
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转3457
仪器信息网认证电话,请放心拨打

氟代烷基化:Togni试剂的拓展
氟代烷基化:Togni试剂的拓展简介在药物和农药候选化合物中引入高度氟化基团是调节其性质强有力的策略。引入含氟侧链可以调节酸碱性和亲脂性,改变偶极矩,锁定确认,减轻母体化合物的不良代谢降解。由于合成途径的局限性,含氟基团通常仅限于单个氟或三氟甲基。通过开发一套新的试剂,现在可以进行更精细的氟代烷基化,目前的氟烷基化工具包括Togni试剂、高价碘全氟烷基化试剂、氟烷基溴化物、硅烷类、羧酸盐类以及通用于后期氟烷基化的磺酰氟类,与此同时还可与多种敏感的官能团兼容,适用性十分广泛。高价碘试剂在过去十年中,Togni试剂已成为一种标准工具,为药物和农药研发提供了化合物三氟甲基化的可行途径[1]。这些高价碘化合物是直接、温和和高效三氟甲基化的亲电CF3转移试剂,并且在许多情况下通过三氟甲基自由基作为关键反应中间体。最近,水解稳定性和热稳定性均有提升的其他环状高价碘试剂已被开发为温和且方便处理的亲电氯化、氟化和叠氮化试剂。易储藏的氟碘烷试剂可在温和条件下对烯烃进行良好的氟官能团化和氟环化,而叠氮碘烷试剂可以用作烯醇化物叠氮化的亲电叠氮化试剂。Togni全氟烷基试剂使用CF3-Togni试剂也可以进行许多类型的转化,从而获得稀有且具有潜在吸引力的氟化化学空间[2]。拥有一套“扩展的Togni试剂”,先导化合物可以在合成的最后阶段变得多样化,以提供难以获得的氟烷基修饰衍生物。2CF2-“扩展的Togni试剂”与烯烃和乙炔进行自由基环化反应,从而获得罕见的四氟杂环官能团[3]。将-CF2CF2-部分结合到环状结构中,赋予分子一种称为“极性疏水性”的独特性质组合——永久电偶极子与四氟乙烯单元的疏水性相结合。氟代烷基溴化物被取代的氟烷基溴化物在被异丙基氯化镁-氯化锂复合物(Turbo-Grignard)金属化后,变成强大的亲核性氟烷基化试剂[4]。原位生成的氟烷基氯化镁中间体在-40℃以下都是适度稳定的,并且可以有效地与各种亲电物截留,得到-CF2CF2-连接的产物。氟代烷基硅烷氟取代的烷基硅烷用作氟代烷基合成子的传统亲核源。在用催化氟化物或醇盐活化时,它们可以使一系列醛、反应性酮或亚胺氟烷基化。硅烷还可以参与过渡金属催化的R-CF2CF2取代的芳族化合物的形成。四氟丙酸酯β-取代的四氟丙酸铯是便于构建氟代烷基甲酰胺的原料。这类酰胺基团的pKa值显著低于其非氟化对应物,大大提升了该化合物作为候选药物分子的生理和药理潜力。与极易吸潮的游离羧酸相比,铯盐的吸潮性降低,因此铯盐的相关反应在空气条件下也可操作。氟代烷基磺酰氟氟代烷基磺酰氟化物可作为反应活性适中的亲电氟代烷基磺酰化试剂。尽管与其同属性的氟代烷基磺酰氯也可以作为亲电氯化试剂对胺起作用,提供不需要的N-氯代胺,但是氟代烷基磺酰氟化物作为反应物参与时,氮磺酰化反应反应速率缓慢且反应体系干净,从而得到相应的磺酰胺。氨基氮原子的氟代烷基磺酰化大大降低了NH基团的pKa值,用以调节候选药物分子的行为或在高酸性氟代烷基磺酰胺周围构建附加的分子复杂性。结论由于有机氟化学对生物制药领域和农作物保护行业的意义越来越大,越来越依赖于新型试剂和反应的发展。这一类基于超价碘支架的新型官能化四氟乙基试剂,可以将有效地氟原子转移到大量的有机分子上,这一概念促进了对其拓展到更复杂的(功能化)氟代烷基的发展。参考文献1.Charpentier J, Früh N, Togni A. 2015. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev.. 115(2):650-682. https://doi.org/10.1021/cr500223h2.Matoušek V, Václavík J, Hájek P, Charpentier J, Blastik ZE, Pietrasiak E, Budinská A, Togni A, Beier P. 2016. Expanding the Scope of Hypervalent Iodine Reagents for Perfluoroalkylation: From Trifluoromethyl to Functionalized Perfluoroethyl. Chem. Eur. J.. 22(1):417-424. https://doi.org/10.1002/chem.2015035313.Charpentier J, Früh N, Foser S, Togni A. 2016. Tandem Radical Fluoroalkylation?Cyclization: Synthesis of Tetrafluoro Imidazopyridines. Org. Lett.. 18(4):756-759. https://doi.org/10.1021/acs.orglett.6b000184.Budinská A, Václavík J, Matou?ek V, Beier P. 2016. Nucleophilic Tetrafluoroethylation Employing in Situ Formed Organomagnesium Reagents. Org. Lett.. 18(22):5844-5847. https://doi.org/10.1021/acs.orglett.6b02890
应用实例
2023.03.31

阿拉丁热聘----欢迎加入阿拉丁大家庭
加入阿拉丁 并与我们一起探索 一种包容、成长和创新的企业文化 伯乐识良马,明主觅良才! 你是否还在寻找平台施展才华,却仍没有收获? 来阿拉丁,让你找到风向标。 你能飞得比自己相像的更高,更远! 你,就是我们一直寻找的人! 阿拉丁热聘职位加入阿拉丁!这里又无限的可能性和宽广的平台。只要你有必胜的决心,这里会成就不一样的你!
企业动态
2023.03.28

阿拉丁产品新标签——您的试剂使用指南
•更温馨、更自然新标签上彩色圆圈的点缀、黄蓝红的撞色,简约清新、活泼轻快,为阿拉丁产品更添一抹灵动。•更全面、更专业标签设计举例:1)常规产品——a——产品中英文名称;b——产品货号,字母&数字-包装规格;c——产品纯度、等级、含量等核心参数;d——产品主要组分的CAS号(CAS Registry Number或称CAS Number, CAS Rn, CAS #,又称CAS登录号或CAS登记号码,是某种物质,化合物、高分子材料、生物序列(Biological sequences)、混合物或合金的唯一的数字识别号码);e——产品化学式;f——产品主要组分的分子量(MW);g——此包装产品的批次号(可根据货号、批次号查询对应的质检参数,下载电子版COA);h——此包装产品批次号二维码,与g同,可在质检、包装、发货环节中减少人工校对环节,通过对应扫码枪扫码录入即可;i——产品存储、运输等过程中的注意事项,如需避光、冷藏、冷冻、惰性气体保护等;j——GHS标签,产品危险等级象形图,共9种,是由联合国开发的,旨在统一不同国家的化学品法规和标准,如下图:k——阿拉丁公司全称、网址、邮箱、地址、电话,及产品仅供研发使用说明;l——产品安全信息。2)具生产许可产品——m——QS生产许可标志(涉及产品包括石油醚、乙酸、苯、苯胺、吡啶、过硫酸铵、过氧化氢溶液、环己酮、甲醇、甲基异丁基甲酮、甲酸、磷酸、二水氯化钡、硼酸、氢氧化钡八水合物、硝基苯、硝酸钡、硝酸银、异丙醇、氟化铵、1,2-二氯乙烷、二氯甲烷、N,N-二甲基甲酰胺、结晶四氯化锡、乙酸乙酯、铬酸钾、高氯酸、氢氧化钾、亚硝酸钠等);n——产品质量执行标准及质控参数;o——产品国标等级;p——产品理化性状。
企业动态
2023.03.27

PANoptosis: 一种促炎性程序性细胞死亡途径
PANoptosis: 一种促炎性程序性细胞死亡途径程序性细胞死亡途径被先天免疫系统激活,以应对微生物感染和其他细胞应激源。焦亡、凋亡和坏死是三种程序性细胞死亡途径,已被广泛研究,并具有很好的特征。圣裘德儿童研究医院的Kanneganti小组的工作表明,这三种途径并不总是相互孤立地运作他们的工作描述了一种新的炎症程序性细胞死亡途径,PAN凋亡。之所以被称为PAN凋亡,是因为它涉及细胞死亡、凋亡和程序性坏死的一系列集体行为。PAN细胞死亡可由多种细菌和病毒病原体诱导,包括单核增生李斯特菌、肠链球菌血清型鼠伤寒、水泡性口炎病毒(VSV)和甲型流感病毒(IAV)全视缺失也可能在重症COVID-19患者的炎症中发挥作用,在本文中后者是由严重急性呼吸综合征冠状病毒2 (SARS-CoV-2)引起的,我们综述了这种新描述的程序性细胞死亡途径的机制和调控,冠状病毒如何触发程序性细胞死亡,并讨论了最近的研究表明PAN细胞凋亡在covid -19相关炎症中的作用,为通过调节这种细胞死亡途径进行治疗干预提供了可能的机会。PAN细胞凋亡由PAN凋亡体调节PAN细胞凋亡的主调控因子是一种称为PAN凋亡体的多聚体细胞质蛋白复合体,由参与死亡、凋亡和程序性坏死的蛋白质组成。1,4它可以包括NOD-、LRR-和含吡啶结构域蛋白3 (NLRP3),含有CARD的凋亡相关斑点样蛋白(ASC),以及细胞凋亡蛋白酶,在死亡和炎症小体中起作用的蛋白质(图1)。凋亡蛋白半胱天冬酶-8抗原和坏死蛋白受体相互作用蛋白激酶1 (RIPK1)和RIPK3也可以被纳入PAN凋亡体。其他蛋白质成分包括半胱胺酸蛋白酶蛋白,作为支架,Z-DNA结合蛋白1 (ZBP1),作为先天免疫传感器,脂肪酸合成酶相关死亡结构域(FADD),作为适配器。通过PAN凋亡体的信号通路激活下游效应子gasdermin D (GSDMD), 半胱天冬酶-3和-7,以及混合谱系激酶结构域样(MLKL),分别执行细胞死亡, 凋亡和程序性坏死。图1。ZBP1依赖的PAN凋亡体的激活在IAV感染时触发死亡、凋亡和坏死。天然免疫传感器ZBP1是PAN凋亡体形成和激活的正向调节因子。1,4,5在IAV感染过程中,ZBP1识别病毒核糖核蛋白并诱导形成ZBP1依赖的泛体。依赖于ZBP1的泛光体由ZBP1(传感器)、RIPK3和RIPK1(坏死蛋白)、NLRP3、ASC、细胞凋亡蛋白酶-1(炎症小体/坏死蛋白)、细胞凋亡蛋白酶-8(凋亡蛋白)和支架细胞凋亡蛋白酶-6(图1)组成。该PAN凋亡体的形成导致RIPK3、细胞凋亡蛋白酶-8和NLRP3炎症小体的激活,导致PAN凋亡。而TGF-β-活化激酶1 (TAK1)则是PAN细胞凋亡的负调控因子。TAK1的抑制与toll样受体(TLRs)或死亡受体(如TNF受体1 (TNFR1)、Fas、TRAIL-R或DR3)的信号耦合,促进ripk1依赖的PAN凋亡体的形成(图2)一种以这种方式诱导泛光症的细菌是耶尔森氏菌。耶尔森氏菌致病菌株可将效应蛋白YopJ分泌到巨噬细胞中抑制TAK1和NF-κB激酶抑制剂(IKK)。2,4这导致PAN凋亡的激活,细胞内病原体清除,以及包括IL-1β和IL-18在内的促炎细胞因子的释放。图2。致病性耶尔森氏菌通过抑制TAK1触发PAN凋亡,从而导致PAN凋亡体激活。如上所述,IAV、VSV、单核增生L.和肠链球菌血清型鼠伤寒都能诱导巨噬细胞PAN细胞死亡仅抑制死亡、凋亡或坏死不足以保护巨噬细胞免受病原体诱导的细胞死亡。只有同时抑制这三种途径,例如通过删除编码细胞凋亡蛋白酶-1和细胞凋亡蛋白酶-11、细胞凋亡蛋白酶-8和坏死蛋白RIPK3的基因,才能保护巨噬细胞免受这些病原体诱导的PAN细胞死亡。这表明PAN凋亡的所有三个分支都是由细胞参与并介导细胞死亡以应对这些病原体。冠状病毒激活程序性细胞死亡途径乙型冠状病毒SARS-CoV、SARS-CoV-2、中东呼吸综合征冠状病毒(MERS-CoV)和小鼠肝炎病毒(MHV)已被证明可激活程序性细胞死亡途径(图3)图3。冠状病毒激活多种程序性细胞死亡途径。SARS-CoV、SARS-CoV-2和MERS-CoV均可致死亡和凋亡在SARS-CoV和SARS-CoV-2的情况下,这些病原体诱导的死亡伴随着促炎细胞因子IL-1β的分泌,以及NLRP3炎症小体的激活。HCoV-OC43是一种可导致普通感冒的冠状病毒,可诱导人类神经细胞坏死。还需要更多的研究来确定SARS-CoV、SARS-CoV-2和MERS-CoV是否也能诱导坏死。然而,小鼠冠状病毒MHV已被证明可以激活小鼠巨噬细胞中的所有三种泛光性细胞死亡途径,并且这种泛光性细胞死亡伴随着促炎细胞因子IL-1β、IL-18、IL-6和TNF.7的释放。TNF-α和IFN-γ诱导PAN细胞死亡和类似COVID-19的炎症Karki等人最近的研究表明,PAN凋亡可能在严重COVID-19.3患者的炎症反应中发挥作用。在小鼠中,促炎细胞因子TNF-α和IFN-γ的联合给药会导致死亡率增加,以及反映严重COVID-19患者观察到的各种表型,包括血清谷丙转氨酶(ALT)、天门冬氨酸转氨酶(AST)、血尿素氮(BUN)和铁蛋白水平升高。还有血小板减少和嗜中性粒细胞与淋巴细胞比例增加。在细胞中,TNF-α和IFN-γ诱导全视凋亡和细胞死亡。缺乏PAN凋亡体成分RIPK3和细胞凋亡蛋白酶-8的小鼠免受TNF-α-和IFN-γ-诱导的死亡。来自这些小鼠的巨噬细胞也受到TNF-α-和IFN-γ-诱导的全视和死亡的保护。这表明PAN凋亡是由TNF-α和IFN-γ联合诱导的,其病理与严重的COVID-19有相似之处。作者接下来试图阐明TNF-α和IFN-γ.3诱导泛光症的信号通路他们发现JAK/STAT1/IRF1信号通路对调节很重要。在这一途径中,Janus激酶2 (JAK2)磷酸化JAK1,然后激活转录因子STAT1,诱导包括IFN调节因子1 (Irf1)在内的基因的转录(图4)。在TNF-α和IFN-γ处理的小鼠巨噬细胞中,JAK2和Irf1基因上调,以及在重症COVID-19患者中。通过删除Irf1或Stat1来破坏这一信号通路,可以保护小鼠巨噬细胞免受TNF-α-和IFN-γ-诱导的细胞死亡,以及在Irf1缺失的情况下诱导全视缺失。类似地,Stat1-/-小鼠免受TNF-α-和IFN-γ-诱导的死亡。图4。TNF-α和IFN-γ诱导JAK/STAT1/IRF1信号通路和PAN凋亡。Karki等人还证明,由JAK/STAT1/IRF1途径诱导的一氧化氮(NO)生成有助于TNF-α-和IFN-γ-诱导的PAN细胞死亡(图4)。3 IRF1 -/-小鼠巨噬细胞可降低诱导型一氧化氮合酶(iNOS)及其编码基因Nos2的表达。与此一致的是,与野生型细胞相比,当TNF-α和IFN-γ刺激时,Irf1-/-和Stat1-/-细胞都减少了NO的产生。通过删除Nos2或使用NO生成抑制剂L-NAME或1400W来干扰NO生成,可以保护细胞免受TNF-α-和IFN-γ-诱导的细胞死亡。鉴于用TNF-α和IFN-γ联合治疗小鼠会诱导与COVID-19患者相似的症状,作者探索了阻断TNF-α和IFN-γ信号通路是否能在小鼠感染模型中预防sars - cov -2诱导的死亡。他们发现,使用针对这两种细胞因子的中和抗体组合来阻断TNF-α和IFN-γ,可以保护小鼠免受SARS-CoV-2引起的死亡。对抗TNF-α和IFN-γ的中和抗体也保护小鼠免受致命剂量的LPS或多聚I:C启动后的LPS组合诱导的死亡,这模拟了严重的全身炎症综合征噬血细胞淋巴组织细胞增多症(HLH)。这些结果表明TNF-α-和INF-γ-介导的泛光缺失参与了包括COVID-19在内的多种炎症疾病的病理,并为这些疾病的治疗提供了新的探索途径。阿拉丁提供了多种工具来研究PAN凋亡和其他程序性细胞死亡途径,包括PAN凋亡体成分和调节剂的小分子抑制剂,其他细胞死亡途径,JAK/STAT信号,和诱导性一氧化氮合酶产生。PAN凋亡体抑制剂组成与调节蛋白PAN凋亡体抑制剂组成/调节蛋白抑制剂PAN凋亡体抑制剂组成/调节蛋白抑制剂NLRP3 NLRP3iRIPK3/RIP3 KinaseGSK872MCC950RIPK1/RIP1 Kinase (±)-Necrostatin-2DapansutrileNecrostatin-1INF39Necrostatin-5Caspase-1 Ac-YVAD-CMKGSK2982772Ac-YVAD-CHOGSK481VX-765TAK1(5Z)-7-OxozeaenolCaspase-8Ac-IETD-CHO (trifluoroacetate salt)TakinibCaspase-6Ac-VEID-CHO (trifluoroacetate salt)其余与细胞死亡、凋亡和坏死有关的抑制剂靶标蛋白抑制剂靶标蛋白抑制剂Pan-细胞凋亡蛋白酶Z-VAD(OMe)-FMK细胞凋亡蛋白酶-3 Z-DEVD-FMKQ-VD-OPHAc-DEVD-CMKZ-VAD(OH)-FMKCaspase-3 Inhibitor VIIEmricasan细胞凋亡蛋白酶-1/3Z-YVAD-CMK (trifluoroacetate salt)Boc-D-FMKMLKLNecrosulfonamideZ-Asp-CH2-DCB程序性死亡Necrostatin-2细胞凋亡蛋白酶-3/7Ac-DEVD-CHONecrostatin-7Caspase-3/7 Inhibitor IJAK 抑制剂iNOS 抑制剂RuxolitinibL-NAME (hydrochloride)Baricitinib1400W (hydrochloride)FilgotinibL-NIL (hydrochloride)TG101348 (Fedratinib)Diphenyleneiodonium (chloride)查看所有 JAK抑制剂产品AMT (hydrochloride)Aminoguanidine (hydrochloride)
应用实例
2023.03.24

砌块丨阿拉丁全方位“氟”务您的科研事业!
含氟砌块是一类含有氟原子或含氟基团的有机化合物。由于氟原子电负性强、原子半径小和极化率低,含氟砌块通常具有独特的物理、化学和生理特性。 因此将氟原子或含氟基团掺入有机分子中,可有效调整分子的物理、化学性质或生物学性质,使得含氟砌块在药物化学、农业化学和材料化学各领域均发挥着日益重要的作用。 (一)氟让药物更高效 目前世界上已商品化和正在开发的含氟药物有百余种,每年上市的新药中大约有20%都含有一个或多个氟原子。含氟药物凭借其高效、低毒、易代谢等特点,在医药领域应用发挥着至关重要的作用。 「 阿拉丁含氟砌块医药中间体应用产品 」 产品名称阿拉丁货号cas号医药应用结构式三氟乙酸乙酯E105562383-63-1 新冠口服药Paxlovid中间体2,4-二氯氟苯D1077541435-48-9新型抗菌剂氟喹诺酮类药物中间体4-氟苯酚F107127371-41-5 抗炎和抗类风湿药物索比尼尔中间体对氟苯甲酰氯F109226403-43-0心脑血管药物氟伐他汀钠中间体3-氯-4-氟苯胺C106553367-21-5 抗菌药物诺氟沙星中间体4-溴-2-氟苯胺B115549367-24-8 抗炎药布洛芬中间体 (二)氟让农业更高产 含氟农药在农药领域占有重要地位,在农药中引入氟进一步提高了其化学稳定性和药效。 (三)氟让材料更高能 含氟材料也具有许多特殊性能,例如被誉为“塑料之王”的聚四氟乙烯(PTFE),也称作特氟隆。应用领域从航空航天、军工等国防领域涵盖到石油化工、电子电器等国民经济领域。 阿拉丁可提供含氟砌块产品近2000种,现货率高达80+%。此外,有机砌块、杂环砌块和卤代杂环砌块等砌块产品50000余种。含氟砌块系列产品阿拉丁有如下三大特点:1、种类丰富:涵盖杂环含氟砌块、脂肪族含氟砌块、芳香族含氟砌块;包含三氟甲基、二氟甲基、二氟甲氧基等各种含氟基团。2、质量卓越:产品纯度高,批次稳定性好。3、规格齐全:包装规格既有小包装,也支持大包装定制。 「 热销 Top 10 产品 」 产品货号产品名称纯度包装规格B102576双三氟甲烷磺酰亚胺锂99%1g/5g/10g/25g/50g/100g/250g/500gT1627091,1,2,2-四氟乙基2,2,3,3-四氟丙醚98%1g/5g/25g/100g/500g/1kgE105562三氟乙酸乙酯99%25g/100g/250g/500g/1kgH1572081H,1H,2H,2H-全氟-1-辛醇>98.0%(GC)5g/25g/100g/500gT1617322,2,2-三氟-N,N-二甲基乙酰胺≥95.0%5g/25g/100gS102743三氟甲磺酸钠98%5g/25g/100g/500gT101190(三氟甲氧基)苯98%5g/25g/100g/500gH106257七氟丁酸98%5g/25g/100g/500gB1203134-溴-2-氟联苯98%5g/25g/100g/250g/500gT107263三乙胺三氢氟酸盐97%5g/25g/100g/500g/2.5kg 文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请在阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
应用实例
2023.03.21

