
视频号

抖音号

哔哩哔哩号

前沿资讯手机看

山东大学药学院教授,博士生导师,齐鲁青年学者,山东省杰出青年基金获得者。从事抗病毒、抗痛风创新药物研究。先后承担国家自然科学基金、山东省重大科技创新工程等项目10余项,发现了一系列具有开发前景的先导化



上期,展鹏教授团队分享并阐述了冠状病毒的基本结构、冠状病毒的生命周期、抗冠状病毒药物的主要靶点等内容,本期将分享靶向冠状病毒刺突蛋白、RdRp、蛋白酶及宿主靶标的一系列冠状病毒广谱抑制剂,以及其对抗击新冠肺炎疫情、预防未来的冠状病毒传播具有的重要意义。
本文讨论的冠状病毒广谱抑制剂是针对冠状 病毒与宿主的关键靶点开发的抗病毒化合物。现 阶段,根据这类化合物靶向的生理过程不同,分别靶向冠状病毒的侵入过程、RNA复制过程、多聚 蛋白裂解过程以及宿主靶标。
4.1靶向冠状病毒侵入过程的抑制剂
在抗病毒药物中,侵入抑制剂可以使病毒的生命周期停止在第一步,使其对宿主的危害最小化。SARS-CoV和SARS-CoV-2是通过刺突蛋白与人类呼吸道上皮细胞的ACE2结合而侵入[16], 而MERS侵入所利用的胞外受体是CD26,也称 作二肽基肽酶(DPP4)。
刺突蛋白是一种I型跨膜蛋白(图3),分子 表面高度糖基化,它组装成三聚体后,分布在病毒颗粒的最外层,形成了冠状病毒独特的外观。所有冠状病毒刺突蛋白的胞外部分都是由两个相同的结构域结合而成:氨基端的S1亚单位与受体结 合相关,含有受体结合域(receptor binding domain,RBD);羧基端的S2亚单位含有融合肽 (fusion peptide),与病毒融合相关。在S1完成结合后,S2被细胞表面的TMPRSS2蛋白酶裂解,该过程是病毒与宿主细胞膜融合所必需的[17]。因此,靶向S蛋白或TMPRSS2的分子可成为有效的冠状病毒侵入抑制剂。
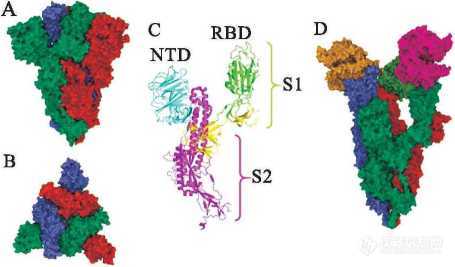
Figure3 (A-B ) Structure of S protein trimer, from different angles of view ( PDB code:6XM5);( C) Structure of S protein monomer and location of NTD and RBD;(D) Binding mode ofSprotein with ACE2 ( PDB code:7KNY)
4.1.1 靶向S蛋白的侵入抑制剂
在S蛋白抑制剂中,肽类具有高效、低毒的优势[18]。基于ACE2胞外序列设计的水溶性肽 作为潜在的侵入抑制剂曾受到重视,但其体内半衰期短,难以转运到肺泡[19]。为提高成药性, Lei[20]将ACE2片段与人免疫球蛋白IgGl的Fc结构域结合,提高了血浆中稳定性并增强了结合力。
目前,已设计并合成了一系列模拟ACE2的N端螺旋结构域的肽类化合物,如Barh[21]通过扫 描现有的抗菌、抗病毒肽类数据库,得到了10个可能有效阻断S蛋白RBD区域与人ACE2作用 的肽类,但其体内外活性有待进一步研究。在此 基础上,Larue[22]设计了一系列针对刺突蛋白的 ACE2多肽类似物(SAP1 ~SAP6,表1),并在编码荧光素酶并负载SARS-CoV-2刺突蛋白的慢病毒侵染HEK293T-ACE2细胞体系中测定各个多 肽对病毒侵入的抑制作用,各物质活性以半数抑 制浓度(IC50)计量,活性最好为SAP6[(1.90 ± 0. 14) mmol • L-1 ]。同时,上述多肽对SARS- CoV-2刺突蛋白RBD区域的亲和力(Kd)最高为 (0.53 ±0.01) mmol-L-1(SAPl)。
Table1Amino acid sequence of ACE2 derivatives targeting S protein
Compd. |
Sequence |
Location |
SAP1 |
27-TFLDKFNHEAEDLFYQ42 |
Helix-1 |
SAP2 |
37-EDLFYQSSLS5 |
Helix-1 |
SAP3 |
79-LAQMYPL-85 |
Helix-3 |
SAP4 |
352-GKGDFRYL-359 |
Helix-11 |
SAP5 |
24-QAKTFLDKFNHEA-36 |
Helix-1 |
SAP6 |
37-EDLFYQ42 |
Helix-1 |
