 ℃条件下放置6 h),至少重复测定3份;(2)并在相同条件下,同步考察3份不加膜的对照溶液,评估与膜吸附无关的药物损失;(3)如果实验结束时,接收液中药物的回收率在100%±5%,可认为在该条件下膜是惰性的。 综上所述,在合成膜的选择过程中,应选择具有适当惰性和商业化的人工膜,不宜采用生物膜或模拟生物膜开发的合成膜。在方法开发过程中,应确定合成膜的种类(包括孔径大小、厚度、材质)、是否需要进行浸泡处理(如需浸泡处理,需确定浸泡介质和浸泡时间),同时需对膜的惰性和非匀速渗透情况进行评估。此外,部分膜有正反面,在试验过程中,应根据厂家说明书进行使用。 2.3 样品分析方法 通常,IVRT样品分析方法可基于成品的含量测定方法建立,但应确保所用的分析方法具有适当的灵敏度,以便可以定量测定从半固体制剂释放到接收介质中低浓度水平的药物量。建议将接收液的选择作为样品分析方法开发的初始步骤,其优点是,在使用接收液对不同膜评估前,可先采用选定的接收液对样品分析方法进行优化。此外应对方法的专属性进行评估,以确认从制剂和/或膜中浸出的其他物质不干扰药物的定量测定;同时,应对样品在最高相关温度的接收液中放置的最长时间进行评估。根据FDA IVRT指南和USP,应采用多点校正曲线法(6~8个)进行测定。 2.4 IVRT方法开发 2.4.1 上样量及上样方式 FDA SUPAC-SS指南[16]建议上样量为300 mg,在上样过程中为防止样品挥发,供给室应保持封闭状态。USP要求拟定的上样量应确保剂量损耗不会影响试验期间的稳态药物释放动力学,正如FDA IVRT指南所述,虽然一些外用制剂在剂量消耗大于30%时也能持续观察到稳态释放动力学,但通常而言,稳态释放动力学应假定剂量消耗低于30%。EMA为伪无限上样,但应确保实际上样量在拟定上样量的±5%范围内。PMDA要求根据释放和渗透的药物量确定。 因此,通常建议上样量在300 mg以上,以避免过量的剂量消耗,或因上样量过低导致上样难度的增加(如需将上样量控制在±5%范围内)以及对检测方法的重复性、灵敏度造成挑战。此外,EMA要求释放量至少为标示量的70%,PMDA要求至少为70%或达到平台期,因此上样量亦不能过高,以免整个试验时间过长。 除上述要求外,在上样过程中应尽量减少和控制剪切应力,采用相同的上样方式(如直接从管中挤出到膜上、用抹刀转移后涂覆到膜上、用外置活塞式移液器或一次性采集器转移后分配)将药物均匀地涂覆到膜上,制成紧凑的圆柱形,且在上样过程中应避免膜与样品之间产生气泡。 综上所述,在IVRT方法开发过程中,应根据样品特性的不同对不同的上样方式进行评估,确保上样的均匀性和一致性,并根据稳定药物释放动力学、释放量、试验时长以及方法的灵敏度和重复性等要求综合考虑确定样品的上样量。 2.4.2 接收介质 Proniuk等[25]分别采用pH7.4磷酸盐缓冲液(pH7.4 PBS)和肉豆蔻酸异丙酯为接收介质,用肉豆蔻酸异丙酯对膜进行浸泡,以及采用pH7.4 PBS为接收介质并用pH7.4 PBS对膜进行浸泡的三种方法,对IVRT从两种密切相关的水凝胶中选择酮洛芬释放速率最快的外用半固体制剂的能力进行评估。结果表明,在产品开发早期进行处方筛选时,只有当接收介质具有与人体皮肤相似的特性时,IVRT才有助于预测可供吸收的药物量,说明接收介质选择的重要性。 pH是接收介质选择时需要考虑的一个重要因素,其选择应基于配方制剂的pH、原料药在不同pH条件下的溶解度以及药物作用部位的pH,通常情况下,皮肤的pH范围在5~6之间[20, 26]。 虽然希望接收介质具有与皮肤相似的生理状态,但也必须确保其能够恰当的测量药物的释放,而选择的最重要因素是药物在其中的溶解度,即在半固体制剂的药物释放过程中,接收介质应可以提供合适的漏槽条件。 EMA指南[7]中未明确说明接受介质的组成,但要求在整个试验期间,接收介质中活性物质的最大浓度不应超过在该接收介质中活性物质最大溶解度的30%。同时要求,应尽量减少接收介质的反向扩散,以避免药物产品的转化,且在整个释放测试期间,应使接收介质的 pH值保持恒定。 PMDA要求采用pH值在5~7的缓冲液(离子强度约为0.05)作为接收介质,但当采用标准制剂进行试验,如果24 h的平均释放量小于20%,则可改变离子强度,如添加表面活性剂、改变pH。在不影响制剂形变的情况下,可以采用水醇或有机溶剂混合液。 FDA IVRT指南和USP指出接收介质通常为水和醇或有机溶剂混合液,要求药物在接收介质中的溶解度应超过IVRT研究中最高样品浓度,理想情况下是一个数量级,同时需要证明在研究期间可达到稳态释放速率。 根据上述分析,在接收介质选择中,可从以下几个方面进行考虑:(1)通常可采用水醇或有机溶剂混合液,但采用该类型的接收介质时,应尽量减少接收介质的反向扩散,以避免药物产品发生形变,如KANFER I等[27]研究表明乙醇可通过聚合物膜反向渗透导致乳膏制剂的完整性发生改变;(2)对于水性缓冲液(pH值在5~7之间,离子强度约为0.05),在整个释放测试期间,应使接收介质的pH值保持恒定,如释放量较低,可添加表面活性剂、改变pH;(3)符合漏槽条件;(4)采用标称规格150%的配方制剂,也可达到稳态释放速率;(5)在整个试验期间,药物在接收介质中稳定;(6)如适用,需对接收介质进行脱气处理,避免试验期间产生气泡。 2.4.3 设备平衡 在IVRT试验开始之前,设备应在预定的试验条件下进行适当的平衡。通常情况下需对接收介质进行脱气处理,以避免在试验前或试验过程中膜下产生气泡。系统温度应保持恒定并进行良好的控制。根据不同的试验方法,IVRT膜可以在扩散池组装之前预先用接收介质浸泡,也可以上样前在扩散池内与接收介质原位平衡(如,在膜温度为32℃下保持30 min)。 2.4.4 试验时长和取样 根据USP,试验时长应适合于表征药物的稳态药物释放动力学,这通常是基于4~6 h的持续稳定释放。在第一个小时获得的数据通常不能代表稳态释放动力学,对于很多IVRT方法,稳态释放动力学是根据1小时之后的时间点计算的(例如,在1、2、3、4和5 h,或在2、3、4、5和6 h)。至少选取5个取样时间点,以获得表征良好的释放率。在稳态释放速率评估中,取样时长至少为4h,较短的取样时长(例如2 h)可能不能有效表征稳态释放动力学。此外,应精确控制样品的取样时间,将其控制在预定取样时间的±15 min或±2%,取较窄的时间间隔范围。FDA SUPAC-SS指南要求至少进行5次取样。在取样后,需要采用预热至系统预定温度的接受介质进行补液,同时需确保,在每次取样和补液后,膜下均无气泡产生。 EMA要求试验时长应足以表征释放曲线,理想情况下,释放量至少为活性物质的70%。在药物释放曲线的线性部分至少选取6个时间点(每小时至少取样一次),包括药物扩散达到稳态后的第一个取样时间点。PMDA要求试验时长应确保释放曲线达到平台期或释放量超过70%,但最长不应超过24 h。 