稳定性同位素的应用
稳定性同位素的应用蛋白质组学在过去的十年中,基于质谱(MS)的蛋白质组学已经成为生物学家的重要工具。质谱仪能够从复杂的生物样本中鉴定出数千种蛋白质,这一能力给科学实验带来巨大的变化。然而,为了充分了解蛋白质组在健康和疾病中的功能,我们必须有能力准确定量许多不同类型的生物样本中的蛋白质。更快和更高分辨率的质谱仪的发明使复杂的蛋白质组动力学定量成为可能。质量更大的稳定性同位素通常用于生成精确和准确的定量蛋白质组学数据。被重稳定同位素标记的肽与“轻”或未标记的肽具有相同的生化特性,只是质量不同。将重肽与轻肽混合后形成的多肽对共洗脱进入质谱仪中,质谱仪可以根据其质量差异轻松区分多肽。当使用重肽作为内标准或对照时,可以实现在不同生物条件或实验下的蛋白质组之间的定量差异。阿拉丁可以提供多种稳定的同位素试剂,用于标记任何用于定量质谱分析的蛋白质组。通过对疾病动物模型的定量生物标志物分析和定量蛋白质组学分析,这些稳定同位素质谱的分析方法使科学家离治愈人类疾病更近了一步。代谢标记一种蛋白质组标记将稳定同位素氨基酸引入细胞生长培养基或啮齿类动物饲料。生长期和摄食期允许同位素标记的稳定氨基酸以代谢方式并入蛋白质组。涉及细胞培养的实验称为SILAC(细胞培养中氨基酸的稳定同位素标记),而哺乳动物系统称为SILAM(哺乳动物的稳定同位素标记)。每种代谢标记技术在下文中会做具体阐述。SILACSILAC是指用重氨基酸标记培养细胞进行定量蛋白质组学分析。用体内重氨基酸标记整个蛋白质组是定量蛋白质组学的理想标准。当重标记的蛋白质组与未标记的蛋白质组混合后进行消化,每一个经质谱鉴定的未标记肽都可以通过其相应的重肽进行定量。在SILAC中,胰蛋白酶氨基酸、精氨酸(R)和赖氨酸(K)均含有重稳定同位素,所以如果用胰蛋白酶消化,每一个肽都被标记。这种代谢标记策略已被多个蛋白质组学研究采用。与体外标记技术相比,代谢标记技术的优点是在样品制备前将重的和未标记的样品混合,防止不同制剂之间的差异影响最终定量结果。当需要大量样品制备(例如分离细胞器)时,这一点尤其重要。SILAMSILAM是指用重稳定同位素标记整个啮齿类动物用于定量蛋白质组学组织分析。体内重同位素标记整个蛋白质组是定量蛋白质组学的理想标准。当重标记的蛋白质组与未标记的蛋白质组混合后进行消化,每一个经质谱鉴定的未标记肽都可以通过其相应的重肽进行定量。与体外标记技术相比,代谢标记技术的优点是在样品制备前将重的和未标记的样品混合,防止不同制剂之间的差异影响定量结果。当需要大量样品制备(例如分离细胞器)时,这一点尤其重要。在SILAM中,啮齿类动物的食物会被改变为含有重赖氨酸或15N-螺旋藻作为唯一的蛋白质来源。重组织被用作基础哺乳动物生理学和疾病动物模型定量蛋白质组学分析的内部标准。酶标记将两个18O原子整合到生物样品的蛋白水解酶的肽的羧基端已成为全球领先的标记策略之一,用于比较定量蛋白质组学。该技术的成功部分归因于18O水相对较低的成本,“重”肽的分子量增加了+4道尔顿质量,以及能够从反相HPLC中共洗脱18O/16O肽对。化学标记用于蛋白质组学测量的同位素标记标准物可以用化学方法制备。这可以在肽或蛋白质水平分别通过固相合成或重组基因表达来实现。为了帮助合成稳定的同位素标记肽,阿拉丁提供了一系列受保护的氨基酸和预加载树脂,除此之外还提供部分全长蛋白(例如CRP、泛素),用于自下而上的LC-MS实验。多肽合成靶向质谱同位素分析(即选定的反应监测或SRM)是用于验证临床相关生物标志物的基于抗体的检测的替代方法,但也被用于基于发现的定量蛋白质组学。这种策略的一个障碍是每一种肽都具有独特的生化特性,其氨基酸组成和可能的翻译后修饰决定了其从液相色谱柱、电离和碎片化的洗脱特性。为了开发诊断性临床质谱测定,必须在对体内分析有效的肽之前用合成肽表征这些肽的性质。还可通过将已知量的重肽添加到生物样品中,利用重稳定同位素合成肽用于绝对定量。这些策略也常用于验证大规模定量蛋白质组学分析的结果。蛋白表达稳定同位素标记的细胞生物量可用于蛋白质组学和代谢组学研究。此外,利用纯化的、标记完整的蛋白质作为内标,定量、蛋白质组学的质谱研究可以大大受益。使用正确折叠、标记完整的蛋白质是理想的内部标准,因为它们将尽可能接近地模拟样品中内源性目标蛋白在消化前、消化中和消化后的物理和化学性质。特别是,它们将经历与未标记的对应物相似的蛋白水解裂解程度,从而提高同位素稀释质谱(IDMS)实验结果的准确性,无论是中向下还是自下而上的方法。化学标记重同位素在蛋白质组中的代谢掺入(如SILAC和SILAM)是一种制备内标或标记对照的常用方法;然而,有些生物和动物并不适合代谢掺入。幸运的是,分析物可以很容易地通过化学标记反应进行修饰。实例包括蛋白质或肽中伯胺的还原以及蛋白质组样本中游离N-连接聚糖的肼标记。代谢研究阿拉丁可以提供稳定同位素标记的代谢底物,这些代谢底物用13C、15N、18O、D等稳定同位素标记。这些材料的一些应用包括利用氨基酸进行蛋白质周转研究、利用碳水化合物进行葡萄糖代谢研究、利用脂肪酸进行脂肪分解研究。这些物质的稳定同位素标记使研究者能够以一种安全、准确和无创的方式研究生命系统中的代谢途径。同位素稀释质谱法(IDMS)是一种准确、灵敏、可重复性强、广泛应用于各种样品类型的中小分子定量分析的方法。稳定同位素富集的化合物成为质谱比较或绝对定量的理想内部标准的一个主要原因是,同一化合物的“重”(同位素富集)和“轻”(天然)形式的分离信号可以同时被检测到。13C和15N核具有核磁共振活性,因此富含这些同位素的化合物可用于磁共振检测。13C核具有大的化学位移范围和良好的弛豫特性,使13C富集底物成为极具价值的细胞化学和代谢探针,尤其是在快速发展的超极化领域。新陈代谢研究人员使用稳定同位素技术研究各种各样的代谢紊乱和疾病,包括阿尔茨海默症、帕金森、癌症、糖尿病和肥胖。同位素是代谢研究中最常用的示踪剂,用于定量体内的生化或代谢反应。它们可以用于研究代谢途径,确定生物标志物,测试药物的效果,以及开发特定状态下生物系统的代谢概况。制药用氘代试剂近年来,一些制药公司已经开始研究氘代分子,这些分子可能具有现有的非氘代分子不具备的优势。此外,目前对新型氘代药物潜在医疗优势的研究也在增加。稳定的同位素标记合成中间体氘代药物的潜在优势包括:•改善代谢特征:改善的代谢特征有可能减少或消除不必要的副作用或不良的药物相互作用;•提高口服生物利用度:一些化合物中的氘化减少了消化道发生的系统前代谢,从而使更多未代谢的药物达到目标;•增加的半衰期:氘代化合物的药代动力学作用较慢,可延长在体内的吸收和分布。与使用非氘代药物的患者相比,这可减少患者在某一时间段内可能需要的剂量数。用于光电的氘代试剂阿拉丁可以提供一系列常用于微电子和OLED制造的氘代有机分子和氘代气体,有助于提升器件的使用寿命。有机发光二极管用氧化氘有机发光二极管(OLED)被广泛应用于电视和手机屏幕等设备。OLED通常由两个电极之间的薄层有机分子构成。当电流通过这些设备时,它们就会发光。直到几年前,OLED最大的技术问题是有机材料的寿命有限,通常是LCD、LED或PDP的一半,因为在运行过程中产生的热量和氧化会导致化学物质的不稳定。这个问题通过对OLED中的一些有机分子进行氘化处理得以解决,这将器件的寿命提高了5到20倍,而不会显著影响器件的其他性能。氘代在该领域的另一个应用是中子反射和特定分子层的氘化,这已成为研究有机薄膜半导体器件的形态、扩散和界面行为的关键方法。光导纤维与传统的铜线相比,光纤被广泛用于长距离传输数据和更高的带宽(数据速率)。然而,在一个互联网驱动的世界中,数据的及时性、实现Gbps范围内的数据传输是至关重要的。传统的玻璃或塑料光纤由于吸收水的峰值在1360 nm和1460 nm之间而速度有限。现在,用氘取代材料中的氢,使得达到与当今需求相适应的更高速度成为可能。
操作维护
2023.03.20

春季乐购 满额焕新礼
企业动态
2023.03.17

含氟氮杂吲哚的高效合成
简介氮杂吲哚及其衍生物具有显著的生物活性,因此在药物优化策略方面受到广泛关注。四种氮杂吲哚位置异构体通过稠合的C-C键将吡啶和吡咯环连接在一起,同时具有成为吲哚或嘌呤体系的生物电子等排体所需的所有标准。使用氮杂吲哚代替其他双环稠合杂环可以调节和微调化合物的溶解度、pKa值和亲脂性、靶结合能力以及毒性等理化性质和药理性质。图1.氮杂吲哚的四种氮杂吲哚位置异构体合成含氟杂环化合物具有适合用于医药和农业领域的独有优势,特别是它们在生物体内对膜、组织的穿透能力的增强,有助于含氟化合物的吸收、传递和扩散。含氟氮杂吲哚合成过程中的挑战是以往是通过Balz-Schiemann反应或通过亲电氟化实现的选择性芳香族氟化。中性芳香族化合物亲电氟化的缺点是它通常会产生单氟化和多氟化产物的混合物。另一方面,Balz-Schiemann反应通过四氟硼酸重氮盐的受控热分解提供区域选择性的单氟芳基氟化物。2003年,C. Thibault等人报告了两种合成4-氟-1H-吡咯并[2,3-b]吡啶(4-fluoro-1H-pyrrolo[2,3-b]pyridine),别名4-氟-7-氮杂吲哚的方法。第一种方法利用了区域选择性的Balz-Schiemann氟化反应,这需要合成中间体胺[1]。在DMF中使用甲磺酰氯在C-4处对1H-吡咯并[2,3-b]吡啶7-氧化物进行区域选择性氯化[2]。使用氯化物和N-烯丙基胺进行Buchwald-palladium催化胺化[3]生成烯丙基胺。随后,在酸性醇溶液中使用碳钯进行脱烯基化,得到1H-吡咯并[2,3-b]吡啶-4-胺[4]。然后将其置于Balz-Schiemann反应条件下[5],热解得到氟化芳香族化合物(方案1)。 方案1.通过Balz-Schiemann反应合成含氟氮杂吲哚上述Balz-Schiemann路线提供了一个四步合成4-氟-1H-吡咯并[2,3-b]吡啶的方法。第二种方法尝试将离去基团从氯化物改变为溴化物,在锂卤素交换反应中使用溴化物,然后用亲电氟试剂处理,生成4-氟-1H-吡咯并[2,3-b]吡啶[6](方案2)。 方案2. 通过亲电氟化反应合成含氟氮杂吲哚在DMF中用甲磺酸酐和溴化四甲基铵处理N-氧化物得到4-溴-1H-吡咯并[2,3-b]吡啶,然后其被保护为N-三异丙基硅基衍生物。在-78°C的THF中使用叔丁基锂进行溴化物的锂-卤素交换[7],然后添加N-氟代双苯磺酰胺得到4-氟-1H-吡咯并[2,3-b]吡啶[8]。总之,以上两种合成4-氟-1H-吡咯并[2,3-b]吡啶的路线:第一种合成方法的特点是在室温下进行Balz-Schiemann反应。第二种方法的特点是相应的溴化物的有效锂-卤素交换,然后用亲电氟源淬灭。应用含氟氮杂吲哚衍生物,特别是含氟氮杂吲哚已被公认为是生物过程调节、药物化学和药物发现计划中的优异结构。它们的商业可用性稳步增加,合成创新也在不断更新。一些含氟氮杂吲哚衍生物已经从药物化学项目中崭露头角,其中一些已经也已经发展成为治疗人类疾病的候选药物分子。参考文献1. (a) Meade, E. A.; Beauchamp, L. M. J. Heterocycl. Chem. 1996, 33,303. (b) Antonini, I.; Claudi, F.; Cristalli, G.; Franchetti, P.; Grifantini,M.; Martelli, S. J. Med. Chem. 1982, 25, 1258. (c) Schneller, S. W.; Luo,J.-K. J. Org. Chem. 1980, 45, 4045. 2. Benoıˆt, S.; Gingras, S. Processes for the preparation of antiviral 7-azaindole derivatives. U.S. Provisional Patent 60/367,401, 2003.3. Wolfe, J. P.; Tomori, H.; Sadighi, J. P.; Yin, J.; Buchwald, S. L. J.Org. Chem. 2000, 65, 1158.4. (a) Jaime-Figueroa, S.; Liu, Y.; Muchowski, J. M.; Putman, D. G.Tetrahetron Lett. 1998, 39, 1313. Other deallylation conditions attempted did not improve yield; see other conditions in ref 9a and: (b) Garro-Helion,F.; Merzouk, A.; Guibe´, F. J. Org. Chem. 1993, 58, 6109.5. On a larger scale, purification could also be done using Dowex 50W X 4 resin.6. Differding, E.; Ofner, H. Synlett 1991, 187. 7. A >1.5 M solution of tert-butyllithium in pentane was used to avoid the precipitation of N-fluorobenzenesulfimide.8. Barnes, K. D.;Hu, Y.; Hunt, D. A. Synth. Commun. 1994, 24, 1749.
应用实例
2023.03.14

高性能锂离子电池硅负极材料研究进展
引言最近人们对电动汽车和混合动力汽车的需求,加上价格的降低,使得锂离子电池(LIB)成为一种越来越受欢迎的可充电电池技术。锂离子电池的市场也在不断增大。然而,作为锂离子电池传统负极材料的石墨(项目号:P196402)由于其有限的理论比容量为~370 mAhg−1[2],无法满足先进电动和混合动力汽车市场的高能量需求,在过去十年中,锂离子电池方向出现了大量具有增强存储容量、高能量密度和改善循环特性的负极材料[3-7]。表1总结了几种不同负极材料的性能。在这些先进的负极材料中,Si作为锂离子电池的替代品引起了大量关注,主要原因是:1) 其比容量为4200 mAhg-1,体积容量为9786 mAhcm-3,是已知的LIB负极中最高的;2) 工作电位相对较低(0.5 V vs. Li/Li+);3) Si元素的自然丰度及其环境良性度[8-10]。表 1 各种负极材料的比较然而,由于三大问题,硅负极的实际应用仍然受阻。首先,硅材料的循环寿命较差。这是由于伴随锂离子插层和脱层的巨大体积波动(>300%)期间的粉碎造成的。其次,在合金化/脱合金过程中,硅负极的机械断裂导致了剧烈的不可逆容量损失和低库仑效率。最后,固体电解质间相(SEI)在脱除过程中随着纳米结构的收缩而断裂。这导致新鲜的硅表面暴露在电解液中,SEI发生重组,导致SEI随着每一次充放电循环而变厚,如图1所示[11,12]纳米硅负极材料为了解决这些问题,已经开发出了几种策略来适应巨大的体积变化。一种有效的策略是将活性颗粒尺寸减小到纳米范围,因为纳米级颗粒可以适应较大的应力而不开裂,同时还可以减小电子和离子之间的距离。此外,纳米材料中晶粒边界的高密度也为锂离子提供了快速扩散路径,并充当了额外的锂存储位点[13-16]。Huang等人通过原位透射电子显微镜(TEM)展示了Si纳米颗粒尺寸对结构应力释放的影响,并提出如果颗粒直径[17]。最近,Kim等报道了在380°C的高压下,通过使用各种表面活性剂,可以合成5、10和20 nm尺寸的Si纳米颗粒[18]。将这些材料以 0.2 C的速率在0-1.5 V之间循环,可实现超过40次充放电循环,容量保持率分别为71,81和67%。图 1 硅在循环过程中受直径大小影响的稳定性示意图Kim等人还报道了一种具有高度互联的多孔结构的3D硅结构[19]。这种Si结构具有40 nm厚的孔壁,即使在100次循环后,也可以承受较大的应力而不变形,并以1 C(2000 mAg-1)的速率保持了大于2800 mA h g-1的电荷容量。尽管纳米结构Si负极具有优势,但纳米颗粒也有缺点,如表面积大,制造成本高,处理困难[22]。即便如此,纳米级别硅材料仍被认为是克服下一代锂离子电池硅负极挑战的最有前途的方法之一。硅基碳复合负极材料克服循环过程中体积变化的另一种方法是形成复合材料[23]。如果基体不会经历明显的体积变化,这可能会缓冲硅的膨胀,保持电极的结构完整性,并通过减少硅聚集或电化学烧结来增强稳定性。硅基碳复合材料是一个很有前景的研究领域,其优点在于提高了导电性和碳基体的膨胀缓冲效应。此外,碳添加剂还具有优异的离子导电性和锂离子存储能力[28,29]。然而,Si活性材料上的保形碳涂层在循环过程中会破裂,从而导致Si暴露在电解质中并额外沉积SEI。因此,一种能够容纳Si的大体积波动的碳涂层形式是必要的。Wang等人通过原位催化生长石墨烯表面纳米硅(Si@Graphene)的方法,成功制备出了一种硅基复合负极催化剂[30]。如图2所示,该材料展现出优异的循环稳定性和速率能力,在0.2 A g-1下经历100次循环后,可保持高达1909 mAhg-1的可逆放电容量。即使在52 Ag-1的大电流下也能提供975 mAhg-1的放电容量。图2 Si@Graphene的制备过程示意图及性能测试图Liu等人报道了一种具有优异容量(在C/10时为2833 mAhg-1)、循环寿命(1000次循环,容量保持74%)和库伦效率(99.84%)的蛋黄-壳结构Si@C(图 3A) [31]。Si纳米颗粒首先被SiO2层包裹,然后再被聚多巴胺层包裹,随后将其碳化以形成氮掺杂碳涂层,经氢氟酸(HF)处理选择性去除SiO2层后获得Si@Void@C“蛋黄-壳”结构。最近,Li等人报道了空心核壳多孔Si-C纳米复合材料,100次循环后可逆容量为650 mAhg-1(电流密度为1Ag-1),对应86%的容量保留率[32]。这些独特结构的优势可以归结为两个方面:1) Si核层与碳壳层之间的空隙空间允许Si纳米颗粒在锂化时膨胀而不破坏壳;2) 碳壳层的电导率和离子电导率阻止了电解质到达Si表面。另一种策略是生产多孔Si@C复合材料。Magasinski等报道了具有高容量(可逆容量:1950 mAhg-1)和长循环寿命的Si@C多孔复合材料[33]。采用分层自下而上的组装方法制备了多孔Si@C结构,其中不规则的通道可以确保锂离子快速进入颗粒体,而颗粒内部的孔隙度可以适应循环过程中Si体积的大变化。石墨烯(项目号:G139798,G139805,G139804)由于其优越的导电性,高表面积(2600m2g-1),优异的化学稳定性和强大的机械强度,也被用于Si负极中以缓冲体积变化和提高电子导电性[34-38]。最近,Wen等人报道了用氨基丙基三甲氧基硅烷(APS)处理Si(项目号:S108980)和用海藻酸钠取代羧甲基纤维素(CMC)可以改善石墨烯封装Si阳极的电化学性能(项目号:C294622,C104978)。这两种方法都可以改善石墨烯键合和封装的Si基与集电极之间的相互作用。这些石墨烯封装的功能化硅纳米颗粒在0.1℃时的容量为2250 mAhg-1,在10℃时的容量为1,000 mAhg-1,即使在120次循环后也能保持85%的初始容量。Zhao等人报道了嵌入3D石墨烯支架中的硅纳米颗粒(图 3B),并表现出约3200 mAg-1(电流密度:1Ag-1)的可逆容量,在150次循环后仍保留其理论容量的83%[39]。在这种情况下,3D导电石墨烯支架是用从剥离的氧化石墨烯中,通过一种简便的湿化学方法构建的。在这种负极材料中保持高容量的能力归因于优异的跨平面离子扩散率,它缩短了锂离子在整个电极中的扩散路径,允许完全进入Si纳米颗粒中的内部和快速锂化。Xin等人还报道了通过一系列化学过程合成了具有 3D多孔结构的Si/石墨烯纳米复合材料[40]。这种架构提供了900 mAhg-1的可逆容量,即使在1 A g-1的充电速率下,30次循环后也几乎没有褪色。由于3D石墨烯网络增强了电极的导电性,3D石墨烯基复合材料显示出优越的循环稳定性和高倍率性能,表现出优于2D纳米结构的倍率特性。图3 A)单个Si@Void@C粒子示意图 (上图)和合成 Si@Void@C在锂化前后的原位TEM图像(下图);B)平面内碳空位缺陷石墨烯支架构造的复合电极材料截面示意图(上图); (Si:大颗粒;Li离子:小球体)和Si–3D石墨烯支架截面的 SEM图像(下图),插图显示Si纳米颗粒均匀嵌入石墨烯薄片之间[39]。未来的发展方向最近,Wu等人报道了理想的3D多孔硅/导电聚合物水凝胶复合电极,具有相对稳定的可逆容量(1000次深度循环后1,600 mAhg-1)和非常稳定的性能(循环5000 次没有明显的容量衰减) [2]。多孔分层水凝胶框架具有显著优势:导电聚合物3D 网络提供了快速的电子和离子转移通道,此外还提供了用于Si颗粒体积膨胀的多孔空间。这种原位聚合的制备方法显示了可扩展性和工业商业化的前景,如图 4所示。图4 3D多孔硅纳米颗粒/导电聚合物水凝胶复合电极示意图:(A)其中每个硅纳米颗粒被封装在导电聚合物表面涂层中,并进一步连接到高多孔水凝胶框架,照片(B-D)显示了电极制造过程的关键步骤。总结与挑战硅是最有前途的锂离子电池负极材料之一,其优点包括已知容量最高和相对较低的工作电位。然而,在硅负极应用于实际锂电池之前,必须克服体积膨胀的问题。本文阐明了各种硅阳极和改善电化学性能的硅基复合负极,展示了两种可行的解决方案来绕过硅负极。还需要进一步的研究来解决Si负极的实际要求,包括高功率密度、长寿命、简单制造和低成本等方面。
应用实例
2023.03.08