Curreli等[23]基于ACE2蛋白结合区中30个 氨基酸残基长度的螺旋结构,以8 ~11碳的不饱 和炷链连接肽链上一定跨度的邻近氨基酸,设计了 4个高度螺旋化的装订肽(stapled peptide) NYBSP-1~NYBSP-4,并在 HT1080/ACE2 细胞 与人肺A549/ACE2细胞系中使用基于假病毒的 单循环方法测定了上述多肽分子的EC50值。其中3 个多肽分子显示出了潜在的抗病毒活性:HT1080/ ACE2 中的 EC50值为(1. 9 ~ 4. 1 )μmol• L-1, A549/ACE2 中 EC50值为(2. 2 ~ 2. 8) μmol • L-1,且在最高测试剂量时,未显示出任何细胞毒性。使用SARS-CoV-2病毒侵染Vero E6细胞时, NYBSP-1显示出了最高的抑制活性,在 17.2 μmol•L-1的浓度完全阻止了细胞病理效应。NYBSP-2和NYBSP-4活性稍低,EC100值为 33 μmol• L-1,NYBSP-4在血浆中的半衰期为289 min,代谢稳定性好。
Glasgow 采用“受体陷阱”,(receptor trap)策略,合成出高亲和性、高溶解性的ACE2胞外部分结构域,阻止病毒刺突蛋白与人体细胞表面的 ACE2的结合与入侵[24]。基于此策略设计的肽类分子使冠状病毒难以产生抗药性,并可以抑制几乎所有通过ACE2侵入细胞的冠状病毒[25]。在进一步研究中,Glasgow[24]利用计算机/实验组合的蛋白质工程方法,重新设计了能与SARS- CoV-2刺突蛋白结合的ACE2胞外可溶性区域 (氨基酸18-614) 。最终得到的ACE2变体对于单体刺突蛋白RBD区域的KD app ( apparent binding affinity)值已接近100 pmol• L-1。同时,最理想的 “受体陷阱”分子抑制SARS-CoV-2假病毒和真正 SARS-CoV-2 病毒的 IC50值已达到(10~100) ng-mL-1的范围。这类多肽分子有望真正实现针对利用ACE2入侵宿主细胞的冠状病毒的广谱抑制。
由于S蛋白分子高度糖基化,可与多糖衍生物产生多种相互作用,引导人们去探索针对S蛋 白的多糖类抑制物。早在2013年,Milewska就证实了N-(2-羟丙基)-3-三甲氨基甲壳素氯化物 (HTCC,1,图4)及其疏水性修饰的同系物(HM- HTCC)是HCOV-NL63的潜在抑制剂[26],并制备 了不同比例的氨基被甲壳素取代的HTCC衍生物, 各自具有对不同种类人冠状病毒的抑制作用[27]。
近期,文献报道了在人呼吸道上皮细胞中,HTCC 具有抑制 SARS-CoV-2 和 MERS-CoV 的 活性。尽管HTCC中单个正电基团对于靶标的作用较弱,但冠状病毒连环化的特性和多聚物分 子中的多个位点协同作用使得HTCC可以稳定 结合S蛋白。目前,虽然HTCC仍未被批准用于 临床,但实验已经证明其在肺部局部给药的可行 性,且毒副作用极低口旳。综合考虑,上述各种甲 壳素衍生物联合使用,有望成为广谱抗人冠状病 毒感染的防治药物。
Griffithsin(2,图4)是由海藻中分离得到的天 然血凝素,可利用糖基结构域结合病毒包膜糖蛋白中特定的寡糖[29]。已有研究表明,griffithsin可以与多种病毒表面的糖蛋白相互作用,包括HIV gpl20 以及 SARS-CoV 的 S 蛋白[30-31]。2016 年,Millet 等[32]报道了 griffithsin 对于 MERS-CoV 的抑制作用。在2μg • mL-1 浓度下,griffithsin抑制了 MERS 病毒对 Huh-7、MRC-5 和 Vero-81 细 胞系90%以上的感染性。针对迅速爆发的新冠 肺炎疫情,一系列针对griffithsin抗新冠病毒活性 的研究正在展开。Xia等[33]首先发现griffithsin 对SARS-CoV-2假病毒侵染呈现剂量依赖性地抑 制作用,EC50值为293 nmol•L-1Cai等[34]网进一 步在体外试验中测定了 griffithsin对SARS-CoV- 2的抑制活性,结果表明,griffithsin对SARS-CoV- 2活病毒的EC50值达63 nmol• L-1,同时对S蛋白 介导的细胞间融合的EC50值为323 nmol-L-1值得注意的是,该研究团队还报道了 griffithsin与肽 类冠状病毒侵入抑制剂EK1的协同作用。未来, griffithsin可以单独或与EK1联合制成鼻喷剂、吸入剂或凝胶,以预防或治疗新冠肺炎。