如上所述,在试验时长和取样时间点方面,各国要求存在差异,其中FDA仅要求对稳态释放动力学(即释放曲线的线性部分)进行评估,EMA除需要对稳态释放动力学进行评估外,还要求在试验结束时释放量至少为70%,PMDA与EMA要求相似,但相对较为宽松(如释放曲线达到平台期或释放量超过70%均可作为释放终点)。 因此,在IVRT研究过程中,对于试验时长和取样,通常应满足以下要求:(1)在稳态药物释放阶段至少选取5个取样点(如申报欧盟,至少选取6个时间点),且取样时长不应少于4 h;(2)将样品的取样时间控制在预定取样时间的±15 min或±2%,取较窄的时间间隔范围;(3)如适用,释放曲线应达到平台期或释放量超过70%,但最长时间不应超过24 h;(4)在取样后,需采用预热至系统预定温度的接受介质进行补液,同时需确保在每次取样和补液后,膜下均无气泡产生。 2.4.5 重复次数 根据USP和FDA SUPAC-SS指南,通常采用6个剂量单位可足以表征该产品的性能(释放速率),但在数据等效性评估中,如不能满足要求,应额外增加12个剂量单位进行评估。EMA要求在首次进行方法验证或等效性评估时,每个批次的样品最少需要12个重复,而在日常放行检测中,每个批次采用6个剂量单位即可。PMDA主要用于上市后处方变更的生物等效性评估,要求每个批次至少采用12个剂量单位。 2.4.6 膜温度 对于皮肤外用制剂,FDA和EMA均要求控制在32 ℃±1 ℃,而PMDA要求将温度控制在32 ℃±0.5 ℃。对于直肠和阴道等内用产品,温度应控制37 ℃±1 ℃。如适用,实验前用非接触式红外温度计对温度进行测定。 2.5 数据统计分析 对于每个扩散池,应测定每个取样点(t1,t2 , etc)的药物释放量(通常以µg×cm-2为单位),并以累积释放量对作图,在稳态药物释放阶段,二者应呈线性关系(r2≥0.97),其斜率为药物释放速率。在IVRT研究中,各国均要求对药物释放速率的一致性进行评估。在理想情况下,EMA和PMDA还要求药物的累计释放量大于70%。下文对FDA、EMA和PMDA指南中推荐的一致性评价方法进行介绍。 2.5.1 FDA 由于普遍存在的测定误差,如气泡和膜缺陷,测定结果常呈非正态分布,推荐采用Mann-Whitney U检验计算受试制剂(T)与参比制剂(S)之间斜率比值的90%置信区间。通过以下示例说明,采用6个剂量单位,分别以RS和TS代表参比制剂和受试批次的斜率,计算所有T/R斜率的比值,如下表2所示。 表2 受试制剂斜率(TS)和参比制剂斜率(RS)比值RS1RS2RS3RS4RS5RS6TS1TS1/RS1TS1/RS2TS1/RS3TS1/RS4TS1/RS5TS1/RS6TS2TS2/RS1TS2/RS2TS2/RS3TS2/RS4TS2/RS5TS2/RS6TS3TS3/RS1TS3/RS2TS3/RS3TS3/RS4TS3/RS5TS3/RS6TS4TS4/RS1TS4/RS2TS4/RS3TS4/RS4TS4/RS5TS4/RS6TS5TS5/RS1TS5/RS2TS5/RS3TS5/RS4TS5/RS5TS5/RS6TS6TS6/RS1TS6/RS2TS6/RS3TS6/RS4TS6/RS5TS6/RS6 在计算出T/R斜率比值后,将36个T/R斜率比值从低至高排序。然后,将第18个和第29个斜率比值转换为百分单位,分别代表90%置信区间的下限和上限,如果二者均在75%~133.33%范围内,则药物释放速率是等效的。如果置信区间在75%~133.33%之外,R和T再各测12个剂量单位,与第1 次实验结果组合,2组各有18个释放速率,按上述进行置信区间计算,如果第110个和第215个斜率比值均在75%~133.33%范围内,可判定药物释放速率是等效的。 2.5.2 EMA 与FDA的统计方法相似,采用12个剂量单位,计算R和T各释放速率的相互比值TS/RS(共计144个),然后对这些TS/RS值进行对数变换,使用下式计算置信区间:其中,X是计算所得的所有TS/RS对数的平均值,n表示样本量,t是n-1个自由度(t)的90%置信区间的t值(n=6,t=2.015;n=12,t=1.796),s表示所有对数转换比率的标准偏差。所得结果取反对数,即为置信区间。如果R和T释放速率几何均值比的90%置信区间在90%~111%范围内,则药物释放速率是等效的。 2.5.3 PMDA 在规定的试验时间或释放曲线达到平台期后的1个时间点,以及和该时间点对应的释放速率的一半所对应的时间点,受试制剂与标准制剂的平均释放速率之比应在0.8~1.2之间。此外,PMDA要求受试制剂释放速率的偏差与标准制剂相比,应相同(可采用F检验进行判断)或更小。 3 结语 本文汇总了FDA、EMA和PMDA等国家关于IVRT研究的政策法规,目前国内陆续发布外用药物制剂相关研究技术指南,但尚未有统一的技术规范对IVRT的研究进行规定。外用药物制剂本身具有工艺复杂性,在IVRT研究中亦有很多技术难点,如合成膜的选择及处理、接收介质的筛选、上样方式的控制等。伴随着我国对外用制剂研究的越加深入,希望结合上述对IVRT研究技术参数的讨论,重点参照2022年USP和FDA IVRT指南,结合我国实际需要,尽早统一IVRT研究技术要求。
企业动态
2024.03.26

IVRT没有区分力怎么办
IVRT没有区分力怎么办一般情况下,在 IVRT 区分力研究中,难点在100%规格和150%规格的区分力研究,能解决这两个规格的区分力问题,通常50%和100%规格均能满足要求。然而,虽然上样量和合成膜的选择比较重要,但二者对区分力的影响远没有接受介质的选择重要。 一、上样量 通常建议上样量在0.3~2g 之间,增加上样量有助于维持试验结果良好的重复性,降低上样难度。对于聚乙二醇基质的软膏剂,一般情况下,药物的释放速率较大,导致药物的剂量损耗较大,呈现100%和150%规格释放均较快现象。此时,可尝试增加上样,降低剂量损耗(稳定药物释放通常假定剂量损耗≤30%)。 对于凡士林基质的软膏剂或难溶性药物,与聚乙二醇基质相反,其释放速率和累积释放量通常情况下较低。此时,可尝试降低样品的上样量,以增加剂量损耗,但应确保上样量的降低不会影响各扩散池之间的重复性或精密度。 二、合成膜 合成膜最大的作用在于防止接收介质的搅拌对样品的机械扰动,起到隔离供给室和接收室的作用。在IVRT研究中,应选择具有适当的惰性和商业化的人工膜(如聚丙烯膜、聚醚砜膜、醋酸纤维素/尼龙混合酯膜、聚四氟乙烯膜、尼龙膜、聚砜膜等)。膜不应限制活性成分释放,应呈非匀速渗透。 通常情况下,商业化的合成膜有不同的厚度、孔径和物理性质,在IVRT研究中常用的膜厚度为70 μm、孔径为0.45 μm。对于脂溶性药物和亲脂性基质,可采用较大孔径的膜(如1.2 μm),此外可能还需要将膜预先在适当的溶剂(如肉豆蔻酸异丙酯)中浸泡,以赋予膜亲脂性,提高药物从亲脂性基质中扩散的速率。如释放速率过快,可采用孔径为0.2 μm的合成膜。 但是,通常情况下,仅通过调整膜的孔径等,对分析方法的区分力影响较小。 三、接收介质 毋庸置信,在IVRT方法开发过程中,接收介质的选择尤为重要,其中对样品的稳定性、方法的区分力选择,都有最要影响,可优先从该方面进行考虑。 