
 关注
关注
已关注
![]() 已认证
已认证
粉丝量 0
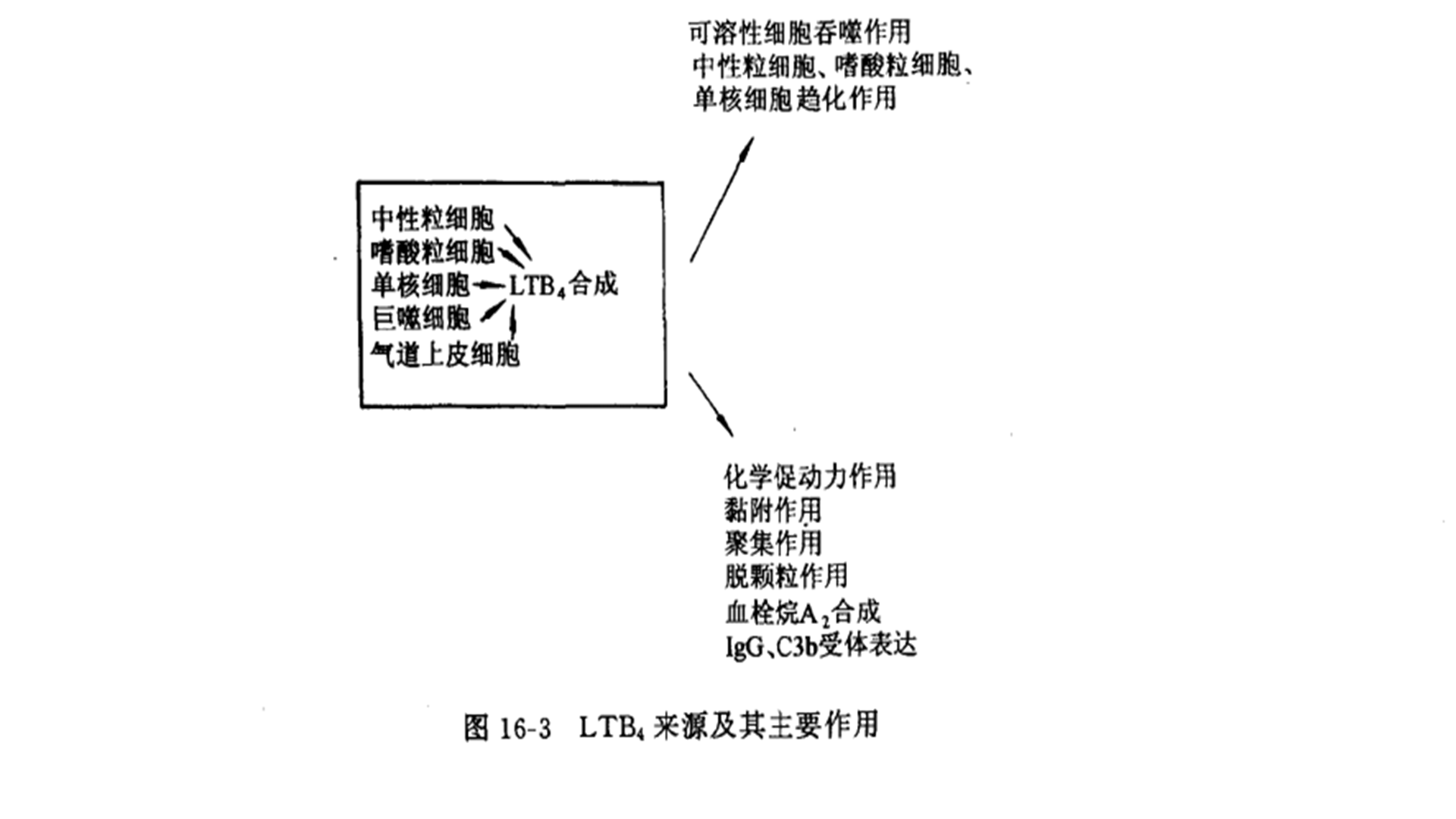
花生四烯酸的脂氧化酶代谢产物
花生四烯酸首先经特异性的脂氧化酶作用,继之经多种酶修饰,如水合过氧化物酶、水解酶、脱水酶、转移酶和肽酶等多种酶族作用,形成三大系列的代谢产物;羟十二碳四烯酸(hydroxyeicosatertraenoic acid,HETE),硫肽白细胞三烯(cysteinyl leukotrienes,LTC4、LTD4、LTE4),非肽白细胞三烯(leukotriene B4,LTB4)及其代谢产物脂氧化酶产物主要由粒细胞、单核-巨噬细胞、浆细胞等炎性细胞合成,其主要作用为白细胞趋化、细胞化学动力作用和增加血管的通透性(图16-3)。LTB4、通过促进黏附和血细胞渗出参与粒细胞与血管内皮细胞之间的作用,它对粒细胞的脱颗粒和毒性产物的生成也有调节作用。白细胞三烯LTC4、LTD4,LTEk4的生物学活性为既往所称的参与过敏反应的“慢反应物质”,这是早在这些白细胞三烯发现之前即早已发现的物质。它们会导致血管尤其是毛细血管后小静脉的过度收缩,由此增加血管内液体外渗,促使水肿形成。因此,在研究ARDS和MOF的机制中血管通透性的改变时,应着重注意这些花生四烯酸的代谢产物。白细胞三烯在肝脏通过N-乙酰化和ω-氧化后通过胆道和肾脏排出。LTB4。则通过细胞内重吸收经ω-氧化迅速灭活。总之,花生四烯酸脂氧化和环氧化两大通路的代谢产物在机体遭受严重创伤、毒血症时可由局部受损器官和组织产生,也可在整个机体的炎症反应过程中形成,粒细胞、单核-巨噬细胞是主要合成场所。这些产物能够导致循环障碍和血管通透性的增加,也会影响肺循环。实验研究显示,使用该代谢途径的抑制剂对机体有益,但是,它们的临床应用尚待时日,初步设想应结合患者的病理生理状况进行进一步的研究。一般认为,使用花生四烯酸主要代谢途径的阻断剂,例如使用环氧化酶和脂氧化酶抑制剂,不仅能够阻断它们的有害作用,也会阻断其有益作用,这种情况下,可能会促使未经阻断的途径产生过多的有害产物,基于这一点,将来的研究会进一步深入至针对各种不同的类二十烷酸的选择性抑制剂或它们的特异性受体方面。
参数原理
2023.05.15
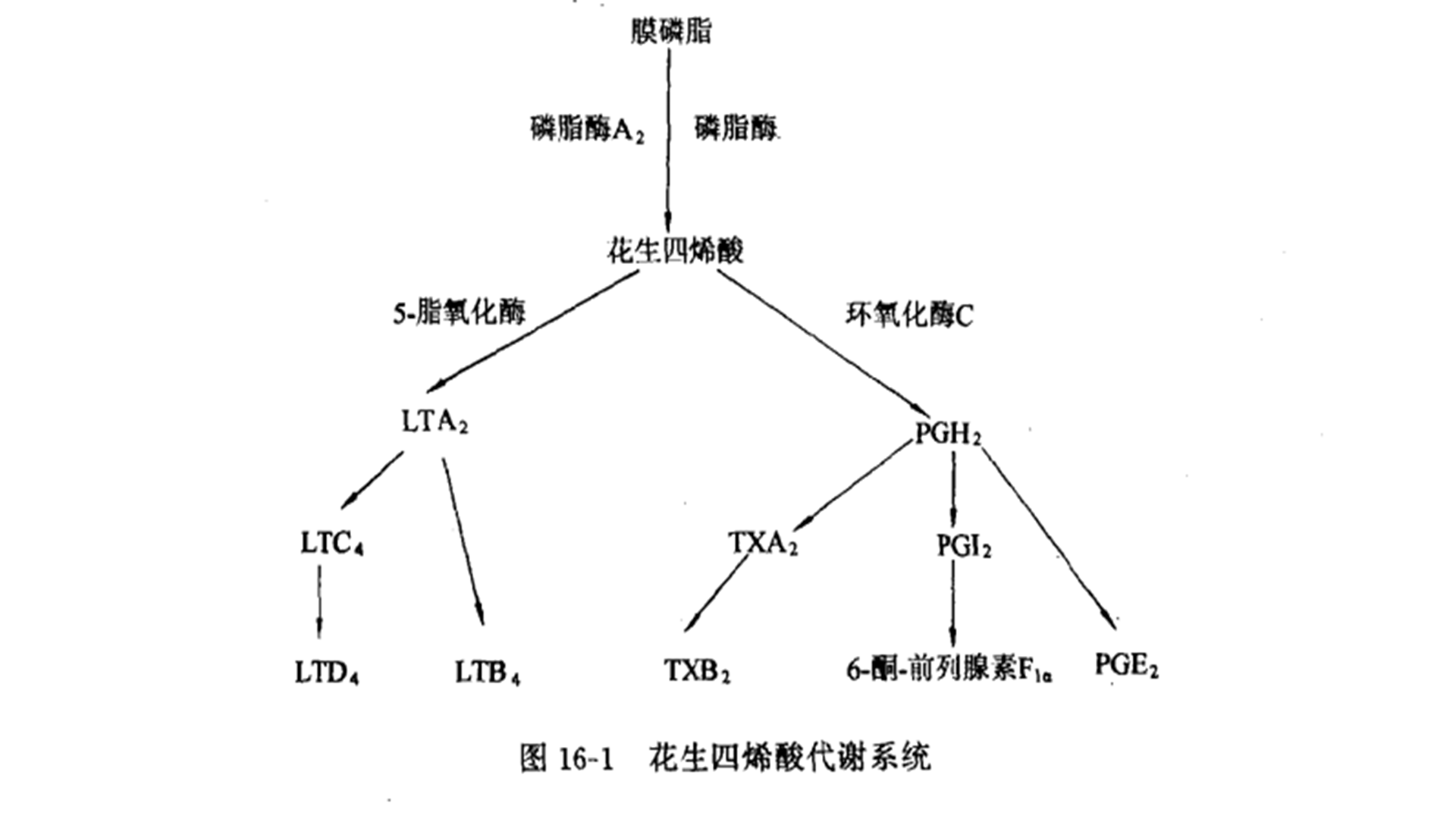
花生四烯酸的环氧化酶产物
花生四烯酸的环氧化酶代谢产物主要是通过作用于血管平滑肌来调节其张力,以及调节血小板活性来产生循环效应(图16-1)。血栓烷A2(TXA2)、前列腺素2α(PGF2α)和前列腺素D2(PGD2)是缩血管物质,特别是TXA2,它具有强烈收缩肺循环的作用。TXA2和PGF2α也有明显的支气管收缩效应。而前列腺素I2(PGI2)和前列腺素E2(PGE2)则具有放松全身血管和松弛气管、支气管平滑肌的效应。TXA2促使血小板积聚,而PGI2和PGE2,则抑制之。PGI主要由血管内皮细胞产生,TXA2由血小板和巨噬细胞合成,而PGD2则是浆细胞的主要环氧化酶产物。PGE2是肾髓质的主要产物,也可由活化的单核细胞和巨噬细胞合成。它通过抑制淋巴细胞合成IL-2来抑制T淋巴细胞的活化。 许多种环氧化酶产物影响胃肠道、泌尿生殖道平滑肌的张力,并与腺体分泌、体温调节、细胞分布和受体表达有关(图16-2)。PGI2和血栓烷在体内通过水解作用在几分钟内即可被灭活,PGI2的半衰期为2~3min,其转化物为TXA2和6-酮基-前列腺素1α,它们的半衰期为30s,然后转化成TXB2。通过测定血液中这些失活的较为稳定的产物,能够反映器官合成PG和TX的情况。PGE和PGF通过主要存在于肺、脾和肾的前列腺素15-羟-脱氢酶快速灭活,通过主要存在于肺、肝和脂肪组织的PG-13-分解酶灭活。在这些器官中,前列腺素的半衰期不足1min,它们进一步经α-、β-、ω-氧化代谢。值得一提的是,在失血性休克的情况下,肺摄取PGE、PGF的速率有所下降。
参数原理
2023.05.15

