
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转4098
仪器信息网认证电话,请放心拨打
米拉贝隆缓释片的体内外相关性溶出方法研究
2024/01/02 16:55
阅读:101
分享:方案摘要:
产品配置单:

华溶活塞泵流池法溶出系统DS-7CP
型号: DS-7CP
产地: 广东
品牌: 华溶仪器
¥100万 - 150万
参考报价
联系电话
方案详情:

作者:刘芳伶、范诗雨、周 群等
米拉贝隆缓释片的体内外相关性溶出方法研究
目的:流通池模拟难溶性药物米拉贝隆缓释制剂的溶出,建立米拉贝隆体内和体外的相关性(IVIVC)模型,开发具有预测能力的体外溶出方法。
方法:Loo-Riegelman法对三种不同释放速率制剂的体内血药浓度进行反卷积分获得相应的累积吸收百分数(Fabs%),建立体外溶出目标曲线。以纯水为试验介质,流速4.0mL·min-1的试验条件下进行制剂R(贝坦利®,50mg)和制剂T1、T2(50mg)的溶出试验,通过高效液相色谱法测定溶出度,梯形面积法获得制剂的累积溶出百分数(Fdiss%)。
结果:立了米拉贝隆缓释制剂体内累积吸收与体外溶出度之间的A级 IVIVC(回归系数大于0.9), 制剂的外部预测误差在规定范围内。
结论:研究建立的米拉贝隆缓释制剂IVIVC模型经验证具有较好的预测能力,该方法拥有的良好区分力及线性模型也可以为质量控。

一、简 介
膀胱过度活动症(OAB)是一种以夜尿、尿失禁、尿急、尿频为主要症状的泌尿系统疾病。OAB不仅会严重影响患者的生活质量,而且也会给患者带来巨大经济负担。一项来自欧洲4国的调查结果显示,过去40年OAB男女发病率分别为13.4% 和14.6%。在美国,每年用于OAB的治疗花费接近十亿美元。米拉贝隆是由日本安斯泰来 ( Astellas) 公司研发的首个用于治疗OAB的选择性β3肾上腺素受体激动剂。
美国食品药品管理局(FDA)指南推荐将体内外相关性(in vivo-in vitro correlation, IVIVC)作为预测性数学模型,用于描述剂型的体外特性(通常是药物溶解或释放的速率或程度)与相关的体内反应(如血浆药物浓度或药物吸收量)之间的关系。20世纪80至90年代,美国药典(USP)/ FDA的研讨会报告确定了IVIVC的发展目标为“使用溶出度试验替代生物等效性试验,并帮助制定溶出度规范”。米拉贝隆缓释片于2017年在中国批准上市,近5年间,国内仅一家企业研发的米拉贝隆缓释片通过一致性评价,且国内关于其溶出度/释放度及IVIVC方面的报道较少,已有的相关研究主要考察自研制剂与参比制剂在四条曲线下释放行为一致性。由于米拉贝隆属于生物药剂学分类系统(biopharmaceutical classification, BCS)Ⅱ类药,低溶解度是药物在体内吸收的限制因素,因此该制剂适合开展制剂IVIVC研究,建立具有预测能力的体外溶出方法。