pH是接收介质选择时需要考虑的一个重要因素,其选择应基于配方制剂的pH、原料药在不同pH条件下的溶解度以及药物作用部位的pH,通常情况下,皮肤的pH范围在5~6之间。 EMA指南中未明确说明接受介质的组成,但要求在整个试验期间,接收介质中活性物质的最大浓度不应超过在该接收介质中活性物质最大溶解度的30%。同时要求,应尽量减少接收介质的反向扩散,以避免药物产品的转化,且在整个释放测试期间,应使接收介质的 pH值保持恒定。 PMDA要求采用pH值在5~7的缓冲液(离子强度约为0.05)作为接收介质,但当采用标准制剂进行试验,如果24 h的平均释放量小于20%,则可改变离子强度,如添加表面活性剂、改变pH。在不影响制剂形变的情况下,可以采用水醇或有机溶剂混合液。 FDA IVRT指南和USP指出接收介质通常为水和醇或有机溶剂混合液,要求药物在接收介质中的溶解度应超过IVRT研究中最高样品浓度,理想情况下是一个数量级,同时需要证明在研究期间可达到稳态释放速率。 根据上述分析,在接收介质选择中,可从以下几个方面进行考虑:(1)通常可采用水醇或有机溶剂混合液,但采用该类型的接收介质时,应尽量减少接收介质的反向扩散,以避免药物产品发生形变;(2)对于水性缓冲液(pH值在5~7之间,离子强度约为0.05),在整个释放测试期间,应使接收介质的pH值保持恒定,如释放量较低,可添加表面活性剂、改变pH;(3)符合漏槽条件;(4)采用标称规格150%的配方制剂,也可达到稳态释放速率;(5)在整个试验期间,药物在接收介质中稳定;(6)如适用,需对接收介质进行脱气处理,避免试验期间产生气泡。 在满足上述条件的情况,其中对区分力影响最大的是接收介质的种类,如在方法开发过程中,发现区分力较差,可优先对不同的接收介质进行筛选,分别采用:PBS、有机溶剂-水混合液(包括甲醇、乙醇、乙腈、异丙醇等);如仍无区分力,对于溶解性较大的API,可采用一些特殊溶剂,如四氢呋喃、二甲基亚砜等;当然,有机溶剂的比例,有时对区分力也有较大的影响
企业动态
2024.03.26

Correlation of Physical and Structural利多卡因/丙胺卡因外用药
Abstract摘要Background and Purpose:In the past few years a collective weight of evidence approach has been recommended to demonstrate bioequivalence (BE) for several topical dermatological drug products.An essential component of this approach is a comprehensive characterization of the physical and structural (Q3) properties of complex topical semisolid dosage forms. The purpose of this study is to determine if comparative Q3 characterization of topical lidocaine and prilocaine products maybe used to predict the comparative product performance, which was evaluated by comparing cutaneous pharmacokinetics (PK) of lidocaine and prilocaine in vitro and in vivo.背景和目的:在过去的几年中,已经推荐了一种集体证据权重法来证明几种局部皮肤病药物的生物等效性。该方法的一个重要组成部分是对复杂局部半固体剂型的物理和结构(Q3)特性的全面表征。本研究的目的是通过比较利多卡因/丙胺卡因在体外和体内的皮肤药代动力学(PK),确定是否可以利用外用利多卡因和丙胺卡因产品的比较Q3特性来预测产品的比较性能。Methodology: The products evaluated in this study were 1) the reference product, EMLA® (lidocaine; prilocaine) cream, 2.5%;2.5% 2) a generic version of EMLA® cream, and 3) Oraqix® (lidocaine; prilocaine) gel, 2.5%;2.5% as a different formulation with the same strength of lidocaine and prilocaine. The comparative Q3 properties of these three drug products were assessed. The cutaneous PK of lidocaine and prilocaine from the gel and cream products were compared by an in vitro permeation test (IVPT). The BE of the generic cream and of Oraqix® gel to EMLA® cream was evaluated based upon cutaneous PK endpoints for both lidocaine and prilocaine. The dermal bioavailability of EMLA® and Oraqix® was also compared in an in vivo pilot study using dermal open flow microperfusion (dOFM) in 6 healthy subjects.方法学:本研究评价的产品为:1)参比产品EMLA®乳膏(2.5%利多卡因/2.5%丙胺卡因),2)EMLA®乳膏的仿制药,3)Oraqix®凝胶(2.5%利多卡因/2.5%丙胺卡因)。比较了这三种药品的Q3性质。通过体外渗透试验(IVPT)比较利多卡因/丙胺卡因凝胶和乳膏产品的皮肤PK。基于利多卡因/丙胺卡因的皮肤PK终点,评估了Oraqix®凝胶与EMLA®乳膏的生物等效性BE。