

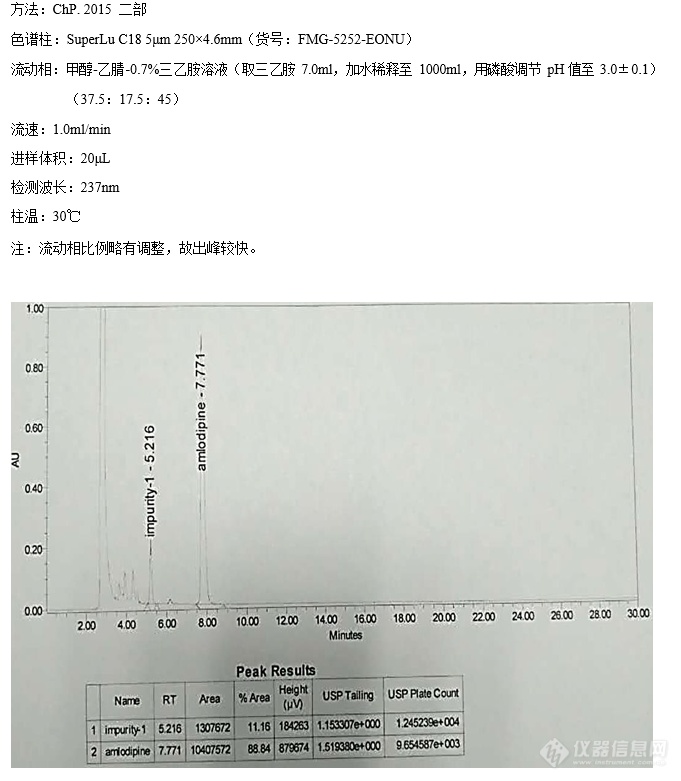
苯磺酸氨氯地平的有关物质
厂商
2017.05.05

动物源食品中氯霉素检测的固相萃取方法
动物源食品中氯霉素检测的固相萃取方法(polyclean x-hlb)一、实验目的本法为以polyclean x-hlb (60mg/3ml)柱开发的针对动物源食品(禽肉、畜肉和水产品)中氯霉素样本前处理净化方法,hplc法作为检测手段。polyclean x-hlb (60mg/3ml)柱净化效果良好,回收率满足测试要求,重现性好,与知名厂商同型号柱性能相当。二、实验目标物氯霉素(cas:56-75-7)三、应用范围本方法适用于动物源性食品(禽肉、畜肉和水产品)中残留药物的检测。四、参考标准《农业部1025号公告-21-2008动物源食品中氯霉素残留检测气相色谱法》五、实验材料nuanalyticalpolycleanx-hlb固相萃取柱60mg/3ml,w品牌hlb固相萃取柱60mg/3ml。六、实验方法1、样品待测溶液制备取5±0.05g猪肉样品,于50ml离心管中,加入20 ml乙酸乙酯混合2min,4000 r/min离心15 min,取上清液于另一离心管中,再用20 ml乙酸乙酯重复提取残渣1次,合并两次提取液,并加入500 μl氨水,于45 ℃旋转蒸发近干,加入500 μl甲醇混合1min,加入10 ml4%nacl涡旋混匀10 s,加入10 ml正己烷摇匀20次,4000r/min离心5 min,弃去上层,再用10 ml正己烷脱脂一次,最后加入500 μl氨水混匀供上柱净化用。2、活化依次用2 ml甲醇和2 ml水活化。3、上样和洗脱取上述备用液重力作用下过柱依次用2 ml水、1 ml甲醇/乙腈/水/氨水(15:15:65:5, v/v)淋洗上样柱并抽干,最后用2 ml 5%氨化甲醇溶液洗脱柱子上的待测成分。4、重新溶解收集洗脱液,于45℃氮气吹干,准确加入1.0 ml甲醇/水(4:6,v/v)溶液溶解残余物混匀,0.22 μm滤膜过滤,供hplc测定。5、hplc条件色谱柱:superlu c18 (250×4.6 mm,5 μm)检测器:uv@278 nm流动相:a:水 b:甲醇洗脱方式:等度洗脱,a:b=60:40柱温:30℃ 流速:1.0ml/min 进样量:20μl七、实验结果1、添加水平为0.5 mg/kg猪肉基质中氯霉素(cap)的添加回收结果表1 添加水平为 0.5 mg/kg 猪肉基质中氯霉素(cap)的添加回收结果 名称 回收率(%) 平均回收率(%)rsd(%) 123 氯霉素 92.6 89 8789.5 3.2 表 2 w 品牌同型号柱,添加水平为 0.5 mg/kg 猪肉基质中氯霉素(cap)的添加回收结果 名称 回收率(%) 平均回收率(%)12 氯霉素90.891.491.12、添加水平为 0.5mg/kg 猪肉基质中氯霉素(cap)检测色谱图1 添加水平为0.5mg/kg猪肉基质中氯霉素(cap)检测色谱图
厂商
2017.05.04