本研究旨在利用流通池法对米拉贝隆缓释片进行IVIVC研究,用以区分空腹状态下制剂间的溶出行为,建立米拉贝隆的A级IVIVC,并评估模型对于药动学参数的预测误差是否在标准范围之内,这可能对监管机构的监管审查、评价制剂间的差异提供帮助。
二、仪器与试药
2.1 仪器
流通池(推荐使用华溶DS-7CP流池法溶出系统);1260 型高效液相色谱仪(Agilent);BSA224S-CW 型万分之一电子天平 ;Vortex-5 型可调式微型涡旋混合器(上海嘉鹏科技有限公司);TGL16M 型高速冷冻离心机;Eso-S15HUVF 型超纯水仪(湖南启沁环保科技有限公司);FE28 型pH 计 ( 梅特勒 - 托利多国际贸易 ( 上海 ) 有限公司 );KS-600VDE/3 型数控超声波清洗器(昆山市超声仪器有限公司)。
2.2 试药
米拉贝隆缓释片(原研制剂,商品名:贝坦利 ®, 制 剂 R), 生 产 厂 家:Avara PharmaceuticalTechnologies Inc.,规格:50mg,批号:20K2349;米拉贝隆缓释片(制剂T1),批号:210906,规格:50mg,由国内某药企提供;米拉贝隆缓释片(制剂T2),批号:20211011,规格:50mg,由国内某药企提供;乙腈(色谱纯,MREDA);甲醇(色谱纯,MREDA);乙酸铵(国药集团化学试剂有限公司);乙酸(国药集团化学试剂有限公司);水为纯化水。
2.3 溶出仪试验装置
全自动流通池溶出仪主要包含溶出主机、计算机精密测控系统、自动取样系统、恒温水浴等组成。溶出主机主要包含6个流通池,其直径约2 cm,高约10 cm,流通池内搭载内循环,以控制介质的涡旋流动,两端均配备由二氧化硅过滤垫片组成的过滤系统,模拟药物的吸收。试验过程中,恒温溶出介质通过操作控制系统集成的精密泵以精确稳定的流速从流通池底部进入,样品溶出液经过滤系统进入自动取样装置,待至取样时间点可自动取样。将药物投放入流通池,计算机精密测控系统设置相应参数,即可开始试验。
三、方法与结果
3.1 体内研究
3.1.1 制剂R与制剂T1的生物等效性试验
采用单中心、随机、开放、两制剂、三周期、三序列、部分重复交叉试验设计。委托海口市人民医院开展试验,并经其生物医学伦理委员会批准 审批件编号:2018-( 伦审 )-144,志愿者对研究知情同意,并自愿签署知情同意书,30例体检状况良好的健康志愿者入组该试验。每周期志愿者分别服用米拉贝隆缓释制剂T1或制剂R1片。分别在给药前0h和给药后0.5,1,1.5,2,2.5,3,3.5,4,4.5,5,6,8,10,12,24,48,72,96h采集血样4mL,并保存在含氟化钠的肝素钠抗凝真空管中。离心(4℃、1700×g、10min)分离血浆后保存至 -80℃超低温冰箱。采用经验证的LC-MS/MS法测定血浆样本中米拉贝隆的浓度。
