【序号】:1【作者】: 彭朋元唯安胡薏慧【题名】:载药医疗器械临床试验质量控制要点【期刊】:药物评价研究. 【年、卷、期、起止页码】:2021,44(02)【全文链接】:https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CJFD&dbname=CJFDLAST2021&filename=YWPJ202102007&uniplatform=NZKPT&v=rN02X8_as4SvGbMhBnHoiLNembr77XHnzUEZj_uYmgzp0lXDalaFhOSxcGcSttlk
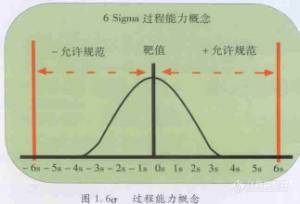
6σ质量标准在临床实验室质量控制的应用(王治国)6σ质量标准 6σ(Sigma)质量标准是摩托罗拉公司在八十年代质量管理策略的基础。其思想是开发的生产过程很完善以至生产出无缺陷的产品。其规定了6σ的过程变异落在产品的"公差"范围(允许规范)之内,如图1所示。摩托罗拉公司把它的质量管理目标称为6σ质量标准,这里σ是正态分布的标准偏差。而6σ质量标准意味着不合格品率是每百万件产品中有3件不合格。换句话说,偏离分布均值超过6标准偏差分布的百分率是0.0003%!在某些公司看来,99%的合格率即每百件产品有一件不合格品已相当完美。可是,让我们看看,在这一质量标准下,像美国邮政服务这样的大量业务每天将会丢失或读寄500万件寄邮件。这还将意味着每一百个病人,将有一被误诊或被错误治疗,或处方开错口在摩托罗拉公司,6σ质量标准是公司努力追求的目标,实际的不合格品率目前比这要高。但是,这一标准确实给我们一个暗示:无论质量水平有多高,总还有进一步改进的余地。[img]http://ng1.17img.cn/bbsfiles/images/2005/11/200511152257_10220_1618686_3.jpg[/img]
求《临床试验控制程序》。谢谢
特殊医学用途配方食品相关配套法规密集的出台,为规范管理工作,加强质量安全监管提供了保障。产品的注册申报只是一种规范手段和流程规则;企业的长远发展和产品持续的生命力关键还是在于研发创新与质量控制,开发高品质特医食品将成为相关特医食品企业今后工作的重中之重。在新的政策法规要求下特医食品企业如何进行产品研发?研发过程中要注意些什么?临床营养支持的重点如何判断?生产工艺如何制定?生产工艺与研发报告中频繁出现的问题如何规避......时间:2017年3月18日-20日(18日全天报到)地点:石家庄(地点确定直接通知报名者)议程安排:(一)、会议拟出席部分主讲嘉宾及议题(排名不分先后) 嘉 宾:《“特殊医学用途配方食品发展与质量安全”目录》课题组 相关专家报告主题:特殊医学用途配方食品的质量标准介绍嘉 宾:中国疾病预防控制中心营养与健康所研究员 杨晓光报告主题:中国特殊医学用途食品法规现状和展望报告大纲:1、特殊医学用途配方食品相关法规解读;2、常见特定全营养配方食 品中可调整的营养素含量及依据;3、产品配方的技术要求;4、特医 食品临床试验的实施;5、如何解决技术审评中遇到的专业问题;嘉 宾:美国外科学院院士(FACS)、中国科学院北京转化医学研究院\航空总 医院肿瘤医学中心主任、北京康爱营养医学研究院院长、中国抗癌协 会肿瘤营养与支持治疗专委会主任委员 石汉平报告主题:肿瘤营养若干问题报告大纲:1、肿瘤患者是否应该进行营养支持?2、如何判断肿瘤患者是否需要营养支持?3、肿瘤患者为什么要营养治疗?4、肿瘤患者需要什么样 的营养支持?5、肿瘤患者怎样实施营养支持?嘉 宾:上海交通大学国家健康产业研究院特殊医学用途配方食品研究所执行 所长、杭州纽曲星生物研究所所长 裘耀东 报告主题:特殊医学用途配方食品研发与临床应用报告大纲:1、FSMP的作用;2、FSMP研发、生产流程;3、 FSMP临床应用; 4、FSMP研发实例嘉 宾:中国医师协会营养医师专业委员会专家,河南临床营养学会副主任委 员、郑州大学第一附属医院营养科主任 陈改云报告主题:FSMP与肿瘤患者的营养支持报告大纲:1、营养治疗概述;2、营养治疗流程;3、FSMP规范应用; 4、临床案例分享嘉 宾:清华大学生物力学与医学工程研究所博士、副教授 赵红平报告主题:肿瘤全营养配方食品的研发与应用报告大纲:1、肿瘤特医食品研发的技术路线;2、基于肿瘤病人的营养代谢规律(以肠内营养支持为例);3、特医食品营养作用的再认识;4、肿瘤特医食品的研制与验证;嘉 宾:西安朗睿生物科技有限公司 佐建峰报告主题:肠内营养产品开发的战略思考报告大纲:1、肠内营养的基本知识;2、肠内营养产品的机遇和挑战;3、肠内 营养的国内外市场状况和趋势;4、肠内营养产品开发的投资;5、肠 内营养产品的市场策略;嘉 宾:西安力邦临床营养有限公司研发总监 杨 宏报告主题:全营养检测技术与配方设计报告大纲:1、全营养配方设计探究;2、特殊医学用途配方食品检测解决方案; 3、全营养检测实验室的建设;4、全营养检测技术设备的配套选型; 嘉 宾:罗特集团有限公司副总裁 狄志鸿报告主题:特医食品研究的理论依据及发展前景报告大纲:1、特医食品是解决临床病人什么问题,和药品,保健食品区别在那里? 2、特医食品与营养医学的关系;3、营养与人体关系;4、研究特医食品技术难点和需要解决的问题;5、前景;相关报告继续预约中,敬请持续关注!(二)、专家对话沙龙环节 针对特殊医学用途配方食品的相关法律法规如何贯通运用、研发过程中的实际问题、临床营养支持的应用以及产业发展面临的机遇与挑战等,参会者与专家进行互动提问解答,并进行深入探讨,探寻产业的健康成长之路。联 系 人: 马超 电话:010―51606934 邮 箱:1683101345@qq..com
众所周知,目前定量检测室内质控的主要工具为质量控制图。工作中经常遇到对质量控制图的理解和应用问题,下面谈一些基本认识,供同道们参考。 一、“事后检查”与“予防为主” 日常工作中,当每批检验结果出来后,都会对检验结果进行复核,检查有无漏项、填错结果等等,并对一些异常结果的可信度进行评估,显然这对保证检验结果是否正确无误有重要作用,但也不能否认,这种复核制度有许多局限性,例如患者间的结果各不相同,检测结果出来前,无法知道每一患者测定值应该是多少,有怀疑时经常进行重复检查,但重复检查也只是检查重复性,如存在系统误差,复查也发现不了问题。 大家知道,质控图法是从工业中引进临床实验室的。1924年W.A.Shewhart发明了质量控制图,直到1951年Levey-Jennings才将Shewhart质控图引入临床实验室,将临床实验室的质量控制推向了一个新阶段,质控图也成为临床实验室内质控的主要方法。但临床检验与企业生产有许多不同,工业生产中,每一批产品的不管数量多大,其规格是事先规定了的,而且都是一致的,但由于临床标本某一成分的含量事先并不知道,检测结果是否正确的评估就带有一定主观性、评估的结果也带有一定不确定性。分析阶段的质量控制是通过检测过程的控制来保证检验质量的。其基本思路是检测条件得到控制,其检验结果的准确性(与真值或理想值的偏倚)及精密度是满足临床要求的话,则检测过程如果是在控制条件下进行的,那么检验结果就应该是可靠的,反之如果检测过程失控,检验结果将是不可靠的。所以质控图法是通过对检测过程是否在控的判断,来推论检验结果是否可靠,这是总体上的判断。这是一个重要的思想,但总体上的判断不能完全代替“个体的判断。”因为一批检验结果中,难免有个别非常“异常”、难以解释的结果,这就需要“个别对待、个别处理”;同时质控图法用来判断检测过程是否在控,并作出该批结果可否发出时,还有一个前提:即送检标本的质量必须是合格的。
