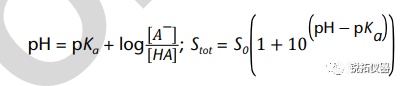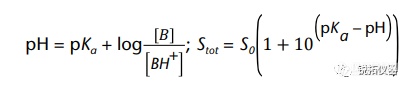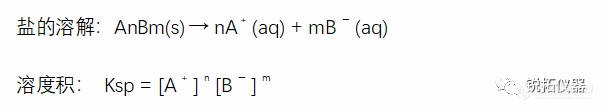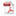摇瓶法基于40年前开发的相溶解度技术,至今仍被大多数人认为是最可靠、最广泛使用的溶解度测量方法(11-18)。当需要测定平衡溶解度时,应使用摇瓶法。其他方法可用于评估表观溶解度,但不适用于评估真实平衡溶解度。
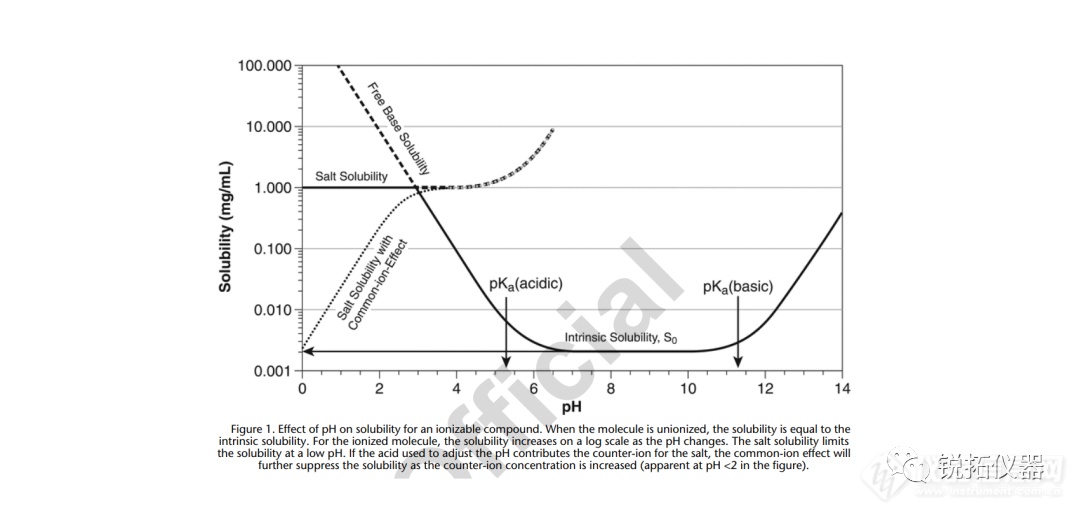
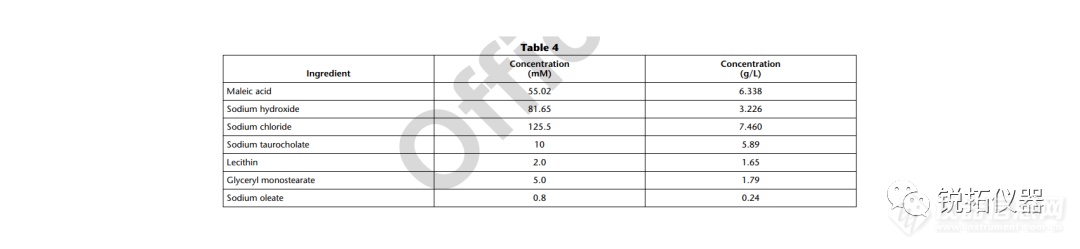
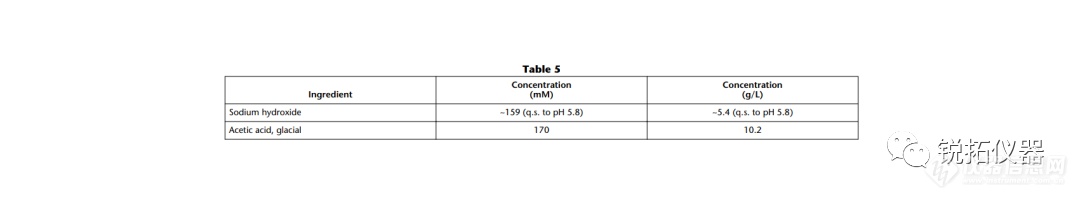
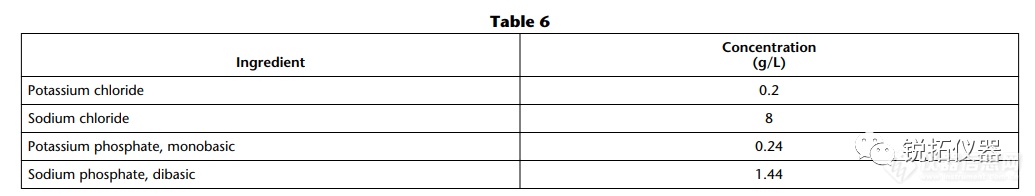
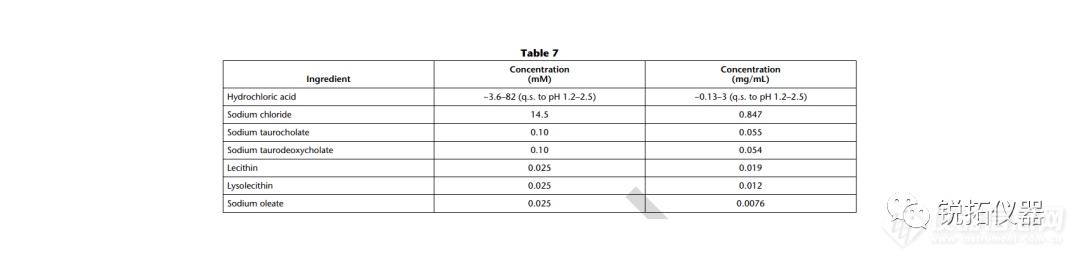
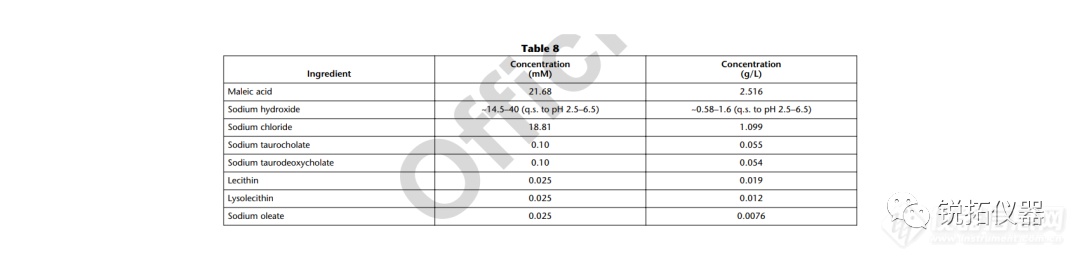
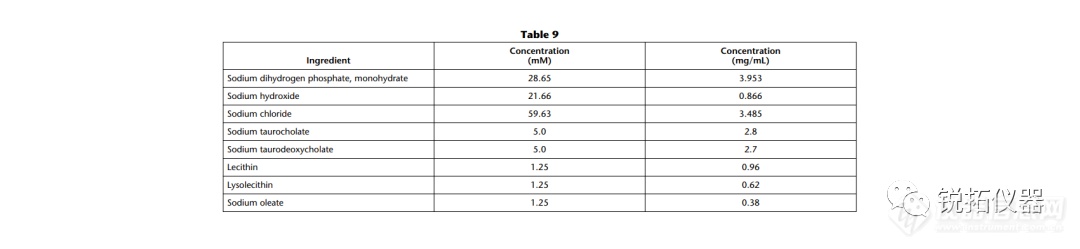
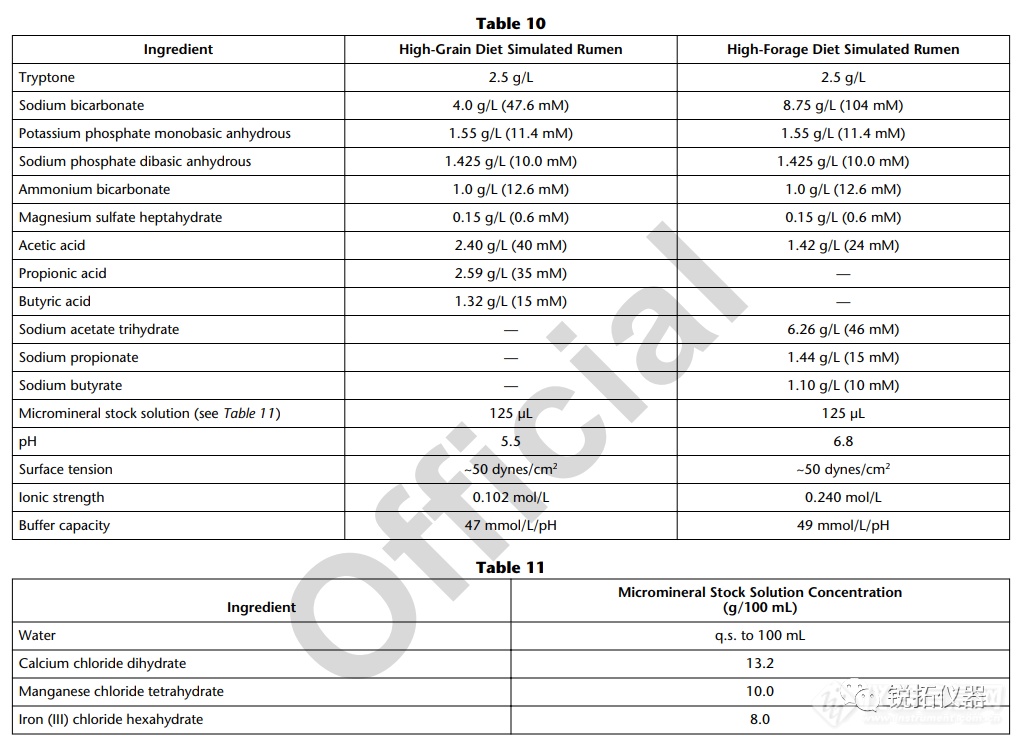
选择用于溶解度测量的溶解度介质应与应用相关,并努力控制表面活性剂的类型和浓度、缓冲液的离子强度以及缓冲液中存在的反离子的类型。当结果用于预测吸收或生物利用度时,建议使用一种生物相关介质溶液(见生物相关介质中的溶解度测量)。当结果旨在支持溶解试验的发展时,建议使用溶解介质。出于研究目的,当评估化合物的pH依赖性时,建议使用一种能够在宽pH范围内控制离子强度和反离子类型的缓冲液(例如,Britton–Robinson或Sörensen缓冲液)。对于将用于BCS分类的溶解度测量,应使用USP推荐的缓冲液(19)。