内毒素与类二十烷酸
内毒素血症能导致机体产生一系列综合征早已为人所共知,但是,其特异性细胞学和分子水平的机制仍待揭示,通常认为是宿主释放的炎症介质而非内毒素本身介导了内毒素血症时的病理生理过程。在内毒素刺激下,机体可释放出多种介质,它们包括许多种细胞因子、花生四烯酸代谢产物以及其他脂类介质,它们共称为类二十烷酸。这些类二十烷酸起源于环氧化酶或脂氧化酶途径两者之一,由血栓烷(TX)、前列腺素(PG)和白细胞三烯(LT)共同组成。本章讨论类二十烷酸在体液和血液中产生的过程,它们的生物学和病理生理作用,研究的根本目的在于为将来的药物干预研究打下基础。在各型休克过程中,除了常规的介质如儿茶酚胺、组胺、5-羟色胺和体液代谢系统的各类产物以外,脂类介质如类二十烷酸在各型休克中也起着重要的作用,并已引起足够的重视。实验和临床研究都有证据表明类二十烷酸参与休克时的微循环障碍和非特异性炎症反应,可导致重要脏器的形态学损害和功能衰竭,如导致急性呼吸窘迫综合征和多器官功能衰竭。类二十烷酸参与在粒细胞、血小板和血管内皮细胞之间的相互作用,在免疫应答过程中也起着重要作用,另外,类二十烷酸也可在局部产生并作用于局部,起局部介质的作用。类二十烷酸的合成和释放相互关联,但它并不贮存于细胞内。由于化学性质不稳定,它常被酶促反应迅速灭活,其生物学活性很大程度上取决于它持续存在的时间和存在的场所,故此类物质适于促进和调节局部反应而并不产生全身和系统性的效应。类二十烷酸是含20个碳原子的不饱和脂肪酸氧化产物的总称,其中花生四烯酸是哺乳动物细胞膜磷脂的主要组成成分,其代谢产物是最主要的类二十烷酸。游离花生四烯酸合成类二十烷酸有环氧化酶和脂氧化酶两条主要代谢途径,依据各种器官和细胞所含的合成酶、同工酶和脂氧化酶的不同,花生四烯酸被代谢为不同产物。例如,肺含有许多种不同的细胞,其酶组成不同,故而能够产生许多种不同的花生四烯酸。类二十烷酸的稳定代谢产物能够从血样中测出。然而在血液循环中的浓度并不能真正反映这些局部介质的局部浓度。特别是休克状态时,由于微循环障碍和细胞间质的存在,其外周血循环中的浓度可能受到血流动力学障碍的影响,但是测定循环血样中的类二十烷酸对于确定它们与血流动力学障碍之间的联系,对于确定其来源器官仍有意义。类二十烷酸的总的合成速率是能够通过测定其尿中代谢终产物来计算的。
参数原理
2023.05.15

脂多糖刺激左旋精氨酸途径的体外实验
体内实验中观察到的内毒素导致的血管对缩血管物质反应性降低的情况在离体动物血管的研究中也可以观察到。这些体外实验表明,血管收缩功能失调与非败血症和内毒素血症时伴随的代谢性酸中毒无关,也与由高循环浓度的儿茶酚胺诱导的受体下调及循环中的扩血管代谢产物无关。这些研究结果的共同特征是:无论内皮细胞存在与否,对多种缩血管物质如去甲肾上腺素、去氧肾上腺素、 indamidine(它促使钙离子通过钙通道内流,也刺激内质网释放钙离子)钙激动剂[Bayk8644和(+)S-202-791]以及钙离子载体A23-187和钙离子本身导致的血管收缩均存在反应性降低,只有对咖啡因及内皮素的反应不受或仅受轻微影响,这些研究表明,血管平滑肌的收缩能力并未被内毒素脂多糖消除,故不存在血管收缩机制的缺陷。阻断花生四烯酸代谢途径后,不管血管内皮存在与否,离体动物的胸主动脉经内毒素处理4h后均表现出对缩血管物质反应性降低。这种低反应性能够被左旋精氨酸和一氧化氮途径的抑制剂如L-NMMA和L-NAME所纠正,因此表明左旋精氨酸和一氧化氮途径参与其中。D-NMMA则不能抑制该反应性,且L-NMME纠正血管低反应性的效应能够被左旋而非右旋精氨酸克服。不管是左旋还是右旋精氨酸本身对未经内毒素处理的离体大鼠的主动脉的缩血管反应均无影响,但是对经内毒素处理的离体鼠的主动脉,左旋精氨酸有抑制作用。上述研究表明,L-NMMA系左旋精氨酸的具有立体结构专一性的拮抗剂:内源性的左旋精氨酸并不能使经过内毒素处理的大鼠血管完全激活以产生足够的一氧化氮,至于左旋精氨酸在对照组和经内毒素处理组效应的差别是否与它组织穿透力的不同有关,仍需进一步研究。左旋精氨酸-一氧化氮途径被脂多糖激活的证据也可由平行测定血管中的环磷酸鸟苷水平,以及通过使用亚甲基蓝、LY-83583等可溶性鸟苷酸环化酶的抑制剂以消除一氧化氮从而抑制可溶性鸟苷酸环化酶来获得支持。实验中,对经内毒素处理组和对照组均测定环磷酸鸟苷水平,然后两组均暴露于去甲肾上腺素。结果显示,不管血管内皮存在与否,经内毒素处理组主动脉环鸟苷酸水平均显著高于对照组,升高的环磷酸鸟苷水平可被L-NAME抵消,进一步加入左旋精氨酸后又能使之恢复。在对照组,左旋精氨酸并不影响环磷酸鸟苷水平。使用鸟嘌呤环化酶抑制剂亚甲基蓝或LY83583可使血管反应性恢复。左旋精氨酸-一氧化氮途径被脂多糖激活的证据也可由平行测定血管中的环磷酸鸟苷水平,以及通过使用亚甲基蓝、LY-83583等可溶性鸟苷酸环化酶的抑制剂以消除一氧化氮从而抑制可溶性鸟苷酸环化酶来获得支持。实验中,对经内毒素处理组和对照组均测定环磷酸鸟苷水平,然后两组均暴露于去甲肾上腺素。结果显示,不管血管内皮存在与否,经内毒素处理组主动脉环鸟苷酸水平均显著高于对照组,升高的环磷酸鸟苷水平可被L-NAME抵消,进一步加入左旋精氨酸后又能使之恢复。在对照组,左旋精氨酸并不影响环磷酸鸟苷水平。使用鸟嘌呤环化酶抑制剂亚甲基蓝或LY83583可使血管反应性恢复。主动脉对血管阻力无影响,有学者对小动脉,即股动脉及第三级肠系膜血管进行了研究,结果认为,只有加入了超氧化物歧化酶并阻止超氧化自由基对一氧化氮的破坏时,内毒素的血管反应性降低才表现出来。然而,去甲肾上腺素所致的小血管收缩可被左旋精氨酸松弛,尤其是当血管内皮存在时更为明显。L-NAME或亚甲基蓝能够阻断上述舒解效应。内毒素脂多糖激活左旋精氨酸-一氧化氮途径的体外实验证据可归纳如下:(1)动物离体主动脉血管平滑肌经内毒素预处理4h后表现出对去甲肾上腺素的低反应性,这与环磷酸鸟苷水平升高有关。使用左旋精氨酸-一氧化氮途径抑制剂L-NMMA、L-NAME或可溶性鸟嘌呤环化酶抑制剂亚甲基蓝或-一氧化氮裂解剂LY83583时方可使血管反应性恢复。(2)内毒素可激活左旋精氨酸-一氧化氮途径的血管阻力,但是依赖于外源性的左旋精氨酸。值得一提的是,所需的左旋精氨酸的浓度与临床上内毒素血症患者的相当。脂多糖可激活内皮细胞外左旋精氨酸-一氧化氮途径,Moncada等研究发现,在内毒素处理的大鼠,其主动脉中层一氧化氮合成酶活力增加,故支持这一结论。
参数原理
2023.05.11

左旋精氨酸-一氧化氮途径在细菌脂多糖诱导缩血管反应的体内实验证据
1990年,Thiemermann和Vane发现,给予实验动物NG-甲基-左旋精氨酸(MeArg),能够使大肠杆菌内毒素造成的低血压得到明显改善,使血压显著升高。这是一氧化氮可能参与脂多糖对循环效应的最初证据,结果如图15-2所示。该结果的难以解释之处在于,给予左旋精氨酸-一氧化氮途径的抑制剂MeArg,能够使血压较对照组有所升高。事实上,给予MeArg较之单独给予内毒素导致的血压下降程度差别不大。Hosford等学者研究了PAF抑制剂对于冲击剂量内毒素静脉滴注造成低血压的缓解作用,结果发现,血压下降程度与上述实验相当,但是没有证据表明PAF参与了内毒素引发的血管低反应性。经静脉注射大剂量的内毒素造成的迅即而显著的低血压与临床上内毒素血症时的血管反应性的丧失并不相干。临床内毒素血症时,血管反应性的丧失可以通过静脉持续缓慢滴注不引起体循环动脉压明显下降的低剂量脂多糖来模拟。有两种动物模型可供验证血管反应性丧失与低血压无关。一种为处于麻醉状态的大鼠,这种情况下去除了内毒素引发的儿茶酚胺作用;另一种为通过脑脊髓刺毁法制成的大鼠模型,它被去除了交感神经的血管支配。Julou-Schaeffer等通过麻醉鼠的实验研究发现,静脉滴注去甲肾上腺素造成的升压反应可被静脉持续滴注大肠杆菌内毒素所抵消,这种内毒素的抑制作用又可被静脉滴注左旋精氨酸-一氧化氮途径的抑制剂如L-NMMA或L-NAME所纠正,而使用D-NMMA则无此效应,若接着补充左旋精氨酸,则血压再次下降,重新表现为对去甲肾上腺素的低反应性。在无内毒素存在的情况下,L-NMMA与左旋精氨酸均不会抑制去甲肾上腺素的升压反应。这种内毒素-内毒素抑制剂和左旋精氨酸之间的相互作用具有特异性,只有左旋体才具有恢复升压反应性的能力;再次恢复的血管反应性只有给予左旋而非右旋精氨酸才能使血管反应性再次受抑。若以吲哚美辛阻断前列腺素合成,则上述L-NAME恢复血管对去甲肾上腺素的升压反应并未受到影响,故推论环氧化酶途径并不参与内毒素血症时的低血管反应。在另一实验中,Gillespie使用脊髓毁损大鼠,这种大鼠对于电刺激引起的脊髓交感神经输出发生频率依赖的升压反应。这种升压反应也可被内毒素抑制,而L-NAME或血管升压素能恢复血管反应性。与前述麻醉大鼠实验结果不同的是,无内毒素存在时,L-NAME和血管升压素可增强血管对去甲肾上腺素的反应性。Guc等推测,可能是这种情况下有更多的钙离子流入,造成血管收缩的结果。L-NAMA也能够抑制由内毒素引发的巨噬细胞释放肿瘤坏死因子,造成低血压反应。与内毒素相似,肿瘤坏死因子能够造成明显的低血压,抗肿瘤坏死因子抗体可阻断这种效应。大鼠实验中,抗肿瘤坏死因子抗体也能对抗由内毒素造成的血管无反应性,肿瘤坏死因子能使内皮细胞释放白细胞介素-1等细胞因子。现在仍不清楚对交感神经刺激或去甲肾上腺素的无反应性发生于血管床的哪一部位,但肾脏及肠系膜血管肯定是受累的。在离体的冠状动脉,由血栓烷类似物U46619导致的缩血管反应可被内毒素抑制,而地塞米松又能消除这种抑制。
参数原理
2023.05.11