食品中喹诺酮检测的固相萃取方法
食品中喹诺酮检测的固相萃取方法(polyclean x-hlb)一、实验目的本研究利用固相萃取法作为样品的前处理方法,hplc法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂的使用,操作简便。二、实验目标物依诺沙星(cas:74011-58-8),恩诺沙星(cas:93106-60-6),培氟沙星(cas:70458-95-6), 丹诺沙星(cas:119478-55-6)三、应用范围本方法适用于食品中喹诺酮的hplc检测及确证。四、参考标准《gb/t 21312-2007动物源性食品中14种喹诺酮药物残留检测方法》五、实验材料nuanalyticalpolycleanx-hlb固相萃取柱200mg/6ml。六、实验方法1、 样品待测溶液制备称取均质牛奶试样5.0 g试样(精确至0.001g)于50 ml离心管中,加入20 mledta-mellvaine缓冲溶液,1000r/min涡旋混合1min,超声提取10min,10000r/min离心5 min(温度低于5 ℃),提取三次合并上清液。2、活化依次用6 ml甲醇和6 ml水活化。3、上样和洗脱将提取液以2 ml/min的速度过柱,弃去滤液,用2 ml5%甲醇水溶液淋洗,弃去淋洗液,将小柱抽干,再用6 ml甲醇洗脱并收集洗脱液。4、重新溶解50℃缓慢氮气流条件下吹至近干,用1 ml 0.2 %甲酸水溶液溶解。1000 r/min涡旋混合1 min,经0.45 μm有机滤膜过滤,供hplc测定。5、hplc条件色谱柱:superlu c18 (250×4.6mm,5μm)检测器:uv@254 nm流动相:a: 乙腈 b:0.1%甲酸的水溶液柱温:室温 流速:1.0ml/min 进样量:20μl洗脱方式:梯度洗脱,洗脱程序如下:时间/mina/%b/%0.00991119912029712537632610003010003199136991七、实验结果1、1mg/kg牛奶基质中喹诺酮的添加回收结果表1 1mg/kg牛奶基质中喹诺酮的添加回收结果名称平均回收率(%)依诺沙星92.8丹诺沙星82.4培氟沙星93.8恩诺沙星81.22、添加水平为1mg/kg牛奶基质中喹诺酮检测色谱图图1 添加水平为1mg/kg牛奶基质中喹诺酮检测色谱图
厂商
2017.05.04

水体中布洛芬检测的固相萃取方法
水体中布洛芬检测的固相萃取方法(polyclean x-hlb)一、实验目的本研究利用固相萃取法作为样品的前处理方法,hplc法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂的使用,操作简便。二、实验目标物布洛芬(cas:15687-27-1)三、应用范围本方法适用于水体中布洛芬的检测的hplc检测及确证。四、参考标准《sn/t 2190-2008进出口动物源性食品中非甾体类抗炎药残留量检测方法 液相色谱-质谱质谱》五、实验材料nuanalytical polyclean x-hlb固相萃取柱60mg/3ml。六、实验方法1、 样品待测溶液制备取1000 ml(准确至0.1 ml)水体,将水体使用中速滤纸过滤,除去悬浮物,加入0.5 g na-edta,并使用甲酸调节ph至4.5。2、活化依次用5 ml甲醇和5 ml水活化。3、上样和洗脱取待测液,通过hlb固相萃取小柱富集,弃去流出液,富集完成后使用3ml水,5 ml5%甲醇水溶液淋洗固相萃取柱,真空抽干5min,弃去流出液。使用7ml甲醇洗脱萃取柱,收集洗脱液。4、重新溶解洗脱液于40 ℃氮气吹干,加乙腈1.0 ml溶解残余物,滤膜过滤,供hplc测定。5、hplc条件色谱柱:superlu c18 (250×4.6mm,5μm)检测器:uv@268 nm流动相:a:乙腈 b:0.1 mol/l甲酸水溶液洗脱方式:等度洗脱,a:b=50:50柱温:室温 流速:1.0ml/min 进样量:20μl七、实验结果1、10ppm水基质中布洛芬的添加回收结果 名称 回收率(%) 平均回收率(%)rsd(%) 123 布洛芬 82.79 81.48 80.85 81.71 1.21表1 10ppm水基质中布洛芬的添加回收结果2、添加水平为10ppm水基质中布洛芬检测色谱图 图1 添加水平为10ppm水基质中布洛芬检测色谱图
厂商
2017.05.04
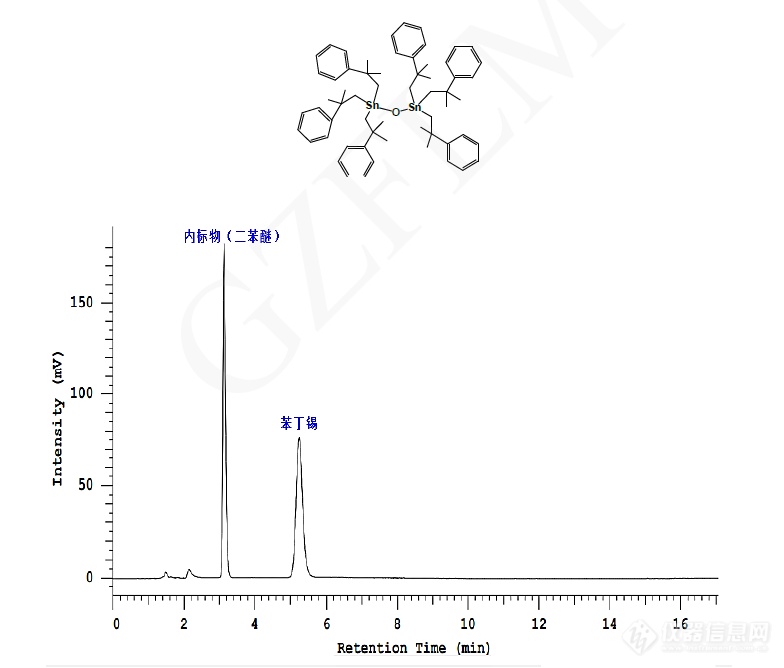
苯丁锡的含量测定
方法:自订色谱柱:ACE Excel C18 5μm 150×4.6 mm(货号:EXL-121-1546U)流动相:甲醇-10g/L 磷酸二氢钠水溶液-磷酸(900:100:0.2)流速:1.0ml/min进样体积:5μL(0.5mg/mL)检测波长:210 nm温度:室温
厂商
2017.05.03
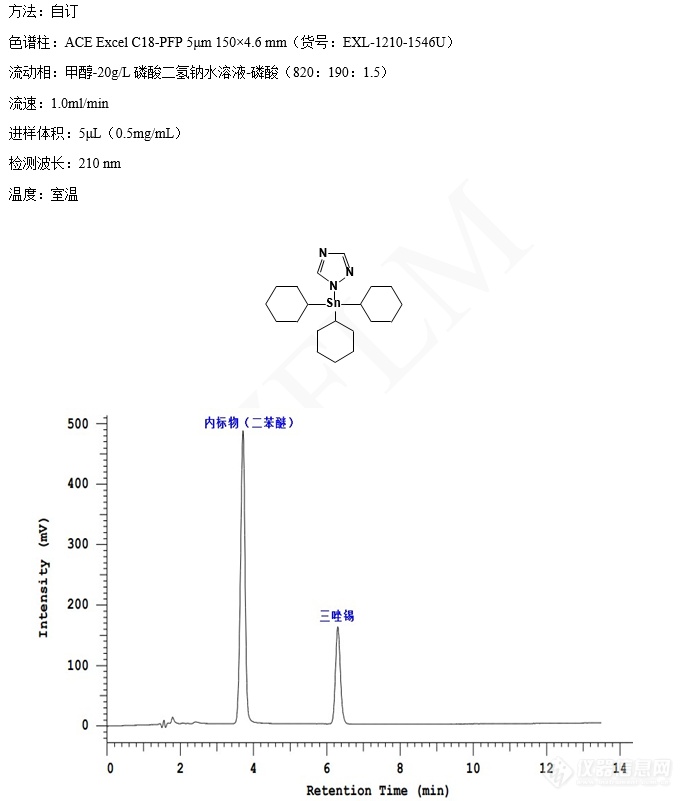
三唑锡的含量测定
厂商
2017.05.03
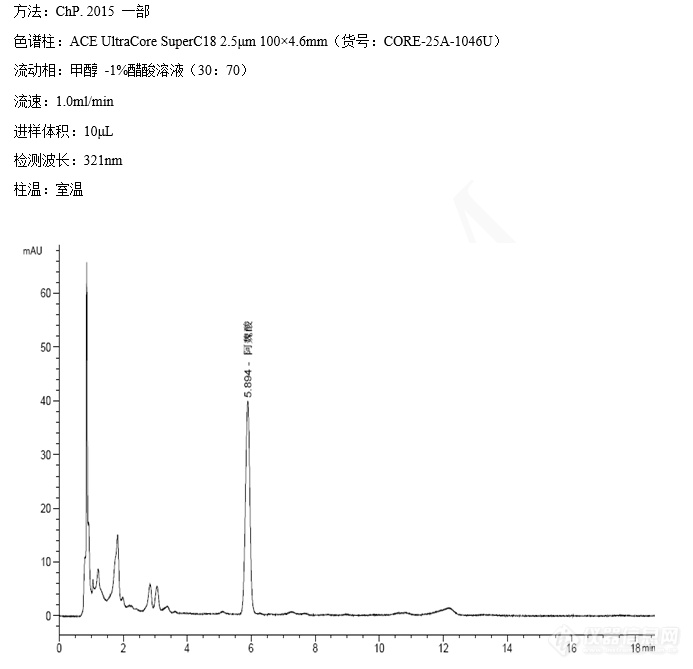
川芎中阿魏酸的测定
厂商
2017.05.03

