视频号

抖音号

哔哩哔哩号

前沿资讯手机看

热爱生命科学


分享到微信朋友圈
打开微信,点击底部的“发现”,
使用“扫一扫”即可将网页分享到朋友圈。
随着三代测序技术(TGS,也即单分子测序技术)的发展,基于大量细胞的三代基因组测序数据被广泛应用于各种复杂大型基因组的组装,由于其读长相比于二代测序(NGS)技术有数百倍的增加,因此基因组中重复序列区域以及染色体重排等复杂结构变异区域都能被更好地组装出来。
对于人类基因组的组装研究,端粒到端粒(T2T)联盟在2022年3月,使用纯合二倍体细胞系CHM13率先发布了首个完整的端粒到端粒的人类基因组参考序列CHM13v1.1。2022年3月,人类泛基因组联盟(HPRC)在预印本平台bioRxiv上发布了首个高质量人类杂合二倍体细胞系HG002的单倍型组装结果。
目前,高质量的基因组组装通常依赖于大量细胞混合样本的三代测序数据,需要大量的基因组DNA(通常需要从数百万个细胞中提取几十微克基因组DNA),然而在基因组组装的实际应用中常常要面对两个困难:
1、细胞群体中存在遗传异质性。基于大量细胞三代测序数据的基因组组装需要确保测序的样本中每个细胞的遗传背景高度一致,否则组装结果将很难区分同一个细胞内的不同单倍型基因组之间的差异和不同细胞亚群之间的基因组差异。只有降低或者消除细胞间的遗传异质性才能确保单倍型组装的准确性。但是,在人体正常组织样本中也常常广泛存在体细胞拷贝数变异(CNA)。与此同时,正常的人类细胞也会不断积累突变,同一块人体组织常常是由很多包含不同突变的细胞克隆组成。在癌症研究中,同一个肿瘤样本中不同癌细胞亚克隆之间的基因组异质性就更为明显。
2、细胞数量稀少。在很多情况下,很难获取上百万个细胞以提取大量(几微克)基因组DNA。例如,在早期胚胎发育研究、司法检验、特别是在癌症基因组研究中(如循环肿瘤细胞、肿瘤活检样本、脑脊液中的肿瘤细胞、以及腹水中的肿瘤细胞等),能够获取的细胞数量常常很稀少,而且这些细胞很难在体外培养和扩增;即使偶尔可以培养扩增,也不能保证在体外培养扩增过程中其基因组不会进一步产生新的遗传变异。
基于二代测序(NGS)平台的单细胞基因测序技术被广泛应用于微生物等简单小型基因组的组装。许多种类的细菌无法在实验室中培养,单细胞基因组测序可以与宏基因组学方法结合起来完成微生物的基因组组装。由于人类基因组结构、大小、以及复杂程度远超细菌等微生物,单纯使用基于二代测序平台的大量细胞基因组测序数据也无法组装出高质量的人类基因组参考序列(NG50很难达到Mb(百万碱基对)级别),那么使用少量DNA甚至单细胞基因组测序数据组装人类基因组则更具挑战性,它不仅需要基于三代测序平台的单细胞基因组长读长测序技术的支持,还需要合适的组装软件以及良好的生物信息学分析策略。
2022年7月12日,北京大学生物医学前沿创新中心(BIOPIC)汤富酬课题组在Nucleic Acids Research发表了题为De novo assembly of human genome at single-cell levels的研究论文。该研究使用优化的SMOOTH-seq单细胞基因组三代测序技术,基于Pacific Biosciences(PacBio)HiFi和Oxford Nanopore Technologies(ONT)两种三代测序平台首次在单细胞水平上完成了Mb级连续性的人类基因组组装,并使用多种评价指标,充分探索了不同测序策略和组装工具对基因组组装结果的影响。