EMLA®和Oraqix®的皮肤生物利用度也在6名健康受试者的皮肤开放流微灌注(dOFM)体内试点研究中进行了比较Results: The Q3 properties of the reference and generic lidocaine/prilocaine topical creams were similar to each other, while Oraqix® gel had a lower pH, a higher evaporative rate, a lower yield stress, and an absence of globules compared to the cream products. The results of IVPT study demonstrated that the cutaneous PK of lidocaine and prilocaine was comparable between the reference and generic creams. By contrast, the maximum flux (Jmax) and area under the curve (AUC) of both lidocaine and prilocaine were lower for Oraqix® gel compared to EMLA® cream and the gel and cream products were not found to be bioequivalent. The results of an in vivo cutaneous PK study using dOFM in healthy subjects were in agreement with the in vitro (IVPT) results.结果:参比型和仿制型利多卡因/丙胺卡因外用乳膏的Q3性质相似,而Oraqix®凝胶与乳膏产品相比具有较低的pH值、较高的蒸发速率、较低的屈服应力,与凝胶产品相比乳膏没有球状物的特点。IVPT研究结果表明,利多卡因/丙胺卡因的皮肤PK在参比乳膏和仿制乳膏之间具有相关性。相比之下,Oraqix®凝胶与EMLA®乳膏相比,利多卡因/丙胺卡因的最大通量(Jmax)和曲线下面积(AUC)都较低,凝胶和乳膏产品没有生物等效性。使用dOFM对健康受试者进行的体内皮肤PK研究结果与体外(IVPT)结果一致。Conclusion:These results demonstrate the correlation between the Q3 similarity or difference of three lidocaine prilocaine topical products used for comparison and the similarity or difference in their product performance (cutaneous PK), both in vitro (IVPT) and in vivo (dOFM). The similarity of Q3 characteristics between the reference and generic creams accurately correlated with and was predictive of comparable bioavailability (and bioequivalence) for both lidocaine and prilocaine, whereas the difference in Q3 characteristics between the reference cream and the gel accurately correlated with and was predictive of differences in bioavailability.结论:本研究结果表明,三种利多卡因/丙胺卡因外用产品的Q3相似性或差异性与其产品在体外(IVPT)和体内(dOFM)性能的相似性或差异性之间存在相关性。参比制剂乳膏和仿制药乳膏Q3特征的相似性与利多卡因丙胺卡因的可比生物利用度(和生物等效性)准确相关并可预测,而参比制剂乳膏和凝胶之间Q3特征的差异与生物利用度的差异准确相关并可预测。Introduction引言In the past few years, a collective weight of evidence approach has been recommended to support a demonstration of bioequivalence (BE) for several topical drug products. An essential component of this approach is a comprehensive characterization of the physico-structural (Q3) properties of complex topical semisolid dosage forms.在过去的几年中,已经推荐了一种集体证据权重方法来支持几种外用药物产品的生物等效性(BE)论证。该方法的一个重要组成部分是对复杂局部半固体剂型的物理结构(Q3)特性的全面表征。The purpose of this study was to determine if comparative Q3 characterization of topical lidocaine and prilocaine products maybe used to predict the comparative product performance, which was evaluated by comparing the cutaneous pharmacokinetics (PK) of lidocaine and prilocaine in vitro and in vivo. The specific objectives of this study include:本研究的目的是通过比较利多卡因和丙胺卡因在体外和体内的皮肤药代动力学(PK),确定是否可以用外用利多卡因/丙胺卡因产品的比较Q3特性来预测产品的比较性能。