
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转4098
仪器信息网认证电话,请放心拨打
体外释放试验(IVRT):原理和应用
2024/02/02 20:17
阅读:177
分享:方案摘要:
产品配置单:

华溶自动透皮扩散系统TD-12AT
型号: TD-12AT
产地: 广东
品牌: 华溶仪器
¥50万 - 80万
参考报价
联系电话
方案详情:

翻译:华溶应用中心
审核:工业药剂发烧友
体外释放试验(IVRT):原理和应用
体外药物释放试验已成为半固体制剂开发和批准过程中最重要的工具之一。体外释放试验(IVRT)能够反映多种理化特性、颗粒或液滴大小、黏度、物质微观结构以及剂型聚集状态的综合作用情况。本文讨论了IVRT的起源、原理及其与药物动力学反应(如血管收缩或皮肤药代动力学(离体皮肤))的等级关系,还讨论了监管审批试验要求的演变状况。
IVRT能反映各种参数,是比较局部给药制剂及以相似速率释放相似量的活性成分的能力的逐级评价方式的重要部分。此外,除了能进行处方成分的定性和定量(Q1和Q2)考察外,IVRT还是评估局部给药药物分类系统(TCS)方法1类和3类药物中提出的微结构排列(Q3)相似性的重要工具。本文讨论了TCS系统以及局部给药皮肤病药物从Q1, Q2, Q3相同到Q1, Q2, Q3相似的发展概念,这个要求变化让处方的调整变动有了更大的空间。
一、简 介
体外药物释放试验目前已经成为半固体制剂开发和批准过程中最重要的工具之一。这是如何发生的呢?体外释放试验(IVRT)的起源是什么? 1987年,著名皮肤科医生Richard Stoughton博士在《皮肤病学档案》上发表了一篇文章,文章质疑了局部给药糖皮质激素仿制药与原研药的等效性。这类出版物令监管机构感到不安,并且立即予以关注。从口服制剂评价方面获得的经验可知,溶出度在剂型的生物利用度中起着重要作用。固体口服制剂溶出度试验在当时已经逐渐得到重视并建立了质控检测,以确保产品性能和批间一致性。溶出度试验的通用原则应适用于局部给药制剂的体外释放。与固体口服制剂的溶出类似,局部给药剂型的药物释放对其质量、安全性和有效性也起着重要作用。对于局部给药制剂,给药后的第一步就是将有效成分从制剂中释放出来,然后药物分布、渗透或穿过角质层。根据活性成分的理化性质、处方因素以及预期作用部位的不同,特定的药效作用可以在皮肤的特定的、最外层或更深层表达。对局部浓度的评估是复杂的,需要在适当的时间间隔下保持足够的且持续的浓度。1989年在华盛顿举行的“体内经皮渗透/吸收”会议上列清了每种可用的方法都有其优点和局限性。因此,研究人员决定探索局部给药剂型的体外释放方法开发。
许多在皮肤医学领域工作的制药行业和药物研究人员都非常熟悉使用尸体皮肤的Franz扩散池(垂直扩散池,VDC)系统。该系统用于评估局部给药药物开发中有效成分的渗透和渗入情况。决定将该系统改良用于开发适应局部给药半固体特性的体外释放。尸体皮肤的使用受到许多变量的影响,不适合在质量控制检验程序中常规使用。因此,有必要开发一种利用VDC和具有更强重复性的市售合成膜的药物释放方法。
使用VDC和合成膜的体外药物释放方法被开发出来,并于1989年首次发表在《国际药学杂志》上。局部用药领域的研究在几个中心进行了探索,包括美国食品药品监督管理局(FDA)实验室,以及法国的Hans Schaefer教授、密歇根大学的Gordon Flynn教授、罗格斯大学的Joel Jatz教授和犹他大学的Lynn Pershing博士,他们都对局部给药制剂的IVRT和生物等效性(BE)的不同方面进行了评估。大部分研究结果随后由所有研究人员发表。由于IVRT采用了一种新技术,FDA在德克萨斯州达拉斯市的实验室为工业科学家们提供了几次实践培训课程。