3.1.2 制剂R与制剂T2的生物等效性试验
采用单中心、随机、开放、两制剂、两周期的自身交叉试验设计。委托长沙泰和医院开展试验,并经其伦理委员会批准 审批件编号:(2021)第(53)号,志愿者确定为体检状况良好的健康男性和女性,对研究知情同意,并自愿签署知情同意书。16例志愿者每周期按随机表单次空腹口服米拉贝隆缓释制剂T2或制剂R1片。分别于给药前0h及给药后 0.5,1,1.5,2,2.5,3,3.5,4,4.5,5,6,8,10,12,24,48,72,96 h分别采集静脉血约4mL并保存在提前预冷30min的 EDTA-K2+氟化钠抗凝管中。离心(4℃、1 700×g、10min)分离血浆后保存至-80℃超低温冰箱。采用经验证的LC-MS/MS法测定血浆样本中米拉贝隆的浓度。
3.1.3 药动学数据分析
人体单剂量口服米拉贝隆缓释制剂R、制剂T1、制剂T2后的平均血药浓度-时间曲线见图1,各项参数比值(ratio)的90%CI 见表1、表2。在 R与T1对比研究中,R与T1的Cmax、AUC0-t和AUC0-∞3个参数几何均值比(geometricmean ratio,GMR)的90%CI均在80%~125%范围内,相较制剂R,制剂T1的吸收速度稍慢、吸收程度稍低。在R与T2对比研究中,所有药动学参数GMR的90%CI均超出了80%~125%范围,提示制剂T2与制剂R未实现生物等效,制剂T2较制剂R的吸收速度更快、吸收程度更高。
3.1.4 目标曲线的确立
采用WinNonlin8.2 软件中的PK模块对米拉贝隆制剂R的人体血浆药物浓度-时间数据进行房室模型拟合,根据其AIC值评估为二室模型。利用Loo-Riegelman法对米拉贝隆的两项生物等效性(BE)研究中制剂R、制剂T1和制剂 T2的血浆药物浓度进行反卷积,解析体内各时间点的累积吸收百分数(Fabs%),即吸收曲线:
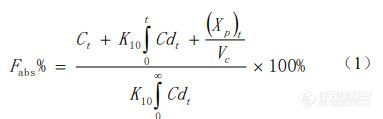
式中,Ct为t时间点的血浆药物浓度,(Xp)t为 t 时间周边室的药物量,Vc为中央室的表观分布容积,K10 为药物从中央室消除的一级速率常数(由文献报告的静脉数据中求算获得)。
如图1-C所示,三制剂在6~8h间吸收完全,制剂T2累积吸收高于制剂R,与体内BE结果相吻合;制剂T1与制剂R的累积吸收接近,说明经L-R计算出的三条累积吸收曲线可作为体外溶出的目标曲线。
3.2 体外研究
3.2.1 制剂R与制剂T1和T2溶出试验
