推荐厂家
暂无
暂无
 400-860-5168转1777
400-860-5168转1777
 留言咨询
400-860-5168转0819
留言咨询
留言咨询
留言咨询
400-860-5168转0819
留言咨询
留言咨询
 400-612-9980
留言咨询
400-612-9980
留言咨询
 400-612-9980
留言咨询
400-612-9980
留言咨询
 400-612-9980
留言咨询
400-612-9980
留言咨询







定量方法可分以下三种: 1、内标准法 取标准被测成分,按依次增加或减少的已知阶段量,各自分别加入各单体所规定的定量内标准物质中,调制标准溶液。分别取此标准液的一定量注入色谱柱,根据色谱图取标准被测成分的峰面积和峰高和内标物质的峰面积和峰高的比例为纵座标,取标准被测成分量和内标物质量之比,或标准被测成分量为横坐标,制成标准曲线。 然后按单体中所规定的方法调制试样液。在调制试样液时,预先加入与调制标准液时等量的内标物质。然后按制作标准曲线时的同样条件下得出的色谱,求出被测成分的峰面积或峰高和内标物质的峰积或峰高之比,再按标准曲线求出被测成分的含量。 所用的内标物质,应采用其峰面积的位置与被测成分的峰的位置尽可能接近并与被测成分以外的峰位置完全分离的稳定的物质。 2、绝对标准曲线法 取标准被测成分 按依次增加或减少阶段法,各自调制成标准液,注入一定量后,按色谱图取标准被测成分的峰面积或峰高为纵座标,而以标准被测成分的含量为横坐标,制成标准曲线。然后按单体中所规定的方法制备试样液。取试样液按制标准曲线时相同的条件作出色谱,求出被测成分的峰面积和峰高,再按标准曲线求出被测成分的含量。3、峰面积百分率法 以色谱中所得各种成分的峰面积的总和为100,按各成分的峰面积总和之比,求出各成分的组成比率。
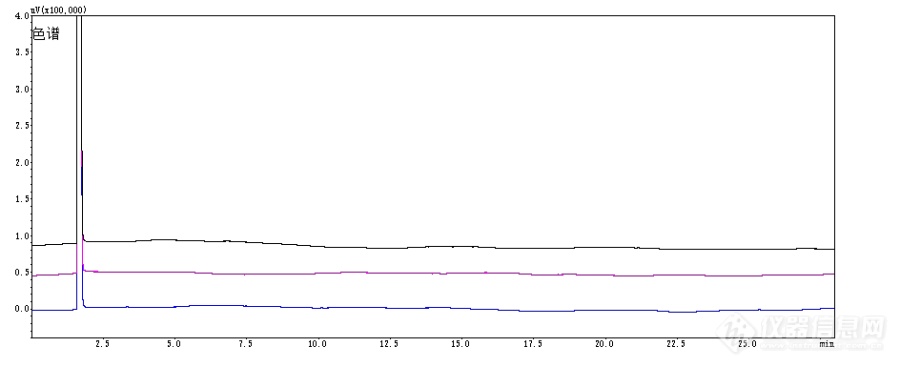
小序,本实验涉及毒麻制品,是与合作单位测样的研究性工作。测定样品过程中,发现完全按照[color=#333333]GA/T 1008.3-2013[/color][color=#333333]标准[/color]的程序升温及分流比三种成分都不出峰。后经过调节柱温进行检测,三种物质出峰且得到有效分离。[b]1 仪器与试药1.1 [/b]仪器 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-2010plus[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]配制自动进样器,FID检测器(日本岛津);Rtx-1701毛细管色谱柱(30 m×0.25 mm×0.25 μm);Rtx-1毛细管色谱柱(30 m×0.25 mm×0.25 μm);Rtx-5毛细管色谱柱(30 m×0.25 mm×0.25 μm);超声波提取仪KQ-500E(昆山);ME204电子天平(万分之一,梅特勒);AUW220D电子天平(十万分之一,日本岛津)。[b]1.2 [/b] 试药 大麻样品(合作公安单位提供),甲醇分析纯(北京北化精细化学品有限责任公司),四氢大麻酚,大麻二酚,大麻酚均由合作单位提供。[b]2 方法与结果2.1[/b] 分析条件 检测器:FID检测器;色谱柱:Rtx-5毛细管色谱柱(30 m×0.25 mm×0.25 μm);程序升温:初始温度100℃,保持2 min,以15℃/min升温至240 ℃,保持18 min,然后以20 ℃/min升温至280 ℃,保持2min;不分流;进样口温度:280 ℃;检测器温度:300 ℃;进样量:1.0 μl;尾吹气流量:30 ml/min,空气流量:400 ml/min,氢气流量:40 ml/min。[b]2.2 [/b]溶液制备[b]2.2.1 [/b]对照品溶液的制备 精确量取各对照品适量,置10 ml量瓶中,加甲醇至刻度,摇匀,然后配制混合对照品溶液,使得每1 ml对照品溶液中含四氢大麻酚、大麻酚和大麻二酚为20 μg/mL。