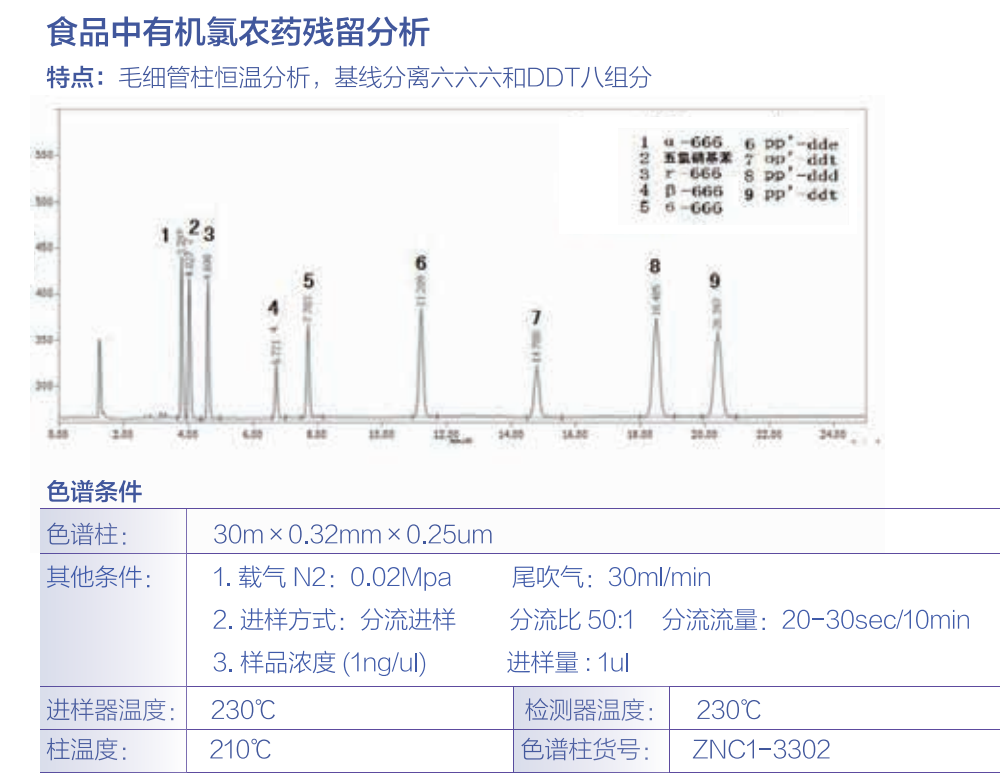
北分三谱食品中有机氯农药多组分残留量的测定1 范围 本标准规定了食品中六六六( HCH) 、滴滴滴( DDD)、六氯苯、灭蚁灵、七氯、氯丹、艾氏剂、狄氏剂、异狄氏剂、硫丹、五氯硝基苯的测定方法。第二法规定了食品中六六六、滴滴涕( DDT) 残留量的测定方法。本标准适用于肉类、蛋类、乳类动物性食品和植 物(含油脂)中a-HCH 、六氯苯、仕HCH 、r- HCH 、五氯硝基苯、o-HCH 、五氯苯胺、七氯、五氯苯基硫酪、艾氏剂、氧氯丹、环氧七氯、反式氯丹、a硫 丹、顺式氯丹、p , p ' -滴滴伊(DDE汃狄氏 剂、异狄氏剂、仕硫丹、p , p ' -DDD、o , p ' -DDT 、异狄氏剂醒、硫丹硫酸盐、p , p' -DDT、异狄氏剂酮、灭蚁灵的分析。第二法适用千各类食品中 HCH 、DDT 残留量的测定。测定的检出限随试样基 质而不同,参见附录 A。第二法的检出限:取 样量 2 g, 终体积为5 rnL, 进样体积为 10 μ.L 时,a- HCH 、/3-HCH 、Y- H C H、o-HCH 依次为 o. 038 μ.g/ kg 、o. 16 μ.g/ kg、o. 047 μ.g/ kg、0. 070μ.g/kg p, p' -DDE、o, p' -DDT 、p , p' -DDD、p , p' -DDT 依次为 o. 23 μ.g/ kg、o. 50 μ.g/ kg、1. 8 μ.g/ kg 、2. 1 μ.g/ kg。 毛细管柱气相色谱-电子捕获检测器法 2 原理 试样中有机氯农药组分经有机溶剂提取、凝胶色谱层析净化,用毛细管柱气相色谱分离,电子捕获 检测器检测,以保留时间定性,外标法定量。3 试剂 3. 1 丙酮( CH3C OCH3) : 分析纯,重蒸。3.2 石油酪:沸程 30 "C ~60c, 分析纯,重蒸。3.3 乙酸乙酷( CH3 CO OC2 比 ):分 析纯,重蒸。3.4 环己烧( C6 H12) : 分析纯,重蒸。3.5 正已烧( n心 H心 :分析纯,重蒸。3.6 氯化钠( NaC l) : 分析纯。3. 7 无水硫酸钠( Na2 S0 4) : 分 析纯,将无水硫酸钠置干燥 箱中,于 120 °C 干燥 4 h, 冷却后,密闭保存。3.8 聚苯乙烯凝胶(Bio-Beads S-X3) : 200 目~ 400 目 ,或同类产品 。3.9 农药标准品: a-六六六(a- HCH ) 、六氯苯( HCB) 、f3-六六六 (/3-H C H) 、Y-六六六 ( Y- HCH ) 、五氯硝基苯(PCNB)、8-六六六 (8- HCH ) 、五氯苯胺( PCA) 、七氯 ( Heptachlor ) 、五氯苯基硫酪 ( PCPs ) 、艾氏剂( Aldrin) 、氧氯丹( Oxychlordane) 、环氧七氯 ( Heptachlor epoxide 入反氯丹( tra ns-chlordane) 、a-硫丹(a-endos