
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转3457
仪器信息网认证电话,请放心拨打

「核酸」「抗原」「抗体」三种新冠检测方式有何差别?
目前主要有三种检测方式:核酸检测抗原检测抗体检测 首先,我们用一张图,来解析新型冠状病毒的组成。如果将可怕的新型冠状病毒比喻成一个橘子?橘子内部的橘子瓣 = 病毒核酸(RNA) 橘子皮/橘瓣表面内皮 = 病毒蛋白质(S蛋白/E蛋白/M蛋白) 1. 核酸检测核酸检测检测的是病毒的遗传物质DNA或RNA,利用PCR(聚合酶链式反应)技术明确样本中是否存在新型冠状病毒,是目前确诊新型冠状病毒感染的肺炎无创诊断的“金标准”。2. 抗原检测3. 抗体检测抗体检测的是患者体内免疫系统为应对新冠病毒感染而产生的抗体,如果检测结果为阳性,说明患者体内存在针对新冠病毒的抗体,患者正处于感染期或已经康复,可作为辅助手段帮助确诊感染。 新冠疫情爆发以来阿拉丁第一时间向医疗卫生及应急部门捐赠防控物资包括口罩、手套、防护服等得到社会积极反馈 阿拉丁官网提供各类防疫产品 项目号内容M7194抗菌洗手液M299596免洗手消毒凝胶E29958575%消毒用酒精喷雾E29957875%消毒用酒精M9432一类隔离服H299581复合过氧化氢消毒液D1863医用级乳胶手套,麻面无粉D5852一次性PVC手套,无粉C1674CPE塑料鞋套A397042镜架式医用防护面罩新冠抗原检测试剂盒原料 我们已经整理在下方表格 本次阿拉丁的小科普就到这里大家要注意多多防护呀!如果喜欢本文的话别忘了分享给你的朋友哦!
企业动态
2022.12.29

用于2D和3D细胞培养的可降解型聚乙二醇水凝胶
简介随着对多样化功能型生物材料需求的不断增加,组织工程和药物输送等生物技术领域也在持续发展着。几十年来,聚合物生物材料的研究一直集中在测试为其他应用或其加工(如:静电纺丝、溶剂浇铸/致孔剂浸出、3D打印)开发的聚合物的生物相容性。而近期,研究人员已将主要关注方向转向合成专门用于生物医学用途的材料,包括合成蛋白质、糖模拟物和与水性介质相容的聚合物,以及天然聚合物的化学改性(如:通过凝胶化来增加物质在体内的稳定性)。在过去的十年中,聚合物化学家为设计的生物材料创造了一个良好的适用场景,用作于细胞支架或者药物递送。水凝胶是长期以来受到人们关注最多的生物材料之一[1],因为其在化学和物理性质上都非常接近细胞的自然环境,因此作为细胞的二维和三维支架被广泛研究[2]。水凝胶可以由合成的(例如,聚(乙二醇)、聚(甲基丙烯酸羟乙酯))与天然存在的聚合物(例如,胶原蛋白、透明质酸、肝素)交联形成,并且由于它们的含水量非常高,可以有效被应用于组织培养的3D模型,不受细胞、蛋白质和DNA的影响。根据不同组成部分的反应性,可以使用pH[3]、温度[4]、库仑相互作用、共价键、非共价相互作用[5]或聚合来诱实现导凝胶化。PEG(聚乙二醇)聚乙二醇(PEG)是一种亲水性聚合物,当形成交联网络时,能够具备很高的含水量。PEG是适用于生物应用的材料,因为它通常不会引起免疫反应[6]。自20世纪70年代以来,PEG已被用于修饰治疗性蛋白质和多肽,以增加它们的溶解度,降低其毒性,并延长其循环半衰期[7]。自20世纪70年代末,研究人员便开始尝试使用PEG水凝胶进行细胞培养。聚乙二醇水凝胶具有良好的化学反应性,多种反应性基团可用于其形成水凝胶并且实现高效的化学修饰。PEG大分子单体通过环氧乙烷的活性阴离子开环聚合能够非常方便地合成聚乙二醇;这些聚乙二醇具有较宽的分子量分布,且包含多种端基(例如:羟基、甲醚基、氨基、N-羟基琥珀酰亚胺(NHS)酯),在众多反应中都可适配。为了形成水凝胶,PEG必须发生交联反应。最开始的时候,PEG是通过电离辐射进行非特异性交联的[8],而现在PEG水凝胶的形成通常是通过共价交联具有反应链端的PEG高分子合成的。具有活性反应末端的PEG高分子聚合物,如丙烯酸酯、甲基丙烯酸酯、烯丙醚、马来酰亚胺、乙烯基砜、NHS酯和乙烯基醚基团(图1)很容易从现成的一般性材料合成。可在碱存在下使用酸性氯化物(例如:丙烯酰氯、甲基丙烯酰氯)使PEG醇链的末端酯化。PEG链末端可在碱性条件下通过与诸如2-氯乙基乙烯基醚或溴烯丙基等烷基卤化物反应而醚化。PEG二乙烯基砜是通过将PEG偶联到大量过量的二乙烯基砜或通过多步骤工艺来制备氯乙基砜链末端,氯乙基砜链末端可以通过基本消除反应形成的乙烯基砜基团[9]来制备。图1:不同PEG大分子的末端基团高分子的两个末端可以是两个相同或者不同的官能团。同双官能高分子通常用于形成网络,而异双官能高分子则可用于将具有治疗性的小分子连接到水凝胶网络中。水凝胶形成机理形成水凝胶的交联机制取决于PEG高分子链末端的特性。在大多数情况下,是与反应性乙烯基链末端聚合的同时发生交联,通常采用的是自由基引发剂。例如,大分子单体的聚合可以使用通过氧化还原反应生成的自由基(比如过硫酸铵和TEMED)或光照产生的自由基(图2中的Irgacure®651,λ=365 nM)来引发,随后通过丙烯酸酯和甲基丙烯酸酯链末端基元反应来发生链增长。在阶梯生长网络的形成中,多官能度(f>2)的交联剂以化学当量与PEG链末端进行反应,或多官能度的PEG(f>2)也可以与双官能团的交联剂发生交联。丙烯酸酯、甲基丙烯酸酯、乙烯基砜、马来酰亚胺、乙烯基醚和烯丙基醚都可以根据反应条件转化为硫醇,形成阶梯生长网络。典型的交联剂可包括巯基或胺的部分。混合模式聚合是在同一反应容器中发生的两种机制的结果;丙烯酸酯和甲基丙烯酸酯基团可以形成混合模式网络。两种水凝胶形成机制均可用于包裹活体细胞,并且两种机制均可使肽、蛋白质或其他治疗药物发生反应性掺入。图2:链式增长和阶梯式增长反应由不同机理反应得到的网格结构如图3所示。在链式增长网络中,交联位点形成了一个动力学链,而在阶梯式生长网络中,交联位点与多功能交联剂具有相同的功能,隐藏了结构上的缺陷。在链式增长和步进增长中,网络结构上的缺陷如闭环、长链缠结和末端链悬挂均有可能存在。不同高分子本身的化学性质的选择和水凝胶不同的形成机理都很重要,因为它们都会影响水凝胶网络的交联密度。材料的性质对于二维和三维培养很重要,也很容易通过水凝胶形成的化学过程来控制。随着交联密度的增加,网格尺寸减小,膨胀率降低,储能模量增加。改变PEG高分子的分子量能够实现对水凝胶性质的大体控制(交联密度差异引起的性质差异)。同时,通过改变用于生产水凝胶的反应机理,可以精细地调控水凝胶的性质(可用于调节体系的交联密度)。图3:不同形成机理导致的对水凝胶网络结构和网络缺陷的影响水凝胶的降解为了使用3D水凝胶支架来研究细胞分化和组织进化,通过空间和时间调控的方式来设计凝胶的物理和化学性质是至关重要的[10]。聚合物材料性质通常借由聚合/交联(键形成)或借由受控降解及/或释放(键断裂)来改变。键的形成通常会用到小分子试剂(引发剂、催化剂,单体、连接到材料的配体),而键断裂则通常不依赖于外源试剂。小分子在体外和体内均具有比聚合物试剂更多的副反应,因此许多研究小组用降解作为原位操作聚合物生物材料的工具。水解降解水凝胶中最常用的降解机理就是水解,即一个水分子加入到聚合物骨架中,导致链断裂。酸酐、酯和酰胺都很容易水解,任何氢化物通常水解得太快,而酰胺类的若未经过催化则水解太慢,因此大多数水凝胶的水解降解都是利用酯键。为了获得具有生理相关时间维度上的可降解水凝胶,研究人员通常使用交酯或乙醛交酯段将具有可降解酯键的PEG功能化。PEG上的醇链末端可引发3,6-二甲基-1,4-二恶烷-2,5-二酮和1,4-二恶烷-2,5-二酮的开环反应,分别生成聚乙二醇交酯和聚乙二醇交酯(图4)[11]。开环反应通常由锡(II)-2-乙基己酸盐催化[12],但使用二甲氨基吡啶作为催化剂也很容易完成反应[13],二甲氨基吡啶可能比残余锡更容易除去。聚乙二醇交酯或聚乙二醇交酯的醇链末端很容易被丙烯酸酯和甲基丙烯酸酯等反应性双键官能化。图4:聚乙二醇乙交酯和聚乙二醇丙交酯的合成酶促降解虽然酯键是可通过酶降解的,但大多数研究人员都会使用具有特异性序列的酶降解掺入水凝胶中的肽,而不是非特异性的酶降解酯和酰胺。Hubbell团队率先使用了这一方法[14],他们通过Michael加成反应把丙烯酸酯、马来酰亚胺和乙烯基砜官能化的半胱氨酸肽,将基质金属蛋白酶(MMP)具有反应敏感型的连接键引入水凝胶(图5)[15]。MMP-可降解键也被用作联结治疗剂与水凝胶的载体。例如,血管内皮生长因子(VEG-F)等生长因子可通过酶降解MMP-敏感性链而释放,从而诱导血管生成[16]。在水解和酶解过程中,降解速率由大分子的化学性质决定。在水解中,材料的降解率是通过其本身的性质(如疏水性或亲水性)和可水解基团的数量预先设计的,并且一旦材料被制造出来,就不能改变。在酶解过程中,降解通常发生在产生酶的细胞局部区域。水解和酶解均是水凝胶缓释和缓释治疗药物的有效方法,但水凝胶制备后无法调节或阻滞其释放速率,且释放不受空间限制。图5:通过Michael加成将含有半胱氨酸的肽添加到含乙烯基砜基团的酶中制备可降解型水凝胶光降解型水凝胶与水解和酶降解相比,光降解允许更加精准的空间和时间控制降解和释放。虽然已有许多研究者报道了光聚合水凝胶和光功能化水凝胶,但关于生物相容性光降解水凝胶的报道很少。Kloxin和Kasko发表了由含2-甲氧基-5-硝基-4-(1-羟乙基)苯氧丁酸的PEG高分子形成的光可降解水凝胶网络(图6)[17];邻-硝基苄基(o-NB)连接基团的光降解行为具有良好的表现。由光降解聚合物形成的水凝胶在光照下表现出体积降解,这与曝光时间、波长和光强度有关。当光线被关闭时,降解停止;在恢复光暴露后,上述样品继续光解。hMSCs(人骨髓间充质干细胞)被包裹在含有光可释放的细胞黏附配体RGDS(精氨酸-甘氨酸-天冬氨酸-丝氨酸)的水凝胶中,当RGD在第10天释放时(与纤维连接蛋白在软骨形成过程中的下调相对应),hMSCs沿成软骨途径分化。这种可降解水凝胶的表面侵蚀和透胶光刻可用于形成从10-7-10-2米或更大范围的特征局部区域[18]。部分降解导致水凝胶的交联密度降低和溶胀程度增加,这提供了一种如何制备柔软性更高的水凝胶的方法。图6:光降解o-NB掺杂的水凝胶骨架用于释放治疗药剂除了单光子光解,含有o-NB的水凝胶也对双光子光解敏感,从而允许被用于3D蚀刻[19-20]。在单光子反应中,任何暴露在光下的区域都会发生反应。相反,多光子光刻应该只发生在多个光子同时被吸收的地方,这发生在光源的焦体积(如图7)。生物材料的单光子光刻的典型波长范围从长波UV(≥365 nm)到可见光区域,而双光子光刻则使用红外光(通常~ 740-800 nm)较多。红外光具有更好的生物相容性,对活体组织的破坏性更小,并能够有更大的穿透深度。发生双光子吸收的区域也被严格限制在了光的焦点上,而不是沿着光的整个路径,提供了对激发3D控制的新思路。单光子和多光子反应都有能力制备出特征点小于500 nm的材料,远小于哺乳动物细胞的大小[21]。这代表了对水凝胶支架结构和化学的空间控制水平达到了前所未有的高度。图7:单光子光解(左)发生在暴露于紫外-可见光的水凝胶的整个区域双光子光解(右)只发生在同时吸收两个红外光光子的区域o-NB连接子还可用于将治疗剂拴系到水凝胶中以实现在活细胞中的递送。Griffin等证明了通过o-NB-PEG高分子栓系到水凝胶中可以实现荧光素的受控释放[22]。在不同波长(365-436 nm)、不同强度(5-20 mW/cm2)和不同时间(0-20分钟)的光照射下,对模型的释放进行定量分析。虽然最快的释放发生在365 nm(这对应于在该波长的o-NB连接剂的较高摩尔吸收率),显著的释放也同样见于405 nm;从分子的物理常数(如摩尔吸收率)上去推测很容易模拟释放。在这些体系中,光的衰减使化学和机械梯度容易形成。小结聚乙二醇是一种易制备、易改性的聚合物。它被广泛应用于水凝胶制造,包括作为组织培养的2D和3D支架。聚乙二醇水凝胶易于引入可降解键。水解可降解凝胶允许持续的材料降解和/或治疗剂释放。酶降解凝胶的降解和释放是由细胞决定的。光降解允许用户对水凝胶的化学和物理性质进行实时定制的外部操作。参考文献1. Drury JL, Mooney DJ. 2003. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 24(24):4337-4351. http://dx.doi.org/10.1016/s0142-9612(03)00340-52. Lee KY, Mooney DJ. 2001. Hydrogels for Tissue Engineering. Chem. Rev.. 101(7):1869-1880. http://dx.doi.org/10.1021/cr000108x3. HOFFMAN AS. Hydrogels for Biomedical Applications. 944(1):62-73. http://dx.doi.org/10.1111/j.1749-6632.2001.tb03823.x4. Hoffman AS. 2002. Hydrogels for biomedical applications. Advanced Drug Delivery Reviews. 54(1):3-12. http://dx.doi.org/10.1016/s0169-409x(01)00239-35. Tibbitt MW, Anseth KS. 2009. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng.. 103(4):655-663. http://dx.doi.org/10.1002/bit.223616. Lin C, Anseth KS. 2009. PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine. Pharm Res. 26(3):631-643. http://dx.doi.org/10.1007/s11095-008-9801-27. Richter A, Paschew G, Klatt S, Lienig J, Arndt K, Adler H. Review on Hydrogel-based pH Sensors and Microsensors. Sensors. 8(1):561-581. http://dx.doi.org/10.3390/s80105618. Ruel-Gariépy E, Leroux J. 2004. In situ-forming hydrogels?review of temperature-sensitive systems. European Journal of Pharmaceutics and Biopharmaceutics. 58(2):409-426. http://dx.doi.org/10.1016/j.ejpb.2004.03.0199. Yang Z, Xu B. 2007. Supramolecular hydrogels based on biofunctional nanofibers of self-assembled small molecules. J. Mater. Chem.. 17(23):2385. http://dx.doi.org/10.1039/b702493b10. Zalipsky S, Harris JM. 1997. Introduction to Chemistry and Biological Applications of Poly(ethylene glycol).1-13. http://dx.doi.org/10.1021/bk-1997-0680.ch00111. Davis FF. 2002. The origin of pegnology. Advanced Drug Delivery Reviews. 54(4):457-458. http://dx.doi.org/10.1016/s0169-409x(02)00021-212. Morpurgo M, Veronese FM, Kachensky D, Harris JM. 1996. Preparation and Characterization of Poly(ethylene glycol) Vinyl Sulfone. Bioconjugate Chem.. 7(3):363-368. http://dx.doi.org/10.1021/bc960022413. Lutolf MP, Hubbell JA. 2005. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 23(1):47-55. http://dx.doi.org/10.1038/nbt105514. Sawhney AS, Pathak CP, Hubbell JA. 1993. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers. Macromolecules. 26(4):581-587. http://dx.doi.org/10.1021/ma00056a00515. Du YJ, Lemstra PJ, Nijenhuis AJ, Van Aert HAM, Bastiaansen C. 1995. ABA Type Copolymers of Lactide with Poly(ethylene glycol). Kinetic, Mechanistic, and Model Studies. Macromolecules. 28(7):2124-2132. http://dx.doi.org/10.1021/ma00111a00416. Kim H, Kim HW, Suh H. 2003. Sustained release of ascorbate-2-phosphate and dexamethasone from porous PLGA scaffolds for bone tissue engineering using mesenchymal stem cells. Biomaterials. 24(25):4671-4679. http://dx.doi.org/10.1016/s0142-9612(03)00358-217. Benoit DS, Nuttelman CR, Collins SD, Anseth KS. 2006. Synthesis and characterization of a fluvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration. Biomaterials. 27(36):6102-6110. http://dx.doi.org/10.1016/j.biomaterials.2006.06.03118. West JL, Hubbell JA. 1999. Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules. 32(1):241-244. http://dx.doi.org/10.1021/ma981296k19. Lutolf MP, Hubbell JA. 2003. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules. 4(3):713-722. http://dx.doi.org/10.1021/bm025744e20. Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proceedings of the National Academy of Sciences. 100(9):5413-5418. http://dx.doi.org/10.1073/pnas.073738110021. Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, et al. 2003. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB j.. 17(15):2260-2262. http://dx.doi.org/10.1096/fj.02-1041fje22. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. 2009. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science. 324(5923):59-63. http://dx.doi.org/10.1126/science.1169494
应用实例
2022.12.23