样品制备:试验物质通常是通过向塞瓶或小瓶中的溶解介质中添加过量固体来制备的。烧瓶或小瓶中的介质量不需要精确测量。建议将固体以超过估计溶解度约1–2 mg/mL的量添加到溶解度介质中。(对于低溶解度化合物,1–2 mg/mL的浓度可能就足够了。)固体的表面积可以通过在加入培养基之前研磨(例如,在研钵和研杵中)样品或在加入溶媒后对样品进行超声处理来增加。[注意:当使用高能方法增加表面积时,建议谨慎使用,因为这可能会改变溶质的固体形式。]建议一式三份进行样品制备,以在每个测试条件下提供至少3个溶解度结果。
溶液的平衡:为了促进固体的溶解,应积极混合或搅拌悬浮液。作为良好的初始孵育时间,建议24小时;然而,必须验证所选平衡时间的适用性。在溶解阶段(±0.5°),应很好地控制悬浮液的温度。在溶解阶段之后,建议允许过量固体完全沉淀。建议将沉淀和倾析作为从饱和溶液中分离固体的最安全方法。对于非澄清胶体溶液,可以使用离心法。上清液的取样应避免加入任何未溶解的固体,因为这将显著影响溶解度结果。转移移液管在使用前需要用样品溶液进行预处理,这样表面吸附不会改变转移的溶液。如果无法避免过滤,则必须选择合适的过滤器类型。对于极性电离物质,建议使用疏水型过滤器(尼龙);而对于未结合的物种,建议使用亲水型过滤器[例如聚偏二氟乙烯(PVDF)或聚醚砜(PES)]。过滤应在沉淀后进行,而不是直接在搅拌后进行。过滤器的预饱和是必要的(即,应丢弃滤液的初始部分)。在沉淀和离心步骤期间,悬浮液的温度也必须得到很好的控制(±0.5°),并且与发生溶解的温度相等。
当在不同的平衡时间段后对多个样品进行分析,得出相同的结果(例如,在24小时内变化小于5%,或小于0.2%/h)时,即达到饱和(平衡)。为了确认表观溶解度为平衡溶解度,建议通过相同的程序对同一悬浮液进行重新平衡(例如,再混合24小时)。