ulfan) 、顺氯 丹 ( cis-chlordane ) 、p , p ' - 滴滴伊 ( p , p ' -DDE ) 、狄氏 剂 ( Dieldrin ) 、异狄氏 剂( Endrin) 、f3-硫丹 ( /3-endos ulf an) 、p , p ' - 滴滴滴( p , p' -DDD) 、a , p ' - 滴滴涕( o, p ' -DDT 汃异狄氏剂陛(Endrin aldehyde入硫丹硫 酸盐( Endos ul fan sul fate) 、p , p ' - 滴滴涕( p , p ' -DDT 入异狄氏剂酮( Endrinketone) 、灭蚁灵( Mirex) , 纯度均应不低于 98 %。3. 10 标准溶液的配制:分别准确称取或量取上述农药标准品适量,用少量苯溶解,再用正已烧稀释成 一定浓度的标准储备溶液。量取适量标准储备溶液,用正已烧稀释为系列混合标准溶液。 4 仪器 4. 1 气相色谱仪(GC) : 配有电子捕获检测器( ECD) 。4.2 凝胶净化柱:长 30 cm, 内径 2. 3 cm~2. 5 cm 具活塞玻璃层析柱,柱底垫少许 玻璃棉。用洗脱剂乙酸乙酣-环己烧Cl+ l) 浸泡的凝胶,以湿法装入柱中 ,柱床高约 26 cm, 凝胶始终保待在洗脱剂中。4.3 全自动凝胶色谱 系统:带有固定波长( 254 nm) 紫外检测器 ,供选择使用。4.4 旋转蒸发仪。4.5 组织匀浆器。4.6 振荡器。4. 7 氮气浓缩器。 5 分析步骤 5. 1 试样制备蛋品去壳,制成匀浆;肉品去筋后,切成小块,制成肉糜;乳品混匀待用。5.2 提取与分配5. 2. 1 蛋类:称取试样 20 g ( 精确到 0. 01 g) 千 200 mL 具塞三角瓶中,加水 5 mL ( 视试样水分含量加水,使总水量约为 20 g。通常鲜蛋水分 含量约 75 % , 加水 5 mL 即可),再加入 40 mL 丙酮,振摇 30 min后,加入氯化钠 6 g, 充分摇匀,再加入 30 mL 石油酪,振摇 30 min。静 置分层后,将有机相全部转移至100 mL 具塞三角瓶中经无水 硫酸钠干燥 ,并歉取 35 mL 于旋转蒸发瓶中,浓缩至约 1 mL, 加入 2 mL 乙酸乙酣-环己烧Cl+ l) 溶液再浓缩,如此重复 3 次,浓缩至约 1 mL, 供凝胶色谱层析净化使用,或将浓缩液转移至全自动凝胶 渗透色谱系统 配套的进样试管中,用乙酸乙酷-环己烧Cl+ l) 溶液洗涤旋转蒸发瓶数次,将洗涤液合并 至试管中,定容至 10 mL。5. 2.2 肉类:称取试样 20 g ( 精确到 0. 01 g), 加水15 mL ( 视试样水分含量加水,使总水量约 20 g ) 。加40 mL 丙酮,振摇 30 min,以下按照 5. 2. 1 蛋类试样的提取、分配步骤处理。5.2.3 乳类:称取试样 20 g( 精确到o. 01 g), 鲜乳不需加水,直接加丙酮提取。以下按照 5. 2. 1 蛋类试样的提取、分配步骤处理。