4. 1.2 TMPRSS2 抑制剂
在SARS-CoV或MERS-CoV的刺突S蛋白 发挥作用之前,要依赖宿主细胞的跨膜蛋白酶TMPRSS2将其裂解为S1和S2亚单位[35]。针对 这类蛋白酶的抑制剂也可用于阻断各种冠状病毒 的入侵过程。
蔡莫司他(nafamostat,3,图5)最初用于治疗 胰腺炎,后发现也是TMPRSS2抑制剂,对MERS-CoV具有拮抗活性[36]。进一步研究发现,蔡莫司 他甲磺酸盐对SARS-CoV-2的EC50值达到了纳摩尔级[37]。同时,在日本批准用于治疗胰腺炎的 药物甲磺酸卡莫司他(camostat mesilate,4,图5) 同样具有抑制TMPRSS2的活性[17],在微摩尔浓度即可有效抑制MERS-CoV感染中合胞体的形成[38],EC50值达到0.11μmol•L-1[39]:对SARS- CoV-2的EC50值为87nmol•L-1[37]o现阶段仍无 法确定该化合物能否在肺部达到抑制病毒的有效浓度[40],但已有临床研究正在评估其对新冠肺炎的治疗作用。
4. 1. 3 宿主细胞激酶抑制剂
病毒在生命周期中利用了宿主细胞的若干信 号通路。冠状病毒以内吞方式入侵宿主细胞的过 程中,除S蛋白与ACE2的作用外,还需要Abel- son激酶(Abl)的介导。Abl是细胞中重要的管 家蛋白,参与正常细胞的多个生理过程,同时也与 病毒的入侵与复制密切联系,是开发广谱冠状病 毒抑制剂的有效靶点[41]。伊马替尼(imatinib,5,图5)是Abl的抑制剂,已被批准用于治疗慢性粒 细胞白血病。已有研究证实,伊马替尼通过阻断病毒颗粒与胞内体膜融合,从而抑制病毒以内吞 路径入胞,并在感染早期抑制SARS-CoV和MERS-CoV的增殖關。据报道,伊马替尼抑制SARS-CoV-2增殖的EC50值达到130 nmol-L-1 ,同时对SARS-CoV-2 S蛋白的RBD区域结合活 性高达2.32pimol-L-1,可通过双靶点作用有效 抑制SARS-CoV-2的侵入關。但在细胞实验中, 其毒性较为明显,用于治疗新冠肺炎或其他冠状 病毒感染前还要经过充分评估。目前,世界范围 内已有多项伊马替尼针对新冠肺炎的临床试验正 在进行(NCT04394416、EudraCT2020-001236-10、NCT04357613)。
4. 1. 4 组织蛋白酶L与Furin蛋白酶抑制剂
组织蛋白酶L位于宿主细胞的胞内体,在无 TMPRSS2表达的细胞中,组织蛋白酶L发挥裂 解活性,介导病毒粒子与胞内体膜融合,从而完成侵入过程[44]。2003年,SARS-CoV疫情引起了人 们对组织蛋白酶L抑制剂研发的重视。随后的十几年内,已发现数种具有抗冠状病毒活性的组 织蛋白酶L抑制剂。其中,K11777(6,图5)是通 过筛选2 000余个人组织蛋白酶抑制剂发现的[45],其对人体或某些寄生虫的半胱氨酸蛋白酶具 有显著抑制作用。K11777抑制SARS-CoV和MERS-CoV感染的EC50值分别达到0.68nmol•L-1与46nmol•L-1,但其不可逆的共价结合机制可能导致较强的毒副作用。目前,K11777仅作为锥虫 病治疗药物进行临床试验M,其针对SARS- CoV-2的抑制作用有待于进一步确证。
SARS-CoV-2 S蛋白的裂解过程也可依赖 Furin蛋白酶进行。Cheng[47]研究了以蔡基荧光 素(naphthofluorescein, 7,图5 )为代表 的数个 Furin蛋白酶抑制剂,证实了此类分子可抑制SARS-CoV-2的感染进程及细胞病理效应。但冠状病毒侵入细胞的不同路径中的关键酶具有互补作用,因此单一种类的蛋白酶抑制剂难以起效[48],而多种抑制剂联用的毒性可能大幅度增加。针对冠状病毒生命周期中宿主蛋白酶的药物应用尚存在一定的风险与挑战。

