推荐厂家
暂无
暂无




 400-860-5168转3614
400-860-5168转3614
 留言咨询
留言咨询
 400-860-5168转5911
留言咨询
400-860-5168转5911
留言咨询
 400-860-5168转1978
留言咨询
400-860-5168转1978
留言咨询



【序号】:1【作者】: 彭朋元唯安胡薏慧【题名】:载药医疗器械临床试验质量控制要点【期刊】:药物评价研究. 【年、卷、期、起止页码】:2021,44(02)【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2021&filename=YWPJ202102007&uniplatform=NZKPT&v=rN02X8_as4SvGbMhBnHoiLNembr77XHnzUEZj_uYmgzp0lXDalaFhOSxcGcSttlk
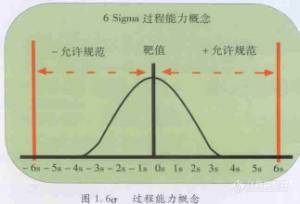
6σ质量标准在临床实验室质量控制的应用(王治国)6σ质量标准 6σ(Sigma)质量标准是摩托罗拉公司在八十年代质量管理策略的基础。其思想是开发的生产过程很完善以至生产出无缺陷的产品。其规定了6σ的过程变异落在产品的"公差"范围(允许规范)之内,如图1所示。摩托罗拉公司把它的质量管理目标称为6σ质量标准,这里σ是正态分布的标准偏差。而6σ质量标准意味着不合格品率是每百万件产品中有3件不合格。换句话说,偏离分布均值超过6标准偏差分布的百分率是0.0003%!在某些公司看来,99%的合格率即每百件产品有一件不合格品已相当完美。可是,让我们看看,在这一质量标准下,像美国邮政服务这样的大量业务每天将会丢失或读寄500万件寄邮件。这还将意味着每一百个病人,将有一被误诊或被错误治疗,或处方开错口在摩托罗拉公司,6σ质量标准是公司努力追求的目标,实际的不合格品率目前比这要高。但是,这一标准确实给我们一个暗示:无论质量水平有多高,总还有进一步改进的余地。[img]http://ng1.17img.cn/bbsfiles/images/2005/11/200511152257_10220_1618686_3.jpg[/img]
求《临床试验控制程序》。谢谢










 我要推广仪器
我要推广仪器
 下载APP
下载APP