碳化硅为光伏系统提供技术解决方案
引言太阳能技术的持续发展对人类的能源利用意义重大。它是目前世界上最清洁和丰富的资源。太阳能可以通过多种方式加以利用,例如光伏转换和太阳能加热。太阳辐射的能量为3.8×1020 MW(3.8×1020 M/s),到达地球的能量为173×106 kw(相当于1360 W/m2)。虽然太阳能能量十分巨大,但是其较低的利用效率一直是一个令人头疼的问题。硅太阳能电池具有理论由于材料特性,最大效率为31-40%,但在实际部署项目中,最大面板效率仅达到15-30%。因此,最大限度地将太阳能转化为电能是一项挑战。相关领域的研究者们已经提出了太阳能电池技术中的各种解决方案以有效利用从太阳接收的总太阳能。其中一些如下:l使用由元素铅(项目号:L121996)和硒(项目号:S105193)制成的纳米晶体作为太阳能电池原料l降低太阳能电池制造成本l用小圆柱体或纳米棒组成太阳能电池的基本单元l使用染色剂修饰的二氧化钛(项目号:T164497)材料增加阳能电池的光吸收效率[1-3]除以上四点之外,还有关于使用半导体材料制造太阳能逆变器的记录,目的是实现高效率和可靠性。碳化硅(项目号:S104650)是第三代半导体材料,由于其优越的材料特性,目前在大功率应用中占有一席之地。与硅相比。碳化硅器件在太阳能逆变器的制造中发挥着至关重要的作用。在光伏能量转换系统中,逆变器的成本、性能和运行是主要关注点。当今的逆变器需要在以下参数方面进行改进,例如高可靠性、高效率、增强的通信、更低的成本和支持专业应用的灵活性。典型的光伏逆变器应用场景如下:l1-10 kW用于生活应用l100 W至 300 kW用于商业应用l10-500 kW(未来将达到2 MW~20 MW) 用于公用工程系统目前的重点是提高体积功率密度(W/m3) 和比功率 (W/kg),从而最大限度地降低成本光伏逆变器。SiC功率半导体器件在光伏电池中的应用,可以帮助解决几个重要问题。SiC用于光伏电池中的逆变器50 kW三相光伏逆变器系统商业光伏装置的额定功率通常为100 kW至1 MW,尤其适用于商业体系。为满足大功率光伏系统的需求,有研究机构开发了50kW光伏逆变器系统样机,是业内第一款比功率为1kW/kg的全SiC逆变器[4]。图1:简化的50kw光伏逆变器电路原理图,显示系统中各种功率转换阶段电源转换过程由4个通道组成(每通道12.5 kW)交错升压转换器和三个相位逆变器。升压转换器由两个20A SiC MOSFET和两个1200 V/10 A SiC肖特基二极管并联组成。升压转换器在75 kHz的切换频率下运行,在不同的输入电压条件下效率超过99%。图2:50kw升压变流器部分的光伏逆变系统硬件单元结构图5 kW三相逆变器除此之外,也有研究者使用额定1200v/160a的XT-1000半桥MOSFET模块研制了一种5 kW三相全SiC逆变器样机[5]。图3显示了最终原型及其内部结构,尽管该逆变器不是专门为光伏应用设计的,但它能够证明SiC功率器件在缩小系统规模的同时提高其效率的能力。该系统的切换频率为50 kHz。将SiC逆变器与商业化的5 kW硅基逆变器[6]进行比较,以量化性能参数。两个系统都使用自然空气对流进行冷却。从图4中可看出,与硅基逆变器相比,SiC基逆变器能够减少27%的损耗。图3:5kW的SiC三相逆变器样机及其内部结构图图4:SiC逆变器相对于商用硅逆变器的优势(基于关键性能参数)SiC用于光伏电池中的转换器因为具备较宽的可调能带带隙,并且易于在较低的衬底温度下合成,无定形非化学计量碳化硅(a-SixC1-x)是光电应用的理想候选材料。通常情况下,化学计量SiC在可见光区吸收系数低,即使掺杂后电学性能也很差。为了克服这些缺点,近年来有很多研究都集中在了制备具有可调能带带隙的非化学计量SixC1-x [7,8]。通过等离子体增强化学气相沉积(PECVD),通过改变生长参数(如衬底温度)合成了非化学计量的SixC1-x[9,10]。该过程类似于合成非化学计量的SiOx和SiNx材料。非化学计量SixC1-x的可调能带带隙与其C/Si成分有很大关系,进而影响吸收光谱。在氢稀释的PECVD过程中,氢载流子可以降低表面缺陷态的密度。然而,在氢稀释下的制备通常需要较高的衬底温度和射频等离子体功率。在无氢PECVD下,非化学计量富Si的SixC1-x可以在较低的衬底温度下合成,显著提高其吸收系数。与结晶Si薄膜相比,非化学计量的富Si的SixCx材料在可见光区(400-600 nm)具有更小的光学带隙和更高的吸收系数。许多研究报道了a-Si和SixC1-x杂化PVSC的实际应用。然而,很少有报道强调所有基于SixC1-x的PVSCs。Gao等人[11]将基于SixC1-x的n-i-p结PVSCs作为半透明太阳能电池应用于光透过调制器中,但报道转换效率xC1-x的p-n结PVSCs的n型SixC1-x薄膜的厚度从150 nm降低到25 nm,这种参数调谐将转换效率从5×10-3%提高到4.7%[12]。图5:用n型(厚度25 ~ 100 nm)和p型(厚度50 nm)富硅SiC薄膜制备了ITO/p-SiC/n-SiC/Al基PVSC结构在石英衬底上生长全SixC1-x基的单p-i-n结半透明PVSC也是一个非常好的思路。Lin等人[13]使用异常无氢PECVD在远低于SiC合成温度1000℃的衬底温度下生长非化学计量的SixC1-x薄膜。本底SixC1-x (i-SixC1-x)薄膜作为吸收层,在生长过程中通过改变硅烷(SiH4)和甲烷(CH4)的通量比来调节其组成比例,以提高光电流响应。此外,研究者还制备了具有不同C/Si组成比的i- SixC1-x层的富硅SixC1-x/a-Si串联太阳能电池。为了优化富Si的SixC1-x/a-Si串联太阳能电池的转换效率,通过在生长过程中改变SiH4和CH4的通量比来调节n-a-SixC1-x层的C/Si组成比,增加n-a-SixC1-x的p-a-Si界面的隧道化概率。图6:SixCi1-x/a-Si串联PVSC的能带结构结论随着全球温室效应日益显著,碳中和势在必行,这给新能源领域的发展注入了巨大动力。太阳是最清洁的能源,这使得光伏材料的发展和应用具有十分重要的意义。以碳化硅等具备优异特性的半导体材料,正在光伏转换器,逆变器等关键器件中发挥重要作用,未来也一定能够持续贡献力量。参考文献[1] El Chaar L, El Zein N. Review of photovoltaic technologies[J]. Renewable and sustainable energy reviews, 2011, 15(5): 2165-2175. https://doi.org/10.1016/j.rser.2011.01.004[2] Parida B, Iniyan S, Goic R. A review of solar photovoltaic technologies[J]. Renewable and sustainable energy reviews, 2011, 15(3): 1625-1636. https://doi.org/10.1016/j.rser.2010.11.032.[3] Behar O, Khellaf A, Mohammedi K. A review of studies on central receiver solar thermal power plants[J]. Renewable and sustainable energy reviews, 2013, 23: 12-39. https://doi.org/10.1016/j.rser.2013.02.017.[4] Mookken J, Agrawal B, Liu J. Efficient and compact 50kW Gen2 SiC device based PV string inverter[C]//PCIM Europe 2014; International Exhibition and Conference for Power Electronics, Intelligent Motion, Renewable Energy and Energy Management. VDE, 2014: 1-7. https://ieeexplore.ieee.org/abstract/document/6841306.[5] Pushpakaran B N, Subburaj A S, Bayne S B, et al. Impact of silicon carbide semiconductor technology in Photovoltaic Energy System[J]. Renewable and Sustainable Energy Reviews, 2016, 55: 971-989. https://doi.org/10.1016/j.rser.2015.10.161.[6] Bhalla A. Market Penetration of Wide-Bandgap SiC and GaN technology in light of Silicon Super junction and IGBT technology evolution[C]//CS MANTECH Conference. 2014: 9-12.[7] Lin G R, Lo T C, Tsai L H, et al. Finite silicon atom diffusion induced size limitation on self-assembled silicon quantum dots in silicon-rich silicon carbide[J]. Journal of The Electrochemical Society, 2011, 159(2): K35.https://iopscience.iop.org/article/10.1149/2.014202jes/meta.[8] Lo T C, Tsai L H, Cheng C H, et al. Self-aggregated Si quantum dots in amorphous Si-rich SiC[J]. Journal of non-crystalline solids, 2012, 358(17): 2126-2129. https://doi.org/10.1016/j.jnoncrysol.2012.01.013.[9] Cheng Q, Tam E, Xu S, et al. Si quantum dots embedded in an amorphous SiC matrix: nanophase control by non-equilibrium plasma hydrogenation[J]. Nanoscale, 2010, 2(4): 594-600. https://pubs.rsc.org/en/content/articlehtml/2010/nr/b9nr00371a.[10] Cheng Q, Xu S, Long J, et al. Homogeneous nanocrystalline cubic silicon carbide films prepared by inductively coupled plasma chemical vapor deposition[J]. Nanotechnology, 2007, 18(46): 465601. https://iopscience.iop.org/article/10.1088/0957-4484/18/46/465601/meta.[11] Gao W, Lee S H, Bullock J, et al. First a-SiC: H photovoltaic-powered monolithic tandem electrochromic smart window device[J]. Solar Energy Materials and Solar Cells, 1999, 59(3): 243-254. https://doi.org/10.1016/S0927-0248(99)00025-2.[12] Lee C T, Tsai L H, Lin Y H, et al. A chemical vapor deposited silicon rich silicon carbide PN junction based thin-film photovoltaic solar cell[J]. ECS Journal of Solid State Science and Technology, 2012, 1(6): Q144. https://iopscience.iop.org/article/10.1149/2.005301jss/meta.[13] Cheng C H, Chang J H, Wu C I, et al. Semi-transparent silicon-rich silicon carbide photovoltaic solar cells[J]. RSC advances, 2015, 5(46): 36262-36269. https://pubs.rsc.org/en/content/articlehtml/2015/ra/c4ra16998k.
应用实例
2022.12.20

三氟甲基在有机合成中的引入
简介三氟甲基在药物化学领域中是重要的化学基团之一,由于自身拥有高脂溶性、优异的代谢稳定性、高电负性以及良好的生物利用度等特点,因而被广泛地应用于生物活性分子中。之所以在药物分子中引入三氟甲基,是因其具有强吸电子诱导效应、亲脂性及稳定的C-F键,可有效延长其在生物体内的作用时间,增强其代谢稳定性;与此同时,三氟甲基的引入还会伴随药物分子脂溶性的增加,因此有助于药物分子在生物体内的吸收、传递和扩散。图1.含有三氟甲基的药物分子1.抗癌药物——索拉非尼(Sorafenib tosylate) 2.抗抑郁药物——氟西汀(Fluoxetine) 3.新型植物广谱杀菌剂——肟菌酯(Trifloxystrobin) 由于自然界中含氟化合物的种类比较稀少,因此如何方便温和高效安全地将含氟基团引入到各种有机化合物中制备特殊的含氟化合物是现今化学界的一个重要研究方向。三氟甲基的引入一般分为两类:一、直接氟化法(三氟甲基)三甲基硅烷(TMSCF3)是最广泛使用的亲核三氟甲基化试剂之一。由于Ruppert于1984年引入该试剂[1],而后Prakash又主要负责推广其使用[2],所以该试剂也被称为Ruppert-Prakash试剂。TMSCF3的广泛适用性使其成为合成各种药物靶点的常用试剂。 合成具有三氟甲基酮基团的谷氨酸和谷氨酰胺肽过程中[3],β-氨基醇分五步合成,其中关键步骤就是利用TMSCF3生成单一非对映体的三氟甲基甲硅烷基醚(方案1)。报告中的两种肽显示出对严重急性呼吸综合征冠状病毒蛋白酶(SARS-CoV 3CLpro)的抑制活性。方案1另一个例子是利用TMSCF3合成了一种非甾体类选择性雄激素受体调节剂,该调节剂在肌肉中表现出优异的口服生物利用度和合成代谢活性(方案2)[4]。该化合物还改善了绝经后骨质疏松症大鼠模型的骨强度。同样,Hudso和他的同事又使用TMSCF3合成出了一种选择性糖皮质激素受体调节剂,显示出与骨髓瘤治疗剂地塞米松相同的抗增殖活性(方案3)[5]。方案2方案3通常,需要额外的氟化物源(TBAF、CsF等)来引发三氟甲基化反应。然而,Prakash最近的一份报告详细介绍了使用一系列不需要额外的氟化物引发剂或无水条件的催化剂进行羰基化合物的三氟甲基化(方案4)[6]。方案4Prakash的另一份报告表明,使用TMSCF3对N-未活化的亚胺进行处理,可以得到相应的三氟甲基胺。只要稍微改变反应条件,也可以通过HF消除和还原制备二氟甲基胺(方案5)[7]。方案5尽管Ruppert-Prakash试剂被广泛使用,但使用该试剂进行立体选择性三氟甲基化仍是一项挑战。Dieter Enders报告了α-烷基二氧杂环己烷的非对映选择性三氟甲基化,产率非常好,ee值也非常高[8]。丙酮基团很容易被去除,生成2-三氟甲基-1,2,3-三醇(方案6)。方案6Shibata和Toru团队最近报告了一种操作简单的方法,基于金鸡纳生物碱的溴化铵(1)和四甲基氟化铵(TMAF)的组合进行对映选择性的三氟甲基化。该反应以中等至优异的产率进行,ee值高达93%(方案7)。方案7二、三氟甲基砌块最经典的三氟甲基砌块非三氟乙酰乙酸乙酯莫属,是含氟杂环化合物合成中重要砌块之一。因其具有多个反应位点,同时具有酮和烯醇性质,因此可以以不同的反应来构建含氟的吲哚、吡唑、嘧啶和其他含氟杂环,且用于医药、农药、染料等应用领域所需的含氟中间体合成[9,10]。图2. 一些重要的三氟甲基杂环以三氟乙酰乙酸乙酯、氰基乙酰胺和芳基醛为原料,在哌啶催化下合成3-酰氨基-4-芳基-6-三氟甲基哌啶-2-酮化合物。在Hofmann重排和芳基迁移后,该化合物在碱性条件下与乙酸碘苯反应,最终生成3-芳基-6-三氟甲基吡啶-2-酮化合物。方案8. 三氟乙酰乙酸乙酯作为含氟砌块这类含氟砌块是由Uneyama Kenji课题组[11]于1992年在日本开发。在四卤化碳和三苯基膦的作用下使用三氟乙酸和胺,从而合成了一系列含氟酰亚胺酰卤化合物。此后,Hao jian课题组在三乙胺、四氯化碳和三苯基膦的反应条件下,含氟碳酸和邻氨基、羟基或巯基取代的苯胺,一锅合成2-氟烷基取代的苯并1,3二唑,并基于此,使用了更多的底物和衍生物,尝试合成出更多具有潜在生物活性的化合物[12]。参考文献:1.Ruppert I, Schlich K, Volbach W. 1984. Die ersten CF3-substituierten organyl(chlor)silane. Tetrahedron Letters. 25(21):2195-2198. https://doi.org/10.1016/s0040-4039(01)80208-22.Prakash GKS, Krishnamurti R, Olah GA. 1989. Synthetic methods and reactions. 141. Fluoride-induced trifluoromethylation of carbonyl compounds with trifluoromethyltrimethylsilane (TMS-CF3). A trifluoromethide equivalent. J. Am. Chem. Soc.. 111(1):393-395. https://doi.org/10.1021/ja00183a0733.Sydnes MO, Hayashi Y, Sharma VK, Hamada T, Bacha U, Barrila J, Freire E, Kiso Y. 2006. Synthesis of glutamic acid and glutamine peptides possessing a trifluoromethyl ketone group as SARS-CoV 3CL protease inhibitors. Tetrahedron. 62(36):8601-8609. https://doi.org/10.1016/j.tet.2006.06.0524.Martinborough E, Shen Y, vanOeveren A, Long YO, Lau TLS, Marschke KB, Chang WY, López FJ, Vajda EG, Rix PJ, et al. 2007. Substituted 6-(1-Pyrrolidine)quinolin-2(1H)-ones as Novel Selective Androgen Receptor Modulators. J. Med. Chem.. 50(21):5049-5052. https://doi.org/10.1021/jm070231h5.Hudson AR, Roach SL, Higuchi RI, Phillips DP, Bissonnette RP, Lamph WW, Yen J, Li Y, Adams ME, Valdez LJ, et al. 2007. Synthesis and Characterization of Nonsteroidal Glucocorticoid Receptor Modulators for Multiple Myeloma. J. Med. Chem.. 50(19):4699-4709. https://doi.org/10.1021/jm070370z6.Prakash GKS, Panja C, Vaghoo H, Surampudi V, Kultyshev R, Mandal M, Rasul G, Mathew T, Olah GA. 2006. Facile Synthesis of TMS-Protected Trifluoromethylated Alcohols Using Trifluoromethyltrimethylsilane (TMSCF3) and Various Nucleophilic Catalysts in DMF. J. Org. Chem.. 71(18):6806-6813. https://doi.org/10.1021/jo060835d7.Prakash GKS, Mogi R, Olah GA. 2006. Preparation of Tri- and Difluoromethylated Amines from Aldimines Using (Trifluoromethyl)trimethylsilane. Org. Lett.. 8(16):3589-3592. https://doi.org/10.1021/ol061357w8.Enders D, Herriger C. 2007. Asymmetric Synthesis of 2-Trifluoromethyl-1,2,3-triols. Eur. J. Org. Chem.. 2007(7):1085-1090. https://doi.org/10.1002/ejoc.2006008959. Zhu Shizheng, Wang Yanli, Jin Guifang. Research on fluorine-containing active methylene compounds [J]. Journal of Chemistry, 2002 (04): 555-565+75810. Zhao Min, Yang Xiangmin. Synthesis of a new class of trifluoromethyl ketones [J]. Journal of East China University of Technology, 1999 (04): 111-11211. Tamura, K.; Mizukami, H.; Maeda, K.; Watanabe, H.; Uneyama, K., J. Org. Chem.1993,58, 32.12. Ge, F.; Wang, Z.; Wan, W.; Lu, W.; Hao, J., Tetrahedron Lett.2007,48, 3251.
应用实例
2022.12.20

钙离子指示剂与离子载体使用指南
钙离子指示剂与离子载体使用指南阿拉丁帮助您寻找成像Ca2+和细胞功能的最佳指标和电离层。钙(Ca2+)是一种重要的无处不在的第二信使,参与调节多种细胞过程,包括细胞增殖、基因转录、肌肉收缩和内吞作用。使用本指南,我们可以为您的实验找到最佳的Ca2+指示器,螯合剂和离子团。Ca2+指标一目了然指示剂激发波长(nm)发射波长(nm)Kd (nM)Fura-2340/380505145Indo-1346475/405230Fluo-4488520345Fluo-8490520390Fluo-54945162300Rhod-2552581570BAPTABAPTA是一种非荧光Ca2+螯合剂,从EGTA中提取,对Ca2+的选择性为Mg2+的105倍。它通常用于缓冲,在没有Mg2+的情况下,Kd为160 nM。也可提供AM形式。Fura-2Fura-2是第一种商业化的荧光钙指示剂。激发比为340 nm/380 nm,发射波长为505 nm。在科学文献中有成千上万的Fura-2参考文献,它是迄今为止最重要的Ca2+指示染料之一。Indo-1Indo-1与Fura-2在1986年同时推出。它也是比例的,但模式不同。当在346 nm激发时,它在475 nm/405 nm处表现出发射比,而不是激发比。与Fura-2不同,它有光漂白的倾向。它也被用于各种各样的应用,以大量的文献出版物为例。Fluo-8自1989年引入以来,荧光成像揭示了Ca2+信号通路中许多基本过程的空间动力学。在细胞实验中发现,Fluo-8(或Fluo-2介质亲和力)比Fluo-4更亮(1.5倍)。它改善了细胞负载和Ca2+响应,同时保持方便的Fluo-3和Fluo-4光谱波长,在490 nm处最大激发,在520 nm处最大发射。Fluo-8加载可在室温下进行。Rhod-2Rhod-2于1989年首次问世,其荧光激发极值在552 nm,发射极值在581 nm。在Ca2+结合之前,Rhod-2本质上是不荧光的,随着Ca2+浓度的增加,Rhod-2变得更加荧光。Rhod-2较长的激发和发射使该指示物在具有高水平自身荧光的细胞和组织的实验中非常有用,并可与其他波长较短的荧光染料进行多路复用。这些基于罗丹明的指示物的AM酯形式是阳离子的,这可以导致电位驱动的摄取进入线粒体。Fluo-4 AMFluo-4是Fluo-3的类似物,两个氯取代基被氟取代,这导致荧光激发增强在488 nm,因此更高的荧光信号水平。Fluo-5 AMFluo-5 AM是Fluo-4的类似物,但其钙结合亲和力降低,因此适合检测细胞内钙水平,这将饱和Fluo-4 AM指示剂。钙离子载体A23187(钙霉素)具有可被紫外光激发的固有荧光,使其对可被紫外光激发的Ca2+指示剂(如Fura-2)不太有用,但对长波长Ca2+指示剂(如Fluo-2)仍然有用。4-溴A-23187是非荧光的,因此与所有Ca2+指示剂兼容。它通常用于荧光Ca2+指标的原位校准,以平衡细胞内和细胞外Ca2+浓度,并允许Mn2+进入细胞,淬灭细胞内的染料荧光。离子霉素是一种有效的Ca2+离子胞体,通常用于校准荧光Ca2+指标和修改细胞内Ca2+浓度,并用于研究Ca2+在细胞过程中的调节特性。参考文献1.Erdahl WL, Chapman CJ, Taylor RW, Pfeiffer DR. Ca2+ transport properties of ionophores A23187, ionomycin, and 4-BrA23187 in a well defined model system. Biophys J. 1994 May;66(5):1678-93. 2. Drummond IA, Lee AS, Resendez E Jr, Steinhardt RA. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J Biol Chem. 1987 Sep 15;262(26):12801-5.
参数原理
2022.12.12