第一章 总则第一条 为加强药物Ⅰ期临床试验的管理,有效地保障受试者的权益与安全,提高药物Ⅰ期临床试验的质量,根据《中华人民共和国药品管理法》、《药品注册管理办法》、《药物临床试验质量管理规范》等相关规定,参照国际通则,制定本指导原则。第二条 本指导原则适用于药物Ⅰ期临床试验,旨在为药物Ⅰ期临床试验的组织管理和实施提供原则性指导。人体生物利用度或生物等效性试验可参考本指导原则。第二章 职责要求第三条 申办者应建立筛选国家药物临床试验机构和研究者的程序和标准,选择、委托获得相关专业资格认定的药物临床试验机构进行药物Ⅰ期临床试验。 第四条 申办者应建立质量保证体系,对临床试验的全过程进行监查和稽查,确保临床试验的质量,保障受试者的权益与安全。申办者可以委托合同研究组织执行临床试验中的某些工作和任务。申办者对临床试验的真实性及质量负最终责任。第五条 国家药物临床试验机构的药物Ⅰ期临床试验研究室负责药物Ⅰ期临床试验的实施。研究者应遵循《药物临床试验质量管理规范》,并严格执行临床试验方案,对临床试验的真实性和质量负责,保护受试者的权益与安全。第六条 临床试验生物样本分析应在符合国家有关管理规定的实验室进行。第七条 伦理委员会在审查试验方案和知情同意书等资料的基础上,应针对药物Ⅰ期临床试验的特点,重点关注试验风险的管理及控制、试验方案和知情同意书的修改等,对临床试验进行必要的监督检查,加强对受试者的权益与安全的保护。
多年来临床检验质量一直是检验工作者关注的核心问题,如何做好临床实验室的质量管理,特别是在与国际临床实验室质量管理模式接轨的同时,提出适合于我国经济发展现状、适合我国国情的实验室基本资格要求、适用于不同级别临床实验室的质量管理方案已成为我们面临的重要任务。 根据国际标准化组织ISO 15189:2003《医学实验室-质量和能力的专用要求》中的定义,以诊断、预防、治疗人体疾病或评估人体健康提供信息为目的,对取自人体的材料进行生物学、微生物学、免疫学、化学、血液免疫学、血液学、生理学、细胞学、病理学或其它检验的实验室统称为临床实验室(以下简称实验室),也称之为医学实验室。针对临床实验室质量管理,一些发达国家和国际组织已经出台了一些法律和标准供我们借鉴。美国国会于1967年就通过了专门针对临床实验室质量管理的法律,即临床实验室改进法案(Clinical Laboratory Improvement Act 1967,简称CLIA 67)。在实行此法案20年后,1988年又通过了对CLIA 67的修正案临床实验室改进法案修正案(Clinical Laboratory Improvement Amendment 88,简称CLIA 88),并于1992年正式实施。法国政府也于1999年11月26日发布了NOR:MESP9923609A《关于正确实施医学生物分析实验的决议》。1999年,国际标准化组织制订了医学实验室的管理标准,即ISO 15189《医学实验室-质量和能力的专用要求》。目前国际上对临床实验室的质量管理主要分为以CLIA 88为代表的法律文件和ISO发布的推荐标准两种形式。ISO 15189主要强调实验室内部质量体系的建立,在此基础上建立的实验室认可制度是一种自愿行为,是实验室质量保证的较高标准;CLIA 88着眼于政府对临床实验室质量的外部监控,是政府对实验室强制执行的资格要求,两者存在互补性。这些文件对我们制定临床检验质量保证方案提供了很好的参考作用。
国食药监注483号各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后卫生部药品监督管理局: 为加强药物Ⅰ期临床试验的管理,有效的保障受试者的权益与安全,提高药物Ⅰ期临床试验的研究质量与管理水平,根据《中华人民共和国药品管理法》、《药品注册管理办法》和《药物临床试验质量管理规范》等有关规定,国家局组织制定了《药物Ⅰ期临床试验管理指导原则(试行)》,现予印发。请你局组织本行政区域内药物临床试验机构学习,参照执行。 附件:《药物Ⅰ期临床试验管理指导原则(试行)》起草说明 国家食品药品监督管理局 二O一一年十二月二日药物Ⅰ期临床试验管理指导原则(试行)第一章 总则 第一条 为加强药物Ⅰ期临床试验(以下简称I期试验)的管理,有效地保障受试者的权益与安全,提高Ⅰ期试验的研究质量与管理水平,根据《中华人民共和国药品管理法》、《药品注册管理办法》、《药物临床试验质量管理规范》等相关规定,参照国际通行规范,制定本指导原则。 第二条 本指导原则适用于Ⅰ期试验,旨在为Ⅰ期试验的组织管理和实施提供指导。人体生物利用度或生物等效性试验应参照本指导原则。第二章 职责要求 第三条 申办者应建立评价药物临床试验机构的程序和标准,选择、委托获得资格认定的I期试验研究室进行Ⅰ期试验。 第四条 申办者应建立质量保证体系,对I期试验的全过程进行监查和稽查,确保临床试验的质量,保障受试者的权益与安全。 第五条 申办者可以委托合同研究组织(CRO)执行I期试验中的某些工作和任务。委托前对合同研究组织的研究条件、能力、经验以及相应的质量管理体系进行评价。当合同研究组织接受了委托,则本指导原则中规定的由申办者履行的责任,合同研究组织应同样履行。申办者对临床试验的真实性及质量负最终责任。 第六条 Ⅰ期试验研究室负责Ⅰ期试验的实施。研究者应遵循临床试验相关法律法规、规范性文件和技术指导原则,执行临床试验方案,保护受试者的权益与安全,保证临床试验结果的真实可靠。 第七条 药物临床试验生物样本分析应在符合《药物临床试验生物样本分析实验室管理指南》(以下简称《实验室管理指南》)的实验室进行。从事药物临床试验生物样本分析的实验室均应接受药品监督管理部门的监督检查。 第八条 伦理委员会应针对Ⅰ期试验的特点,加强对受试者权益与安全的保护,重点关注:试验风险的管理与控制,试验方案设计和知情同意书的内容,研究团队的人员组成、资质、经验,受试者的来源、招募方式,实施过程中发生的意外情况等。
中药临床试验申报资料中存在的问题SFDA药品审评中心审评一部许青峰一、对药品注册临床要求的理解: 总体要求、分类要求、受试例数二、试验常见问题试验设计、安全性数据总结分析三、对药品注册临床要求的理解(一)总体要求1、药物临床研究必须经国家药品监督管理局批准后实施,必须执行《药物临床试验质量管理规范》(GCP)2、药物的临床研究包括临床试验和生物等效性试验。3、申请新药注册、应当进行临床试验和生物等效性试验,申请已有国家标准的药品注册(仿制药),一般不进行临床研究,需用工艺和标准控制药品质量的中成药,应当进行临床试验。4、临床试验分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期。申请新药注册应当进行Ⅰ、Ⅱ、Ⅲ期临床试验。有些情况下可仅进行Ⅱ期和Ⅲ期。或者Ⅲ期临床试验。5、药物临床研究的受试例数应当根据临床研究的目的,符合相关统计学的要求和本办法所规定的最低临床研究病例数要求。
