推荐厂家
暂无
暂无
 金牌5年
金牌5年
 400-860-5168转4265
400-860-5168转4265
 留言咨询
留言咨询
 银牌14年
400-860-5168转2060
留言咨询
留言咨询
银牌14年
400-860-5168转2060
留言咨询
留言咨询
 400-612-9980
留言咨询
400-612-9980
留言咨询
 400-629-8889
留言咨询
400-629-8889
留言咨询
 400-629-8889
留言咨询
400-629-8889
留言咨询


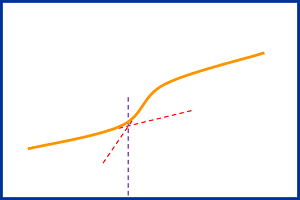


[size=3][font=宋体][color=#fe2419]维权声明:本文为[/color][color=#0365bf]wuweicheng85[/color][color=#fe2419]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/color][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]是我们化验室最重要最常用的仪器之一,也是化验室最昂贵的仪器之一,我们的工作中经常要用到[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],但是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]也是比较复杂的仪器,不是一天两天就能精通的仪器,所以我们在使用的过程中会经常出现很多的问题,而我们在遇见这些问题后,有的通过同事间的探讨,得到了解决。有些可能就一直没有解决。今天我们就大家在工作中遇到的色谱仪基线不稳,保留时间改变方面的问题展开讨论,将自己在工作中遇到的,或者是查取资料得到的这些方面的东西说出来供大家分享,促进大家的共同提高。[/font][/size][b][size=3][font=宋体]一、基线不稳及处理办法。[/font][/size][/b][size=3][font=宋体]所谓基线不稳定,也就是基线的噪音、漂移以及该检测器的基流等超出了仪器所要求的最低指标。[/font][/size][size=3][font=宋体]引起基线不稳的因素很多,如外围条件、工作站、电路、气路都有可能。我们就我们所知道的列出以下几种。[/font][/size][size=3][font=宋体]1[/font][/size][size=3][font=宋体]、在我们做煤气时曾经遇见过由于载气的纯度不够而产生基线不稳,通过更换载气问题解决。[/font][/size][size=3][font=宋体]2[/font][/size][size=3][font=宋体]、有一次工作中我们发现基线不稳,我们通过检查发现载气压力开的太小,我们通过调整问题得以解决,后来经过上网查证,发现载气总压力小于0.2Mpa就可能引起基线不稳定。另外[/font][/size][size=3][font=宋体]载气压力若不稳定、载气流速过高、以及稳定阀、稳流阀控制精度差,也会导致基线不稳,稳定载气压力后可解决。[/font][/size][size=3][font=宋体][/font][/size][size=3][font=宋体]3[/font][/size][size=3][font=宋体]、由于仪器接地不好导致基线不稳。我们的有些仪器非常灵敏,记得我们以前的高麦色谱,因为接地线没有弄好,导致基线很乱。还有放置仪器的地方机械振动太大导致基线不稳。[/font][/size][size=3][font=宋体]将仪器放到合适的地方解决。[/font][/size][size=3][font=宋体][/font][/size][size=3][font=宋体]4[/font][/size][size=3][font=宋体]、系统某处泄漏导致基线不稳。检查隔垫、柱子连接处看是否有漏气。[/font][/size][size=3][font=宋体]5[/font][/size][size=3][font=宋体]、隔垫用的时间过长,[/font][/size][size=3][font=宋体]被污染[/font][/size][size=3][font=宋体]会有残渣而产生噪声,从而基线不稳,更换隔垫或者清洗[/font][/size][size=3][font=宋体]衬管[/font][/size][size=3][font=宋体]可解决。[/font][/size][size=3][font=宋体]6[/font][/size][size=3][font=宋体]、FID气体配比不合适导致火焰不稳,从而反映出基线不稳。调整气体配比可解决此问题。