在选择纯水为溶出介质、流速为4 mL·min-1、1.5 h自然溶胀后以 100mL·min-1 循环 6.5 h 的溶出条件下,分别于30,60,90,120,150,180,210,240,300,360,480 min 时间点采集样本。利用 HPLC法在柱温30℃,Agilent TC-C18 柱(4.6 mm×150mm,5μm,Agilent,USA),以乙腈 -50 mmol·L-1 的醋酸铵(3∶7,v/v)为流动相,流速为 1 mL·min-1,检测波长为248nm的条件下测定样品各个时间点的浓度。
3.2.2 溶出数据处理及分析
本研究采用的流通池法属于开环系统,经开环系统溶出所得到米拉贝隆的浓度 - 时间曲线为浓度 - 时间曲线,经数值积分转换为累积溶出百分数 - 时间曲线,其公式 如下:
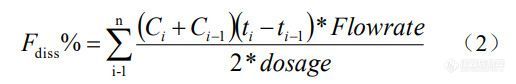
式中,Fdiss% 为累积溶出百分数,n 为体外溶
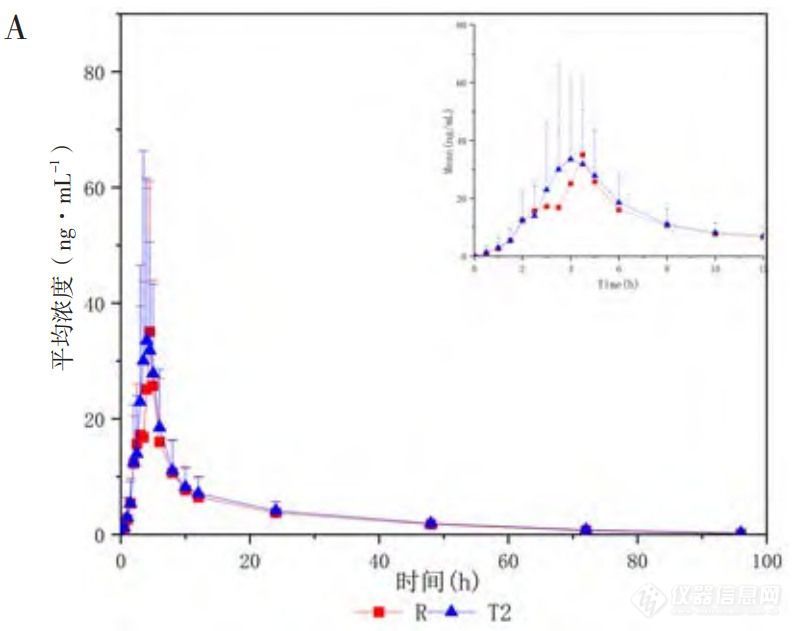
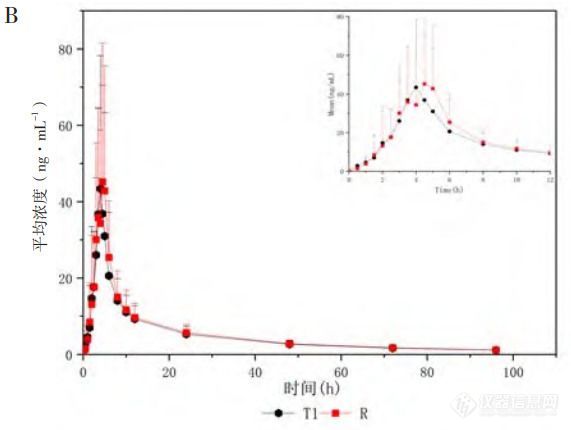
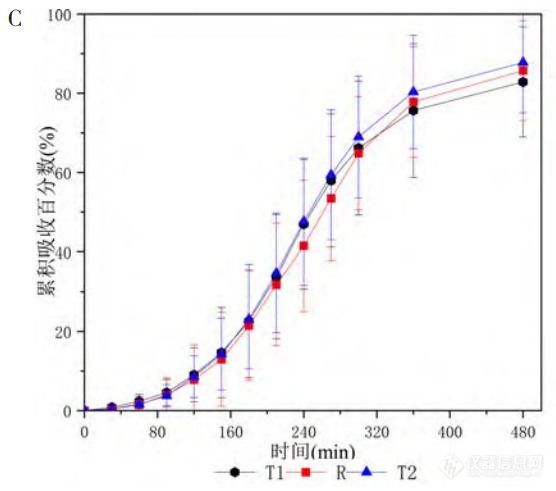
图1 米拉贝隆缓释制剂R、制剂T1和制剂T2的体内血药浓度-时间曲线及体内吸收分数-时间曲线
注:A.口服制剂R与制剂T1后的平均血药浓度-时间曲线(n=30,Mean±SD);B.口服制剂R与制剂T2后的平均血药浓度-时间曲线(n=16,Mean±SD);C.三制剂(R、T1和T2)的体外溶出目标曲线
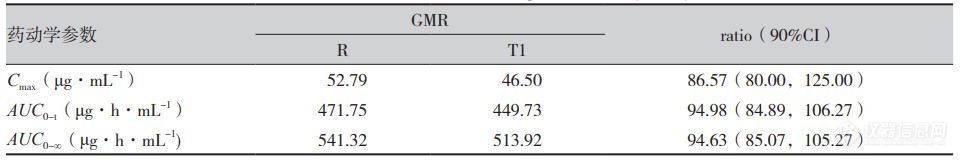
表1 空腹条件下米拉贝隆缓释制剂R与制剂T1几何均值比的90%可信区间(n=30)
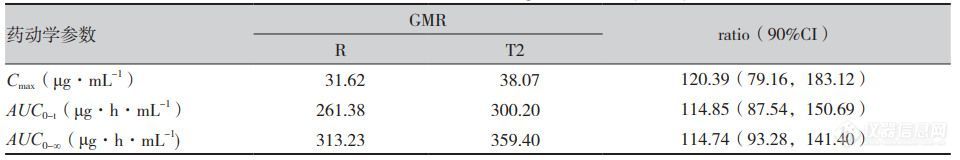
表2 空腹条件下米拉贝隆缓释制剂R与制剂T2几何均值比的90%可信区间(n=16)
出试验取样点数量,ti为0~n中的第i个整数时间点,ti-1为0~n中的第i-1个整数时间点,Ci为ti时间点浓度,Ci-1为ti-1时间点浓度,Flowrate为溶出介质流速,dosage为制剂规格。
以制剂R溶出曲线作为对照,微分溶出曲线(图2-A)表明,制剂R与制剂T1在整个试验过程中溶出速度变化趋势基本相似,2h后溶出过程较为平稳,实现了药物从制剂中的缓慢释放,展现出缓释制剂的缓释特性,而制剂T2在3h浓度达最大值时高出制剂R最大浓度的29%,溶出行为较制剂R差异大。同时,累积溶出分数-时间曲线(图2-B)表现出制剂间良好的区分力,制剂T2在180min后累积溶出分数高出制剂R约5%~10%,溶出速度快。三制剂在此溶出方法下所表现出的快慢趋势与目标曲线趋势一致。
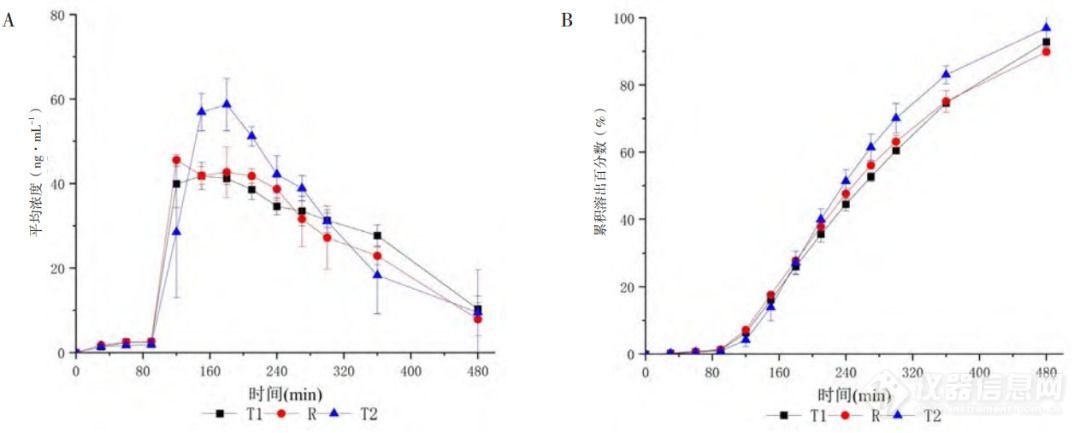