材料 | 听100遍反方向的钟不能回到过去,但PEG水凝胶可以
水凝胶是长期以来受到人们关注最多的生物材料之一,它们在化学和物理性质上都非常接近细胞的自然环境,因此作为细胞的二维和三维支架被广泛研究[1-2]。水凝胶可以由人工合成的聚合物(如聚(乙二醇)、聚(甲基丙烯酸羟乙酯)等)与天然存在的聚合物(如胶原蛋白、透明质酸等)交联形成,并且由于它们的含水量非常高,可以有效被应用于组织培养的3D模型,不受细胞、蛋白质和DNA的影响。根据不同组成部分的反应性,可以通过pH、温度、库仑相互作用、共价键、非共价相互作用或聚合来诱实现凝胶化[3-5]。 通过环氧乙烷的活性阴离子开环聚合能够非常方便地合成聚乙二醇;这些聚乙二醇具有较宽的分子量分布,且包含多种端基,在众多反应中都可适配。为了形成水凝胶,PEG必须发生交联反应。最开始的时候,PEG是通过电离辐射进行非特异性交联的[6],而现在PEG水凝胶的形成通常是通过共价交联具有反应链端的PEG高分子合成的。具有活性反应末端的PEG高分子聚合物,如丙烯酸酯、甲基丙烯酸酯、烯丙醚、马来酰亚胺、乙烯基砜、NHS酯和乙烯基醚基团(图1)很容易通过直接购得的试剂合成[7]。 图1:不同PEG大分子的末端基团 高分子的两个末端可以是两个相同或者不同的官能团。同双官能团高分子通常用于形成网络,而异双官能团高分子则可用于将具有治疗性的小分子连接到水凝胶网络中。 形成水凝胶的交联机制取决于PEG高分子链末端的特性。在大多数情况下,是与反应性乙烯基链末端聚合的同时发生交联,通常采用的是自由基引发剂。例如,大分子单体的聚合可以使用通过氧化还原反应生成的自由基(比如过硫酸铵和TEMED)或光照产生的自由基(图2,λ=365 nM)来引发,随后通过丙烯酸酯和甲基丙烯酸酯链末端基元反应来发生链增长。在阶梯生长网络的形成中,多官能度(f>2)的交联剂以化学当量与PEG链末端进行反应,或多官能度的PEG(f>2)也可以与双官能团的交联剂发生交联。丙烯酸酯、甲基丙烯酸酯、乙烯基砜、马来酰亚胺、乙烯基醚和烯丙基醚都可以根据反应条件转化为硫醇,形成阶梯生长网络。典型的交联剂有包含巯基或胺官能团的结构。混合模式聚合是在同一反应容器中发生的两种机制的结果;丙烯酸酯和甲基丙烯酸酯基团可以形成混合模式网络。两种水凝胶形成机制均可用于包裹活体细胞,并且两种机制均可使肽、蛋白质或其他治疗药物发生反应性掺入。 图2:链式增长和阶梯式增长反应 为了使用3D水凝胶支架来研究细胞分化和组织进化,通过空间和时间调控的方式来设计凝胶的物理和化学性质是至关重要的[8]。聚合物材料性质通常借由聚合/交联(键形成)或借由受控降解及/或释放(键断裂)来改变。键的形成通常会用到小分子试剂(引发剂、催化剂,单体、连接到材料的配体),而键断裂则通常不依赖于外源试剂。小分子在体外和体内均具有比聚合物试剂更多的副反应,因此许多研究小组用降解作为原位操作聚合物生物材料的工具。 1.水解降解 水凝胶中最常用的降解机理就是水解,即一个水分子加入到聚合物骨架中,导致链断裂。酸酐、酯和酰胺都很容易水解,氢化物通常水解得太快,而酰胺类的若未经过催化则水解太慢,因此大多数水凝胶的水解降解都是利用酯键。为了获得具有生理相关时间维度上的可降解水凝胶,研究人员通常使用交酯或乙醛交酯段将具有可降解酯键的PEG功能化(图3)[9]。 图3:聚乙二醇乙交酯和聚乙二醇丙交酯的合成 2.酶促降解 虽然酯键是可通过酶降解的,但大多数研究人员都会使用具有特异性序列的酶降解掺入水凝胶中的肽,而不是非特异性的酶降解酯和酰胺。Hubbell团队[10-11]率先通过Michael加成反应把丙烯酸酯、马来酰亚胺和乙烯基砜官能化的半胱氨酸肽,将基质金属蛋白酶(MMP)具有反应敏感型的连接键引入水凝胶(图4)。 图4:通过Michael加成将含有半胱氨酸的肽添加到含乙烯基砜基团的酶中制备可降解型水凝胶 MMP-可降解键也被用作联结治疗剂与水凝胶的载体。例如,血管内皮生长因子(VEG-F)等生长因子可通过酶降解MMP-敏感性链而释放,从而诱导血管生成[12]。 在水解和酶解过程中,降解速率由大分子的化学性质决定。在水解中,材料的降解率是通过其本身的性质(如疏水性或亲水性)和可水解基团的数量预先设计的,并且一旦材料被制造出来,就不能改变。在酶解过程中,降解通常发生在产生酶的细胞局部区域。水解和酶解均是缓释治疗药物的有效方法,但水凝胶制备后无法调节或阻滞其释放速率,且释放不受空间限制。 3.光致降解 与水解和酶降解相比,光降解允许更加精准的空间和时间控制降解和释放。虽然已有许多研究者报道了光聚合水凝胶和光功能化水凝胶,但关于生物相容性光降解水凝胶的报道很少。Kloxin和Kasko发表了由含PEG高分子形成的光可降解水凝胶网络(图5)[13];邻-硝基苄基(o-NB)连接基团的光降解行为具有良好的表现。由光降解聚合物形成的水凝胶在光照下表现出体积降解,这与曝光时间、波长和光强度有关。当光线停止照射时,降解停止;在恢复光照后,上述样品继续光解。部分降解导致水凝胶的交联密度降低和溶胀程度增加,这提供了一种如何制备柔软性更高的水凝胶的方法。 图5:光降解o-NB掺杂的水凝胶骨架用于释放治疗药剂 除了单光子光解,含有o-NB的水凝胶也对双光子光解敏感,从而允许被用于3D蚀刻[14-15]。在单光子反应中,任何暴露在光下的区域都会发生反应。相反,多光子光刻应该只发生在多个光子同时被吸收的地方,这发生在光源的焦体积。生物材料的单光子光刻的典型波长范围从长波UV(≥365 nm)到可见光区域,而双光子光刻则使用红外光(通常~ 740-800 nm)较多。红外光具有更好的生物相容性,对活体组织的破坏性更小,并能够有更大的穿透深度。发生双光子吸收的区域也被严格限制在了光的焦点上,而不是沿着光的整个路径,提供了对激发3D控制的新思路。单光子和多光子反应都有能力制备出特征点小于500 nm的材料,远小于哺乳动物细胞的大小[16]。这代表了对水凝胶支架结构和化学的空间控制水平达到了前所未有的高度。 聚乙二醇是一种易制备、易改性的聚合物。它被广泛应用于水凝胶制造,包括作为组织培养的2D和3D支架。聚乙二醇水凝胶易于引入可降解键——水解可降解凝胶允许持续的材料降解和/或治疗剂释放;酶降解凝胶的降解和释放是由细胞决定的;光致降解允许使用者对水凝胶的理化性质进行实时定制的外部操作。 产品货号产品名称包装规格 P103730 聚乙二醇 100g/250g/500g/1kg/5kg P109710 聚(乙二醇)甲基丙烯酸酯 25mL/100mL/500mL/2.5L P109711 聚乙二醇甲醚甲基丙烯酸酯 100mL/500mL/2.5L P133766 聚乙二醇二甲基丙烯酸酯 50mL/250mL/1L/2.5L M130065 聚乙二醇单甲醚 100g/250g/500g/1kg P274350 聚乙二醇8000 500g/1kg/2.5kg P303204 聚对苯二甲酸乙二醇酯 25g/100g/500g/2.5kg M109437 甲氧基聚乙二醇胺 250mg/500mg/1g/5g/25g A163261 氨基 PEG, mPEG-NH2 100mg/1g/5g T164391 巯基 PEG, mPEG-SH 200mg/1g/5g 阿拉丁技术文章:用于2D和3D细胞培养的可降解型聚乙二醇水凝胶 ——点击查看详情 1. Drury JL, Mooney DJ. 2003. Hydrogels for tissue engineering: scaffold design variables and applications.Biomaterials.24(24):4337-4351. http://dx.doi.org/10.1016/s0142-9612(03)00340-52. Lee KY, Mooney DJ. 2001. Hydrogels for Tissue Engineering.Chem. Rev..101(7):1869-1880. http://dx.doi.org/10.1021/cr000108x3. HOFFMAN AS. Hydrogels for Biomedical Applications. 944(1):62-73. http://dx.doi.org/10.1111/j.1749-6632.2001.tb03823.x4. Hoffman AS. 2002. Hydrogels for biomedical applications.Advanced Drug Delivery Reviews.54(1):3-12. http://dx.doi.org/10.1016/s0169-409x(01)00239-35. Tibbitt MW, Anseth KS. 2009. Hydrogels as extracellular matrix mimics for 3D cell culture.Biotechnol. Bioeng..103(4):655-663. http://dx.doi.org/10.1002/bit.223616. Ruel-Gariépy E, Leroux J. 2004. In situ-forming hydrogels?review of temperature-sensitive systems.European Journal of Pharmaceutics and Biopharmaceutics.58(2):409-426. http://dx.doi.org/10.1016/j.ejpb.2004.03.0197. Yang Z, Xu B. 2007. Supramolecular hydrogels based on biofunctional nanofibers of self-assembled small molecules.J. Mater. Chem..17(23):2385. http://dx.doi.org/10.1039/b702493b8. Zalipsky S, Harris JM. 1997. Introduction to Chemistry and Biological Applications of Poly(ethylene glycol).1-13. http://dx.doi.org/10.1021/bk-1997-0680.ch0019. Davis FF. 2002. The origin of pegnology.Advanced Drug Delivery Reviews.54(4):457-458. http://dx.doi.org/10.1016/s0169-409x(02)00021-210. Sawhney AS, Pathak CP, Hubbell JA. 1993. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers.Macromolecules.26(4):581-587. http://dx.doi.org/10.1021/ma00056a00511. Du YJ, Lemstra PJ, Nijenhuis AJ, Van Aert HAM, Bastiaansen C. 1995. ABA Type Copolymers of Lactide with Poly(ethylene glycol). Kinetic, Mechanistic, and Model Studies.Macromolecules.28(7):2124-2132. http://dx.doi.org/10.1021/ma00111a00412. Kim H, Kim HW, Suh H. 2003. Sustained release of ascorbate-2-phosphate and dexamethasone from porous PLGA scaffolds for bone tissue engineering using mesenchymal stem cells.Biomaterials.24(25):4671-4679. http://dx.doi.org/10.1016/s0142-9612(03)00358-213. Benoit DS, Nuttelman CR, Collins SD, Anseth KS. 2006. Synthesis and characterization of a fluvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration.Biomaterials.27(36):6102-6110. http://dx.doi.org/10.1016/j.biomaterials.2006.06.03114. Lutolf MP, Hubbell JA. 2003. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition.Biomacromolecules.4(3):713-722. http://dx.doi.org/10.1021/bm025744e15. Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics.Proceedings of the National Academy of Sciences.100(9):5413-5418. http://dx.doi.org/10.1073/pnas.073738110016. Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, et al. 2003. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth.FASEB j..17(15):2260-2262. http://dx.doi.org/10.1096/fj.02-1041fje 文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请在阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
应用实例
2022.12.12

EGF信号:追踪癌症的路径
表皮生长因子(EGF)家族是一组结构相关的蛋白质,通过靶细胞上的酪氨酸激酶受体调节细胞增殖、迁移和分化。EGF受体有一个细胞质酪氨酸激酶结构域,一个跨膜结构域和一个与EGF结合的细胞外结构域。配体与EGF受体结合导致其二聚、自磷酸化和激活。一旦被激活,EGF受体通过几个蛋白质的磷酸化传递细胞内信号。Ras被EGF受体激活是EGF信号转导的重要组成部分。鸟嘌呤核苷酸交换因子SOS激活Ras, Ras进而触发丝裂原激活蛋白(MAP)激酶通路。MAP激酶磷酸化转录因子,如激活蛋白1(AP-1;Fos-Jun二聚体)和Elk-1,导致细胞生长和发育。EGFR对Janus激酶(JAK)的磷酸化导致转录蛋白信号换能器和激活器(STATs)的激活,最终导致细胞的生长和分化。EGF信号的另一个关键方面涉及磷脂酶c - γ1 (PLCγ1),它将PIP2裂解为IP3和DAG。IP3的产生导致内质网钙的释放,而DAG促进蛋白激酶C (PKC)的激活。PKC反过来磷酸化并激活转录因子Elk-1,导致细胞增殖。已知EGFR的突变影响其表达或活性,这使EGFR成为重要的药物靶点。该通路强调了EGF信号转导的重要组成部分。几个重要的表皮生长因子信号通路参考文献1. Corbalan-Garcia S, Margarit SM, Galron D, Yang S, Bar-Sagi D. 1998. Regulation of Sos Activity by Intramolecular Interactions. Mol. Cell. Biol.. 18(2):880-886. https://doi.org/10.1128/mcb.18.2.8802. Russell M, Lange-Carter CA, Johnson GL. 1995. Direct Interaction between Ras and the Kinase Domain of Mitogen-activated Protein Kinase Kinase Kinase (MEKK1). Journal of Biological Chemistry. 270(20):11757-11760. https://doi.org/10.1074/jbc.270.20.117573. Ackerman P, Glover CV, Osheroff N. 1990. Stimulation of casein kinase II by epidermal growth factor: relationship between the physiological activity of the kinase and the phosphorylation state of its beta subunit.. Proceedings of the National Academy of Sciences. 87(2):821-825. https://doi.org/10.1073/pnas.87.2.8214. Andl CD, Mizushima T, Oyama K, Bowser M, Nakagawa H, Rustgi AK. 2004. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. American Journal of Physiology-Gastrointestinal and Liver Physiology. 287(6):G1227-G1237. https://doi.org/10.1152/ajpgi.00253.20045. Baker SJ, Kerppola TK, Luk D, Vandenberg MT, Marshak DR, Curran T, Abate C. 1992. Jun is phosphorylated by several protein kinases at the same sites that are modified in serum-stimulated fibroblasts.. Mol. Cell. Biol.. 12(10):4694-4705. https://doi.org/10.1128/mcb.12.10.46946. Takekawa M, Tatebayashi K, Saito H. 2005. Conserved Docking Site Is Essential for Activation of Mammalian MAP Kinase Kinases by Specific MAP Kinase Kinase Kinases. Molecular Cell. 18(3):295-306. https://doi.org/10.1016/j.molcel.2005.04.0017. Chong M, Barritt G, Crouch M. 2004. Insulin potentiates EGFR activation and signaling in fibroblasts. Biochemical and Biophysical Research Communications. 322(2):535-541. https://doi.org/10.1016/j.bbrc.2004.07.1508. Krug AW, Schuster C, Gassner B, Freudinger R, Mildenberger S, Troppmair J, Gekle M. 2002. Human Epidermal Growth Factor Receptor-1 Expression Renders Chinese Hamster Ovary Cells Sensitive to Alternative Aldosterone Signaling. Journal of Biological Chemistry. 277(48):45892-45897. https://doi.org/10.1074/jbc.m2088512009. Lim CP, Cao X. 1999. Serine Phosphorylation and Negative Regulation of Stat3 by JNK. Journal of Biological Chemistry. 274(43):31055-31061. https://doi.org/10.1074/jbc.274.43.3105510. Diakonova M, Payrastre B, van Velzen AG, Hage W, van Bergen en Henegouwen PM, Boonstra J, Cremers F, Humbel B. 1995. Epidermal growth factor induces rapid and transient association of phospholipase C-gamma 1 with EGF-receptor and filamentous actin at membrane ruffes of A431 cells.. J Cell Sci..(108):2499–2509.11. Eldar H, Zisman Y, Ullrich A, Livneh E. 1990. Overexpression of protein kinase C alpha-subtype in Swiss/3T3 fibroblasts causes loss of both high and low affinity receptor numbers for epidermal growth factor.. Journal of Biological Chemistry. 265(22):13290-13296. https://doi.org/10.1016/s0021-9258(19)38297-312. Weston CR, Wong A, Hall JP, Goad MEP, Flavell RA, Davis RJ. 2004. The c-Jun NH2-terminal kinase is essential for epidermal growth factor expression during epidermal morphogenesis. Proceedings of the National Academy of Sciences. 101(39):14114-14119. https://doi.org/10.1073/pnas.040606110113. Carpenter G, Cohen S. 1990. Epidermal growth factor.. Journal of Biological Chemistry. 265(14):7709-7712. https://doi.org/10.1016/s0021-9258(19)38983-514. Hu, Bowtell D. 1996. Sos1 rapidly associates with Grb2 and is hypophosphorylated when complexed with the EGF receptor after EGF stimulation. Oncogene.. 12(9):1865–72.15. Ueno H, Sasaki K, Miyagawa K, Honda H, Mitani K, Yazaki Y, Hirai H. 1997. Antisense Repression of Proto-oncogene c-Cbl Enhances Activation of the JAK-STAT Pathway but Not the Ras Pathway in Epidermal Growth Factor Receptor Signaling. Journal of Biological Chemistry. 272(13):8739-8743. https://doi.org/10.1074/jbc.272.13.873916. Cummins AB, Palmer C, Mossman BT, Taatjes DJ. 2003. Persistent Localization of Activated Extracellular Signal-Regulated Kinases (ERK1/2) Is Epithelial Cell-Specific in an Inhalation Model of Asbestosis. The American Journal of Pathology. 162(3):713-720. https://doi.org/10.1016/s0002-9440(10)63867-917. Yoshikawa S, Tanimura T, Miyawaki A, Nakamura M, Yuzaki M, Furuichi T, Mikoshiba K. 1992. Molecular cloning and characterization of the inositol 1,4,5-trisphosphate receptor in Drosophila melanogaster.. Journal of Biological Chemistry. 267(23):16613-16619. https://doi.org/10.1016/s0021-9258(18)42047-918. Mahimainathan L, Ghosh-Choudhury N, Venkatesan BA, Danda RS, Choudhury GG. 2005. EGF stimulates mesangial cell mitogenesis via PI3-kinase-mediated MAPK-dependent and AKT kinase-independent manner: involvement of c-fos and p27Kip1. American Journal of Physiology-Renal Physiology. 289(1):F72-F82. https://doi.org/10.1152/ajprenal.00277.200419. Xia Y, Makris C, Su B, Li E, Yang J, Nemerow GR, Karin M. 2000. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proceedings of the National Academy of Sciences. 97(10):5243-5248. https://doi.org/10.1073/pnas.97.10.524320. Chen D, Davis JS. 2003. Epidermal growth factor induces c-fos and c-jun mRNA via Raf-1/MEK1/ERK-dependent and -independent pathways in bovine luteal cells. Molecular and Cellular Endocrinology. 200(1-2):141-154. https://doi.org/10.1016/s0303-7207(02)00379-9
参数原理
2022.12.02

热门半导体材料砷化镓应用揭秘
背景介绍砷化镓(GaAs)(项目号:G119227)是镓和砷两种元素所合成的化合物,两者是重要的IIIA族、VA族半导体材料。因此,GaAs也是重要的化合物半导体材料。GaAs外观呈亮灰色,具金属光泽、性脆而硬。常温下比较稳定,不与盐酸、硫酸、氢氟酸等反应,但能与浓硝酸反应,也能与热的盐酸(项目号:H399545)和硫酸(项目号:S399850)作用。当其被加热到873K时,外表开始生成氧化物,形成氧化膜。除此之外,GaAs还具备较好的电子特性,如较高的饱和电子速率及电子迁移率,较快的切换速度,抗天然辐射等一系列独特性质。GaAs天然存量稀少,通常采用镓和砷直接化合的方法,其中水平区域熔炼法是普遍采用的方法。通过区域提纯便可获得单晶。采用间接的方法也可获得GaAs。如通过砷蒸气(项目号:A110123)将GaCl(项目号:G345285)还原来制备GaAs(式一),或者通过Ga(CH3)3和AsH3在一定温度下,发生热分解得到GaAs(式二)。4GaCl + 2H2 + As4 → 4GaAs + 4HCl (一)Ga(CH3)3 + AsH3 → GaAs + 3CH4 (二)应用GaAs具备许多优异的特性,其应用也十分广泛,具体大致可以分为RF(射频), PHOTONICS(光电子), LED(发光二极管)和PV(光伏发电)四大领域。射频器件射频器件主要功能是实现信号发送和接收,由功率放大器(Power Amplifier,PA)、射频开关、滤波器、数模/模数转换器等器件组成。GaAs应用于射频领域,主要环节是PA。经过PA放大的信号,最终从设备中发射出去,属于通信设备高能耗环节。最近,Nguyen等人[1]采用0.15 μm GaAs设计了一种具有谐波调谐输出匹配网络的叠置场效应晶体管PA。所制备的PA具有28.5 dBm的输出功率,12 dB增益和38.4%的功率附加效率(PAE)。这是第一次将叠置场效应晶体管技术与谐波调谐输出网络相结合,实现了GaAs PA中的高PAE和高功率密度。图1:功率放大器芯片照片(1.1 mm × 0.8 mm)[1]此外,GaAs在5G手机PA领域也占据了主导地位。GaAs具备较高的饱和电子速率及电子迁移率,使得其适合应用于高频场景,在高频操作时具有较低的噪声;同时因为GaAs有比Si更高的击穿电压,所以GaAs更适合应用在高功率场合。因为这些特性,GaAs在5G时代,仍然将是功率放大器及射频开关等手机射频器件的主要材料。光电器件GaAs的另一个优点是其具备的直接能隙,使其拥有了较好的光电性能,可以用于制作光电子器件。使用GaAs衬底制造的红外激光器、传感器因其具备高功率密度、低能耗、抗高温、高发光效率、高击穿电压等特点受到了广泛关注。高效的光电转化是人们不懈追求的目标,对推动新能源和信息领域的发展和应用具有十分重要的意义。光子功率转换器或光电传感器可以吸收通过多模光纤传输的红外激光功率,并将其转换为电能以供远程使用。为了将此功率转换为有用的电压,Hinzer等人[2]设计和制造了一种GaAs光伏光电传感器,该光电传感器使用单片、晶格匹配、垂直堆叠的单电池器件产生>5 V,从而消除了复杂的制造和组装步骤。实验测量结果也表明,在波长为835 nm、光照强度为11 W/cm2的条件下,转换效率可达60.1%。图2:实验装置示意图[2]LED器件发光二极管(light-emitting diode,LED)是由化合物半导体(GaAs、GaN等)组成的固体发光器件,可将电能转化为光能。不同材料制成的LED会发出不同波长、不同颜色的光。LED根据芯片尺寸可以区分为常规LED、Mini LED、Micro LED等类型,其中Mini LED、Micro LED应用于新一代显示。电致发光(在LED中电子到光子的转换)可以用作制冷机制,前提是LED具有较高的量子效率。Xiao等人[3]通过优化GaAs/GaInP双异质结构LED,研究了电致发光技术在制冷领域的实际应用。研究首先基于精细平衡物理和统计射线光学方法建立了设计模型,并预测了263 K时的外部发光效率ηext为97.7%。其次,为了提高冷却系数的性能,研究者进一步将冷却的LED与光伏电池配对,光伏电池可以将部分发出的光能转化为电能。对于接近室温和中等功率密度(1.0-10 mW/cm2)的应用,使用现有GaAs器件中的材料质量,电致发光制冷器可以以较高的性能系数运行(预计为原来的1.7倍)。图3:用于电致发光的Ga0.51In0.49P/GaAs双异质LED结构示意图[3]光伏发电在光伏发电技术中,太阳能电池是其中最重要的元件,目前主要应用于光伏发电的电池大都是基于半导体技术。GaAs属于III-V族化合物半导体材料,其能隙与太阳光谱的匹配较适合,且能耐高温。与硅太阳电池相比,GaAs太阳电池具有较好的性能。如图4,Sheng及其同事[4]制备了一种微型薄膜双结砷化镓光电二极管器件(double junction GaAs photodiode ), 并研究了其在不同波长和强度激发光下的光子和载流子传输行为。实验测试表明,光子回收效应与激发光波长和功率等参数密切相关,在蓝紫色光(400~480 nm)和近红外光(~800 nm)照射下,双结电池产生的光电流与激发光功率分别呈现超线性和线性的特性。同时,在高强度激发光照射下,光子回收效应可以显著改善子电池间的电流匹配情况,实现宽波段、高效率的光电响应(波长400~800 nm,外部量子效率接近50%)。图4:在475 nm光照下,GaAs双结光电二极管内部的光学过程示意图[4]纳米流体近年来被认为是PV/T体系中一种良好的冷却液。Samir及其同事[5]提出了一种新的、具有独立通道的级联纳米流体PV/T配置,其中一个通道控制光学特性,而另一个通道增强PV电池的热排出。在第一种情形中,光学纳米流体在PV电池上方充当液体光学带通滤波器。在第二种情形中,热纳米流体从PV电池背面除去热量。对不同浓度比下的GaAs和Si基光伏电池进行了模拟。仿真结果表明,最佳的光学纳米流体滤光片可向GaAs或Si光伏电池传输的能量约为理想光谱的82%。在集中系统中,GaAs (C¼45)和Si (C¼30)的电效率方面,分离通道系统(D-1)比双通道设计(D-2)高出8.6%。将热纳米流体的体积分数从0.001提高到1.5%,加入GaAs (C¼160)和Si (C¼100)的D-1体系的整体效率分别提高了约5.8%和约4.6%。结果表明,具有单独通道的光伏/T配置在高浓度(C > 100)太阳能系统中有进一步发展的潜力。图5:PV/T异质结示意图:(a) 分离通道系统;(b) 双通道系统[5]在商业应用方面,由于砷化镓高昂的制造成本,地面光伏电站极少使用。但目前新的生长技术大大缩短了制作太阳能电池的时间,有望带来工艺成本的大幅下降,使砷化镓电池大规模商用成为可能。参考文献:1.Nguyen D P, Pham T, Pham B L, et al. A high efficiency high power density harmonic-tuned Ka band stacked-FET GaAs power amplifier[C]//2016 IEEE compound semiconductor integrated circuit symposium (CSICS). IEEE, 2016: 1-4. https://doi.org/10.1109/CSICS.2016.77510202. Valdivia C E, Wilkins M M, Bouzazi B, et al. Five-volt vertically-stacked, single-cell GaAs photonic power converter[C]//Physics, Simulation, and Photonic Engineering of Photovoltaic Devices IV. SPIE, 2015, 9358: 48-55. https://doi.org/10.1117/12.20798243. Xiao T P, Chen K, Santhanam P, et al. Electroluminescent refrigeration by ultra-efficient GaAs light-emitting diodes[J]. Journal of Applied Physics, 2018, 123(17): 173104. https://doi.org/10.1063/1.50197644. Ding H, Hong H, Cheng D, et al. Power-and spectral-dependent photon-recycling effects in a double-junction gallium arsenide photodiode[J]. ACS Photonics, 2019, 6(1): 59-65. https://doi.org/10.1021/acsphotonics.8b014045. Hassani S, Taylor R A, Mekhilef S, et al. A cascade nanofluid-based PV/T system with optimized optical and thermal properties[J]. Energy, 2016, 112: 963-975. https://doi.org/10.1016/j.energy.2016.06.142
参数原理
2022.12.02