[align=center][img=,549,205]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221504444576_3444_1858223_3.png!w549x205.jpg[/img][/align][align=center]图1 大麻酚标准品谱图[/align][align=center][img=,527,198]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221505508187_8222_1858223_3.png!w527x198.jpg[/img][/align][align=center]图2 大麻二酚标准品谱图[/align][align=center][img=,554,182]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221506039107_4535_1858223_3.png!w554x182.jpg[/img][/align][align=center]图3 四氢大麻酚标准品谱图[/align][b]2.2.2[/b] 供试品溶液的制备 分别取研磨样品粉末约10 mg,精密称定,置具塞三角瓶中,[color=#333333]加入[/color][color=#333333]甲醇[/color]10 ml,称定重量,浸泡30min,超声处理(功率600 W,频率40 kHz)10 min,放冷,再称定重量,用甲醇补足失重,摇匀,滤过,取续滤液,过0.45 μm滤膜即得。[align=center][img=,543,199]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221506337296_3413_1858223_3.png!w543x199.jpg[/img][/align][align=center]图4 送检大麻样品色谱图[/align]3 结果与讨论3.1 程序升温条件选择 按照公安部标准规定程序升温条件是初始温度200℃,保持2 min,以10℃/min升温至240 ℃,保持18 min,然后以20 ℃/min升温至280 ℃,保持2min,根据条件我们换了三根色谱柱都没有出峰。[align=center][img=,534,229]https://ng1.17img.cn/bbsfiles/images/2019/07/201907221508057570_4858_1858223_3.png!w534x229.jpg[/img][/align][align=center]图5 三种色谱柱按照公安部标准程序升温色对比色谱图[/align]3.2 排除其他原因之后,发现国标的初温比较高,然后将其初温改为100℃,选择不分流条件进样,三种成分出峰正常且分离度良好。 小结:标准的适应性根据实验室的情况而定,很多时候我们发现重复标准重复不出来,就需要我们技术人员做更多的思考和实验去调整方法,最终都能找到合适的方法和条件进行实验。
粘度计粘度测定有:动力粘度、运动粘度和条件粘度三种测定方法。 (1)动力粘度:ηt是二液体层相距1厘米,其面积各为1(平方厘米)相对移动速度为1厘米/秒时所产生的阻力,单位为克/厘米秒。1克/厘米秒=1泊一般:工业上动力粘度单位用泊来表示。 (2)运动粘度:在温度t℃时,运动粘度用符号γ表示,在国际单位制中,运动粘度单位为斯,即每秒平方米(m2/s),实际测定中常用厘斯,(cst)表示厘斯的单位为每秒平方毫米(即1cst=1mm2/s)。 运动粘度广泛用于测定喷气燃料油、柴油、润滑油等液体石油产品深色石油产品、使用后的润滑油、原油等的粘度,运动粘度的测定采用逆流法 (3)条件粘度:指采用不同的特定粘度计所测得的以条件单位表示的粘度,各国通常用的条件粘度有以下三种:①恩氏粘度又叫思格勒(Engler)粘度。是一定量的试样,在规定温度(如:50℃、80℃、100℃)下,从恩氏粘度流出200毫升试样所需的时间与蒸馏水在20℃流出相同体积所需要的时间(秒)之比。温度to时,恩氏粘度用符号Et表示,恩氏粘度的单位为条件度。 ②赛氏粘度,即赛波特(sagbolt)粘度。是一定量的试样,在规定温度(如100oF、F210oF或122oF等)下从赛氏粘度流出200毫升所需的秒数,以“秒”单位。赛氏粘度又分为赛氏通用粘度和赛氏重油粘度(或赛氏弗罗(Furol)粘度)两种。③雷氏粘度即雷德乌德(Redwood)粘度。是一定量的试样,在规定温度下,从雷氏度流出50毫升所需的秒数,以“秒”为单位。雷氏粘度又分为雷氏1号(Rt表示)和雷氏2号(用RAt表示)两种。 上述三种条件粘度测定法,在欧美各国常用,我国除采用恩氏粘度计测定深色润滑油及残渣油外,其余两种粘度计很少使用。三种条件粘度表示方法和单位各不相同,但它们之间的关系可通过图表进行换算。同时恩氏粘度与运动粘度也可换算,这样就方便灵活得多了。 粘度的测定有许多方法,如转桶法、落球法、阻尼振动法、杯式粘度计法、毛细管法等等。对于粘度较小的流体,如水、乙醇、四氯化碳等,常用毛细管粘度计测量;而对粘度较大流体,如蓖麻油、变压器油、机油、甘油等透明(或半透明)液体,常用落球法测定;对于粘度为0.1~100Pa?s范围的液体,也可用转筒法进行测定








 我要推广仪器
我要推广仪器
 下载APP
下载APP