本研究的具体目标包括:•Characterize and compare the Q3 properties of cream and gel products, each containing both lidocaine and prilocaine•表征并比较含有利多卡因/丙胺卡因的乳膏和凝胶产品的Q3特性•Compare the performance of lidocaine/prilocaine cream and gel products using in vitro and in vivo cutaneous PK studies•通过体外和体内皮肤PK研究比较利多卡因/丙胺卡因乳膏和凝胶产品的性能Materials and Methods材料与方法The products evaluate in this study were 1) the reference product, EMLA® (lidocaine; prilocaine) topical cream, 2.5%;2.5% 2) a generic version of EMLA® cream, and 3) Oraqix® (lidocaine; prilocaine) dental gel, 2.5%;2.5% as a different formulation with the same strength of lidocaine and prilocaine. The comparative Q3 assessment of these three drug products included microscopic examination, pH, evaporative rate, and rheological behavior. The cutaneous PK of lidocaine and prilocaine from the gel and cream products were compared by an in vitro permeation test (IVPT) with a replicate study design (six skin donors with six replicates per donor) using heat separated human epidermis and a flow through diffusion system. The BE of the generic cream and of Oraqix® gel to EMLA® cream was evaluated based upon cutaneous PK endpoints for both lidocaine and prilocaine, using a reference scaled average BE (SABE) analysis and evaluation of the 90% confidence interval (CI). The dermal bioavailability of EMLA® and Oraqix® was also compared in an in vivo pilot study using dermal open flow microperfusion (dOFM) in 6 healthy subjects. The dose of all products used in the IVPT and dOFM studies was 10 mg product/cm2 .本研究评价的产品有:1)参比产品EMLA®乳膏(利多卡因/丙胺卡因);2) EMLA®乳膏的仿制药,以及3)Oraqix®凝胶(2.5%利多卡因/2.5%丙胺卡因)。这三种药品的Q3比较评价包括显微镜检查、PH值、蒸发速率和流变行为。采用体外渗透试验(IVPT),采用重复研究设计(6个皮肤供体,每个供体6个重复),利用热分离的人体表皮和流动扩散系统,比较利多卡因和丙胺卡因凝胶和乳膏产品的皮肤PK。使用参考标度平均BE (SABE)分析和90%置信区间(CI)评估,基于利多卡因/丙胺卡因的皮肤PK终点,对Oraqix®凝胶与EMLA®乳膏进行BE评估。EMLA®和Oraqix®皮肤生物利用度也在6名健康受试者的皮肤开放流微灌注(dOFM)体内试点研究中进行了比较。在IVPT和dOFM研究中使用的所有产品的剂量为10mg/cm2。Figure 1. Light microscopy images: (a) EMLA® cream and (b) generic lidocaine and prilocaine cream showing globules, vs. a homogenous globule-free matrix in the (c) Oraqix® gel. The scale bars are 20 µm in images a and b, and 100 µm in image c.图1所示.显微镜图像:(a) EMLA®乳膏和(b)仿制药利多卡因和丙胺卡因乳膏显示球状,(c) Oraqix®凝胶中均匀无球状基质。图a和图b的比例尺为20µm,图c的比例尺为100µm。Figure2.Cryo-SEM images at 3000X magnification depicting the internal microstructures of (a) EMLA® cream (b) generic lidocaine prilocaine cream and (c) Oraqix® gel. Scale bar - 1µm图2. 3000倍放大的冷冻扫描电镜图像描绘了(a) EMLA®乳膏(b)通用利因双卡因乳膏和(c) Oraqix®凝胶的内部微观结构。比例尺- 1µmResults and Discussion结果与讨论Quality tests and Q3 propertie质量测试和Q3属性The Q3 properties of the reference and generic lidocaine and prilocaine topical creams were similar to each other and different from those of Oraqix® gel:•The average pH values measured for the reference lidocaine/prilocaine cream, the generic lidocaine/prilocaine cream, and Oraqix®gel were 9.10, 8.90 (i.e., 9.0 ± 0.1) and 7.65, respectively.•The microscopic images of the cream products showed the presence of globules with a diameter of 1-3 µm, while the Oraqix® gel appeared to be a homogenous globule-free system. The cream products also showed a different microstructure than the gel product under cryo-scanning electron microscopy (cryo-SEM) (Figure 2).与Oraqix®凝胶相比,参比型和仿制药利多卡因/丙胺卡因外用乳膏的Q3性质相似,但与Oraqix®凝胶的Q3性质不同:•参比型利多卡因/丙胺卡因乳膏、仿制型利多卡因/丙胺卡因乳膏和Oraqix®凝胶的平均pH值分别为9.10、8.90(即9.0±0.1)和7.65。•凝胶产品的显微图像显示直径为1-3 μ m的小球的存在,而Oraqix®凝胶似乎是一个均匀的无小球系统。