阿拉丁连续11年荣膺“最受用户欢迎试剂品牌”!
企业动态
2022.12.01

烯基含氟砌块研究
简介 由于氟原子半径小,且具有最强的电负性(4.0),引入氟原子后对C-F键极性方向和整个分子的电子云分布均会发生变化,影响分子的偶极矩、酸碱性等,进而影响整个分子的物理性质和化学性质。氟原子或含氟基团的引入通常还会增加分子的脂溶性和疏水性,提高药物分子的溶解度,促进药物的传导和吸收。所以很多含氟药物或者农药相对具有用量少、毒性低、药效高等特点,这使得含氟药物或农药所占比例越来越高。在各种含氟砌块中,烯基含氟砌块是非常重要的生物活性分子结构,具有各种药理活性(如抗癌、抗菌、抗艾滋病毒、抗糖尿病),还可以作为合成砌块用于制备不同的含氟官能团。图1. 具有生物活性的烯基含氟砌块1.HDAC抑制剂(histone deacetylase inhibitors)——抗癌2.DPP-4抑制剂(Dipeptidyl peptidase 4 inhibitors)——抗2型糖尿病3.HIV融合抑制剂(HIV fusion inhibitor)——抗艾滋病毒药物4.蛋白合成抑制剂 (Protein synthesis inhibitors)——抗菌药物5.视黄醇X受体调节剂(Retinoid X receptor regulator )——抗2型糖尿病6.核糖核苷酸还原酶抑制剂(Ribonucleotide reductase inhibitor)——抗癌7.DNA回旋酶抑制剂(DNA gyrase inhibitor)——抗菌药物合成方法1. 烯化反应2-氟-2-膦酰基乙酸三乙酯(Triethyl 2-fluoro-2-phosphonoacetate)和氟甲基苯基砜(Fluoromethyl phenyl sulfone)是比较经典的单氟砌块。2-氟-2-膦酰基乙酸三乙酯通常由膦酰基乙酸三乙酯通过亲电氟化试剂氟化合成,其可以在碱作用下与醛酮化合物经由膦叶立德中间体制得α-氟-α,β-不饱和羧酸酯 。其中,醛羰基反应活性高于酮羰基反应活性,同时还具有更好的顺反选择性。(方案1)方案1. 2-氟-2-膦酰基乙酸三乙酯作为含氟砌块1994年,Patrick课题组报道甘油醛和2-氟-2-膦酰基乙酸三乙酯在丁基锂作用下E式单一产物[1]。(方案2)方案2. 2-氟-2-膦酰基乙酸三乙酯作为含氟砌块Gernert课题组也在2003年使用2-氟-2-膦酰基乙酸三乙酯与酮羰基化合物反应合成氟化产物,但产物是不具有顺反选择性的[2]。(方案3)方案3. 使用含氟砌块2-氟-2-膦酰基乙酸三乙酯合成氟化烯烃上文提到的氟甲基苯基砜近年来也被作为被经常使用的含氟砌块。在1985年,McCarthy课题组早在1985年研究了其制备方法,反应性质以及反应机理。氟甲基苯基砜首先会与丁基锂反应生成氟甲基苯基砜锂盐中间体,然后直接与芳香醛反应得到相应的α-氟-β-羟基砜化合物。该化合物再通过脱去砜基以形成单氟末端烯烃[3-5]。(方案4)方案4. 氟甲基苯基砜作为含氟砌块2.消除反应消除反应是制备α-取代的α-氟烯烃的常用方法。为了将末端烯烃转化为它们的氟化当量,卤化反应和消除反应被广泛使用[6]。(方案5)。这种消除过程通常被认为是SN2反应的竞争过程,特别是当存在强碱时。它可以发生在一级、二级和三级底物上。除非使用特殊的非亲核胺碱,否则强碱存在下的初级底物通常只会产生取代。用具有强碱和胺碱的二级和三级底物很容易消除。消除是二级的,取决于底物和碱浓度。碱需要在一步过程中攻击β-氢。大多数E2反应提供取代最多的烯烃作为主要异构体,但可能使用的条件会改变区域选择性。形成更多取代的烯烃称为Saytzeff消除。反应的区域选择性受碱的性质影响很大。大的叔丁醇碱太大而不能攻击内部CH2基团,从而攻击CH3基团。甲醇比叔丁醇更小,对CH2基团的攻击更大,但主要产物仍然是1-烯烃。几乎所有的消除反应都产生烯烃混合物,而不是一种清洁的产物。反应由产物的稳定性(和过渡态)决定,因此烯烃的稳定性差异很小。取代度高的烯烃比取代度低的烯烃更稳定,但能量差很小。此外,当E和Z烯烃异构体可能存在时,主要异构体是E异构体,但Z异构体通常存在一定程度。3.交叉偶联反应在金属催化交叉偶联领域,Heck反应是烯烃芳基化的一种非常方便的方法。在大多数情况下,乙烯基的氢原子被有机卤化物的有机残基取代。当使用不对称取代的烯烃时,由于空间位阻决定的高区域选择性,导致在较少取代位点发生烯烃芳基化。反应的关键步骤之一是β-氢化物消除。1991年,Heitz和Knebelkamp试图从芳基碘化物和偏二氟乙烯合成β,β-二氟苯乙烯。在乙酸钯催化作用下,如果β-氢化物消除是必要的反应步骤,β,β-二氟苯乙烯应该是唯一的反应产物。而β,β二氟苯乙烯在这些反应中仅作为副产物形成。第一次发现了在Pd(OAc)2催化条件下使芳基碘化物与1,1-二氟乙烯反应来获得α-氟乙烯衍生物的一步反应[7]。(方案6)4.亲电氟化Pacheco和Gouverneur研究了烯丙基甲基硅烷在Selectfluors反应条件下的反应性,以便在不使用氟化构建块的情况下进行亲电氟脱硅反应制备氟二烯怕[8]。(方案7)其中,Selectfluor氟化试剂(N-氟-N'-(氯甲基)三乙二胺双(四氟硼酸盐)或F-TEDA)是一种用户友好性的、温和的、在空气和水分状况下稳定的、非挥发性的亲电氟化反应剂。Selectfluor氟化试剂能够在一个步骤内将氟引入有机底物,具有非常广泛的反应范围[9]。这些反应中,绝大多数都表现出了优异的区域选择性。烯丙基氟化物可以通过交叉复分解反应/亲电氟脱硅路线制备(方案8)。这种途径避免了使用DAST进行亲核置换或开环反应时烯丙基转位产生的副产物的形成[10]。应用目前含氟砌块已经在医药、农药、染料、表面活性剂、氟碳材料、航空航天等各种化工领域得到广泛应用。其中,烯基含氟砌块在材料科学和合成有机化学中具有潜在的应用,可以用作氟化合成子进行进一步的功能化。此外,氟化烯基可以作为肽键同工酶用于药物化学,这为寻找新的生物活性化合物开辟了新的机会。参考文献1.Patrick, T. B.; Lanahan, M. V.; Yang, C.; Walker, J. K.; Hutchinson, C. L.; Neal, B. E., J. Org. Chem.1994,59, 1210.2. Gernert, D. L.; Ajamie, R.; Ardecky, R. A.; Bell, M. G.; Leibowitz, M. D.; Mais, D. A.; Mapes, C. M.; Michellys, P. Y.; Rungta, D.; Reifel-Miller, A.; Tyhonas, J. S.; Yumibe, N.; Grese, T. A., Bioorg. Med.Chem. Lett.2003,13, 3191.3. McCarthy, J. R.; Peet, N. P.; LeTourneau, M. E.; Inbasekaran, M., J. Am. Chem. Soc.1985,107, 735.4. McCarthy, J. R.; Peet, N. P.; LeTourneau, M. E.; Inbasekaran, M., J. Am. Chem. Soc.1985,107, 735.5. McCarthy, J. R.; Huber, E. W.; Le, T.-B.; Mark Laskovics, F.; Matthews, D. P., Tetrahedron 1996,52, 45.6. Takeuchi Yoshio,Yamada Asuka,Suzuki Takanori,Koizumi Toru. Synthetic studies towards proline amide isosteres, potentially useful molecules for biological investigations[J]. Tetrahedron,1996,52(1).7. Walter Heitz,Arno Knebelkamp. Synthesis of fluorostyrenes via palladium‐catalyzed reactions of aromatic halides with fluoroolefins[J]. Macromolecular Rapid Communications,1991,12(2).8. Furuya Takeru,Ritter Tobias. Fluorination of boronic acids mediated by silver(I) triflate.[J]. Organic letters,2009,11(13).9. Singh, R. P. , Shreeve, J. M.. 2004. For a review of recent highlights: Acc. Chem. Res..37, 31.10. Thibaudeau S, Gouverneur V. 2003. Sequential Cross-Metathesis/Electrophilic Fluorodesilylation:? A Novel Entry to Functionalized Allylic Fluorides. Org. Lett.. 5(25):4891-4893.
参数原理
2022.11.29
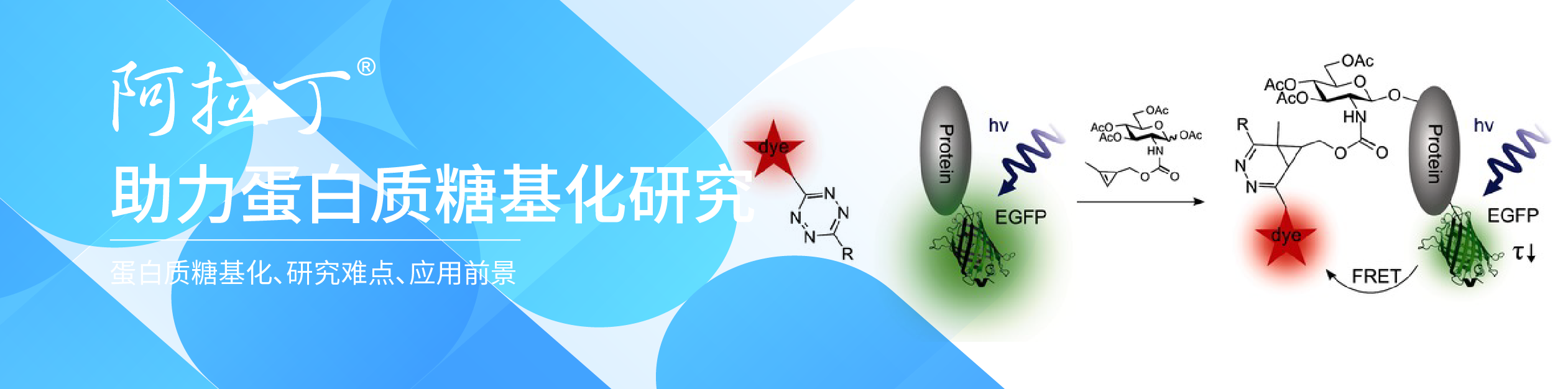
用糖苷酶编辑治疗性蛋白质上的糖链
什么是蛋白质糖基化真核生物蛋白质最常见的翻译后修饰之一是糖基化。蛋白质的糖基化可以影响许多生物活性。对于治疗性糖蛋白,它可以改变生物活性、靶向性、转运、血清半衰期、清除性和受体的识别[1,2]。出于这样的原因,生物制造厂商必须监测和表征他们的重组治疗蛋白的糖基化模式>[3,4]。治疗蛋白有两种主要的糖基化类型:N-连接的糖链和O-连接的糖链[5]。N-糖链的附着始于内质网(ER),在内质网中,核心的新生多糖通过蛋白质的特定天冬酰胺(产品号:F141104)残基上的侧链酰胺氮连接在具有NXS/T序列的位置,其中X可以是除脯氨酸(产品号:P111001)以外的任何氨基酸残基。当糖蛋白通过内质网和高尔基体时,N-糖链被修剪和进一步修饰。宿主细胞和细胞培养条件可以改变糖蛋白上存在的N-糖基化类型(从高甘露糖到复杂和杂交的N-糖链)。O-糖基化发生在高尔基体中。N-葡聚糖有一个共同的核心,由两个N-乙酰氨基葡萄糖(产品号:N355747)(GlcNAc)残基和三个甘露糖(Man)残基组成。但O-葡聚糖唯一的共同核心是N-乙酰半乳糖胺(产品号:N389642)(GalNAc)残基,它通过蛋白质的丝氨酸(产品号:S329534)或苏氨酸残基的侧链上的氧原子连接。糖苷酶:糖基化是复杂的和异质性的,所以必须使用多种分析方法来确定糖链的结构和它们在糖蛋白上的位置。糖苷酶是重要的工具,通常与其他分析方法一起使用,以去除、修剪或修饰多糖。糖苷酶(糖苷水解酶)(产品号:G128643、N159659、A109181)是一种能分解复合糖的糖苷键的酶。这些酶应用于治疗性糖蛋白的三个主要分析领域:去除用于分析的葡聚糖、修剪用于测序的葡聚糖和在糖工程中修饰葡聚糖。在这里,我们描述了在每个领域中使用的不同的酶,以及它们的应用的具体例子。提取用于分析的多聚糖从糖蛋白中去除N-糖链最常用的酶是多肽-N-糖苷酶F(PNGase F)(产品号:P420186)。它是一种酰胺酶,在N-糖链最里面的GlcNAc残基和天冬酰胺残基之间裂解,释放N-糖链,从而在蛋白质上产生天冬氨酸(产品号:A329587)残基而不是天冬酰胺残基。糖苷酶如此频繁使用的一个主要原因是它们具有非常广泛的特异性,可以裂解高甘露糖、复杂的和混合的N-糖链。这种酶的唯一限制是,如果有α1-3岩藻糖连接到核心GlcNAc残基(蛋白质旁边的糖残基),就不能切割。这种修饰只发生在植物、昆虫、软体动物和寄生蠕虫中。含有这种修饰的N-葡聚糖可以被PNGase A酶去除。一旦N-葡聚糖被酰胺酶释放,就可以对其进行流动标记,以供液相色谱-荧光检测(LC-FLD)或质谱(LC-MS)或毛细管电泳联用激光诱导荧光检测(CE-LIF)分析。图1说明了PNGase F从Erbitux治疗性抗体(西妥昔单抗)中释放的N-糖链的种类,并通过LC-MS分析进行了检测。除了PNGase F,许多不同的内切-β-N-乙酰氨基葡萄糖苷酶也从糖蛋白中释放N-糖链。大多数内切糖苷酶仅限于它们能识别和切割的N-糖链类型。表1显示了大多数商业上可获得的内切-β-N乙酰氨基葡萄糖苷酶的不同特性。所有这些内切糖苷酶都能水解N,N’二乙酰壳寡糖部分(位于N-糖核上的两个GlcNAc残基之间),使GlcNAc残基附着在蛋白质上。因此,它们经常被用来确定N-聚糖在特定部位的占有率或存在情况。这对具有多个N-糖基化位点的糖蛋白特别有用。通常情况下,糖蛋白先用内切糖苷酶消化,然后用蛋白酶(产品号:P298993)裂解。然后用MS分析这些多肽片段。任何肽片段上单个GlcNAc残基的额外质量证实该位置被N-糖链占据[6]。由于内切-β-N-乙酰氨基葡萄糖苷酶的水解点远离蛋白质骨架,因此在非变性条件下,这些酶通常能裂解N-糖链。即使这些位点在相似的自然条件下不能切割PNGase F,也会发生这种情况。对于Endo S尤其如此,它专用于对免疫球蛋白Fc区N-糖链的水解,并且偏爱非变性条件。当目标是保存蛋白质的结构时,这种内切糖苷酶在自然条件下水解糖的能力特别强。目前,还没有已知的广谱特异性内切糖苷酶能将所有O-糖链从糖蛋白中分离出来。因此,O-糖链的分析比N-糖链的分析要困难得多。有两种O-糖苷酶可以在市场上买到。一种来自肺炎链球菌,可以释放核心1二糖,由连接β1-3的半乳糖(Gal)和N-乙酰半乳糖胺(GalNAc)组成,其中GalNAc残基与蛋白质的丝氨酸或苏氨酸(产品号:T108221、T100459)残基相连[7]。另一种O-糖苷酶来自粪肠球菌,具有稍宽的专一性,将核心1二糖和核心3二糖都裂解,其中核心3二糖由β1-3连接到GalNAc[8]。如果双糖被唾液酸或其他糖进一步修饰,两种酶都不能裂解。使用β消除法可以化学释放完整的O-葡聚糖,但必须注意这种方法不能降解释放的葡聚糖(产品号:D140049等)。这种方法的另一个主要缺点是蛋白质被破坏,不能再分析其结构或活性。与O-葡聚糖的完整化学释放相比,可以使用外切糖苷酶和O-糖苷酶的组合来修剪O-葡聚糖(图2)。虽然这种方法可以保留蛋白质的结构和活性,但它会降解多糖,因此无法对其进行表征。此外,一些O-葡聚糖含有糖类修饰(例如,硫化或乙酰化),使修饰后的糖不被外切糖苷酶切割。修剪糖链以进行测序胞外糖苷酶是一种重要的酶,它可以一次一个糖残留物地将多聚糖从多聚糖的非还原末端去除。商业上有大量不同的外切糖苷酶。胞外糖苷酶对糖的类型及其异构体(α或β)都是特异的。一些外切糖苷酶比较普遍,可以分解许多不同的连接。例如,广特异性神经氨酸酶(产品号:N128387)可以去除连接到糖链上的α2-3、α2-6、α2-8或α2-9上的唾液酸残基。其他外切糖苷酶对特定的连锁更具专一性。例如,β1-4半乳糖苷酶只能去除β1-4连接的半乳糖。由于这些酶的先天专一性,外切糖苷酶对修剪糖链和确定糖链的序列都很有用。基本的LC-MS或基质辅助激光解吸/电离(MALDI)分析只能确定多糖的大小。然而,为了确定多糖上存在的单糖的连接或类型,有必要进行串联质谱仪(MS-MS),例如碰撞诱导解离(CID)或使用外切糖苷酶,然后进行CE或MS分析。当使用外切糖苷酶时,重要的是要考虑酶制剂的质量。从天然来源分离的酶制剂往往会被其他糖苷酶活性污染,这使得多糖结构的阐明变得更加困难。最常用的外切糖苷酶的重组测序级版本现在可以在商业上用于敏感的葡聚糖分析工作流程。图3显示了糖苷酶如何对从Enbrel治疗蛋白(依那西普)释放的多糖进行测序。胞外糖苷酶也可以用来对仍然附着在糖蛋白上的糖链进行测序。这些酶对于分析和检测糖链上潜在的抗原结构特别有用。例如,使用通用的α-半乳糖苷酶,如来自绿咖啡豆的酶来处理治疗性蛋白质,可以帮助识别低水平的半乳糖α1-3Gal(α半乳糖表位),这些半乳糖苷酶在人类中不存在,并且可以产生免疫原性[9]。葡聚糖的改性除了去除完整的糖链和进行糖链测序外,糖苷酶还可用于修饰糖蛋白上的糖链[10](称为糖工程)。一种方法是使用内切糖苷酶去除不需要的N-连接的多糖。然后,通过内切糖苷酶裂解留下的GlcNAc残基,可以将通过化学酶合成产生的所需的均匀的葡聚糖连接到糖蛋白上。可以使用内切糖苷酶(自然地转糖基化),如Endo-M[11],或使用内切糖苷酶突变体来实现所需的葡聚糖的连接,该酶突变体可以驱动反应朝向添加糖而不是去除糖的方向发展[12](图4)。糖工程的另一种方法是使用外切糖苷酶将糖蛋白上的糖链修剪成统一的大小。然后,可以使用特定的糖基转移酶(产品号:G293642)来重建这种多糖,这种转移酶将单糖从核苷酸糖供体转移到糖链上。这会产生一种糖蛋白,具有更均一的糖链结构(图5)。未来的应用到目前为止,这些体外糖工程方法大多是小规模使用的。用于治疗溶酶体储存疾病的糖蛋白酶是个例外。这些酶中的几种已经被糖工程利用外糖苷酶暴露蛋白质上的甘露糖结构,以通过受体介导的内吞作用改善酶向溶酶体的运输[13]。此外,宿主细胞中糖基化途径的修饰正被用于生产具有所需糖基化的治疗性蛋白质。这些细胞系的治疗性蛋白质生产正在顺利进行,有几种治疗性蛋白质正在进行临床试验。科学家们预计,随着对高通量方法的需求增加,葡聚糖分析的速度和简单性将随着酶和分析的进一步改进而进步。新的抗体特异性蛋白水解酶,如IDES和IDEZ,是对典型的抗体胰酶/溶菌酶C或木瓜酶(产品号:P128675、P128674)消化抗体的改进,因为它们特异性地切割在抗体的铰链区域,而不是次级位点切割。使用这些酶和改进的软件,可以对抗体的恒定区进行MS分析。这使得能够确定FC上存在的多糖的类型,而不需要移除、纯化和标记N-聚糖。其他方面的改进可能会导致发现新的酶特异性。例如,一种对复杂O-葡聚糖具有广泛专一性的酶可以更好地分析这些类型的蛋白质修饰。随着更多的治疗性蛋白在转基因植物和昆虫细胞系中表达,可以去除所有N-糖链的PNGase将非常有用。最后,随着葡聚糖分析方法变得更精细,更高的吞吐量和更高的灵敏度,在开发过程中应该可以选择克隆,或者使用选择的糖工程表达宿主来产生具有所需糖基化模式的抗体。这样的速度和灵敏度还将允许对发酵过程进行持续监测,这将提高治疗性糖蛋白的生产。参考文献:1. Ohtsubo K,Marth JD.Glycosylation in Cellular Mechanisms of Health and Disease.Cell 126(5)2006:855–867;doi:10.1016/j.cell.2006.08.0192. Walsh G.Posttranslational Modifications of Protein Biopharmaceuticals.Drug Discovery Today 15(17–18),2010:773–780;doi:10.1016/j.drudis.2010.06.009.3. Beck A,et al.Trends in Glycosylation,Glycoanalysis,and Glycoengineering of Therapeutic Antibodies and Fc-Fusion Proteins.Curr.Pharm.Biotechnol.9(6),2008:482–501.4. Sethuraman N,Stadheim TA.Challenges in Therapeutic Glycoprotein Production.Curr.Op.Biotech.17(4)2006:341–346;doi:10.1016/j.copbio.2006.06.010.5. Spiro RG.Protein Glycosylation:Nature,Distribution,Enzymatic Formation,and Disease Implications of Glycopeptide Bonds.Glycobiology 12(4)2002:43R–56R.6. Wang L,et al.Structural Analysis of a Highly Glycosylated and Unliganded gp120-Based Antigen Using Mass Spectrometry.Biochem.49,2010:9032–9045;doi:10.1021/bi1011332.7. Fujita K,et al.Identification and Molecular Cloning of a Novel Glycoside Hydrolase Family of Core 1 Type O-Glycan-Specific Endo-Alpha-N-Acetylgalacto-saminidase from Bifidobacterium longum.J.Biol.Chem.280(45)2005:37415–37422;doi:10.1074/jbc.M506874200.8. Koutsioulis D,Landry D,Guthrie EP.Novel Endo-N-Acetylgalactosaminidases with Broader Substrate Specificity.Glycobiol.18(10).2008:799–805;doi:10.1093/glycob/cwn069.9. Bosques CJ,et al.Chinese Hamster Ovary Cells Can Produce Galactose--1,3-Galactose Antigens on Proteins.Nat.Biotechnol.28(11)2011:1153–1156;doi:10.1038/nbt1110-1153.10. Rich JR,Withers SG.Emerging Methods for the Production of Homogeneous Human Glycoproteins.Nat.Chem.Biol.5(4)2009:206–215;doi:10.1038/nchembio.148.11. Yamamoto K,et al.Transglycosylation Activity of Mucor hiemalis Endo-Beta-N-Acetyl-Glucosaminidase Which Transfers Complex Oligosaccharides to the N-acetylglucosamine Moieties of Peptides.Biochem.Biophys.Res.Comm.203(1)1994:244–252.12. Huang W,et al.Chemoenzymatic Glycoengineering of Intact IgG Antibodies for Gain of Functions.J.Am.Chem.Soc.134(29)2012:12308–12318;doi:10.1021/ja3051266.13. SoláRJ,Griebenow K.Glycosylation of Therapeutic Proteins.BioDrugs.24(1)2010:9–21;doi:10.2165/11530550-000000000-00000.14. Trimble RB,Tarentino AL.Identification of Distinct Endoglycosidase(Endo)Activities in Flavobacterium meningosepticum:Endo F1,Endo F2,and Endo F3.Endo F1 and Endo H Hydrolyze Only High Mannose and Hybrid Glycans.J.Biol.Chem.266(3)1991:1646–1651.15. Tarentino AL,et al.Multiple Endoglycosidase F Activities Expressed By Flavobacterium meningosepticum Endoglycosidases F2 and F3:Molecular Cloning,Primary Sequence,and Enzyme Expression.J.Biol.Chem.268(13)1993:9702–9708.16. Collin M,Olsén A.Endo S:A Novel Secreted Protein from Streptococcus pyogenes with Endoglycosidase Activity on Human IgG.EMBO J.20(12)2001:3046–3055.17. Sjögren J,et al.EndoS 2 Is a Unique and Conserved Enzyme of Serotype M49 Group A Streptococcus That Hydrolyses N-Linked Glycans on IgG and1-Acid Glycoprotein.Biochem.J.455(1)2013:107–118;doi:10.1093/emboj/20.12.3046.18. Mizuochi T,Amano J,Kobata A.New Evidence to the Substrate Specificity of Endo-Beta-N-Acetlylglucosiamindase D.J.Biochem.95(4)2008:1209–1213.19. Yamamoto K,et al.Novel Specificities of Mucor hiemalis Endo-Beta-N-Acetylglucosaminidase Acting Complex Asparagine-Linked Oligosaccharides.Biosci.Biotechnol.Biochem.58(1)1994:72–77.标签:葡聚糖 糖基化 天冬氨酸 糖苷酶 蛋白酶
参数原理
2022.11.24