图2 米拉贝隆缓释制剂R、制剂T1和制剂T2的浓度-时间曲线(A)及体外累积溶出百分数-时间曲线(B)(n=4,Mean±SD)
3.3 体内外相样性研究
3.3.1 IVIVC模型的建立
将任意两种不同速率制剂的体外累积溶出分数(Fdiss%,mean)作为自变量, 经解析获得的体内累积吸收分数(Fabs%,mean)作为因变量, 利用最小二乘法进行线性回归建立A级IVIVC(见 图 3-A、图3-B、 图3-C)。制剂R与制剂T2建立的IVIVC方程为:Fabs%=0.9463Fdiss%+0.2451,R2=0.9906(P=0.940> 0.05)。制剂T1与制剂T2建立的 IVIVC方程为 Fabs%=0.9381Fdiss%+1.5570,R2=0.9879(P=0.931> 0.05)。制剂R与制剂T1建立的IVIVC 方程为:Fabs%=0.9681Fdiss%+0.4491,R2=0.9842(P=0.991> 0.05)。采用SPSS21软件进行独立样本t检验,计算95%CI。结果表明:单个制剂由两两制剂建立的 IVIVC 模型方程所获得的 Fabs% 与目标 Fabs%间差异无统计学意义(P>0.05)。为了验证已经建立的A级IVIVC,将比较三种制剂 Fabs% 实测值与预测值之间的差异,结果见图3-D、图3-E、图 3-F。所有模型的回归系数均大于 0.9,拟合优度接近于1,满足点对点的A级水平相关。制剂R与制剂T1和 T2间的预测吸收曲线与实测吸收曲线几乎相吻合,表明建立的A级IVIVC具有良好的预测能力。
3.3.2 IVIVC模型预测能力评价
BCSⅡ类药物体内药物的吸收主要受限于药物的溶出,认为药物进入胃肠道内的溶出过程等价于吸收过程 。根据FDA指南,单个制剂的药动学参数预测误差小于10%时,表明模型的预测能力较好。依据制剂R与制剂T2所建立的IVIVC模型获得制剂T1相应时间点预测的Fabs%,再利用如下卷积分公式获得制剂T1各时间点药动学参数:
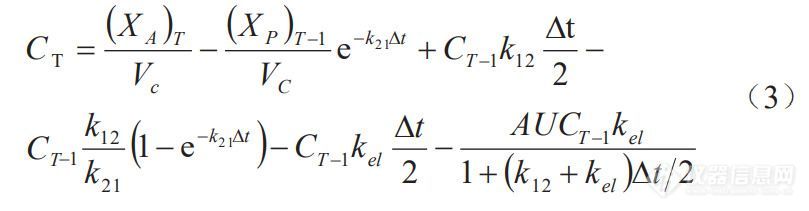
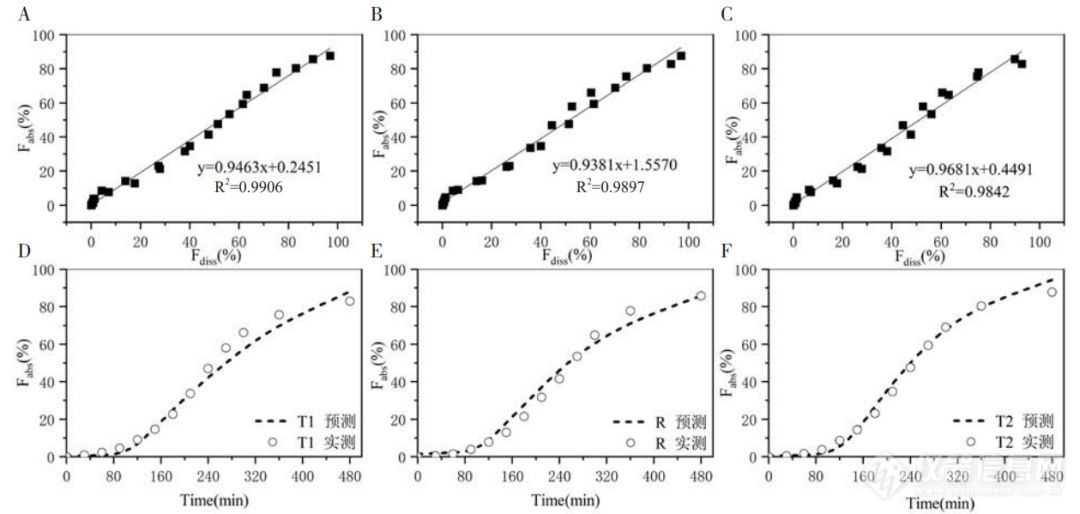
图3 制剂R(制剂T1和制剂T2)预测吸收曲线与目标曲线对IVIVC模型验证
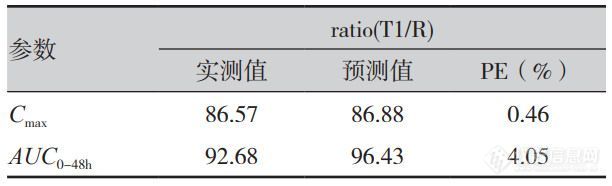
式中,CT为T时间点预测血药浓度,CT-1为T-1时间点浓度,(XA)T为T时间药物吸收的总量,(XP)T-1为T-1时间周边室的药量,Vc为中央室表观分布容积,K12与K21为药物在中央室与周边室间转运的一级速率常数(由静脉数据求算获得),Kel 为药物从中央室消除的一级速率常数;△t为t时间与t-1时间的差值。如表3所示,制剂T1的 Cmax、AUC0-48h 的预测误差(PE)分别为0.46%,4.05%,预测误差小于10%,IVIVC模型符合要求。