酶|阿拉丁带您科学认识“新冠诱发的心肌炎”
心肌炎更容易袭击身体健康的年轻人,并且可能致命,成了很多人担忧的对象。那到底该如何识别和应对呢?近期,由新冠引发的“心肌炎”多次引发关注。尤其是具备营养心肌作用的辅酶Q10(C111044)受到格外关注。 心肌炎,顾名思义就是心肌的炎症性病变,可由感染(如病毒、细菌或真菌)或非感染因素(如自身免疫疾病等)引起。临床上,和其他病毒感染一样,奥密克戎感染后也有一定概率诱发心肌炎,但心肌炎通常发生在病毒感染后期或1~2周后,这和病毒相关免疫反应有关。 心肌炎可对各年龄段、不同性别人群发起“无差别攻击”,不过20~40岁青壮年比老人、儿童更易中招。病毒进入血液后,随着血液循环到达心脏,如果病人的抵抗力弱,就有可能引起心肌炎。病毒性心肌炎是指病毒感染引起的心肌局限性或弥漫性的急性或慢性炎症病变,属于感染性心肌疾病。 通常心肌炎的检测是通过查心肌酶五项的数据来判断的。心肌酶是存在于人体心肌细胞中的一种特有的物质,当人体心肌受到损伤时,心肌酶就会释放出来。在临床中,通过抽取一定量的血液,检查其心肌酶各项指标的含量,一旦发现指标数值有异常,就说明患者可能发生了心肌损害,如心肌坏死、心肌梗塞、心肌炎等。心肌酶种类比较多,但是常用的就只有五种,包含AST(谷草转氨酶-天门冬氨酸氨基转移酶)、α-HBDH(α-羟丁酸脱氢酶)、CK(磷酸肌酸激酶 C486345)、CK-MB(磷酸肌酸激酶同功酶)以及LDH(乳酸脱氢酶)。 另外,肌钙蛋白也是反映心肌损伤的指标,虽不属于酶类,但是现在更常用来检查是否有心肌损害,是目前首选的反映心肌损伤的指标。 心肌酶检测主要包含以下五项:检测项目正常值肌酸激酶(CK)40~200 IU/L肌酸激酶同工酶(CK-MB)正常值<24 IU/L天门冬氨酸氨基转移酶(谷草转氨酶(AST))正常值13~35 IU/L乳酸脱氢酶(LDH)正常值120~250 IU/Lα—羟丁酸脱氢酶(α-HBDH)正常值72~182U/L (A)肌酸激酶(CK)分布情况:CK存在于肌肉及心肌细胞中的细胞质和线粒体中,平滑肌中也有,主要作用与肌肉收缩及能量代谢、ATP再生有关。CK有多种亚型,其中的CK-MB主要存在于心肌细胞内。结果解释:心肌梗死时血清CK水平明显升高,以肌酸激酶同工酶(CK-MB)为主,对急性心肌梗塞(AMI)早期诊断的灵敏度明显高于总CK,其阳性检出率可达100%,且具有较高特异性。其他原因的心肌损伤,如病毒感染后发生的心肌损伤、病毒性心肌炎,不稳定性心绞痛、各类心脏手术包括射频消融、安装心脏起搏器等,CK-MB也可以增高。运动到出现肌肉酸痛时可以有轻度血清CK升高,有的人挤公交车亦有出现升高的情况肌肉疾病如进行性肌营养不良、多发性肌炎、骨骼肌损伤和全身性惊厥时CK增高。甲状腺激素可抑制CK的活性,甲状腺功能减退的患者甲状腺激素分泌减少,他们的血清CK可升高,甲状腺功能亢进时血清CK可降低。 (B)乳酸脱氢酶(LDH)分布情况:乳酸脱氢酶(L139685)见于全身各个器官,占比顺序为 LDH2>LDH1>LDH3>LDH4>LDH5,虽然总乳酸脱氢酶中包括所有的同工酶,但LDH-2所占比例最大。分布如下:LDH-1,心脏,红细胞,肾脏,生殖细胞LDH-2,心脏,红细胞,肾脏(较LDH -1少)LDH-3,肺和其他组织LDH-4,白细胞,淋巴结,肌肉,肝脏(较LDH-5 少)LDH-5,肝脏,骨骼肌结果解释:在各类疾病中都有乳酸脱氢酶水平的升高,这反映了它在组织中广泛的分布。总LDH升高,主要见于心脏与肝脏疾病,乳酸脱氢酶水平的升高和乳酸脱氢酶同工酶之间比例的变化通常提示有某种类型的组织的损伤。心脏病发作时,血液中总乳酸脱氢酶的水平将在24至48小时内升高,在2至3天达到高峰,在10到14天内又恢复正常。一般情况下,当细胞的破坏开始时,乳酸脱氢酶的水平将增加,经过一段时间的高峰期,然后开始下降。剧烈运动亦可出现LDH升高,血小板高者的血LDH的检测结果偏高,检测结果不能反映其真实水平。 (C)天门冬氨酸氨基转移酶(AST)分布情况:主要分布在心肌,其次是肝脏、骨骼肌和肾脏等组织中。正常时血清中的AST含量较低,但相应细胞受损时,细胞膜通透性增加,胞浆内的AST释放入血,故其血清浓度可升高,临床一般常作为心肌梗死、心肌炎、肝炎或其它肝脏疾病的辅助检查。结果解释:在急慢性肝脏疾病,AST可以升高,ALT同时升高,肝硬化、肝癌、肝淤血、胆道梗阻可正常或轻度升高,对肝有毒性作用的药物如鲁米那、安定、非那西汀(P109420)、呋喃(F116392)类等可使AST浓度升高。AST在心肌细胞中含量最高,心肌梗死时血清AST活性增高,在发病后6~8小时血清AST活性开始上升,18~24小时达高峰,AST活性峰值与梗死灶大小成正比。若无新的梗死发生,4~5天后酶活性恢复正常;若再次上升则提示梗死灶扩大或有新的梗死部分。另外,肌炎、挤压综合征、肌肉损伤、肾炎及肺炎等也可引起血清AST活性升高。 (D)α-羟丁酸脱氢酶(α-HBDH)分布情况:它存在于人体各组织中,以心肌组织含量最多,约为肝脏的2倍,其活性达总酶活力的一半以上。当心肌受损时,α-羟丁酸脱氢酶就会释放到血中,所以血清α-羟丁酸脱氢酶在发生心肌疾病时明显增高。结果解释:α-羟丁酸脱氢酶(α-HBDH)与乳酸脱氢酶(LDH)、肌酸激酶(CK)、天门冬氨酸氨基转移酶(AST)共同构成了心肌酶谱,用于心肌梗死和心肌炎的诊断。血清α-羟丁酸脱氢酶增高主要见于:心肌梗死、活动性风湿性心肌炎、急性病毒性心肌炎、溶血性贫血等。肝脏和心脏疾病均可引起α-HBDH活性增高,但α-HBDH活性在肝脏疾病时变化不是很大,而在心脏疾病时有明显增高,故α-HBDH可用于肝病和心肌梗死的鉴别诊断。 (E)肌钙蛋白(cTnI、cTnT)分布情况:肌钙蛋白(cTn)(T469327)主要存在于心肌肌原纤维细胞中,其主要包括三个亚基:肌钙蛋白I、肌钙蛋白T和肌钙蛋白C,是维持心肌纤维收缩与舒张的重要功能蛋白。结果解释:正常情况下,外周血中几乎没有肌钙蛋白,当心肌细胞因缺氧或其他因素引起细胞破损时而释放入血,肌钙蛋白I和肌钙蛋白T均具有相同的心肌特异性,是诊断心肌梗死的首选标志物。目前认为cTnI和cTnT对急性心肌梗死、不稳定性心绞痛、围术期心肌损伤等疾病的诊断、病情监测、疗效观察及预后评估都有较高的价值,特别是对微小的、小灶性心肌梗死的诊断具有重要价值。 产品货号产品名称包装规格C111044辅酶Q10250mg/1g/5g/25g/100gL139685乳酸脱氢酶5KU/25KU/100KUP109420非那西汀25g/100g/500g/25kgF116392呋喃100mL/500mL/10L/25LT469327肌钙蛋白1g/5gC486345肌酸激酶活性测定试剂盒1kitD128544心肌黄酶2KUD128545肌黄酶1KUS128537超氧化物歧化酶2mg/5mg/10mgT128536酪氨酸酶25KU/100KU/500KU 文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请在阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
应用实例
2023.02.27

明星系列产品(一)| 阿拉丁色谱溶剂
企业动态
2023.02.22

类器官新用途
为什么在药物研发中选择类器官?缺乏能够准确代表特定组织和疾病状态的合适体外模型是基础研究和转化研究的重大障碍。这导致了3D类器官的发展,它提供了比2D模型更大的复杂性,并建立了稳定的、与生理相关的模型,可以长时间培养。类器官已经被用来模拟多种组织类型,包括胰腺、肝脏、肾脏、视网膜、大脑和肿瘤,并且已经证明了这些系统在促进我们对复杂系统生物学的理解方面的广泛潜力。类器官在药物筛选、毒性试验、疾病建模和研究胚胎发育方面具有潜力。什么是类器官?类器官是干细胞衍生的自组装3D结构,可以复制 器官的某些特征。类器官由成体干细胞或诱导多能干细胞(iPSCs)产生,诱导分化依赖于细胞粘附分子的不同表达谱和空间限制的谱系承诺。在组织中限制细胞空间或使用生物支架促进干细胞的进一步分化,在类器官的生成中至关重要。来自Engelbreth-Holm-Swarm (EHS)小鼠肉瘤细胞的生物支架,如基底膜提取物,最常用于实验室,并提供环境线索,包括生长因子,鼓励细胞附着并形成类器官结构。小分子在培养基中也被广泛用于指导类器官的生长和分化。利用类器官筛选新化合物类器官可能是药物发现的一个特别有用的领域。从患者来源的iPSCs中产生的类器官已被发现可重现疾病特征,并在新疗法的临床前筛查中有用,以在细胞水平上建立疗效。然而,Pellegrini等人最近的一篇论文对类器官如何用于药物筛选提供了不同的视角。脉络膜丛(ChP)由围绕毛细血管和结缔组织的一层上皮细胞组成,负责产生脑脊液(CSF)。它还在血液和脑脊液之间形成一道屏障,防止循环中的有毒物质到达大脑。ChP位于大脑深处,迄今为止,这使得其结构和功能难以研究。佩莱格里尼和他的团队通过调整从人类iPSCs中生成脑类器官的方案,将BMP-4和CHIR 99021添加到成熟培养基中,建立了ChP类器官。所形成的类器官在立方上皮中富集,并形成含有无色液体的充满液体的腔室,在结构和功能上与脉络膜丛相似。对这种无色液体的分析显示,它与脑脊液非常相似。在结构上,ChP类器官有紧密连接,初级纤毛,广泛的微绒毛,多泡体,以及细胞外囊泡,这些都是ChP的特征。这些类器官有可能预测正在开发的新疗法的中枢神经系统渗透性,以确定一种化合物治疗神经疾病的潜力或其可能的毒性。在体内,脉络膜丛对左旋多巴和多巴胺的通透性不同。ChP类器官也表现出对这些化合物的不同渗透性,前者被运输到类器官中,而后者则没有,这证明了这个3D系统可以用于模拟药物的CNS渗透性的原理证明。2016年,BIA 10-2474的临床试验正在法国进行,BIA 10-2474是一种脂肪酸酰胺水解酶抑制剂,可用于各种神经系统疾病的治疗。试验中有5名参与者患上了严重的急性神经中毒,其中一人死亡。该化合物尚未在动物身上表现出神经毒性作用,但来自人类iPSCs的ChP类器官表现出BIA 10-2474的毒性积累,揭示了该系统在新疗法毒性测试中的潜在用途。类器官和癌症治疗多种不同癌症类型的肿瘤类器官,如乳腺癌、前列腺癌、结肠癌和子宫内膜癌,已经从患者的癌症活组织检查中生成。最近Maenhoudt等人报道了从高级别严重卵巢癌(HGSOC)患者中生成卵巢癌(OC)类器官的方法,该方法采用了Boretto等人(2019)的方法,以改善类器官的建立和生长。这些活检来源的肿瘤类器官表现出与原发肿瘤和复发性疾病相同的表型。它们为OC的发展研究提供了一个有用的模型,还可以用于筛选新的治疗方法,以确定它们对这种类型的癌症的有效性。然而,它们还有另一个潜在用途,那就是个性化医疗。来自不同患者的OC类器官对常规化疗药物如紫杉醇、卡铂、吉西他滨和阿霉素表现出不同的敏感性。因此,它们可以在临床实践中发挥效用,使临床医生能够为个别患者选择最有效的治疗方法。更新:类器官和COVID-19研究自发表原创论文以来,佩莱格里尼和团队使用ChP类器官来了解更多关于COVID-19的信息。这种病毒感染的特征是严重的呼吸道症状,但一些患者也会出现神经系统症状,如头痛、精神错乱和癫痫发作。佩莱格里尼的团队因此检查了SARS-CoV-2或携带SARS-CoV-2刺突蛋白的假病毒粒子对ChP类器官的影响,发现病毒主要感染脉络膜丛上皮屏障细胞,而不是神经元或胶质细胞。这导致上皮细胞损伤,并导致血- csf屏障渗漏。参考文献1.Boretto et al. (2019) Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat Cell Biol 21, 1041. PMID: 313718242.Maenhoudt et al. (2020) Developing organoids from ovarian cancer as experimental and preclinical models. Stem Cell Rep 14, 717. PMID: 322438413.Pellegrini et al. (2020) Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 369, eaaz5626. PMID: 325279234.Pellegrini et al. (2020) SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell 27 951. PMID: 33113348这有帮助吗? 是 否 第一个投票!标签:类器官相关产品列表A274862 A 83-01,ALK4、5和7激酶抑制剂 ,≥98%SDS| 价格C125082 CHIR-99021,GSK-3抑制剂 ,≥98%SDS| 价格D126677 DAPT,γ-分泌酶抑制剂 ,≥98%SDS| 价格D133402 地诺前列酮 ,98%SDS| 价格D139352 吗啡肽 ,≥98%SDS| 价格F127328 佛司可林 ,≥98%SDS| 价格G127588 GDC-0068,Akt1 / 2/3抑制剂 ,≥98%SDS| 价格G118956 Gastrin Ⅰ, 人 ,≥97% (HPLC)SDS| 价格A105422 N-乙酰-L-半胱氨酸 ,用于细胞培养,≥99.0%SDS| 价格R106320 维生素A酸 ,98%SDS| 价格S134307 SB 202190,p38 MAPK抑制剂 ,99%SDS| 价格S125924 SB431542,ALK抑制剂 ,≥98%SDS| 价格Y125330 Y-27632 ,98%SDS| 价格
应用实例
2023.02.20

NMR用溶剂的储存和使用信息指南
溶剂的储存注意:某些溶剂的包装尺寸可能需要经过特殊处理,因此在下面的举例中没有具体给出。使用前应仔细检查瓶或安瓿瓶包装信息,以获得进一步更准确的使用说明。乙酸-d4/丙酮-d6/苯-d6/环己烷-d12/重水/N, N-二甲基甲酰胺-d7/二甲亚砜-d6/1,4-二氧六环-d8(对二氧六环)/乙醇-d6/甲醇-d4/二氯甲烷-d2/吡啶-d5/1,1,2,2-四氯乙烷-d2/甲苯-d8/三氟乙酸-d/ 2,2,2-三氟乙醇-d3储存在室温下,远离阳光和潮湿环境。上述产品在常规条件下性质是稳定的。乙腈-d3储存在室温下,远离阳光和环境。在上述条件下(未开封),该产品在检测后的一年内保持稳定;一年后,使用前应重新进行检定纯度等参数。三氯甲烷-d/四氢呋喃-d8冷藏保存(-5℃至5℃),避光防潮。在上述条件下(未开封),该产品在检测后的6个月内保持稳定;6个月后,使用前应重新进行检定纯度等参数。含残留D2O的氘化溶剂中活性质子的氘交换一些氘化溶剂是通过质子化溶剂与氧化氘的催化交换而得到的,并通过精馏加以提纯。残留水(与D2O平衡交换的水)保持在约20-200 ppm的最小值,值越高,其在吸湿溶剂中的含量越高。水的活性氘核(和质子)可与样品中的活性质子交换,从而产生不准确的积分比。下面的例子表明(图1),在研究分析物的稀溶液时,仅100 ppm的D2O就会导致分析结果出现问题。取5 mg有机化合物(MW~200)溶于含100 ppm D2O的1 g DMSO-d6的样品中,可以观察到1个活性质子积分数值显著下降(图2),同时随着分析物分子量的增加,准确性下降得会更多。(X代表残余溶剂;*代表残余水分)图1:5.0 mg 2,6-二叔丁基-4-甲基苯酚(分子量为220.36 g/mol)在干燥的DMSO-d6中的1H-NMR谱图。注意:峰面积的积分比为18:3:1:2(t-丁基:甲基:环-H:-OH),3.3 ppm处的峰为单峰H2O。图2:加入100 ppm的D2O后的DMSO-d6中5.3 mg 2,6-二叔丁基-4-甲基苯酚的1H-NMR谱图。注意:还原酚的峰面积的积分比变成了18:3:2:0.47(t-丁基:甲基:环-H:-OH),并且HOH和HOD的峰在波谱图中是分离的。解决方案水(包括H2O、HOD或D2O)可以通过添加分子筛到溶剂中,搅拌该混合物并将其静置几个小时来去除。通过这种方式,水含量可以减少到大约10-20 ppm。如果这样的水分去除方式仍然不能达到目的使得实验结果不准确,建议使用吸湿性较差的溶剂,如氯仿、二氯甲烷或乙腈等。溶剂的使用ALD提供了广泛的质量控制方案,以评估我们相关溶剂的化学纯度和同位素丰度。如今高场的核磁共振波谱仪的灵敏度和分辨率的提高都需要依赖于具有最高化学纯度和高同位素富集的溶剂,我们的每一批核磁共振溶剂在出货前都经过了严格的质量控制检测。所有安瓿和瓶上都清楚地标明了产品货号和生产批号,以便在极小概率发生问题的情况下进行溯源。水峰水的存在导致氘化核磁共振溶剂被污染是普遍存在的问题,有几种方法可以减少/消除水峰——• 可以考虑使用一次性安瓿瓶包装的溶剂;许多易吸水的溶剂采用的是0.25-3 mL的一次性安瓿瓶包装;• 在干燥的环境中处理溶剂;• 需要用于样品制备的核磁共振管和移液管在干燥箱中预先干燥一夜,并且使用前在含有干燥剂的环境中冷却;• 用D2O预先对核磁共振管进行冲洗;先用甲醇- d4或丙酮- d6淋洗,再用选择的溶剂漂洗去除残留的D2O。这个过程中虽然不会去除水,但它会将H质子交换为氘,并使水峰最小化。“100%” D2O为了避免由于与环境水分交换而造成富集损失,血清瓶中储存的“100%”D2O应使用已用干燥的氮气预吹气的注射器取样。此外,在吸取D2O之前,应向血清瓶中注入与D2O去除量相等的干燥氮气。TMS蒸发在室温下储存(除非特殊注明)并用恰当的方式密封保存时,含有TMS的溶剂一般不会发生TMS蒸发。然而,在这些溶剂发生转移时,可能会导致一些TMS的损失。储存所有血清瓶应竖直存放于冰箱中;不建议放在冷冻层;同时建议将氯仿、乙醚、二甘醇二甲醚、四氢呋喃、TMS等均存放在冰箱中。以氘代氯仿为例ALD的氘代氯仿是以最高的化学纯度标准生产的。随着时间的推移,无论储存的容器或条件如何,氯仿都会分解。在室温下(例如在储藏室)储存几个月后,氘代氯仿会变成酸性。然而,如果瓶子是在避光冷藏的环境中保存的,它的分解就会被最小化。ALD在氯仿-d的生产和包装过程中采取了一些预防措施,以进一步减少其分解变为酸性。通过在氘代氯仿的生产和包装过程中使用氩气,使其暴露在氧气中的程度降到最低;琥珀色的瓶子用来保护产品不受光线照射;最后,在溶剂中加入银箔作为自由基清除剂,这有助于随着时间的推移提升溶剂的稳定性。氘代氯仿的质量控制为了确保最高的质量,ALD会定期检测每批溶剂的化学和同位素纯度。化学纯度会在生产和包装过程中进行监测,会使用到1H-NMR、GC、卡尔费休滴定法测定总含水量,以及其他化学湿选法测定酸度和各种杂质。氘代氯仿的储存和使用未开封的氘代氯仿应该在冷藏(-5°C至5°C)的条件下储存,以最大限度地延长保质期。首次使用后,水分和氧气将通过打开瓶子时进入的空气被引入到溶剂中,分解后会导致氘氯变为酸性,用下面的方法可以测试溶剂的酸度——测试氘代氯仿的酸度将1 mL溶剂加入到含有1 mL蒸馏水(pH 5.0-7.0)和2滴溴百里酚蓝(0.04% w/v)的试管中,将混合溶液的颜色与2 mL空白溴百里酚蓝指示剂溶液(pH 5.0-7.0)比较,如果样品溶液相对于空白溶液(蓝绿色)变色(黄色),则氘氯呈酸性。已变为酸性的氘代氯仿样品可通过下列步骤中和:• 将3-5 g 5Å分子筛放入50或100克的溶剂瓶中;• 稍稍旋转摇晃一下试剂瓶,随后静置一晚上,多余的水分和酸性物质会被去除;这也是储存氘代氯仿溶剂瓶的首选方式,因为它能够保持溶剂干燥和稳定更长的时间;• 在瓶中保持惰性气体(氩气或氮气)氛围;• 小的灰尘或粉末颗粒可能从分子筛上脱落进入到溶剂中。然而,这些颗粒可以简单地通过在玻璃移液管中插入一小塞的玻璃棉或棉花来过滤除去。要求超干无酸氘代氯仿的特殊应用对于涉及高度酸敏感或湿敏感化合物的应用,氘代氯仿在使用前需要进一步纯化,按以下方式可将溶剂处理得特别干燥和无酸——• 将玻璃棉塞入一次性玻璃移液管(直径约7mm);• 向移液管中加入干燥的氧化铝粉末至3-4 cm高;• 将溶剂通过小氧化铝层再流入装有待分析产品的样品容器;• 尽快分析样品。此操作步骤将确保氘氯是干燥的,并且在与样品接触之前不会含有额外的微量酸;注意,氯仿会与碱性化合物反应,如生物碱或胺;如果要回收产品,应尽快进行回收,以尽量减少可能发生的反应。
参数原理
2023.02.16

芳基氟化反应
芳基氟化反应简介芳基含氟砌块是指一系列拥有一个或几个直接连接到芳环骨架上的氟原子的芳香族化合物。当氟引入芳烃时,可以提供许多优异的特性。首先,氟化芳烃比非氟化芳烃亲脂性更强,这可以在药物开发中得到利用,提高药物分子的脂溶性,可增强含氟药物分子在生物体内对膜、组织的穿透能力,从而提高生物体内的吸收和传输速度。此外,在药物化学中,氟有时被用作氢的电子等排体(氢和氟的空间参数相似,范德华半径分别为1.2 Å和1.35 Å)。除此之外,含氟化合物还可以战略性地被用作过渡态抑制剂图1.芳基含氟砌块由于氟自身特殊的性质,无论采用什么方法,任何生成C-F键的反应都是一个重大的挑战。氟化物阴离子,由于其电负性和小的离子半径(1.33 Å),可以与各种氢键供体如水、醇、胺和酰胺形成强氢键。氟离子在水介质中的高溶解性会导致其周围有一个紧密结合的水分子水合层。因此,在有氢键供体的情况下,氟化物通常只有微弱的亲核性,而氟化物的这种弱亲核性限制了通过亲核取代反应生成C-F键。芳烃氟化方法传统的亲核芳烃氟化反应关于传统亲核芳烃氟化反应的一个主要挑战是对相对简单的底物的限制,这是由苛刻的反应条件和亲核氟化物的强毒性造成的。1927年,Balz和Schiemann通过伯芳香胺通过四氟硼酸重氮中间体经过热分解转化为芳基氟化物[1,2],开辟了芳烃的亲核氟化反应(图2)。图2.芳基重氮盐的亲核氟化该反应在概念上类似于Sandmeyer反应,将重氮盐转化为其他芳基卤化物(ArCl、ArBr)[3]。然而,尽管Sandmeyer反应涉及铜试剂/催化剂和自由基中间体,但四氟硼酸重氮的热分解在没有助催化剂的情况下进行,生成高度不稳定的芳基阳离子(Ar+),从而提取来自BF4−的F−,得到氟芳烃(ArF)以及作为副产物的三氟化硼。亲核氟化反应亲核氟化是一种简单而有效的芳烃亲核氟化的方法,排除了水和其他会减弱氟化物亲核性的氢键供体。四丁基氟化铵(TBAF)是一种商业易得,易溶于有机溶剂的亲核试剂,可作为三水合物使用(图3)。图3.使用TBAF的亲核芳烃氟化酚类的亲核脱氧氟化除了芳基卤化物,苯酚是合成芳基氟化物更易反应且更易获得的底物。例如,邻苯二酚可以先进行脱氧氟化,然后用硼氢化钠还原(图4)。图4.通过一锅反应对邻苯二酚进行脱氧氟化亲电氟化反应:与上述芳基氟化互补的方法是使用芳基亲核试剂和亲电氟化试剂。N-F氟化试剂在形式上可以作为氟离子的来源,常用的亲电氟化试剂如图5所示。图5.常用的亲电氟化试剂其中,Selectfluor(N-氟-N'-(氯甲基)三乙二胺双(四氟硼酸盐)或F-TEDA)是一种用户友好、温和、空气和水分稳定、非挥发性的亲电氟化试剂。Selectfluor试剂能够在一个步骤内将氟引入有机底物,同时具有非常广泛的反应性[4]。此外,这些反应均表现出良好的区域选择性。使用Selectfluor氟化试剂完成了一种强效和无细胞毒性的丙型肝炎病毒RNA复制核苷抑制剂的合成(图6)。与母体2'-C-甲基腺苷相比,这种核糖苷显示出明显的酶学稳定性[5]。图6.使用Selectfluor的亲电氟化试剂应用芳基含氟砌块被广泛用作合成药物的中间体或药物分子,其中氟取代可以通过影响pKa、调节构象、疏水相互作用和亲脂性或这些属性的叠加来提高药效和对靶标的选择性。除此之外,芳基含氟砌块还可以应用于杀虫剂、塑料和与液晶技术有关的分子中。参考文献1.Balz, Günther; Schiemann, Günther (1927). "Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung" [Aromatic fluorine compounds. I. A new method for their preparation.]. Chemische Berichte (in German). 60 (5): 1186–1190. https://doi.org/10.1002/cber.192706005392.Furuya, Takeru; Klein, Johannes E. M. N.; Ritter, Tobias (2010). "C–F Bond Formation for the Synthesis of Aryl Fluorides". Synthesis. 2010 (11): 1804–1821. https://doi.org/10.1055/s-0029-12187423.Swain, C. G.; Rogers, R. J. (1975). "Mechanism of formation of aryl fluorides from arenediazonium fluoborates". J. Am. Chem. Soc. 97 (4): 799–800. https://doi.org/10.1021/ja00837a0194.Singh, R. P. , Shreeve, J. M.. 2004. For a review of recent highlights: Acc. Chem. Res..37, 31.5.Eldrup AB, Prhavc M, Brooks J, Bhat B, Prakash TP, Song Q, Bera S, Bhat N, Dande P, Cook PD, et al. 2004. Structure?Activity Relationship of Heterobase-Modified 2’-C-Methyl Ribonucleosides as Inhibitors of Hepatitis C Virus RNA Replication. J. Med. Chem.. 47(21):5284-5297.https://doi.org/10.1021/jm040068f
应用实例
2023.02.16