内毒素激活左旋精氨酸途径的早期证据
早期的研究通过将15NH3掺入硝酸盐,测定尿中硝酸盐及亚硝酸盐的排泄量来获得间接证据。研究确实发现,内毒素血症患者以及给予内毒素的动物的尿中硝酸盐排量增加。为使左旋精氨酸-一氧化氮途径参与该过程的结论成立,则须证实以下假设:①在患者身体中,硝酸盐不是来源于侵入的细菌。②所测定的产物仅来自于一氧化氮,而且可精确反映其产量。已有实验证据表明,精氨酸水平在内毒素血症患者确实有所下降,特别是那些最终死亡的患者。然而一些更为直接的证据显示,一氧化氮的产量依赖于其底物的供量,而且由于精氨酸在免疫反应中担当非特异性的免疫调节剂,因此有理由认为在内毒素血症患者中,它的含量的下降可能仅为免疫反应的结果。1989 年,Vane等发现了细菌脂多糖(LPS)刺激一氧化氮产生的第一个直接证据。实验将小牛的主动脉内皮细胞与脂多糖孵育,并加入吲哚美辛以抑制前列腺素合成,此时释放出一种能够抑制血小板聚集的物质,且超氧化物歧化酶SOD和左旋精氨酸能够加强其作用,但右旋精氨酸不能增强其作用;而L-NMMA和氧合血红蛋白却能够抑制该反应。这些实验表明,脂多糖可诱导培养内皮细胞中一氧化氮的生成增加。在此基础上,得出内皮细胞释出一氧化氮参与早期休克的结论。第二项直接证据曾于1989年伦敦生理学协会上作交流,该报道揭示,离体大鼠血管经内毒素预处理后,肾上腺素对它的缩血管效应明显降低,然而,L-NMMA可使其缩血管效应恢复,因此血管反应性的失调与左旋精氨酸-一氧化氮途径有关。由于该实验采用剥除了内皮细胞的血管,所以这种反应并不依赖于血管内皮细胞。上述的两项证据表明,脂多糖能够刺激血管内皮细胞及血管壁的其他组成细胞产生一氧化氮或一氧化氮样物质,从而证实了内毒素诱导的血管低反应性的根本原因在于左旋精氨酸-一氧化氮途径的激活。
参数原理
2023.05.10

一氧化氮与EDRF的关系
早在1987年,Palmer 等人即揭示了EDRF的许多生物活性是由一氧化氮承担的,E-DRF与一氧化氮两者在溶液中有相似的半衰期,两者均在酸性环境中稳定,在氧原子及超氧化离子存在的情况下自动激活;两者均为亲脂性,易于渗透过生物膜;均与血红蛋白发生反应,并激活鸟嘌呤环化酶从而提高环磷酸鸟苷的水平;抑制血小板聚集和血小板与内皮细胞表面的黏附,扩张动静脉平滑肌和表现出细胞毒活性。因此推断两者是同一物质。由左旋精氨酸合成一氧化氮和左旋瓜氨酸的过程广泛存在于血管内皮细胞、活化的巨噬细胞、中性粒细胞和血小板等许多种细胞中。其前体可由食物摄取或经过肝脏鸟氨酸循环和内皮细胞从左旋瓜氨酸合成,提示这是个循环过程。该过程受制于左旋谷氨酸盐,由左旋精氨酸合成NO的途径如图15-1所示。它涉及末端胍基氮的氧化,受NO合成酶控制。这些酶是根据它们是诱导酶还是合成酶以及它们的辅因子来分类的。合成酶存在于内皮细胞和脑组织,但不存在于巨噬细胞和中性粒细胞,必须与钙离子、钙调蛋白、NADPH同时存在才能发挥作用。一氧化氮合成的激活可发生在合成细胞本身或远离合成细胞的其他细胞,激活的结果是可溶性鸟嘌呤环化酶的激活使cGMP产量增加,通过刺激内质网摄取钙离子或增加细胞膜钙离子ATP酶活性和(或)抑制钙离子内流入胞使钙离子排出增加,从而降低细胞质钙离子浓度,以及激活蛋白激酶,使血管松弛。左旋精氨酸-一氧化氮途径可被左旋精氨酸含NG的类似物抑制,这些类似物包含NG-甲基-左旋精氨酸(L-NMMA);NG-硝基-左旋精氨酸(L-NA)及其甲基脂化物NG-硝基-左旋精氨酸甲基脂化物(L-NAME),一般认为它们具有立体结构及专一性,是浓度依赖的竞争性拮抗剂,尽管目前有证据表明在一些细胞其抑制作用已不可逆,但它们仍未被广泛应用于研究该途径在细胞功能中的作用。由于L-NMMA在内皮细胞中可被转化为左旋瓜氨酸从而成为合成左旋精氨酸的底物,因而使情况变得更为复杂。一氧化氮的产生对血管反应性的丧失起主要作用,最后不可避免地造成多器官功能衰竭直至死亡。
参数原理
2023.05.10

血管内皮细胞松弛血管平滑肌因子
本章讲述的是内毒素脂多糖激活各种细胞中的盐酸精氨酸途径导致临床上中毒性休克患者死亡的证据。人们早就知道血管内皮细胞可以生成具有松弛血管平滑肌和抑制血小板聚集的物质。这些物质中最重要的为前列腺素,它是花生四烯酸的衍生物,可激活腺苷环化酶,提高血小板和血管平滑肌细胞中的cAMP水平。前列腺素的释放伴随着内皮细胞衍生舒张因子(endothelium derived relaxing factor,EDRF)的释放。EDRF的活化可继而激活磷酸酯酶-磷酸肌醇系统产生两种第二信使,即二酰甘油(DAG)和肌醇三磷酸(IP3),以激活细胞内钙离子。前列环素和其他前列腺素虽不在本章讨论范围之内,但也很重要,因为它们与EDRF存在显著的协同作用,并影响着靶细胞中cAMP和cGMP的生成。1980年,Furchgott和Zawadzki 揭示内皮细胞中存在着非前列腺素性扩血管物质,它可经乙酰胆碱刺激而释放。事实上,离体制备的动脉血管经乙酰胆碱刺激而松弛的反应依赖于完整的内皮层,因为实验发现,当内皮层被剥离后,血管松弛反应即受到抑制。这说明乙酰胆碱松弛血管平滑肌的效应是间接的,依赖于它刺激并促发EDRF的作用。许多其他的内源性扩血管物质如缓激肽也是依赖于它刺激EDRF的产生和释放来起作用的。EDRF在内皮细胞中播散,通过与血液中酶分子的铁原子作用,刺激可溶性鸟嘌呤环化酶,使细胞内环磷酸鸟苷水平增高,从而松弛血管平滑肌。凭借瀑布式的生物系统的分析发现,EDRF的半衰期从4~50s不等,而且这种EDRF系统的快速激活部分地归因于超氧自由基。
参数原理
2023.05.10

内毒素与左旋精氨酸途径
在败血症导致的多器官功能衰竭中,无反应性低血压或称顽固性低血压是一个造成许多患者死于外周血管衰竭的重要因素。有学者研究发现,有57%的患者存在持续性的外周血管扩张和(或)过高的心指数。这种持续性的血管壁张力的丧失归因于血管平滑肌的瘫痪,此时血管平滑肌对于缩血管剂量的儿茶酚胺类激素产生抵抗。外周血管壁张力的持久低下可能是由于前列腺素、组胺、血小板活化因子(platelet activating factor,PAF)、血浆激肽和各种细胞因子等强烈的扩血管物质释放的结果。然而对离体动物血管的实验提示,导致平滑肌收缩功能缺陷可能是更为基本的机制。这些离体血管取自经过盲肠结扎、穿孔或直接给予活的细菌/内毒素的动物。尽管上述已提及的代谢产物及通常伴随的代谢性酸中毒可能都对内毒素血症早期的高动力循环的形成有作用,但离体动物血管即使脱离了上述环境其收缩功能缺陷仍然存在,提示存在血管内皮或平滑肌细胞水平本身的缺陷,而且上述缺陷为局部现象,而非由于存在循环扩血管物质所致。研究表明,该种缺陷在很大程度上可归因于左旋精氨酸途径激活继而产生的一氧化氮的作用。故一氧化氮成为中毒性休克和内毒素血症时重要的血管运动失调介质。
参数原理
2023.05.09

补体在内毒素休克病理生理过程中发挥保护作用
虽然内毒素血症时补体被明显激活可导致致命的结果,但在内毒素休克的病理生理过程中,补体依然发挥着保护作用。补体成分C3或C4缺陷的小鼠对内毒素的敏感性增高,但清除内毒素的能力降低,所以完整的补体系统有助于机体清除内毒素,保护机体免于陷入内毒素休克。C3和C4缺陷小鼠清除循环中内毒素能力的下降,最终导致内毒素休克和死亡。反过来长时间的内毒素血症可消耗C1-in(前述C1-in是一种补体调节因子,是补体和血凝级联反应的蛋白酶抑制剂),而补充外源性C1-in则可挽救这种补体缺陷小鼠的内毒素血症,提示C1-in对内毒素血症有奇特的治疗效果。此外,补体在免疫防御机制中也起着重要作用,如在腹膜炎时,腹膜通过激活细胞和体液免疫防御机制局部防御细菌入侵,而补体激活途径的终产物C5b~9以及趋化因子C3a、C5a和C5~7在这种防御机制中起关键作用,它可通过TCC溶破靶细胞从而将入侵的细菌清除。同时,入侵细菌含有的内毒素激发免疫防御机制可促使巨噬细胞产生细胞因子和促使补体激活,结果促使粒细胞从血管间隙移至腹膜腔,从而参与腹膜腔局部的杀菌作用。许多关于内毒素血症,败血症休克以及其他疾病与补体激活关系的研究中均提示内毒素可引起补体激活,释放过敏毒素和末端C5b~9补体复合物。败血症休克和死于循环衰竭的患者入院时虽已有明显的补体激活,但如果通过测定各种补体成分来预测是否发展为ARDS或MOF则是不可能的。临床实验提示,补体激活及其激活产物如C5a在败血症相关的脏器衰竭和死亡中起重要作用。但目前尚缺乏灵长目动物的实验,因此不能完全证明补体系统在败血症相关脏器衰竭中的最重要地位。因关于补体的作用目前尚无最后的结论,故在败血症休克患者中,目前亦无针对补体激活的特殊治疗。所以,目前在内毒素休克中最重要的治疗是去除能激活补体的感染原,如应用足够的抗生素治疗及脓肿外科引流治疗﹐从而减少补体的激活,使循环中的过敏毒素和末端C5b~9补体复合物水平下降。皮质激素治疗并不能改善预后。目前动物实验已发现抗C5a抗体可抑制中性粒细胞趋化因子,保护E.coli引起的动物ARDS和败血症休克。体外实验显示,抗B因子单克隆抗体可阻断补体激活,从而保护内毒素休克。但这些作用尚需进一步研究。
参数原理
2023.05.09

补体水平与内毒素血症的预后
关于败血症患者的补体激活与发生ARDS,MOF的相关性,研究结果各异。一些研究者发现,患者进入ICU时的补体蛋白水平与发生ARDS或死亡的危险性密切相关,而另一些研究则未发现有任何相关。Hach等证明,过敏毒素C3a、C4a的水平升高与败血症死亡相关,败血症休克患者的C3a水平比患败血症而血压正常的患者高,但在发展为ARDS的患者与无ARDS的患者之间过敏毒素水平并无差异。Weinburg等也认为,去精C3a,或去精C5a,或白细胞聚集能力与急性肺损伤之间并无关系。Slotman等发现,补体激活后释放去精C3a和去精C5a,且败血症休克患者的血浆去精C3a浓度比患败血症但不伴低血压的患者以及低血容量性休克的患者高,但去精C5a的浓度无显著差异。Kellerman发现,患败血症的ARDS患者的去精C3a浓度比患败血症但无肺部病变的患者高。但Parsons和Giclas发现,患败血症伴ARDS的患者和患败血症但不伴ARDS的患者之间补体水平并无差异。另一些研究证实,如果动态随访补体活性,则可发现ARDS的发展和补体激活之间密切相关。而Langlois更认为,末端补体复合物可用作败血症患者是否发展为ARDS的预测因素。败血症伴MOF的患者以及败血症康复的患者入院时,末端C5b~9补体复合物的浓度均升高,但两者之间却无显著差异。而随访1周后发现,前者血浆末端C5b~9补体复合物水平仍高,而后者则已趋正常。所以,补体活性可作为败血症伴MOF的随访指标。Parsons等研究了补体激活和循环中的内毒素水平与发展为ARDS的关系。他将患者分为ARDS组和ARDS高危组,发现C5a分裂产物的释放与ARDS之间无任何关系,约89%的ARDS组患者C3分裂产物增加,但62%的ARDS高危组患者C3分裂产物也增高;74%的ARDS患者血中可检出内毒素,而仅22%的ARDS高危组患者血浆中有内毒素。提示内毒素和补体分裂产物在ARDS的发展中起重要作用。即使低浓度的脂多糖也能增加C5a刺激的中性粒细胞释放过氧化酶和弹性酶,提示补体激活和内毒素的存在呈正相关。
参数原理
2023.05.08