关于印发药物临床试验伦理审查工作指导原则的通知 国食药监注436号 2010年11月02日 发布 各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后卫生部药品监督管理局: 为加强药物临床试验的质量管理和受试者的保护,规范和指导伦理委员会的药物临床试验伦理审查工作,提高药物临床试验伦理审查工作质量,根据《药品注册管理办法》和《药物临床试验质量管理规范》的有关规定,国家局组织制定了《药物临床试验伦理审查工作指导原则》,现予印发。请你局指导本辖区内药物临床试验机构学习,参照执行。 附件:药物临床试验伦理审查工作指导原则起草说明 国家食品药品监督管理局 二○一○年十一月二日
室内质量控制(IQC,internal quality control,简称“室内质控”):就是用一种稳定物质,通过您自己重复检测得到这种物质在您检测系统上的一个检测值,通过统计方法建立质控范围,考察检测结果是否准确和稳定,决定了当批实验测定是否有效,检验报告能否发出。也就是客观评价“稳不稳”。▋ 室间质量评价(EQA,external quality assessment,简称“室间质评”):用一种稳定物质,分发到各个实验室,用各种仪器分别检测该物质,得到一组检测值。通过统计方法得到该物质的检测值可接受范围。用来评判您的试验系统是否与其他试验系统结果一致的质控方法。也就是客观评价“准不准”。▋ 试点单位要根据相关技术规范,制订全过程标准操作程序(SOP),在基因测序技术操作过程中遵照执行。要按规定开展实验室室内质量控制,参加国家卫生计生委临床检验中心组织的室间质量评价,或定期进行有效的实验室室间比对。(来源:《高通量基因测序技术临床应用试点工作方案》)
尿液质量控制的一般情况在常规试验中,虽然尿液分析仪的使用一般不受人为因素的影响,但尿液分析准确与否却受许多因素的影响。这些影响因素可以出现在分析前、中、后三个环节。分析前的质量控制(简称质控)主要包括样品的标签、收集样吕使用的容器和样品收集的时间、尿标本新鲜程度等。一般均应在取样后2小时内完成检测,否则可影响模块上所有检查项目结果的准确性。分析后的质控主要包括参考值范围的认可,判定试验结果是否受药物的干扰和病理物质的影响,报告单结果的书写和报告单回报时间等方面。分格中的质控主要包括化妆品和试带条的准确度、试带条的效期、仪器的校正、仪器操作正确程度和尿液标本中影响因素及处理等方面。
上海疾控中心与辉瑞中国签合作协议,建设疫苗临床试验基地。(新浪)
2010年回顾之九,临床试验失败录 http://img2.jiansuo.net/blog/upload/74/75/t_51484.jpg http://www.dxyer.cn/images/zoomin.gif 2010年大药厂经历不少重大临床试验失败或挫折(健康博客)新药研发处处有风险,过临床试验一期,也许比例最高,但进入临床II和III时,风险就明显加大.尤其在FDA提高对新药审批的门槛,对安全和疗效优越性特别看重时,药厂的临床试验就需要精心设计,必须拿出过硬数据,否则FDA拒绝批准可能性很大,至少会要求补充更多临床试验数据,拖延额二年以上时间.在过去的一年中,我们既看到让人鼓舞的临床研究进展和21种NCE新药上市,同时也看到不少令人可惜和遗憾的临床后期失败或重大挫折,在以下的失败产品名录中,不乏有曾经抱很大希望,甚至很有可能成为重磅药. 有的项目真的很可惜,比如罗氏德糖尿病药,临床效果比默克的药还好,就是因为安全因素,需要重新考虑新剂量,估计未来凶多吉少. 阿斯利康与雅培合作开发复方药,并不是完全没戏,但因为最佳上市时机错过,从商业角度考虑,只能放弃.所有这些项目的失败对这些大药厂打击很大.裁人成了减少损失的必然选择.临床试验失败之后,礼来决定结束糖尿病新药, 10月阿斯里康终止骨质疏松药开发项目,核销4.4亿研发损失 12月礼来皮肤癌药物III期临床试验受挫 患者死亡后期试验停止12 月 因为临床IIb失败,西雅图遗传公司放弃AML新药开发 9月罗氏阿瓦斯丁治疗胃癌临床III期失败 2月,临床效果欠佳,辉瑞终止骨关节疼抗体药研发项目 6月礼来老年痴呆药物 semagacestat临床试验失败 8月 诺华在二度遭遇临床III失败后,被迫放弃肺癌ASA404药物开发项目 11月辉瑞因III期临床试验年老年痴呆药 Dimebon失败而放弃 3月罗氏糖尿病新药taspoglutide因为有高血压风险被迫延迟申报甚至放弃 6月葛兰素发现其收购的新药临床试验疗效很弱,肾毒风险大而彻底放弃 12月强生/阿斯利康由于安全因素终止双方合作项目 12月FDA拒绝批准强生与其合作伙伴合作开发的抗生素ceftobiprole 1月因被FDA拒绝,错失商机,阿斯利康决定放弃与雅培合作开发的降血脂复方药 12月评论: 做新药研发,临床试验风险很大, 国内很少有公开报道和承认新药临床试验失败的消息, 究竟是我们的临床试验百发百中, 还是为了保护民族工业, 让效果一般的新药过关? . 如果未来还是这样的局面,那说明国家的钱被大忽悠了. 老百姓就更被蒙了.
[center]微生物实验室规范化质量控制 [/center]微生物实验室规范化质量控制 卫生部北京医院 质量是临床微生物实验室的生命,正确的鉴定和药敏报告对医生的诊断和治疗有重要的参考价值,反之会误导医生。所以实验室应建立完善的质量控制制度以保证检验质量的正确和稳定。一、临床微生物实验室要建立质量控制的基本范围和内容室内质控是室间质控的基础,而室间质控是对室内质控的验证,质量是临床微生物实验室的生命。在新发感染性疾病(emerging infectious disease)不断发生的形势下,临床微生物室的检验质量保证尤其重要。(一)建立标本接种前的质量控制制度 没有好质量的标本,实验室采取任何先进的分离、鉴定和药敏技术都是无济于事的。建议成立由医生、护士和细菌室参与的监督责任程序,对不同的标本规定不同的接种时间范围,如:痰标本应规定在留取标本后1 小时内送至实验室,细菌室应在收到标本后30分钟内接种完毕;粪便标本应采集后立即接种;血液标本种入培养血瓶后立即送检或于室温下保存,切不能放冰箱;尿标本应于2 小时内送实验室,实验室收到标本后应于30分钟内接种完;拭子标本采集后应全部采用输送培养基送到实验室检查。 (二)建立细菌分离率质控系统呼吸道标本或可能含嗜血杆菌的标本一定要接种巧克力培养基;含5%脱纤维绵羊血琼脂;采用5%-10%的CO2培养系统;培养周期24~72 h。 考察指标:嗜血杆菌属在常规送检标本中的分离率应不低于30%~50%;肺炎链球菌的分离率不低于5%~10%;卡他莫拉菌的分离率不低于2%~5%。如果在1周中未分离出上述苛养菌应认真查找原因。(三)药敏质控系统的规范化质控的抗菌药物种类应包括全部常用抗菌药物;正确选择质控菌株,如金黄色葡萄球菌ATCC25923 用于纸片法药敏的质控,而金葡菌ATCC29213用于稀释法药敏质控;建立3种系统药敏质控:30 d初始质控,在30 d内失控不超过3次时可进入1周质控。如失控超过3次应进入5 d的纠控系统,经5 d纠控,合格后方可进入1周质控。1周质控是维持日常质控的形式,一旦失控应进入纠控系统,按纠控规则处理。(四)鉴定质控系统 包括自动化设备鉴定系统、商品鉴定系统如API、单一的生化反应管及原材料,原则上应进行每换批号时都要有质控这也称为批批检。特别容易被忽略的是羊血的质量,如羊血中是否含抑制细菌生长的物质;M—H琼脂中胸腺嘧啶含量。