材料|阿拉丁锂离子电池材料新鲜出炉!
电池的发展给人类的生活带来了巨大的便利,锂离子电池更是电池领域中的佼佼者,其足迹遍布我们生活的各个角落。 一、消费电子产品 随着人们对科技生活和快节奏生活的追求,对消费电子产品中的电池能源有了更高的要求。以锂离子电池为驱动电源的消费类电子产品正广泛应用于生活的方方面面。 二、新能源汽车 锂离子动力电池作为新能源汽车最关键的核心部件,直接影响着新能源汽车的性能,包括新能源汽车的续航里程、安全性、使用寿命、充电时间、高低温适应性等多个方面。 三、储能产品 锂电池储能系统的发展具有增长的前景。未来,下游可再生能源并网、电动汽车、5G基站等应用场景将有助于锂离子电池解决方案的发展。 四、其他产品应用 动力电池是轻型动力汽车的心脏,是新能源汽车产业发展的关键。近年来,中国提出积极开展电动汽车、汽车动力电池等新型动力的研究和产业化,加快了动力锂电池在轻功率领域的推广。 阿拉丁拥有种类丰富的锂离子电池相关产品。其中锂离子电池材料产品有近400种,包括正极材料,负极材料,电解液,隔膜材料四大类。此外,锂离子电池材料测试辅助试剂产品有900余种。 正极&负极材料产品货号产品名称试剂规格或纯度CAS包装规格L107451钴酸锂99.5% metals basis12190-79-3500g/100g/25g/25kgL302983磷酸铁锂98%15365-14-7500g/100g/25g/25kgM103247N-甲基吡咯烷酮(NMP)standard for GC,≥99.9%(GC)872-50-45mlP169015聚偏二氟乙烯average Mw ~400,00024937-79-95g/500g/100g/25g/25kg/1gP104272聚丙烯酸average Mv ~450,0009003-01-45g/100g/25g/1gP104268聚丙烯酸 50%水溶液平均分子量M.W ~3,0009003-01-4500g/100g/25kg/10kg/25kgC124533单壁碳纳米管≥95%,直径:<2nm,长度:0.3-5 μm, 移动催化法308068-56-6250mg/1gC121255双壁碳纳米管>50%,直径:<5nm,长度:<50μm308068-56-6250mg/1gC313046多壁碳纳米管>95%, 外径:8-15nm, 长度:~50μm, SSA:>140m2/g308068-56-65g/100g/25g/1gC283946羟基化多壁碳纳米管>95%,外径:8-15nm,长度: ~50μm308068-56-65g/25g/1gI489798工业级氧化石墨烯溶液Single layer ratio >95% ,0.8~1.2 nm7782-42-525mlG139803氧化石墨烯粉末>99%7782-42-5250mg/1gG139802氮掺杂石墨烯>98%7782-42-5250mg/1gG302114石墨烯高纯级,>98%1034343-98-0500mg/250mg/1gS100128海藻酸钠AR9005-38-3500g/100g/25g/25kg/10kg/25kgP105128聚乙烯醇0588低粘度型(PVA-205)醇解度:87.0~89.0(mol/mol),CPS:4.6-5.49002-89-5500g/100g/1kg/25kgP141443聚(甲基丙烯酸甲酯)耐热型射出级9011-14-7500g/100g/1kg/5kgP301928聚四氟乙烯微粉平均粒径: 25μm9002-84-0500g/100g/25g/25kgG170696石墨氟化聚合物>61 wt. % F51311-17-25g/1gG123646石墨粉99.95% metals basis,≥100目7782-42-5500g/100g/25kgG299101纳米石墨粉D50<600nm,99.9% metals basis7782-42-5100g/25gC294622羧甲基纤维素DS=0.7 ,200-500mPa.s9004-32-4500g/100gP104268聚丙烯酸 50%水溶液平均分子量M.W ~3,0009003-01-4500g/100g/25kg/10kg/25kgO111912草酸 二水合物GR,99.8%6153-56-6500g/25kgO291734草酸溶液10% (w/v)144-62-7500ml/1L 隔膜材料项目号产品名称规格或纯度Cas编号包装P110852聚丙烯熔融指数 2.2g/10min9003-07-0500g/100g/25kgB302379勃姆石3.4μm1318-23-6500g/100g/25kgH102613氧化铝99.99% metals basis,晶型α,0.20μm1344-28-1500g/100g/25kg/10gA102091纳米氧化铝99.99% metals basis,γ相,20nm1344-28-1500g/100g/1kg/50g/5kg/10gP141443聚(甲基丙烯酸甲酯)耐热型射出级9011-14-7500g/100g/1kg/5kgE111993乙醇色谱级,≥99.8%64-17-5500ml/25L/4L/4×4LE111989乙醇AR,水分≤0.3%64-17-5500ml/5L/25L/25L/4×5L/12×500ml/20×500ml/10LE118433乙醇工业级,99.5%64-17-55L/25LG116205甘油≥99.5%(GC)56-81-5500ml/5L/12×500ml 电解液材料项目号产品名称规格或纯度Cas编号包装D119695碳酸二甲酯无水级,≥99%616-38-6500ml/100ml/1LD105557焦碳酸二乙酯(DEPC)98%1609-47-85g/500g/100g/25gP301659聚碳酸酯抗紫外线级,melt index:5 g/10 min (300°C/1.2kg)25037-45-01kg/250gL157770六氟磷酸锂97%21324-40-35g/500g/100g/25gL107398四氟硼酸锂99.9% metals basis14283-07-95g/100g/25gL120347双乙二酸硼酸锂≥99.0% metals basis244761-29-35g/500g/100g/25g/1gL303675二氟草酸硼酸锂≥99%409071-16-55g/500g/100g/25g/1gB398978双三氟甲烷磺酰亚胺锂≥99.9%90076-65-65g/25g/250gL157764双(氟磺酰)亚胺锂>98.0%(T)171611-11-35g/500g/100g/25g 测试辅助试剂项目号产品名称规格或纯度Cas编号包装H399657盐酸(易制毒)37%7647-01-0500ml/25L/4×25L/12×500ml/1LN116238硝酸(易制爆)AR,65-68%7697-37-2500ml/5L/12×500mlN116243硝酸(易制爆)电子级,70%7697-37-2500mlT1281241,1,1-三氯乙烷标准溶液2000ug/ml in Purge and Trap Methanol71-55-61ml/2mlD1173872,4-二甲酚标准溶液1000μg/ml,溶剂:甲醇105-67-912mlS111504氢氧化钠标准溶液0.5000mol/L(0.5N)1310-73-21LP119551氯化钾电导率标准溶液147.4μs/cm(25°C)7447-40-7100mlM115418汞标准溶液100ug/ml in 3%HNO37439-97-620mlX128150邻二甲苯标准溶液2000ug/ml in Purge and Trap Methanol95-47-61mlS196953醋酸钠标准溶液0.3M127-09-31LP112173重铬酸钾标准溶液1/60mol/L(0.1N)7778-50-91LS108761钠标准溶液100ug/ml in 1%HCl7440-23-550ml/80mlP103824钾标准溶液500mg/L in water7440-09-7(water)20mlP112168重铬酸钾(易制爆)99.99% metals basis7778-50-9100g/25gP196945重铬酸钾标准液Analysis of standard solution, 0.02 M7778-50-91LD100399二苯胺磺酸钠≥97.0% (HPLC)6152-67-65g/100g/25g/1gP112025磷酸色谱级, 85-90%7664-38-2500ml/1L/4LP291679磷酸溶液75% w/w7664-38-21LS399876硫酸(易制毒)99.999% metals basis7664-93-9500ml/25L/100mlN291980硝酸溶液(易制爆)20% (v/v)7697-37-2500ml/1LW120477ACS级水ACS 级,用于痕量分析7732-18-51LW119425色谱质谱级水用于UPLC, LC-MS,LC-MS/MS痕量和超痕量分析7732-18-56×1L/1L/4LA103533抗坏血酸AR,>99.0%(T)50-81-7500g/100g/25kg/10kg/5kgI115411铁标准溶液100ug/ml in 5%HCl7439-89-6(Hydrochloricacid solution)20ml 阿拉丁技术文章:如何提高锂离子电池中电解液的安全性?——点击查看详情 文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请在阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
应用实例
2023.02.14

生物分子NMR:蛋白质动力学研究的同位素标记法
长期以来,液相核磁共振光谱测定蛋白质结构都是依赖于13C和15N均匀稳定的同位素富集,以减少共振重叠,并允许在尽可能多的原子位点上进行多距离和多角度的限制,以方便计算最佳的三维结构模型[1]。近年来,这些标记技术的优化增加了可修改的蛋白质尺寸范围,提升了三维结构的质量,简化了实验数据的分析过程[2]。同时,蛋白质动力学领域的发展也得益于同位素标记技术的进步,该技术使研究人员能够在更广阔的时间尺度范围内研究更大体积的蛋白质运动特性,同时更准确地描述蛋白质的运动轨迹。在许多方面,动力学同位素标记技术的进步映射了结构研究中所使用的技术的进展。然而,为研究蛋白质动力学而设计的自旋弛豫实验对残留标记有独特的要求,需要同位素富集技术的极速发展来更好地应用于这些先进的研究课题。动力学中孤立自旋系统的需求液相核磁共振是一种强大的方法,通过测量所需核的弛豫率来表征蛋白质在大范围时间尺度上的运动。如果将目标蛋白质的位置视为一个孤立的自旋对,那么这些弛豫实验的设计以及数据的分析和解释都将大大简化。在这种情况下,脉冲序列设计不需要考虑和操作多个不需要的相干路径,得到的弛豫率可以直接从峰值强度的单指数衰减剖面测量。然而,多个大型单键耦合的存在会通过多指数弛豫路径和信号噪声退化,使实验结果复杂化。正因为如此,常规的标记方法大多都是通过提供一种标记蛋白质中不同的孤立自旋对的同位素的方法,这样单键标量(J)耦合就不会构成实验数据分析上的困扰了。15N-标记迄今为止,大多数动力学研究都是通过单一的15N的富集来实现的。15N是一个很好的研究方向,细胞生长所需的必要氮元素可以通过易于获得的富含15N的微量或富含氮元素的生长培养基来控制,使样品制备变得容易。单一的15N标记构成了一个孤立的自旋系统(1H-15N),使得其很适合进行弛豫实验。无论在蛋白质的主链还是侧链中,每一个15N的位置都与另一个15N的原子通过至少两个键分开。因此,没有能导致复杂的多指数弛豫行为的1JNN耦合,这类耦合将很难精确测量,并会影响对相关运动的解释。然而,15N的富集本身并不能提供蛋白质运动的完整图像。氮只占蛋白质主链的1/3,而且在20个氨基酸中也仅有6个侧链中含有氮元素。因此,除了少数可选择的位置(Asn天冬酰胺、Gln谷氨酰胺、His组氨酸、Trp色氨酸、Lys赖氨酸和Arg精氨酸),15N弛豫实验不允许大量在蛋白质尺寸范围内的动态覆盖,以防遮挡主、侧链的运动全貌,尤其是酰胺弛豫可能对蛋白质疏水核心的运动相对不敏感。同时人们还注意到,通过监测酰胺位置的弛豫,蛋白质主干的某些运动行为是不会被检测到的。尽管如此,15N蛋白质标记的便捷性、现有的强大实验积累以及氮元素对结构、静电和氢键效应的敏感性使15N成为现代动力学研究的重要组成部分。13C-标记从表面上看,碳元素的弛豫实验为利用核磁共振光谱进行分子动力学研究提供了许多额外的机会。每个氨基酸中碳的丰度为酶动力学的研究提供了更多的探针。这些碳包含在主链和侧链中,能够提供整个蛋白质的动态信息。甲基残基通常埋在疏水核心中,特别适合提供蛋白质折叠和稳定性的动态过程提供信息。13C的化学位移,特别是Cα,对蛋白质二级结构的依赖比酰胺中的氮更加明显,这使得从某些自旋弛豫实验中获得的化学位移变化更容易被理解。不幸的是,理想的碳标记方法并不像氮标记方法那样直接。蛋白质中大量的碳元素能够覆盖在蛋白质动力学研究中的绝大多数位置,这也是给其研究带来的最大阻碍;大量的碳原子意味着几乎所有的碳原子都与另一个碳原子相邻。因此,统一标记的13C蛋白样品导致许多残基存在大量的13C-13C偶联,使得本应能够直接解释弛豫行为的数据在许多情况下变得难以处理。唯一不受上述1JCC偶联影响的位置是蛋氨酸上的甲基,它被硫原子与蛋白质的其他部分隔开来。因此,在统一的13C标记蛋白中,对动力学的研究受到很多限制。虽然蛋氨酸的弛豫数据可能非常有用,但它不能提供从碳标记方案中获得更加完整的动态覆盖水平。正因为如此,许多利用已知细菌代谢途径的同位素标记方法已经被开发出来。一种用于分离13C标志物的方法是使用15%的13C-醋酸盐进行部分标记,其余保留为12C[3]。这种方法了可以将13C标志物稀释到一定的水平,使松弛实验可行,但由于标记蛋白的比例减少,信噪比也会随之降低。采用[3-13C]丙酮酸作为唯一的碳来源,在>90%掺入水平下,可以实现Leuδ、Valγ、 和Ileγ的13C标记[4]。更重要的是,同位素标记在很大程度上没有打乱直接成键的碳,从而使得我们可以对这些残基进行弛豫测量。因此,在这些甲基的位置就能观察到良好的单噪声和单指数弛豫行为了。和蛋氨酸一样,这些残基可以让我们了解蛋白质在疏水核中的运动轨迹。α-酮酸的使用也为生产13C-甲基标记的氨基酸提供了一种经济有效的方法,这种氨基酸也与其他位置上高水平氘化的碳兼容[5,6]。采用氘化方法可以在比其他方法更大的蛋白质系统上进行动力学研究。这种方法的另一个好处是13C标志物的有序性最高,从而能最大限度地减少上述由于13C-13C偶联导致的问题。在营养缺陷型细胞系中使用1或2位置标记的甘油允许在整个蛋白质侧链的大多数碳上掺入交替标记。通常,需要两个蛋白质样品才能尽可能完整地覆盖原子位置[7]。芳香族残基可以补充甲基基团获得的数据,因为它们通常也存在于疏水核心中。鉴于存在的强J偶联以及小范围的化学位移,这些残基中的特定标记尤为重要。早期研究表明,在[2-13C]位置甘油上的生长将导致大多数氨基酸中交替碳的同位素富集,包括 Phe、Tyr 和 Trp 中的孤立芳香碳。 在[1,3-13C]位置甘油上的生长将发生相反的标记模式。对此的替代方法是使用[1-13C]-葡萄糖作为唯一的碳元素来源,芳香环会被标记在Pheδ、Tyrδ、Hisδ2/ε1和Trpδ1/ε3这几个位置上[8]。[1-13C]-葡萄糖标记法是非常实用且高效的,因为它不仅更经济实惠,并且能够得到更高的蛋白质产量。最近的研究则表明,[1-13C]-葡萄糖还会导致Ala丙氨酸、Val缬氨酸、Leu亮氨酸、Met甲硫氨酸和Ile异亮氨酸的甲基残基富集率达到约45%,这些残基会被两个或多个键与其他13C标记的原子隔开[9]。同时还发现,使用[2-13C]-葡萄糖作为唯一碳元素来源的表达显示会导致在Cα位点富集率达到20-45%,不会标记 C'位点,仅仅会标记Leu、Val和Ile的Cβ位点。这种标记允许CPMG弛豫实验在Cα位置上进行,提供更多的数据来补充常见15N CPMG实验得到的结论。最后,同位素标记可以通过将所需的标记物氨基酸直接引入生长培养基中来精准地掺入[10,11]。通常,为了避免标记物扰乱或者被稀释,所需的氨基酸会被包含在含有所有其他未被标记的氨基酸混合物中,只是考虑到被标记的同位素的位置,合成过程可能非常耗时。该技术已被证明在动力学和结构研究中很有用,并且最近已用于大体积蛋白质的NMR结构测定[2]。前景与展望随着越来越多的细菌代谢途径被用于提供特定的同位素标记,通过开发利用这些技术进行溶液NMR弛豫实验,可以实现对各种残基类型的蛋白质动力学行为的深入研究。参考文献1. Muchmore DC, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW. 1989. [3] Expression and nitrogen-15 labeling of proteins for proton and nitrogen-15 nuclear magnetic resonance.44-73. https://doi.org/10.1016/0076-6879(89)77005-12. Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Mei Ono A, Güntert P. 2006. Optimal isotope labelling for NMR protein structure determinations. Nature. 440(7080):52-57. https://doi.org/10.1038/nature045253. Wand A, Bieber R, Urbauer J, Mcevoy R, Gan Z. 1995. Carbon Relaxation in Randomly Fractionally 13C-Enriched Proteins. Journal of Magnetic Resonance, Series B. 108(2):173-175. https://doi.org/10.1006/jmrb.1995.11194. Lee AL, Urbauer JL, Wand AJ. 1997. 9(4):437-440. https://doi.org/10.1023/a:10183110133385. Gardner KH, Kay LE. 1997. Production and Incorporation of15N,13C,2H (1H-?1 Methyl) Isoleucine into Proteins for Multidimensional NMR Studies. J. Am. Chem. Soc.. 119(32):7599-7600. https://doi.org/10.1021/ja97065146. Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. 1999. 13(4):369-374. https://doi.org/10.1023/a:10083932012367. LeMaster DM, Kushlan DM. 1996. Dynamical Mapping ofE. coliThioredoxin via13C NMR Relaxation Analysis. J. Am. Chem. Soc.. 118(39):9255-9264. https://doi.org/10.1021/ja960877r8. Teilum K, Brath U, Lundström P, Akke M. 2006. Biosynthetic13C Labeling of Aromatic Side Chains in Proteins for NMR Relaxation Measurements. J. Am. Chem. Soc.. 128(8):2506-2507. https://doi.org/10.1021/ja055660o9.Lundström P, Teilum K, Carstensen T, Bezsonova I, Wiesner S, Hansen DF, Religa TL, Akke M, Kay LE. 2007. Fractional 13C enrichment of isolated carbons using [1-13C]- or [2-13C]-glucose facilitates the accurate measurement of dynamics at backbone C? and side-chain methyl positions in proteins. J Biomol NMR. 38(3):199-212. https://doi.org/10.1007/s10858-007-9158-610. LeMaste D, Cronan J. 1982. Biosynthetic production of 13C-labeled amino acids with site-specific enrichment. Journal of Biological Chemistry. 257(3):1224-30.11. LeMaster DM, Richards FM. 1982. Preparative-scale isolation of isotopically labeled amino acids. Analytical Biochemistry. 122(2):238-247. https://doi.org/10.1016/0003-2697(82)90275-5
参数原理
2023.02.14

引入二氟甲基的研究进展
简介将氟原子引入生物活性有机化合物已经成为药物设计和先导化合物优化的重要策略之一。在药物合成策略中,氟经常被用来改变生物相关特性,如代谢稳定性、碱性、形成氢键(HB)能力、亲脂性和生物利用度。此外,化合物与生物目标的结合亲和力也可能在氟化后得到改善,因此,氟被认为是羟基、硫醇或胺基团的生物电子等排体的优秀候选。图1.二氟甲基基团上市药物中的大多数氟化合物由含有单个氟原子或三氟甲基,由于二氟甲基自身特殊的化学和物理特性从而具备的一些独特的优势,如今人们对含有二氟甲基的有机化合物越来越感兴趣。二氟甲基作为亲脂性氢键供体,其规模与硫酚、苯胺和胺基相似,但与羟基不同。特别是,相较于三氟甲基取代甲基时亲脂性急剧改变这一情况,当二氟甲基取代时,其亲脂性只是适度增加,同时还可以具有氢键供体的性质,实例表明二氟甲基能够与靶点形成氢键,可以加强药物分子与靶点的相互作用[1]。例如,一系列ArOCF2H化合物表现出高亲脂性以及氢键的酸性(图2)。图2.二氟甲基基团(CF2H)的理化性质二氟甲基的引入1.直接引入二氟甲基Hartwig课题组通过使用硼氢化钠还原将著名的Rupert-Prakash试剂(TMSCF3)制备成了一种新型二氟甲基化试剂TMSCF2H[2],该试剂可以实现铜盐催化下的碘苯二氟甲烷化,产率高,操作简单(方案1)。然而,该反应仅仅适用于富电子型碘苯底物,且需要向反应中加入大过量的二氟甲基化试剂(5倍当量)。此外,该反应不能与醛和酮等官能团兼容,否则羰基会发生亲核加成。方案1另一方面,Prakash课题组在铜盐催化下使用三丁基二氟甲基锡试剂n-Bu3SnCF2H完成了碘苯底物的二氟甲基化,反应温度为100至120°C[3]。(方案2)不同于TMSCF2H,二氟甲基锡试剂即可以与含吸电子基团的底物反应,而且还能与醛和酮等官能团兼容。方案22.含氟砌块引入二氟甲基将含氟中间体作为合成砌块,再经由适当的反应路线将其合成含氟的目标化合物,是合成含氟有机化合物的重要方法之一。一方面,利用含氟砌块有利于目标分子进行工业化合成;另一方面,含氟砌块相较于氟化剂而言,其反应中不涉及C-F键的断裂和生成,而仅涉及普通官能团转化和C-C键的生成,因此具有反应温和、选择性好和产率高的优点。在含二氟甲基化合物的合成中,二氟乙酸酯和二氟乙酸衍生物类的砌块由于具备羰基和烯基等可以作为反应位点的官能团,可使二氟甲基基团高效地引入到化合物中,且更适合后续的工业化生产,所以受到了越来越多的关注。二氟甲基吡唑酰胺的关键中间体3-(二氟甲基)-1-甲基-1H-吡唑-4-羧酸,可以用含氟砌块二氟乙酸乙酯作为原料进来合成。在碱性条件下含氟砌块二氟乙酸乙酯进行克莱门森缩合反应,然后与原甲酸乙酯进行缩合消除反应,下一步在低温条件下与甲基肼进行环合反应并水解得到产物[4] (方案3)。方案3使用含有二氟甲基的丁烯酮砌块在钠醇条件下与脒类化合物反应,待反应结束后,加入水并冷却、再通过滤和干燥,即可得到相应的含二氟甲基的嘧啶类化合物[5]。(方案4)方案4药物设计中的应用二氟甲基具有非凡的化学和物理性质,可用于药物设计。根据分子中所需的变化,还可以设置更精确的设计规则用XCF2H替换XCH3。此外,在药物设计中二氟甲基的作用与其连接的官能团密切相关,该官能团不仅是氢键供体官能团,同时调节分子的亲脂性。除了单氟原子和三氟甲基之外,这在药物结构中引入含氟基团方面提供了额外的选择。参考文献[1]Zafrani Yossi,Yeffet Dina,Sod-Moriah Gali,Berliner Anat,Amir Dafna,Marciano Daniele,Gershonov Eytan,Saphier Sigal. Difluoromethyl Bioisostere: Examining the "Lipophilic Hydrogen Bond Donor" Concept.[J]. Journal of medicinal chemistry,2017,60(2). https://doi.org/10.1021/acs.jmedchem.6b01691[2]Fier P S, Hartwig J F. Copper-mediated difluoromethylation of aryl and vinyl iodides[J]. J. Am. Chem. Soc., 2012(134): 5524.[3]Prakash G K S, Ganesh S K, Jones J, et al. Copper-mediated difluoromethylation of (hetero)aryl iodides and beta-styryl halides with tributyl(difluoromethyl)stannane[J]. Angew. Chem., Int. Ed., 2012(51): 12090.[4]Stierli D, Walter H, Rajan R, et al. Novel microbiocides:WO, 2009127726[P]. 2009-10-22.[5]Daniel R, Fandrick D R, Desrosiers J, et al. General and rapid pyrimidine condensation by addressing the rate limiting aromatization[M]. Org. Lett., 2014, 16: 2834-2837.
参数原理
2023.02.10