内毒素与补体激活
补体激活是机体的一种防御机制,如通过补体可杀灭细菌,但补体在激活的同时也引起机体的一系列级联反应,形成多种补体分裂产物如C5a等,后者可增加血管通透性,释放多种生物活性物质,趋化白细胞聚集,从而引起脏器功能损害。在难治性感染性休克中,因有高浓度的内毒素导致的难以控制的补体激活,可严重威胁生命。体外实验证明,血浆与脂多糖一起孵育可激活补体级联反应,形成C3a和末端补体复合物。Roeise等发现,在含有内毒素的血浆中,C3a和TCC浓度明显增高。不同类型的细菌均可引起补体激活,并形成过敏毒素和末端补体复合物。将不同浓度的大肠杆菌在正常人新鲜血清中孵育发现,血清C3a、C5a和末端C5b~9补体复合物的形成和释放量与大肠杆菌的量相关。而在人类实验性内毒素血症中发现循环的TNF水平增加,随即出现中性粒细胞减少,而无补体级联反应的激活。动物实验亦证实,内毒素可引起补体的激活,并引起循环障碍和ARDS。现已证明,在多种动物模型中输入细菌或内毒素均可引起补体激活,形成过敏毒素C3a,C5a和末端补体复合物,且C5a的量与内毒素量成正相关。实验证明,过敏毒素可增加平滑肌收缩,增加血管通透性,并使肥大细胞释放组胺。C3a和C5a分子一旦在血中形成,就转变为致痉挛的去精C3a(C3a desArginine)和去精C5a(C5a desArginine)。C5a是白细胞的潜在激动剂,具有趋化作用,并促使巨噬细胞和中性粒细胞分泌溶酶体酶,使巨噬细胞产生白细胞介素和前列腺素;而去精C5a能刺激中性粒细胞趋化因子。Smedegard等证明,给小鼠注入内毒素,其血中去精C3a和去精C5a增加;注入内毒素和(或)去精C5a后可导致循环出现低血压,低灌注,血循环中多形核白细胞、单核细胞和血小板数量减少。Haugen等将E.coli注入猕猴体内引起败血症,发现其血中C3、C4、C5水平减少,而血中去精C3a和去精C5a水平增加,引起严重的败血症休克和肺水肿,75%动物死亡。这些实验证实,C5a和去精C5a 在败血症呼吸衰竭和休克的发展中起重要作用。Otson等通过结扎小肠和盲肠诱导败血症,并评价,C5健全的小鼠和C5缺陷小鼠之间在生存时间、动脉血氧分压、毛细血管内粒细胞捕获(trapping)、气-血屏障厚度上存在不同:C5缺陷小鼠的气-血屏障厚度,血氧分压正常,生存时间较长;而C5健全小鼠气-血屏障厚度增加,血氧分压水平较低,毛细血管内白细胞捕获增多。这些现象都提示,C5和C5裂解产物(如C5a及去精C5a等)在败血症肺损伤的病理过程中起重要作用。补体激活也常见于多种疾病的患者中。补体激活后可形成过敏毒素和末端补体复合物,最明显的补体激活见于败血症、败血症休克、多发创伤、急性胰腺炎和进行心肺旁路手术的患者。败血症患者存在内毒素血症,多数研究均证明败血症患者血浆补体蛋白C3、C4和总补体活性降低与败血症休克相关。最近10年来的几个研究亦证明,败血症和败血症休克患者有补体激活,C3a、C4a、C5a和末端C5b~9补体复合物的形成与败血症和败血症休克相关。Weinburg 等观察了25名血培养阳性的败血症患者发现,其血浆过敏毒素水平均升高,且去精C3a和去精C5a水平亦升高。Solomkin在研究腹腔内感染的患者时发现其血浆C5a水平比对照组高;而Brandtzaeg 等则发现末端补体复合物的形成与患者入院时的血浆内毒素血症相关。一些研究还发现,败血症休克患者的补体蛋白水平明显下降。如Sprung等就发现,败血症休克患者的C3、C4及B因子水平比正常对照组低,且其中死亡患者的C3及B因子的水平比存活的患者低。而另一些研究者则发现,败血症休克患者血中的补体蛋白浓度并无下降,且在死于败血症和败血症后恢复的患者之间血中的补体蛋白浓度亦无差异。如Leon等发现,败血症休克患者血浆CH50、C3、C4 水平明显下降,但死于败血症的患者和康复患者之间的补体水平无差异。
参数原理
2023.05.06

内毒素与补体:补体级联反应的介绍
内毒素与补体:补体级联反应的介绍补体的级联反应传统分为经典途径和旁路途径,近年来按照其功能将补体级联反应分为三个部分。1.第一前端反应第一前端反应指由C1到C3的反应顺序,也称C1途径,即传统的或经典的补体激活途径的前端部分。参与的补体成分为C1(C1q、C1r、C1s)、C4、C2及C3,受到C1抑制物(C1-in)等因子的调节。2.第二前端反应第二前端反应由C3、C3b、B因子,D因子等几个成分参与,受备解素(P)等的调节,也称备解素系统、C3旁路、替代途径等。由于第一和第二两个前端反应序列在C3交汇,因此C3常被称为补体系统的中心分子。3.末端补体复合物(terminal complement complex,TCC)由第一前端反应形成的C3转化酶C4b2a,以及由第二前端反应途径形成的C3转化酶C3bBb,在另一些C3b分子存在的情况下,可分别形成第一途径的C5转化酶(C4b2a3b)及第二途径的C5转化酶(C3bBb3b或C3bnBb)。这两种酶可将C5裂解为大、小两个片段C5b及C5a,C5b上有C6受体,可结合成C5b6,以后陆续与C7、C8及多分子C9结合,最后形成C5b~9,即TCC。TCC若在膜上有多个C9分子参与,则形成(C5b~C8)-(C9)n,即膜攻击复合体(membrane attack complex,MAC),可使靶细胞溶破从而被消除(图14-1)。血浆中的补体级联反应激活后,血浆中的补体固有成分被消耗,活性降低,或补体分裂产物增加,故检测补体各种成分及分裂产物的活性可反映补体系统激活的程度。补体活性常用以下几种方法来检测,需要注意的是备测的血浆标本必须为新鲜的或保存在-80~-70℃的温度下,如保存温度过高、再溶化或使用血清标本均可引起补体级联反应的自发激活,造成补体分裂产物的假性高值以及补体固有成分的假性低值。总补体测定可采用滴定的方法,其活性以每毫升未稀释血清的溶血单位的50%来表示。在补体中加入结合了抗羊红细胞抗体的羊红细胞,应用光度检测法估计部分溶血的程度作为完全溶血的百分比。血浆或血清中各种补体固有蛋白(如B因子,D因子、C1,C3、C4,C5、C6、C7、C8和C9)和调节因子(如因子1、C1-in)的浓度可用免疫技术测定,如放射免疫扩散、火箭免疫电泳沉淀、双向火箭免疫电泳沉淀等,过敏毒素(C3a、C4a和C5a)亦可用放射免疫检测方法测定,其改良方法的试剂盒有市售,这种方法是将EDTA-血浆与盐酸混合,因为C3或C5在酸性环境下变性沉淀,而对酸稳定的过敏毒素仍保持溶解状态。通过检测C3a或C5a可分析补体激活的程度,而测定C4a则可鉴别经典抑或旁路激活途径。亦有采用ELISA方法检测过敏毒素的。近20年来开展了多种用以定量测定血浆末端C5b~9补体复合物的免疫测定方法研究。补体的级联反应是一个连续的过程,而补体的第一前端反应一般需要激活才能顺利进行,即C1r与C1s从酶原形式转变为有活性的蛋白酶C1r和C1s,这种激活分为自发性激活和激活物激活。自发性激活的速率较慢;而激活物激活反应迅速,反应量大,其激活物又可分为免疫性激活物与非免疫性激活物。免疫性激活物一般指免疫复合物,非免疫性激活物种类多样,按其化学性质可分为蛋白质类、糖类、脂类等。目前常依据其与C1q相互作用抑或与非C1q相互作用而分为两大类,革兰阴性细菌的内毒素LPS等就是已知的与C1q反应的非免疫激活物,纤维蛋白溶酶则是与非Clq反应的非免疫性激活物。下面介绍一下内毒素激活补体级联反应的过程和结果。现已知道,C1由C1q、C1r、C1s三种亚成分组成。其中,C1q是补体激活途径的识别单位,是补体激活剂的受体,C1r 与C1s则为酶原。内毒素就是通过与C1q的胶原样区相结合,进而激活C1r、C1s酶原变为C1s的。后者为一种丝氨酸蛋白质,催化C4b和C2成为C4b2,经过如图14-2所示的途径形成过敏毒素(C3a、C5a)和末端补体复合物。
参数原理
2023.05.06

内毒素与补体之补体的概况
内毒素等可激活补体级联反应,补体激活则形成过敏毒素C3a,C5a,以及末端补体复合物。多数研究表明,高浓度的过敏毒素与急性呼吸窘迫综合征(ARDS)或多器官功能衰竭有关。亦已发现补体固有成分C3和C4对内毒素休克有保护作用,末端补体复合物可溶破和消耗靶细胞,从而起到保护机体的作用。补体是一类重要的免疫分子。19世纪80年代,Grohman和Buchner发现血浆和新鲜血清能杀灭细菌,并命名具有这种杀菌作用的物质为防御素。1894年,Pfeiffer在豚鼠体内发现溶菌现象,并鉴定防御素对热敏感,55℃30min即被灭活,故认为这种溶菌活性是血清酶的作用。其后,Bordet发现在加热灭活的血清中加入新鲜血清可恢复杀菌作用,并认为血清的杀菌作用需要两种不同的物质:一种是耐热的,因免疫而作用增强,并特异性地与免疫原发生反应,即现在所知的抗体;另一种不耐热,在免疫与不免疫血清中均存在,提示是一种非特异的补充成分,被命名为补体。以后又证明,防御素与补体为同一成分,统称为补体。各种动物红细胞抗体加补体可引起免疫溶血现象,所以认为补体为血清的单一成分。直到20世纪初,Ferrata首先发现补体成分并不单一,透析新鲜血清可得到非水溶性的优球蛋白部分和水溶性的假球蛋白部分。这两部分单独都不能使已结合抗体的红细胞溶解,如先与优球蛋白部分结合,再与假球蛋白部分作用可导致溶血;如次序相反,则无溶血现象。由此可见,补体的溶血作用是由两种成分依次反应引起的,优球蛋白和假球蛋白部分分别被命名为补体第一成分(C1)和补体第二成分(C2)。后来又陆续提出了补体第三成分和补体第四成分,直至20世纪50年代才确定这四个补体的反应顺序为C1→C4→C2->C3。50年代至60年代发现C3也并非单一成分,而是由六个因子组成的复合体,即由C5、C6、C7、C8与两个C9组成的补体末端复合物。60年代,研究者发现C1也非单一成分,是由C1q、C1r及C1s三个亚成分组成的。以上所述即为补体经典途径的11种成分。1954年,Pillemer等在血清中发现一种非抗体成分,命名为备解素(即现在所称的B因子),它可与一些多糖类物质作用,不经补体前段成分C1、C4 及C2而直接活化C3,并经补体末端效应成分C5~9,即通过补体活化的第二途径而溶破靶细胞。以后,研究者又逐渐认识了C1r、C1s及C4在本质上属于酶成分,发现了补体调节因子如C1抑制物(C1-in)、1因子、备解素(P)以及补体成分的cDNA克隆等。补体的成分是蛋白质,补体的激活需经蛋白水解发挥作用。补体通过肽链的有限蛋白水解,产生一些新的片段,其中有些直接表现某种功能,有些则再行组装,形成一些新的复合酶,再进一步进行有限的蛋白水解,从而形成复杂的级联反应。
参数原理
2023.05.06

内毒素与补体的关系
内毒素等可激活补体级联反应,补体激活则形成过敏毒素C3a、C5a,以及末端补体复合物。过敏毒素可引起平滑肌痉挛收缩,增加血管通透性,促使细胞释放多种生物活性物质,如组胺、前列腺素(PG)、白细胞三烯(LT)、5-羟色胺、白细胞介素-6、溶酶体酶等,并促进白细胞黏附与聚集,促进白细胞与血管内皮的黏附、游离出血管及局部集中,并具有强力的趋化作用,其中尤以C5a作用最为强烈。多数研究表明,高浓度的过敏毒素与急性呼吸窘迫综合征(ARDS)或多器官功能衰竭有关。亦已发现补体固有成分C3和C4对内毒素休克有保护作用,末端补体复合物可溶破和消耗靶细胞,从而起到保护机体的作用。所以,内毒素可激活补体,并与补体激活后引起的脏器功能损害及衰竭有关,同时补体亦具有杀菌和保护内毒素休克的作用。
参数原理
2023.05.05