4.2靶向冠状病毒RNA复制过程的抑制剂
针对冠状病毒另一类极为重要的治疗靶标是 RNA依赖的RNA聚合酶(RdRp),由非结构蛋白 nspl2、nsp7与nsp8结合构成。其活性位点高度保守,包括在一个β转角中突出的两个连续的天 冬氨酸残基样[49],在不同的正链RNA病毒如冠状病毒和HCV中结构相似[50]。RdRp作为RNA复 制的工具,在病毒的复制中具有重要作用[51]。同 时该酶结构高度特异化,人体无同源酶,是药物开 发的优良靶点。
4. 2. 1 RNA依赖的RNA聚合酶抑制剂
瑞德西韦(remdesivir ,8,图6-A)是一种腺昔 酸类似物,作为RNA聚合酶的广谱抑制剂,能够抑制人与鼠冠状病毒[52]。更为重要的是,研究证明瑞德西韦在体外针对SARS-CoV-2具有抑制活性, 其抑制 SARS-CoV-2 的 EC50值为 0.77μmol•L-1, 且CC50值大于100 μmol•L-1[53]。基于“老药新用”的原则,2020年10月23日,瑞德西韦获得美 国FDA的正式使用批准,用于治疗12岁以上的新冠肺炎患者[54]。
作为一种核昔类似物,瑞德西韦可以与 SARS-CoV、MERS-CoV 和 SARS-CoV-2 RdRp 的 NTP结合位点相互作用。其代谢后以核昔母体9 (GS-441524,图6-A)的形式掺入新生的子代 RNA链中,但允许子链RNA的进一步延长。瑞 德西韦在新生链中移动到-4位时,分子中1,-氰基 与RdRp侧链的Ser861残基发生空间上的碰撞,阻碍了 RdRp在RNA链上的进一步移动,进而导致RNA复制终止(图6-B)。由于终止作用是在瑞德西韦结合RdRp后发生的,该过程称为延迟链终止[54]。