菲罗门将出席第七届药安大会
会议背景:会议背景:为配合国家飞行检查及《中国药典》2015年版的宣传贯彻工作,进一步提升药品质量人员的管理能力,建立与完善质量体系,深入了解新政下相关法规与飞行检查工作流程及重点,掌握最新技术应用,搭建药品质量管理与控制专业技术人才学术交流和信息共享的桥梁,促进我国药品质量控制技术发展水平可持续创新,由中国社科院食品药品发展与监管研究中心主办,北京中培科检信息技术中心承办的“2017第七届中国药品质量安全大会”,将于2017年5月25-27日在广州花都皇冠假日酒店召开。届时,主委会将邀请国内外有关专家领导、企业界代表、技术厂商代表出席2017中国药品质量安全大会,就目前我国药品安全领域普遍关注的重要话题、领先技术、最佳实践、以及制药行业未来发展趋势进行深入探讨交流。主办方:中国社科院食品药品产业发展与监管研究中心承办单位:北京中培科检信息技术中心
厂商
2017.05.02

进出口食品中罗丹明 B 的检测的固相萃取方法
进出口食品中罗丹明b的检测的固相萃取方法(superclean al-n)一、实验目的 本研究利用固相萃取法作为样品的前处理方法,hplc法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂的使用,操作简便。二、实验目标物 罗丹明b(cas:81-88-9)三、应用范围 本方法适用于进出口食品中罗丹明b的检测的hplc检测及确证。四、参考标准 《sn/t 2430-2010 进出口食品中罗丹明 b 的检测方法》nuanalyticalsuperclean al-n中性氧化铝固相萃取柱1g/6ml。 六、实验方法1、 样品待测溶液制备称取辣椒粉2 g(精确至0.01)于50 ml离心管中,准确加入20 ml 20%丙酮正己烷,于涡旋混匀器上混合提取2 min,再超声提取15 min,于4000 r/min离心5 min,上清液过滤待净化。2、活化依次用6 ml正己烷活化。3、上样和洗脱取待净化液10 ml加入中性氧化铝小柱,1滴/秒流速通过中性氧化铝小柱后,弃去溶液,加入10 ml丙酮-正己烷溶液(20:80,v/v)淋洗,弃去淋洗液,抽干小柱。取10 ml 2%氨水甲醇溶液洗脱小柱,收集洗脱液,洗脱液于50 ℃氮吹至干。4、重新溶解用1 ml甲醇定容,0.22μm微孔滤膜过滤,供hplc测定。5、hplc条件色谱柱:superlu c18 (250×4.6mm,5μm)检测器:waters 2475 荧光检测器,λex=550 nm,λem=580 nm流动相:a:水 b:甲醇洗脱方式: 等度洗脱,a:b=25 : 75柱温:室温 流速:1.0ml/min 进样量:20μl七、实验结果1、 25μg/kg辣椒粉中罗丹明b的添加回收结果名称 1 2 3平均回收率(%)rsd(%)罗丹明b86.088.088.087.331.32 表1 25μg/kg辣椒粉中罗丹明b的添加回收结果回收率(%)图1 添加水平为25μg/kg辣椒粉中罗丹明b检测色谱图