在冷冻扫描电镜(cryo-SEM)下,乳霜产品也显示出与凝胶产品不同的微观结构(图2)。Figure 3.Rate of evaporation of volatile components from lidocaine; prilocaine topical cream and gel products measured gravimetrically at 32ºC. Data are expressed as Mean ± SD (n=3).图3.在32℃利多卡因/丙胺卡因挥发性成分的蒸发速率;重量法测量,数据以Mean±SD (n=3)表示。Figure 4. Left: Viscosity as a function of shear stress for lidocaine and prilocaine cream and gel products.Right: Strain sweep for all three products. Closed symbols (G’) represent the storage modulus and the open symbols represent loss modulus (G”). The yield stress was determined to be 110 for the cream products and 11 for Oraqix®gel.图4.左图:利多卡因/丙胺卡因乳膏和凝胶产品的粘性随剪切应力的变化。右图:对所有三种产品进行应变扫描。闭合符号(G’)表示存储模量,开放符号(G”)表示损耗模量。凝胶的屈服应力为110,Oraqix®凝胶的屈服应力为11。Performance tests性能测试In vitro cutaneous PK study using IVPT1.体外应用IVPT研究皮肤PKFigure 5. Cutaneous PK (flux profile) of lidocaine and prilocaine in vitrofrom topical applications of the same dose of EMLA® cream, the genericcream, and Oraqix®gel.Data are shown as Mean ± SEM from 6 donorsand 6 replicates.图5.相同剂量的EMLA®乳膏、仿制药乳膏和Oraqix®凝胶对体外利多卡因/丙罗卡因皮肤PK(通量Jmax)的影响。数据显示为来自6个供体和6个重复的平均值±SEMTable 1. BE analysis results for lidocaine (in orange); prilocaine (in yellow) based on PK endpoints of area under the curve (AUC ) and maximum flux (Jmax) and within donor variability associated with EMLA® cream (Swr)表1.利多卡因BE分析结果(橙色);基于曲线下面积(AUC)和最大通量(Jmax)的PK终点以及与EMLA®乳膏(Swr)相关的供体变异性,丙胺卡因(黄色)2.In vivo cutaneous PK study using dOFM 使用dOFM研究体内皮肤PKFigure6.Mean lidocaine and prilocaine concentration-time profiles (±SE) for EMLA® cream and for Oraqix® gel following application of 10 mg/cm2 of products.Data are shown as Mean±SEM from six subjects.图6.EMLA®乳膏和Oraqix®凝胶在给药10mg /cm2产品后的平均利多卡因/丙胺卡因浓度-时间曲线(±SE)。数据以6名受试者的平均值±SEM表示。Conclusion结论These results demonstrate the correlation between the Q3 similarities (or differences) of three comparator products and their corresponding cutaneous PK, both in vitro (IVPT) and in vivo (dOFM). The similarity of Q3 characteristics between the reference and generic creams accurately correlated with and was predictive of comparable bioavailability (and bioequivalence) for both lidocaine and prilocaine between the two creams, with the exception of prilocaine AUC in the (underpowered) IVPT study. The difference in Q3 characteristics between the reference cream and the gel accurately correlated with and was predictive of differences in bioavailability.这些结果证明了三种比较产物的Q3相似性(或差异)与其相应的皮肤PK之间的相关性,无论是体外(IVPT)还是体内(dOFM)。仿制和参比制剂乳膏Q3相似性与生物利用度(和生物等效性)准确相关,并可预测两种乳膏之间的一致性,(underpowered)IVPT研究中丙胺卡因AUC除外。参比制剂乳膏和凝胶之间Q3差异与生物利用度的差异准确相关,并可预测。Acknowledgement and Disclaimer: Funding for this work was made possible, in part, by the U.S. Food and Drug Administration through award U01FD005226 and award 1U01FD005861. The views expressed here do not reflect the official policies of the U.S . Food and Drug Administration or the U.S. Department of Health and Human Services; nor does any mention of trade names, commercial practices, or organization imply endorsement by the United States Government.确认和免责声明:本工作的资金部分由美国食品和药物管理局通过奖励U01FD005226和奖励1U01FD005861提供。这里表达的观点不反映美国食品和药物管理局或美国卫生与公众服务部的官方政策;任何提及商品名称、商业惯例或组织也不意味着得到美国政府的认可。