延迟链终止机制的RdRp抑制剂针对冠状病 毒具有一定的抗耐药性。包括SARS-CoV-2在内 的冠状病毒会编码具有核酸外切酶活性的nspl4,该酶可以在3,端切除掺入RNA链的异常 碱基,并重启正确的RNA合成[56]。在此机制下, 导致RNA合成即时终止的分子,如去除3,羟基 的核甘类似物,在插入后会被nspl4切除。相对地,在一定延迟后使RNA链合成终止的RdRp抑制剂可有效逃脱nspl4的校对。但研究证实,核酸外切酶仍会识别并切除部分含有瑞德西韦的子 链RNA,并重启RNA复制[57]。同时,病毒体外 传代实验中发现了针对瑞德西韦的耐药现象。与 SARS-CoV-2相似的鼠肝炎病毒(MHV)传代培 养至23代后,其RdRp中出现了不利于瑞德西韦 结合的氨基酸突变[58]。
一系列瑞德西韦的临床试验也引起了研究人 员对其临床疗效的争议。2020年5月,原研公司 吉利德发布了适应性试验的“最终报告” (NCT04280705)[59],称瑞德西韦在临床中可缩短住院时间,改善呼吸系统症状。但WHO在2020 年12月2日发表的“团结实验” (NCT04315948) 结果显示,瑞德西韦无法显著改善总体死亡率、通气时间与住院时间,疗效仍待改进[60]。Spin-ner[61]在为期11天的周期内研究了瑞德西韦针 对新冠肺炎轻中症患者的疗效(NCT04292730), 结果表明,在治疗期间,虽然患者的某些临床数 据出现显著改变,但并不表示任何程度的病情改善。
近H,Li[62]在一系列细胞实验中比较了瑞德 西韦与核昔母体GS-441524在体外细胞中的抗病毒能力。结果显示,GS-441524在Vero E6细胞 系中对SARS-CoV-2的抑制能力略强于瑞德西韦,但在Calu-3和Caco-2细胞系中活性稍弱。GS-441524亦可显著提高感染鼠肝炎病毒 (MHV)小鼠的生存率,初步展示出广谱抗病毒作用。由于GS-441524合成方便、成本低、可口服, 同样有望成为治疗SARS-CoV-2的候选药物。
法匹拉韦(favipiravir, 10,图7)最早在日本上 市,用于治疗流感,其通过与RdRp活性位点结合 发挥抑制活性[63],对所有种类及亚型的流感病毒均有拮抗作用,具有治疗多种RNA病毒感染的 潜力。此外,法匹拉韦在抑制病毒RdRp的同时, 不对哺乳动物机体的RNA及DNA合成路径产生影响[64-65]。虽然法匹拉韦在体外试验中对 SARS-CoV-2的抗病毒活性较低(EC50= 62μmol•L-1),但在两次临床试验中均显示出良 好的效果3项7]。
利巴韦林(ribavirin, 11,图7)是已上市的广谱抗病毒药物,已被批准用于治疗丙型肝炎与呼吸道合胞病毒感染。其作用机制是通过靶向病毒 RdRp而使病毒基因组RNA中出现多位点突变, 最终导致病毒mRNA加帽终止,进而抑制病毒 RNA合成[68]。利巴韦林的疗效已经在SARS- CoV和MERS感染者中得到了证实,但严重的不 良反应限制了其临床应用[69]。且在体内外实验中,利巴韦林对SARS-CoV-2感染的疗效约为瑞德西韦的1 /100[53]。综合考虑,利巴韦林治疗 SARS-CoV-2感染的药效、安全性及潜在的毒性 作用有待在临床试验中进一步研究。
Galidesivir( BCX4430,12,图 7 )也是腺昔酸 类似物,最初为病毒RNA聚合酶抑制剂,曾被用 来治疗丙型肝炎,且对多种RNA病毒如SARS- CoV,MERS-CoV, Ebola 病毒和 Marburg 病毒具 有广谱抑制活性。在生物体内,galidesivir首先被 转化成相应的三磷酸核昔,再以此形式插入病毒 新合成的RNA链中,导致RNA转录或复制的提 前终止[70]。因此,其有望成为治疗新冠肺炎的候 选药物[71]。
阿兹夫定(azvudine,FNC,13,图7)是首个核 首类双靶点HIV抑制剂,针对多种HIV耐药毒株有良好的抑制活性[72]。新冠肺炎疫情爆发后,在我国进行的一项临床试验(CTR2000029853)显 示,阿兹夫定可以显著缩短新冠肺炎轻中症状患 者的核酸转阴时间,对重症患者也具有潜在的治 疗作用。同时临床上未观察到任何与药物有关的 不良反应,安全性有充分保障。目前针对阿兹夫 定更大样本的临床试验正在进行中[73]。