核酸电泳工作流程-5大主要步骤
参数原理
2022.11.22
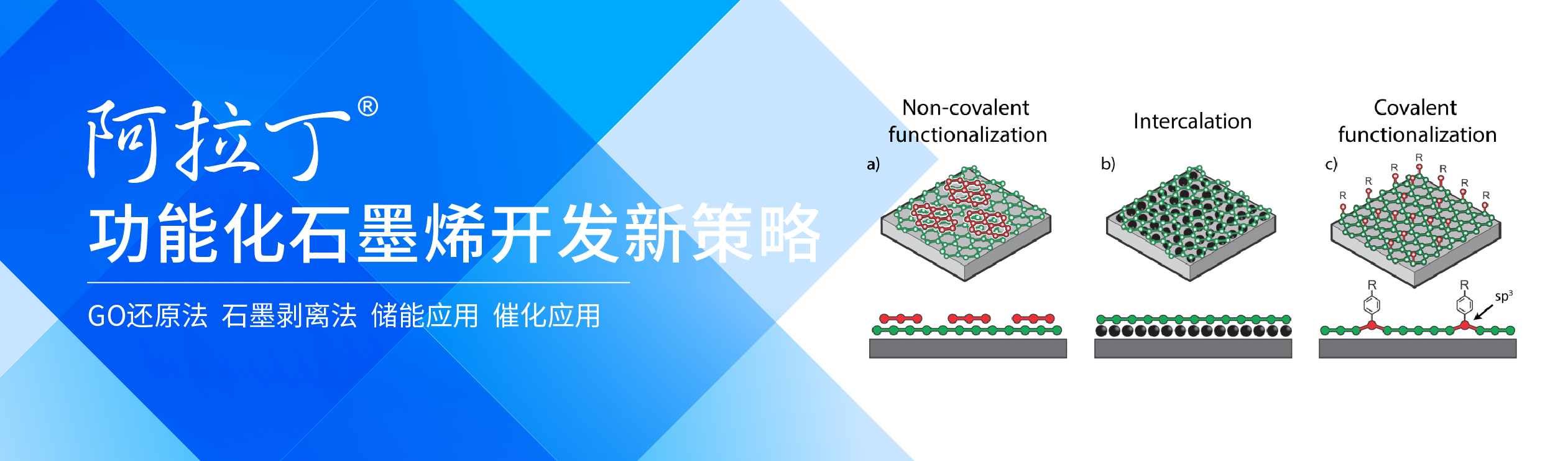
新型石墨烯基纳米结构的制备及功能化设计
应用实例
2022.11.21

阿拉丁高纯科研试剂研发中心奠基仪式圆满举行!
阿拉丁高纯科研试剂研发中心项目正式启动 2022年11月16日,阿拉丁高纯科研试剂研发中心项目奠基典礼在上海化学工业区奉贤分区圆满举行! 本次活动得到了上海市奉贤区政府、杭州湾经济开发区的大力支持。活动开始,奉贤区政府副区长厉蕾发表开场致辞,对阿拉丁高纯科研试剂研发中心奠基表达了祝贺。 奉贤区政府副区长厉蕾 接着,杭州湾开发区党委书记、董事长蒋祎青和阿拉丁公司实际控制人、副总经理招立萍相继发表致辞。 杭州湾开发区党委书记、董事长蒋祎青 奉贤区委副书记、区长袁泉上台宣布阿拉丁高纯科研试剂研发中心项目启动开工。 奉贤区委副书记、区长袁泉 礼炮响起,奉贤区委副书记、区长袁泉与副区长厉蕾,杭州湾开发区党委书记、董事长蒋祎青,杭州湾开发区党委副书记、总经理王雪忠,阿拉丁公司实际控制人、副总经理招立萍,阿拉丁公司董事会秘书赵新安进行培土奠基仪式。 培土奠基仪式 此次奠基仪式的举行,标志着阿拉丁高纯科研试剂研发中心的建设正式启动! 阿拉丁迈上新征程 阿拉丁高纯科研试剂研发中心项目总投资1亿多元,总建筑面积达1.2万平方米。新的研发中心将现代化、信息化等要素完美结合。研发中心将配备研发大楼、甲类仓库、乙类仓库,配套建设供电、给排水、空调等系统。 阿拉丁高纯科研试剂研发中心蓝图 仪式上,阿拉丁公司副总经理招立萍回顾了阿拉丁的发展历程,并表示:“阿拉丁高纯科研试研发中心项目的完成,有利于提高公司技术成果转化能力和开发效率,增强新产品的整体竞争力,积累更多科研成果,并实现技术升级,同时将进一步提升公司仓储的容纳能力。” 对于阿拉丁来说,新研发中心并不是简单的规模扩张,它将成为阿拉丁发展过程中新的里程碑,对科研试剂行业的技术发展也将起到积极的促进作用! 阿拉丁公司副总经理招立萍 阿拉丁科研试剂发展新机遇 “十三五”期间是我国科研试剂发展的重要机遇期,生物医药、新材料、新能源、节能环保、航空航天等下游战略新兴产业的快速发展,对高纯试剂等提出了新的需求。高纯试剂不只是纯度的概念,而是综合概括了高性能、高洁净度、高批次一致性、低紫外吸收、低荧光残留、低水分含量等特点,其在色谱分析、光谱分析、质谱分析、农药残留检测、有机合成和组合化学、DNA 与 RNA 合成等领域得到了越来越广泛的应用。 以进口替代为己任让科研创新更便捷 创新是公司进步和保持核心竞争力的源泉,技术创新能力的大小直接决定公司成长和发展速度。阿拉丁在纯化、合成技术领域已获得12 项国家发明专利授权,同时还有17 项发明专利申请已被受理。阿拉丁依靠掌握的制备方法,实现了科研试剂常备库存产品超过 53.5 万多种,是国内品种最齐全的企业之一,部分试剂产品技术指标达到或超过国际先进企业同类产品。 未来,阿拉丁将以差异化、精细化、系列化为目标,持续加大研发投入及技术升级,向专业化发展,为适应用户的多样化需求而努力,矢志成为国际一流的科研试剂生产商!
企业动态
2022.11.17

酶与膳食抗氧化剂
什么是抗氧化剂?抗氧化剂可以保护生物系统免受含氧自由基和氧化还原过渡金属离子(如铁、铜和镉)产生的氧化损害[1]。线粒体中的葡萄糖的氧化代谢过程中,超氧阴离子(O2-)作为辅酶Q复合体III还原的副产品产生。超氧化物歧化酶(产品号:S128537)将超氧阴离子转化为过氧化氢(H2O2),而过氧化氢又可以转化为过氧自由基(RO2-)、羟自由基(OH-)或次氯酸(ClO-)离子。超氧阴离子还可以与一氧化氮(NO)反应,形成高活性的过氧亚硝基阴离子(ONOO-)[2]。在正常情况下,这些细胞氧化剂被细胞内的抗氧化剂和抗氧化酶还原或清除,其中最重要的是谷胱甘肽(产品号:L274260)、硫氧还蛋白、超氧化物歧化酶、过氧化氢酶(产品号:C163049)和过氧化物酶(产品号:P105528)。饮食中的抗氧化剂,如抗坏血酸(维生素C)、维生素E、β-胡萝卜素和其他类胡萝卜素,以及硒,已被认为是细胞和血浆总抗氧化能力的重要成分。类胡萝卜素、叶黄素和玉米黄素是眼睛和视网膜的重要抗氧化剂[3]。维生素E是生育酚和生育三烯酚的混合物,其中α-生育酚(产品号:T105540)是主要的抗氧化剂[4],是细胞中主要的脂溶性抗氧化剂,在保护膜脂质过氧化方面起着重要作用。低密度脂蛋白(LDL)携带维生素E进入细胞,随后通过向脂肪过氧自由基提供氢气来防止LDL的过氧化反应[5]。多酚类化合物,特别是黄酮类化合物,最近已被证明是培养细胞中强有力的抗氧化剂。对黄酮类化合物的人体研究也表明,其效果可部分归因于其抗氧化作用[6]。抗氧化剂可以直接作为还原剂,将质子氢提供给未配对的氧电子,或通过稳定或转移自由基电子而发挥作用[7]。在这个过程中,还原剂被氧化;例如,两个谷胱甘肽分子的半胱氨酸巯基被氧化,形成氧化谷胱甘肽的分子间胱氨酸(图1)。图1.氧化的谷胱甘肽的结构膳食抗氧化剂的研究硫辛酸是一种内源性抗氧化剂,最近作为膳食补充剂受到关注,因为它不仅可以清除自由基,而且它的二氢脂酸盐形式也是非常有效的还原剂。硫辛酸能减少其他抗氧化剂的氧化形式,并最终能保持组织中还原谷胱甘肽的浓度[8]。一些抗氧化剂捕获或清除自由基,并在这个过程中成为自由基本身。当类胡萝卜素虾青素(图2)、叶黄素和玉米黄素清除氧自由基时,未配对电子的电荷在分子的整个多烯链上变得分散[9]。黄酮醇槲皮素被氧化为醌,可与硫醇反应[10]。 黄酮类化合物也是很好的金属离子螯合剂,可以防止铜催化的LDL过氧化[1,4,11]。在螯合铜和防止低密度脂蛋白过氧化方面,白藜芦醇(产品号:R107315)比黄酮类化合物更有效力[12]。这可能有助于白藜芦醇和黄酮类化合物的抗动脉硬化作用。图2.虾青素的结构最新研究进展Moskaug等人推测,黄酮类化合物通过诱导细胞中的慢性低水平氧化应激来增强细胞的抗氧化系统,从而提高细胞的抗氧化防御系统(激素原理)[15]。 相反,Halliwell等人认为,未被吸收的微摩尔浓度的黄酮类化合物和其他酚类化合物仍然留在小肠和结肠中,它们的抗氧化、金属螯合和其他作用对结直肠癌的发生有保护作用。最近的研究证实,多酚类物质除了清除自由基和螯合金属离子外,还具有许多细胞内效应。活性氧(ROS)和活性氮(RNS)也是潜在的信号分子。它们通过一类对氧化还原敏感的转录因子调节基因表达的研究,包括诱导抗氧化/解毒酶表达的Nrf2,以及诱导产生炎症细胞因子、细胞粘附分子、急性期蛋白和抗凋亡的NFkB和AP-1[1,2,16]。这些转录因子在应对氧化应激时被激活;持续升高的活性氧(ROS)水平通过诱导其抑制性亚基IkB的磷酸化和分离而激活NFkB。据报道,水飞蓟素(产品号:S408123)和水飞蓟宾(产品号:S1098009)[17]、儿茶素和原花青素(产品号:P413229),[1,11]以及其他黄酮类化合物[16]可以阻止NFkB的激活。Forman等人引用证据表明,过氧化氢和超氧阴离子通过介导氧化还原信号反应表现出第二信使的特性。他们假设过氧化物与存在于信号蛋白活性部位的关键半胱氨酸硫酸盐可逆地相互作用[18]。已知蛋白酪氨酸磷酸酶和硫氧还蛋白具有硫酸盐形式的活性部位半胱氨酸,而转录因子AP-1和NFkB以及一些半胱天冬酶具有可能是硫酸盐形式的氧化还原敏感半胱氨酸。一些蛋白激酶c(产品号:P343699)异构体调节部位的锌结合的半胱氨酸也可能被过氧化氢氧化。受体刺激产生的过氧化物也导致所有有丝分裂原激活蛋白激酶途径(ERK、JNK和p38 MAPK)的激活。其他信号蛋白和酶是一氧化氮、过氧化物或两种氧化剂的目标(表1)。因此,清除活性氧中间物的抗氧化剂多酚可能对介导细胞对氧化应激反应的细胞内信号转导途径有深远的影响。信号蛋白调节剂氧化剂PTP1BNO, H₂O₂SHP-2H₂O₂LMW-PTPNO, H₂O₂PTENH₂O₂TrxNO, H₂O₂SrcH₂O₂RasNO, H₂O₂GSTp/JNKH₂O₂Gi/GoH₂O₂NMDANO表1.氧化还原信号的其他靶点参考文献1. Frei B, Higdon JV. 2003. Antioxidant Activity of Tea Polyphenols In Vivo: Evidence from Animal Studies. The Journal of Nutrition.133(10):3275S-3284S. https://doi.org/10.1093/jn/133.10.3275s2.Chew BP, Park JS. 2004. Carotenoid Action on the Immune Response. The Journal of Nutrition.134(1):257S-261S. https://doi.org/10.1093/jn/134.1.257s3.Ribaya-Mercado JD, Blumberg JB. 2004. Lutein and Zeaxanthin and Their Potential Roles in Disease Prevention. Journal of the American College of Nutrition. 23(sup6):567S-587S. https://doi.org/10.1080/07315724.2004.107194274.Halliwell B, Rafter J, Jenner A. 2005. Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: direct or indirect effects? Antioxidant or not?. The American Journal of Clinical Nutrition.81(1):268S-276S. https://doi.org/10.1093/ajcn/81.1.268s5.Chattopadhyay A, Bandyopadhyay D. 2006. . Vitamin E in the prevention of ischemic heart. . Pharmacological reports . 58(179):179-187. . https://pubmed.ncbi.nlm.nih.gov/16702619/6.Williamson G, Manach C. 2005. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. The American Journal of Clinical Nutrition.81(1):243S-255S. https://doi.org/10.1093/ajcn/81.1.243s7.Grajek W, Olejnik A, Sip A. Probiotics, prebiotics and antioxidants as functional foods.. Acta Biochim Pol. 52(3):665-671. https://doi.org/10.18388/abp.2005_34288.Bilska A, Wlodek L. 2005. . Lipoic acid-the drug of the future. . Pharmacol Rep . 57(5):570-577.. https://pubmed.ncbi.nlm.nih.gov/16227639/9.Liu RH. 2004. Potential Synergy of Phytochemicals in Cancer Prevention: Mechanism of Action. The Journal of Nutrition.134(12):3479S-3485S. https://doi.org/10.1093/jn/134.12.3479s10.Moskaug JØ, Carlsen H, Myhrstad MC, Blomhoff R. 2005. Polyphenols and glutathione synthesis regulation. The American Journal of Clinical Nutrition.81(1):277S-283S. https://doi.org/10.1093/ajcn/81.1.277s11.Keen Cea. 2005. Cocoa antioxidants and cardiovascular health. Am. J. Clin. Nutr.The American Journal of Clinical Nutrition. 81, 298S-303S .https://doi.org/10.1093/ajcn/81.1.298S12.Pervaiz S. 2003. Resveratrol: from grapevines to mammalian biology.The FASEB journal. 17(14):1975-1985. https://doi.org/10.1096/fj.03-0168rev13.Crespy V, Williamson G. 2004. A Review of the Health Effects of Green Tea Catechins in In Vivo Animal Models. The Journal of Nutrition.134(12):3431S-3440S. https://doi.org/10.1093/jn/134.12.3431s14.Schroeter H, Heiss C, Balzer J, Kleinbongard P, Keen CL, Hollenberg NK, Sies H, Kwik-Uribe C, Schmitz HH, Kelm M. 2006. (-)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proceedings of the National Academy of Sciences. 103(4):1024-1029. https://doi.org/10.1073/pnas.051016810315.Scalbert A, Johnson IT, Saltmarsh M. 2005. Polyphenols: antioxidants and beyond. The American Journal of Clinical Nutrition.81(1):215S-217S. https://doi.org/10.1093/ajcn/81.1.215s16.Surh Y, Kundu JK, Na H, Lee J. 2005. Redox-Sensitive Transcription Factors as Prime Targets for Chemoprevention with Anti-Inflammatory and Antioxidative Phytochemicals.The Journal of Nutrition. 135(12):2993S-3001S. https://doi.org/10.1093/jn/135.12.2993s17.Kren V, Walterova D. 2005. Silybin and silymarin - new effects and applications. BIOMED PAP. 149(1):29-41. https://doi.org/10.5507/bp.2005.00218.Forman HJ, Fukuto JM, Torres M. 2004. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. American Journal of Physiology-Cell Physiology. 287(2):C246-C256. https://doi.org/10.1152/ajpcell.00516.2003
参数原理
2022.11.15
寡核苷酸合成
多年来,寡核苷酸的合成方法发展历程:,从 Michelson和Todd 早期的H-膦酸酯和磷酸三酯法[1],以及Khorana在 1950 年代的磷酸二酯法[2],再到1960年代磷酸三酯法的重新研究和1970年代的亚磷酸三酯法[3]。这些方法中的每一种都存在其弊端。亚磷酰胺法由Marvin Caruthers在1980年代初期开创,并通过固相技术和自动化的应用得到加强,现已成为首选方法。亚磷酰胺法亚磷酰胺寡核苷酸的合成沿3'至5'方向进行(与DNA 复制中生物合成的5'至3'方向相反)。每个合成周期添加一个核苷酸[4]。图1.亚磷酰胺寡核苷酸合成循环DNA合成的亚磷酰胺单体主要由碱基、脱氧核糖、5'端的DMT和3'端的2-氰乙基以及二异丙基胺基组成。此外,由于腺嘌呤、胞嘧啶和鸟嘌呤和一些人工碱基存在伯氨基,因此也需要在单元中用适当的保护基团对其保护。在寡核苷酸合成之前必须从载体结合的核苷中脱去DMT保护基团。步骤1:活化和偶联在进行偶联之前,活化剂四唑提供一个质子给亚磷酰胺单体3'磷酸上二异丙胺基的氮原子,质子化的二异丙胺是一个良好的离去基团,与四唑形成亚磷酰胺四唑这种活性中间体,这个过程被称为活化。脱保护的5'端-OH会与亚磷酰胺四唑活性中间体偶联,形成新的磷氧键,并脱去四唑。在这一步核苷酸链得到延伸,需要注意的是:单体及活化剂中水分超标导致偶联效率低,原料中杂质较多也会导致偶联效率低。图2.亚磷酰胺偶联反应机理核苷亚磷酰胺在惰性气体氛围中相当稳定,可以用于制备与运输,并可以在使用前以干燥固体形式储存数月。只有在质子化作用时,核苷亚磷酰胺才具有反应性。步骤2:加帽在每个偶联步骤中使用大大过量的四唑和亚磷酰胺单体可以将每一步偶联的效率提高到99.5%以上,但即使使用最有效的化学方法和纯度最高的试剂,也不可能实现100%的核苷都参与反应。这意味着核苷酸上会有一些未反应的 5'-羟基;如果不加以控制,这些 5'-羟基将可用于参与下一个偶联步骤,并与引入的亚磷酰胺反应,导致得到的寡核苷酸会缺少一个碱基,并且对应于所需寡核苷酸的缺失突变。如果不检查这种缺失突变,它们会随着每个连续的循环而累积,最终产物将是寡核苷酸的复杂混合物,其中大部分将携带不正确的遗传信息,并且难以纯化。这还会导致后续生化实验的失败。图3.正确寡核苷酸的缺失突变序列(上);和一个包含缺失突变的失败序列(下),对应于第 6 位胸腺嘧啶碱基的缺失。通过在偶联反应后引入“加帽”步骤来避免缺失突变,以阻断未反应的5'端-OH。合成器上使用了两种封端溶液:乙酸酐和N-甲基咪唑 (NMI)。这两种试剂(溶解在四氢呋喃中并添加少量吡啶)在输送到合成柱之前在 DNA 合成仪混合。亲电子混合物快速乙酰化,吡啶确保 pH 值保持碱性,以防止由乙酸酐与 NMI 反应形成的乙酸对核苷亚磷酰胺进行脱保护。 5'端-OH的乙酰化即可使它们对随后的反应呈惰性。图4.亚磷酰胺寡核苷酸合成中加帽步骤的机理步骤3:氧化偶联反应后新加上的核苷酸通过亚磷酯键(磷为三价)与寡核苷酸链相连,此亚磷酯键不稳定,易被酸、碱水解,因此需将此处三价磷氧化为五价的磷。被氧化后形成的磷酸二酯键实际上就是DNA链中的磷酸二酯键外加一个2-氰乙基保护,该保护基可以使磷酸二酯键在后续的合成中更稳定。这一步常用的氧化剂为碘的四氢呋喃溶液,此步反应速度亦很快。图5.亚磷酰胺寡核苷酸合成中氧化步骤的机理在一些 DNA 合成仪上,碘氧化后还有第二个加帽步骤。这样做的目的是干燥磷酸酯,因为氧化混合物中的残留水会持续存在并抑制下一次偶联反应。过量的水与酰化剂反应形成乙酸,该乙酸会在THF/吡啶溶剂混合物中被除去。步骤4:脱保护在磷酰胺偶联、封端和氧化后,必须脱去核苷酸5'端的DMT保护基,以便伯羟基可以与下一个核苷酸亚磷酰胺进行偶联反应。在二氯甲烷中用三氯乙酸脱保护是快速且定量的。注意太浓缩的TCA溶液或太长的脱三苯甲基化时间会导致脱嘌呤,因而会降低最终寡核苷酸的总产率。裂解产生的DMT碳阳离子显示橙色,它在495nm处有可见光吸收,可通过检测其吸光度监测偶联效率。这一步结束后,延伸后的寡核苷酸链进入下一循环,然后不断重复循环直至链延伸至所需长度。参考文献:1. A. M. Michelson,Alexander R. Todd. Nucleotides part XXXII. Synthesis of a dithymidine dinucleotide containing a 3′: 5′-internucleotidic linkage[J]. Journal of the Chemical Society (Resumed),1955,0(0). https://doi.org/10.1039/JR95500026322. P. T. Gilham,H. G. Khorana. Studies on Polynucleotides. I. A New and General Method for the Chemical Synthesis of the C5’-C3’ Internucleotidic Linkage. Syntheses of Deoxyribo-dinucleotides1[J]. J. Am. Che m. Soc.,2002,80(23). https://doi.org/10.1021/ja01556a0163. Reese Colin B..The chemical synthesis of oligo-and poly-nucleotides by the phosphotriester approach[J]. Tetrahedron,1978,34(21). https://doi.org/10.1016/0040-4020(78)87013-64. 4.Beaucage S.L.,Caruthers M.H.. Deoxynucleoside phosphoramidites--A new class of key intermediates for deoxypolynucleotide synthesis[J]. Tetrahedron Letters,1981,22(20). https://doi.org/ 10.1016/S0040-4039(01)90461-7
参数原理
2022.11.11