四、讨 论
在米拉贝隆缓释制剂的溶出行为研究中,倪冬胜的溶解度实验表明米拉贝隆的溶解度呈pH 依赖,随pH 增加而减小,0.1 mol·L-1的盐酸条件下,原研制剂可在2h 内溶出约40%,与目标曲线中前120min 的累积吸收百分率未超过10% 的结果有一定差距,由此推测米拉贝隆可能主要不在胃部释放。此外,欧洲公开审评资料也显示米拉贝隆主要释放部位在肠道,于是本研究选择了pH较高的介质。并在进行一系列条件摸索实验后发现在纯水条件下,制剂R显示出良好的相关性,这一结果也与文献一致。故本研究选择纯水作为释放介质。米拉贝隆缓释制剂是一种亲水凝胶骨架型缓释制剂,主要由亲水性聚合物控制药物的释放。文献表明,基质吸水后会在基体表面形成凝胶层,折叠的聚合物逐渐打开,凝胶层逐步溶蚀,一般情况下,药物主要通过聚合物溶胀和侵蚀的方式实现药物的缓慢释放。据此估计该制剂前期可能存在吸水溶胀的过程,所以本研究在经时8h 试验过程中先进行了1.5h无循环的自然溶胀过程。试验结果显示在此条件下,制剂R 的累积溶出曲线整体呈现先慢后快的趋势,可与解析出的目标曲线相吻合,表明该方法下体外溶出曲线可良好地反映出药物在体内的药动学过程。米拉贝隆属于BCSⅡ类药物,以流通池为溶出装置,难溶性药物的漏槽问题可在持续更换新鲜介质的过程中得到解决。开环式流通池序贯性模拟胃肠道的液体流动状态,可真实客观反映药物在体内的溶出行为。生物等效评价是以R与T1的药动学参数的几何均值比的90%CI落在 80%~125%范围内为标准。本研究因客观条件限制,两项研究分别在不同的临床试验机构进行,所获得的药动学参数有一定的差异。在模型能力评价时采用比值比较,一定程度上可减小误差,故本研究选择对制剂T1的AUC与Cmax的预测ratio值与实测ratio值进行比较。本研究中,建立的米拉贝隆缓释片溶出方法可以实现A级相关性,可以显著区分米拉贝隆缓释片不同制剂间的溶出能力差异。该方法可以为米拉贝隆缓释片的制剂开发、处方优化提供有力支持。
五、参考文献
略
下载本篇解决方案:
更多![]()



好文推荐 |依匹哌唑原位凝胶植入剂的体外释放研究
目的 建立一种依匹哌唑原位凝胶植入剂的体外释放方法,研究处方在体外的释放行为和缓释机制。 方法 以抗精神病依匹哌唑为模型药物,制备以聚乳酸⁃羟基乙酸共聚物( polylactide gly⁃ colic acid,PLGA)/醋酸异丁酸蔗糖酯( sucrose acetate isobutyrate,SAIB)为基质的原位凝胶植入剂。开发体外释放检测方法,采用多种体外释放装置研究依匹哌唑原位凝胶植入剂的释放差异, 考察体外释放的影响因素。
制药/生物制药