厂商
2017.05.02
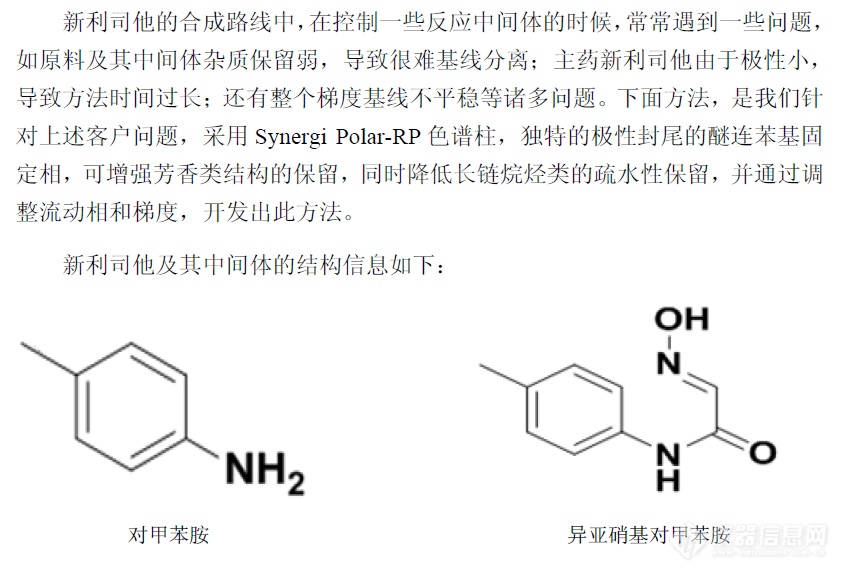
新利司他中控方法
厂商
2017.05.02

食品添加剂-甜菊糖苷
厂商
2017.04.28
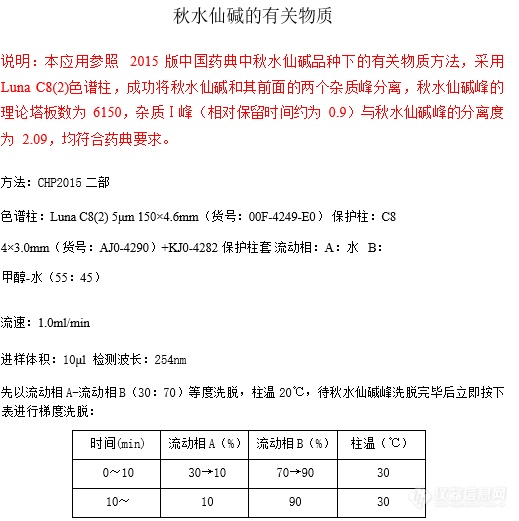
秋水仙碱的有关物质
厂商
2017.04.28
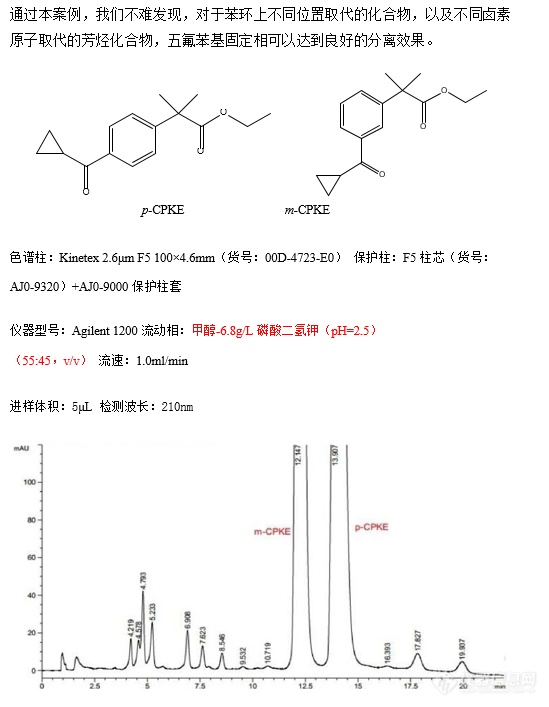
p-CPKE 和m-CPKE 的分离
厂商
2017.04.28
皮革中六价铬含量的测定
一、实验目的本实验利用固相萃取法作为皮革样品的前处理方法,对PolyClean X-PAmide小柱进行评价。 二、实验目标物六价铬(Cr6+) 三、应用范围本方法使用于皮革样品的脱色以及六价铬含量的测定。 四、参考标准《GB/T 22807-2008《皮革和毛皮 化学实验 六价铬含量的测定》和《EN ISO 17075-2007 皮革--化学试验--铬(VI)含量的测定》。五、实验材料PolyClean X-PAmide小柱 500mg/6mL(9B-P006-06500) 。 六、实验方法1、样品提取(1)称取2 g剪碎的皮革样品,吸取100 mL脱气后的萃取液(22.8 g三水磷酸氢二钾溶于1 L水中,用磷酸调节pH至8 ± 0.1,再用氮气或氩气排出氧气) 加入皮革样品,盖上瓶塞。(2) 在 机 械 震 荡 器 上 震 荡 3h, 萃 取 Cr6+ 。 (3)萃取完成后,通过薄膜滤器将锥形瓶中的溶液过滤,待净化。检查溶液的pH值,应为7.5 - 8.0,如果不在此范围,则需重新提取。 2、SPE柱净化(1)活化:5 mL甲醇,5 mL水,10 mL萃取液。(2)上样:取10 mL待净化液过柱,收集上样液于25 mL容量瓶,用10 mL萃取液淋洗,一并收集,用萃取液定容至刻度。 3、六价铬含量测定(1)移取净化后的溶液于25 mL容量瓶中,用萃取液稀释至容量瓶的3/4,加入0.5 mL磷酸溶液和0.5 mL二苯卡巴肼溶液(1%,酸性) 用萃取液定容,摇匀,静置15 min。(2)以空白溶液按上述操作显色,作为参比溶液。 (3)每次测试样品,同时取净化后的溶液于25 mL容量瓶,不加显色剂,直接用萃取液定容。(4)将上述溶液用1 cm比色皿在540 nm处测量吸光度。七、脱色效果图1 菲罗门色素小柱PolyClean X-PAmide脱色效果如图1所示, PolyClean X-PAmide专用柱没有被色素穿透,净化后样品几乎无色。八、实验结果1、校正曲线绘制配制1 μg/mL的六价铬标准溶液,使用7个不同浓度的溶液绘制校正曲线。移取一定体积的标准溶液,加入显色剂并用萃取液定容,15 min后比色。同时以空白溶液按同样操作进行显色,作为参比。以六价铬浓度(1 μg/mL)作为X轴,以吸光度作为Y轴绘制标准曲线。(图2)
厂商
2017.04.27