内毒素血症致氧自由基产生的机制介绍
一、儿茶酚胺增加内毒素血症或败血症时,交感-肾上腺髓质-儿茶酚胺轴亢进,分泌大量的儿茶酚胺,目的是重新恢复血流动力学的稳定。但体内过多的儿茶酚胺可自动氧化,提供电子,产生氧自由基。在生理情况下,儿茶酚胺自动氧化的速度很慢,故对机体影响不大。内毒素血症时,儿茶酚胺释放增多,特别是伴有休克时,儿茶酚胺释放大大增加,并伴有组织缺血缺氧、酸中毒,以及儿茶酚胺氧化速度加快、氧自由基大量形成等。二、补体的作用对补体与自由基的生成无系统的研究,但部分资料表明,补体可影响活性氧的产生。某些补体成分可改变细胞的代谢,如果影响到线粒体的呼吸,氧自由基的产生就会增多。研究认为,通过血清加热处理灭活补体,可使中性粒细胞产生氧自由基的量较对照组降低90%以上。如血清中缺乏C3和B因子,可使氧自由基的产生分别降低56%和68%,而其他因子如C19及C2、C4~7等的缺乏则对氧自由基的产生没有影响。因此,补体与氧自由基产生的关系密切。相反,氧自由基也可激活补体,形成互为因果的关系,内毒素血症及败血症时,补体系统被激活。在内毒素血症时,内毒素的功能类似一个垫木,C3和B因子以及A蛋白在上面发生复合物的互相反应,产生能裂解C3的复合物,并由此引发补体替代途径。C3a和C5a的释放使中性粒细胞在内脏血管中聚集,引起组织缺氧,氧自由基产生。中性粒细胞的呼吸爆发过程也能产生大量自由基。三、多型核白细胞的作用血液和组织的吞噬细胞在使机体免受各种毒素及微生物侵入时起着非常重要的防御作用,当G-杆菌及其代谢产物(内毒素)侵入机体时,可激活这些吞噬细胞(中性粒细胞、单核细胞、巨噬细胞及其他多型核白细胞)。这些细胞在吞噬过程中能产生大量的氧自由基,其目的是杀死侵入的微生物,但若侵入的细菌或异物数量较大,就会导致细胞死亡,损伤组织细胞。四、黄嘌呤氧化酶体系内毒素血症或休克时,组织血中缺氧,组织细胞有氧氧化受抑制,而无氧酵解增加,这与内毒素抑制有氧氧化关键酶及促进无氧酵解酶有关,最后使ATP生成减少。同时,由于机体处于应激状态,代谢亢进,所需ATP增多,因此消耗也增多,ATP也不断分解为ADP、AMP、腺苷和次黄嘌呤。由于能量缺乏,各种需能活动受影响,钠泵、钙泵功能障碍,并造成Na+、Ca2+内流增加。细胞内钙离子增加可激活蛋白水解酶,后者可将组织细胞中的黄嘌呤脱氢酶转为黄嘌呤氧化酶。五、磷脂-花生四烯酸途径内毒素可直接激活磷脂酶A2,内毒素血症时,胞质中Ca²+内流,也可激活此酶,最后使磷脂酶A2活性增加,膜磷脂降解,花生四烯酸释放增多,同时在环氧化酶作用下生成前列腺素(PGE2、PGH2、PGI2等)。在这个过程中伴有大量自由基的产生。花生四烯酸在脂加氧酶作用下生成白细胞三烯等,在12-脂加氧酶作用下生成羟十二碳四烯酸(12-HETE)。另外,还有许多途径影响自由基的产生。总之,内毒素血症时,可通过吞噬细胞、黄嘌呤氧化酶及磷脂酶以及补体等使组织及血流内的氧自由基含量增加,脂过氧化反应加剧,最后损伤组织细胞,产生严重后果。
参数原理
2023.05.05

内毒素血症时氧自由基产生的证据
自由基在内毒素血症中对各组织细胞损伤的作用已被广泛而深入地研究。尽管对内毒素血症时自由基损伤的程度尚未得出最后结论,但许多研究已证实,自由基确实参与了内毒素血症的组织损伤作用。目前研究自由基是否参与某一疾病的措施主要有两个,即:①直接分析组织和血浆中的自由基。②采用自由基清除剂及抗氧化剂。一、直接测定由于氧自由基的半衰期甚短,因而使直接测定组织或血浆中的自由基非常困难。Baldwin报道,采用直接测定法测得急性肺损伤患者组织H2O2水平明显升高。之后,Sznajder等也报道脑损伤及败血症患者血浆中的H2O2水平也增高。二、间接测定由于直接测定自由基困难复杂,因此人们常应用间接测定法来研究氧自由基。因为体内到处均存在着抗氧化系统,而自由基的产生必须伴有体内抗氧化剂的减少,因此分析体内抗氧化剂的含量同样能推断自由基产生的量。1.抗氨化剂分析结果最有代表性的抗氧化剂是维生素E,抗休克成功者休克模型中发现维生素E水平可回升至正常水平。Takeda等对重危手术患者的研究表明,患者血浆维生素E水平为5~6mg/L,与维生素E缺乏患者的水平相似。另外,研究者还发现﹐硫巴比妥酸反应物(thiobarbituric-acid-reactive substances,TBAR-S)的血浆水平与脂过氧化反应一致。急性肺损伤患者中也发现了类似情况。有报道,严重烧伤患者血浆中的5,5-二硫-2-硝基苯酸反应物(5,5-dithio-nitrobenzoic-acid-reactive substance,DINB)疏基及维生素E水平均下降。败血症及MODS患者的血浆酮蛋白水平降低、TBAR-S水平增加。2.脂过氨化产物测定氧自由基能攻击生物膜磷脂中的多不饱和脂肪酸(polyunsaturated fatty acid,PUFA)而引发脂质过氧化作用,并形成脂质过氧化物(lipid peroxide,ROOH),其中最主要的是氢过氧化物和内过氧化物。在过氧化条件下,脂质过氧化物极不稳定,能分解成一系列复杂产物,包括氧自由基。因此,应同时监测数种产物。除乙醛外,估计磷脂上的羟基也是脂质过氧化的指标之一。创伤、烧伤患者及动物模型的血及组织中的脂过氧化产物可有增加。大鼠注射内毒素后,血浆中TBAR-S水平暂时升高。Peavy及Fairchild发现,在内毒素注射后,小鼠有TBAR-S水平增高,其呼出的气体中有乙烷。内毒素血症绵羊的动脉血中有结合双烯(conjugated dienes)水平增加,但在静脉血中却未见有双烯水平增加。注射高剂量内毒素后,绵羊肺组织中有TBAR-S的增加,但低剂量及中等剂量内毒素则不引起肺组织TBAR-S水平的变化。重危患者,特别是伴有ARDS的患者血中H2O2浓度则增加。三、氧自由基清除剂应用后的证据由于直接或间接测定氧自由基困难,多数提示氧自由基参与各疾病的组织损伤证据均来自氧自由基清除剂,即抗氧化剂及离子鳌合剂,因为这两种酶可防止自由基的连锁反应,但由于其半衰期较短,应用时困难较多。研究表明,SOD不能减少低血容量性休克动物的维生素E及疏基水平,不能使增加的TBAR-S水平降低,也不能阻止各组织的损伤。SOD对内毒素血症及败血症患者应用的结果不一,许多研究者报道SOD对猪、兔及大鼠的呼吸衰竭无作用,而在绵羊中有进一步使病情恶化的趋势。相反,另一部分报道认为SOD对内毒素诱导的肺水肿大鼠及小鼠有用。过氧化氢酶的作用也是如此,其对绵羊有保护作用,但对山羊则无作用。尽管上述的研究结果不同,但对脂质过氧化产物的检测则发现应用SOD或过氧化氢酶后可减少脂质过氧化反应。四、离子螯合剂应用后的证据由于·OH的生成是金属依赖性的,因此金属离子鳌合剂主要应用于阻止·OH的产生。尽管许多研究表明离子螯合剂(deferoxamine)对失血性休克有益,能增加实验狗的生存率,且对其他创伤所致的休克也有一定保护作用,但对内毒素血症及败血症的动物则无效。五、黄嘌哈氧化酶阻滞剂及合成抗氧化剂证据研究显示,别嘌醇(allopurinal)及氧嘌呤醇(oxypurinol)对缺血-再灌注动物有明确的保护作用,可能是减少了氧自由基对肺组织的损伤或阻止了十二烷基释放的结果。N-乙酰半胱氨酸(N-acetylcysteine,NAC)能保护内毒素诱发的肝损伤,也可能与减少自由基有关。总之,氧自由基参与了内毒素血症的组织损伤作用,但其在组织损伤作用中的地位还不甚清楚。
参数原理
2023.05.05

科德角国际 | 细菌内毒素检测技术应用及PKF型细菌内毒素定量检测系统实操培训
多年来,科德角国际生物医学科技(北京)有限公司始终专注于细菌内毒素检测服务,积累了丰富的细菌内毒素检测经验,为了进一步帮助药品检验检测机构和相关制药生产企业提升细菌内毒素检测能力,我司于2023年5月30日-2023年5月31日举办“科德角国际细菌内毒素检测技术应用及PKF型细菌内毒素定量检测系统实操培训”。一、培训组织主办单位:科德角国际生物医学科技(北京)有限公司协办单位:北京阿克庇斯医药有限公司二、培训对象(一)各省 (区、市)药品审评中心、核查中心、药检(院)所相关人员;(二)制药企业、研发公司、CRO 公司、高等院校、科研院所等相关专业人员。三、培训时间报名时间:2023年5月4日-2023年5月29日报到时间:2023年5月29日培训时间:2023年5月30日-2023年5月31日四、培训地点科德角国际生物医学科技(北京)有限公司北京市大兴区中关村科技园区大兴生物医药产业基地华佗路50号院18幢五、培训内容※本次培训结业学员,将由科德角国际生物医学科技(北京)有限公司颁发培训合格证书。六、培训讲师范玉明科德角国际资深技术总监【专业及专长】药理学、毒理学及药事管理擅长细菌内毒素检测领域的研究医学硕士研究生,执业药师,编辑,GLP 、GMP 、GCP 培训证书七、公司荣誉细菌内毒素检测实验室ILPQ国际能力认证 中国食品药品检定研究院能力验证结果报告通知单 2022年度细菌内毒素LGC能力验证八、实验环境九、报名方式扫描下方二维码进行报名▲扫描二维码进入报名页面十、培训费用2000元/人(包括资料费、培训费、证书费、午餐费,其他费用自理)。地址:北京市大兴区中关村科技园区大兴生物医药产业基地华佗路50号院18幢科德角国际生物医学科技(北京)有限公司北京市大兴区中关村科技园区大兴生物医药产业基地华佗路50号院18号楼2层
企业动态
2023.05.04

去离子LPS的特性
脂多糖(lipopolysaccharide,LPS)在电透析后以酸性形式存在。这种酸性LPS在双蒸水中溶解度极低,如其是R-LPS可表现为完全不溶。酸性LPS经加热(100℃)后LPS发生裂解,多糖与类脂A分子间的糖苷键可发生断裂,上述两个部分便可得到分离。去离子后形成的酸性LPS可直接以碱中和而制成各种不同的LPS 盐。LPS电透析时透析出的主要物质为无机离子及胺类。无机离子为Na+,K+ ,Ca2+,Mg2+,Fe2+及Fe3+,其中Na+的含量最高,而仅发现有微量的Fe2+及Fe3+析出。胺类物质有乙醇胺、腐胺、精胺、亚精胺及尸胺,但一般未发现有携带负电荷的物质被析出。
参数原理
2023.04.28

LPS盐的水溶性介绍
电透析后直接得到的酸性脂多糖(lipopolysaccharide,LPS)可分别以不同的碱中和而取得各种相应的LPS盐。这些LPS盐在双蒸水中的溶解度差异很大。迄今为止,LPS的三乙基乙胺或四乙基乙胺盐在水中的溶解度最大。R-IPS的三乙胺盐在水中的溶解度可达20mg/ml,此时形成一种透明、无粘度的溶液。S-LPS的三乙胺盐溶解度更大,可高达400mg/ml。LPS钠盐的溶解度稍差,特别是R-LPS钠盐。形成溶液的透明度亦差于三乙胺型LPS盐,另外,LPS钠盐的粘度亦增加。LPS钾盐的溶解度稍好于钠盐,而LPS吡啶盐及铵盐的溶解度则差于LPS的钠或钾盐。LPS的钙及镁盐表现出极低的溶解度,这一点尤以R-LPS为明显。LPS的镁盐常常是不溶解性的。另外,LPS的有机盐,如1LPS的腐胺或精胺盐亦表现出极低的溶解性。
参数原理
2023.04.28