2024/06/24

好文推荐 | 水杨酸片在流池法溶出仪性能验证试验中的应用
流池法溶出仪是控释剂型、难溶性药物和许多特殊剂型(如混悬液、软胶囊、植入剂、微球和脂质体)溶出度测试的首选仪器。虽然流池法溶出仪已列入药典多年,但一直没有正式的性能验证试验(PVT)方法。在这项研究中,水杨酸片被用来开发流池法的PVT。水杨酸片在篮法和桨法装置上相同的溶蚀和零级释放机制,可作为流池法装置中PVT的潜在参考标准品。在第一阶段,采用实验设计法(DoE)系统考察了四个参数对水杨酸片溶出度的影响。片剂装载方式是影响溶出度的最重要参数;流速和池体内径(ID)也有显著影响,温度对溶出的影响可以忽略不计。在第二阶段,由四名不同的分析人员在不同的流池法装置上(即四名合作者)进行溶出试验,以进行重复性和重现性评估,并确定PVT的初步可接收标准。第二阶段的实验条件是将片剂放置在带有玻璃珠的片剂支架上,池体ID为12 mm,流速为16 mL/min,温度为37℃,90分钟采集样品。第五个合作者的数据证实了PVT的可重复性。
制药/生物制药
2024/06/14

介质脱气小能手 | 华溶DGU-900在线溶媒脱气机,只为溶出仪而生
在进行溶出实验过程中,介质中溶解气体的释放是影响溶出结果的主要变动因素之一。因此,对溶出介质进行脱气处理变得尤为重要,并应将具体脱气程序写入溶出检测操作的标准操作规程。
制药/生物制药
2024/04/15
好文推荐 | 奈帕芬胺眼用混悬液的体外释放试验方法
体外释放试验(IVRT)方法对于监测制药生产过程中批次间的质量变化以及表现仿制药与原研药的药物等效性非常重要。为了满足批准眼用混悬液仿制药的监管要求,需要进行体外释放研究。目前尚无药典或非药典方法用于奈帕芬胺眼用混悬液的体外释放研究。 目前的研究旨在筛选使用不同的常规和非常规的各种方法,以建立最合适的奈帕芬胺眼用混悬液的技术,然后优化方法参数并进行验证。试验使用带透析袋的桨法装置(USP 2型)、流通池装置(USP 4型)、水浴转瓶装置和Franz扩散池装置。使用USP 4型装置,在pH 7.4的模拟泪液(STF)中,药物在120分钟内的释放度约为83%,添加表面活性剂月桂醇硫酸酯钠(SLS)后,释放度增加到约97%。使用USP 2型和Franz扩散池装置,药物释放缓慢或未接近完全释放。然而,在水浴转瓶装置的情况下,观察到了突释曲线。药物释放量的估计是通过HPLC方法进行的,所有方法验证参数,如专属性、准确度、线性和精密度都在可接受标准范围内。
制药/生物制药
2024/04/02
公司名称: 深圳市华溶分析仪器有限公司
公司地址: 深圳市宝安区沙井街道马安山鞍胜路31号第5栋 联系人: 余丽梅 邮编: 518104 联系电话: 400-860-5168转4098
仪器信息网APP

展位手机站