普洱中农药多残留检测的固相萃取方法
普洱中农药多残留检测的固相萃取方法一、实验目的(superclean gcb/nh2)本研究利用固相萃取作为样品前处理方法,gc-ecd 和 lc-ms/ms 作为分析方法,检测普洱中的农药残留水平。该方法操作简便,可简化样品前处理过程,减少有机溶剂的使用。二、应用范围本方法适用于茶叶中有机磷类、有机氯类、拟除虫菊酯类和氨基甲酸酯类农药多残留的测定。三、实验材料nuanalytical superclean gcb/nh2 固相萃取柱 500 mg/500 mg/6 ml。四、实验方法1、样品提取称取粉碎好的普洱 2 g(精确到 0.001 g),加入 50 ml 离心管中,加入 10 ml 乙腈,剧烈振荡 1 min,静置 30min,4000 r/min 离心 5 min。上清液待净化。2、spe 柱活化gcb/nh2 固相萃取柱中加入约 2 cm 高无水硫酸钠,使用前使用 10 ml 乙腈-甲苯(3:1,v/v)活化。3、上样和洗脱当溶液液面到达柱吸附层表面时,立即倒入上述待净化溶液 4 ml, 用鸡心瓶接收流出液,逐步加入 25 ml 乙腈-甲苯(3:1,v/v)洗涤小柱,收集上述所有流出液于鸡心瓶中。4、重新溶解流出液于 40 ℃水浴中旋蒸至 1 ml 左右,加入 2 ml 乙腈转移至 10 ml 试管中,于40 ℃下氮气吹干,加入 1 ml 乙腈溶解残渣,0.22 μm 微孔滤膜过滤,分别供 gc-ecd 和lc-ms/ms 上机测试。5、仪器条件(1)、 gc-ecd 条件气相仪器:agilent 7890a 色谱柱:fb-5, 30 m×0.32 mm, 0.25 μm进样口温度:220 ℃ 检测器温度:300 ℃升温程序:180 ℃(保持 2 min);以 10 ℃/min 升温到 230 ℃(保持 2 min);以 2 ℃/min升温到 260 ℃(保持 2 min);以 25 ℃/min 升温到 270 ℃(保持 1.6 min)载气:氦气 流速:1.6 ml/min 进样方式:分流进样(分流比 10:1)(2)、lc-ms/ms 条件质谱仪:api 4000 色谱柱:superlu c18(2.0 mm×150 mm, 5 μm)流动相:a: 0.1%甲酸+10 mm 乙酸铵(1 ml 甲酸+0.77 g 乙酸铵溶于 1 l 水中);b: 甲醇洗脱方式:梯度洗脱,洗脱程序如下: 时间/mina(%)b(%)0.09551.509556.059511.059511.0195515955流速:0.35 ml/min 柱温: 40 ℃ 进样体积:5 μl离子源:电喷雾(esi) 扫描模式:正离子模式 检测方式:多反应监测(mrm) 质谱仪离子源参数如下: source/gascollision gas (cad)6curtain gas (cur)12ion source gas 1 (gs1)50ion source gas 2 (gs2)50ion spray voltage (is)5500temperature (tem)550interface heater (ihe)on氨基甲酸酯类农药各组分名称、保留时间及母离子和子离子检测离子对如下: 物质名称保留时间/min检测离子对dpepcecxp涕灭威7.06208.1>89.1208.1>1163030101022101212克百威7.13222.3>123.1222.3>165.24848101016311212涕灭威砜6.25223.1>86.2223.1>148.46969101021131212涕灭威亚砜6.10207.1>132.2207.1>89.16060101013221212啶虫脒6.83223.4>126.1223.4>907070101029461212五、实验结果1、普洱中农药多残留的添加回收结果表 1 0.25 mg/kg 普洱中有机氯和拟除虫菊酯类农药多残留的添加回收结果 回收率(%)名称平均回收率(%)rsd (%)123乙烯菌核利84.576.080.080.25.30腐霉利110.5102.0105.0105.84.07异菌脲112.0107.5119.0112.85.14联苯菊酯94.587.590.590.83.87甲氰菊酯109.5100.0106.5105.34.61高效氟氯氰菊酯84.079.582.582.02.79氟氯氰菊酯86.586.894.189.14.83氟氰戊菊酯120.5114.0120119.23.06氰戊菊酯95.585.092.991.16.00氟胺氰菊酯70.472.7581.074.77.45表 1 0.05 mg/kg 普洱中氨基甲酸酯类农药多残留的添加回收结果 回收率(%)名称平均回收率(%)rsd (%)123涕灭威95.687.290.090.94.70克百威84.478.082.281.53.99涕灭威砜77.483.081.480.63.58涕灭威亚砜70.074.475.273.13.73啶虫脒82.494.088.488.36.572、普洱中农药多残留检测色谱图图 1 添加水平为 0.25 mg/kg 普洱中有机氯和拟除虫菊酯类农药多残留检测色谱图 图 2 添加水平为 0.0625 mg/kg 普洱中氨基甲酸酯类农药多残留检测色谱图
厂商
2017.04.27