LPS的电透析原理介绍
电透析(electrodialysis)的原理在于:在电场作用下,脂多糖(lipopolysaccharide,LPS)溶液中存在的小分子量带电物质(离子或分子),可以从LPS中离解出来,从而达到去除这类物质的目的。目前所常用的电透析仪包括两个组成部分:一为市售的电泳仪;二为自制或市售的电透析槽。电透析槽由3个相互分隔开的小槽组成,即中间槽及两个侧槽。两个侧槽中装有铂电极而与电泳仪相连,中间槽装置有冷却系统以保证在电透析条件下LPS溶液控制在10℃以下。中间槽放置人一定浓度的LPS溶液,而两个侧槽注入双蒸馏水。3个槽之间以透析膜隔开。在电流作用下,LPS中存在的带电小分子物质通过透析膜而分别趋向两个侧槽中的正负电极。电透析时观察到阴极pH值上升,而阳极pH值不变或仅有轻微的下降,证明LPS中含有多量带正电荷的物质和很少或不含带负电荷的物质。当阳极pH值上升至9~10时,调换侧槽中的双蒸馏水。这一步骤在整个电透析过程中需重复数次。在电透析过程中LPS溶液的pH值常常下降,同时LPS使形成不溶性物质。为了很好地达到去离子的目的,LPS可以用三乙胺或NaOH中和至pH为7.0左右,而使LPS重新溶解,这一步亦需重复2~3次。电透析结束时,LPS一般以酸性的形式存在。此时可以不同的碱中和而取得所需的LPS盐,如以NaOH中和则取得LPS钠盐,以三乙胺中和则取得LPS三乙胺盐。电透析后LPS可直接进行冷冻真空干燥,此后保存在-4~-20℃,每次使用前再转化成需要的盐。
参数原理
2023.04.28

内毒素信号转导分子诱导性改变的介绍
(一)TLR4分子表达下调将小鼠腹腔巨噬细胞用内毒素预先处理后,再次用内毒素攻击,则此时细胞因子的分泌显著减少,表现出时间和剂量依赖性的特点。在耐受的巨噬细胞中证实,依赖于TLR4-MyD88信号途径的近侧信号转导分子受到影响。用小剂量内毒素刺激巨噬细胞后数小时内,TLR4 mRNA表达显著下调,24h后才恢复正常水平,而膜表面上TLR4分子在1h开始表现出渐进式下降,其抑制性状态持续超过24h。此时的细胞因子分泌下降与TLR4表达下调有关,也是内毒素耐受的发生机制之一。在内毒素耐受中,TLR4的基本调控因子PU.1和干扰素基因序列结合蛋白(interferon consensus sequence binding protein,ICSBP)是如何相互作用影响Tlr4基因转录的目前还不清楚。(二)IRAK分子改变IRAK为IL-1受体的信号转导分子,现证实其也参与TLR家族的信号转导。过量表达IRAK的显性失活形式能抑制LPS的信号转导,而且在lRAK基因缺陷的293细胞中转染野生型IRAK能使细胞对LPS发生反应。Li等对THP-1细胞进行内毒素攻击时,发现内源性IRAK能够被快速激活,初次内毒素刺激时,LPS可促使IRAK与MyD88迅速结合;在内毒素耐受的THP-1细胞中发现,IRAK表达数量显著下降,只有正常水平的20%,在再次内毒素攻击时,无法诱导出IRAK的酶学活性;同时,IRAK与MyD88发生分离不能结合,无法转导LPS的跨膜信号。可见,IRAK从量和质的两个方面下调内毒素的激活效应。(三)NF-κB复合物分子组成的改变内毒素耐受细胞若再次受到内毒素刺激,则不能有效激活NF-κB。未激活的巨噬细胞、NF-κB组成异源二聚体(p50和p65)形式,并与抑制性IκB结合,滞留在胞质内。当细胞初次受到内毒素刺激后,IκB迅速被IκB激酶(IKK)磷酸化,并经泛蛋白-蛋白质酶体的途径降解。在内毒素耐受细胞中,NF-κB复合物主要为p50/p50,后者缺乏反式转录活性,并能抑制基因表达。p50的前体蛋白为p105,经过酶切生成。在内毒素耐受细胞中,由于p105合成显著增加,p50与p50形成二聚体,而p65 mRNA无改变,故不能诱导p65蛋白表达增加,所以p50/p50占优势,p65/ p50比例下降,并抑制相关基因表达。(四)IκB激酶的改变内毒素耐受的细胞中IKK不能被激活,结果IκB无法降解,持续与NF-κB结合,而NF-κB复合物不能从胞质转位进入胞核内使其调控基因表达。可见,IκB激酶也参与了内毒素耐受的发生。(五)蛋白激酶C的改变内毒素可以激活不同的致分裂原活化蛋白激酶(rmitogen-activated protein kinase,MAPK)的级联反应,包括细胞外信号调节蛋白激酶、JNK(c-Jun N-terminal kinase),p38MAPK/反应激酶途径(p38 MAPK/reactivating kinase pathway)。MAPK可以使下游分子的丝氨酸/苏氨酸发生磷酸化。有活性的细胞外信号调节蛋白激酶使下游分子磷酸化并调节其活性,其中包括其他蛋白激酶、细胞骨架、磷脂酶A2和核转录因子(如Elk1/TCF及c-Jun),调节即刻早期基因的表达。内毒素可激活PI-3K,后者分解膜上的脂质后产生DAG和IP3,IPs进一步激活PKC,并发生多种效应。在内毒素耐受细胞中,使用PKC的激活剂如佛波酯,能恢复细胞因子的合成和分泌,可见PKC也参与内毒素耐受效应。(六)G蛋白与内毒素的耐受用百日咳毒素使巨噬细胞G蛋白亚单位Gi的近C端Cys残基发生核糖基化,修饰后的Gi对受体介导的信号转导无反应而处于持久失活状态,此时用内毒素进行刺激可显著降低细胞因子的合成和分泌。可见G蛋白也参与了机体对内毒素反应的调节。总之,在天然内毒素耐受之外,宿主作为一个整体,其中有多种成分共同参与内毒素耐受的发生,而并非某一个成分单独发挥作用,这也表现出了机体反应的协调性。一旦某个成分逃脱抑制的束缚,则会破坏整个耐受的平衡状态,使耐受现象消失,并摆脱原有的耐受状态,继而下传LPS信号转导,对机体产生效应。
参数原理
2023.04.27

内毒素信号转导概述
内毒素信号转导的主要途径为:G-菌在崩解或繁殖时释放出脂多糖(lipopolysaccharide,LPS),进入宿主血液或细胞培养基中,并与血浆脂蛋白结合(HDL,乳铁蛋白等)或与脂多糖结合蛋白(LBP)结合。LBP的主要功能是使LPS聚集体解离为LPS单体,形成LBP-LPS复合物,然后将LPS 转递给单核-巨噬细胞、中性粒细胞等细胞膜上的mCD14(membrane-bound CD14)受体,并与之结合形成LBP-LPS-mCD14复合物,或者与游离的sCD14(soluble CD14)形成LBP-LPS-sCD14复合物,后者再将LPS转递给mCD14受体,或转递给无mCD14的细胞,如上皮细胞、内皮细胞。mCD14是以糖化磷酸肌醇(glycosylphosphatidylinositol,GPI)锚定在膜上的糖蛋白,其在单核-巨噬细胞中的表达极为丰富,每个细胞表面的分子数约为106~109个,其主要功能是结合和浓集各种LPS分子,但缺乏配体结合的特异性。跨膜型Toll样受体4(TLR4)在单核-巨噬细胞中约分布有103个分子,mCD14能催化LPS与TLR4的胞外亮氨酸富集重复体(leucine-rich repeats,LRR)结构发生物理接触,形成CD14-LPS-TLR4三元复合物,并诱导TLR4的胞内结构域发生空间构象改变,使TLR4受体二聚化。TLR4胞质区的TIR结构域可募集胞内衔接蛋白(adaptor)髓样分化因子88(MyD88)和白细胞介素-1受体相关激酶(interleukin-1 receptor-associatedkinase,IRAK),并锚定到TLR4的TIR结构上。MyD88的N端为一个死亡结构域(D-D),C端为一个TIR结构域,后者能与TLR4的TIR发生蛋白质-蛋白质相互作用,同时其D-D结构则与IRAK的D-D结构发生蛋白质-蛋白质相互作用,使IRAK发生自身磷酸化并因而具有酶学活性。后者作用于其下游衔接蛋白TNF受体相关因子6(TNF receptor-associated factor 6,TRAF6),出现信号分流,既可通过TGF-β激活激酶(TGF-β-activated kinase 1,TAK1),也可通过ECSIT,分别引发酶学级联反应,激活NF-κB、AP-1、Jun等多个转录因子,并促使相关基因表达。在低内毒素浓度时,MyD88的信号转导效应对CD14具有依赖性;而在高浓度时,则不依赖CD14,此时内毒素能通过CD11/CD18、衰变加速因子,膜外突蛋白(moesin)等受体进行信号转导。TLR4受体突变可导致内毒素耐受,使整个机体的细胞对内毒素的反应均显著低下,并只能诱导出微量细胞因子的表达。TLR4是内毒素进行信号转导的关键性受体,由于TLR4的胞外结构域在进化上具有多态性,故理论上能够区别不同的LPS 分子,从而对不同的LPS分子产生不同的效应。另外,当吞噬细胞吞噬G-菌或LPS聚集体时,可以在胞质内进行降解并释放少量LPS,也能与胞质内受体Nod1中的LRR结合,诱导其构象改变,也能引发酶学级联反应,激活NF-κB,使细胞因子表达。后者的途径为:LPS→Nod1→RICK-→IKK→NF -κB→基因表达。在小鼠Lps 基因的位点上,目前已鉴定出四个结构基因,包括TLR4和Ran(Ras-likenuclear G protein),Ran也可能涉及内毒素信号转导信号效应。其他受体如嘌呤受体P2X7以及G蛋白,K+通道蛋白、天然耐受相关巨噬细胞蛋白1(natural resistance-associated macrophage protein 1,Nramp1)等均以不同形式参与内毒素的信号转导,如此才使细胞因子的分泌达到最大化。
参数原理
2023.04.24

脂多糖(LPS)的提取与纯化
脂多糖(lipopolysaccharide,LPS)根据О-特异性侧链的存在与否可分为光滑型(smooth-form,S-LPS)用粗糙型(rough-form,R-LPS)两大类,前者的组成成分有类脂A、核心寡聚糖及О-特异性侧链,而后者仅含有类脂A及完整或部分核心成分。这两类LPS分子在某些物理特征上表现出显著的差异性,主要表现为表观分子量及水溶性方面。纯度对于研究LPS的物理、化学及生物学特性是至关重要的,因为细菌其他成分的污染可影响LPS的物理特性及生物学活性。LPS制剂中最常见的污染物质是核酸、蛋白质及多聚糖,这类污染物质存在的量取决于LPS的提取和纯化方法。70年代以前提取LPS最常用的方法为Westphal等于1952年介绍的酚水法(phenol water,PW),用这一方法提取的LPS纯度较高。污染蛋白质含量仅为1~3%,而在此以前所应用的方法(如三氯醋酸法,超声波法、正丁醇提取法等)所提取得到的LPS蛋白污染量可超过10%,因而PW方法是一种较为有效的方法。自1969年Galanos 等介绍苯酚-氯仿-石油醚(phenol-chloro-form-petroleum ether,PCP)法后,LPS的纯度又得到了显著的提高。一方面,经PW法提取得到的S-LPS可以PCP法纯化,另一方面,R-LPS由于脂溶性成分的含量较高,因而可直接应用PCP法进行提取与纯化。PCP法可去除LPS中常见的污染物质。最终得到的纯化LPS不含核酸及多聚糖,蛋白含量亦可控制在1%以下。LPS中存在的另一类污染物质为无机金属离子及小分子量多胺成分,它们的存在可影响LPS的物理性状及生物学活性。应用电透析可将这类成分除去。
参数原理
2023.04.23