可通过平皿抑制试验检测羊血是否含抗生素,在涂有细菌的平皿不同方位滴入羊血数滴,35℃培养过夜观察结果。要求在滴有羊血的部位不出现抑菌环;用粪肠球菌ATCC29212 或ATCC33186质控菌株测试MH琼脂上氨苄西林的抑菌环,若产生20mm或更大的抑菌环,表示合格。 (五)质控记录要真实,要有失控(超过质控允许范围的结果称为失控)记录,并认真的寻找原因和解决问题。(六)质量控制应该是每个有发报告检验人员的职责,不要把质控工作让一个人去做,在实验室要树立质量意识。 (七)室间质量控制 二级或以下医院应参加本地区或本市的质量控制活动;三级医院除参加本地区的质控活动外还应该参加全国或全球的质量控制活动。二、细菌分离和鉴定质量控制实施 细菌鉴定的质量由多种因素组成,所以细菌鉴定的质量控制应包括细菌分离的质量控制、染色液的质量控制、鉴定试剂的质量控制、鉴定设备的质量控制等。(一)分离培养质量控制用血琼脂和巧克力培养基接种肺炎链球菌、B-溶血和草绿色溶血的链球菌、流感嗜血杆菌等苛养的标准菌株或经标准化试剂已证实结果正确的菌株进行测试,判断实验室的分离能力。(二)染色[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]量控制 每天在开始用染色液前应检查染色液的质量和检测有无污染。(三) 鉴定试剂质量控制购入新的试剂;试剂更换批号;更换生产厂家;结果发生异常等情况时均应选择相应标准菌株进行质量控制。(四)自动化设备试剂质控要求同鉴定试剂;设备软件升级、修改版本、或大的维修时均应进行质量控制程序。三、药敏试验的质量控制实施 (一)建立合理的常规药敏种类 不同抗生素的抗菌谱不同、抗菌强度不同、细菌耐药谱不同、对不同的病原菌建立正确的常规药敏种类是非常必要的,错误的药敏种类的报告会误导医生,按CLSI要求和根据本单位用药习惯建立药敏常规试验的种类。(二)M-H琼脂平皿定量在琼脂扩散法药敏试验中,M-H琼脂厚度是严重影响纸片药敏试验结果的重要因素,琼脂培养基越厚抑菌环直径越小、反之则越大;克服的办法是要在保持水平的平台上(水平仪)定量倾注培养基,这个问题至今未被充分的重视。10cm的平皿应倾注23-25ml的培养基。 (三)抗菌药纸片贮藏纸片失效是造成药敏结果错误的最重要的原因,必须在-20℃或更低温度的低温冰箱中保存纸片,近期使用的纸片只需放4℃冰箱,一周后弃取,更换新纸片。(四)特殊菌药敏规范化(1)万古霉素中介菌株的报告: 中介肠球菌药敏试验应用含6mg/l万古霉素的琼脂,点种0.5麦氏单位菌液,35℃培养18~24h,点种菌生长者示为耐药,不生长者示为敏感, 葡萄球菌对万古霉素中介(中度敏感)时,应再测定其最低抑菌浓度(MIC)确定。(2) 葡萄球菌苯唑西林中介株: 用含4%NaCL和每升含6 mg 笨唑西林的琼脂,点种0.5麦氏单位的菌液,置35℃培养24h,生长者为耐药株。不生长者为敏感株。(3)克林霉素报告:必须在“D”试验的基础上,“D“试验是在琼脂或血琼脂平皿上均匀涂被测菌,稍后,在 相距15~26mm分别贴15ug/片红霉素纸片和2ug 克林霉素纸片,培养18~24h后,在克林霉素侧出现切痕为“D”试验阳性,应报告克林霉素耐药,无切痕可报告敏感。 (4)耐甲氧西林金葡菌(MRSA):用头孢西丁纸片代替苯唑西林纸片,试验结果仍应报告为苯唑西林敏感或耐药。(5)肠外标本中分离的沙门菌属:当环丙沙星MIC=0.12~1.0mg/L低耐时用奈啶酸测试MIC,当MIC=2mg/l为中介度(I),MIC≥4 为耐药(R)。 四、其他仪器设备质量控制实施(一)培养箱的质量控制 每天第一个开箱和最后离开实验室的人员均应观察培养箱的温度并作质量控制记录 (二)CO2培养箱的质量控制 每天应记录CO2的含量并在质量控制图上作记录。(三)冰箱温度记录按每一冰箱存放的物品不同设置不同温度范围,一般在5?2℃每天观察并记录。(四)低温冰箱 每天观察并记录一次,要求温度变化不超过设定范围的上下2℃。五、真菌检测的质量保证的实施低免疫人群的增长、超广谱抗生素的广泛应用使真菌感染迅速增加,侵袭性念珠菌及曲霉感染使重症监护病房(ICU)的病死率明显上升,50-75%的病例不能在生前获得细菌学的阳性结果,因此加强真菌检测的规范化操作,提高检测阳性率及药敏试验的正确率十分必要。 (一) 真菌涂片的规范化 真菌涂片是真菌鉴定最重要的方法,真菌检验人员应熟悉真菌的形态特点;无菌体液标本涂片应离心浓缩以增加阳性率,并一定要检测菌丝;对痰及口腔标本要进行定量(菌落计数)或半定量(用加号表示,在分区划线的3个区都丰富生长报告为+++、依次类推2个区生长为++、1个区生长为+);组织标本涂片要研磨后涂片但要保护菌丝体的形态;在标本量许可的前题下涂片不应少于3张。 (二) 真菌鉴定的规范化对酵母样真菌如念珠菌或隐球菌在常规临床细菌室已有较成熟的方法学,可用显色培养基或自动化系统鉴定真菌;常规鉴定丝状真菌主要依赖于其生长过程中形态变化和特点,所以要求从事丝状真菌检测的检验人员应作专业短期培训;任何实验室都应建立规范的涂片鉴定方法,要正确区分酵母样真菌或丝状真菌、毛霉菌和曲霉(有顶囊),对不常见的真菌不要轻易作为污染菌报告,(三) 真菌药敏的质量保证建立稀释法药敏试验,报告MIC值的报告对临床更有参考价值;必须正确掌握终点判断标准,氟康唑、伊曲康唑、氟胞嘧定等药物终点允许有20%真菌生长而两性霉素B应无真菌生长作为终点;对无折点的抗真菌药物药敏试验报告可只报告MIC,不能报告敏感度。鉴定真菌到种水平是临床用药的重要的参考。(四) 要重视组织标本的培养首先要无菌采集组织标本,接种前要无菌作适当粉碎将组织内的菌丝体暴露。每一份组织标本必须同时做涂片和培养。(五)开展非培养方法的真菌检查试验,如-D葡聚糖和或诊断曲霉菌的GM试验,用于区别真菌、曲霉菌或细菌感染。
化学药临床试验申报资料中存在的问题卓宏目录:一、临床研究在新药开发过程中的地位二、临床研究的基础三、临床研究的必要条件四、临床研究的定义及内容五、各类新药临床研究的要求六、各期临床试验的目的七、临床研究应注意问题一、临床研究在新药开发过程中的地位临床前研究:化学合成,动物试验临床研究:Ⅰ期临床、Ⅱ期临床、Ⅲ期临床上市:Ⅳ期临床二、临床研究的基础1、药学基础2、药理毒理基础3、临床方案设计的基础考虑:有动物试验依据,可根据国外资料、文献,适应症不超出已上市的范围,安全性要注意动物试验结果及常规试验。