开发有意义的半固体体外释放试验的研究工作继续进行,重点是开发一种简单、可重复和灵敏的方法,在可行的情况下,具有一些已知的临床相关性。在IVRT的初始开发过程中获得的经验表明,在保持方法简单和可重复性的同时,很难模拟所有导致体内过程复杂性的因素。作为生理屏障,皮肤的厚度、结构、渗透性和对局部给药处方的反应在个体内和个体间都是多变的。此外,在体内给药后,剂型会发生形态变化、不同程度的组成和结构转变,这取决于剂量、方法和给药部位等因素。因此,很难开发一种能够模拟临床环境的标准化测试。与固体口服剂型相比,IVRT和局部给药制剂建立体内外相关性被证明更具挑战性。然而,两种不同的氢化可的松乳膏的体外药物释放率(Caron et al, 1990)、两种戊酸倍他米松乳膏产品的皮肤漂白和药物释放(Shah等,1992)、类固醇的效力和药物释放(Shah et al, 1999)与体内皮肤药代动力学(DPK、胶带剥离)和药效学(血管收缩试验、VCA或漂白效果)参数之间至少有一定的顺序对应关系。很明显,使用非皮肤的人工膜,惰性是关键要求,并且在封闭条件下使用高剂量(伪无限),而释放的药物在满足漏槽条件的接收池中收集,这将无法预测生物利用度。在最初的工作中,重点是了解IVRT所反映的内容,比较已知成分和/或效力存在差异的产品的体外行为。体内数据对于证实一种简单的方法至关重要,尽管临床复杂性过于简化,但能够通过不相似的体外行为确认这些差异。IVR已经发展成为一种比较测试,它应该对局部作用产品的特性敏感,这对体内结果至关重要。这进一步意味着了解体外比较的产品(均质或异质体系,活性物质完全溶解或部分悬浮,其在体系组成相中的分布,释放机制和限速步骤等),识别可能与临床相关的差异,并应用适当的相似性标准。规格区分是首要要求之一。IVRT必须在给定制剂中显示不同药物浓度的不同释放(Pillai et al, 2001)。IVRT受到了许多研究者、产业界和学术界的挑战、反对和抵制。它并没有被轻易接受。1997年,在SUPAC-SS指南中引入了IVRT,作为扩大规模和批准后变更产品一致性的衡量标准(FDA, 1997)。1998年举行了FDA /美国制药科学家协会研讨会,讨论IVRT的利弊。研讨会总结了IVR方法的有用性(Flynn et al, 1999)。1998年第一个FDA局部给药制剂指南草案强调了IVRT的重要性和价值(FDA, 1998)。因此,IVRT慢慢开始被接受。关于局部给药制剂的指南草案后来因IVRT以外的原因被撤回(FR, 2002)。
二、体外释放
体外释放(IVR)方法的发展与体外溶出度试验类似,应用于研究和开发阶段,稳定性研究或变更或变异分析。它的作用是基于假定的能够综合反映诸如颗粒或液滴大小、药物溶解度、流变性等几个因素的影响。IVRT被纳入FDA发布的非无菌半固体制剂规模扩大和批准后变更指南(FDA, 1997),用于评估已批准的成分或生产工艺的明确的二级(中等)变更。这一首次正式申请标志着一种谨慎的方法,在这种方法中,首先对两种比较产品之间的变更进行定义、分析和分类,并根据它们在体内的潜在影响进行分类,然后通过能够评估性能显著变化的适当方法进行评估。上市的、变更前的产品作为参比,而自制的、变更后的制剂在组分或生产工艺都一定程度的不相似。1998年指南草案试图扩大其在开发低规格局部给药产品中的作用。对于新药申请(NDA)的提交,指南草案指出,在“BA(生物利用度)文件”被认为重要的情况下,对于较低规格“可以执行IVR”。一旦仿制药最高规格的生物等效性在体内得到证明,IVRT就有可能被用于免除较低规格的体内评估(FDA, 1998),前提是:i)两种规格的组成的唯一区别是活性成分和相应稀释剂的量;Ii)生产工艺和设备相同;Iii)参比制剂有相应的两种规格上市。应用相同的方法获得的IVR速率将在产品和规格之间进行比较。该指南草案于2002年被撤回(FR, 2002),原因是两位研究者在DPK研究结果中观察到不一致。然而,后来人们注意到,两位研究者采用了不同的方案。指南草案中描述的IVRT相关问题没有争议,因为它的地位已经在SUPAC-SS中确立。