内毒素作为一种特征成分在细菌外膜功能中所起的作用
我们已经越来越清楚外膜结构对于革兰氏阴性菌起着十分重要的生理作用。由于外膜的存在,使得细菌对机体的防御性体液因子,如溶菌酶、β-溶解素及许多白细胞毒性蛋白具有抵抗作用,而这类物质一般讲对于革兰氏阳性菌具有高度的毒性。存在于人或动物肠道中的革兰氏阴性肠杆菌其外膜得到了高度的发达,并成为一种有效的屏障结构,这样,这类细菌不易受到胆盐的去垢作用及消化酶的降解作用。另外,肠道及其它的一些革兰氏阴性菌的外膜,对于许多抗菌素如大内环类抗生素、新菌素,利福平、林可霉素、柯林达霉素及梭链孢酸等具有屏障作用。即使有微量的抗菌素进入细胞,细菌体内的酶系统亦较易将抗生素灭活,因而革兰氏阴性菌经常易建立对抗生素的高度抵抗性。细菌外膜这些功能与现代医院中革兰氏阴性菌的感染流行是密切相关的。细菌外膜的另一个重要的功能就是提供细菌表面高度的亲水性,这一点在防御机体吞噬作用、补体的攻击作用,以及通过膜表面抗原的改变而抵抗特异性免疫攻击等作用中有着十分重要的意义。当然,细菌外膜的这些功能与其中存在的内毒素亦有密切的关系,这也就是内毒素在外膜中所发挥的主要作用。
参数原理
2023.04.23

去垢剂对细菌外膜的作用
1、EDTA的作用:EDTA可减弱外膜中LPS分子间的相互作用从而损伤细菌外膜。实验证明,EDTA在Tris缓冲液存在下,可使外膜约50%的LPS分子释放出来,但并不影响细菌外膜的其它构成部分。这一处理的结果可使细菌对某些疏水性抗生素如放线菌素D、新菌素(novobiocin)等形成高度敏感状态,由于部分LPS被游离出来,外膜外叶中将出现原先由LPS占据的“空白”区域,这些空间可被磷脂所替代。这些填补的磷脂分子来源于外膜的内叶或细胞膜,这样所形成的外膜就成为一种类似于真核细胞膜的双层磷脂结构。这种膜结构的通透性大大地高于由LPS/磷脂所形成的膜结构。另一方面,由于EDTA可去除两价阳离子,从而减弱了LPS与磷脂分子间的相互作用,这样LPS分子间以及LPS与磷脂分子间便出现静电排斥反应,从而形成不稳定的外膜结构。另外,EDTA处理的细菌发现有游离的脂肪酸被释放出来,这是由于内源性磷脂酶受EDTA激活所致。2.多价阳离子的作用:多粘菌素B(polymyxin B,PMB)携带一个脂肪酸“尾巴”,整个分子携带5个正电荷但不带负电荷。PMB分子量太大不能通过外膜的透道,故而首先必须损伤外膜结构,才能进入到细胞膜而发挥抗菌作用。实验证明PMB与外膜结合后可在显微镜下发现外膜有广泛的结构变化,此时外膜对许多疏水性抗生素、去垢剂及溶菌酶的通透性增加。鼠伤寒PmrA突变株由于在外膜中LPS分子上含有多量的4-氨基阿拉伯糖及乙醇胺(均携带正电荷),可与PMB发生静电排斥反应,故而PMB不易透过外膜结构。这类突变株便成为多粘菌素抵抗菌株。寡聚赖氨酸(Lys20)及PMB的一种九肽衍生物(PMBN,PMB经胃蛋白酶处理后失去N-末端的二氨基丁酸及脂肪酸尾巴)对于野型鼠伤寒及大肠杆菌均没有明显的抗菌作用,但可以增加这些细菌对于疏水性物质如新菌素、梭链孢酸、红霉素、柯林达霉素、利福平、放线菌素D等的敏感性。Lys20的作用机理与EDTA相似。经Lys20处理的菌细胞可失去20%左右的LPS分子,从而影响了LPS/磷脂双层膜的结构与功能,但这一改变过程需时较长。而PMBN可直接与LPS结合(通过异性电荷的吸引)而变成LPS-PMBN复合物,这一复合物的形成增加了LPS在外膜中所占据的空间,从而影响外膜的结构及功能。这一过程的发生极为迅速。体内正常防御机制中存在的某些因子,亦可改变细菌外膜的通透性。一种为富含精氨酸的鱼精蛋白,微量时可增加外膜对去垢剂去氧胆酸及新菌素的通透性。另一种为从多形核白细胞颗粒中提取得到的“杀菌性通透性增加蛋白”(bactericidal permeability-increasing protoin,BPI),这一蛋白为强碱性蛋白,等电点为9.6。BPI可与外膜中的LPS结合从而增加外膜对抗生素的通透性。BPI对于粗糙型细菌的作用要比对野型细菌强得多,其原因可能是后者所含的LPS携带长链О-特异性多糖﹐从而BPI不易与LPS中的类脂A结合。
参数原理
2023.04.21
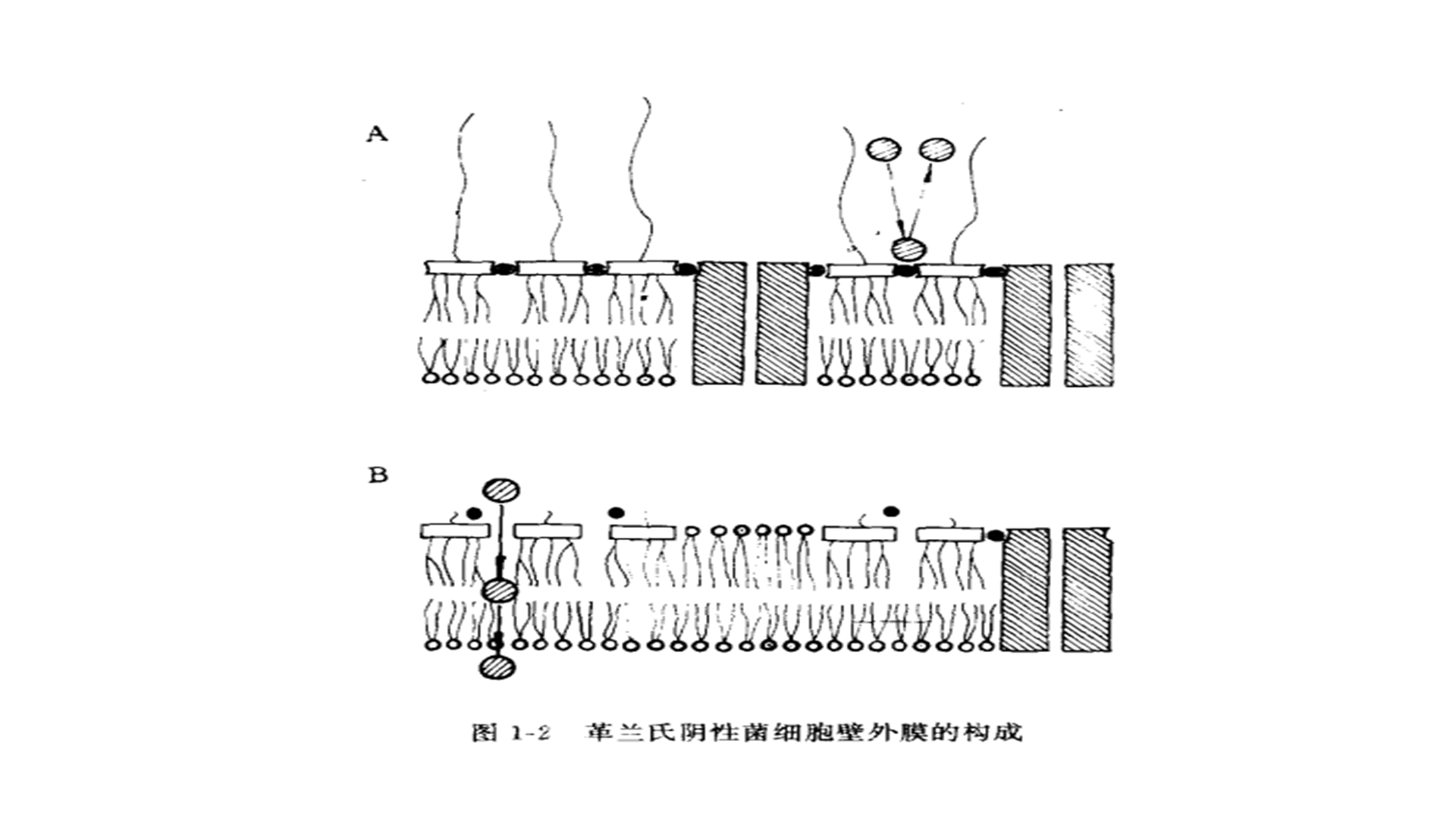
细菌外膜的分子构成
(一)内毒素在外膜中的位置细菌外膜与真核细胞膜一样,亦是由不对称的磷脂双层构成基本骨架。内毒素作为一种特殊的磷脂,全部存在于外膜的外叶中,应用抗体标记技术可观察到内毒素抗体仅结合于外叶上,而不与内叶结合。应用酶降解技术亦能证明这一点。对于单个菌细胞来讲,内毒素与磷脂具有相同数目的轻碳链(脂肪酷基)数目,因而它们在外膜中占据相同大小的区域。因此理论计算亦可证明外膜中的外叶全部由 LPS(及蛋白质)组成,而基本上不含磷脂。反之外膜的内叶则全部由磷脂所占据。这至少对于野型(光滑型)细菌来讲是完全可以肯定的。但是粗糙型细菌外膜的构成与光滑型菌株不一样,主要表现在粗糙型菌株外膜的外叶并不完全是由内毒素组成,而是由内毒素与磷脂共同组成,而粗糙型细菌外膜的内叶则仍然全部由磷脂(及蛋白质)组成(图1-2)。 A:光滑型(野型)菌的外膜,外膜外叶全部由LPS(及蛋白质)组成,而内叶则全部由磷脂(及蛋白质)组成。B:粗糙型菌的外膜,外膜外叶由LPS和磷脂(及蛋白质)共同组成而内叶则全部由磷脂(及蛋白质)组成。事实上,细菌外膜的这种非对称性结构在细菌的生理学上具有十分重要的意义。肠道菌多存在于含高浓度胆盐的环境中,而由内毒素组成的外膜外叶可阻止胆盐的进入而免遭破坏。而肠道以外的细菌则不需要具备对脂溶性物质高度的不通透性,从而这些细菌一般形成更为对称性的外膜结构。粗糙型细菌由于其外膜的外叶中存在一定量的磷脂,故而易受外环境的影响,因而其外膜不如野型细菌外膜稳定。(二)LPS分子间的相互作用LPS 是在细胞浆内合成的,最终被转运到外膜而成为细菌外膜的特征性成分。由于LPS仅存在于外膜的外叶中,故其间必定有什么特异性的机制参与而使LPS转运到外叶而不是内叶,这种机制可能涉及 LPS与磷脂及它们各自分子间的相互作用。已知LPS与磷脂有强烈的相互作用,但比较起来,LPS与LPS,磷脂与磷脂,亦即同源性物质间的相互作用要更为强烈,化学提纯的LPS由于其分子间具有强烈的相互作用,故而在一定条件下可形成LPS单层膜结构,如果加入生理比例量的磷脂,则便可形成+分稳定的双层膜区域结构。当然LPS分子间的相互作用还需两价阳离子(如 Ca2+,Mg2+)的参与。LPS远端的糖残基部分含有多量的负电荷,因而如果没有对应离子的介入,LPS分子间可发生静电排斥反应。天然条件下存在的LPS中含有许多阳离子(Na+、Mg2+、Ca2+及携带正电荷的胺类物质(尸胺、腐胺、精胺、亚精胺及乙醇胺)。应用电透析去除这些无机及有机物质,可使LPS的物理学性状乃至生物学性状发生改变。这类阳离子的存在对于外膜的构成起着十分重要的作用,如果应用鳌合剂去除外膜中阳离子则外膜即可遭到破坏。另一方面,加入Mg2+则可起到稳定外膜的作用。因而当培养基中加入适量Mg2+(0.15mmol/L)可抑制粗糙型及脂蛋白缺陷菌间周酶的漏出。(三)LPS与蛋白质的相互作用细菌外膜中存在的LPS与蛋白质分子可发生强烈的相互作用。应用酚水法提取到的LPS含有相当多量的蛋白质成分(可高达10%左右)使再通过凝胶过滤亦不能将这类蛋白完全去除这些蛋白质分子先前曾称为“LPS相关蛋白”(LPS-associate proteins),其主要成分为透道蛋白,或OmpA蛋白。另一方面OmpA蛋白可作为TuⅡ噬菌体的受体,但是在体外实验中,噬菌体并不能识别并与OmpA蛋白结合,仅在加入了定的LPS后噬菌体才能与这种蛋白结合,说明LPS是OmpA蛋白作为Tu菌体的受体所必需的。加入一定量的Mg2+和磷脂可促进 OmpA 蛋白与噬菌体结合。
参数原理
2023.04.21