药物临床试验生物样本分析实验室管理规定(征求意见稿)第一章 总 则第一条 为提高临床试验生物样本分析实验室的分析质量,确保所产生的数据和结果的可靠性、完整性和科学性,根据《药品注册管理办法》、《药物临床试验质量管理规范》、《药物非临床研究质量管理规范》,参照国际通则,制定本规定。第二条 临床试验生物样本是指按照临床试验方案的要求、从临床试验受试者采集的需要加以分析的材料(如血浆、血清、尿液、粪便、组织和细胞等)。临床试验生物样本分析实验室(以下简称“实验室”)是指对临床试验生物样本中药物及其代谢物分析,以及对临床试验生物样本进行血液学、生物化学、生物物理学等学科的检测或评估,为药品注册申请提供数据支持的机构。第三条 所有经过国家食品药品监督管理局批准进行临床试验生物样本分析的实验室,均须遵循本规定。第二章 组织机构和人员第四条 实验室应建立完善的组织管理体系,任命负责人、质量保证部门负责人、分析负责人,并配备相应的实验人员。第五条 所有人员应符合以下要求:(一)具备严谨的科学作风和良好的职业道德以及相应的学历,经过专业培训,具备所承担的工作需要的理论知识、工作经验和业务能力;(二)严格履行职责,熟练掌握并严格执行与所承担工作有关的标准操作规程;(三)及时、完整、准确和清晰地进行实验记录,对实验中发生的可能影响实验结果的任何情况应及时报告;(四)对涉及保密的技术资料、受试者信息等履行其保密责任;(五)根据工作岗位的需要着装,遵守健康检查制度。第六条 实验室负责人应具备相应专业的理论知识,具备有效组织、指导和开展实验室业务工作的能力。实验室负责人的职责为:(一)负责或协调所在机构法人与申办者签订书面合同;(二)建立有效的联系机制,以保证与申办者、药物临床试验机构之间可以及时、有效地沟通;(三)建立完善的教育培训和考核制度,确保实验人员具有相应专业的理论知识、技能和经验,保存实验人员的学历资质、工作经历、专业培训以及工作性质描述等材料; (四)全面负责实验室的建设,确保实验室具有满足工作要求的各项条件;(五)组织制定和修改管理制度、技术规范和标准操作规程;(六)制定主计划表,掌握各项分析工作的进展;(七)聘任质量保证部门负责人;(八)在每项实验开始前,指定分析负责人,试验过程中确需更换时,应记录更换的原因和时间,并保留相关记录;(九)审查、批准实施方案、分析结果或分析报告;(十)指定专人负责档案资料的管理。第七条 实验室应设立独立的质量保证部门,其人员的数量应与其开展的分析工作相适应。质量保证负责人的职责为:(一)负责质量保证部门的工作安排和运行;(二)审核实施方案、实验记录、分析结果或分析报告;(三)根据每项分析工作的内容和持续时间制定稽查计划并实施稽查,详细记录稽查的内容、发现的问题、采取的措施等,并向实验室负责人和/或分析负责人报告;(四)定期或不定期检查实验室环境、设施、仪器设备和档案管理等;(五)参与标准操作规程的制定、审核标准操作规程,并保存标准操作规程的副本;第八条 每项分析工作必须指定分析负责人。分析负责人的职责包括:(一)制定该项目的实施方案;(二)全面负责该项目的运行管理、组织实施;(三)建立并验证分析方法;(四)确保所有参与该项目的实验人员明确各自所承担的工作,并掌握和执行相关的标准操作规程;(五)掌握工作进展,确保实验记录及时、完整、准确和清晰; (六)确保实验中偏离试验方案的情况及采取的措施均有详细记录;(七)整理、分析实验数据和结果,撰写分析报告;(八)及时处理质量保证部门的报告。
3月25日,由南开大学药学院团队自主研发的化药1类新药CP0119片取得国家药品监督管理局签发的《药物临床试验批准通知书》,获准用于治疗结肠慢传输的临床试验。该项目是南开大学作为申报单位的第二项获得临床批件的新药项目,团队曾在2017年获批了南开大学首个新药临床批件。流行病学调查结果显示,我国结肠慢传输发病率达7.3%~20.39%,且近年来发病率呈逐年上升趋势,严重影响患者生活质量,损害患者身心健康,而且可能引发多种严重并发症。现有治疗方法主要分为基础治疗和药物治疗,基础治疗包括调整生活方式和认知治疗,经过4-8周的基础治疗无效可选用药物治疗。但目前现有的治疗手段与治疗效果存在局限性,可选用的治疗方法常常不能令人满意,结肠慢传输的治疗仍然具有挑战性。因此,研发一种高效低毒的治疗药物具有重要意义。[align=center][img=,600,598]https://img1.17img.cn/17img/images/202404/uepic/f56ec165-88cb-40a1-854c-0c4d27555528.jpg[/img][/align][align=center]CP0119靶向Transgelin治疗结肠慢传输性便秘的作用机制[/align]基于此,南开大学药学院院长领衔成立了CP0119创新药物研发团队,项目负责人为周红刚、孙涛和杨光教授。据项目负责人介绍,CP0119是一种新型小分子胶转蛋白(Transgelin)激动剂,Transgelin是钙调蛋白家族中的一种肌动蛋白结合蛋白,相对分子量为22 kDa,主要分布于平滑肌细胞中。同时,CP0119能够激动肠道平滑肌细胞上的Transgelin,促进G激动蛋白(G-actin)向F激动蛋白(F-actin)的聚集,增加肠平滑肌细胞中应力纤维束的形成,进而增强细胞收缩能力,促进肠道蠕动,最终达到改善结肠慢传输的效果。目前没有以Transgelin为靶点治疗便秘的同类药物上市,CP0119的成功开发属于全新靶点的化药1类创新药物。项目骨干艾笑羽副教授和谷小婷老师主要负责推动项目的临床前研究工作。该项目历时4年完成了原料药的中试放大、制剂开发、系统化药理和药代研究以及标准化临床前GLP安全性评价,充分证明了CP0119质量可控、安全有效。因此,CP0119的开发有望成为结肠慢传输患者的新治疗选择,具有较高的经济效益和社会效益。该项目成果还得到了天津医科大学总医院消化科王邦茂主任等临床专家的高度评价。CP0119项目的研发由南开大学药学院和药物化学生物学全国重点实验室(全重)的创新药物研发团队承担,也是与华润医药中国医药研究开发中心和天津国际生物医药联合研究院合作的重要成果,是践行产学研和全重共建单位之间合作的实例。在国家大力发展新质生产力和鼓励高校科研成果转化的大背景下,CP0119片临床试验的成功获批,不仅进一步助力南开大学创新药物研发能力,同时也对培养我校创新药学人才和助力健康中国建设具有重要意义。[来源:南开大学][align=right][/align]
国食药监注482号各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后卫生部药品监督管理局: 为加强药物临床试验生物样本分析实验室的管理,提高生物样本分析数据的质量和管理水平,根据《药品注册管理办法》、《药物临床试验质量管理规范》、《药物非临床研究质量管理规范》,参照国际规范,国家局组织制定了《药物临床试验生物样本分析实验室管理指南(试行)》,现予印发。请你局组织本行政区域内有关单位学习,参照执行。 附件:《药物临床试验生物样本分析实验室管理指南(试行)》起草说明 国家食品药品监督管理局 二O一一年十二月二日药物临床试验生物样本分析实验室管理指南(试行)第一章 总 则 第一条 为加强药物临床试验生物样本分析实验室的管理,提高生物样本分析数据的质量和管理水平。根据《药品注册管理办法》、《药物临床试验质量管理规范》、《药物非临床研究质量管理规范》,参照国际规范,制定本指南。 第二条 药物临床试验生物样本(以下简称生物样本)是指按照药物临床试验方案的要求、从临床试验受试者采集的需要进行分析的材料(如血浆、血清、尿液、粪便、组织和细胞等)。药物临床试验生物样本分析实验室(以下简称实验室)是指对生物样本中药物、药物代谢物及生物标志物等进行分析,为药品注册申请提供数据支持的机构。 第三条 凡为提交药品监督管理部门作为药品注册数据而进行生物样本分析的实验室,均须遵循本指南,并接受药品监督管理部门的监督检查。第二章 组织机构和人员 第四条 实验室应建立完善的组织管理体系,任命实验室负责人和项目负责人,并配备相应的实验人员。隶属于药物I期临床试验研究室(以下简称研究室)的实验室,应纳入研究室的质量保证体系;独立的实验室应建立质量保证部门,并任命质量保证部门负责人。 