PD-1/PD-L1 信号通路
背景介绍程序性细胞死亡蛋白1,又称PD-1和CD279,是一种细胞表面受体,通过抑制T细胞炎症活性,在下调免疫系统和促进自我耐受方面发挥重要作用。在人类中,它由PDCD1基因编码。PD-1是免疫球蛋白超家族的一员,在t细胞和前b细胞上表达。PD-1作为受体,有两个配体PD-L1 (B7-H1)和PD-L2 (B7-DC)。当PD-1与配体结合时,可诱导淋巴结内抗原特异性t细胞发生凋亡(程序性细胞死亡)的双重机制,同时减少调节性t细胞(抗炎、抑制性t细胞)的凋亡。 根据之前的研究,我们发现PD-L1负责肿瘤免疫调节。因为PD-1与PD-L1的结合亲和力是PD-1对肿瘤细胞和造血细胞中PD-L2和PD-L1表达的亲和力的3倍,这是由促炎因子如IFN-γ和TNF-α的刺激所决定的。PD-L1在造血细胞和非造血细胞中广泛表达,而PD-L2在分泌IL-4和IFN -γ的巨噬细胞、树突状细胞(dc)和肥大细胞上表达受限。最近有报道称PD-L2与巨噬细胞(M8)蛋白质的排斥引导分子B (RGMB)相互作用。虽然有一些关于PD-L2的报道,但关于其在癌症免疫抑制中的作用的信息很少。PD -1和PD-L1/L2介导的免疫抵抗机制PD-1/PD-L信号通路调节外周耐受的诱导和维持。PD-L通过在耐药树突状细胞中表达实现诱导T细胞耐受的功能。此外,PD-L1在APCs和树突状细胞上组成性表达,并被促炎细胞因子进一步上调。当T细胞被激活时,PD-L1被上调。在最初的T细胞激活后,PD-1/PD-L相互作用可以限制自反应性T细胞增殖和细胞因子的产生。在肿瘤微环境中,PD-1及其配体PD-L1逃避肿瘤中和免疫监视,在肿瘤进展和存活中发挥着重要作用。研究发现PD-1在多种免疫细胞、肿瘤浸润淋巴细胞(TILs)和肿瘤细胞上表达,PD-L1与T细胞PD-1的结合会导致T细胞功能障碍、衰竭、中和和肿瘤肿块中白细胞介素-10 (IL-10)的产生。因此,肿瘤过度使用PD-L1的功能是保护自身免受细胞毒性T细胞(CD8+)介导的细胞杀伤。由于CD8+ T细胞的耗尽,肿瘤细胞变得非常具有侵略性,并分泌几种促炎细胞因子。这些细胞因子进一步上调PD-L1。另一种亚型T细胞,如调节性T细胞(Treg, CD4+ Foxp3+)通过维持其表面PD-1的表达,创造了高度免疫抑制的肿瘤环境。我们观察到,在CD3和TGF-β存在的情况下,调节性T细胞(Treg细胞)的PD-1受体增加了原始CD4+ T细胞向Treg细胞的新生转化,从而减弱了免疫应答。这种转化通过抑制哺乳动物雷帕霉素靶蛋白(mTOR) -Akt信号级联增加Treg的表达和CD4+ T细胞的免疫抑制功能。由此可见,PD-1的表达不仅抑制了效应t细胞的功能,而且增强了免疫抑制Treg细胞群的转化。尽管PD-1在t细胞中已被广泛研究,但它在b细胞中的功能在肿瘤免疫抑制中也已变得明显。PD-1的表达在B细胞分化过程中受到高度调控,但在pro-B细胞中PD-1的表达不显著,且随B细胞分化而升高。此外,PD-1激活的toll样受体9 (TLR9)激动剂可以显著促进b细胞的成熟。因此,抑制PD-1在B细胞上的功能已被证明可以增强抗原特异性抗体反应,表明PD-1在抑制B细胞介导的t细胞激活中发挥作用。癌症免疫疗法在癌症免疫治疗中,抗原呈递细胞(APCs)与肿瘤细胞和T细胞释放的抗原(Ag)结合,激活T细胞受体(TCR)和MHC结合。肿瘤间质PD-L1与T细胞PD-1相互作用抑制T细胞介导的肿瘤细胞毒性;如果使用抗PD-1和PD-L1药物,通过阻断PD-1或PD-L1可增加T细胞介导的肿瘤细胞毒性,并最终杀死肿瘤细胞。与早期激活的CTLA-4不同,PD-1检查点只调节细胞毒性T淋巴细胞迁移到肿瘤的活性。PD-L1配体的表达具有选择性,在正常炎症组织中没有过表达。这使得阻断PD-1途径药物的生物学效应成为可能,PD-1途径药物比抗ctla -4毒性小得多。抗pd -1药物nivolumab的首次小规模试验非常成功。在接下来的临床试验中,相当一部分顽固性黑色素瘤、肾癌和肺癌也观察到肿瘤消退。以前被认为是非免疫原性(不能引起免疫排斥反应)的肺癌被证明对PD-1具有耐药性。这一发现立即改变了PD-1应用的视野,研究人员对抗PD-1和抗pd - l1药物在各种癌症类型中进行了多次临床试验。自2014年初针对晚期黑素瘤的抗pd -1单克隆抗体首次获批以来,已有6种不同的药物进入临床,用于各种适应症。这些适应症包括非小细胞肺癌、霍奇金淋巴瘤和皮肤梅克尔细胞癌、肾癌、膀胱癌、头颈部癌,以及被称为微卫星不稳定性的基因标记具有高突变负担的肿瘤。这说明抗pd -1和抗PD-L1药物可能在表达PD-L1的肿瘤中作为广谱抗肿瘤药物。图1. PD-1/PD-L1信号通路参考文献1. Francisco L M., et al. The PD-1 pathway in tolerance and autoimmunity. Immunological Reviews.2010, 236: 219–42.2. Arlene H S., et al. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. NATURE IMMUNOLOGY. 2007, 8: 239-245.3. Hashem O A., et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Frontiers in Pharmacology. 2017, 8: 1-15.
参数原理
2023.02.07

负型光刻胶光刻流程
光刻工艺是半导体制造中最为重要的工艺步骤之一。主要作用是将掩膜板上的图形复制到硅片上,为下一步进行刻蚀或者离子注入工序做好准备。光刻的成本约为整个硅片制造工艺的1/3,耗费时间约占整个硅片工艺的40~60%。详细步骤1. 基材清洗经过不同工序加工后,基材表面受到了严重的沾污,常见的有:有机杂质沾污,颗粒沾污,金属离子沾污等等。利用乙醇(项目号:E130059)或三氯乙烯(项目号:T119718)作为溶剂清洗基材,可以有效去除沾污。经过清洗,漂洗后的基材,要在120-200°C的温度下将基板烘烤20分钟。最终基材上不再有任何有机杂质和表面颗粒残留。2. 底胶涂覆使用旋转涂层系统可以达到均匀的高度和一致的涂层厚度。在旋涂开始之前,基材应涂上光刻胶。旋涂时千万不要涂光刻胶,因为可能会导致分布不均。方形或矩形衬底最好在低转速(50-1000转)下涂覆。75转时未稀释的抗蚀剂可产生2.5μM左右的涂层,基材的边缘和角落更厚。抗蚀剂的厚度取决于自旋速率、加速度和抗蚀剂的粘度。圆形基材最好在高转速(2000-5000转)下涂覆。基材在> 5000 rpm的转速下旋转对阻片厚度影响不大。随着粘度的降低,厚度对自旋速率的依赖性也随之降低。为了获得更均匀、更薄的薄膜,并避免基材边缘增厚,加速到峰值转速0.1秒是最佳的。使用旋涂系统,薄膜厚度从0.3到2μM不等。为了避免蚀刻穿透和针孔的形成,通常首选厚膜(1-2μM)。然而,厚的薄膜会导致分辨率的降低。为了获得更好的效果,可以涂两层薄薄的涂层。3. 预烘为了使抗蚀剂和基材之间产生较大的附着力,需要通过蒸发去除所有残留的溶剂和挥发性成分。未能彻底烘烤抗蚀剂会影响抗蚀剂工艺所必需的交联。但也应注意避免过度烘烤,否则会导致抗蚀剂起雾或分解。一般推荐在82°C下预烘20分钟即可。4. 光致抗蚀剂曝光任何有近紫外辐射的光源都可以用来曝光光刻胶。大面积光源应仅用于粗线(50 μM 或更大)。对于细线图案,必须使用漫射较小的光源。推荐的细纹光源有碳、高压汞蒸汽或氙气闪光灯。适当的曝光需要约100 MW/cm2的光强。厚度和加工变量会影响曝光。只要光源产生10 MW/cm2的最小辐照量在衬底表面,1-10秒的曝光时间即可。曝光能的变化不应超过最优值的10%,否则会失去细线清晰度和再现性。光刻胶的衍射效应可引起掩模下的交联,导致线展宽达2.5μM。5. 显影将显影剂喷在涂覆的基材上10-60秒,然后用异丙醇(项目号:I292350)冲洗几次。然后用氮气或纯净的压缩空气吹干表面。6. 后烘需要对光刻胶进行烘干处理,以去除残留的溶剂和挥发性成分,并有助于增强光刻胶的化学稳定性和附着力。建议后焙时间为10-20分钟,温度为120°C,绝不能超过148°C。7. 刻蚀对于大多数刻蚀步骤,晶圆上层的部分位置都会通过“罩”予以保护,这种罩不能被刻蚀,这样就能对层上的特定部分进行选择性地移除。在有的情况中,罩的材料为光阻性的,这和光刻中利用的原理类似。而在其他情况中,刻蚀罩需要耐受某些化学物质,氮化硅就可以用来制造这样的“罩”。8. 光刻胶去除光刻胶的主要功能是在整个区域进行化学或机械处理工艺时,保护光刻胶下的衬底部分。所以当以上工艺结束之后,光刻胶应全部去除,这一步骤简称去胶。常用的去胶方式有两种:湿法去胶和干法去胶。湿法去胶又可以细分成有机溶剂去胶(利用有机溶剂除去光刻胶)和无机溶剂(通过使用一些无机溶剂,将光刻胶这种有机物中的碳元素氧化为二氧化碳,进而而将其除去)。干法去胶则是利用等离子体将光刻胶剥除。注意事项1. 注意避免灰尘和杂质污染;2. 相对湿度应控制在30-50%之间;3. 适当的照明,最好是金色荧光,黄色白炽灯,或白色荧光带黄色或橙色滤镜;4. 保持适当的通风环境。
参数原理
2023.02.06
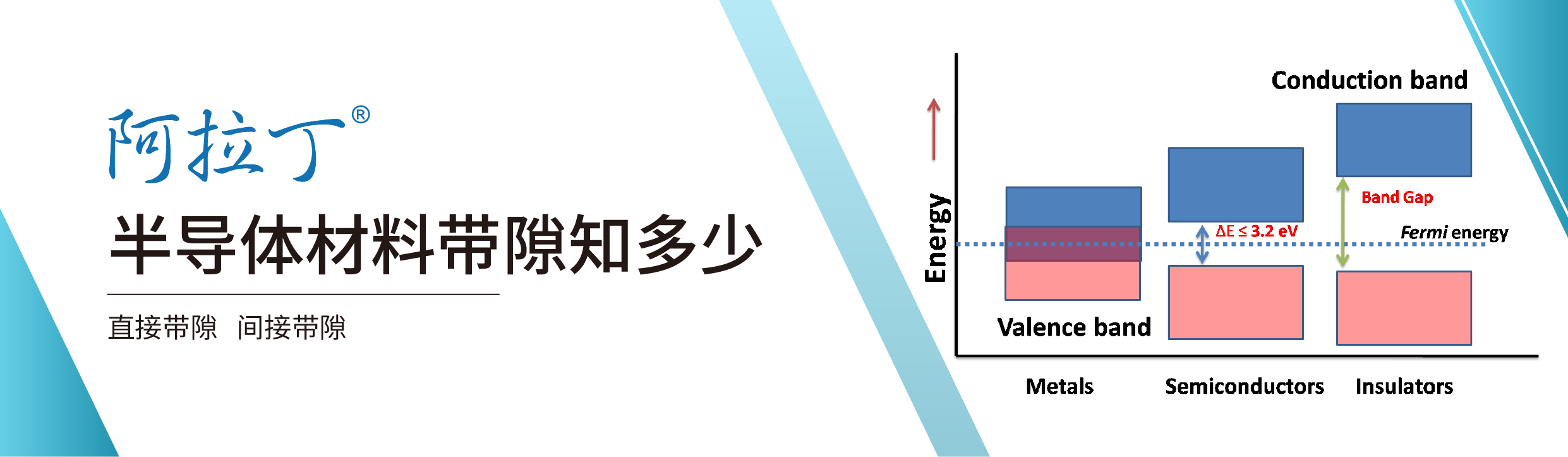
半导体材料带隙知多少
什么是半导体材料带隙?在原子或分子中,电子通常占据不同的能级。然而,在固体中,这些能级合并在一起形成能带。能带分成价带和导带两个部分。其中,用于将原子结合在一起的电子存在于价带中,而其余未成对电子则存在于导带中[1]。导带和价带之间的区域被称为能带带隙。它是激发电子从价带(束缚态)跃迁到导带(自由态)所需要的最小能量。带隙的大小对材料的某些性质有很大的影响。例如,如果给一个用于成键的电子提供足够的能量,它就可以被激发跃迁至导带,从而可以自由导电。如何根据带隙对材料进行分类?对于导体来说,导带和价带之间的带隙为零,因此导带充满电子,材料的导电性很强。相反地,绝缘体的价带和导带之间有很大的带隙,使得价带的电子几乎不可能跃入导带,材料体现为不导电。半导体的带隙则介于两者之间,一般体现为不导电,但是通过注入能量的形式(光激发,热激发等),可以使其价带上的电子发生跃迁,从而具备导电能力。图1:导体、半导体和绝缘体的能带示意图根据电子跃迁所需的能量和动量条件,又可以将半导体分为直接带隙半导体和间接带隙半导体。在直接带隙半导体中,电子要跃迁到导带上产生导电的电子和空穴(形成半满能带)只需要吸收能量。代表有GaN(项目号:G119228),GaAs(项目号:G119227), InAs(项目号:I119233),GaSb(项目号:G119230)等III-V族半导体。对于间接带隙半导体,电子要跃迁到导带上产生导电的电子和空穴(形成半满能带)不仅需要吸收能量,还要改变动量。代表有Si(项目号:S108980), Ge(项目号:C105165)等元素半导体。如何计算半导体材料带隙?一般而言,判断半导体材料带隙大小有两种方法:方法一:截线法。截线法是一种简易的求取半导体禁带宽度的方法,依据原理是半导体的吸收阈值λg和其禁带宽度Eg成反比,两者之间关系式如下:Eg (eV)=1240/λg (nm)因此,可以通过求取λg来得到Eg。从UV-vis DRS(紫外可见漫反射)谱图中可以得到材料在不同波长下的吸收。对波长-吸收曲线求一次微分,之后在极值点做截线(斜率为极值点纵坐标数值),截线与横坐标交点即为λg。代入上式可得材料的禁带宽度Eg。方法二:Tauc plot法。此方法由Tauc, Davis和Mott[4]等人推导出,具体表达式如下:(αhv)1/n=A(hv-Eg)hv=hc/λ其中,α为吸光指数,h为普朗克常数,c为光速,λ为光的波长。V为频率,A为常数,Eg为半导体禁带宽度。指数n与半导体类型相关:其中直接带隙半导体为1/2,间接带隙半导体为2。需要注意的是,读取的UV-vis DRS谱图纵坐标应为吸收值Abs,如果是透过率T%,可以通过公式Abs=-lg(T%)进行换算。宽带隙半导体的应用宽频带隙(WBG或“宽频带隙”)半导体是各种电子器件的关键,如透明触点、p-n结和薄膜晶体管。自20世纪50年代,氧化物宽带隙半导体得到了广泛的研究,特别是其高透明度和高导电性的矛盾特性。透明导电氧化物(TCO)掺锡的In2O3,即ITO,在过去的几十年里一直是各种商业设备的关键可能的替代品,如f掺杂SnO2(FTO)[5]和al掺杂ZnO(AZO)[6],已经得到了深入的研究和应用。在21世纪,多透明非晶氧化物半导体(TAOS),如氧化铟镓锌(IGZO),由于其高透明度、高迁移率和良好的均匀性,也被广泛研究为薄膜晶体管(TFT)中的通道层[7]导致其在液晶显示器(lcd)中的商业应用。图3:柔性TTFTs a)在塑料片材上制备的TTFT结构;b)柔性TTFT板材弯曲到R=30 mm的照片;c)柔性TTFT片材的照片[7]虽然这些n型TCOs表现出优异的性能,但p型掺杂和氧化物中的高孔洞迁移率在实践中被证明要困难得多。这主要是由于(1)色散中定位价带空穴的内在限制和(2)通过p型掺杂引入空穴和最小化补偿缺陷的挑战。以脱钙基CuAlO2为初步实例,提出的“价带化学调制”(CMVB)策略取得了突破[8]。这种方法使用O 2p态与金属3d态在价带最大值(VBM)处的杂化,增加色散。随后,这种策略和其他方法的使用导致了各种基于铜的p型TCOs,尽管p型TCOs具有光电性能,但仍然不能与n型TCOs相比。由于这些挑战,只有一小部分p型TCO材料已被纳入商业器件应用,例如Cu2O和SnO,它们主要用于薄膜晶体管。参考文献[1] Kittel C. and McEuen P. (2019), Introduction to Solid State Physics, 8th Ed. Hoboken, NJ: John Wiley & Sons.[2] Tsagli K, Dordevic S V. Temperature Dependence of Photoluminescence Spectra in Polystyrene[J]. Materials Performance and Characterization, 2020, 9(1): 675-681. https://www.astm.org/mpc20200093.html[3] Semiconductors W B. Fundamental Properties and Modern Photonic and Electronic Devices/Eds[J]. K. Takahashi, A. Yoshikawa, A. Sandhu. New York, 2007.[4] Tauc J, Menth A. States in the gap[J]. Journal of non-crystalline solids, 1972, 8: 569-585. https://doi.org/10.1016/0022-3093(72)90194-9[5] Rakhshani A E, Makdisi Y, Ramazaniyan H A. Electronic and optical properties of fluorine-doped tin oxide films[J]. Journal of applied physics, 1998, 83(2): 1049-1057.https://doi.org/10.1063/1.366796[6] Jiang X, Wong F L, Fung M K, et al. Aluminum-doped zinc oxide films as transparent conductive electrode for organic light-emitting devices[J]. Applied Physics Letters, 2003, 83(9): 1875-1877. https://doi.org/10.1063/1.1605805[7] Nomura K, Ohta H, Takagi A, et al. Room-temperature fabrication of transparent flexible thin-film transistors using amorphous oxide semiconductors[J]. nature, 2004, 432(7016): 488-492. https://www.nature.com/articles/nature03090?free=2[8] Kawazoe H, Yasukawa M, Hyodo H, et al. P-type electrical conduction in transparent thin films of CuAlO2[J]. Nature, 1997, 389(6654): 939-942. https://www.nature.com/articles/40087
参数原理
2023.02.01

喜报!阿拉丁荣登“上海生产性服务业品牌价值榜”!
阿拉丁荣登上海生产性服务业品牌价值榜 上海阿拉丁生化科技股份有限公司品牌价值超1亿元荣登2022“SFEO上海生产性服务业品牌价值榜” “SFEO上海生产性服务业品牌价值榜”由上海市工业经济联合会、上海市经济团体联合会、联手上海企业文化与品牌研究所打造。此次荣登榜单是对阿拉丁一直坚持走品牌化道路的肯定,同时也充分说明了阿拉丁在区域内和行业内的影响能力。 图片1:阿拉丁张江生物研发基地(细胞融合实验室)图片2: 阿拉丁奉贤化工厂(色谱分析仪器) 品牌是提升企业附加值的有效途径,也是衡量经济转型成效的重要指标。品牌价值榜也是各地区、各行业引导品牌经济发展、反映品牌建设成果的手段之一。未来,阿拉丁将坚持走品牌经营之路,打造高质量发展引擎,继续为客户提供高效的产品和服务,不断提高企业的品牌价值和影响力,为行业发展做出贡献! 图片3: 阿拉丁物联化仓库 图片4 : 阿拉丁产品新包装
企业动态
2023.01.29

阿拉丁客服质量再提升
近日阿拉丁在针对客户的售后及售前问题上,为了使公司会员(登录用户)可以更方便快捷的解决其相关疑惑,对阿拉丁网站的相关功能模块进行了优化升级!将“服务单”体系与真人客服“阿拉丁铁蛋”进行关联,更好的诠释了阿拉丁“便、捷、快、优”的一站式服务特色。 高效 | 官网“服务单”再升级 阿拉丁已实行用“服务单”管理售前售后售中,一切以客户需求为导向,将您的问题形成“服务单”,以便客服跟踪,直至解决。 登录用户:可直接我的账号查看提交服务单及我司专业人员的答复;未登录用户:可通过官网的联系我们或者客户支持填写相对应的问题,从问题类型下拉框选择不同类型的问题,我们后台会据此分配相应的专业解答人员进行解惑。 便捷 | 客服“铁蛋”真人沟通 将原48小时提交服务单升级为客户与客服机器人“阿拉丁铁蛋”进行实时聊天,该优化基于您的需求指派真人为您服务—— 可根据您的问题相关性指派对应领域专业客服进行实时有效沟通,退出即可开始新的对话;也可根据您的选择在1-2分钟内快速生成服务单(在您的账号下查找到相关联问题的服务单),以便对应问题客服及时高效的跟踪您的需求,并将在1-2个工作日内彻底解决。 声明:文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请移步至阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
企业动态
2023.01.13