阿拉丁荣获上海市企业技术中心认定
企业动态
2022.11.08

阿拉丁诚邀您的莅临 | 第十七届有机合成化学研讨会
企业动态
2022.11.08

PEG-N3与炔基的生物正交和点击化学
生物正交化学简介生物正交化学是指在不影响生物分子或干扰生化过程的情况下,在生物环境中发生的一系列反应。为达到此目的,反应必须满足快速、高效和特异性的要求。• pH值:反应必须在生理环境的温度和pH值下发生;• 精准:反应必须具有选择性并且高效得到目标产品,且须不受水或内源性亲核试剂、亲电试剂、还原剂或复杂生物环境中氧化剂的影响;• 快速:即便在低浓度下,反应也必须是快速的,并且须生成稳定的反应产物;• 特异性:反应应涉及生物系统中非天然存在的官能团。生物正交反应的优点和缺点生物正交反应优点缺点Staudinger连接反应叠氮化物和膦化物具有生物相容性,生成的产物是以稳定的酰胺键连接的。反应速度慢,磷化氢容易被氧化。CuAAC(叠氮+炔基)反应迅速,在20 μM Cu(I)的催化下k值可达~10-100(M-1S-1);具有良好的区域选择性。尽管有些配体如THPTA比较稳定,但是铜催化剂的铜毒性仍需考虑在内。SPAAC(叠氮+DBCO)无需铜催化,k值约~1-60 (M-1S-1)。1. 反应慢于CuAAC,大块的环状砌块难以融入生物分子;2. 所需溶剂:10%-40%的乙醇溶液或DMSO(至多60%)/PBS缓冲液;3. pH 4. 巯基、叠氮化钠都会与DBCO发生反应。IEDDA (TCO + Tz)反应非常迅速,k值约~1-106 (M-1S-1)TCO在含水环境中稳定性差。• CuAAC:铜催化的叠氮-炔基环加• SPAAC:叠氮-烷烃环化加成反应 • IEDDA:逆电子需求的D-A反应。表1:各类生物正交反应的优缺点小结表叠氮-炔基点击化学简介经典的点击化学反应会用到铜作为催化剂,一般是一价铜催化1,3-偶极环加成炔和叠氮化物形成1,2,3-三唑[1,2]。一价铜的来源包括碘化铜、溴化铜、铜屑或硫酸铜与还原剂发生反应生成的产物[3]。然而,一价铜具有热力学不稳定性,容易被氧化成无活性的二价铜,通常需要铜催化剂与合适的螯合配体共同制备。优化后的点击化学反应是使用了用抗坏血酸钠和一价铜稳定的配体三(苄基三唑基甲基)胺(TBTA)还原硫酸铜来原位制备一价铜Cu(I)。通过这个方法,可以避免溶解氧氧化催化一价铜,因此能够使得点击化学反应更为有效。在典型的点击化学中,硫酸铜是与TBTA预络合的,现将这种复合催化剂与炔和叠氮化物混合,随后再加入抗坏血酸钠来引发点击反应。TBTA覆盖了在点击化学领域的大量实际应用,除了含水环境下的共轭反应。而水溶性的THPTA点击配体则可以适用于水相反应,进一步简化点击化学步骤,为点击化学反应提供具生物相容性的THPTA配体来结合Cu(I),阻断Cu(I)的生物利用度,改善其潜在的毒性作用,保持催化效率[4]。同时THPTA配体可以有效地被用于高效标记活细胞,并维持细胞活性[5]。CuSO复合物在至少一个月后仍能保持原有的活性。实操案例寡核苷酸类和DNA的标记1. 配制以下原液:• 200 mM THPTA配体的水溶液• 100 mM硫酸铜水溶液• 炔基标记的水相寡聚物• 100 mM抗坏血酸钠水溶液• 10 mM在DMSO/叔丁醇或水中的含叠氮化合物2. 在反应前将硫酸铜和THPTA配体以1:2的当量比混合静置几分钟,这样的溶液在冷冻后可以稳定储存数周;3. 向寡聚糖/DNA溶液中添加过量4~50倍当量的叠氮化物(如PEG-N3等);4. 添加25当量的THPTA/CuSO4;5. 添加40当量的抗坏血酸钠;6. 在室温下静置反应30~60分钟;7. 在乙醇中沉淀纯化寡聚物。细胞裂解物的标记1. 配制以下原液:• 100 mM THPTA配体的水溶液或缓冲溶液• 20 mM硫酸铜水溶液• 300 mM抗坏血酸钠水溶液• 2.5 mM在水或DMSO中的炔基或叠氮标记试剂2. 对每个叠氮化物或炔基修饰的蛋白质裂解物样品,添加以下物质至1.5 mL的离心管中,然后简单涡旋混合:• 50 µL存放于蛋白质萃取缓冲液中的蛋白质裂解产物(1-5 mg/mL)• 90 µL PBS缓冲液• 20 µL在水中或DMSO中2.5 mM相应叠氮化物(或含炔基化合物)的检测试剂3. 加入10 µL的100 mM THPTA溶液并简单涡旋混合;4. 加入10 µL的20 mM硫酸铜溶液并简单涡旋混合;5. 加入10 µL的300 mM 抗坏血酸钠溶液来引发点击化学反应并简单涡旋混合; 6. 保护反应避光,并在室温下反应30分钟;7. 现在在裂解物中的蛋白质已通过点击化学反应被标记了,可以进行下一步的处理和分析了。铜催化的炔基-叠氮化物点击化学标记活细胞图1:(上图)细胞标记的步骤(下图)炔基探针试剂和催化剂添加剂叠氮聚糖在HeLa和CHO细胞表面的标记及共聚焦显微镜成像将细胞以1 × 105个/mL的浓度置于35 mm玻璃底培养皿中培育,并在含有或不含50 μM Ac4ManNAz的生长培养基(含10%胎牛血清、1%谷氨酰胺和1%青链霉素溶液的MEM培养基)中在37 ℃和5% CO2的条件下过夜反应。轻轻地抽吸培养基将多余的溶液移除,用1 mL DPBS洗涤细胞2次。在微量离心管中,4 ℃下以1:5的摩尔当量比将硫酸铜和THPTA添加到含有染料-1炔基或2炔基(浓度:25 μM)的DPBS和氨基胍(浓度:1 mM)中。配制新鲜制备的抗坏血酸钠原液(100 mM)便于后续配制最终浓度为2.5 mM的抗坏血酸钠溶液。将该反应混合物在4 ℃条件下冰浴保温10分钟,然后添加到细胞中。将此混合物再次在4 ℃保温5分钟之后,洗涤细胞,室温下用3%多聚甲醛、0.3%戊二醛和1 mM的氯化镁混合溶液在DBPS中固定10分钟。加入4', 6-二脒基-2-苯基吲哚(DAPI)对细胞核进行染色,并在每一步之间,用DPBS冲洗玻片3次,玻片采用Vecta Shield介质来安装;使用Bio-Rad 2100共聚焦显微镜和60×的油镜来进行切片成像,用ImageJ(http://rsbweb.nih.gov/ij/)来分析和处理得到的数据及图片。对于双标记的实验,在标记反应后,用1 mL的生长培养基洗涤细胞两次,然后重新置于含有50 μM的Ac4ManNAz的培养基放置继续培育20 小时。细胞表面标记的最佳条件为在4 ℃的培养基中,用25 μM的炔基化合物溶液、硫酸铜溶液(50 μM)、THPTA (250 μM)、氨基胍(1 mM)和抗坏血酸钠(2.5 mM)标记1-5分钟。参考文献1. Polytriazoles as Copper(I)-Stabilizing Ligands in Catalysis; T. R. Chan, R. Hilgraf, K. B. Sharpless, V. V. Fokin, Org. Lett. 2004, 6, 2858. https://pubs.acs.org/doi/10.1021/ol04930942. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation; V. Hong, S. I. Presolski, C. Ma, M. G. Finn, Angew. Chem. Int. Ed. 2009, 48, 9879. https://onlinelibrary.wiley.com/doi/10.1002/ange.2009050873. Stabilization of Virus-like Particles with Poly(2-oxazoline)s; F. Manzenrieder, R. Luxenhofer, M. Retzlaff, R. Jordan, M. G. Finn, Angew. Chem. Int. Ed. 2011, 50, 2601. https://onlinelibrary.wiley.com/doi/10.1002/ange.2010061344. Synthesis of Cyclic Peptide Disulfide–PHPMA Conjugates via Sequential Active Ester Aminolysis and CuAAC Coupling; K. A. Günay, H. Klok, Polym. Chem. 2016, 7, 970. https://pubs.rsc.org/en/content/articlelanding/2016/py/c5py01817j5. Cellular Consequences of Copper Complexes used to Catalyze Bioorthogonal Click Reactions; D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester, J. P. Pezacki, J. Am. Chem. Soc. 2011, 133, 17993. https://pubs.acs.org/doi/10.1021/ja2083027
应用实例
2022.11.04