[/font][/size][size=3][font=宋体]7[/font][/size][size=3][font=宋体]、气路出口管道中有冷凝物或异物,通过清理解决。[/font][/size][size=3][font=宋体][/font][/size][size=3][font=宋体]8[/font][/size][size=3][font=宋体]、柱箱或者检查器温度不稳定导致基线不稳,通过调整柱箱或者检测器温度解决,若是温度控制出现问题,进一步修理可解决问题。[/font][/size][size=3][font=宋体]9[/font][/size][size=3][font=宋体]、柱填充物松动,柱中固定相流失导致基线不稳,更换色谱柱解决。[/font][/size][size=3][font=宋体][/font][/size][size=3][font=宋体]10[/font][/size][size=3][font=宋体]、热导池污染,清理检测器。[/font][/size][size=3][font=宋体]11[/font][/size][size=3][font=宋体]、样品太脏导致基线不稳。过滤或者蒸馏处理样品后再做。[/font][/size][size=3][font=宋体]12[/font][/size][size=3][font=宋体]、电源电压太低或波动太大,通过调整可解决(网上查取)。[/font][/size][size=3][font=宋体]13[/font][/size][size=3][font=宋体]、桥路配置电位器接触不良,修理检测器解决(网上查取)。[/font][/size][size=3][font=宋体][/font][/size][size=3][font=宋体]14[/font][/size][size=3][font=宋体]、仪器环境空气中尘埃太多(网上查取)。[/font][/size][b][size=3][font=宋体]二、保留时间改变及处理办法[/font][/size][/b][size=3][font=宋体]导致保留时间改变(推迟或者提前)的原因很多,今天就我们平时遇到或者查资料的原因总结如下:[/font][/size][size=3][font=宋体]1[/font][/size][size=3][font=宋体]、色谱条件改变导致保留时间提前或者推迟,如改变载气流速、色谱柱温度等条件都能使保留时间推迟或者提前,通过调节到合适的条件可解决。[/font][/size][size=3][font=宋体]2[/font][/size][size=3][font=宋体]、气路泄漏导致保留时间提前或者推迟,检查进样隔垫、进样口和色谱柱接口。从理论上说,如果载气泄漏恒定,对保留时间的影响不会太大。通过重新安装可解决。[/font][/size][size=3][font=宋体]3[/font][/size][size=3][font=宋体]、我们在拆卸过色谱后某个部件没有安装好导致保留时间改变。重新安装可解决。[/font][/size][size=3][font=宋体]4[/font][/size][size=3][font=宋体]、样品中所测成分含量改变较大,也会使保留时间稍微的推迟或者提前。[/font][/size][size=3][font=宋体]5[/font][/size][size=3][font=宋体]、进样量不合适导致保留时间改变,通过进正确合适的进样量解决。[/font][/size][size=3][font=宋体]6[/font][/size][size=3][font=宋体]、色谱柱变短,或者固定相流失导致保留时间提前,通过更换新的色谱柱可解决此问题。[/font][/size][size=3][font=宋体]7[/font][/size][size=3][font=宋体]、手动进样时,进样方法不规范也会导致保留时间推迟或者提前,规范进样方法可解决此问题。[/font][/size][size=3][font=宋体]8[/font][/size][size=3][font=宋体]、进样隔垫或者衬管过脏也可导致保留时间改变,适时清洗或者更换可以解决此问题。[/font][/size][size=3][font=宋体]9[/font][/size][size=3][font=宋体]、色谱柱时间用长以后,柱头被污染也会导致保留时间推迟。通过将柱头切除一截可解决。[/font][/size]