1、全面优化了SMOOTH-seq单细胞基因组三代测序技术,使其同时适用于PacBio和ONT两种主流单分子测序平台。此前的SMOOTH-seq技术只适用于PacBio单分子测序平台,使用场景有较大的局限性。优化后的SMOOTH-seq技术既可以用于PacBio单分子测序平台,也可以用于ONT单分子测序平台,使用场景更加灵活,可以兼顾测序数据准确性和测序成本。
2、使用hifiasm,Hicanu,wtdbg2等主流组装工具和95个单细胞的三代基因组测序数据(Pacbio HiFi平台),对人类慢性粒细胞性白血病(CML)细胞系K562进行了高质量基因组组装。组装出的主要叠连群(primary contig)的NG50(可覆盖50%的已知基因组区域的最短叠连群的长度)可达2.11Mb,也就是说在这个组装出的参考序列中,人类基因组中一半(15亿碱基对)以上的区域都被至少2.11Mb以上的叠连群覆盖了。最长叠连群可达14.12Mb,完整的通用单拷贝同源基因基准(Complete BUSCOs)比例接近95%,且大部分组织相容性复合体(MHC)位点(基因组上的一个有代表性的复杂区域,全长约6Mb)被成功组装出来(如图1所示)。
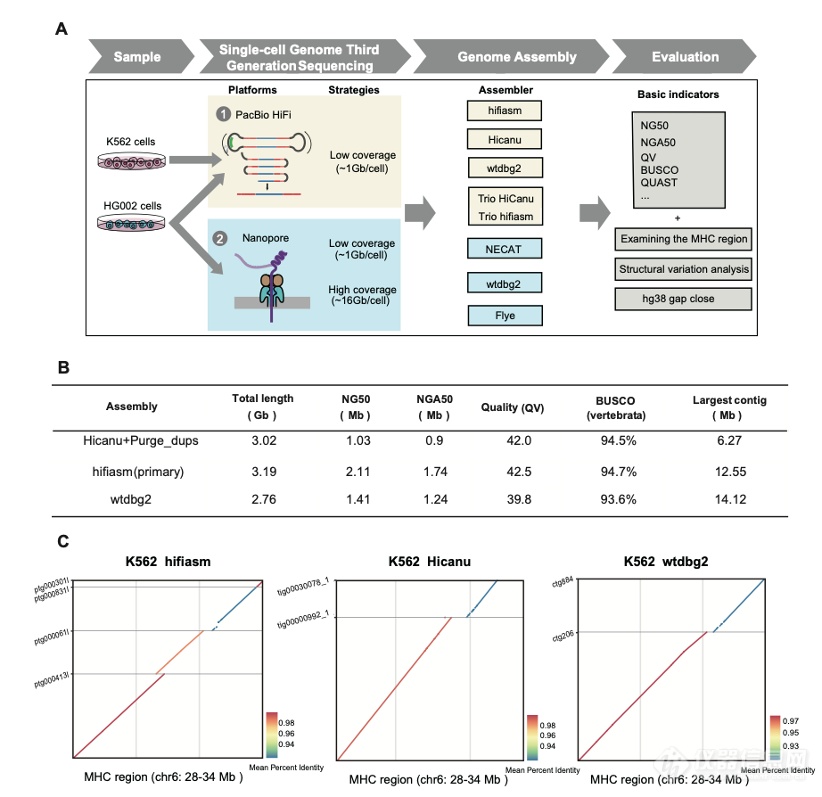
图1. 95个K562细胞的基因组组装结果(Pacbio HiFi)
3、使用hifiasm,Hicanu,wtdbg2等主流组装工具和人类正常二倍体细胞系HG002的157个单细胞的基因组三代测序数据(Pacbio HiFi平台)对人类基因组进行了高质量组装。组装出的主要叠连群(primary contig)的NG50可达0.65Mb,最长的叠连群可达6.82Mb,完整的通用单拷贝同源基因基准(Complete BUSCOs)比例接近91%。在使用此数据进行HG002的单倍型组装的过程中该研究发现经过指数扩增的基因组数据的k-mer分布会发生偏移,因此使用有双亲二代测序数据作为辅助的Trio-binning模式进行基因组单倍型组装结果更为准确。因此该研究分别使用Trio hifiasm和Trio Hicanu两种组织工具进行单倍型组装,得到的亲本叠连群的NG50可达0.3Mb左右,完整的通用单拷贝同源基因基准(Complete BUSCOs)比例均超过84%。通过比较HG002亲本六种经典人类白细胞抗原(HLA)位点的组装分型结果,Trio Hicanu能够正确组装出HLA区域的两个亲本的大部分基因位点(如图2所示)。
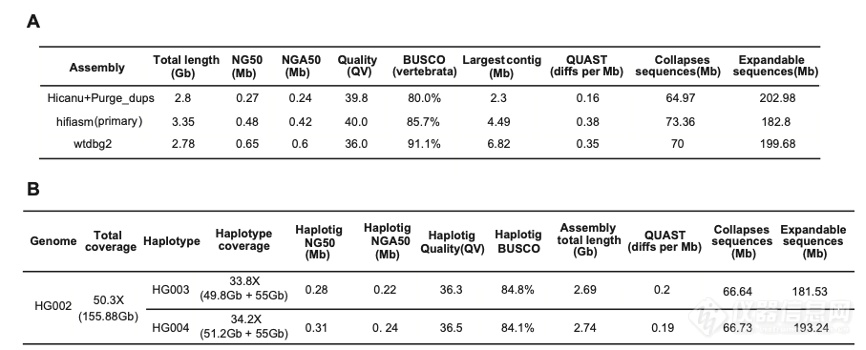
图2. 157个HG002细胞的基因组组装结果(Pacbio HiFi)