低规格的生物豁免标准现已包含在欧洲药品监督管理局(EMA)于2018年发布的关于局部用药质量和等效性的新指南草案中(EMA, 2018)。这说明了与体外溶出试验的类似性,以及在局部半固体制剂的开发和评价中日益重要的作用。美国药典(USP, 2013)第1724章首次描述了各种类型的扩散池。美国FDA发布了越来越多的产品个例指南(PSG)文件草案,其中包括体外选择作为生物豁免的一种手段。被比较的两种产品(试验制剂和参比制剂)应具有相同的定性组成和相似的定量组成。根据剂型的复杂程度,IVRT是主要测试之一(1%磺胺嘧啶银乳膏或5%阿昔洛韦软膏的PSG;FDA, 2017年,2019年),或者在许多情况下,是更复杂的比较方法的一部分,该方法结合了几种物理化学评估和通过人体皮肤样品的体外渗透试验(IVPT) (5%阿昔洛韦乳膏的PSG;FDA,2016)。只有IVRT有明确的接受标准,长期实施的SUPAC-SS指南验证了这一点(FDA, 1997)。
三、IVRT的现状
体外释放试验当前被认为是保证产品质量的有价值的工具,并在PSG文件中找到了它的位置。体外释放度反映原料药和制剂的几种理化性质的综合作用,包括活性成分的溶解度和粒径;这也是半固体制剂的组成和微观结构特性综合影响的一个很好的指标。生产方式和工艺可能改变制剂属性,从而影响药物释放速率和药物的生物利用度。当前版本的EMA局部给药药品质量和等效性指南草案(EMA, 2018)中指出,需要IVRT来支持扩展药物等效性的概念。其他监管机构建议将其用于选择进行体内测试的代表性批次药品(NIHS, 2003)。
现已提供IVR方法开发和验证的建议(FDA, 2016)。一般情况下,释放介质的选择是基于漏槽条件的要求,膜的选择必须证明其惰性,人工膜作为被测半固体的机械支撑。介质和膜的组合应与制剂和活性成分相容,在整个试验期间防止任何明显的降解或结合现象。此外,它应该提供一个与半固体基质适当的接触角,从而允许明显释放。具有已知优点和局限性的各种各样的扩散池和现有的溶出装置适应装置是可用的。方法验证包括规格-释放速率关系的评估以及微观结构差异的区分。
对于目前的所有应用,必须开发并使用IVRT作为比较评估,即比较速率,在某些情况下描述扩散过程的附加参数。生成的数据的相关性总是依赖于参比制剂的存在,需要对参比制剂进行适当的描述。两种半固体药品之间的相似性和差异性应根据其对质量、安全性和有效性的潜在影响进行识别和理解。比较表征的方案应从成分组成开始,然后应根据剂型的复杂性进行理化特性和微观结构测试。值得注意的是,局部给药半固体在整个保质期内不断变化。随着时间的推移,这种变化可能会给仿制药的开发带来额外的问题,参比制剂是一个移动的质量目标。IVRT由于其对一系列物理化学和微观结构参数的敏感性,也应考虑这些动态变化。
四、应 用