5.2.4 大豆油:称取试样 1 g ( 精确到 0. 01 g), 直接加入 30 mL 石油酪,振摇 30 min 后,将有机相全部转移至旋转蒸发瓶 中,浓缩至约 1 mL, 加 2 mL 乙酸乙酣-环己烧Cl+ l) 溶液再浓缩,如此重复 3 次,浓缩至约 1 mL, 供凝胶色谱层析净化使用,或将浓缩液转移至全自动凝胶渗透色谱系统配套的进样试管中,用乙酸乙百护环己 烧Cl+ l) 溶液洗涤旋转蒸发瓶数次,将洗涤液合并 至试管中,定容至 10 mL 。5.2.5 植物类:称取试样匀浆 20 g, 加水 5 mL ( 视其水分含量加水,使总 水量约 20 mL), 加丙酮40 mL, 振荡 30 min, 加氯化钠 6 g, 摇匀。加石油酪 30 mL, 再振荡 30 min , 以下按照 5. 2. 1 蛋类试样的提取、分配步骤处理。5.3 净化选择手动或全自动净化方法的任何一种进行。5. 3. 1 手动凝胶色谱柱净化:将试样浓缩液经凝胶柱以乙酸乙百昔环己 烧Cl+ D 溶液洗脱,弃去 0 mL~ 35 mL 流分 ,收集 35 mL~70 mL 流分。将其旋转蒸发浓缩至约 1 mL, 再经凝胶柱净化收集 35 mL~ 70 mL流分,蒸发浓缩,用氮气吹除溶剂 ,用正已烧定容至 1 mL, 留待 GC 分析。5.3.2 全自动凝胶渗透色谱系统净化:试 样由 5 mL 试样环注入凝胶渗透色谱( GPC) 柱,泵流速5. 0 mL/min, 以乙酸乙酣-环己烧Cl+ D 溶液洗脱,弃去 0 min~7. 5 min 流分,收集 7. 5 min~15 min流分,1 5 min~20 min 冲洗 GPC 柱。将收集的流分旋转蒸发浓缩至约 1 mL, 用氮气吹至近干,用正已烧定容至 1 mL, 留待 GC 分析。5.4 测定5. 4. 1 气相色谱参考条件5. 4. 1. 1 色谱柱: DM-5 石英弹性毛细管柱 ,长 30 m、内径0. 32 mm 、膜厚0. 25 p.m 或等效柱。5. 4. 1. 2 柱温:程序升温90°CC1 min)40 • c /m in l 70 • c2 3 • c / m in 230 °C (l 7 min)40• c/min280°C(5 min)5. 4. 1. 3 进样口温度: 280 "C 。 不分流进样 ,进样量 1 μ.L。5. 4. 1. 4 检测器:电子捕获检测器 ( ECO) , 温度 300 c 。5. 4. 1. 5 载气流速:氮气C N2 汃流速 1 mL/min 尾吹,25 mL/ min 。5. 4. 1. 6 柱前压: 0. 5 MPa。5.4.2 色谱分析分别吸取 1 μ.L 混合标准液及试样净 化液注入气相色谱仪中,记录色谱图 ,以保留时间定性,以试样和标准的峰高或峰面积比较定量。
 留言咨询
留言咨询

 400-860-5168转2141
400-860-5168转2141














 我要推广仪器
我要推广仪器
 下载APP
下载APP