水产品中孔雀石绿检测的固相萃取方法
水产品中孔雀石绿检测的固相萃取方法(superclean al-n)一、实验目的本研究利用固相萃取法作为样品的前处理方法,hplc 法作为分析方法,检测水产品中孔雀石绿含量。该方法可简化样品的前处理过程,节省有机溶剂的使用,操作简便。二、实验目标物孔雀石绿(cas:569-64-2)三、应用范围本方法适用于鲜活水产品中孔雀石绿的检测的 hplc 检测及确证。四、参考标准《gb/t 19857-2005 水产品中孔雀石绿和结晶紫残留量的测定》五、实验材料nuanalytical superclean alumina-n 固相萃取柱 1000mg/3ml。六、实验方法1、 样品待测溶液制备准确称取鱼肉样品 5.0 g 于 50 ml 离心管中,加入 10 ml 乙腈,超声波振荡提取 5 min,涡旋 1 min,4000r/min 离心 5 min,上清液转移至 50 ml 离心管中,残渣按照上述步骤重复提取一次;合并两次提取液,用乙腈定容至 25 ml。2、活化依次用 5 ml 乙腈活化3、上样和洗脱在中性氧化铝固相萃取柱上加入 5 ml 待净化样品,收集流出液,然后用 4 ml 乙腈洗脱,合并收集流出液。4、重新溶解流出液 45 ℃氮吹至近干,1 ml 乙腈定容,过 0.45 μm 滤膜,,供高效液相色谱测定。5、hplc 条件色谱柱:superlu c18 (250×4.6mm, 5μm) 检测器:uv-vis@618 nm流动相:a-乙腈 b-0.05 mol/l 乙酸铵缓冲溶液流速:1.2 ml/min 进样体积: 50 μl洗脱方式:梯度洗脱,洗脱程序如下: 时间/mina/%b/%0.060403.0060403.10802010.00802010.10604013.006040七、实验结果1、0.5 mg/kg 鱼肉中孔雀石绿的添加回收结果表 1 0.5 mg/kg 鱼肉中孔雀石绿的添加回收结果 名称123平均回收率(%)rsd(%)孔雀石绿91.091.887.290.02.732、添加水平为 0.5 mg/kg 鱼肉中孔雀石绿检测的液相色谱图图 1 添加水平为 0.5 mg/kg 鱼肉中孔雀石绿检测的液相色谱图
厂商
2017.04.27

盐酸吡格列酮片的含量测定和有关物质
盐酸吡格列酮片的含量测定和有关物质方法:jp色谱柱:titank c18 5μm 150×4.6mm(货号:fmf-5560-eonu) 流动相:乙腈-100mm 乙酸铵-乙酸(25:25:1)流速:0.8ml/min进样体积:(a)20μl; (b)40μl检测波长:269nm温度:25℃
厂商
2017.04.26

染发剂中禁限用成分的检测
染发剂中禁限用成分的检测方法:《化妆品中 32 种禁限用染料成分的检测方法》色谱柱:ace excel c18-amide 5μm 250×4.6mm(货号:exl-1212-2546u) 流动相:磷酸溶液(1+9):吸取磷酸(ρ20=1.69g/ml)10ml,加水 90ml磷酸盐混合溶液:称取十二水合磷酸氢二钠 1.8g,磷酸二氢钾 2.8g 和庚烷磺酸钠 1.0g,用水稀释至 1l,混匀,配制成含庚烷磺酸钠(1g/l)的磷酸盐缓冲液,加入磷酸溶液(1+9),调节 ph 至 6磷酸盐混合溶液-乙腈(60:40) 流速:1.0ml/min检测波长:280nm温度:图 1.:60℃;图 2.:25℃图 1.图 2.
厂商
2017.04.26