IVRT最重要的应用是在局部皮肤科药物审批过程中的地位。在某些情况下,它是仿制药批准的基础,在许多情况下,它是批准要求的一部分。目前,IVRT是多方面方法的一部分,经常结合根据剂型的特殊性和复杂性选择的一系列比较的物理化学评估。当要比较多源产品时,对IVR数据的充分解释取决于定性和定量组成的相似程度(由限制性标准定义)。根据EMA指南草案(EMA, 2018),这种由+/-5%差异定义的限制性(在某些情况下为+/-10%)可以通过对质量、安全性和有效性的潜在影响来解释。进一步扩大IVRT作用的主要问题是,单独的这项测试可能无法适当地反映超出这些限制的差异的临床影响,特别是当注意到不同辅料会对体内结果产生影响时。IVR速率取决于测试条件,不反映渗透或渗入特性。然而,该速率从未被用作安全性和有效性的直接预测指标。IVRT一直是一种比较评价,它被认为反映了半固体基质的成分组成(辅料的种类、质量和用量)和成分排列的相似性和差异性的综合效应。对这些差异的潜在后果进行深入分析,无法充分解释IVRT结果。根据这一基本原理,提出了局部给药药物分类系统(TCS),如果被接受,将为许多仿制局部给药产品提供生物豁免(Shah等人,2015)。
五、局部给药药物分类系统(TCS)
局部给药药物分类系统(TCS)是基于SUPAC-SS、成分定性(Q1)和定量(Q2)的一致性、非活性成分的作用、微观结构排列(Q3)和IVRT对局部给药仿制药进行分类的框架。在TCS 1类仿制药中,Q1和Q2与参比制剂(RLD)相同,Q3和IVRT与RLD相同,反映了SUPAC-SS的1级变更。这些仿制药将有资格获得生物豁免。在TCS 2类的情况下,仿制药与RLD相比具有Q1和Q2,但不具有Q3,并且具有不同的IVRT特性。这可能是因为辅料来源和质量特性不同,也可能是由于生产过程中使用的方法和参数不同。在这些情况下,仿制药不符合生物豁免资格,必须按照监管机构的要求进行BE研究。在TCS 3类中,当测试产品与RLD进行比较时,Q1和Q2是不同的,但具有功能相似的非活性成分,导致Q3和IVR与RLD相似。这些仿制药将有资格获得生物豁免。我们发现处方/产品流变学的变化总是反映IVR的变化,从而支持了确保变化下产品一致性的有效性和重要性。TCS 1类和3类仿制药和生物豁免概念与公认的生物药剂学分类系统(BCS)概念相当。在这两种情况下,生物豁免是基于剂型的体外释放或溶出结果提供的,并且对3类生物豁免的要求比1类更严格。针对TCS 3类产品,提出了一种基于风险的辅料评估方法。TCS分类不排除任何额外的物理化学测试要求。如果是TCS 4类,仿制药的成分不同,导致微观结构不同,IVR也不同,不符合生物豁免条件。简而言之,根据TCS提案,1类和3类局部给药仿制药具有与RLD相似的释放特性(通过应用SUPAC-SS定义的数据分析、比较程序和验收标准)将有资格获得生物豁免。2类和4类仿制药将需要按照监管机构的要求进行体内BE研究。
仿制局部给药皮肤科药品现在被归类为复杂仿制药。这些产品的批准多年来一直在不断发展,从要求仿制药与RLD达到Q1、Q2并要求进行比较临床终点研究(糖皮质激素除外),到Q3的一致性,与RLD相比,具有不同非活性成分的仿制药只要不显着影响药物的局部或全身生物利用度就可以接受。在最近的监管报告中反映的2019年的这一变化是一个受欢迎的变化(Raney, 2019)。然而,应该指出的是,“相似性”的概念在2015年的TCS分类TCS 3类中被描述和提出(Shah et al, 2015)。TCS 3类中“类似Q3”的概念符合当前Q1、Q2、Q3相同向Q1、Q2、Q3相似的演变思维(Raney, 2019)。我们对已上市的5%阿昔洛韦乳膏产品的广泛研究也支持了这一概念(Miron等,2021b)。
图1显示了TCS 3类和Q3相似概念的比较。局部皮肤科药物概念的变化,即Q3相同演变为Q3相似,解释为 Q1和Q2可能存在差异,但与参比制剂相比,它不应显著影响局部或全身生物利用度。这显然符合提出的TCS 3类的分类。

六、Q1,Q2,Q3,TCS和SUPAC-SS