[align=center][size=18px][b]气相色谱仪峰形不规则的原因和处理方法[/b][/size][/align][align=left][b] 在使用气相色谱仪的过程中,有时会出现峰形不规则的现象,如出现拖尾峰或平顶形或锯齿形峰。关于这个问题,分析仪器工程技术人员就和大家共同探讨一下,希望大家在今后具体操作这类仪器时能很好的处理此类故障。 1、检测器所造成的影响 我们以热导检测器TCD为例判断,TCD通过载气和被测样品气体的热导率不同, 在检测桥路中所产生不平衡电压与被测组分的浓度成正比, 从而实现被测组分的分析测量。 (1)由于样品的多样性,首先我们考虑的是TCD 检测器被污染,这时会造成基线漂移或者出现台阶型基线现象, 并可能导致出现高噪音。 (2)TCD 热阻丝被烧断, 基线降为零点。 (3)电源电压不稳定使热导检测器TCD,出现不规则的脉冲干扰峰或规则的脉冲峰。 2、载气的影响 载气携带分析样品流经色谱柱,经固定相分离后的气体随时间先后逐一被载气携带出色谱柱,送往检测部分检测。载气的流量、载气的性质及载气压力的影响等操作条件都会影响色谱分离效能。 (1)载气流量偏低, 会引起保留时间增长, 灵敏度降低或出现圆顶峰、拖尾峰。 (2)载气流量偏高过大, 会引起高噪音或组分分离不开。 (3)载气流量阀控制不稳, 造成不规则基线漂移或波状基线漂移。 以上情况应检查气体发生器运行状况是否良好,使用钢瓶应看减压阀是否超过使用范围, 必要时应更换减压阀, 然后再检查载气气路是否存在漏气等情况。 3、电路问题 电路故障一般较容易判断, 如电源不启动, 检测器、进样口不加热, 热导池恒流源电路故障等。若基线出现周期性正弦波, 则是由于放大电路版故障引起 处理方法一般更换损坏的电子元件。 以上就是气相色谱仪峰形不规则的一些原因和处理方法。相信大家通过这方面知识的了解,能清楚这些故障的排除方法。[/b][/align]
[align=center][size=18px][b]气相色谱仪常见问题及处理方法[/b][/size][/align] 气相色谱仪在石油、化工、生物化学、医药卫生、食品工业、环保等方面应用很广,它除用于定量和定性分析外,还能测定样品在固定相上的分配系数、活度系数、分子量和比表面积等物理化学常数。今天我们主要来介绍一下气相色谱仪常见故障问题及处理方法,希望可以帮助到大家。 气相色谱仪在进样后检测信号没有变化,也不出峰,输出仍为直线。遇到这种情况时,应按从样品进样针、进样口到检测器的顺序逐一检查。 1、首先检查注射器是否堵塞,如果没有问题, 2、再检查进样口和检测器的石墨垫圈是否紧固、不漏气, 3、然后检查色谱柱是否有断裂漏气情况, 4、最后观察检测器出口是否畅通。 5、检测器出口的畅通是很重要的,有人在工作中会遇到这样的问题:前一天仪器工作还一切正常,第二天开机后却无响应峰信号。检查进样口、注射器、垫圈和色谱柱都正常,可就是不出峰,无意中发现进样口柱头压达不到设定值,总是偏高,这时才怀疑是ECD检验器出口不畅通。由于ECD的排放物有一定的放射性,所以ECD出口是引到室外的。当时是秋冬之交,雨水进入到ECD排出口之后冻住了,因此造成仪器ECD的出口堵塞,柱头压居高不下,气体在气路中无法流动,也就无法载样品到检测器,所以不出峰。 二、基线问题 气相色谱基线波动、飘移都是基线问题,基线问题可使测量误差增大,有时甚至会导致仪器无法正常使用。 1、遇到基线问题时应先检查仪器条件是否有改变,近期是否新换气瓶及设备配件。 2、如果有更换或条件有改变,则要先检查基线问题是不是由这些改变造成的,一般来说,这种变化往往是产生基线问题的原因。有些人在工作中就遇到过这种情形:新载气纯度不够,换过载气之后,基线逐渐上升(由于载气净化管的原因,基线不是马上变化的)。第二天开机之后,基线非常高,并伴有基线强烈抖动,所有峰都湮没在噪音中,无法检测。经过检查,问题出现在新换的载气上,重新更换载气后,立即恢复了正常。 3、当排除了以上可能造成基线问题的原因后,则应当检查进样垫是否老化(应养成定期更换进样垫的好习惯) 4、石英棉是不是该更换了 5、衬管是否清洁。值得一提的是,清洗衬管时可先用试验zui后定容的溶剂充分浸泡,再用超声波清洗几分钟,然后放入高温炉中加热到比工作温度略高的温度,最后再重新安装。 6、此外,检测器污染也可能造成基线问题,其可以通过清洗或热清洗的方法来解决。 三、造成峰丢失的故障 造成峰丢失的原因有两种:一是气路中有污染,另一可能是峰没有分开。 (一)、这种情况可通过多次空运行和清洗气路(进样口、检测器等)来解决。 1、为了减少对气路的污染,可采用以下的措施:程序升温的zui后阶段应有一个高温清洗过程 2、注入进样口的样品应当清洁 3、减少高沸点的油类物质的使用 4、使用尽量高的进样口温度、柱温和检测器温度。 (二)、峰丢失的第二种情况是峰没有分开,除了以上原因外,其也有可能是因系统污染造成的柱效下降造成,或者是由于柱子老化导致的,但柱子老化所造成的峰丢失是渐进的、缓慢的。 假峰一般是由于系统污染和漏气造成的,其解决方法也是通过检查漏气和去除污染来解决。在平时的工作中应当记录正常时基线的情况,以便在维护时作参考。








 我要推广仪器
我要推广仪器
 下载APP
下载APP