第五条 实验室负责人应具备相关专业本科以上学历,熟悉业务,能有效组织、指导和开展实验室业务工作。对分析工作的实施和结果负责。其职责包括: (一)全面负责实验室的建设,确保实验室具有满足工作要求的各项条件。 (二)组织制定和修改管理制度、技术规范和标准操作规程;定期审阅所有管理制度、技术规范和标准操作规程文件,确保所有文件适时更新。 (三)制定主计划表,掌握各项分析工作的进展。 (四)确保质量保证工作的开展。 (五)建立有效的沟通交流机制,以保证与申办者、药物临床试验机构及研究者之间可以及时、有效地沟通。 (六)建立完善的教育培训和考核制度。 (七)在每项实验开始前,指定项目负责人,试验过程中确需更换项目负责人时,应记录更换的原因和时间,并保留相关记录。 (八)审查、批准实验方案、标准操作规程、结果或报告。 (九)指定专人负责档案资料与生物样本的管理。 第六条 质量保证部门应配备与其开展的工作相适应的人员。质量保证部门负责人的职责为: (一)负责质量保证部门的工作安排和运行; (二)审核分析实验方案、实验记录、结果或报告; (三)根据每项工作的内容和持续时间制订稽查计划并实施稽查,详细记录稽查的内容、发现的问题、采取的措施等,并向实验室负责人和项目负责人报告; (四)检查实验室环境、设施、仪器设备和档案管理等; (五)参与标准操作规程的制订和审核,并保存标准操作规程的副本。 第七条 项目负责人具体负责某项临床试验生物样本的分析工作,具备相应专业本科或以上学历,两年以上生物样本分析工作经验,能够独立进行生物样本分析方法的建立和验证,并对所承担项目的分析方法、分析结果和分析报告负直接责任。项目负责人的职责包括: (一)制订该项目的实验方案; (二)全面负责该项目的运行管理、组织实施; (三)建立并验证分析方法,撰写验证分析报告; (四)确保所有参与该项目的实验人员明确各自所承担的工作,并掌握和执行相关的标准操作规程; (五)掌握工作进展,确保实验记录及时、完整、准确和清晰; (六)确保实验中偏离方案的情况及采取的措施均有详细记录; (七)整理、分析实验数据和结果,撰写分析报告; (八)及时处理质量保证部门的报告。 第八条 实验室工作人员应符合以下要求: (一)具备严谨的科学作风和良好的职业道德以及相应的学历,经过专业培训与考核,并保存个人的培训与考核记录,具备相应的经验和能力并取得上岗资格; (二)熟悉本指南要求,掌握并严格执行相关的标准操作规程; (三)及时、完整、准确和清晰地进行实验记录,对实验中发生的可能影响实验结果的任何情况应及时报告给项目负责人; (四)对涉及保密的技术资料、受试者信息等履行其保密责任; (五)根据工作岗位的需要着装,保持工作环境正常有序,遵守健康检查制度,确保实验样本不受污染。
一直以来,大型制药公司研发药物的临床试验数据都作为公司的高度机密,而保存在公司的数据库中。葛兰素史克(GSK)决定打破常规,公开公司的“高度机密”。讨论公布在研药物所有临床试验数据利弊.
11月11日晚,食药总局终于使出了杀手锏,第一次曝光了涉嫌临床试验数据造假的八家企业名单,并表示对其涉及的11个药品注册申请不予批准。 “其实对于上述处罚,医药企业并不害怕,他们现在都与临床试验机构互相推诿责任。”一位业内人士向记者透露,医药企业真正害怕的是食药总局的“三年禁令”。“不能申报新药,企业如何发展、业绩如何保证,尤其是上市公司。”他说。 药企最怕“三年禁令” 国家食药总局相关人士对记者表示,这11个药品注册申报资料的临床试验数据存在擅自修改数据、瞒报数据以及数据不可溯源等涉嫌弄虚作假问题。“食药总局下一步还将继续开展临床试验数据现场核查工作。”他说。 药物临床试验是指任何在人体(病人或健康志愿者)进行的药物的系统性研究,以证实或发现试验药物的安全性和有效性。药物临床试验一般分为Ⅰ、Ⅱ、Ⅲ、Ⅳ期临床试验。 食药总局相关人士表示,将在查清事实的基础上,明确注册申请人、药物临床试验机构和合同研究组织的相关人员的责任,涉及医疗机构的相关责任人由卫生计生部门处理,涉嫌犯罪的移交公安机关。 食药总局之前称,对核查中发现临床试验数据真实性存在问题的相关申请人,三年内不受理其药品申请。将弄虚作假的申请人、临床试验机构、合同研究组织以及相关责任人员等列入黑名单。弄虚作假的直接责任人参与研究或组织研究的临床试验资料十年内不予受理。 制剂出口巨头华海药业中枪 记者12日分别致电上述企业。华海药业相关人士表示,公司正在研究出问题的环节,可能是临床试验机构的问题,不是企业的问题。“至于出口制剂是否存在临床数据造假问题,我们没有收到任何信息”。 作为我国知名制剂出口企业华海药业在此次事件中也中枪。食药总局称,华海药业坎地沙坦酯片原始记录缺失、分析测试系统无稽查轨迹、试验用药品未留样、部分图谱真实性高度存疑,甚至隐瞒弃用试验数据、修改调换试验数据。 今年8月,华海美国和普洛康裕签署战略合作协议,双方将展开化学原料药及制剂产品在美国市场的业务合作。目前,该公司有30多个ANDA申请文号,预计未来每年有5至10个ANDA 文号获批,海外制剂业务将长期受益。 华海药业三季报显示,公司2015年前三季度实现营业收入24.21亿元,同比增长34.09%;实现归母净利润3.35亿元,同比增长65.73%。中国证券金融股份有限公司持股1646.28万股,占比2.08%;中央汇金投资有限责任公司持股1177.33万股,占比1.65%。 华海药业指出,上述增长归结于原料药和中间体业务营收稳定增长,前三季度实现营业收入约14.53亿元,同比增长约30%,主要是沙坦类产品和神经类产品销售大幅增长。而国际制剂业务营收大幅增长,制剂业务实现营业收入约9.68亿元,其中国际制剂业务同比增长43%,预计全年制剂业务有望增长40%左右。 八家企业宽限期内未主动撤回申请 记者了解到,食药总局清理临床数据造假行动是从7月22日开始,食药总局副局长吴浈明确表态“药物临床试验中的问题是比较严重的,不规范、不完整的问题非常普遍,不可靠、不真实、弄虚作假的问题确实存在,已经严重影响了药品审评审批的正常进行,严重干扰了上市药品有效安全的科学评价,严重破坏了审评审批的正常秩序。” 随后,食药总局出了一份企业核查清单,医药界哗然了。自查核查品种清单1622个受理号中,进口有171个受理号,新药948个受理号,已有国家标准503个受理号。受理号中有309个受理号分别属于国内103家医药类上市公司,包括华海药业、恒瑞医药(51.16, 0.00, 0.00%)等国内外制药龙头企业。其中,化药需要自查1283个受理号,占79.1%,中药大多是2005年至2006年的受理号,共237个,占14.6%。生物制品共102个受理号。 “我们非常重视这个事情,董事长、总经理全部开会研究。”国内一家知名研发企业副总告诉记者。另一家上市药企老总说,公司有6个品种在名单上,现在已经撤回了3个药品申请。 为了给予药企业充分的自查时间,食药总局给出了一个多月的宽限期,允许企业自动撤回有问题的申请。8月28日,食药总局公布,此次自查结果中,申请人提交自查资料的注册申请为1094个,占 67%;主动撤回的注册申请317个,占20%;申请减免临床试验等不需要提交的注册申请193个,占12%。但上述八家企业的11个品种并不在企业主动撤回之列。 土豆点评:这个发现数据涉及造假,是在审查资料的时候发现啊?还是去现场进行注册核查的时候发现的啊?现场注册核查是不是开始严格查电子数据的真实性了?