回顾SUPAC-SS指南成功应用超过25年,并作为当前局部给药半固体性能测试应用的基础,使用IVRT来评估TCS提出的Q3相似性或差异性是合理的。SUPAC-SS指南中描述的二级变更及其转到潜在的Q1/Q2/Q3或TCS分类中表明,随着成分和组成的变更或生产和工艺的变更,如果变更前和变更后的产品之间的IVRT相似,那么变更后的产品不怀疑对产品的体内性能有任何重大影响。表1总结了SUPAC-SS 2级成分组成变更与TCS 1、3类的关系。
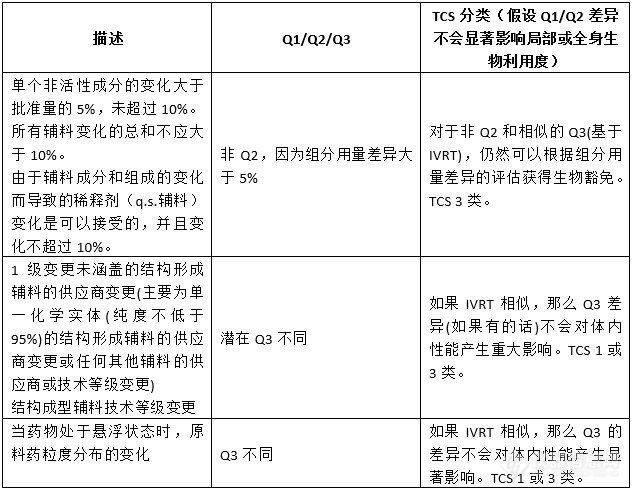
七、扩展药物等效的概念
EMA基于比较仿制药和参比药的分步方法,阐述了扩展药物等效性的新概念(EMA, 2018)。EMA表示,应该从深入了解局部给药产品提供预期安全性和有效性的方式开始,制定具体的方案,而不是由US-FDA发布的PSG。它是从剂型的复杂性、给药部位、相似的定性和定量组成的重要性、详细的和产品特异性的比较理化测试、IVRT和IVPT,以及渗透动力学和药效学甚至临床治疗等效研究(如适用)开始的逐步方法。没有这些评估应说明理由。鼓励采用新的方法。如前所述,“需要经过验证的体外释放试验来支持扩展的药物等效性”。
在当前版本的EMA指南草案(2018)中,与美国FDA相比,IVRT的一般原则相似。然而,在样本量(每批产品进行比较评估的批次数、每批重复的次数)、试验期结束时的剂量消耗、实验数据与Higuchi模型拟合的标准、比较参数(累积释放量、释放率和滞后时间)、统计比较(包括IVR数据的假设分布)和相似性的接受标准等方面仍然存在显著差异。
八、USP药典第1724章的修订和扩展
USP药典第1724章的修订版于2022年5月2日在药典论坛上发表。在简介部分,文件总结了一般测试原则以及将IVRT和IVPT作为产品性能测试的正确使用方法。列出了IVRT的局限性,以及当前的扩展应用,例如将“预期的仿制(测试)产品”与“可以在某些情况下支持生物等效性证明”的参比产品进行比较,或将“经过验证的IVRT方法(…)作为将来产品参考的基础”的体外释放速率的潜在使用。典型的精度和可重复性对于性能测试来说是一个相当大的优势,假设是“反映了处方中物质的物理化学性质和/或结构排列的差异”。IVRT的微观结构敏感性在目标的表格摘要中提到,我们增加了预期的效用或相关性(表2)。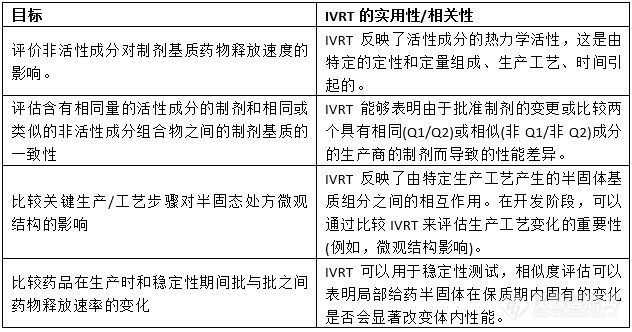
九、展 望
IVRT已经慢慢进入局部给药半固体的开发和评价领域。当前版本的监管文件,无论是PSG还是指南草案,都证明了谨慎使用这种体外性能测试。它是作为一个复杂的、产品适合的、比较评估的一部分,在许多情况下,IVPT被认为是一个更相关的体内反映的方法。IVRT的更广泛应用受到了批评。假设高度复杂的体内递送和渗透过程的过度简化和标准化不可能具有生物相关性。那么,IVRT是一种不反映体内性能的质量测试,还是一种只反映药物质量的性能测试??SUPAC-SS指南从一开始就将IVRT与体内性能的潜在影响联系起来。另一方面,将IVRT纳入多源产品的比较评估表明,它不是一个多余的质量测试,而是一种评估,可以解决具有相似成分和微观结构的产品之间生物不等效的额外风险。
在局部给药药品、生物等效性和IVRT的监管方面做出了重大努力。IVRT可能会成为半固体制剂的强制性规定,类似于口服固体制剂的溶出,以确保批间产品质量。IVRT作为一种产品开发工具和质量控制工具可以获得更多的重要性,尽管在许多情况下,半固体基质在保质期内的内在变化是复杂和显著的,这使得接受限度的设置特别具有挑战性。最后,IVRT将成为生物豁免的工具:(i)低规格半固体制剂的批准和(ii)基于局部给药药物分类系统(TCS)的生物豁免,类似于BCS。