溶液分析:用于定量溶质浓度的分析方法的要求和所需的分析验证水平应与溶解度数据的预期用途相称。一般来说,该方法应该是线性的和具体的。在分析之前可能需要稀释上清液,以使其在分析方法的线性范围内,并避免可能的沉淀。溶液可通过紫外-可见光谱法或液相色谱法进行分析,以确定可溶性浓度。高效液相色谱的优点是,它可以通过分解与药物有关的杂质来检测不稳定性(13,20)。
建议在溶解度测量结束时分析悬浮液中过量的固体,以验证固体形式没有改变。在固体形式已经改变的情况下,新的固体形式可能具有比初始固体形式更低的溶解度,并且观察到的溶解度是由于新的更低的溶解度形式;然而,这应该根据具体情况进行评估。粉末X射线衍射(PXRD)、拉曼或近红外(NIR)光谱法或通过差示扫描量热法(DSC)评估熔点是可用于评估固体形式的技术的实例。在平衡时间内不稳定(化学或物理)的溶质不适合通过摇瓶法进行平衡溶解度测量。例如,将转化为低溶解度盐或多晶型物的无定形药物应使用表观溶解度方法之一进行分析。
溶解度结果的报告:如果在溶解度测定中使用了介质的非标准成分,则应报告成分的详细信息。溶解度测定中使用的介质的离子强度应与溶解度结果一起计算和报告。当提取样品进行分析时,应记录上清液的pH值(在溶解度测量的温度下)。当使用定义明确的标准介质时,建议不要调整介质的pH值以补偿溶解物质对pH值的改变;相反,应在平衡步骤结束时观察到的pH值和温度下报告溶解度值(12,18)。如果介质的pH值受到溶解物质的显著影响,并且需要在特定pH下的溶解度,则建议在更高缓冲容量的介质中进行额外的溶解度测量。报告平衡过程中的平均温度和温度控制精度
报告的平衡溶解度的精度应反映测量值之间的一致性水平,而不是溶解度分析的精度。应包括测量溶解度的标准偏差(基于3个或更多独立样品的平均值)。

 关注
关注