4、使用Flye,Necat,wtdbg2等主流组装工具和人类正常二倍体细胞系HG002的192个单细胞的三代基因组测序数据(ONT平台,低测序深度)对人类基因组进行高质量组装。研究发现,不同的组装工具对最终组装结果有很大影响,Flye展现出更为适合单细胞ONT三代测序数据的特性,组装出的叠连群的NG50可达1.38Mb,最长叠连群可达11.42Mb,完整的通用单拷贝同源基因基准(Complete BUSCOs)比例超过93%,多项指标都远超另外两个组装工具。同时组装结果能够补齐39个hg38版本的人类参考基因组中未组装出的缺口(gap)区域,其中14个区域在hg38中注释的长度超过50Kb(如图3所示)。
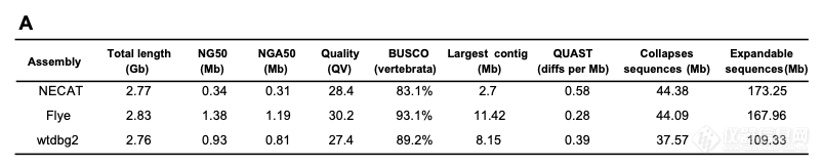
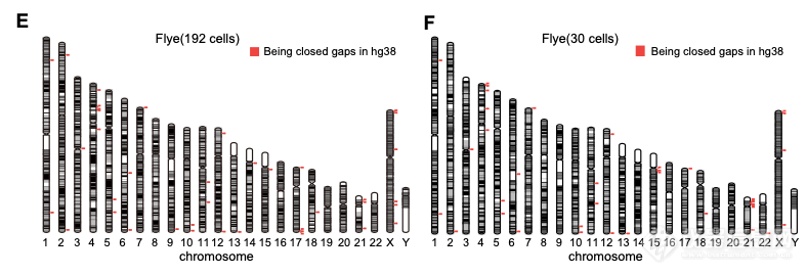
图3. 192个HG002细胞以及30个HG002细胞的基因组组装结果(ONT)
5、使用Flye,wtdbg2等组装工具和人类正常二倍体细胞系HG002的30个单细胞的三代基因组测序数据(ONT平台,高测序深度)对人类基因组进行高质量组装。为了探究仅使用极少量单细胞的基因组测序数据进行人类基因组组装的极限情况,该研究分别使用1个、10个、20个和30个单细胞尝试进行人类基因组组装,发现仅需要高测序深度的30个单细胞的基因组测序数据(平均基因组覆盖度~41.7%)就能完成叠连群 NG50高达1.34Mb连续性的组装。同时组装结果能够补齐38个hg38版本的人类参考基因组未组装出的gap区域,其中15个区域在hg38注释的长度超过50Kb(如图4所示)。
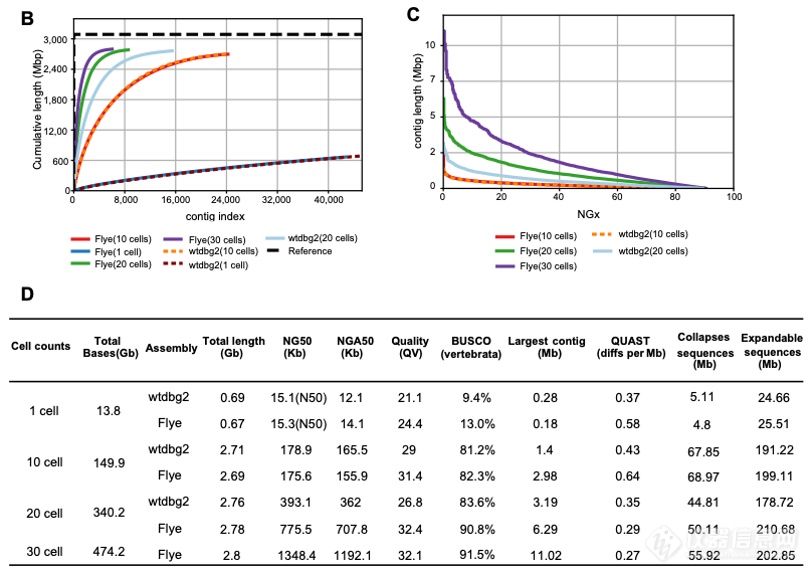
图4. 30个基因组高覆盖度HG002细胞的基因组组装结果(ONT)
6、通过对K562细胞系基因组的从头组装,该研究相比于使用原始单细胞基因组三代测序数据能更精准地鉴定出更多的基因组插入事件和复杂结构变异事件。对于K562这样的白血病细胞系,基因组从头组装之后是否能更好地鉴定出基因组结构变异(SV)事件是癌症研究中的重要问题。该研究分别使用hifiasm和Hicanu组装出的主要(primary)叠连群和替代(alternate) 叠连群来进行结构变异鉴定。发现组装后的叠连群比起原始单细胞数据直接比对能更准确地鉴定出基因组插入事件,召回率达到70%以上,精确度达到90%以上。同时,K562中的三对经典融合基因:CDC25A-GRID1、BCR-ABL1和NUP214-XKR3都能被精准地鉴定出来,而CDC25A-GRID1融合在原始单细胞基因组数据直接比对到参考基因组时是无法被发现的 (如图5所示) 。为了进一步验证基因组从头组装后找到的结构变异事件的准确性,该研究挑选了20个(14个插入事件,6个缺失事件)在组装后的叠连群中被鉴定到、但是在单细胞基因组原始测序数据直接比对到参考基因组时没有被鉴定出来的结构变异事件进行了PCR验证,准确率高达80%,证明了组装后的叠连群对结构变异事件的鉴定是精准可靠的(如图6所示)。
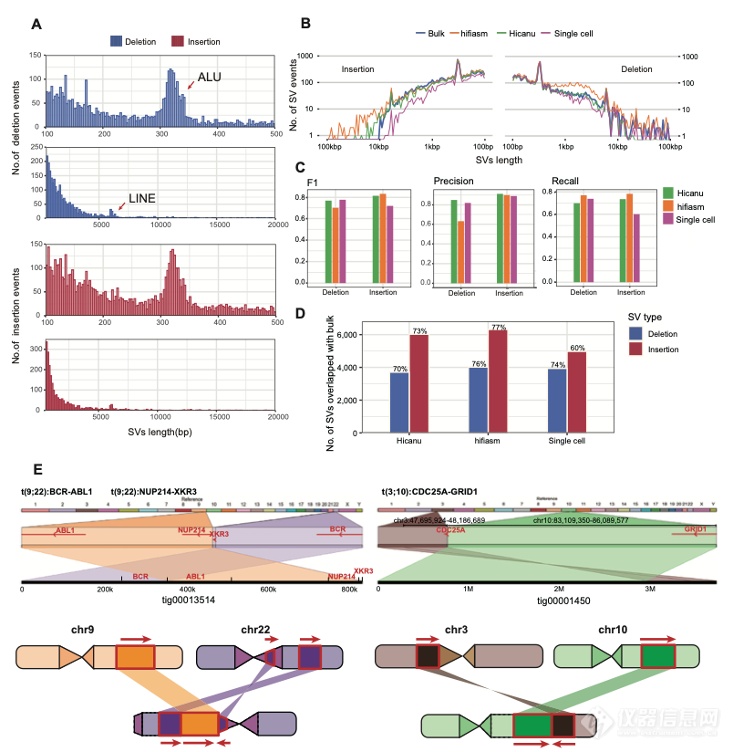
图5. 组装后叠连群(contig)中结构变异事件检测的准确性