阿拉丁小分子抑制剂,助力新冠药物研发!
阿拉丁小分子抑制剂,助力新冠药物研发!截至2022年9月30日根据FDA数据统计目前有700多个药物开发项目处于规划阶段 450多个试验 已被FDA审查 13种治疗药物 获得紧急使用许可 2种新药 获得FDA批准用于治疗COVID-19 新型冠状病毒治疗方法 抗病毒药物免疫调节剂中和抗体疗法细胞疗法基因疗法 阿拉丁COVID-19抑制剂产品系列 CAS号项目号产品名称中文名称83905-01-5A134451Azithromycin (CP-62993)阿奇霉素95233-18-4A151027Atovaquone阿托伐醌394730-60-0B12549Boceprevir博赛泼维190786-44-8B129218Bepotastine Besilate苯磺酸贝托司汀1611493-60-7B413365Bictegravir比克替拉韦125602-71-3B413372Bepotastine贝他斯汀868540-17-4C127870Carfilzomib (PR-171)卡非佐米59721-29-8C129276Camostat Mesilate甲磺酸卡莫司他50-63-5C129284Chloroquine diphosphate二磷酸氯喹61422-45-5C153374Carmofur卡莫氟126544-47-6C287438Ciclesonide环索奈德23541-50-6D122335Daunorubicin (RP 13057) HCl道诺霉素206361-99-1D125841Darunavir达芦那韦850876-88-9D127014Danoprevir (ITMN-191)丹诺瑞韦635728-49-3D129325Darunavir Ethanolate地瑞那韦乙醇盐259793-96-9F303252Favipiravir (T-705)法匹拉韦747-36-4H141480Hydroxychloroquine Sulfate (NSC 4375)羟氯喹70288-86-7I141334Ivermectin (MK-933)伊维菌素, 依维菌素157810-81-6I303016Indinavir Sulfate硫酸茚地那韦1256388-51-8L125620Ledipasvir (GS5885)雷迪帕韦192725-17-0L129320Lopinavir (ABT-378)洛匹那韦134678-17-4L129788Lamivudine (BCH-189)拉米夫定, 拉美呋定, 拉米呋啶82586-52-5M129328Moexipril HCl盐酸莫西普利70476-82-3M129460Mitoxantrone (NSC-301739) 2HCl米托蒽醌二盐酸盐2349386-89-4M406650Molnupiravir (EIDD-2801)莫纳皮拉韦82956-11-4N129262Nafamostat mesilate (FUT-175)甲磺酸萘莫司他159989-65-8N137745 Nelfinavir (AG 1343) Mesylate甲磺酸奈非那韦147127-20-6T125736Tenofovir替诺福韦, 泰诺福韦327036-89-5T126722TDZD-8402957-28-2T126775Telaprevir (VX-950)特拉匹韦865854-05-3T127039Tideglusib (NP031112)泰格列净1377049-84-7V173757Velpatasvir维帕他韦
企业动态
2023.01.13

通过生物正交点击反应制备多功能细胞培养支架
简介在组织工程和再生医学领域,设计能够以三维方式再现活组织复杂细胞外本质的生物材料支架是一项具有挑战性的要求。水凝胶是一类被广泛研究和开发的合成型细胞外基质模拟物(ECM),由于其高含水量、足够的弹性和易于转移的特性,因此适合在2D和3D环境中细胞培养,并作为在模拟软组织环境中一些关键力学性能测试的载体[1-5]。一般来说,水凝胶可以通过各种天然和合成的前驱体来制备得到,这些前驱体可以通过物理、离子或共价相互作用交联[2]。当细胞在这样的体系中被培养时,由天然聚合物形成的水凝胶能够为其提供大量的生物信号(例如:细胞粘附蛋白、生长因子库、蛋白质降解域),但在其结构上通常缺乏灵活性,以重复单元的结构组成,来控制最终获得材料的理化性质。此外,由合成大分子形成的水凝胶可以精确控制在组织工程上的一些关键参数,如力学、膨胀和降解,但通常这些化学成分是细胞无法识别的。因此,人们对开发合成凝胶作为细胞载体和培养体系的研究方向越来越感兴趣,这样的体系能够有效整合合成材料和天然材料的优点。本文将基于聚乙二醇(PEG)水凝胶的研究进展讨论其中涉及到的一些概念。聚乙二醇水凝胶已被广泛应用于2D和3D细胞培养,各种方法和理论被用于控制聚乙二醇凝胶的性质和整合细胞信息[3-5]。更重要的是,聚乙二醇材料能够有效减少非特异性蛋白质的相互作用,使研究人员能够更好地了解细胞如何从细胞外部环境接收特定信号。聚乙二醇水凝胶合成的化学和分子设计原理通常在含水条件下,PEG水凝胶是由共价或非共价交联的线性或支链/星型聚合物链形成的。物理交联的聚乙二醇(PEG)和聚环氧丙烷嵌段共聚物由于具有较低的临界溶解温度(LCST)(≈37℃),通过原位形成水凝胶而得到了广泛的应用[6];然而,这些通过非共价键组装而成的水凝胶的力学强度通常较弱。为此,研究者们发现了一种新的替代方法,即通过共价交联法来构建高稳定性的PEG水凝胶[2]。为了建立用于细胞培养的共价交联网络,还必须注意所选取的交联方法的细胞相容性。在这项研究中,两种聚合机理被广泛使用:(i)链式增长;(ii)阶梯式增长[4,5]。末端官能化的PEG的链聚合是稳定且简单的[4],但是它的网络构成复杂,结构具有一定的异构性,动力学链长度分布宽,且它的降解仅限于网络交联点。近期,聚乙二醇的阶梯式增长聚合机理成为一种更为简便的方法,可以实现可控的、更均匀的交联聚合物网络,同时提供了引入生物信号的简单途径。通常,对基于PEG的阶梯增长水凝胶,会采用以化学计量的方式使用双官能化的分子交联多官能化的分子系统;一般来说,形成凝胶的基本条件是其平均官能度必须大于2。根据所需的网格大小或交联密度,这些分子中的任何一种或两种都可以从基于PEG的聚合物中衍生出来(图1)。针对3D细胞培养和组织再生的水凝胶,能够适用于生理条件和高效、无毒的化学交联方法都是关键的必要条件,因为凝胶通常需要在有细胞和蛋白质存在的情况下形成。这些复杂性促使生命科学家们探索各种化学反应,以形成温和和具特异性的水凝胶,但其反应条件仍需保持在适当的生理pH值和温度下进行。图1:4臂和线性PEG前驱体的结构;以及通过这些PEG大分子形成的阶梯式生长水凝胶的示意图。在结构复杂但明确的支架中的点击化学反应最近在生物材料领域带来重大进展的化学策略之一是“点击化学”,由Sharpless和他的同事提出,并获得了2022年的诺贝尔化学奖,用于描述在水溶液条件下连接两个分子组分的快速、高选择性和高产率的反应[7]。虽然铜催化的叠氮炔环加成反应(CuAAC)(图2A)是最早被发现的点击反应,但后来发现了多种化学反应,包括Michael加成反应(图2B)、光引发的硫醇-烯反应(图2C)、Staudinger连接反应和应变促进的叠氮化物-炔基环加成反应(SPAAC),以实现点击化学的概念[7-9]。由于这组反应不仅相互正交,而且在生物系统中与功能基团正交,它们已经成为生物材料科学家构建控制良好、高度明确的复杂材料支架的优越化学工具。图2:常见的点击反应:A)铜催化叠氮炔环加成;B)碱催化硫醇与马来酰亚胺的Michael加成反应;C)光引发硫醇-烯偶联反应。在以上对聚乙二醇水凝胶和点击化学的介绍之后,本文的剩余部分将集中在:(i)利用各种点击反应构建用于细胞培养的聚乙二醇水凝胶,以及(ii)如何利用点击反应的正交性,通过选择性生物表达精确功能化这些材料,用以指导细胞功能。MICHAEL加成反应由于反应条件温和、反应速度快,迈克尔型的加成反应是在细胞存在的情况下形成水凝胶材料的绝佳点击反应。这种类型的反应是由于硫醇盐阴离子(迈克尔供体)亲核进攻α,β-不饱和酮(迈克尔受体)的缺电子双键而进行的(图2B)。乙烯基砜、马来酰亚胺和丙烯酸酯都是广泛用于形成水凝胶的迈克尔受体。Hubbell和他的团队[10,11]是第一批使用4臂聚乙二醇乙烯基砜和半胱氨酸侧翼肽(图3)开发阶梯式增长水凝胶并且用于细胞培养的团队之一。由于硫醇到硫醇盐阴离子的去质子化对这些反应至关重要,需要通过添加碱来拔氢,Hubbell团队通过简单地调节缓冲液的pH值来解决这个问题。这种合成路径使他们能够在不需要额外修饰的情况下合并半胱氨酸多肽。通过这种方法,他们证明了许多肽表位可以控制细胞粘附(例如:Arg-Gly-Asp-Ser (RGDS)、Ile-Lys- Val-Ala-Val (IKVAV)),以及一种纤溶蛋白敏感肽交联剂,可以被细胞分泌的蛋白酶降解。Garcia团队报道了马来酰亚胺功能化的多臂PEG,使用了极低浓度的碱基交联二巯基肽交联剂,具有更快的反应动力学,这在封装更敏感的细胞类型时提供了额外的帮助。图3:通过Michael加成,在4臂聚乙二醇四聚乙烯基砜和半胱氨酸末端功能化的MPP可切割肽之间形成阶梯生长的水凝胶。依序点击反应在许多生物学应用中,人们通常希望在特定的时间点以及空间限定的区域引入相关功能信号线索。这些信号可能包括生存线索(例如整合素结合分子)、生长因子(例如隔离细胞因子的亲和配体)或细胞特异性降解位点(例如用于释放信号或允许细胞运动的MMP可裂解连接子)。这导致了连续点击反应的出现:使用一次点击化学反应形成凝胶,随后使用另外的点击化学反应以一种时空控制的方式去修饰它。由于Thiol-ene化学反应是由光驱动的,因此它已成为一种强大而通用的方法,以实现在PEG水凝胶细胞壁龛中明确和高度复杂结构的引入。利用SPAAC和Thiol-ene点击反应之间的正交性:该研究小组已经证明了可以通过SPAAC形成PEG水凝胶(图6A),然后使用Thiol-ene点击化学来控制发生反应的时间和空间位置,在这种情况下,肽交联剂携带一个垂悬的烯丙基(图6A),以允许凝胶后结构的光照反应仍然是有效的[19],在这种水凝胶基质中包裹的细胞不仅可以表达>90%的细胞活力,而且对图形化的生化线索有反应。例如,细胞形态和黏附空间的控制是通过在凝胶的特定位置绘制RGD肽图建立的(图6A)。同样的,当自我淬灭的二荧光素胶原酶裂解肽(裂解后荧光增强)经光照射后,在胶原酶活性高的区域即细胞周围能够观察到更强的荧光,从而可以实现实时观察到细胞的局部活性。近期,该研究团队同样利用正交光耦联和光裂解反应实现了生物相关线索的依序模式化和去模式化(图6B和6C)[21]。通过使用含有硝基苯醚连接体[21-23]的巯基化RGD肽,利用不同波长的光对偶联和裂解行为进行控制,从而实现光裂解(图6D),随后我们可以在水凝胶内特定位置对人间充质干细胞(hMSC)群进行外部粘附和分离(图6B和6C)。这种对水凝胶支架生物化学线索的动态控制不仅使人们能够在空间上控制特定信号和细胞类型的释放,用于再生医学中的细胞递送应用,而且还提供了通过在时空上控制特定和多个线索的呈现来调节干细胞分化的行为。图6:SPAAC水凝胶化学:A)通过利用4臂PEG四叠氮和二环基化的基质金属蛋白酶(MMP)可降解肽通过SPAAC点击化学通过逐步增长的机理形成的3D水凝胶(*显示裂解位点);B)Thiol-ene光修饰SPAAC形成的水凝胶和细胞黏附;C)模式线索的空间移除和由此产生的细胞脱离;D)硝基苯醚光裂解反应。小结在过去的十年中,点击化学凭借其便捷、多功能性和生物正交性的特点,在构建用于再生医学应用的复杂材料支架方面起到了重要的作用,彻底改变了用于细胞培养和传递的生物材料支架领域的反应体系[24]。其不仅能够创造多功能性的聚乙二醇水凝胶,并且还开辟了创新的反应途径,如依序点击反应,以时间控制的方式将生化信号嵌入水凝胶支架的特定位置。然而,在这个对于化学背景要求相对较高的领域,特别是在生物材料支架设计中,许多材料开发的创新点还没有出现。例如,虽然这些高效的化学方法已被大量用于从根本上理解和控制细胞特性,包括3D环境中的形态和粘附性,但它们如何用于更好地理解干细胞微环境中的作用机理,并以受控的方式指导其在体外更有效的分化,仍是生物科学家们面临的重大机遇和挑战。参考文献1. Peppas N, Hilt J, Khademhosseini A, Langer R. 2006. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater.. 18(11):1345-1360. https://doi.org/10.1002/adma.2005016122. Lee KY, Mooney DJ. 2001. Hydrogels for Tissue Engineering. Chem. Rev.. 101(7):1869-1880. https://doi.org/10.1021/cr000108x3. Tibbitt MW, Anseth KS. 2009. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng.. 103(4):655-663. https://doi.org/10.1002/bit.223614. Kloxin AM, Kloxin CJ, Bowman CN, Anseth KS. Mechanical Properties of Cellularly Responsive Hydrogels and Their Experimental Determination. Adv. Mater.. 22(31):3484-3494. https://doi.org/10.1002/adma.2009041795. Zhu J. 2010. Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials. 31(17):4639-4656. https://doi.org/10.1016/j.biomaterials.2010.02.0446. Jeong B, Kim SW, Bae YH. 2002. Thermosensitive sol?gel reversible hydrogels. Advanced Drug Delivery Reviews. 54(1):37-51. https://doi.org/10.1016/s0169-409x(01)00242-37. Kolb H, Finn M, Sharpless K. 2001. Angew. Chem. Int.. 40. 2004-2021. https://onlinelibrary.wiley.com/doi/10.1002/1521-3773(20010601)40:11%3C2004::AID-ANIE2004%3E3.0.CO;2-58. Hoyle C, Bowman C. 2010. Thiol-Ene Click Chemistry. Angewandte Chemie International Edition. 49(9):1540-1573. https://doi.org/10.1002/anie.2009039249. Sletten E, Bertozzi C. 2009. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed.. 48(38):6974-6998. https://doi.org/10.1002/anie.20090094210. Pratt AB, Weber FE, Schmoekel HG, Müller R, Hubbell JA. 2004. Synthetic extracellular matrices for in situ tissue engineering. Biotechnol. Bioeng.. 86(1):27-36. https://doi.org/10.1002/bit.1089711. Lutolf MP, Hubbell JA. 2003. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules. 4(3):713-722. https://doi.org/10.1021/bm025744e12. Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, García AJ. 2012. Maleimide Cross-Linked Bioactive PEG Hydrogel Exhibits Improved Reaction Kinetics and Cross-Linking for Cell Encapsulation and In Situ Delivery. Adv. Mater.. 24(1):64-70. https://doi.org/10.1002/adma.20110357413. Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. 2009. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Adv. Mater.. 21(48):5005-5010. https://doi.org/10.1002/adma.20090180814. Lin C, Anseth KS. 2009. Controlling Affinity Binding with Peptide-Functionalized Poly(ethylene glycol) Hydrogels. Adv. Funct. Mater.. 19(14):2325-2331. https://doi.org/10.1002/adfm.20090010715. Lin C, Anseth KS. 2011. Cell-cell communication mimicry with poly(ethylene glycol) hydrogels for enhancing -cell function. Proceedings of the National Academy of Sciences. 108(16):6380-6385. https://doi.org/10.1073/pnas.101402610816. Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, et al. 2006. Synthesis of well-defined hydrogel networks using Click chemistry. Chem. Commun..(26):2774. https://doi.org/10.1039/b603438a17. Liu SQ, Rachel Ee PL, Ke CY, Hedrick JL, Yang YY. 2009. Biodegradable poly(ethylene glycol)-peptide hydrogels with well-defined structure and properties for cell delivery. Biomaterials. 30(8):1453-1461. https://doi.org/10.1016/j.biomaterials.2008.11.02318. Polizzotti BD, Fairbanks BD, Anseth KS. 2008. Three-Dimensional Biochemical Patterning of Click-Based Composite Hydrogels via Thiolene Photopolymerization. Biomacromolecules. 9(4):1084-1087. https://doi.org/10.1021/bm701263619. DeForest CA, Polizzotti BD, Anseth KS. 2009. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nature Mater. 8(8):659-664. https://doi.org/10.1038/nmat247320. DeForest CA, Sims EA, Anseth KS. 2010. Peptide-Functionalized Click Hydrogels with Independently Tunable Mechanics and Chemical Functionality for 3D Cell Culture. Chem. Mater.. 22(16):4783-4790. https://doi.org/10.1021/cm101391y21. DeForest CA, Polizzotti BD, Anseth KS. 2009. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nature Mater. 8(8):659-664. https://doi.org/10.1038/nmat247322. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. 2009. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science. 324(5923):59-63. https://doi.org/10.1126/science.116949423. DeForest CA, Polizzotti BD, Anseth KS. 2009. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nature Mater. 8(8):659-664. https://doi.org/10.1038/nmat247324. Nimmo CM, Shoichet MS. 2011. Regenerative Biomaterials that “Click”: Simple, Aqueous-Based Protocols for Hydrogel Synthesis, Surface Immobilization, and 3D Patterning. Bioconjugate Chem.. 22(11):2199-2209. https://doi.org/10.1021/bc200281k
应用实例
2022.12.30

「核酸」「抗原」「抗体」三种新冠检测方式有何差别?
目前主要有三种检测方式:核酸检测抗原检测抗体检测 首先,我们用一张图,来解析新型冠状病毒的组成。如果将可怕的新型冠状病毒比喻成一个橘子?橘子内部的橘子瓣 = 病毒核酸(RNA) 橘子皮/橘瓣表面内皮 = 病毒蛋白质(S蛋白/E蛋白/M蛋白) 1. 核酸检测核酸检测检测的是病毒的遗传物质DNA或RNA,利用PCR(聚合酶链式反应)技术明确样本中是否存在新型冠状病毒,是目前确诊新型冠状病毒感染的肺炎无创诊断的“金标准”。2. 抗原检测3. 抗体检测抗体检测的是患者体内免疫系统为应对新冠病毒感染而产生的抗体,如果检测结果为阳性,说明患者体内存在针对新冠病毒的抗体,患者正处于感染期或已经康复,可作为辅助手段帮助确诊感染。 新冠疫情爆发以来阿拉丁第一时间向医疗卫生及应急部门捐赠防控物资包括口罩、手套、防护服等得到社会积极反馈 阿拉丁官网提供各类防疫产品 项目号内容M7194抗菌洗手液M299596免洗手消毒凝胶E29958575%消毒用酒精喷雾E29957875%消毒用酒精M9432一类隔离服H299581复合过氧化氢消毒液D1863医用级乳胶手套,麻面无粉D5852一次性PVC手套,无粉C1674CPE塑料鞋套A397042镜架式医用防护面罩新冠抗原检测试剂盒原料 我们已经整理在下方表格 本次阿拉丁的小科普就到这里大家要注意多多防护呀!如果喜欢本文的话别忘了分享给你的朋友哦!
企业动态
2022.12.29