企业动态
2024.03.26

不同厚度皮肤对体外渗透实验(IVPT)的影响
不同厚度皮肤对体外渗透实验(IVPT)的影响 目的:研究不同厚度皮肤的电阻值对药物体外渗透实验的影响。方法:本文使用合邦VDC12扩散仪,采用两组经皮失水合格、不同厚度区间的皮肤:L(左)侧厚度:0.68-0.75mm,TEWL:2.3-5.3g/m2/h;R(右)侧厚度:1.07-1.17mm,TEWL:2.7-7.6g/m2/h。测量其皮肤电阻值同时进行IVPT实验。结果:在相同实验条件下0.7mm的皮肤IVPT各取样点结果RSD在1.9%-11.7%之间,1mm的皮肤IVPT各取样点结果RSD在44.3%-67.9%之间。结论:0.7mm皮肤IVPT累计渗透量平行性更好。 1.目的研究不同厚度皮肤对IVPT累计渗透量平行性影响。 2.仪器及材料 2.1仪器经皮失水测量仪(瑞士SKT)、电阻仪(瑞士SKT )、VDC12扩散仪(北京合邦兴业科学仪器有限公司)、液相色谱-紫外检测器(Hero LC100,北京合邦兴业科学仪器有限公司) 2.2试药利多卡因凝胶贴膏(某企业用户提供) 2.3皮肤巴马小香猪皮肤 3.实验方法 3.1透皮实验条件介质:pH7.2 PBS+0.01%庆大霉素;扩散池体积:10ml;温度:32℃;转速:600rpm;取样体积:1.5ml;取样时间点:2h、4h、6h、8h、12h、16h、20h。 3.2实验步骤先将裁切好的猪皮放置在充满介质的扩散池上,介质温度达到32℃±1℃,孵育30min后,用经皮失水测量仪测定每个皮肤的经皮失水值。测定经皮失水值后,在皮肤上方再加入少量介质(温度达到32℃±1℃),用电阻仪测量每个皮肤的电阻值(如表1),测量结束后取下皮肤贴上药贴,用吸水纸吸干皮肤表面介质,左侧(L)和右侧(R)同时进行透皮实验。表1. 皮肤电阻值基础电阻(kΩ)0.22扩散池编号123456L(左侧)8.88.4178.46.415R(右侧)238.4191.33.611 3.3液相方法参数 色谱柱:C18(150mm x 4.6mm,4μm);检测波长:220nm;流动相:甲醇-0.3%磷酸氢二铵(67:33);流速:1.0ml/min;柱温:30℃;进样量:20μl;运行时间:10min。 4.实验结果表2. L(左)侧累计渗透量(ug/cm²)注:Cell1上样后拧紧时皮褶皱变小,导致药贴边缘直接接触介质,发现后及时重新安装,所以,整体浓度偏高。数据结果整理未纳入cell1数据. 表3. R(右)侧累计渗透量(ug/cm²) 图1 L(左)侧累计渗透量曲线 图2 R(右)侧累计渗透量曲线5.结论由以上结果可知,本次实验通过比较各取样点RSD值,0.7mm厚度皮肤的IVPT结果平行性好于1mm厚度皮肤的IVPT结果平行性。比较两组数据累计渗透量的中位数最终点0.7mm厚度结果为48.54ug/cm²,1mm厚度结果为47.11ug/cm²。 6.讨论:本次实验我们同时也探索电阻值与渗透量的关系,我们发现所有皮肤TEWL均合格的前提下,0.7mm厚度皮肤的电阻值有差异的情况下IVPT累计渗透量结果比较接近;1mm厚度皮肤电阻值差异比较大的情况下IVPT累计渗透量结果也有较大波动。此结果是在样本量相对局限的条件下得出,后续会进行更多的实验以近一步考察。就以上结果可以考虑在进行IVPT实验时,为提高实验结果平行性,应尽可能选择较薄的皮肤。
企业动态
2024.03.26

突破性技术OFM(微灌流):合邦科仪携手JOANNEUM RESEARCH开启新纪元
北京合邦兴业科学仪器有限公司&JOANNEUM RESEARCH Forschungsgesellschaft mbH(JOANNEUM RESEARCH)奥地利格拉兹大学生物医学技术研究所(格拉兹研究所)达成战略合作,北京合邦科仪成为其在大中华区的授权总代理。JOANNEUM RESEARCH(格拉兹研究所)总部位于奥地利,OFM(微灌流)为其全球独特创新产品。JOANNEUM RESEARCH与全球包括FDA在内的多家制药企业及机构,就OFM产品进行临床前和临床合作。近期通过与美国食品和药物管理局(FDA)的合作,JOANNEUM RESEARCH公司成功建立了一种新方法来确定外用制剂在皮肤中的生物等效性。该项目得到了美国卫生与公众服务部 (HHS) 食品和药物管理局 (FDA) 的支持,作为由 FDA/HHS 资助的总额为 1,500,000 美元的财政援助奖励的一部分U01FD007669。内容是作者的内容,并不一定代表FDA/HHS或美国政府的官方观点,也不一定代表其认可。https://www.fda.gov/drugs/regulatory-science-action/impact-story-developing-new-ways-evaluate-bioequivalence-topical-drugs 稳定的流速,确保数据准确性OFM 采样时,探头灌注液与周围间质液之间的样品保持稳定浓度平衡但不带来其他影响。这种独特的特性使得其可以对靶组织间质液中的内源性和外源性物质进行定量,而不会有改变研究样品的风险。无膜交换取样技术,大小分子/亲水亲脂成分无限制OFM(开流微灌注)采样技术在神经科学、皮肤病学、肿瘤学和生物标志物研究中发挥着重要作用。无膜 OFM 探针提供了从大脑和外周组织获取间质液的全新微创方式,OFM也可以从皮下和脂肪组织进行采样,尤其在亲脂性或大分子量样品的获取上有突出优势。为临床前和临床药物测试提供了新的视角。持续监测,严谨的数据保障OFM作为一种突破性采样技术,可以连续监测皮肤组织中药物的药代动力学和药效学,监测时间长达48小时,为药品开发提供了关键信息。该技术的有效性通过早期临床试点研究得到验证,为制药公司提供了预测主要临床研究结果的重要依据。合邦科仪和 JOANNEUM RESEARCH 的合作将为中国市场带来领先的 OFM 采样技术,为临床前和临床研究提供创新的解决方案,加速新药上市进程,为患者带来更及时的治疗选择。美国食品和药物管理局(FDA)最近发布了一段视频,展示了真皮开流微灌注(dOFM)在仿制药开发中的潜力。FDA对大多数外用仿制药的批准需要进行临床终点研究,以将治疗效果与原始产品进行比较。JOANNEUM RESEARCH HEALTH 开发了一种新的方法和策略 (dOFM),无需临床终点研究即可申请仿制药的批准。现在,FDA发布了一个简短的 视频 ,说明了使用dOFM来评估外用药物的生物等效性。视频地址:
企业动态
2024.03.13

重要通知! 诚招水活度仪省级总代理!