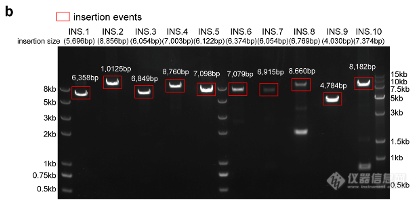
图6. PCR验证基因组结构变异事件的结果
综上,为了解决基因组从头组装在实际应用中遇到的细胞遗传异质性和细胞稀缺性的问题,该研究使用优化的SMOOTH-seq技术在两种不同的主流三代测序平台上,采用不同的测序策略(高通量、低深度测序策略(multi-cells with low sequencing depth)和低通量、高深度测序策略(few-cells with high sequencing depth)),使用多种不同组装软件(hifiasm,Hicanu,wtdbg2, Flye,Necat等)、多个评价指标、以及不同组装策略,探讨了利用单细胞测序数据从头组装人类基因组的可行性,并确定了影响组装结果的主要因素,将基因组组装的分辨率提高到单细胞水平(少至30个单细胞)。未来随着单细胞测序技术和基因组组装策略的进一步发展,最终必将实现只用一个单细胞的测序数据就能组装出Mb级连续性的人类参考基因组的梦想。
北京大学生命科学学院博士生谢昊伶以及北京大学前沿交叉学科研究院博士生李文为该论文的并列第一作者。北京大学生物医学前沿创新中心汤富酬教授为该论文的通讯作者。该研究项目得到了北大-清华生命科学联合中心、国家自然科学基金委、北京市科技委和北京未来基因诊断高精尖创新中心的支持。
论文链接:
https://doi.org/10.1093/nar/gkac586
汤富酬研究员简介:

汤富酬,博士,北京大学BIOPIC/ICG研究员,国家“优青”(2013)、“杰青”(2016)。1998年本科毕业于北京大学,2003年在北大获得细胞生物学博士学位,2004-2010年间在英国剑桥大学Gurdon研究所从事博士后研究, 2010年回到北京大学组建实验室,主要从事人类早期胚胎发育的单细胞功能基因组学研究。在国际上率先系统发展了单细胞功能基因组学研究体系,并利用一系列技术体系对人类早期胚胎发育进行了深入、系统的研究,揭示了人类早期胚胎DNA去甲基化过程的异质性以及其他表观遗传学关键特征,发现了人类早期胚胎中基因表达网络的重要表观遗传学调控机理,为人们提供了一个全面分析人类早期胚胎表观遗传调控网络的研究框架,加深了对人类原始生殖细胞的发育以及表观遗传重编程过程的认识。
[来源:北京大学生物医学前沿创新中心]

2022.07.12

2024.11.04

从尿液中窥探健康,为个性化医学开辟新道路——百名学者联名倡议启动“中国尿计划”
2024.10.30

新疆22家重点实验室获批,涉及生物育种、基因编辑与种质创新等领域
2024.10.28

拥有"绝对色感",光谱流式无惧多色挑战——第六届流式网络大会光谱流式报告推荐
2024.10.28
品牌合作伙伴






























版权与免责声明:
① 凡本网注明"来源:仪器信息网"的所有作品,版权均属于仪器信息网,未经本网授权不得转载、摘编或利用其它方式使用。已获本网授权的作品,应在授权范围内使用,并注明"来源:仪器信息网"。违者本网将追究相关法律责任。
② 本网凡注明"来源:xxx(非本网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,且不承担此类作品侵权行为的直接责任及连带责任。如其他媒体、网站或个人从本网下载使用,必须保留本网注明的"稿件来源",并自负版权等法律责任。
③ 如涉及作品内容、版权等问题,请在作品发表之日起两周内与本网联系,否则视为默认仪器信息网有权转载。
![]() 谢谢您的赞赏,您的鼓励是我前进的动力~
谢谢您的赞赏,您的鼓励是我前进的动力~
打赏失败了~
评论成功+4积分
评论成功,积分获取达到限制
![]() 投票成功~
投票成功~
投票失败了~