皮革中六价铬含量的测定
皮革中六价铬含量的测定(菲罗门色素小柱polyclean x-pamide)一、实验目的本实验利用固相萃取法作为皮革样品的前处理方法,对polyclean x-pamide小柱进行评价。二、实验目标物六价铬(cr6+)三、应用范围本方法使用于皮革样品的脱色以及六价铬含量的测定。四、参考标准《gb/t 22807-2008《皮革和毛皮 化学实验 六价铬含量的测定》和《en iso 17075-2007 皮革--化学试验--铬(vi)含量的测定》。五、实验材料polyclean x-pamide小柱 500mg/6ml(9b-p006-06500) 。六、实验方法1、样品提取(1)称取2 g剪碎的皮革样品,吸取100 ml脱气后的萃取液(22.8 g三水磷酸氢二钾溶于1 l水中,用磷酸调节ph至8 ± 0.1,再用氮气或氩气排出氧气) 加入皮革样品,盖上瓶塞。(2) 在机械震荡器上震荡3 h, 萃取 cr6+ 。 (3)萃取完成后,通过薄膜滤器将锥形瓶中的溶液过滤,待净化。检查溶液的ph值,应为7.5 - 8.0,如果不在此范围,则需重新提取。2、spe柱净化(1)活化:5 ml甲醇,5 ml水,10 ml萃取液。(2)上样:取10 ml待净化液过柱,收集上样液于25 ml容量瓶,用10 ml萃取液淋洗,一并收集,用萃取液定容至刻度。3、六价铬含量测定(1)移取净化后的溶液于25 ml容量瓶中,用萃取液稀释至容量瓶的3/4,加入0.5 ml磷酸溶液和0.5 ml二苯卡巴肼溶液(1%,酸性) 用萃取液定容,摇匀,静置15 min。(2)以空白溶液按上述操作显色,作为参比溶液。 (3)每次测试样品,同时取净化后的溶液于25 ml容量瓶,不加显色剂,直接用萃取液定容。(4)将上述溶液用1 cm比色皿在540 nm处测量吸光度。七、脱色效果图1 菲罗门色素小柱polyclean x-pamide脱色效果如图1所示, polyclean x-pamide专用柱没有被色素穿透,净化后样品几乎无色。八、实验结果1、校正曲线绘制配制1 μg/ml的六价铬标准溶液,使用7个不同浓度的溶液绘制校正曲线。移取一定体积的标准溶液,加入显色剂并用萃取液定容,15 min后比色。同时以空白溶液按同样操作进行显色,作为参比。以六价铬浓度(1 μg/ml)作为x轴,以吸光度作为y轴绘制标准曲线。(图2) 2、0.4ppm加标回收结果表1皮革样品中0.4ppm加标回收结果名称回收率(%)平均回收率(%)rsd(%)123六价铬87.9087.1587.3487.460.45
厂商
2017.04.26

丙烯酰胺迁移量的测定
丙烯酰胺迁移量的测定方法:gb 31604.18-2016色谱柱:mars cis 5μm 250×4.6mm(货号:fmg-1038-eonu) 流动相:3.5mm h2so4-乙腈(93:7)流速:0.25ml/min 进样体积:20μl 检测波长: 202nm 柱温:55℃样品:丙烯酰胺
厂商
2017.04.25

腐胺、尸胺、精胺和亚精胺的分离
腐胺、尸胺、精胺和亚精胺的分离方法:自订色谱柱:titank f5 5μm 150×4.6mm(货号:fmf-5565-eonu) 流动相:甲醇/水(30/70),含 10mm 甲酸流速:1.0ml/min进样体积:1μl(25μg/ml) 检测:cad温度:25℃
厂商
2017.04.25

二甲双胍和苯乙双胍的分离---HPLC 到 UHPLC 的方法转移
二甲双胍和苯乙双胍的分离---hplc 到 uhplc 的方法转移方法:自订流动相:100mm 乙酸-乙酸铵(ph 3.7)/乙腈(10/90)检测波长:235nm温度:35℃样品:1.盐酸苯乙双胍; 2.盐酸二甲双胍.
厂商
2017.04.25

果汁中水溶性维生素分析的快速 LC-MS 方法
果汁中水溶性维生素分析的快速 lc-ms 方法方法:自订时间(min)流动相a(%)流动相 b(%)0.009911.009913.009283.1075256.0050506.5050506.519919.00991色谱柱:ace excel c18-pfp 3μm 100×2.1mm(货号:exl-1110-1002u) 流动相:a:15mm 甲酸,用氨水溶液调节 ph 值至 3.8b:甲醇流速:0.4ml/min检测:esi 正离子模式(维生素 c 和柠檬酸为 esi 负离子模式)dl 温 度 :250℃ 加热块温度:400℃柱温:30℃
厂商
2017.04.24

人造甜味剂---甜菊糖苷的快速分析
人造甜味剂---甜菊糖苷的快速分析方法:gb 8270-2014 相似方法,用于 uhplc色谱柱:ace excel superc18 2μm 150×2.1mm(货号:exl-1011-1502u) 流动相:a:10mm 磷酸二氢钠的水溶液,ph 2.8 b:10mm 磷酸二氢钠的水/乙腈(80:20) 流速:0.6ml/min进样体积:1μl 检测波长:200nm 温度:50℃时间(min)流动相a(%)流动相 b(%)060.539.545248瑞鲍迪苷 a 和甜菊苷达到基线分离!
厂商
2017.04.24