阿拉丁张江生物研发中心掠影!
企业动态
2022.11.03

免疫印迹膜剥离和重新检测
免疫印迹是研究蛋白质功能和定位的常用技术。通常,蛋白质样品可在SDS-PAGE上分离并转印至硝酸纤维素膜(NC膜)或PVDF膜上,然后用特异性抗体进行检测。与可重复利用Southern和Northern印迹的核酸技术不同,免疫印迹很难重复使用。免疫印迹的剥离和重新检测技术具有以下几个优点:1.有利于昂贵或量少或珍稀样本的多次杂交;2.有利于用不同抗体分析单次印迹上的不同蛋白,比如不同亚型的抗体;3.有利于对异常结果进行重分析或确认;4.有利于纠正孵错的抗体;5节约试剂成本,并节约时间。目前已经公布了几种用于从免疫印迹上剥离抗体的方案,包括低pH、加热和去垢剂以及离液剂等方法。以下是三种推荐方案。第一种方案适用于任何化学发光底物系统,即通过加热和去垢剂的组合来解离抗体。第二种常用于抗体必须与抗原分离的应用,即采用低pH值来改变抗体的结构,使结合位点失活。第三种方案利用具有特殊配方的Western Blot Stripping Buffer(蛋白质印迹剥离缓冲液)从免疫印迹膜上剥离抗体。该方法的优势在于:避免吸入与β-巯基乙醇有关的刺激性气味。可在室温条件下处理。可在15分钟内剥离抗体。这些方法都不能去除显色检测系统所产生的有色沉淀(如BCIP、4CN、DAB和TMB)。但是,仍可以利用靶向不同目标蛋白的另一种抗体来分析印迹。通常,这些方案仅能用于定性用途,除非已经确定剥离过程不会影响指定抗原的定量结果。许多抗原将经历至少5次剥离循环,具体取决于所用的方法和膜类型。但是,应该考虑到在每个剥离循环中,都将洗去少量的膜固定蛋白。如果需要连续检测几种抗原,建议从预期丰度较低或产生信号较少的抗原开始。在设计具有一轮或多轮抗体剥离的免疫印迹实验时,可参考以下建议。PVDF膜比硝酸纤维素膜更坚韧,因此推荐用于任何涉及抗体剥离的方案。从SDS-PAGE凝胶转印完成后立即干燥PVDF膜,可改善蛋白质与膜的结合,特别推荐用于涉及多次剥离的实验。在第一轮免疫检测之前,必须使用乙醇将干燥的PVDF膜润湿。优先检测低丰度抗原。先使用亲和力低的抗体,再使用高亲和力抗体。重要提示:尽管建议在转印后立即干燥PVDF印迹,但不可在两轮免疫检测之间干燥印迹,否则,任何残留的抗体分子将与膜永久结合。方案1:通过加热和去污剂剥离所需设备和溶液标准印迹或印迹条,在硝酸纤维素或PVDF膜上。封闭液。剥离溶液:100 mM 2-巯基乙醇 (M301574)、2% (w/v) SDS (S108346)、62.5 mM Tris-HCl (T105291), pH 6.7。磷酸盐缓冲盐(PBS):10 mM 磷酸钠、pH 7.2,0.9%(w/v)NaCl。浅托盘,能容纳膜阳性和阴性剥离对照。操作流程1.在通风橱内,将印迹放入剥离溶液中,50 °C摇动孵育30分钟。2.将印迹放入缓冲液中,摇动10分钟。使用新鲜的缓冲液重复一次。3.可选: 重复初始检测操作(省略一抗步骤),4.以确保抗体已失活或从膜上剥离。5.将印迹放入缓冲液中,摇动10分钟。6.继续进行封闭步骤,进行下一轮的免疫检测。方案2:通过低PH值剥离所需设备和溶液标准印迹或印迹条,在硝酸纤维素或PVDF膜上。封闭液。剥离溶液:25 mM 甘氨酸-HCl (G156794), pH 2,1% (w/v) SDS (S108346)。磷酸盐缓冲盐(PBS):10 mM 磷酸钠、pH 7.2,0.9%(w/v)NaCl。浅托盘,能容纳膜阳性和阴性剥离对照。操作流程1.将印迹放入剥离溶液中,摇动孵育30分钟。2.将印迹放入缓冲液中,摇动10分钟。使用新鲜的缓冲液重复一次。3.继续进行封闭步骤,进行下一轮的免疫检测。方案3:利用WESTERN BLOT STRIPPING BUFFER(蛋白质印迹剥离缓冲液)抗体剥离液对硝酸纤维素和PVDF膜均具有良好的剥离效果。但在待剥离膜具有较高信号或抗体剥离液体处理不充分时效果更好。所需设备和溶液标准印迹或印迹条,在硝酸纤维素或PVDF膜上。封闭液。保鲜膜,用于保存不会立即再次检测的印迹。Western Blot Stripping Buffer(蛋白质印迹剥离缓冲液)蒸馏水,用于稀释试剂。浅托盘,能容纳膜阳性和阴性剥离对照。操作流程注意:若要重复使用印迹或单个印迹条,应在第一次使用后立即剥离。如果不能立即剥离,应将膜包在保鲜膜中并置于PBS中,保存在4°C。切勿将印迹干燥保存。1.在塑料托盘中加入适量的1X Antibody Stripping Solution(缓冲液套餐中提供)2.用镊子将印迹或印迹条浸入剥离液中。在室温下,轻轻混合孵育15分钟。在一个干净的塑料托盘中装入相同量的封闭液。3.可使用传统的封闭液,如20 mM Tris HCL、pH 8.0,150 mM NaCl,0.1% 吐温20,5%奶粉,或者类似溶液。4.使用封闭液清洗印迹2次,每次5分钟。5.现在,可使用抗体再次检测印迹。阿拉丁相关产品参考文献1.Lioubin MN, Myles GM, Carlberg K, Bowtell D, Rohrschneider LR. 1994. Shc, Grb2, Sos1, and a 150-kilodalton tyrosine-phosphorylated protein form complexes with Fms in hematopoietic cells.. Mol. Cell. Biol.. 14(9):5682-5691. http://dx.doi.org/10.1128/mcb.14.9.56822.Legocki RP, Verma DPS. 1981. Multiple immunoreplica technique: Screening for specific proteins with a series of different antibodies using one polyacrylamide gel. Analytical Biochemistry. 111(2):385-392. http://dx.doi.org/10.1016/0003-2697(81)90577-73.Harlow E, Lane D. 1988. A Laboratory Manual. 579. New York: Cold Spring Harbor Laboratory.4.Kaufmann SH, Ewing CM, Shaper JH. 1987. The erasable Western blot. Analytical Biochemistry. 161(1):89-95. http://dx.doi.org/10.1016/0003-2697(87)90656-75.Yeung Y, Stanley ER. 2009. A solution for stripping antibodies from polyvinylidene fluoride immunoblots for multiple reprobing. Analytical Biochemistry. 389(1):89-91. http://dx.doi.org/10.1016/j.ab.2009.03.017
参数原理
2022.11.03
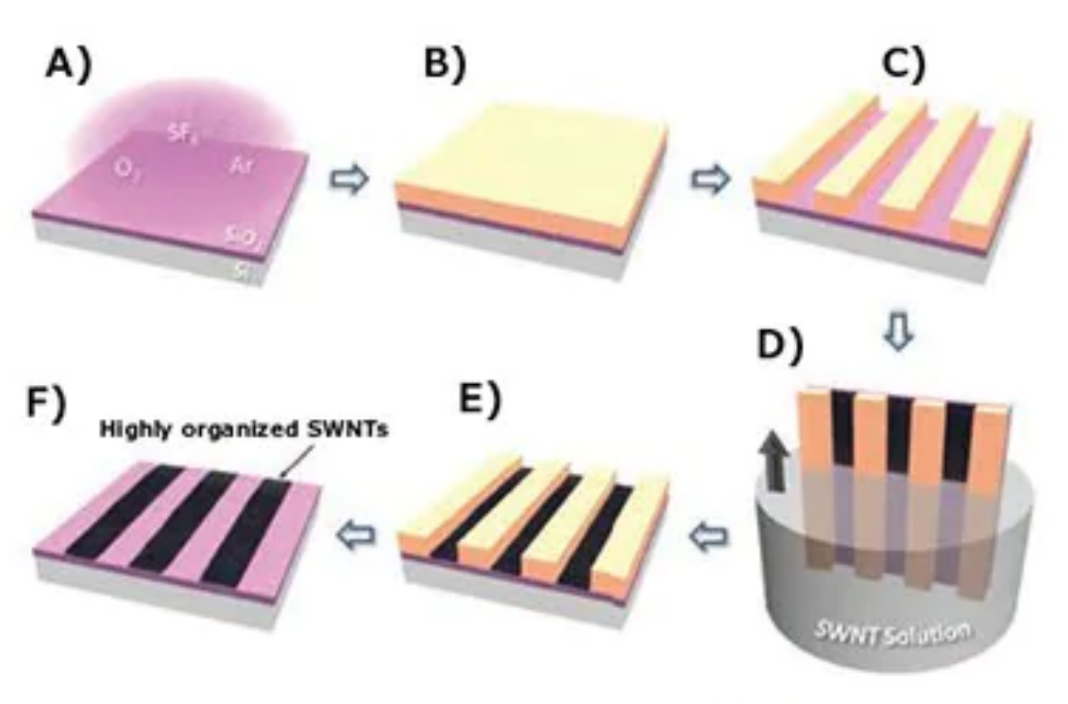
单壁碳纳米管网络的构建及其应用
背景介绍自从饭岛澄男教授发现碳纳米管(CNT)以来,单壁碳纳米管(SWCNT)在过去的20年里一直吸引着大量的研究兴趣[1]。它们的分子结构可以用石墨烯卷成一维(1-D)无缝圆柱体来描述。独特的准一维结构和芳香单层[2,3]表面提供了优异的电学性能(高载流能力,~109Acm-2) [4,5],热导率(~3500Wm-1K-1) [6],和机械性能(杨氏模量,~1-2TPa) [7]。这些特性使SWCNT成为各种应用的理想候选者,包括传感器[8]、能量存储设备[9]、场发射设备[10]、和药物输送剂[11]。对于使用SWCNT 作为主要组件的设备,开发制造工艺非常重要:i)在所需的位置、方向和尺寸上可控地组装SWCNT;ii)将它们集成到功能强大的设备中;iii) 设计它们的分子结构以进一步增强它们的物理性能。本文介绍了一种先进的自组装方法,用于在各种基板上制造可调节的微纳米级SWCNT网络[12-14]。其次,作者展示了这些有组织的架构的集成,以应用于通过创建高度可控的SWCNT和硅基异质结来实现光电器件。这代表了一种新型的基于光电二极管的逻辑器件,它可以由光和电两种输入控制,具有高电压可切换光电流响应性(>1 A/W),光电压响应性(>105 V/W),以及良好的电和光开关比(电:>105和光:>104)[15]。第三,作者还通过用2,2,6,6-四甲基哌啶-官能化证明了这些SWCNT 网络用于高性能硫化氢(H2S)检测1-oxyl(TEMPO)[16]。最后,介绍了SWCNT网络的结构转化到连续的多壁碳纳米管,然后到石墨/多层石墨烯纳米带结构,使用一种新开发的电压控制方法,称为“纳米管聚变”,这不仅证明了工程碳纳米管的sp2结构新方法,而且大大提高了其电和热输运性能[17]。单壁碳纳米管组装 作者使用模板导向流体组件的方法来制备高度组织和对齐的SWCNTs薄膜,它采用了光刻图案模板辅助浸渍涂层[12–14],[18]。SWCNTs在预先设计的光刻胶通道之间直接组装在亲水性表面上,形成微和纳米尺度的高起源SWCNTs侧网络。等离子体处理可以提高流体组装的质量,增加悬浮键和表面亲水氢氧根官能团的数量。图1显示了模板引导射流组装方法的详细程序。图1A-C显示了SiO2基板经过等离子体处理,然后用光刻胶旋涂,并通过光刻技术(分别用于微纳米级图案的光刻和电子束光刻)进行图案化。图1D展示了使用浸涂机将预先图案化的基板首先垂直浸入SWCNT去离子(DI)水溶液中,然后以恒定的拉速度逐渐从溶液中提起的过程。图1E和1C显示了在去除光刻胶后,在SiO2衬底上的微/纳米级沟槽和SWCNT网络之间形成的SWCNT网络图案。图1G-J显示了SiO2/Si衬底上的厘米级、毫米级、微米级和纳米级组装的SWCNT网络。图1 模板辅助流体SWCNT组装示意图和SWCNT网络的SEM图像 A)通过等离子蚀刻对SiO2衬底进行表面处理;B-C)光刻胶涂层和模板图案化;D-E)通过浸涂形成SWCNT条带并组装具有光刻胶图案的SWCNT条带;F)去除光刻胶后高度组织的SWCNT条带;G-H)3英寸上组装的SWCNT条带的照片图像,100μm宽组装的SWCNT网络的晶圆和显微图像;I-J)4 μm宽和200 nm宽组装的SWCNT网络的SEM图像[14,18]。基于光电应用的SWCNTS和Si异质结开发基于硅的光子电路组件,例如片上源、调制器、存储等,是解决传统硅电路传输速度和集成度瓶颈的有希望的方法[19,20]。在这里,作者展示了SWCNT和硅的高度集成和控制异质结可以展示一个完全非常规的、急剧非线性的、反偏相关的光电流。这种新现象为获得高开关比的多功能模拟和混合数字光电运算提供了新的途径。可以通过电压的微小变化获得大的光电流切换,从而使光电门/器件具有逻辑输出,具体取决于光学和电子输入的逻辑状态。我们在厘米尺度的晶圆上展示了许多新型光电开关/器件和大量SWCNT架构器件的光刻组装。图2A和2E显示了高密度SWCNT/Si异质结图案的示意图和数字照片。SWCNTs通过模板流体法和常规光刻法在轻掺杂p的硅表面组装为微/纳米级器件结构,如图2F所示。图2B显示了SWCNT-Si结的暗和亮I-V曲线,以及具有相似尺寸的金属硅结中的光电流响应。虽然SWCNT-Si结中的暗I-V遵循传统二极管整流行为,但电流明显偏离了传统行为,即在反偏压Vr的几伏内急剧上升几个数量级,这与相同光源下同等尺寸的传统金属-Si肖特基结的发光I-V不同。提出的这些异质结能带结构的半定量模型表明,急剧的非线性光电流行为可能与SWCNT带中可用状态的反向偏置可调总数n(ε=eVr)有关。图2C展示了SWCNT组件Si传感器阵列的照片。图2G显示了连接到源极和漏极电极的叉指型SWCNT网络,这相当于两个背靠背光电二极管形成一个双向光电晶体管。图2H显示了一个混合输入的光电和门,光和施加的电压是输入,测量的电流是输出。此外,图2I-J分别显示了2位和4位数字光输入、电压可切换模拟输出加法器电路;当施加反向电压时,输出显示数字和的模拟等效值。因此,SWCNT-Si结是光电传感器、光转换器、光度测量和成像等光电应用的通用平台。图2 新型光电器件示意图和SWCNT-Si传感器阵列图像 A)带有电极的 SWCNT-Si异质结测试结构(2cm×2cm);B)典型SWCNT-Si异质结的暗和亮I-V 曲线;C)SWCNT-Si传感器:0.25兆像素SWCNT-Si传感器阵列的数码照片(阵列面积,12mm×12mm)和D)传感器“核心”的SEM图像;E)SWCNT-Si异质结结构的数码照片;F)组装的SWCNT带SEM 图像;G)使用连接到源漏引线的叉指型SWCNT指的双向光电晶体管。该器件的有源区面积为3mm×200mm;H)具有光和电输入以及电输出的与门。结的有源区面积为3mm×100mm,插图:一组典型的工作条件,确定输入和输出条件的“低”和“高”逻辑状态;I)具有2个光学输入和一个电气输入和输出的2位加法器/或门;J)4位数模转换器带SWCNT器件的高性能H2S检测器SWCNT具有独特的纳米结构、高迁移率、高电流密度、高效电化学表面积等优点,是各种化学传感器积极研究的对象[8]。由于原始的基于CNT的化学传感器仅利用其固有的为了克服其在选择性和灵敏度方面的局限性,用共价或非共价材料对SWCNT进行功能化已被用于进一步提高基于SWCNT的化学传感器的灵敏度。需要注意的是,因为其一维纳米结构,SWCNT对湿度和周围温度等环境变化非常敏感。在开发H2S气体传感器时,我们使用TEMPO分子掺杂作为SWCNT表面的催化剂,以提高室温下通过氧化还原反应检测H2S的灵敏度。图3A-D显示了高度组织的微传感器器件的SEM图像,这些器件通过模板导向流体组装方法,然后涂上TEMPO涂层,制备了大规模的SWCNT网络图案。图3E显示了用于功能化SWCNTs的TEMPO的化学结构。这种化合物能够氧化气态的H2S,可以用作H2S传感器的传感分子。图3F显示了功能化SWCNT传感器在不同浓度的H2S气体下的实时电流变化。图3G显示了器件的固有灵敏度和TEMPO功能化后的优势。另外,根据我们的结果,TEMPO可以被氧化为TEMPO+,这是H2S解离的一个非常重要的产物。TEMPO+到TEMPO-H的还原反应与H2S到S+2H++2e-的解离过程相耦合,因为在空气中,SWCNT器件由于氧离子(O2)的作用而表现出p型特性。新产生的电子被捐赠给SWCNT,使其不再具有P型特性,这使得功能化SWCNT比纯SWCNT更敏感。此外,我们发现湿度通过改善TEMPO到TEMPO+的变化来提高H2S的灵敏度。最后,本文还证明了用TEMPO功能化的SWCNT器件对随着湿度增加的H2S气体高度响应能力。半单壁碳纳米管尤其如此,它在60%的湿度下表现出高达420%的高灵敏度。图3 基于SWCNT网络的高性能H2S检测器 A-D)组装好的SWCNT器件阵列的SEM图像;E)TEMPO分子的化学结构;F)功能化SWCNT传感器的实时电流随 H2S气体浓度的变化情况;G)无修饰的SWCNT器件和TEMPO功能化SWCNT器件的灵敏度随湿度的变化情况[16]。SWCNT网络的电融合:同素异构体间转化sp2结构碳纳米管的改性通常需要极端且难以控制的条件,例如高温、高压和高能辐照。本文提出了一种简单但功能强大的电学方法,通过施加受控的交流电压脉冲来实现碳纳米管从SWCNT到多壁碳纳米管(MWCNT)或多层石墨烯/石墨纳米带结构(MGNR)的结构转变。通过控制电压脉冲的大小、源时间和应用于SWCNT网络的开关周期数,可以在不产生任何结构缺陷的情况下获得目标转化纳米结构(MWCNTs或GNRs)。另外还发现,经过结构改造后,SWCNT网络的电导率(高达35%的电阻降)和热导率(高达6-7倍的热导率,300 W/mK)都大大提高。图4A显示了在TEM网格上与金属电极组装的SWCNT网络结构。图4B显示了SWCNT网络的I-V 曲线、最大电流密度和击穿电压(Vb)。图4C显示了施加在SWCNT器件上的与时间相关的交流电压脉冲。图4D-F显示了电融合过程后原始SWCNT、MWNT(0.6Vb,3,000次循环)和GNR(0.8Vb,3,000次循环)的TEM图像。图4G-H显示了SWCNT器件在相同的3,000 次循环中在不同施加电压下的电阻变化和热导率。图4 脉冲交变电压诱导SWCNT的同素异构体转化及其电学和热学性能 A) 集成在特殊设计的TEM网格上的电极的组装SWCNT网络的SEM图像;B)显示电击穿行为的原始SWCNT网络设备的典型I-V特性,其中Vb是击穿电压;C)随时间变化的交流电压脉冲,用于SWCNT网络的结构转换;D)原始SWCNT的TEM图像;E)施加电压Va=0.6Vb,循环3000次后的多壁结构TEM图像; F)经Va=0.8Vb循环3000次后的多层石墨纳米带TEM图像;G-H)SWCNT网络在不同施加电压下 3,000次循环次数的电学和热学特性[17]。结论在这篇文章中,作者提出了一种高度组织SWCNT网络的模板引导流体组装方法。使用光刻图案化模板辅助浸涂,使用光刻图案模板辅助浸涂方法,SWCNTs直接组装在预先设计的微纳米级光刻胶通道之间,形成密集排列的、具有不同几何形状的SWCNTs侧网络,其特征尺寸从150纳米到数百微米尺度不等。这些高度组织的微纳米级SWCNT网络结构可以集成到各种传感器件架构中。模板导向流体装配技术的室温和晶圆级缩放兼容性提供了在大范围内可复制的可能性。在这方面,本文展示了一种高性能光电探测器和一种新型逻辑器件,基于SWCNT和硅的可扩展异质结,其输出电流可以完全由光和电输入控制。我们还介绍了一种基于SWCNT的化学传感器,该传感器可以通过使用TEMPO作为催化剂功能化的SWCNT网络上发生的氧化还原反应有效地检测H2S气体。最后,作者使用受控的交流电压脉冲证明了在大面积SWCNT网络上精确控制和明确定义的同素异形转变和分子结的形成。使用这种新开发的电学工艺,小直径SWCNT可以选择性地转化为其他同素异形sp2纳米结构,例如多壁碳纳米管、多层石墨烯纳米带和具有sp3键的结构。参考文献1.Iijima S. 1991. Helical microtubules of graphitic carbon. Nature. 354(6348):56-58. http://dx.doi.org/10.1038/354056a02.Avouris P. 2002. Molecular Electronics with Carbon Nanotubes. Acc. Chem. Res. 35(12):1026-1034. http://dx.doi.org/10.1021/ar010152e3.Dai H. 2002. Carbon Nanotubes: Synthesis, Integration, and Properties. Acc. Chem. Res. 35(12):1035-1044. http://dx.doi.org/10.1021/ar01016404.Yao Z, Kane CL, Dekker C. High-Field Electrical Transport in Single-Wall Carbon Nanotubes. Phys. Rev. Lett. 84(13):2941-2944. http://dx.doi.org/10.1103/physrevlett.84.29415.White CT, Todorov TN. 1998. Carbon nanotubes as long ballistic conductors. Nature. 393(6682):240-242. http://dx.doi.org/10.1038/304206.Pop E, Mann D, Wang Q, Goodson K, Dai H. 2006. Thermal Conductance of an Individual Single-Wall Carbon Nanotube above Room Temperature. Nano Lett. 6(1):96-100. http://dx.doi.org/10.1021/nl052145f7.Lu JP. 1997. Elastic properties of single and multilayered nanotubes. Journal of Physics and Chemistry of Solids. 58(11):1649-1652. http://dx.doi.org/10.1016/s0022-3697(97)00045-08.Kong J. 2000. Nanotube Molecular Wires as Chemical Sensors. 287(5453):622-625. http://dx.doi.org/10.1126/science.287.5453.6229.Wen L, Li F, Cheng H. 2016. Carbon Nanotubes and Graphene for Flexible Electrochemical Energy Storage: from Materials to Devices. Adv.Mater. 28(22):4306-4337. http://dx.doi.org/10.1002/adma.20150422510.Fan S. 1999. Self-Oriented Regular Arrays of Carbon Nanotubes and Their Field Emission Properties. 283(5401):512-514. http://dx.doi.org/10.1126/science.283.5401.51211.Shi Kam NW, O'Connell M, Wisdom JA, Dai H. 2005. Carbon nanotubes as multifunctional biological transporters and near-infrared agents for selective cancer cell destruction. Proceedings of the National Academy of Sciences. 102(33):11600-11605. http://dx.doi.org/10.1073/pnas.050268010212.Jaber-Ansari L, Hahm MG, Somu S, Sanz YE, Busnaina A, Jung YJ. 2009. Mechanism of Very Large-Scale Assembly of SWNTs in Template Guided Fluidic Assembly Process. J. Am. Chem. Soc. 131(2):804-808. http://dx.doi.org/10.1021/ja807652313.Xiong X, Chen C, Ryan P, Busnaina AA, Jung YJ, Dokmeci MR. 2009. Directed assembly of high density single-walled carbon nanotube patterns on flexible polymer substrates. Nanotechnology. 20(29):295302. http://dx.doi.org/10.1088/0957-4484/20/29/29530214.Jaber-Ansari L, Hahm MG, Kim TH, Somu S, Busnaina A, Jung YJ. 2009. Large scale highly organized single-walled carbon nanotube networks for electrical devices. Appl. Phys. A. 96(2):373-377. http://dx.doi.org/10.1007/s00339-009-5194-215.Kim YL, Jung HY, Park S, Li B, Liu F, Hao J, Kwon Y, Jung YJ, Kar S. 2014. Voltage-switchable photocurrents in single-walled carbon nanotube? silicon junctions for analog and digital optoelectronics. Nature Photon. 8(3):239-243. http://dx.doi.org/10.1038/nphoton.2014.116.Jung HY, Kim YL, Park S, Datar A, Lee H, Huang J, Somu S, Busnaina A, Jung YJ, Kwon Y. 2013. High-performance H2S detection by redox reactions in semiconducting carbon nanotube-based devices. Analyst. 138(23):7206. http://dx.doi.org/10.1039/c3an01762a17.Jung HY, Araujo PT, Kim YL, Jung SM, Jia X, Hong S, Ahn CW, Kong J, Dresselhaus MS, Kar S, et al. 2014. Sculpting carbon bonds for allotropic transformation through solid-state re-engineering of sp2 carbon. Nat Commun. 5(1): http://dx.doi.org/10.1038/ncomms594118.Kim YL, Li B, An X, Hahm MG, Chen L, Washington M, Ajayan PM, Nayak SK, Busnaina A, Kar S, et al. 2009. Highly Aligned Scalable Platinum-Decorated Single-Wall Carbon Nanotube Arrays for Nanoscale Electrical Interconnects. ACS Nano. 3(9):2818-2826. http://dx.doi.org/10.1021/nn900775319.Almeida VR, Barrios CA, Panepucci RR, Lipson M. 2004. All-optical control of light on a silicon chip. Nature. 431(7012):1081-1084. http://dx.doi.org/10.1038/nature0292120.Liu L, Kumar R, Huybrechts K, Spuesens T, Roelkens G, Geluk E, de Vries T, Regreny P, Van Thourhout D, Baets R, et al. 2010. An ultra-small, low-power, all-optical flip-flop memory on a silicon chip. Nature Photon. 4(3):182-187. http://dx.doi.org/10.1038/nphoton.2009.268
操作维护
2022.11.02

