推荐厂家
暂无
暂无

 400-860-5168转3452
400-860-5168转3452
 留言咨询
留言咨询
 400-889-1769
留言咨询
400-889-1769
留言咨询
 400-860-5168转4930
留言咨询
400-860-5168转4930
留言咨询



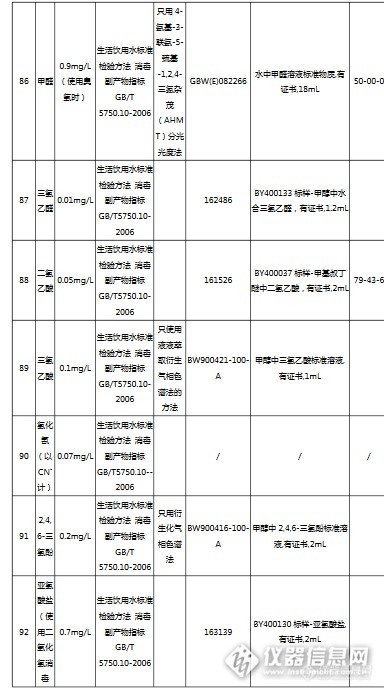
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。http://ng1.17img.cn/bbsfiles/images/2017/04/201704141630_01_3793_3.png坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。坛墨质检是国内唯一提供标准溶液定制服务的标准物质研制单位,定制范围:特殊浓度定制、特殊溶剂定制、混标定制。
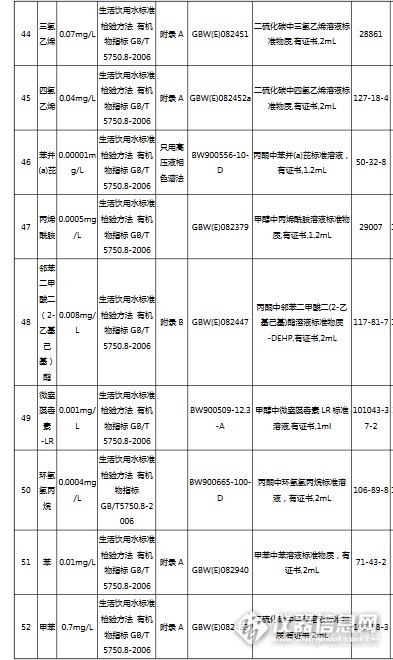
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。http://ng1.17img.cn/bbsfiles/images/2017/04/201704131618_02_3793_3.png坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。坛墨质检是国内唯一提供标准溶液定制服务的标准物质研制单位,定制范围:特殊浓度定制、特殊溶剂定制、混标定制。
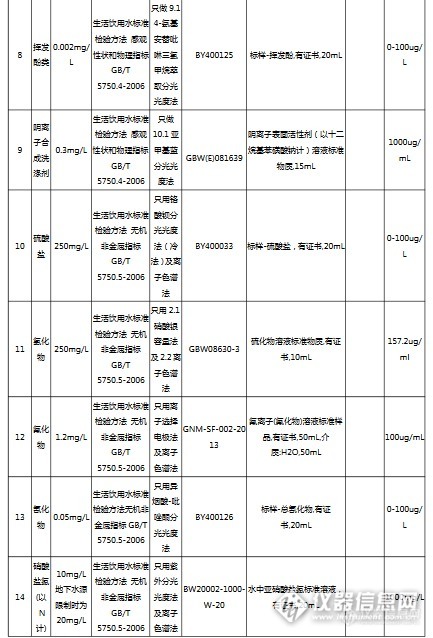
国内最大最专业的国家标准物质服务平台坛墨质检-国家标准物质中心(北京坛墨质检科技有限公司),是国家质检总局指定的国家标准物质研制单位,是国内最大最专业的食品、环境、职业卫生标准物质生产商和服务商。http://ng1.17img.cn/bbsfiles/images/2017/04/201704111647_01_3793_3.png坛墨质检现有员工79人,办公室面积450平米,实验室1650平米;销售、客服、财务及行政人员35人,实验室工作人员21人,库房14人,市场部8人。实验仪器设备:气相色谱、液相色谱、气质联用、液质联用、离子色谱、紫外分光光度计,原子吸收、ICP-OES和ICP-MS;库房面积450平米,库房工作人员12人,现货产品5万个,坛墨质检自主研发的产品近3000个,已申报国标345项,填补国内空白的产品达到65项。坛墨质检是国内唯一提供标准溶液定制服务的标准物质研制单位,定制范围:特殊浓度定制、特殊溶剂定制、混标定制。











 我要推广仪器
我要推广仪器
 下载APP
下载APP