核苷类似物B-D-N4-羟基胞昔(14,NHC/EI- DD-1931,图8)针对多种RNA病毒具有广泛抑 制作用[74]。研究已证明,NHC可有效抑制α属 冠状病毒HCoV-NL63和β属冠状病毒SARS- CoV、MERS-CoV[75-76],且针对 SARS-CoV-2 感染,其在 Vero E6( EC50 =0. 3μmol•L-1)和 Calu-3(EC50=0.08μmol•L-1)细胞中作用显著如。 同时化合物14的酯类前药莫那匹韦(molnupira- vir,15,图8)针对SARS-CoV-2的EC50值也达到 0. 22 μmol•L-1[77]。与其他的核昔类似物相同, NHC或莫那匹韦在细胞内代谢为三磷酸核昔,并作为假底物与RdRp结合。由于NHC的碱基存 在互变异构形式,两种异构体分别可与腺喋吟 (A)及鸟喋吟(G)配对结合(图8),插入病毒 RNA后可导致由G到A和由C到U的碱基突变。突变积累至一定程度即产生功能错误或丧失 的子代RNA,且无法被核酸外切酶校正,最终导 致病毒增殖活动终止[74,78]。
虽然细胞水平研究显示NHC有对哺乳动物 造成突变的风险[79],但NHC的前药莫那匹韦已 在治疗SARS-CoV-2的I期临床试验中充分证明 其安全性,m期临床评估正在展开「"°此外, NHC 口服吸收好,给药方便,有望使发病早期居 家隔离的患者显著降低恶化率与住院率。

4. 2. 2 DHODH 抑制剂
二氢乳清酸脱氢酶(DHODH)是哺乳动物体内嚅嚏生物碱合成的关键酶病毒的增殖必须依赖宿主的核昔酸等物质,因此该酶的抑制剂具有开发为广谱抗RNA病毒药物的潜力。来氟米特(leflunomide, 16,图9)与其体内代谢物特立氟胺(teriflunomide, 17,图9)是目前仅有的FDA批 准上市的DHODH抑制剂,用于治疗自身免疫性疾病[77]。李洪林团队的研究结果表明[83],在Veto E6细胞系中,来氟米特与特立氟胺针对SARS- CoV-2 的 EC50 值 分别为 26. 06μmol•L-1和 63. 56μmol•L-1该团队基于靶标结构,进一步设计了一系列DHODH抑制剂,其中S312(18,图9)与S416(19,图9)在相同条件下对 SARS-CoV-2 的 EC50 值分别为(1. 56 ± 0. 32 )μmol•L-1 和(0.017 ±0.002)μmol•L-1。特别是 S416的选择指数达到10 000以上,且无激酶抑制 活性,在治疗浓度下对宿主细胞毒性极小,基本克 服了脱靶效应,作为广谱抗冠状病毒抑制剂具有 极大的开发潜力。此外,DHODH抑制剂有望在 新冠肺炎的治疗中发挥免疫抑制作用,降低“细 胞因子风暴”产生的炎症损伤。

参考文献见 中国药物化学杂志 第31卷 第9期,2021年9月总173期
[来源:仪器信息网] 未经授权不得转载

2022.01.13
媒体报道| 人民日报:我国研发的核酸联检新方法 可联合检测7种冠状病毒
2022.01.11
2021.12.31

2021.08.11

2024.07.30

2024.07.29
品牌合作伙伴






























版权与免责声明:
① 凡本网注明"来源:仪器信息网"的所有作品,版权均属于仪器信息网,未经本网授权不得转载、摘编或利用其它方式使用。已获本网授权的作品,应在授权范围内使用,并注明"来源:仪器信息网"。违者本网将追究相关法律责任。
② 本网凡注明"来源:xxx(非本网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,且不承担此类作品侵权行为的直接责任及连带责任。如其他媒体、网站或个人从本网下载使用,必须保留本网注明的"稿件来源",并自负版权等法律责任。
③ 如涉及作品内容、版权等问题,请在作品发表之日起两周内与本网联系,否则视为默认仪器信息网有权转载。
![]() 谢谢您的赞赏,您的鼓励是我前进的动力~
谢谢您的赞赏,您的鼓励是我前进的动力~
打赏失败了~
评论成功+4积分
评论成功,积分获取达到限制
![]() 投票成功~
投票成功~
投票失败了~