阿拉丁产品焕新颜!
阿拉丁启用新标签生物试剂标签化学试剂标签 清新靓丽更加亲民 新标签上彩色圆圈的点缀,黄蓝红的撞色简约清新,活泼轻快为阿拉丁产品更添一抹灵动 01ALADDIN 圈圈点点宛若繁星 采用可持续创新与技术产品开发过程中引入可持续发展标准考虑整个价值链 02ALADDIN 更低碳,更环保采用环保材料 采用可持续产品包装: 减少:减少包装 避免:实现零毁林 再利用:提高塑料的可持续性 回收:最大限度地回收 03ALADDIN友好标签温馨自然 采用可持续产品设计研发、产品管理、质量、采购在流程的每一步都进行合作用心提高生命科学产品的可持续性 04ALADDIN 避光包装确保密封 在产品安全与质量上通过创新的解决方案和数字通信工具我们不断努力提高产品安全性减少业务对环境的影响 05ALADDIN 做研究不能只严肃也需要放轻松 我们的产品战略旨在以经济可行和可持续的方式围绕生命科学领域开发一系列产品 变的是包装不变的是质量 从研究成果到将研究成果转化为产品提供最高质量的产品是我们始终如一的目标 更重要的是在产品研发、生产和质量的所有过程中都实施了客户反馈以确保每个产品满足客户对高质量的需求 可以说质量贯穿着阿拉丁整个产品生命周期 ▻▻▻
企业动态
2022.10.23

阿拉丁当选上海市新材料协会副会长单位!
阿拉丁当选上海市新材料协会副会长单位基于阿拉丁在材料科学的影响力和研发能力,阿拉丁当选上海市新材料协会副会长单位。在此,非常感谢新材料协会对阿拉丁的肯定!新材料产业是战略性、基础性产业,也是当前国际竞争的重点领域之一,而阿拉丁的材料科学产品有着丰富的产品线。阿拉丁能源材料 相关产品阿拉丁阿拉丁电子材料相关产品相关产品阿拉丁生物医学材料相关产品上海新材料协会上海新材料协会成立于2000年,协会由中国宝武、上海华谊、上海建材、上海建工等企业集团和上海交通大学、复旦大学、同济大学、中科院上海分院、上海科学院、上海材料研究所等60多家联合发起创建,是中国第一家新材料协会。阿拉丁将在新材料协会的支持和帮助下,将继续坚持新材料产业健康可持续发展。阿拉丁建设新材料研发定制服务和产业化平台,加速公司产品创新升级,加强产业间相互合作,提高企业核心科技竞争力,推进上海新材料健康有序发展,为先进制造业的发展提供材料保障!
企业动态
2022.10.17

缓冲液参考中心
缓冲液参考中心常见生物缓冲液的pH范围Tris缓冲液的制备 - pH vs. 温度磷酸盐缓冲液的制备 - 0.2M溶液柠檬酸 - 磷酸氢二钠缓冲液的配置,pH2.6 - 7.6柠檬酸 - 柠檬酸钠缓冲液的配置,pH3.0 - 6.2乙酸钠 - 醋酸缓冲液的配置,pH3.7 - 5.6磷酸氢二钠 - 磷酸二氢钠缓冲液的配置,pH5.8 - 8.0 (25℃)咪唑 - 盐酸盐缓冲液的配置,pH6.2 - 7.8 (25℃)碳酸钠 - 碳酸氢钠缓冲液的配置,pH6.2 - 7.8 (25℃)缓冲液配置公式选择正确的缓冲液基于酸碱性、解离常数(衡量溶液酸碱性、浓度以及温度的系数)、规格、纯度以及精确的应用领域可帮助您选择一款合适的缓冲液。以下表格通过pH与pKa数据的参考,为您提供一些常用缓冲液的配置方案。缓冲液级别为满足各种形式的实验需求,我们提供不同纯度与杂质含量的缓冲液试剂。目前有7种不同级别的试剂可以满足基本的实验室需求、帮助建立药品配方与生产、检测痕量金属含量以及生产指定纯度级别的材料。常用生物缓冲的有效pH范围(25℃, 0.1M)TRIS缓冲液配置指导 - pH vs.温度磷酸盐缓冲液配置指导 - 0.2M 溶液醋酸-磷酸氢二钠缓冲液配置,pH 2.6-7.6一水柠檬酸,C6H8O7 • H2O,MW 210.14;0.1M 含量为21.01 g/L。磷酸氢二钠,MW 141.98; 0.2M 含量为28.40 g/L,或者磷酸氢二钠二水合物,MW 178.05;0.2M 含量为35.61 g/L。当X mL 0.1M-醋酸 和Y mL 0.2M-磷酸氢二钠混合时醋酸-柠檬酸钠缓冲液配置,PH 3.0-6.2一水柠檬酸,C6H8O7 • H2O,MW 210.14;0.1M 含量为21.01 g/L。柠檬酸三钠二水合物,C6H5O7Na3•2H2O,MW 294.12;0.1M 含量为29.41 g/L。 当X mL 0.1M-醋酸 和Y mL 0.1M-柠檬酸三钠混合时醋酸钠-醋酸缓冲液配置,PH 3.7 - 5.6醋酸钠,CH3COONa • 3H2O,MW 136.09;0.2M 含量为27.22 g/L。X mL 0.2M-NaOAc 和Y mL 0.2M-HOAc 混合时碳酸氢二钠-碳酸二氢钠缓冲液配置,PH 5.8-8.0 25℃Na2HPO4 • 2H2O, MW 178.05;0.2M 含量为35.61 g/L。Na2HPO4 • 12H2O,MW 358.22; 0.2M 含量为71.64 g/L。NaH2PO4 • H2O,MW 138.01;0.2M 含量为27.6 g/L。NaH2PO4 • 2H2O,MW 156.03;0.2M 含量为31.21 g/L。 X mL 0.2M-磷酸氢二钠,Y mL 0.2M-磷酸二氢钠;用水稀释至100mL咪唑-HCl 缓冲液配置,PH 6.2 - 7.8 25℃咪唑,C3H4N2,MW 68.0825mL 0.2M-咪唑(13.62 g/L),X mL 0.2M-HCl,用水稀释至100mL碳酸钠-碳酸氢钠缓冲液配置,PH 9.2 -10.8Na2CO3 • 10H2O,MW 286.2;0.1M 含量为28.62 g/L。NaHCO3, MW 84.0;0.1M 含量为8.40 g/L。X mL 0.1M-碳酸钠和Y mL 0.1M-碳酸氢钠混合时缓冲液制备的配方与方式质量分数(w/v)(缓冲液所需百分比/100)× 最终缓冲液体积(mL)= 最初所需原料的质量(g)配置摩尔浓度所需的物质的量浓度 × 分子量 × 溶液最终体积(L) = 所需的质量(g)亨德森-哈塞尔巴尔赫方程参考文献1. Dawson R, Elliot D, Elliot W, Jones KM. 1986. Data for Biochemical Research. 3rd ed.. Oxford Science Publ..
参数原理
2022.10.14

没有金刚钻,就不揽瓷器活!
企业动态
2022.10.11
阿拉丁碳纳米材料系列产品介绍
碳纳米材料是一个庞大的碳同素异形体家族,按材料尺寸划分,可分为零维碳纳米材料,一维碳纳米材料,二维碳纳米材料系列产品。图 1 不同维度的碳纳米材料示意图 [1]零维碳纳米材料零维碳纳米材料是指在三个维度上均处于纳米尺度的碳材料,主要有富勒烯及类富勒烯的Cn结构碳纳米材料、纳米金刚石、以及碳纳米点等。富勒烯是最早发现的零维碳纳米材料。由于其尺度的特殊性,富勒烯表现出了许多不同于宏观碳材料的性能。例如由于其具有缺电子烯烃的性质,其在催化领域有着重要的作用,不论是自身作为催化剂[2-3],还是经其他物质修饰、掺杂之后作催化剂或是其衍生物做催化剂[4-5],均有良好的催化作用。图 2 C60修饰氧化石墨烯催化膜用于水处理 [6]阿拉丁提供的富勒烯是一系列纯碳组成的原子簇的总称。它们是由非平面的五元环、六元环等构成的封闭式空心球形或椭球形结构的共轭烯。现已分离得到其中的几种,如C60和C70等。阿拉丁富勒烯的成员还有C78、C82、C84、C90、C96等也有管状等其他形状。一维碳纳米材料一维碳纳米材料是指在两个维度上、均在纳米尺度范围内的碳材料,包括但不局限于碳纳米管、碳纳米纤维、碳纳米卷、碳纳米棒等材料。碳纳米管(Carbon Nanotubes, CNTs)是由碳原子形成的石墨烯片层卷成的无缝、中空的管体,其具备许多优异的物理和化学特性,被广泛应用于诸多领域。例如在能源领域,碳纳米管作为锂离子电池负极材料,具有较高比容量、较好循环性能[7],常被用于电池材料相关研究中。在催化领域,碳纳米管及表面官能团处理的碳纳米管是很好的多相催化、光电催化以及光催化剂[8]。此外,碳纳米管还在降解工业污染物、费托合成、光电分解水等方面都具有重要的作用。图 3 基于原位生长CNT制备的大面积数字电子设备 [9]阿拉丁不仅提供单壁碳纳米管、多壁碳纳米管等一系列产品。还提供包括丙烯腈碳纤维和沥青碳纤维两类碳纤维。碳纤维质轻于铝而强力高于钢,它的比重是铁的1/4,强力是铁的10倍,除了有高超的强力外,其化学性能非常稳定,耐腐蚀性高,同时耐高温和低温、耐辐射、消臭。阿拉丁碳纤维也有众多选择。二维碳纳米材料二维碳纳米材料是指仅有一个维度在纳米尺寸范围内的碳材料,最典型的二维碳纳米材料即为石墨烯。石墨烯独特的结构使其显示出许多特殊的物理和化学性能,例如由于其结构稳定,电子传输速率快,电学性质优异,因此可以作为一种很好的电极材料,对锂离子电池、锂硫电池、超级电容器等化学电源的容量等性能有重要的影响[10]。近年来,人们已经很少使用纯石墨烯作为电极材料,而将其进行氧化或者是与金属氧化物进行复合[11],例如将氧化石墨烯与Mn3O4复合制备的多孔结构的电极材料用于超级电容器,所制备的电容器不仅可以弯折,能量密度也得到了提升[12]。图 4 氧化石墨烯制备电极材料用于微型超级电容器[12]阿拉丁石墨烯相关产品包含石墨烯和氧化石墨烯两种,产品形态包含固体及各类溶剂分散液,可以满足各类需求。其他相关产品除以上较为典型的碳纳米材料之外,阿拉丁还提供纳米多孔碳材料,后者可应用于电化学双层电容器、催化载体、有机生物分子吸附载体、高灵敏生物传感器电极、太阳能电池等前沿领域研究。项目号产品名称Cas编号包装F141063富勒烯C6099685-96-8100mg/250mg/1g/5gF166920富勒石131159-39-2100mg/500mgF141064富勒醇1gC139819低纯羟基化单壁碳纳米管308068-56-6250mg/1gC124534单壁碳纳米管308068-56-6250mg/1gC121255双壁碳纳米管308068-56-6250mg/1gC140995大比表面积单壁碳纳米管308068-56-6250mg/1gC299129工业级多壁碳纳米管308068-56-650g/250g/1kgC139885工业级羟基化多壁碳纳米管308068-56-650gC139887工业级羧基化多壁碳纳米管308068-56-650g/250g/1kgC140994氨基化单壁碳纳米管308068-56-6250mg/1gC306012氨基化多壁碳纳米管308068-56-61g/5g/10gC139980氮掺杂碳纳米管1g/5g/25gC139953碳纳米管NMP分散液308068-56-6500mLC139960碳纳米管水分散剂308068-56-6100gG302114石墨烯1034343-98-0250mg/500mg/1gG405797氧化石墨烯5mL/25mL/100mLG196555巯基化石墨烯7782-42-5100mgG139812氧化石墨烯溶胶7782-42-525g/100g/500gG139803氧化石墨烯粉末7782-42-5250mg/1gG196559氨基化石墨烯(纳小尺寸)7782-42-5100mgG293467氨基化石墨烯(纳小尺寸)7782-42-5100mgG196546氮掺杂氧化石墨烯7782-42-51gG139802氮掺杂石墨烯7782-42-5250mg/1gG139801石墨烯NMP浆料7782-42-525g/100g/500gG139799石墨烯分散液7782-42-525g/100g/500gG139800石墨烯水性浆料7782-42-525g/100g/500gG196554羟基化石墨烯(纳小尺寸)7782-42-5100mgG196556羧基化石墨烯(纳小尺寸)7782-42-5100mgG196547试剂级氧化石墨烯溶液7782-42-520mL
应用实例
2022.08.25

不对称催化反应好帮手—Sadphos手性膦配体
手性,指物体无法与其镜像重合,例如我们的双手(图1)。在一次化学合成中,产物分子的两种手性结构:R-对映体和S-对映体出现的比例是相同的,但是不同手性的分子之间存在性能差异,有可能导致只有一半的产品能够使用,另外一半则没有利用价值,甚至可能会造成负面影响。图1 手性结构示意图(来源:搜狗百科)为了解决这类问题,相关研究机构开始寻找方法转换手性分子。不对称催化合成反应是常见的用于产生手性专一分子的方法。在已经开发出的各类手性催化剂中,手性膦具备易于修饰(可充当过渡金属配体),适用范围广等优势,因此逐渐成为研究者们关注的焦点。Sadphos是一类新型的手性叔丁基亚磺酰胺类膦配体/催化剂,其具备多种不同软硬配位原子的特征,使其可以分别作为单膦配体、N,P-配体、P,S-配体、P,O-配体与多种过渡金属以及不同价态金属灵活配位(图2),是一类配位自适应性较强的手性配体。图2 Sadphos配体结构及其特点图3 复旦大学张俊良教授组发展的Sadphos系列配体[1]声明:文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请在阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
应用实例
2022.07.26

阿拉丁IVD原材料家族成员:异硫氰酸胍/盐酸胍
扫码登记、鼻拭子、咽拭子采样、保存、转运、检测-核酸检测流程随着疫情常态化防控为大众所熟悉。有效的病毒保存是准确核酸检测结果的重要保障,构建稳定高效的病毒保存液,作为病毒的采集、运输保存及检测的溶液介质是其中关键。1.病毒保存液种类目前病毒保存液分为两种,灭活型及非灭活型,两种方式各有特点,非灭活型不含裂解盐,但是保存的病毒完整性更好,检出率更高,除了核酸检测外还可以用于其他研究;灭活型病毒保存液则使用上更安全,操作环境要求也不用那么严格。· 非灭活型病毒保存液目前研究的非灭活病毒保存液的成分主要为Hank's液基础,添加BSA、氨基酸、维生素等病毒所需的营养成分以及生物缓冲剂、冷冻保护剂、甘油等多种组分,可在较宽的温度范围内维持病毒的活性,保持样本的原始性,可以用于病毒的核酸提取检测、病毒的培养和分离。· 灭活型病毒保存液添加有裂解盐的病毒保存液就是灭活型的病毒保存液,通过将采集到的样本与病毒裂解液和病毒核酸保存液进行充分混合,实现对样本中病毒的灭活,同时有效保证样本中的病毒核酸完整性,采集后的标本能够在常温条件下运输和长期保存。保存的病毒RNA样本可广泛应用于基因检测、酶联免疫法检测(ELISA)、PCR检测等。灭活保存液有含胍盐、非胍盐及直扩之分,其中以胍盐型为最常用。2.胍盐裂解保存原理高浓度的胍盐,是一类强力的蛋白质变性剂,可以破坏核酸与蛋白质之间的次级键,使核蛋白解联从而把病毒核酸释放出来。可以直接裂解灭活病毒,不需要加热处理样本,简便快捷,目前最常用于病毒裂解的胍盐是异硫氰酸胍、盐酸胍,他们无RNA酶和DNA酶活性,能够破坏RNA酶的分子结构,使样本中可能存在的核酸酶失活,起到稳定保存病毒样本的作用。3.阿拉丁胍盐产品系列阿拉丁生化科技是服务于科研和应用领域的知名供应商,以丰富的产品种类和规格、便捷的电商服务平台、先进的质量控制体系、健全的服务网络服务于广大的科学工作者。订购号产品名称规格G108673盐酸胍25g,100g,500gG110925异硫氰酸胍5g,25g,100g,250g,500g,2.5kg更多产品和服务欢迎您登陆阿拉丁官网或者致电阿拉丁客服中心了解
应用实例
2022.07.19
阿拉丁IVD原料家族成员--ProClin防腐剂
抗原检测、核酸检测,作为常态化疫情防控的工具,逐渐为大众所熟知。全球试剂研发和生产机构均积极应对,开发生产的快速、准确、便捷的IVD试剂盒成为常态化疫情防控的重要保障。生产高品质的体外诊断试剂盒,保证其准确性和重复性,需要各种各样高质量的原材料。如何保证研制出来的试剂盒运输方便、存储简单,并具备足够的保质期,是体外诊断试剂能否被广泛应用的关键。防腐剂就是实现这些需求的小秘密。时代在发展,体外诊断试剂常用的防腐剂也经历了更新换代。曾经的生物防腐剂硫酸汞(thiomersalatum)硫酸汞作为防腐剂已有60多年历史,广泛用于生物制品中的疫苗及体外诊断试剂。虽然它对革兰阳性和阴性菌均有很强的抑菌能力,为广谱抑菌剂。但其作用机理为重金属离子与菌体中酶蛋白的巯基结合而使酶失去活性,对细菌抗原和血清蛋白的损害极其微弱,并不能与酸、碘、铝等重金属盐或生物碱配伍。由于少量有机汞在理论上具有神经毒性,因此硫柳汞的使用安全问题也受到人们的关注。叠氮化钠(sodiumazide.NaN3)作为一种在体外诊断行业广泛使用的防腐原料,它可抑制细菌生长,易溶于水,在绝大多数中性和碱性环境中使用很适合,遇酸会放出有毒的HN3(氢叠氮酸)。它的用量一般为0.1%~0.5%,多数情况下效果不错,但由于叠氮键会干扰辣根过氧化物酶(HRP)、超氧化物歧化酶、触酶等多种酶,所以一般含有HRP的试剂都不建议使用。同理,一般不用于抗体及酶免实验中的标本保存。它既属于易制爆,也属于剧毒品,出于安全方面的考虑,叠氮钠的使用日益受限,会碰到购买、运输、存储、使用等种种限制。抗生素类庆大霉素(gentamicin,C21H43N507)等庆大霉素是一类氨基糖苷类抗生素,是由22-脱氧链霉胺、绛红糖胺、加拉糖胺组成的多组分混合物,并以C族复合物为主。其作用机理是直接与30S核糖体亚单位16S rRNA解码区的A部位结合,从而导致菌体死亡。它的抗菌谱较广,对革兰阴性菌有普遍的抗菌作用,对革兰阳性菌中的金黄色P葡萄球菌也有很强的抗菌作用。但最大的缺点是易产生抗药性。ProClin系列防腐剂ProClin系列防腐剂是体外诊断试剂常用的新型防腐剂,主要成分包含甲基氯异噻唑啉酮(5-Chloro-2-methyl-4-isothiazolin-3-one, CMIT)和甲基异噻唑啉酮(2-Methyl-4-isothiazolin-3-one, MIT),这两种主要活性成分在与细胞膜接触几分钟后,立即可以渗透到膜内并抑制细胞内酶的活性,作用于Krebs循环的四个关键位置:丙酮酸脱氢酶、α-酮戊二酸脱氢酶、琥珀酸脱氢酶和NADH脱氢酶,抑制细胞的生长以及促使细胞凋亡的原理相同。所有的细菌及真菌至少拥有部分KREBS循环,所以ProClin活性具有广谱适用性。广泛应用于生化试剂、免疫试剂、凝血试剂、分子生物试剂、电泳液、PCR/DNA检测相关的缓冲剂。ProClin防腐剂系列产品特点1.具有广谱抑菌能力,对细菌(包括G阳性和阴性)、真菌和酵母菌均有效。2.能快速消除微生物的活性,消除生物膜并能抑制生物膜的再形成。3.不含甲醛,其抑菌作用不是依靠甲醛的释放,低使用剂量,低成本。4.在低浓度下,也能发挥良好的抑菌效果,且药效持续时间长。5.可与广泛的配方组分如阴离子、阳离子、非离子型表面活性剂配伍。6.优异的化学稳定性,与大多数原料或配方有很好的配伍性,不含金属离子。7.所用的溶剂对试剂反应体系没有干扰,和关键的酶(Enzymes)相容。8.不影响抗体结合,对离子选择电极(ISE)没有干扰(氯电极除外)。9.产品无色,不影响反应体系中指示物质吸光度的变化。10.在推荐的温度、pH值下,未开封产品稳定性其长。11.与水完全兼溶,适用于水溶液配方,在试剂盒中的稳定性好 。12.在推荐的浓度下使用无危害化学品,废弃物安全。13.具有良好的生物降解能力,不会对环境造成污染。阿拉丁ProClin系列防腐剂阿拉丁生化科技是服务于科研和应用市场领域的知名企业,致力于为客户提供品种齐全,品质优秀的IVD原材料,为各位研发和生产厂商提供质量保证、价格低廉、响应快速的原料保障方案。订购号产品描述规格P406411ProClin 150 防腐剂50Ml、400Ml、1L、15LP406395ProClin 300防腐剂400Ml、1L、5L、10L、20LP406415ProClin 200 防腐剂50Ml、400Ml、15LP407596ProClin 8020 防腐剂400Ml、15L
应用实例
2022.07.08
企业名称: 上海阿拉丁生化科技股份有限公司
企业地址: 上海市新金桥路36号财富中心16楼 联系人: 阿拉丁客服 邮编: 201206 联系电话: 400-860-5168转3457
仪器信息网APP

展位手机站