蔬菜中农药多残留检测的固相萃取方法
蔬菜中农药多残留检测的固相萃取方法一、实验目的(superclean nh2)本研究利用固相萃取法作为水果蔬菜中农残检测的前处理方法,lc-ms/ms 法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂的使用,操作简便。二、实验目标物四种农残标准品:克百威(cas:1563-66-2),灭多威(cas:16752-77-5),残杀威(cas:114-26-1),多菌灵(cas:63090-40-4)三、应用范围本方法适用于蔬菜和水果中氨基甲酸酯类农药多残留的测定四、参考标准《ny/t 761-2008 蔬菜水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定 第三部分》五、实验材料nuanalytical superclean nh2 固相萃取柱 1 g/6 ml。六、实验方法1、 样品待测溶液制备称取 10.0g韭菜(精确至 0.01 g)于 50 ml 离心管中,加入 20 ml 乙腈,均质 2 min,加入 5-7 g 氯化钠,盖上盖子剧烈震荡 5 min,在室温下静置 10 min,5000 r/min 离心 4 min, 使乙腈和水相分层,取乙腈层待净化。2、活化依次用 5 ml 甲醇-二氯甲烷(1:99, v/v)活化,弃去流出液3、上样和洗脱加入待净化液,流速控制在 1 ml/min 内,收集流出液;用 5 ml 甲醇-二氯甲烷(1:99, v/v)洗脱,接收流出液;合并流出液。4、重新溶解40 ℃缓慢氮气流条件下吹至近干(约 0.5 ml)后挥干,用 1 ml 乙腈-0.1%甲酸水溶液(10:90,v/v)定容至 1 ml,过 0.45 μm微孔滤膜,供 lc-ms/ms 测定。5、lc-ms/ms 条件质谱仪:api 4000 色谱柱:superlu c18(2.0 mm×150 mm, 5 μm)流动相:a: 乙腈 b: 0.1%甲酸水溶液流速:0.3 ml/min 柱温: 30 ℃ 进样体积:2 μl离子源:电喷雾(esi) 扫描模式:正离子模式 检测方式:多反应监测(mrm)洗脱方式:梯度洗脱,洗脱程序如下:时间/mina(%)b(%)0.0010906.0090108.0090108.01109015.001090七、实验结果1、韭菜中农药多残留 0.5 mg/kg 加标回收结果表 1 韭菜中农药多残留 0.5 mg/kg 加标回收结果名称添加水平(mg/kg)平均回收率(%)克百威0.594.25灭多威0.594.38残杀威0.5103.86多菌灵0.5101.792、添加水平为 0.5 mg/kg 韭菜基质中农残检测色谱图图 1 四种农残待测物总离子流图图 2 灭多威(methomyl)(163.1/88.1)质谱图 图 3 多菌灵(carbendazim)(192.1/160.1)质谱图 图 4 克百威(carbofuran)(222.1/123.1)质谱图 图 5 残杀威(propoxur)(210.2/186.3)质谱图
厂商
2017.04.24

培美曲塞对映体纯度检测
培美曲塞对映体纯度检测方法:european pharmacopoeia 9.0色谱柱:ace excel 5μm c18 100? 250 x 4.6 mm (货号:exl-121-2546u)流动相:a(8g β-环糊精加入 900ml 水中,加入 15ml 三乙胺,6ml 磷酸,调节 ph=6.0,加水稀释至 1000ml):b(乙腈)=95:5(v/v) 流速:1.0 ml/min进样体积:20 μl(供试品中培美曲塞浓度 0.24 mg/ml,异构体杂质 e 浓度 0.16 μg/ml)检测波长:230 nm温度:室温 备注:按照欧洲药典标准,客户原先所使用的色谱柱为 phenomenex ib-sil 5um c18 120?(250 x 4.6 mm),据客户反映,在用该类型色谱柱分析培美曲塞这个品种时,色谱柱耐用性很差,寿命短;另外,色谱峰的重现性也不是很好。现我司采用 ace excel c18 色谱柱,完全替代原方法中的 phenomenex ib-sil c18 色谱柱,分析时间更短,分离度更好,对称性良好,性价比更高,ib-sil c18 附图如下
厂商
2017.04.21

维生素 C 钠的有关物质
维生素 c 钠的有关物质方法:ep. 9.0色谱柱:ace excel nh2 5μm 250×4.6mm(货号:exl-1214-2546u)流动相:磷酸盐缓冲液(6.8g kh2po4 溶于 175ml 水中,溶解并用水稀释至 1000ml)-乙腈(25:75)流速:1.0ml/min进样体积:20μl(50mg/ml)检测波长:210nm柱温:室温
厂商
2017.04.21

培美曲塞对映体纯度检测
培美曲塞对映体纯度检测方法:ep. 9.0色谱柱:ace excel c18 5μm 250×4.6mm(货号:exl-121-2546u)流动相:a(8g β-环糊精加入 900 ml 水中,加入 15ml 三乙胺,再加入 6ml 磷酸,调节 ph 值至 6.0,最后用水稀释至 1000ml)-乙腈(95:5)流速:1.0ml/min进样体积:20μl(供试品中培美曲塞浓度为 0.24mg/ml,对映体杂质浓度为 0.16μg/ml)检测波长:230nm柱温:室温 图 1. 培美曲塞二钠供试品图谱
厂商
2017.04.21

菲罗门 FB-Wine 酒类分析
菲罗门现正式推出新型的聚乙二醇型酒类专用柱 fb-wine。此柱不仅具有相比于同类产品超长纯水相进样的稳定性和寿命,适合各类酒成份的分析外,同时解决了白酒检测中的乙酸乙酯与乙缩醛的分离问题。给各类白酒样品分析者提供了直接解决方案。 gc: agilent 7890 w/ fid p/n no: 30g-l101-025 fb-wine 30m*0.25mm*0.25umoven: 48℃ 5mincarrier: hydrogen, 0.9ml/mininlet: split, 240 ℃, split flow 50ml/mindetector: fid 260 ℃samples: 100ppm acetal and ethyl acetate in aqueous solution (including 0.5%ipa and 1%ethanol) inject volume: 1ul 结论:白酒中一项主要考察指标,乙酸乙酯与乙缩醛在 fb-wine 气相柱上可以很容易在 50 ℃左右达到基线分离.说明:1、 在一般的不复杂酒样分析且同时需要对乙酸乙酯与乙缩醛实现分离时,可采用货号:30g-l101-025,描述:fb-wine 30m*0.25mm*0.25um 的毛细管柱;2、 在对非常复杂的酒样分析(30种成份以上)且同时需要对乙酸乙酯与乙缩醛实现分离时,可采用货号:60g-l101-025,描述:fb-wine 60m*0.25mm*0.25um的毛细管柱以确保各种成份一次性达到有效分离;感谢对菲罗门产品的关注和信赖!
厂商
2017.04.20

烟草粗提物中烟碱的制备方法
方法:自订色谱柱:titank c18 5μm 250×4.6mm(货号:fmg-5560-eonu)流动相:20mm 碳酸氢铵溶液-乙腈(80:20)流速:1.0ml/min进样体积:5μl检测波长:260nm柱温:室温
厂商
2017.04.20