用于2D和3D细胞培养的可降解型聚乙二醇水凝胶
简介随着对多样化功能型生物材料需求的不断增加,组织工程和药物输送等生物技术领域也在持续发展着。几十年来,聚合物生物材料的研究一直集中在测试为其他应用或其加工(如:静电纺丝、溶剂浇铸/致孔剂浸出、3D打印)开发的聚合物的生物相容性。而近期,研究人员已将主要关注方向转向合成专门用于生物医学用途的材料,包括合成蛋白质、糖模拟物和与水性介质相容的聚合物,以及天然聚合物的化学改性(如:通过凝胶化来增加物质在体内的稳定性)。在过去的十年中,聚合物化学家为设计的生物材料创造了一个良好的适用场景,用作于细胞支架或者药物递送。水凝胶是长期以来受到人们关注最多的生物材料之一[1],因为其在化学和物理性质上都非常接近细胞的自然环境,因此作为细胞的二维和三维支架被广泛研究[2]。水凝胶可以由合成的(例如,聚(乙二醇)、聚(甲基丙烯酸羟乙酯))与天然存在的聚合物(例如,胶原蛋白、透明质酸、肝素)交联形成,并且由于它们的含水量非常高,可以有效被应用于组织培养的3D模型,不受细胞、蛋白质和DNA的影响。根据不同组成部分的反应性,可以使用pH[3]、温度[4]、库仑相互作用、共价键、非共价相互作用[5]或聚合来诱实现导凝胶化。PEG(聚乙二醇)聚乙二醇(PEG)是一种亲水性聚合物,当形成交联网络时,能够具备很高的含水量。PEG是适用于生物应用的材料,因为它通常不会引起免疫反应[6]。自20世纪70年代以来,PEG已被用于修饰治疗性蛋白质和多肽,以增加它们的溶解度,降低其毒性,并延长其循环半衰期[7]。自20世纪70年代末,研究人员便开始尝试使用PEG水凝胶进行细胞培养。聚乙二醇水凝胶具有良好的化学反应性,多种反应性基团可用于其形成水凝胶并且实现高效的化学修饰。PEG大分子单体通过环氧乙烷的活性阴离子开环聚合能够非常方便地合成聚乙二醇;这些聚乙二醇具有较宽的分子量分布,且包含多种端基(例如:羟基、甲醚基、氨基、N-羟基琥珀酰亚胺(NHS)酯),在众多反应中都可适配。为了形成水凝胶,PEG必须发生交联反应。最开始的时候,PEG是通过电离辐射进行非特异性交联的[8],而现在PEG水凝胶的形成通常是通过共价交联具有反应链端的PEG高分子合成的。具有活性反应末端的PEG高分子聚合物,如丙烯酸酯、甲基丙烯酸酯、烯丙醚、马来酰亚胺、乙烯基砜、NHS酯和乙烯基醚基团(图1)很容易从现成的一般性材料合成。可在碱存在下使用酸性氯化物(例如:丙烯酰氯、甲基丙烯酰氯)使PEG醇链的末端酯化。PEG链末端可在碱性条件下通过与诸如2-氯乙基乙烯基醚或溴烯丙基等烷基卤化物反应而醚化。PEG二乙烯基砜是通过将PEG偶联到大量过量的二乙烯基砜或通过多步骤工艺来制备氯乙基砜链末端,氯乙基砜链末端可以通过基本消除反应形成的乙烯基砜基团[9]来制备。图1:不同PEG大分子的末端基团高分子的两个末端可以是两个相同或者不同的官能团。同双官能高分子通常用于形成网络,而异双官能高分子则可用于将具有治疗性的小分子连接到水凝胶网络中。水凝胶形成机理形成水凝胶的交联机制取决于PEG高分子链末端的特性。在大多数情况下,是与反应性乙烯基链末端聚合的同时发生交联,通常采用的是自由基引发剂。例如,大分子单体的聚合可以使用通过氧化还原反应生成的自由基(比如过硫酸铵和TEMED)或光照产生的自由基(图2中的Irgacure®651,λ=365 nM)来引发,随后通过丙烯酸酯和甲基丙烯酸酯链末端基元反应来发生链增长。在阶梯生长网络的形成中,多官能度(f>2)的交联剂以化学当量与PEG链末端进行反应,或多官能度的PEG(f>2)也可以与双官能团的交联剂发生交联。丙烯酸酯、甲基丙烯酸酯、乙烯基砜、马来酰亚胺、乙烯基醚和烯丙基醚都可以根据反应条件转化为硫醇,形成阶梯生长网络。典型的交联剂可包括巯基或胺的部分。混合模式聚合是在同一反应容器中发生的两种机制的结果;丙烯酸酯和甲基丙烯酸酯基团可以形成混合模式网络。两种水凝胶形成机制均可用于包裹活体细胞,并且两种机制均可使肽、蛋白质或其他治疗药物发生反应性掺入。图2:链式增长和阶梯式增长反应由不同机理反应得到的网格结构如图3所示。在链式增长网络中,交联位点形成了一个动力学链,而在阶梯式生长网络中,交联位点与多功能交联剂具有相同的功能,隐藏了结构上的缺陷。在链式增长和步进增长中,网络结构上的缺陷如闭环、长链缠结和末端链悬挂均有可能存在。不同高分子本身的化学性质的选择和水凝胶不同的形成机理都很重要,因为它们都会影响水凝胶网络的交联密度。材料的性质对于二维和三维培养很重要,也很容易通过水凝胶形成的化学过程来控制。随着交联密度的增加,网格尺寸减小,膨胀率降低,储能模量增加。改变PEG高分子的分子量能够实现对水凝胶性质的大体控制(交联密度差异引起的性质差异)。同时,通过改变用于生产水凝胶的反应机理,可以精细地调控水凝胶的性质(可用于调节体系的交联密度)。图3:不同形成机理导致的对水凝胶网络结构和网络缺陷的影响水凝胶的降解为了使用3D水凝胶支架来研究细胞分化和组织进化,通过空间和时间调控的方式来设计凝胶的物理和化学性质是至关重要的[10]。聚合物材料性质通常借由聚合/交联(键形成)或借由受控降解及/或释放(键断裂)来改变。键的形成通常会用到小分子试剂(引发剂、催化剂,单体、连接到材料的配体),而键断裂则通常不依赖于外源试剂。小分子在体外和体内均具有比聚合物试剂更多的副反应,因此许多研究小组用降解作为原位操作聚合物生物材料的工具。水解降解水凝胶中最常用的降解机理就是水解,即一个水分子加入到聚合物骨架中,导致链断裂。酸酐、酯和酰胺都很容易水解,任何氢化物通常水解得太快,而酰胺类的若未经过催化则水解太慢,因此大多数水凝胶的水解降解都是利用酯键。为了获得具有生理相关时间维度上的可降解水凝胶,研究人员通常使用交酯或乙醛交酯段将具有可降解酯键的PEG功能化。PEG上的醇链末端可引发3,6-二甲基-1,4-二恶烷-2,5-二酮和1,4-二恶烷-2,5-二酮的开环反应,分别生成聚乙二醇交酯和聚乙二醇交酯(图4)[11]。开环反应通常由锡(II)-2-乙基己酸盐催化[12],但使用二甲氨基吡啶作为催化剂也很容易完成反应[13],二甲氨基吡啶可能比残余锡更容易除去。聚乙二醇交酯或聚乙二醇交酯的醇链末端很容易被丙烯酸酯和甲基丙烯酸酯等反应性双键官能化。图4:聚乙二醇乙交酯和聚乙二醇丙交酯的合成酶促降解虽然酯键是可通过酶降解的,但大多数研究人员都会使用具有特异性序列的酶降解掺入水凝胶中的肽,而不是非特异性的酶降解酯和酰胺。Hubbell团队率先使用了这一方法[14],他们通过Michael加成反应把丙烯酸酯、马来酰亚胺和乙烯基砜官能化的半胱氨酸肽,将基质金属蛋白酶(MMP)具有反应敏感型的连接键引入水凝胶(图5)[15]。MMP-可降解键也被用作联结治疗剂与水凝胶的载体。例如,血管内皮生长因子(VEG-F)等生长因子可通过酶降解MMP-敏感性链而释放,从而诱导血管生成[16]。在水解和酶解过程中,降解速率由大分子的化学性质决定。在水解中,材料的降解率是通过其本身的性质(如疏水性或亲水性)和可水解基团的数量预先设计的,并且一旦材料被制造出来,就不能改变。在酶解过程中,降解通常发生在产生酶的细胞局部区域。水解和酶解均是水凝胶缓释和缓释治疗药物的有效方法,但水凝胶制备后无法调节或阻滞其释放速率,且释放不受空间限制。图5:通过Michael加成将含有半胱氨酸的肽添加到含乙烯基砜基团的酶中制备可降解型水凝胶光降解型水凝胶与水解和酶降解相比,光降解允许更加精准的空间和时间控制降解和释放。虽然已有许多研究者报道了光聚合水凝胶和光功能化水凝胶,但关于生物相容性光降解水凝胶的报道很少。Kloxin和Kasko发表了由含2-甲氧基-5-硝基-4-(1-羟乙基)苯氧丁酸的PEG高分子形成的光可降解水凝胶网络(图6)[17];邻-硝基苄基(o-NB)连接基团的光降解行为具有良好的表现。由光降解聚合物形成的水凝胶在光照下表现出体积降解,这与曝光时间、波长和光强度有关。当光线被关闭时,降解停止;在恢复光暴露后,上述样品继续光解。hMSCs(人骨髓间充质干细胞)被包裹在含有光可释放的细胞黏附配体RGDS(精氨酸-甘氨酸-天冬氨酸-丝氨酸)的水凝胶中,当RGD在第10天释放时(与纤维连接蛋白在软骨形成过程中的下调相对应),hMSCs沿成软骨途径分化。这种可降解水凝胶的表面侵蚀和透胶光刻可用于形成从10-7-10-2米或更大范围的特征局部区域[18]。部分降解导致水凝胶的交联密度降低和溶胀程度增加,这提供了一种如何制备柔软性更高的水凝胶的方法。图6:光降解o-NB掺杂的水凝胶骨架用于释放治疗药剂除了单光子光解,含有o-NB的水凝胶也对双光子光解敏感,从而允许被用于3D蚀刻[19-20]。在单光子反应中,任何暴露在光下的区域都会发生反应。相反,多光子光刻应该只发生在多个光子同时被吸收的地方,这发生在光源的焦体积(如图7)。生物材料的单光子光刻的典型波长范围从长波UV(≥365 nm)到可见光区域,而双光子光刻则使用红外光(通常~ 740-800 nm)较多。红外光具有更好的生物相容性,对活体组织的破坏性更小,并能够有更大的穿透深度。发生双光子吸收的区域也被严格限制在了光的焦点上,而不是沿着光的整个路径,提供了对激发3D控制的新思路。单光子和多光子反应都有能力制备出特征点小于500 nm的材料,远小于哺乳动物细胞的大小[21]。这代表了对水凝胶支架结构和化学的空间控制水平达到了前所未有的高度。图7:单光子光解(左)发生在暴露于紫外-可见光的水凝胶的整个区域双光子光解(右)只发生在同时吸收两个红外光光子的区域o-NB连接子还可用于将治疗剂拴系到水凝胶中以实现在活细胞中的递送。Griffin等证明了通过o-NB-PEG高分子栓系到水凝胶中可以实现荧光素的受控释放[22]。在不同波长(365-436 nm)、不同强度(5-20 mW/cm2)和不同时间(0-20分钟)的光照射下,对模型的释放进行定量分析。虽然最快的释放发生在365 nm(这对应于在该波长的o-NB连接剂的较高摩尔吸收率),显著的释放也同样见于405 nm;从分子的物理常数(如摩尔吸收率)上去推测很容易模拟释放。在这些体系中,光的衰减使化学和机械梯度容易形成。小结聚乙二醇是一种易制备、易改性的聚合物。它被广泛应用于水凝胶制造,包括作为组织培养的2D和3D支架。聚乙二醇水凝胶易于引入可降解键。水解可降解凝胶允许持续的材料降解和/或治疗剂释放。酶降解凝胶的降解和释放是由细胞决定的。光降解允许用户对水凝胶的化学和物理性质进行实时定制的外部操作。参考文献1. Drury JL, Mooney DJ. 2003. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 24(24):4337-4351. http://dx.doi.org/10.1016/s0142-9612(03)00340-52. Lee KY, Mooney DJ. 2001. Hydrogels for Tissue Engineering. Chem. Rev.. 101(7):1869-1880. http://dx.doi.org/10.1021/cr000108x3. HOFFMAN AS. Hydrogels for Biomedical Applications. 944(1):62-73. http://dx.doi.org/10.1111/j.1749-6632.2001.tb03823.x4. Hoffman AS. 2002. Hydrogels for biomedical applications. Advanced Drug Delivery Reviews. 54(1):3-12. http://dx.doi.org/10.1016/s0169-409x(01)00239-35. Tibbitt MW, Anseth KS. 2009. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng.. 103(4):655-663. http://dx.doi.org/10.1002/bit.223616. Lin C, Anseth KS. 2009. PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine. Pharm Res. 26(3):631-643. http://dx.doi.org/10.1007/s11095-008-9801-27. Richter A, Paschew G, Klatt S, Lienig J, Arndt K, Adler H. Review on Hydrogel-based pH Sensors and Microsensors. Sensors. 8(1):561-581. http://dx.doi.org/10.3390/s80105618. Ruel-Gariépy E, Leroux J. 2004. In situ-forming hydrogels?review of temperature-sensitive systems. European Journal of Pharmaceutics and Biopharmaceutics. 58(2):409-426. http://dx.doi.org/10.1016/j.ejpb.2004.03.0199. Yang Z, Xu B. 2007. Supramolecular hydrogels based on biofunctional nanofibers of self-assembled small molecules. J. Mater. Chem.. 17(23):2385. http://dx.doi.org/10.1039/b702493b10. Zalipsky S, Harris JM. 1997. Introduction to Chemistry and Biological Applications of Poly(ethylene glycol).1-13. http://dx.doi.org/10.1021/bk-1997-0680.ch00111. Davis FF. 2002. The origin of pegnology. Advanced Drug Delivery Reviews. 54(4):457-458. http://dx.doi.org/10.1016/s0169-409x(02)00021-212. Morpurgo M, Veronese FM, Kachensky D, Harris JM. 1996. Preparation and Characterization of Poly(ethylene glycol) Vinyl Sulfone. Bioconjugate Chem.. 7(3):363-368. http://dx.doi.org/10.1021/bc960022413. Lutolf MP, Hubbell JA. 2005. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 23(1):47-55. http://dx.doi.org/10.1038/nbt105514. Sawhney AS, Pathak CP, Hubbell JA. 1993. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers. Macromolecules. 26(4):581-587. http://dx.doi.org/10.1021/ma00056a00515. Du YJ, Lemstra PJ, Nijenhuis AJ, Van Aert HAM, Bastiaansen C. 1995. ABA Type Copolymers of Lactide with Poly(ethylene glycol). Kinetic, Mechanistic, and Model Studies. Macromolecules. 28(7):2124-2132. http://dx.doi.org/10.1021/ma00111a00416. Kim H, Kim HW, Suh H. 2003. Sustained release of ascorbate-2-phosphate and dexamethasone from porous PLGA scaffolds for bone tissue engineering using mesenchymal stem cells. Biomaterials. 24(25):4671-4679. http://dx.doi.org/10.1016/s0142-9612(03)00358-217. Benoit DS, Nuttelman CR, Collins SD, Anseth KS. 2006. Synthesis and characterization of a fluvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration. Biomaterials. 27(36):6102-6110. http://dx.doi.org/10.1016/j.biomaterials.2006.06.03118. West JL, Hubbell JA. 1999. Polymeric Biomaterials with Degradation Sites for Proteases Involved in Cell Migration. Macromolecules. 32(1):241-244. http://dx.doi.org/10.1021/ma981296k19. Lutolf MP, Hubbell JA. 2003. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition. Biomacromolecules. 4(3):713-722. http://dx.doi.org/10.1021/bm025744e20. Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proceedings of the National Academy of Sciences. 100(9):5413-5418. http://dx.doi.org/10.1073/pnas.073738110021. Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, et al. 2003. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB j.. 17(15):2260-2262. http://dx.doi.org/10.1096/fj.02-1041fje22. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. 2009. Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science. 324(5923):59-63. http://dx.doi.org/10.1126/science.1169494
应用实例
2022.12.23

碳化硅为光伏系统提供技术解决方案
引言太阳能技术的持续发展对人类的能源利用意义重大。它是目前世界上最清洁和丰富的资源。太阳能可以通过多种方式加以利用,例如光伏转换和太阳能加热。太阳辐射的能量为3.8×1020 MW(3.8×1020 M/s),到达地球的能量为173×106 kw(相当于1360 W/m2)。虽然太阳能能量十分巨大,但是其较低的利用效率一直是一个令人头疼的问题。硅太阳能电池具有理论由于材料特性,最大效率为31-40%,但在实际部署项目中,最大面板效率仅达到15-30%。因此,最大限度地将太阳能转化为电能是一项挑战。相关领域的研究者们已经提出了太阳能电池技术中的各种解决方案以有效利用从太阳接收的总太阳能。其中一些如下:l使用由元素铅(项目号:L121996)和硒(项目号:S105193)制成的纳米晶体作为太阳能电池原料l降低太阳能电池制造成本l用小圆柱体或纳米棒组成太阳能电池的基本单元l使用染色剂修饰的二氧化钛(项目号:T164497)材料增加阳能电池的光吸收效率[1-3]除以上四点之外,还有关于使用半导体材料制造太阳能逆变器的记录,目的是实现高效率和可靠性。碳化硅(项目号:S104650)是第三代半导体材料,由于其优越的材料特性,目前在大功率应用中占有一席之地。与硅相比。碳化硅器件在太阳能逆变器的制造中发挥着至关重要的作用。在光伏能量转换系统中,逆变器的成本、性能和运行是主要关注点。当今的逆变器需要在以下参数方面进行改进,例如高可靠性、高效率、增强的通信、更低的成本和支持专业应用的灵活性。典型的光伏逆变器应用场景如下:l1-10 kW用于生活应用l100 W至 300 kW用于商业应用l10-500 kW(未来将达到2 MW~20 MW) 用于公用工程系统目前的重点是提高体积功率密度(W/m3) 和比功率 (W/kg),从而最大限度地降低成本光伏逆变器。SiC功率半导体器件在光伏电池中的应用,可以帮助解决几个重要问题。SiC用于光伏电池中的逆变器50 kW三相光伏逆变器系统商业光伏装置的额定功率通常为100 kW至1 MW,尤其适用于商业体系。为满足大功率光伏系统的需求,有研究机构开发了50kW光伏逆变器系统样机,是业内第一款比功率为1kW/kg的全SiC逆变器[4]。图1:简化的50kw光伏逆变器电路原理图,显示系统中各种功率转换阶段电源转换过程由4个通道组成(每通道12.5 kW)交错升压转换器和三个相位逆变器。升压转换器由两个20A SiC MOSFET和两个1200 V/10 A SiC肖特基二极管并联组成。升压转换器在75 kHz的切换频率下运行,在不同的输入电压条件下效率超过99%。图2:50kw升压变流器部分的光伏逆变系统硬件单元结构图5 kW三相逆变器除此之外,也有研究者使用额定1200v/160a的XT-1000半桥MOSFET模块研制了一种5 kW三相全SiC逆变器样机[5]。图3显示了最终原型及其内部结构,尽管该逆变器不是专门为光伏应用设计的,但它能够证明SiC功率器件在缩小系统规模的同时提高其效率的能力。该系统的切换频率为50 kHz。将SiC逆变器与商业化的5 kW硅基逆变器[6]进行比较,以量化性能参数。两个系统都使用自然空气对流进行冷却。从图4中可看出,与硅基逆变器相比,SiC基逆变器能够减少27%的损耗。图3:5kW的SiC三相逆变器样机及其内部结构图图4:SiC逆变器相对于商用硅逆变器的优势(基于关键性能参数)SiC用于光伏电池中的转换器因为具备较宽的可调能带带隙,并且易于在较低的衬底温度下合成,无定形非化学计量碳化硅(a-SixC1-x)是光电应用的理想候选材料。通常情况下,化学计量SiC在可见光区吸收系数低,即使掺杂后电学性能也很差。为了克服这些缺点,近年来有很多研究都集中在了制备具有可调能带带隙的非化学计量SixC1-x [7,8]。通过等离子体增强化学气相沉积(PECVD),通过改变生长参数(如衬底温度)合成了非化学计量的SixC1-x[9,10]。该过程类似于合成非化学计量的SiOx和SiNx材料。非化学计量SixC1-x的可调能带带隙与其C/Si成分有很大关系,进而影响吸收光谱。在氢稀释的PECVD过程中,氢载流子可以降低表面缺陷态的密度。然而,在氢稀释下的制备通常需要较高的衬底温度和射频等离子体功率。在无氢PECVD下,非化学计量富Si的SixC1-x可以在较低的衬底温度下合成,显著提高其吸收系数。与结晶Si薄膜相比,非化学计量的富Si的SixCx材料在可见光区(400-600 nm)具有更小的光学带隙和更高的吸收系数。许多研究报道了a-Si和SixC1-x杂化PVSC的实际应用。然而,很少有报道强调所有基于SixC1-x的PVSCs。Gao等人[11]将基于SixC1-x的n-i-p结PVSCs作为半透明太阳能电池应用于光透过调制器中,但报道转换效率xC1-x的p-n结PVSCs的n型SixC1-x薄膜的厚度从150 nm降低到25 nm,这种参数调谐将转换效率从5×10-3%提高到4.7%[12]。图5:用n型(厚度25 ~ 100 nm)和p型(厚度50 nm)富硅SiC薄膜制备了ITO/p-SiC/n-SiC/Al基PVSC结构在石英衬底上生长全SixC1-x基的单p-i-n结半透明PVSC也是一个非常好的思路。Lin等人[13]使用异常无氢PECVD在远低于SiC合成温度1000℃的衬底温度下生长非化学计量的SixC1-x薄膜。本底SixC1-x (i-SixC1-x)薄膜作为吸收层,在生长过程中通过改变硅烷(SiH4)和甲烷(CH4)的通量比来调节其组成比例,以提高光电流响应。此外,研究者还制备了具有不同C/Si组成比的i- SixC1-x层的富硅SixC1-x/a-Si串联太阳能电池。为了优化富Si的SixC1-x/a-Si串联太阳能电池的转换效率,通过在生长过程中改变SiH4和CH4的通量比来调节n-a-SixC1-x层的C/Si组成比,增加n-a-SixC1-x的p-a-Si界面的隧道化概率。图6:SixCi1-x/a-Si串联PVSC的能带结构结论随着全球温室效应日益显著,碳中和势在必行,这给新能源领域的发展注入了巨大动力。太阳是最清洁的能源,这使得光伏材料的发展和应用具有十分重要的意义。以碳化硅等具备优异特性的半导体材料,正在光伏转换器,逆变器等关键器件中发挥重要作用,未来也一定能够持续贡献力量。参考文献[1] El Chaar L, El Zein N. Review of photovoltaic technologies[J]. Renewable and sustainable energy reviews, 2011, 15(5): 2165-2175. https://doi.org/10.1016/j.rser.2011.01.004[2] Parida B, Iniyan S, Goic R. A review of solar photovoltaic technologies[J]. Renewable and sustainable energy reviews, 2011, 15(3): 1625-1636. https://doi.org/10.1016/j.rser.2010.11.032.[3] Behar O, Khellaf A, Mohammedi K. A review of studies on central receiver solar thermal power plants[J]. Renewable and sustainable energy reviews, 2013, 23: 12-39. https://doi.org/10.1016/j.rser.2013.02.017.[4] Mookken J, Agrawal B, Liu J. Efficient and compact 50kW Gen2 SiC device based PV string inverter[C]//PCIM Europe 2014; International Exhibition and Conference for Power Electronics, Intelligent Motion, Renewable Energy and Energy Management. VDE, 2014: 1-7. https://ieeexplore.ieee.org/abstract/document/6841306.[5] Pushpakaran B N, Subburaj A S, Bayne S B, et al. Impact of silicon carbide semiconductor technology in Photovoltaic Energy System[J]. Renewable and Sustainable Energy Reviews, 2016, 55: 971-989. https://doi.org/10.1016/j.rser.2015.10.161.[6] Bhalla A. Market Penetration of Wide-Bandgap SiC and GaN technology in light of Silicon Super junction and IGBT technology evolution[C]//CS MANTECH Conference. 2014: 9-12.[7] Lin G R, Lo T C, Tsai L H, et al. Finite silicon atom diffusion induced size limitation on self-assembled silicon quantum dots in silicon-rich silicon carbide[J]. Journal of The Electrochemical Society, 2011, 159(2): K35.https://iopscience.iop.org/article/10.1149/2.014202jes/meta.[8] Lo T C, Tsai L H, Cheng C H, et al. Self-aggregated Si quantum dots in amorphous Si-rich SiC[J]. Journal of non-crystalline solids, 2012, 358(17): 2126-2129. https://doi.org/10.1016/j.jnoncrysol.2012.01.013.[9] Cheng Q, Tam E, Xu S, et al. Si quantum dots embedded in an amorphous SiC matrix: nanophase control by non-equilibrium plasma hydrogenation[J]. Nanoscale, 2010, 2(4): 594-600. https://pubs.rsc.org/en/content/articlehtml/2010/nr/b9nr00371a.[10] Cheng Q, Xu S, Long J, et al. Homogeneous nanocrystalline cubic silicon carbide films prepared by inductively coupled plasma chemical vapor deposition[J]. Nanotechnology, 2007, 18(46): 465601. https://iopscience.iop.org/article/10.1088/0957-4484/18/46/465601/meta.[11] Gao W, Lee S H, Bullock J, et al. First a-SiC: H photovoltaic-powered monolithic tandem electrochromic smart window device[J]. Solar Energy Materials and Solar Cells, 1999, 59(3): 243-254. https://doi.org/10.1016/S0927-0248(99)00025-2.[12] Lee C T, Tsai L H, Lin Y H, et al. A chemical vapor deposited silicon rich silicon carbide PN junction based thin-film photovoltaic solar cell[J]. ECS Journal of Solid State Science and Technology, 2012, 1(6): Q144. https://iopscience.iop.org/article/10.1149/2.005301jss/meta.[13] Cheng C H, Chang J H, Wu C I, et al. Semi-transparent silicon-rich silicon carbide photovoltaic solar cells[J]. RSC advances, 2015, 5(46): 36262-36269. https://pubs.rsc.org/en/content/articlehtml/2015/ra/c4ra16998k.
应用实例
2022.12.20