各省、自治区、直辖市食品药品监督管理局(药品监督管理局),总后勤部卫生部药品监督管理局: 为加强药物I期临床试验的管理,提高临床试验生物样本分析实验室的分析质量,有效地保障受试者的权益与安全,确保所产生的数据和结果的可靠性、完整性和科学性,根据《药物临床试验质量管理规范》,我司组织起草了《药物Ⅰ期临床试验管理指导原则(征求意见稿)》、《药物临床试验生物样本分析实验室管理规定(征求意见稿)》。现公开征求意见,请于2011年4月30日前将修改意见反馈我司。(征求意见稿可在国家食品药品监督管理局网站下载) 联系人:蓝恭涛,唐慧鑫 电 话:010—88330742,0722 传 真:010—88363228 地 址:北京市西城区宣武门西大街26号院2号楼 邮 编:100053 E-mail:yjjdc@sda.gov.cn 附件:1.《药物Ⅰ期临床试验管理指导原则(征求意见稿)》 2.《药物临床试验生物样本分析实验室管理规定(征求意见稿)》 3.起草说明 国家食品药品监督管理局药品注册司 二○一一年三月十八日
近日,在中国心脏大会“牛津—阜外心血管病研究热点论坛”上,有学者披露,我国将参与全球大规模多中心药物临床试验,增强他汀类药物控制心血管发病风险的作用。 通过抑制胆固醇酯转运蛋白(CETP)而升高高密度脂蛋白、降低低密度脂蛋白,是在使用他汀类药物基础上力求使心血管发病风险进一步下降的新希望。然而,首个CETP抑制剂Torcetrapib因导致心血管事件的增加而提前终止。之后的医学分析发现,该抑制剂自身产生醛固酮水平增高、电解质紊乱的副作用,是导致心血管事件增加的罪魁祸首。目前,降脂强度最强的CETP抑制剂是Anacetrapib,临床3期研究显示,该抑制剂可以降低40%低密度脂蛋白,升高140%高密度脂蛋白,而对醛固酮水平、电解质均无影响,也未发现有明显副作用。 中国医学科学院阜外心血管病医院中国牛津国际医学研究中心主任蒋立新教授说,CETP抑制剂Anacetrapib研究由牛津大学设计并组织实施,即将在全球范围内遴选医疗机构开展大规模多中心临床试验,该试验将入选3万名高危心血管疾病患者,而中国作为重要参与者,将入选9400名患者,整个计划预计持续4年。 蒋立新介绍,中国牛津国际医学研究中心负责中国区域实施,目前已基本确定协作单位80余家,登记病人近万名,预计今年年底正式启动该研究
请问做1期临床试验的老师,建立1期临床试验分析试验室的要求有2004年版,有无新的版本?若你是认证专家,你在认证中你的意见呢?
食品安全一直广受诟病!如果我们对其有临床实验的要求,严格监管控制,那我们还会如此担心食品安全吗?先前我们都佩服先吃螃蟹的人,从结果上来看,我们知道螃蟹很美味。但从科学的角度来讲并不是每个人都适合吃,螃蟹是凉性,女性朋友最不宜常吃,影响意义深远哦!而销售商没有必要的提示,只宣传其美味而不顾对女性之不良影响。田螺一样美味,但寄生虫不可忽视。还有非洲人吃大腥腥使人类得了艾滋。以上事例都给我们带来严重后果,确实是这样,不是能吃的食品就无毒。是药三分毒大家都知道,所以有临床志愿者为我们实验。作一种未知的尝试。以上实例等同于我们的实验志愿者,以临床发现为我们发现问题而提供依据。我们现在发现N多种通过转基因或是其他技术而生产我们未知食品,依人群喜好而变种,这样的风险有多大!是否应该建立临床实验数据而被广泛食用才对!
我一直从事血铅分析,用石墨炉[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度计。临床大夫要求我用标准血样进行质量控制,他们知道美国CDC提供这种标准血样,但他们只提供给美国的检测机构。在中国是否有机构提供或出售这种标准血样?
[color=#c00000][i]同济大学附属东方医院再生医学研究所何志颖研究员表示:项目研究从2018年开始,2021年4月Pre-IND,2024年1月18日IND获批!历时6年艰辛。[b]这是东方医院首个干细胞成药临床试验项目,希望能造福于心衰患者![/b][/i][/color]据报道1月18日,天士力公告其一款创新干细胞药物获得国家药监局批准进入临床试验,主治慢性心力衰竭。公告称,公司收到国家药监局核准签发关于[b]人脐带间充质干细胞注射液(B2278注射液)[/b]项目的《药物临床试验批准通知书》,同意开展伴冠状动脉旁路移植术指征的慢性缺血性心肌病导致的慢性心力衰竭的临床试验。[b]人脐带间充质干细胞注射液[b](B2278注射液)[/b]由上海市东方医院(同济大学附属东方医院)研发,2022年8月天士力与东方医院签署《技术转让(合作)合同》受让相关技术及成果,并在全球范围内,优先在中国开展药品注册申报及后续临床试验开发。[/b]临床前研究证明,B2278注射液可通过旁分泌作用调控心肌组织微环境,对于缺血性心肌病中的心肌细胞组织损伤有明显抑制作用,增加动物心功能,促进血管再生,减少心肌凋亡。心力衰竭是由于心脏结构和/或功能异常导致心室充盈和/或射血能力受损的一组临床综合征,是大部分心血管疾病发展的最终阶段,随着年龄增长,心衰患病率和发病率均明显增加。目前对于心力衰竭的治疗主要包括药物治疗、血运重建、细胞和基因治疗,其中冠状动脉旁路移植术(CABG)是常用的血运重建治疗方式,《2022年中国心血管外科手术和体外循环数据白皮书》显示,2022年CABG占心外科手术总量21.1%。上述治疗手段可以在一定程度上延缓心力衰竭的进展,但不能使死亡心肌再生。伴随CABG手术的心肌局部注射干细胞有望通过刺激心脏细胞的增殖和分化、抑制心肌细胞损伤及免疫调节等作用,修复心肌细胞使心肌收缩增强从而对心力衰竭发挥治疗作用。目前国际上获批的干细胞品种已达十余种,但是尚无治疗心衰的干细胞产品上市。[来源:仪器信息网] 未经授权不得转载[align=right][/align]
关注实验室,关注第三方检测市场研究(微信号test2005)。感谢各位同行朋友的关注,截止今天本公众微信的粉丝数已经超过500,这让我有点受宠若惊,因为开通还不到1个月。还是那句话,本账号会不遗余力的为各位提供实验室各类原创信息,包括技术、管理、市场、资质认可等相关信息。 前些天,有个微信好友询问关于实验室质量控制方面,问有没有最好的质控计划或方案。当时想到的第一句话就是:没有最好只有更好!——开个玩笑。 如果严格按ISO/IEC17025相关标准执行的话,质量控制对实验室是相当重要的,但是中国的老板们往往忽视这一块,甚至有些认为这是浪费成本。从监管单位的政策来看,这块的要求是越来越严格了,像CNAS在2012年就发布了CNAS-CL10:2012《检测和校准实验室能力认可准则在化学检测领域的应用说明》替代原来的CNAS-CL10:2006。CNAS-CL10:2012的重点内容就是加强实验室的质量控制和质量保证,所以建议大家条件允许的情况下尽量去做好。 检测都是相通的,质量控制也是相通的,只是具体操作的层面存在差异。如果说有没有最好的质量控制计划或方案,我觉得其实都差不多,重要的是落实在操作环节。再完美的计划不落实到执行,也是空谈。 大家认为质量控制计划难吗?其实我觉得不难,难就难在老板能提供多少资源支持,以及实验室是否真正执行好。