重要通知! 诚招水活度仪省级总代理携手未来,共创辉煌!北京合邦兴业科学仪器有限公司诚邀您的加入!北京合邦兴业科学仪器有限公司为美国 Meter 在中国 制药行业 总代理,将在全国范围内诚招省级总代理,共同推广和销售水活度仪。水活度仪是一款高效、精准检测物质中游离水分含量的仪器,广泛应用于制药、生物科技、食品饮料、环保监测等领域。合邦科仪作为Meter在中国 制药行业 的总代理,我们拥有丰富的行业经验和强大的渠道资源,为代理商提供全方位的支持和服务。产品特点:Meter水活度仪采用USP/EMA水活度检测金标准-镜面露点法设计,突破性的可调激光计数,设计符合美国药典USP第一法,高精度测量所有样品,包括挥发性样品,测量时间小于5min。可以全程监控生产过程微生物生长。是业内公认具有潜力的微生物检测新方法。快速-测量时间小于5分钟!产品应用:用于评估微生物风险的重要工具,控制微生物生长。迅速提高微生物检测质控能力,提高效率,减轻药品的微生物质控负担。降低原料药的水解反应,提高药品的稳定性。决定水分迁移的方向--包装材料(如:胶囊壳)筛选。产品市场:Meter水活度系列产品在食品、生物科技、制药等行业均有大量全球用户。国内外市场占比高达80%,是公认的成熟品牌。水活度是国家局《非无菌药品微生物控制中水活度应用指导原则》公示稿中新提出的微生物控制指标,因而水活度仪拥有可观的市场前景。目前包括中检院在内多家省药监院以及市药检所都拥有Meter水活度仪。以下列举为全球部分制药行业用户名单: “镜面冷凝露点法”是水分活度的金标准,是所有标准化组织推荐的方法。针对挥发性成分可选激光法或电容法水活度仪,本公司能提供所有产品型号。代理优势独家代理权: 我们为您提供了在指定区域内的独家代理权,确保您的商业利益。强大品牌支持: 借助Aqualab的知名品牌和声誉,提升您在市场上的竞争力。丰富的产品线: 我们的产品线不仅限于水活度仪,还有其他先进的科学仪器,满足不同客户需求。技术支持和培训: 我们提供完善的技术支持和培训体系,确保您和您的团队能够熟练操作产品,提高工作效率。优惠政策和市场推广: 您将享受到丰厚的销售额和市场推广优惠政策,增加您的利润空间。此次招商活动的有效期为两个月,机会难得,不容错过!如果您有意加入我们的代理团队,请立即联系我们。北京合邦兴业科学仪器有限公司期待与您携手合作,共创美好未来!
经销代理
2024.01.22

展会回顾|北京合邦中国制剂大会-透皮技术大会
2023年8月3-4日,中国制剂大会-透皮技术大会在苏州成功举办。本次会议旨在解决国内透皮制剂行业面临的起步晚、立项周期长、耗材和设备依赖进口等问题。中国透皮制剂联盟发起人北京合邦兴业科学仪器有限公司联合药事纵横(北京)公司共同组织了会议,就透皮制剂行业的发展机遇、挑战以及法规和注册等话题展开了深度交流。合邦兴业总经理汤宏敏先生在本次大会上就透皮制剂实验室仪器一站式解决方案进行深度演讲。这个方案的引入将大大提升实验室的效率和准确性,为药物研发和生产过程带来变化。合邦兴业的参与,为本次活动增添了亮点,为医药行业的发展和创新贡献了力量。参展人员对仪器设备很感兴趣,纷纷向合邦兴业的工作人员咨询了相关信息。合邦兴业的工作人员耐心解答了他们的问题,并为他们提供了详细的技术支持和服务。PART 1明星产品旋转流变仪《皮肤外用化学仿制药研究技术指导原则》、《化学仿制药透皮贴剂药学研究技术指导原则》都有强调:皮肤外用制剂的关键质量属性(CQAs),对于流变特性需进行对比研究。粒度分析仪《皮肤外用化学仿制药研究技术指导原则》、《化学仿制药透皮贴剂药学研究技术指导原则》表示:皮肤外用制剂的关键质量属性(CQAs)等,需对粒度和液滴粒径进行考察。自动取样透皮扩散系统FDA-IVRT、USP、FDA-IVPT、等法规或指导原则,对自动取样透皮扩散设备的仪器性能、池型要求,都有相关设定和推荐。水分活度仪《非无菌药品微生物控制中水分活度应用指导原则》(征求意见稿)提出:可以从水分活度等方面关注生产过程中的微生物控制。自动取样透皮扩散系统皮肤电阻率仪中表示,在使用皮肤进行IVPT实验前后需要测量皮肤的完整性,且皮肤电阻率测定法是推荐方法之一。皮肤均质器
企业动态
2024.01.15
公司名称: 北京合邦兴业科学仪器有限公司
公司地址: 北京市丰台区南四环西路128号院3号楼8层907 联系人: 汤先生 邮编: 000000 联系电话: 400-860-5168转6127
仪器信息网APP

展位手机站