十、结 论
IVRT已经走过了漫长的道路,它在药物开发和局部皮肤病药物的评估中确立了自己的地位。IVRT作为一种生物豁免工具可能适用于评估剂型的低规格和TCS 1类和3类药物。
十一、参考文献
略
下载本篇解决方案:
更多![]()



好文推荐 |依匹哌唑原位凝胶植入剂的体外释放研究
目的 建立一种依匹哌唑原位凝胶植入剂的体外释放方法,研究处方在体外的释放行为和缓释机制。 方法 以抗精神病依匹哌唑为模型药物,制备以聚乳酸⁃羟基乙酸共聚物( polylactide gly⁃ colic acid,PLGA)/醋酸异丁酸蔗糖酯( sucrose acetate isobutyrate,SAIB)为基质的原位凝胶植入剂。开发体外释放检测方法,采用多种体外释放装置研究依匹哌唑原位凝胶植入剂的释放差异, 考察体外释放的影响因素。
制药/生物制药
2024/06/24

好文推荐 | 水杨酸片在流池法溶出仪性能验证试验中的应用
流池法溶出仪是控释剂型、难溶性药物和许多特殊剂型(如混悬液、软胶囊、植入剂、微球和脂质体)溶出度测试的首选仪器。虽然流池法溶出仪已列入药典多年,但一直没有正式的性能验证试验(PVT)方法。在这项研究中,水杨酸片被用来开发流池法的PVT。水杨酸片在篮法和桨法装置上相同的溶蚀和零级释放机制,可作为流池法装置中PVT的潜在参考标准品。在第一阶段,采用实验设计法(DoE)系统考察了四个参数对水杨酸片溶出度的影响。片剂装载方式是影响溶出度的最重要参数;流速和池体内径(ID)也有显著影响,温度对溶出的影响可以忽略不计。在第二阶段,由四名不同的分析人员在不同的流池法装置上(即四名合作者)进行溶出试验,以进行重复性和重现性评估,并确定PVT的初步可接收标准。第二阶段的实验条件是将片剂放置在带有玻璃珠的片剂支架上,池体ID为12 mm,流速为16 mL/min,温度为37℃,90分钟采集样品。第五个合作者的数据证实了PVT的可重复性。
制药/生物制药
2024/06/14

介质脱气小能手 | 华溶DGU-900在线溶媒脱气机,只为溶出仪而生
在进行溶出实验过程中,介质中溶解气体的释放是影响溶出结果的主要变动因素之一。因此,对溶出介质进行脱气处理变得尤为重要,并应将具体脱气程序写入溶出检测操作的标准操作规程。
制药/生物制药
2024/04/15
好文推荐 | 奈帕芬胺眼用混悬液的体外释放试验方法
体外释放试验(IVRT)方法对于监测制药生产过程中批次间的质量变化以及表现仿制药与原研药的药物等效性非常重要。为了满足批准眼用混悬液仿制药的监管要求,需要进行体外释放研究。目前尚无药典或非药典方法用于奈帕芬胺眼用混悬液的体外释放研究。 目前的研究旨在筛选使用不同的常规和非常规的各种方法,以建立最合适的奈帕芬胺眼用混悬液的技术,然后优化方法参数并进行验证。试验使用带透析袋的桨法装置(USP 2型)、流通池装置(USP 4型)、水浴转瓶装置和Franz扩散池装置。使用USP 4型装置,在pH 7.4的模拟泪液(STF)中,药物在120分钟内的释放度约为83%,添加表面活性剂月桂醇硫酸酯钠(SLS)后,释放度增加到约97%。使用USP 2型和Franz扩散池装置,药物释放缓慢或未接近完全释放。然而,在水浴转瓶装置的情况下,观察到了突释曲线。药物释放量的估计是通过HPLC方法进行的,所有方法验证参数,如专属性、准确度、线性和精密度都在可接受标准范围内。
制药/生物制药
2024/04/02
公司名称: 深圳市华溶分析仪器有限公司
公司地址: 深圳市宝安区沙井街道马安山鞍胜路31号第5栋 联系人: 余丽梅 邮编: 518104 联系电话: 400-860-5168转4098
仪器信息网APP

展位手机站