三氟甲基在有机合成中的引入
简介三氟甲基在药物化学领域中是重要的化学基团之一,由于自身拥有高脂溶性、优异的代谢稳定性、高电负性以及良好的生物利用度等特点,因而被广泛地应用于生物活性分子中。之所以在药物分子中引入三氟甲基,是因其具有强吸电子诱导效应、亲脂性及稳定的C-F键,可有效延长其在生物体内的作用时间,增强其代谢稳定性;与此同时,三氟甲基的引入还会伴随药物分子脂溶性的增加,因此有助于药物分子在生物体内的吸收、传递和扩散。图1.含有三氟甲基的药物分子1.抗癌药物——索拉非尼(Sorafenib tosylate) 2.抗抑郁药物——氟西汀(Fluoxetine) 3.新型植物广谱杀菌剂——肟菌酯(Trifloxystrobin) 由于自然界中含氟化合物的种类比较稀少,因此如何方便温和高效安全地将含氟基团引入到各种有机化合物中制备特殊的含氟化合物是现今化学界的一个重要研究方向。三氟甲基的引入一般分为两类:一、直接氟化法(三氟甲基)三甲基硅烷(TMSCF3)是最广泛使用的亲核三氟甲基化试剂之一。由于Ruppert于1984年引入该试剂[1],而后Prakash又主要负责推广其使用[2],所以该试剂也被称为Ruppert-Prakash试剂。TMSCF3的广泛适用性使其成为合成各种药物靶点的常用试剂。 合成具有三氟甲基酮基团的谷氨酸和谷氨酰胺肽过程中[3],β-氨基醇分五步合成,其中关键步骤就是利用TMSCF3生成单一非对映体的三氟甲基甲硅烷基醚(方案1)。报告中的两种肽显示出对严重急性呼吸综合征冠状病毒蛋白酶(SARS-CoV 3CLpro)的抑制活性。方案1另一个例子是利用TMSCF3合成了一种非甾体类选择性雄激素受体调节剂,该调节剂在肌肉中表现出优异的口服生物利用度和合成代谢活性(方案2)[4]。该化合物还改善了绝经后骨质疏松症大鼠模型的骨强度。同样,Hudso和他的同事又使用TMSCF3合成出了一种选择性糖皮质激素受体调节剂,显示出与骨髓瘤治疗剂地塞米松相同的抗增殖活性(方案3)[5]。方案2方案3通常,需要额外的氟化物源(TBAF、CsF等)来引发三氟甲基化反应。然而,Prakash最近的一份报告详细介绍了使用一系列不需要额外的氟化物引发剂或无水条件的催化剂进行羰基化合物的三氟甲基化(方案4)[6]。方案4Prakash的另一份报告表明,使用TMSCF3对N-未活化的亚胺进行处理,可以得到相应的三氟甲基胺。只要稍微改变反应条件,也可以通过HF消除和还原制备二氟甲基胺(方案5)[7]。方案5尽管Ruppert-Prakash试剂被广泛使用,但使用该试剂进行立体选择性三氟甲基化仍是一项挑战。Dieter Enders报告了α-烷基二氧杂环己烷的非对映选择性三氟甲基化,产率非常好,ee值也非常高[8]。丙酮基团很容易被去除,生成2-三氟甲基-1,2,3-三醇(方案6)。方案6Shibata和Toru团队最近报告了一种操作简单的方法,基于金鸡纳生物碱的溴化铵(1)和四甲基氟化铵(TMAF)的组合进行对映选择性的三氟甲基化。该反应以中等至优异的产率进行,ee值高达93%(方案7)。方案7二、三氟甲基砌块最经典的三氟甲基砌块非三氟乙酰乙酸乙酯莫属,是含氟杂环化合物合成中重要砌块之一。因其具有多个反应位点,同时具有酮和烯醇性质,因此可以以不同的反应来构建含氟的吲哚、吡唑、嘧啶和其他含氟杂环,且用于医药、农药、染料等应用领域所需的含氟中间体合成[9,10]。图2. 一些重要的三氟甲基杂环以三氟乙酰乙酸乙酯、氰基乙酰胺和芳基醛为原料,在哌啶催化下合成3-酰氨基-4-芳基-6-三氟甲基哌啶-2-酮化合物。在Hofmann重排和芳基迁移后,该化合物在碱性条件下与乙酸碘苯反应,最终生成3-芳基-6-三氟甲基吡啶-2-酮化合物。方案8. 三氟乙酰乙酸乙酯作为含氟砌块这类含氟砌块是由Uneyama Kenji课题组[11]于1992年在日本开发。在四卤化碳和三苯基膦的作用下使用三氟乙酸和胺,从而合成了一系列含氟酰亚胺酰卤化合物。此后,Hao jian课题组在三乙胺、四氯化碳和三苯基膦的反应条件下,含氟碳酸和邻氨基、羟基或巯基取代的苯胺,一锅合成2-氟烷基取代的苯并1,3二唑,并基于此,使用了更多的底物和衍生物,尝试合成出更多具有潜在生物活性的化合物[12]。参考文献:1.Ruppert I, Schlich K, Volbach W. 1984. Die ersten CF3-substituierten organyl(chlor)silane. Tetrahedron Letters. 25(21):2195-2198. https://doi.org/10.1016/s0040-4039(01)80208-22.Prakash GKS, Krishnamurti R, Olah GA. 1989. Synthetic methods and reactions. 141. Fluoride-induced trifluoromethylation of carbonyl compounds with trifluoromethyltrimethylsilane (TMS-CF3). A trifluoromethide equivalent. J. Am. Chem. Soc.. 111(1):393-395. https://doi.org/10.1021/ja00183a0733.Sydnes MO, Hayashi Y, Sharma VK, Hamada T, Bacha U, Barrila J, Freire E, Kiso Y. 2006. Synthesis of glutamic acid and glutamine peptides possessing a trifluoromethyl ketone group as SARS-CoV 3CL protease inhibitors. Tetrahedron. 62(36):8601-8609. https://doi.org/10.1016/j.tet.2006.06.0524.Martinborough E, Shen Y, vanOeveren A, Long YO, Lau TLS, Marschke KB, Chang WY, López FJ, Vajda EG, Rix PJ, et al. 2007. Substituted 6-(1-Pyrrolidine)quinolin-2(1H)-ones as Novel Selective Androgen Receptor Modulators. J. Med. Chem.. 50(21):5049-5052. https://doi.org/10.1021/jm070231h5.Hudson AR, Roach SL, Higuchi RI, Phillips DP, Bissonnette RP, Lamph WW, Yen J, Li Y, Adams ME, Valdez LJ, et al. 2007. Synthesis and Characterization of Nonsteroidal Glucocorticoid Receptor Modulators for Multiple Myeloma. J. Med. Chem.. 50(19):4699-4709. https://doi.org/10.1021/jm070370z6.Prakash GKS, Panja C, Vaghoo H, Surampudi V, Kultyshev R, Mandal M, Rasul G, Mathew T, Olah GA. 2006. Facile Synthesis of TMS-Protected Trifluoromethylated Alcohols Using Trifluoromethyltrimethylsilane (TMSCF3) and Various Nucleophilic Catalysts in DMF. J. Org. Chem.. 71(18):6806-6813. https://doi.org/10.1021/jo060835d7.Prakash GKS, Mogi R, Olah GA. 2006. Preparation of Tri- and Difluoromethylated Amines from Aldimines Using (Trifluoromethyl)trimethylsilane. Org. Lett.. 8(16):3589-3592. https://doi.org/10.1021/ol061357w8.Enders D, Herriger C. 2007. Asymmetric Synthesis of 2-Trifluoromethyl-1,2,3-triols. Eur. J. Org. Chem.. 2007(7):1085-1090. https://doi.org/10.1002/ejoc.2006008959. Zhu Shizheng, Wang Yanli, Jin Guifang. Research on fluorine-containing active methylene compounds [J]. Journal of Chemistry, 2002 (04): 555-565+75810. Zhao Min, Yang Xiangmin. Synthesis of a new class of trifluoromethyl ketones [J]. Journal of East China University of Technology, 1999 (04): 111-11211. Tamura, K.; Mizukami, H.; Maeda, K.; Watanabe, H.; Uneyama, K., J. Org. Chem.1993,58, 32.12. Ge, F.; Wang, Z.; Wan, W.; Lu, W.; Hao, J., Tetrahedron Lett.2007,48, 3251.
应用实例
2022.12.20

钙离子指示剂与离子载体使用指南
钙离子指示剂与离子载体使用指南阿拉丁帮助您寻找成像Ca2+和细胞功能的最佳指标和电离层。钙(Ca2+)是一种重要的无处不在的第二信使,参与调节多种细胞过程,包括细胞增殖、基因转录、肌肉收缩和内吞作用。使用本指南,我们可以为您的实验找到最佳的Ca2+指示器,螯合剂和离子团。Ca2+指标一目了然指示剂激发波长(nm)发射波长(nm)Kd (nM)Fura-2340/380505145Indo-1346475/405230Fluo-4488520345Fluo-8490520390Fluo-54945162300Rhod-2552581570BAPTABAPTA是一种非荧光Ca2+螯合剂,从EGTA中提取,对Ca2+的选择性为Mg2+的105倍。它通常用于缓冲,在没有Mg2+的情况下,Kd为160 nM。也可提供AM形式。Fura-2Fura-2是第一种商业化的荧光钙指示剂。激发比为340 nm/380 nm,发射波长为505 nm。在科学文献中有成千上万的Fura-2参考文献,它是迄今为止最重要的Ca2+指示染料之一。Indo-1Indo-1与Fura-2在1986年同时推出。它也是比例的,但模式不同。当在346 nm激发时,它在475 nm/405 nm处表现出发射比,而不是激发比。与Fura-2不同,它有光漂白的倾向。它也被用于各种各样的应用,以大量的文献出版物为例。Fluo-8自1989年引入以来,荧光成像揭示了Ca2+信号通路中许多基本过程的空间动力学。在细胞实验中发现,Fluo-8(或Fluo-2介质亲和力)比Fluo-4更亮(1.5倍)。它改善了细胞负载和Ca2+响应,同时保持方便的Fluo-3和Fluo-4光谱波长,在490 nm处最大激发,在520 nm处最大发射。Fluo-8加载可在室温下进行。Rhod-2Rhod-2于1989年首次问世,其荧光激发极值在552 nm,发射极值在581 nm。在Ca2+结合之前,Rhod-2本质上是不荧光的,随着Ca2+浓度的增加,Rhod-2变得更加荧光。Rhod-2较长的激发和发射使该指示物在具有高水平自身荧光的细胞和组织的实验中非常有用,并可与其他波长较短的荧光染料进行多路复用。这些基于罗丹明的指示物的AM酯形式是阳离子的,这可以导致电位驱动的摄取进入线粒体。Fluo-4 AMFluo-4是Fluo-3的类似物,两个氯取代基被氟取代,这导致荧光激发增强在488 nm,因此更高的荧光信号水平。Fluo-5 AMFluo-5 AM是Fluo-4的类似物,但其钙结合亲和力降低,因此适合检测细胞内钙水平,这将饱和Fluo-4 AM指示剂。钙离子载体A23187(钙霉素)具有可被紫外光激发的固有荧光,使其对可被紫外光激发的Ca2+指示剂(如Fura-2)不太有用,但对长波长Ca2+指示剂(如Fluo-2)仍然有用。4-溴A-23187是非荧光的,因此与所有Ca2+指示剂兼容。它通常用于荧光Ca2+指标的原位校准,以平衡细胞内和细胞外Ca2+浓度,并允许Mn2+进入细胞,淬灭细胞内的染料荧光。离子霉素是一种有效的Ca2+离子胞体,通常用于校准荧光Ca2+指标和修改细胞内Ca2+浓度,并用于研究Ca2+在细胞过程中的调节特性。参考文献1.Erdahl WL, Chapman CJ, Taylor RW, Pfeiffer DR. Ca2+ transport properties of ionophores A23187, ionomycin, and 4-BrA23187 in a well defined model system. Biophys J. 1994 May;66(5):1678-93. 2. Drummond IA, Lee AS, Resendez E Jr, Steinhardt RA. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J Biol Chem. 1987 Sep 15;262(26):12801-5.
参数原理
2022.12.12

材料 | 听100遍反方向的钟不能回到过去,但PEG水凝胶可以
水凝胶是长期以来受到人们关注最多的生物材料之一,它们在化学和物理性质上都非常接近细胞的自然环境,因此作为细胞的二维和三维支架被广泛研究[1-2]。水凝胶可以由人工合成的聚合物(如聚(乙二醇)、聚(甲基丙烯酸羟乙酯)等)与天然存在的聚合物(如胶原蛋白、透明质酸等)交联形成,并且由于它们的含水量非常高,可以有效被应用于组织培养的3D模型,不受细胞、蛋白质和DNA的影响。根据不同组成部分的反应性,可以通过pH、温度、库仑相互作用、共价键、非共价相互作用或聚合来诱实现凝胶化[3-5]。 通过环氧乙烷的活性阴离子开环聚合能够非常方便地合成聚乙二醇;这些聚乙二醇具有较宽的分子量分布,且包含多种端基,在众多反应中都可适配。为了形成水凝胶,PEG必须发生交联反应。最开始的时候,PEG是通过电离辐射进行非特异性交联的[6],而现在PEG水凝胶的形成通常是通过共价交联具有反应链端的PEG高分子合成的。具有活性反应末端的PEG高分子聚合物,如丙烯酸酯、甲基丙烯酸酯、烯丙醚、马来酰亚胺、乙烯基砜、NHS酯和乙烯基醚基团(图1)很容易通过直接购得的试剂合成[7]。 图1:不同PEG大分子的末端基团 高分子的两个末端可以是两个相同或者不同的官能团。同双官能团高分子通常用于形成网络,而异双官能团高分子则可用于将具有治疗性的小分子连接到水凝胶网络中。 形成水凝胶的交联机制取决于PEG高分子链末端的特性。在大多数情况下,是与反应性乙烯基链末端聚合的同时发生交联,通常采用的是自由基引发剂。例如,大分子单体的聚合可以使用通过氧化还原反应生成的自由基(比如过硫酸铵和TEMED)或光照产生的自由基(图2,λ=365 nM)来引发,随后通过丙烯酸酯和甲基丙烯酸酯链末端基元反应来发生链增长。在阶梯生长网络的形成中,多官能度(f>2)的交联剂以化学当量与PEG链末端进行反应,或多官能度的PEG(f>2)也可以与双官能团的交联剂发生交联。丙烯酸酯、甲基丙烯酸酯、乙烯基砜、马来酰亚胺、乙烯基醚和烯丙基醚都可以根据反应条件转化为硫醇,形成阶梯生长网络。典型的交联剂有包含巯基或胺官能团的结构。混合模式聚合是在同一反应容器中发生的两种机制的结果;丙烯酸酯和甲基丙烯酸酯基团可以形成混合模式网络。两种水凝胶形成机制均可用于包裹活体细胞,并且两种机制均可使肽、蛋白质或其他治疗药物发生反应性掺入。 图2:链式增长和阶梯式增长反应 为了使用3D水凝胶支架来研究细胞分化和组织进化,通过空间和时间调控的方式来设计凝胶的物理和化学性质是至关重要的[8]。聚合物材料性质通常借由聚合/交联(键形成)或借由受控降解及/或释放(键断裂)来改变。键的形成通常会用到小分子试剂(引发剂、催化剂,单体、连接到材料的配体),而键断裂则通常不依赖于外源试剂。小分子在体外和体内均具有比聚合物试剂更多的副反应,因此许多研究小组用降解作为原位操作聚合物生物材料的工具。 1.水解降解 水凝胶中最常用的降解机理就是水解,即一个水分子加入到聚合物骨架中,导致链断裂。酸酐、酯和酰胺都很容易水解,氢化物通常水解得太快,而酰胺类的若未经过催化则水解太慢,因此大多数水凝胶的水解降解都是利用酯键。为了获得具有生理相关时间维度上的可降解水凝胶,研究人员通常使用交酯或乙醛交酯段将具有可降解酯键的PEG功能化(图3)[9]。 图3:聚乙二醇乙交酯和聚乙二醇丙交酯的合成 2.酶促降解 虽然酯键是可通过酶降解的,但大多数研究人员都会使用具有特异性序列的酶降解掺入水凝胶中的肽,而不是非特异性的酶降解酯和酰胺。Hubbell团队[10-11]率先通过Michael加成反应把丙烯酸酯、马来酰亚胺和乙烯基砜官能化的半胱氨酸肽,将基质金属蛋白酶(MMP)具有反应敏感型的连接键引入水凝胶(图4)。 图4:通过Michael加成将含有半胱氨酸的肽添加到含乙烯基砜基团的酶中制备可降解型水凝胶 MMP-可降解键也被用作联结治疗剂与水凝胶的载体。例如,血管内皮生长因子(VEG-F)等生长因子可通过酶降解MMP-敏感性链而释放,从而诱导血管生成[12]。 在水解和酶解过程中,降解速率由大分子的化学性质决定。在水解中,材料的降解率是通过其本身的性质(如疏水性或亲水性)和可水解基团的数量预先设计的,并且一旦材料被制造出来,就不能改变。在酶解过程中,降解通常发生在产生酶的细胞局部区域。水解和酶解均是缓释治疗药物的有效方法,但水凝胶制备后无法调节或阻滞其释放速率,且释放不受空间限制。 3.光致降解 与水解和酶降解相比,光降解允许更加精准的空间和时间控制降解和释放。虽然已有许多研究者报道了光聚合水凝胶和光功能化水凝胶,但关于生物相容性光降解水凝胶的报道很少。Kloxin和Kasko发表了由含PEG高分子形成的光可降解水凝胶网络(图5)[13];邻-硝基苄基(o-NB)连接基团的光降解行为具有良好的表现。由光降解聚合物形成的水凝胶在光照下表现出体积降解,这与曝光时间、波长和光强度有关。当光线停止照射时,降解停止;在恢复光照后,上述样品继续光解。部分降解导致水凝胶的交联密度降低和溶胀程度增加,这提供了一种如何制备柔软性更高的水凝胶的方法。 图5:光降解o-NB掺杂的水凝胶骨架用于释放治疗药剂 除了单光子光解,含有o-NB的水凝胶也对双光子光解敏感,从而允许被用于3D蚀刻[14-15]。在单光子反应中,任何暴露在光下的区域都会发生反应。相反,多光子光刻应该只发生在多个光子同时被吸收的地方,这发生在光源的焦体积。生物材料的单光子光刻的典型波长范围从长波UV(≥365 nm)到可见光区域,而双光子光刻则使用红外光(通常~ 740-800 nm)较多。红外光具有更好的生物相容性,对活体组织的破坏性更小,并能够有更大的穿透深度。发生双光子吸收的区域也被严格限制在了光的焦点上,而不是沿着光的整个路径,提供了对激发3D控制的新思路。单光子和多光子反应都有能力制备出特征点小于500 nm的材料,远小于哺乳动物细胞的大小[16]。这代表了对水凝胶支架结构和化学的空间控制水平达到了前所未有的高度。 聚乙二醇是一种易制备、易改性的聚合物。它被广泛应用于水凝胶制造,包括作为组织培养的2D和3D支架。聚乙二醇水凝胶易于引入可降解键——水解可降解凝胶允许持续的材料降解和/或治疗剂释放;酶降解凝胶的降解和释放是由细胞决定的;光致降解允许使用者对水凝胶的理化性质进行实时定制的外部操作。 产品货号产品名称包装规格 P103730 聚乙二醇 100g/250g/500g/1kg/5kg P109710 聚(乙二醇)甲基丙烯酸酯 25mL/100mL/500mL/2.5L P109711 聚乙二醇甲醚甲基丙烯酸酯 100mL/500mL/2.5L P133766 聚乙二醇二甲基丙烯酸酯 50mL/250mL/1L/2.5L M130065 聚乙二醇单甲醚 100g/250g/500g/1kg P274350 聚乙二醇8000 500g/1kg/2.5kg P303204 聚对苯二甲酸乙二醇酯 25g/100g/500g/2.5kg M109437 甲氧基聚乙二醇胺 250mg/500mg/1g/5g/25g A163261 氨基 PEG, mPEG-NH2 100mg/1g/5g T164391 巯基 PEG, mPEG-SH 200mg/1g/5g 阿拉丁技术文章:用于2D和3D细胞培养的可降解型聚乙二醇水凝胶 ——点击查看详情 1. Drury JL, Mooney DJ. 2003. Hydrogels for tissue engineering: scaffold design variables and applications.Biomaterials.24(24):4337-4351. http://dx.doi.org/10.1016/s0142-9612(03)00340-52. Lee KY, Mooney DJ. 2001. Hydrogels for Tissue Engineering.Chem. Rev..101(7):1869-1880. http://dx.doi.org/10.1021/cr000108x3. HOFFMAN AS. Hydrogels for Biomedical Applications. 944(1):62-73. http://dx.doi.org/10.1111/j.1749-6632.2001.tb03823.x4. Hoffman AS. 2002. Hydrogels for biomedical applications.Advanced Drug Delivery Reviews.54(1):3-12. http://dx.doi.org/10.1016/s0169-409x(01)00239-35. Tibbitt MW, Anseth KS. 2009. Hydrogels as extracellular matrix mimics for 3D cell culture.Biotechnol. Bioeng..103(4):655-663. http://dx.doi.org/10.1002/bit.223616. Ruel-Gariépy E, Leroux J. 2004. In situ-forming hydrogels?review of temperature-sensitive systems.European Journal of Pharmaceutics and Biopharmaceutics.58(2):409-426. http://dx.doi.org/10.1016/j.ejpb.2004.03.0197. Yang Z, Xu B. 2007. Supramolecular hydrogels based on biofunctional nanofibers of self-assembled small molecules.J. Mater. Chem..17(23):2385. http://dx.doi.org/10.1039/b702493b8. Zalipsky S, Harris JM. 1997. Introduction to Chemistry and Biological Applications of Poly(ethylene glycol).1-13. http://dx.doi.org/10.1021/bk-1997-0680.ch0019. Davis FF. 2002. The origin of pegnology.Advanced Drug Delivery Reviews.54(4):457-458. http://dx.doi.org/10.1016/s0169-409x(02)00021-210. Sawhney AS, Pathak CP, Hubbell JA. 1993. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(.alpha.-hydroxy acid) diacrylate macromers.Macromolecules.26(4):581-587. http://dx.doi.org/10.1021/ma00056a00511. Du YJ, Lemstra PJ, Nijenhuis AJ, Van Aert HAM, Bastiaansen C. 1995. ABA Type Copolymers of Lactide with Poly(ethylene glycol). Kinetic, Mechanistic, and Model Studies.Macromolecules.28(7):2124-2132. http://dx.doi.org/10.1021/ma00111a00412. Kim H, Kim HW, Suh H. 2003. Sustained release of ascorbate-2-phosphate and dexamethasone from porous PLGA scaffolds for bone tissue engineering using mesenchymal stem cells.Biomaterials.24(25):4671-4679. http://dx.doi.org/10.1016/s0142-9612(03)00358-213. Benoit DS, Nuttelman CR, Collins SD, Anseth KS. 2006. Synthesis and characterization of a fluvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration.Biomaterials.27(36):6102-6110. http://dx.doi.org/10.1016/j.biomaterials.2006.06.03114. Lutolf MP, Hubbell JA. 2003. Synthesis and Physicochemical Characterization of End-Linked Poly(ethylene glycol)-co-peptide Hydrogels Formed by Michael-Type Addition.Biomacromolecules.4(3):713-722. http://dx.doi.org/10.1021/bm025744e15. Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. 2003. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics.Proceedings of the National Academy of Sciences.100(9):5413-5418. http://dx.doi.org/10.1073/pnas.073738110016. Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, et al. 2003. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth.FASEB j..17(15):2260-2262. http://dx.doi.org/10.1096/fj.02-1041fje 文中图片来源于参考文献或网络,若有来源标注错误或侵犯了您的合法权益,请在阿拉丁微信公众号下方留言或私信,我们会及时纠正或删除,非常感谢!
应用实例
2022.12.12
企业名称: 上海阿拉丁生化科技股份有限公司
企业地址: 上海市新金桥路36号财富中心16楼 联系人: 阿拉丁客服 邮编: 201206 联系电话: 400-860-5168转3457
仪器信息网APP

展位手机站