其他不多说,这里大概讲讲质量控制计划或方案怎么写: 首先必须结合实验室自身的情况,比如说你的专业、仪器设备、人员等等条件,然后参考相关标准,我列出一些标准,可以作为参考: CNAS-CL10:2012检测和校准实验室能力认可准则在化学检测领域的应用说明 GB/T 20468-2006临床实验室定量测定室内质量控制指南 GB/T 27401-2008实验室质量控制规范 动物检疫 GB/T 27402-2008实验室质量控制规范 植物检疫 GB/T 27403-2008实验室质量控制规范 食品分子生物学检测 GB/T 27404-2008实验室质量控制规范 食品理化检测 GB/T 27405-2008实验室质量控制规范 食品微生物检测 GB/T 27406-2008实验室质量控制规范 食品毒理学检测 GB/T 27407-2010实验室质量控制 利用统计质量保证和控制图技术 评价分析测量系统的性能 GB/T 27408-2010实验室质量控制 非标准测试方法的有效性评价 线性关系 GB/T 27413-2012石油产品检测实验室质量控制与质量评估 GB/T 29605-2013感官分析 食品感官质量控制导则 GB/T 5750.3-2006生活饮用水标准检验方法 水质分析质量控制 HJ/T 373-2007固定污染源监测质量保证与质量控制技术规范(试行) MT/T 895-2000煤炭实验室测试质量控制导则 SN/T 3590-2013化学分析实验室中的职责和质量控制指南 GB 17378.2-2007海洋监测规范 第2部分:数据处理与分析质量控制 GB/T 22275.1-2008良好实验室规范实施要求 第1部分: 质量保证与良好实验室规范 GBZ/T 173-2006职业卫生生物监测质量保证规范 GB/T 4091-2001常规控制图 ………… 还有,真正要做好质控,最好是有独立的质量部门,且在某些事宜上予以特别的权限,否则根本很难执行。质量控制到底要做什么?下面将本人5年前在深圳工作撰写的一个《质量控制工作实施细则》简单分享下,篇幅比较长,有兴趣的可以耐心看完:1 目的依据公司质量目标、年度质量保证和质量控制计划,为明确各类质量控制活动的策划,以确保检测结果的质量,特制定本细则。2 范围适用于公司开展的所有检测项目的质量控制活动的策划、组织、实施、管理和评价。3 职责3.1 实验室、取样部负责相应质控活动的实施与协作。3.2 办公室负责相应质控活动的协作与支持工作。3.3 质量部负责所有质控活动的策划、组织、管理和评价。4 实验室内/间质量控制 质量控制的目的是把检测工作中的误差,减小到一定的限度,以获得准确可靠的结果;质量控制是发现和控制检测过程产生误差的来源,用以控制和减小误差的措施。 实验室内质量控制可通过空白实验、检出限实验、加标回收实验、平行样测定、标准样品测定等手段实施。 实验室间质量控制可通过组织或参加实验室间比对实验,参加能力验证或测量审核的方式实施。4.1 空白实验每批次检测应做全程序空白(溶解氧、pH值等特殊项目除外)。若全程序空白样品有检出,应查找原因,予以纠正。可根据分析方法的需要,在分析结果中扣除全程序空白值对监测结果进行修正。4.1.1 试剂空白是指除用纯水代替样品外,其所加试剂和操作步骤与样品测定的操作步骤完全相同。试剂空白往往用于检验试剂的纯度,但不能消除样品中可能存在在干扰物质的干扰。4.1.2 容器空白 是指用纯水注入或流经该采样容器后作为一个样品,然后分析所需要的各个参数,检验采样器周期性使用后所引起的空白变化。主要检查容器清洗、现场环境和容器对样品吸附等作用对空白样品的影响。4.1.3 现场空白 即在采样现场以纯水(气体则以空白采样管或吸收液)作样品,按测定项目的采集方法和要求,与样品同等条件下装瓶、保存、运输和送交实验室分析;通过现场空白样检验,掌握采样过程中操作步骤和环境条件对样品质量影响的状况。4.1.3.1 水质采样现场空白:在采样现场以纯水作样品,按测定项目的采集方法和要求,与样品同等条件下装瓶、保存、运输和送交实验室分析。4.1.3.2 空气(废气)采样现场空白:在采样现场以空白采样管(或吸收液)作为样品,按测定项目的采集方法和要求,与样品同等条件下保存、运输和送交实验室分析。4.1.3.3 现场空白必须是平行双份的,通过现场空白样检验,掌握采样过程中操作步骤和环境条件对样品质量影响的状况。4.1.4 空白平行双样的相对差值不大于50%。4.2 最低检出限(浓度) 检出限为某特定分析方法在给定的置信度内可从样品中检出待测物质的最小浓度或量。所谓“检出”是指定性检出,即判定样品中存有浓度高于空白的待测物质。检出限受仪器的灵敏度和稳定性,全程序空白试验值及其波动性的影响。 以空白试验为基础估算检出限,空白每批做平行双样,分别在一段时间内(隔天)重复测定一批,至少测定5批。对不同的测试方法检出限(检出浓度)有几种求法,如:4.2.1 [color=#00
[center]美国CUPID首项基因治疗临床试验结果喜人[/center]近日,在美国新奥尔良召开的美国心脏协会(AHA)科学大会上,研究人员公布了CUPID试验的Ⅰ期临床试验结果。CUPID试验是由美国13家医疗单位共同参与的一项随机、双盲、安慰剂对照、平行分配设计的Ⅰ/Ⅱ期临床试验,预计将纳入46名晚期心力衰竭患者;此项研究同时也是应用基因替代技术治疗晚期心力衰竭的首项临床试验。 众所周知,钙离子是肌肉收缩的关键影响因素。肌浆网钙ATP酶(SERCA)亦被称为肌浆网钙泵,是参与钙离子调节的主要蛋白;而SERCA2a主要存在于心肌和骨骼肌慢肌纤维中,其功能是ATP依赖性地逆浓度梯度将钙离子由胞浆排入肌浆网内,促进心肌舒张。大量研究表明,在衰竭的心肌细胞中,SERCA2a活性及其mRNA表达量均明显下降,心衰部位心肌SERCA2a表达量较正常心肌降低30%左右;动物实验显示,将SERCA2a基因转导到衰竭的心肌后,SERCA2a活性及其表达量明显增加,心功能指标明显改善。由此可知,心肌SERCA2a的减少与心力衰竭的进程之间存在明显相关性。 因此,许多研究机构开展了应用SERCA2a基因转导技术治疗晚期心力衰竭的研究。MYDICAR是Celladon公司开发的一种用于治疗晚期心力衰竭的基因靶向酶替代治疗药物,是一种由1型腺相关病毒(AAV1)壳体与人SERCA2a cDNA共同组成的重组病毒载体系统。这种药物经导管在左心室前壁及侧壁注射后,可将SERCA2a基因转移至体内,增加心肌中SERCA2a的表达,改善和恢复心脏功能。 在这次Ⅰ期临床试验中,9名心力衰竭患者接受了MYDICAR治疗。其中7名在6个月内出现不同程度的病情好转,包括:有5名患者症状获得改善,有4名患者机体功能得到改善,有2名患者生物标记物指标得到改善,有6名患者左心室功能或左室重构获得改善。另外2名患者因体内已经存在病毒载体的抗体,因而病情未出现好转。更为重要的是,MYDICAR在试验中表现出较好的安全性,获得了负责评估该试验的安全委员会及研究者的肯定。 目前,CUPID试验中的Ⅱ期临床试验已经开始招募受试者,它将对MYDICAR治疗晚期心力衰竭病人的安全性和有效性作出进一步的评价。信息来源:医药经济报
[center]赛诺菲-安万特减肥药Acomplia临床试验全面停止[/center]法国赛诺菲-安万特公司近日宣布,停止Acomplia减肥药在全球所有的临床试验。 该公司发表公报说,这一决定是在法国卫生部门的要求下作出的。法国国家健康制品卫生安全局表示,全球共有1.8万人参与了这种减肥药的临床试验,其中法国有500人。 目前已有6人在服用该药后因精神抑郁而自杀身亡,法国国家健康制品卫生安全局由此认为,这种药具有诱发自杀行为的危险性,因此下令停止其在法国的临床试验。 Acomplia是一种新型减肥药,虽然于2006年获准在欧盟销售,但它的安全性还是受到了质疑。2007年7月,美国食品药品管理局拒绝了其在美国的上市申请,理由是这种药容易诱发精神抑郁和自杀行为。就在上月23日,欧盟药品管理局也作出决定,下令在欧盟国家内部暂停销售这种减肥药。欧盟药品管理局认为,这种药的副作用要大于疗效。 分析人士指出,停止Acomplia的临床试验,意味着对它的未来宣判了“死刑”。







 我要推广仪器
我要推广仪器
 下载APP
下载APP