热烈欢迎旭化成医疗器械(杭州)有限公司来我司审计
4月20日下午,旭化成医疗器械(杭州)有限公司一行三人到我司进行审计。公司各相关部门负责人全程陪同。旭化成医疗器械(杭州)有限公司一行三人参观了我司细菌内毒素检测实验室、P2实验室及仓库实验中心区域等。审计人员仔细询问了我司技术人员及设备的部署安排,对实验室的设备及环境要求等进行指导。随后,审计人员对我司细菌内毒素检测实验室环境、产品质量、出入库台账、收货流程、仓库管理、产品运输等情况进行了全面的安全审计,我司相关部门负责人积极配合客户,不仅按审计要求提供了相应的文件资料,还对审计人员提出的各种审查问题进行了细致解答。经过细致的现场考察、资料审查与深入的沟通交流,旭化成医疗器械(杭州)有限公司审计人员对我公司质量体系运行情况给予肯定,并对本次审计发现的问题提出了相关的整改建议,科德角国际对审计人员的重视和关注表示感谢。欢迎审计人员再次来我司审计,同时也欢迎广大客户来我司进行考察和指导。
企业动态
2023.04.21

白细胞介素-6的生物学作用
白细胞介素-6(interlenkin 6,1L-6)的生物学作用可用表10-1来表示。IL-6主要诱导活化后期的B细胞,使其大量合成分泌型Ig的mRNA从而增加Ig(IgM、IgG和IgA)的分泌。IL-6也能够作用于活化的B细胞,激活Ig重链(CU)启动子抗IL-6的抗体能够抑制美洲商陆有丝分裂原(Pokeweed mitogen,PWM)诱导的Ig产生。下列环节均需IL-6的参与:①在PWM诱导Ig产生的过程中。②在IL-4诱导的IgE产生的过程中。③在IL-2诱导的金黄色葡萄球菌活化B细胞产生Ig时。④在人B细胞产生抗多糖抗体时,以及在小鼠B细胞对流感A病毒的原发性免疫应答时。⑤在初次和再次免疫应答中,小鼠B细胞分化为产生特异性抗体形成细胞时也需要IL-6辅助。IL-6诱导IgA产生的作用不包括诱导抗体的类型转换。IL-6也与T细胞的活化、增殖和分化有关。IL-6启动胸腺细胞和被PHA激活T细胞的增殖和分化,且与IL-1和TNF有协同作用;还增加胸腺细胞对IL-4和PMA的增殖反应。IL-6对T细胞的作用有一部分由内源性IL-2介导,抗IL-2Ra链抗体能够抑制IL-6诱导的T细胞增殖。IL-1与IL-6有协同促进T细胞产生IL-2和IL-2R的作用,这可能是IL-6增强CTL功能的原因之一。IL-6还能够促进更多的丝氨酸酯酶(serine esterases)和穿孔素(perforin)的表达,后两者是CTL发挥毒性效应所必需的。虽然正常淋巴细胞的发育并不一定需要IL-6,但在IL-6缺陷的小鼠中,T细胞依赖性的抗体反应。CTL产生、巨噬细胞功能和急性期反应均明显降低。IL-6和IL-3在诱导小鼠多种造血前体细胞增殖方面有协同作用,其中IL-6诱导这些细胞从休眠期进入细胞周期,而IL-3促使离开G0期的造血前体细胞继续增殖。IL-6和IL-3共同刺激产生的骨髓细胞集落比单独由IL-3诱导的集落更容易在致死性放射线照射小鼠中进行骨髓重建。同样,IL-6也能够协同M-CSF和GM-CSF的作用。Fanconi贫血中出现的造血细胞分化缺陷与机体不能产生IL-6有关。IL-6也可协同IL-3诱导巨核细胞成熟,并能显著增加巨核细胞的体积和细胞内乙酰胆碱酯酶的活性。抗IL-6抗体可抑制体外培养的小鼠骨髓细胞发育成巨核细胞。人巨核细胞表达IL-6R并产生IL-6,可能通过自分泌作用调节其终末期成熟。小鼠或猴接受IL-6以后,体内血小板数量显著升高,合用IL-3则有协同效应。白细胞介素-6(interlenkin 6,1L-6)和病毒能够诱导神经胶质细胞、星型胶质细胞和小胶质细胞产生IL-6。腺垂体细胞和滋养层细胞能够自发产生IL-6。IL-6诱导PC12细胞分化为神经细胞,支持体外培养的胆碱能神经元存活,并直接刺激或通过增加促肾上腺皮质激素释放激素刺激肾上腺皮质激素分泌,刺激腺垂体释放诸多激素(催乳素、生长激素和黄体生成素等)。IL-6能影响与神经递质有关的酶表达。IL-6能保护纹状体神经元抵抗N-甲基-D-天冬氨酸(N-methyl-D-asparate,NMDA)的毒性,保护多巴胺能神经元抵抗1-甲基-4-苯吡啶(1-methyl-4-phenylpyridinium,MPP)的毒性。在神经损伤时,神经细胞表达IL-6和IL-6R,且Schwann细胞表达IL-6增加,但抗IL-6抗体能抑制神经重建。在转基因小鼠,IL-6和IL-6R能够促进切断的末梢神经细胞再生。白细胞介素- 6(interlenkin 6,1L-6)可促进杂交瘤、浆细胞瘤、骨髓瘤、LennertT细胞淋巴瘤、EB病毒转化的B细胞、肾癌细胞瘤和Kaposi肉瘤细胞的生长;抑制乳腺癌、卵巢癌的细胞株生长。IL-6对人和小鼠的某些髓性白血病细胞株有促进生长作用,对另外一些则具有抑制生长作用。IL-1、TNF和IL-6都能够诱导肝细胞分泌急性期蛋白。IL-6诱导HepG2细胞产生的急性期蛋白包括:纤维蛋白原,α1抗胰凝乳蛋白酶、α1酸性糖蛋白和触珠蛋白。IL-6诱导人原代肝细胞分泌血清淀粉样蛋白A(amyloid A),C反应蛋白和α1抗胰蛋白酶等。IL-6可诱导大鼠原代肝细胞产生纤维蛋白原、半胱氨酸蛋白酶抑制剂,α2巨球蛋白和α1酸性糖蛋白。在体内,IL-6可诱导大鼠产生典型的类似松节油诱导的急性期蛋白反应,且比松节油反应更快发生。严重烧伤患者血清中IL-6的水平与C反应蛋白(CRP)和发热显著相关。外科手术后,血清IL-6的增加先于CRP增加,这也支持IL-6诱导的急性期蛋白产生的结果。IL-6还能够诱导肝细胞再生。IL-6能够诱导肝脏中脂肪生成。血管平滑肌细胞产生IL-6并对IL-6发挥上增殖反应,提示IL-6在动脉硬化形成中起作用。IL-6直接或间接抑制破骨细胞,使绝经期后雌激素抑制IL-6基因表达的作用消失,于是破骨细胞发育,并易导致骨质疏松。切除卵巢的小鼠接受抗IL-6抗体后﹐能够防止破骨细胞发育;IL-6缺陷的小鼠在进行卵巢切除后没有破骨细胞发育和骨质疏松的发生。细胞表面的IL-6可能是乙型肝炎病毒(HBV)的受体,能介导病毒进入细胞。多种疾病都可能与IL-6基因调节异常有关,包括炎症、自身免疫性疾病和恶性肿瘤。已知的有IL-6表达异常的疾病状态包括;多克隆B细胞异常、自身免疫性疾病(心脏黏液瘤、类风湿关节炎、酒精性肝硬化、Castleman病、1型糖尿病、甲状腺炎等)慢性增殖性疾病(膜增殖性肾小球肾炎、银屑病、Kaposi肉瘤)、恶性疾病(浆细胞瘤、骨髓瘤、淋巴瘤、白血病、肾细胞癌)、AIDS、脓毒血症、骨质疏松、Fanconni贫血和 HBV感染。在许多自身免疫性疾病中出现的自身抗体可能与IL-6的高表达有关。在肺结核、类风湿关节炎、溃疡性结肠炎、类肉瘤病、麻风病、Castleman病、Takayasu动脉炎、多发性骨髓瘤等都有半乳糖化的Ig出现,而IL-6转基因小鼠中也有半乳糖化的Ig增加。膜增殖性肾小球肾炎患者的肾小球膜细胞能够产生IL-6,患者尿液中也能够检测出IL-6,而且尿液中IL-6的水平与该病的进程有关,而在其他类型的肾小球肾炎中则无此现象。IL-6能促进角质细胞增生,可能参与银屑病的发病机制。IL-6还可能与B细胞增生性白血病有关,用抗IL-6抗体能抑制患者体内骨髓瘤细胞的生长。但单独的IL-6并不足以引起浆细胞瘤。无论如何,IL-6拮抗剂在临床的运用有很大的潜在价值。
参数原理
2023.04.19

内毒素介导因子之IL-6的受体及其信号转导
细胞表面的IL-6受体复合物是异源性二聚体,有一条相对分子质量为80000的α链(gp80)即 IL-6R,和一条相对分子质量为130 000的β链(gp130)。IL-6Rα链单独与IL-6结合的亲和力(K=740 pmol/L)较低,α链与IL-6结合以后再与gp130形成高亲和力受体复合物(Ka=9.8 pmol/L),而gp130本身不同IL-6结合。各种细胞表面的IL-6R都一样,IL-6在不同细胞中诱导的不同效应与细胞本身的特异性分子或信号转导途径有关,如细胞表达IL-6R的类型和数目多少。人IL-6Rα链的基因位在1q21点上,编码468个氨基酸长度的多肽,其中有19个氨基酸残基是信号肽,跨膜区有28个氨基酸残基,胞质区内有82个氨基酸残基。小鼠IL-6Rα链是460个氨基酸的多肽,结构与人α链相同。具有典型的“细胞因子家族结构域”,即4个保守半胱氨酸残基和WSXWS模体(W代表色氨酸、S代表丝氨酸,X代表任何氨基酸),以及其氨基末端具有免疫球蛋白样结构域。另外一个受体为gpl30,也具有“细胞因子家族结构域”特点,以及在胞外区域存在4个纤维连接蛋白3型组件(fibronectin type Ⅲ modules)。由于IL-6R胞内结构很短,无法转导信号,因此需要gpl30受体的参与,其中gp130为IL-6家族中其他成员所共用,如IL-11、LIF、肿瘤静止素、OSM(oncostatin M),CNTF、CT-1(cardiotrophin-1)等。IL-6R以两种方式存在:膜结合型(membrane bound form),即 IL-6R;另一种为可溶型(soluble form,sIL-6R),分布在组织液中,能够促使无IL-6R表达的细胞也能够经IL-6-sIL-6R复合物触发IL-6的生物效应。sIL-6R的产生有两种机制:①通过蛋白酶对膜结合型IL-6受体进行酶切,如ADAM(a disintegrin and metalloproetinase)家族能够切割IL-6R的胞外结构,并产生不同的可溶性IL-6R。②通过IL-6基因转录后进行交替剪接,产生不同的mRNA及可溶性的IL-6R,并分泌到细胞外,但缺乏跨膜段。由于膜结合型的IL-6R在不同组织中的分布不同,有些细胞虽不表达 IL-6R,但均分布有gpl30,因此可以通过IL-6与可溶性IL-6R结合,再将信号通过gp130进行信号转导,所以IL-6有广泛的生物学效应。sIL-6R不发挥受体拮抗效应,而是发挥激动剂的效应,这一点与其他细胞因子有显著差异。IL-6同IL-6R结合后会发生快速内化,并在细胞内降解,促使受体下调。
参数原理
2023.04.17
